

Abstract
With the wide clinical use of the third‐generation epidermal growth factor receptor (EGFR) inhibitor osimertinib for the treatment of EGFR‐mutated non–small cell lung cancer (NSCLC), acquired resistance caused by EGFR C797S tertiary mutation has become a concern. Therefore, fourth‐generation EGFR inhibitors that could overcome this mutation have gained increasing attention in recent years. Here, we identified LS‐106 as a novel EGFR inhibitor against C797S mutation and evaluated its antitumor activity both in vitro and in vivo. In cell‐free assay, LS‐106 potently inhibited the kinase activities of EGFR19del/T790M/C797S and EGFRL858R/T790M/C797S with IC50 values of 2.4 nmol/L and 3.1 nmol/L, respectively, which was more potent than osimertinib. Meanwhile, LS‐106 exhibited comparable kinase inhibitory effect to osimertinib on EGFRL858R/T790M and wild‐type EGFR. Results from cellular experiments demonstrated that LS‐106 potently blocked the phosphorylation of EGFR C797S triple mutations in the constructed BaF3 cells that highly expressed EGFR19del/T790M/C797S or EGFRL858R/T790M/C797S, and thus inhibited the proliferation of these cells. We also constructed tumor cells harboring EGFR19del/T790M/C797S (named PC‐9‐OR cells) using the CRISPR/Cas9 system and found that LS‐106 markedly suppressed the activation of EGFR19del/T790M/C797S and the proliferation of PC‐9‐OR cells. Moreover, cells harboring EGFR19del/T790M/C797S underwent remarkable apoptosis upon LS‐106 treatment. In vivo experiments further demonstrated that oral administration of LS‐106 caused significant tumor regression in a PC‐9‐OR xenograft model, with a tumor growth inhibition rate (TGI) of 83.5% and 136.6% at doses of 30 and 60 mg/kg, respectively. Taken together, we identified LS‐106 as a novel fourth‐generation EGFR inhibitor against C797S mutation and confirmed its preclinical antitumor effects in C797S–triple‐mutant tumor models.
Keywords: C797S, epidermal growth factor receptor, fourth‐generation EGFR TKI, non–small cell lung cancer, osimertinib resistance
In this study, we identified LS‐106 as a novel inhibitor against C797S–triple‐mutant epidermal growth factor receptor (EGFR) (EGFR19del/T790M/C797S and EGFRL858R/T790M/C797S), which showed great in vitro and in vivo antitumor activity in EGFR‐C797S–triple‐mutant osimertinib‐resistant tumor models.
Abbreviations
- AUC
area under the curve
- CCK8
Cell Counting Kit‐8
- Cmax
maximum concentration
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- ELISA
enzyme‐linked immunosorbent assay
- ESI‐MS
electrospray ionization mass spectrometry
- ESILC‐MS/MS
liquid chromatography coupled with electrospray ionization tandem mass spectrometry system
- EMT
epithelial‐to‐mesenchymal transition
- FDA
Food and Drug Administration
- HRMS
high resolution mass spectromete
- IHC
immunohistochemistry
- LC/MS
liquid chromatography–mass spectrometry
- MQS
multiplicative quick score method
- MRT
mean residence time
- NMPA
National Medical Products Administration
- NMR
nuclear magnetic resonance
- PI
Propidium Iodide
- NSCLC
non–small cell lung cancer
- p.o.
oral administration
- RTV
relative tumor volume
- SRB
sulforhodamine B
- TGI
tumor growth inhibition rate
- TKI
tyrosine kinase inhibitor
- WB
Western blot
1. INTRODUCTION
Lung cancer is one of the most common cancers worldwide and is the main cause of cancer mortality in men and women. 1 , 2 , 3 Of all lung cancer incidents, approximately 85% are subtypes of non–small cell lung cancer (NSCLC). 4 Epidermal growth factor receptor (EGFR) mutations exist in nearly 50% of Asian patients and 15% of Caucasian patients with advanced NSCLC, and pharmacological targeting of mutant EGFR has become one of the most successful therapeutic strategies for the treatment of NSCLC. 5 , 6 To date, three generations of EGFR tyrosine kinase inhibitors (TKIs) have been developed, bringing huge clinical benefits to EGFR‐mutant NSCLC patients. 7 , 8 , 9
First‐generation EGFR inhibitors, such as gefitinib and erlotinib, were approved by the US Food and Drug Administration (FDA) in 2003 and 2004, respectively, for the treatment of NSCLC patients with EGFR‐sensitive mutations, such as E746‐A750 deletions in exon 19 (19del) and L858R point mutations in exon 21. 10 , 11 , 12 However, acquired resistance limits their clinical application, and the T790M mutation in EGFR exon 20 has been found to be the primary mechanism of resistance of first‐generation EGFR TKIs, accounting for 60%‐70% of resistant cases. 13 , 14 Therefore, second‐generation EGFR TKIs (such as afatinib) targeting both wild‐type EGFR (EGFRwt) and EGFRT790M were developed for overcoming this resistance. 15 , 16 However, side effects due to the poor selectivity over the EGFRwt make it difficult to reach the effective clinical concentration against EGFRT790M. 17 , 18 Consequently, third‐generation EGFR inhibitors that inhibit EGFRT790M while sparing EGFRwt were developed. 19 , 20 , 21 , 22 Osimertinib (AZD9291) was the first approved third‐generation EGFR inhibitor. The response rate of osimertinib in EGFRT790M patients was 61%, with great improvement in patients' prognosis and quality of life. 23 Recently, another two third‐generation EGFR TKIs, almonertinib (HS‐10296) and furmonertinib (AST2818), have been approved by the National Medical Products Administration (NMPA) in China, and several inhibitors such as BPI‐D0316 and ASK120067 are now under the late‐stage clinical development. 20 , 24 , 25
Despite the promising results obtained with third‐generation EGFR TKIs in NSCLC patients, resistance ultimately develops due to the treatment selection pressure and the inherent heterogeneity of NSCLC. 26 , 27 , 28 , 29 Among all resistance mechanisms reported so far, C797S point mutation in EGFR exon 20, in which cysteine at codon 797 with the ATP‐binding site is substituted by serine, has been validated to account for 10% to 26% of all osimertinib‐resistant cases. 30 The C797S mutation results in the loss of the covalent bond between third‐generation EGFR TKIs and the mutant EGFR. 28 , 31 , 32 , 33 Therefore, the development of fourth‐generation EGFR TKIs overcoming C797S mutation is the focus of many studies. EAI045, an EGFR allosteric inhibitor, is the first reported fourth‐generation EGFR TKI. EAI045 has been found to be effective against EGFRL858R/T790M/C797S–triple‐mutant cells when given in combination with the anti‐EGFR antibody cetuximab. 32 JBJ‐04‐125‐02, another novel EGFR allosteric inhibitor, has been reported to inhibit EGFRL858R/T790M/C797S signaling in vitro and in vivo, and the combination of JBJ‐04‐125‐02 with osimertinib proved more effective than either single agent alone. 34 But it is worth noting that both EAI045 and JBJ‐04‐125‐02 are not effective against EGFR19del/T790M/C797S–triple‐mutant cells due to their different structure at the allosteric pocket compared with the EGFRL858R/T790M/C797S variant. 31 , 34 Brigatinib, a dual‐target ALK‐EGFR inhibitor, has been proved to be effective against EGFR19del/T790M/C797S–triple‐mutant cells in vitro and in vivo. 35 Two ATP‐competitive inhibitors, TQB3804 and BPI‐361175, have been reported to show potent antitumor activity in the EGFR–triple‐mutant model and have entered clinical studies recently. TQB3804 possessed effective inhibition against both EGFR19del/T790M/C797S and EGFRL858R/T790M/C797S triple mutations. 36 In addition, there are some other fourth‐generation EGFR TKIs developed in preclinical studies. 31 , 37 As there is no such inhibitor approved for marketing up to now, it is still an urgent clinical need to develop novel fourth‐generation EGFR TKIs.
Herein, we identified compound LS‐106 as a novel fourth‐generation EGFR inhibitor targeting both EGFR19del/T790M/C797S and EGFRL858/T790M/C797S with selectivity over EGFRwt. Moreover, oral administration of LS‐106 led to significant tumor regression in a C797S‐mutant xenograft model.
2. MATERIALS AND METHODS
2.1. Compounds
LS‐106 was designed and synthesized by Ke Ding's group. 1H and 13C NMR spectra were recorded on a Bruker AV‐400 spectrometer at 400 MHz and Bruker AV‐600 spectrometer at 151 MHz, respectively. The high resolution ESI‐MS result was recorded on an Applied Biosystems Q‐STAR Elite ESILC‐MS/MS mass spectrometer. The purity of compound was determined by reverse‐phase high‐performance liquid chromatography (HPLC) analysis, using an Agilent 1260 system with an YMCTriart C18 reversed‐phase column (250 mm × 4.6 mm, 5 μm) at 254 nm. Elution was MeOH in water (containing 0.1% Et3N), and flow rate was 1.0 mL/min. 1H NMR (400 MHz, CDCl3) δ 10.95 (s, 1H), 8.57 (dd, J = 8.5, 4.4 Hz, 1H), 8.09 (s, 1H), 7.88 (d, J = 2.5 Hz, 1H), 7.54 (t, J = 7.9 Hz, 1H), 7.34 (dd, J = 8.6, 2.5 Hz, 1H), 7.29 (d, J = 7.7 Hz, 1H), 7.17‐7.08 (m, 1H), 7.02 (d, J = 8.4 Hz, 1H), 6.84 (s, 1H), 3.04 (s, 4H), 2.63 (s, 4H), 2.37 (s, 3H), 1.84 (d, J = 13.2 Hz, 6H). 13C NMR (151 MHz, CDCl3) δ 157.47 (s), 155.99 (s), 154.84 (s), 145.74 (s), 143.69 (d, J = 2.5 Hz), 135.83 (s), 132.91 (d, J = 1.8 Hz), 129.60 (d, J = 10.9 Hz), 125.22 (s), 122.58 (d, J = 1.9 Hz), 122.52 (d, J = 7.4 Hz), 120.84 (s), 120.09 (s), 120.08 (s), 120.06 (d, J = 95.5 Hz), 106.98 (s), 55.36 (s), 51.92 (s), 46.13 (s), 18.66 (d, J = 71.6 Hz). HRMS (ESI) calculated for C23H28BrClN6OP [M + H]+ 549.0934; found 549.0900. HPLC analysis: MeOH‐H2O (0.1% Et3N) (70:30), RT 8.59 minutes, 100% purity. Osimertinib (AZD9291, #T2490) was purchased from TargetMol.
2.2. Kinase assays
EGFRwt (#14‐531) and EGFRL858R/T790M (#14‐721) kinase proteins were purchased from Eurofins Scientific. EGFRL858R/T790M/C797S (#40351) and EGFR19del/T790M/C797S (#79434) kinase proteins were purchased from BPS Bioscience. EGFR19del (#PV6178) kinase protein was purchased from Thermo Fisher. EGFR19del/T790M (#E10‐122KG) kinase protein was purchased from SignalChem. The ability of LS‐106 to inhibit the activity of these kinases was evaluated by an enzyme‐linked immunosorbent assay (ELISA) according to standard procedures. The reaction was measured using a multiwell spectrophotometer (VERSA max™, Molecular Devices).
2.3. Cell lines and cell culture
BaF3‐EGFR19del/T790M/C797S, BaF3‐EGFRL858R/T790M/C797S, and BaF3‐EGFR19del/T790M cell lines were built by Jian Ding's laboratory 37 and cultured in RPMI‐1640 (Gibco, #C11875500BT) supplemented with 10% fetal bovine serum (Gibco, #10091‐148) and 1 μg/mL puromycin (Selleck, #S7417). PC‐9 (EGFR19del) cell lines were purchased from the European Collection of Authenticated Cell Cultures (ECACC) and cultured in RPMI‐1640 supplemented with 10% fetal bovine serum. All cell lines were maintained in 5% CO2 at 37°C with a humidified atmosphere.
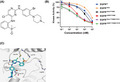
Tumor cell line containing EGFR19del/T790M/C797S mutation was constructed by using CRISPR technology to knock in T790M and C797S mutations into PC‐9 cells. CRISPR Cas9 plasmid lenti‐CRISPRv2 (#52961) was purchased from Addgene, and the sgRNA vector plasmid pGL3‐U6‐sgRNA‐PGK‐puromycin was a kind gift from Xingxu Huang Laboratory of Shanghai University of Science and Technology. Under the guidance of sgRNA, Cas9 cut the DNA at the target location, and at the same time a replacement template was introduced containing double mutations (T790M and C797S) for homology replacement repair (Figure 3A). After introducing the target mutation into the cells, the selection of single clones and multiple levels of verification were carried out (Figure 3B, C), and these results confirmed that we obtained tumor cells containing EGFR19del/T790M/C797S, which were named PC‐9‐OR.
FIGURE 3.
Construction of PC‐9‐OR cells and detection of their sensitivity to osimertinib and LS‐106. A, Schematic diagram of the construction principle of PC‐9‐OR. B, The EGFR mRNA sequence of PC‐9‐OR. C, Sensitivity of PC‐9‐OR and PC‐9 cells to osimertinib. D, The activation of EGFR and the downstream signal molecule AKT in PC‐9 and PC‐9‐OR was measured after LS‐106 treatment. E, The proliferative inhibition of LS‐106 was detected in PC‐9 and PC‐9‐OR. Data are shown as mean ± SEM
2.4. Western blot (WB)
The activation of EGFR and downstream signaling molecules in cells was examined by WB. PC‐9‐OR cells were plated in six‐well plates at a density of 3 × 105 cells per well overnight. Cells were washed three times with PBS after adherence to remove serum protein; then, cells were starved with serum‐free medium for at least 12 hours. BaF3‐EGFR19del/T790M/C797S and BaF3‐EGFRL858R/T790M/C797S cells were directly plated in 12‐well plates at a density of 2 × 106 cells per well with serum‐free medium for at least 6 hours. After starvation, different concentrations of indicated drugs were incubated with cells for 2 hours, and then the cells were stimulated with 50 ng/mL EGF per well for 15 minutes. Cells were lysed in SDS lysis buffer for 15 minutes at 100℃. The following antibodies were used: p‐EGFR (#3777), EGFR (#4267), p‐AKT (#4060S), AKT (#9272S), and ACTIN (#3700S), which were purchased from Cell Signaling Technologies. The recombinant human EGF (#PHG0311) was purchased from Thermo Fisher. WB analysis was subsequently performed with standard procedures. Image J was used for quantitative analysis of WB bands. Gray values of p‐EGFR were normalized with total‐EGFR as control, and then statistical analysis was conducted after three independent repetitions.
2.5. Proliferation assays
BaF3‐EGFR19del/T790M/C797S, BaF3‐EGFRL858R/T790M/C797S, or BaF3‐EGFR19del/T790M cells were plated in 96‐well plates at a density of 5000 cells per well and cultured overnight. Different concentrations of compounds were added, and the cells were incubated at 37°C in a CO2 incubator for 72 hours. Then, the cell growth was measured using Cell Counting Kit‐8 (CCK8; #AC11L057, Life iLab) and multiwell spectrophotometer (VERSA max™, Molecular Devices) at an absorbance of 450 nm.
PC‐9 cells or PC‐9‐OR cells were plated in a 96‐well culture plate at a density of 2000 cells per well and cultured overnight. The compounds in different concentrations were added, and then the cells were cultured for 72 hours. Then, sulforhodamine B (SRB, #S9012, Sigma‐Aldrich) assay was performed according to standard protocols, and the results were acquired using a multiwell spectrophotometer (VERSA max™, Molecular Devices) at an absorbance of 560 nm.
2.6. TOPO TA cloning
EGFR mRNA was prepared from PC‐9 and PC‐9‐OR, respectively, using the RNA Purification Kit (#B0004DP, EZBioscience) and then converted to cDNA using a reverse transcription kit (#R223‐01, Vazyme). The nucleotide sequences including exon 19 and 20 were amplified using Taq DNA polymerase (#10103ES03, Yeasen) and the following primers: 5‐EGFR‐F: GTGGAGAAGCTCCCAACCAA, 5‐EGFR‐R: GCGGTGTTTTCACCA‐ GTACG. To quantify the allele frequency, we ligated the PCR product with the vector TOPO TA (#10907ES20, Yeasen). The ligation products were transferred to competent cells and plated. In principle, each bacterial colony should carry a plasmid containing DNA which was a single PCR product at the beginning. Plasmid DNA was isolated from approximately 100 bacterial colonies and sequenced. Sequencing data were analyzed, and the frequency of 19del, T790M, and C797S mutations in EGFR mRNA were quantified. 38
2.7. Flow cytometry analysis
Cell apoptosis was investigated using an Annexin V‐FITC/PI apoptosis detection kit (#A211‐01/02, Vazyme). BaF3‐EGFR19del/T790M/C797S cells were plated in a 12‐well plate at a density of 3 × 105 cells per well and incubated in gradient concentrations of LS‐106 for 48 hours. PC‐9‐OR cells were plated in a six‐well plate at a density of 2 × 105 cells per well and incubated with gradient concentrations of LS‐106 for 48 hours. Cells were washed twice with PBS and resuspended in 500 µL binding buffer containing 5 µL Annexin V‐FITC and 5 µL PI staining solution for 30 minutes. Flow cytometry analysis was performed by a BD Aria Ⅱ flow cytometer (BD Biosciences), and data were analyzed by FlowJoVX software.
2.8. Animal study
Male Sprague‐Dawley rats were used to perform the pharmacokinetics study. The rats were fasted for 12 hours before administration of LS‐106 at the dose of 25 mg/kg p.o. and remained fasting for 2 hours. Concentrations (ng/mL) of LS‐106 in rat plasma were measured by LC/MS analysis at 0.25, 0.5, 1, 2, 4, 8, 12, and 24 hours after administration. Blank rat blood was used to resolve compounds for LC/MS analysis to obtain a standard curve, and the blood drug concentration in the measured blood samples was calculated according to the standard curve. The nonatrioventricular model was analyzed by Innaphase Kinetica 2000TM software. C max was the maximum (peak) concentration of compound observed in blood serum, and the area under the concentration‐time curve AUC (0‐∞) was calculated using the trapezoidal rule. T 1/2 is an estimate of the period of time that it takes for the concentration or amount of that compound in the body to be reduced by exactly one half (50%). MRTINF_obs means residence time based on time zero extrapolated to infinity. The animal room environment was controlled (target conditions: temperature 18 to 29°C, relative humidity 30% to 70%). Temperature and relative humidity were monitored daily. An electronic time‐controlled lighting system was used to provide a 12‐hour light/12‐hour dark cycle.
Nude mice were cultivated by the Shanghai Institute of Medicine. Animal experiments were performed according to the institutional ethical guidelines of animal care. PC‐9‐OR cells were injected subcutaneously into the right flank of each mouse at a density of 5 × 106 in 200 μL PBS. When the tumor volume reached around 70 mm3, the mice were randomly assigned into control and treatment groups (n ≥ 6 each group). LS‐106 was orally administrated daily at dose of 30 or 60 mg/kg. Tumor sizes was measured once per week using a microcaliper. The tumor volume (V) was calculated as follows: V = 0.5 × [length (mm) × width2 (mm2)]. The individual relative tumor volume (RTV) was calculated as follows: RTV = V t/V 0. The calculation formula of the tumor volume growth inhibition rate (TGI) was as follows: TGI = [1‐(TVt‐TV0)/(CVt‐CV0)] × 100%, where TVt is the tumor volume measured each time in the treatment group, TV0 is the tumor volume of the treatment group obtained at the first administration, CV t is the tumor volume measured each time in the control group, and CV0 is the tumor volume of the control group obtained at the first administration.
2.9. Immunohistochemistry (IHC)
Tumor samples were fixed in formalin for over 24 hours, transferred to 70% ethanol, and embedded in paraffin wax. The progress of IHC was completed by Shanghai Zuo Cheng Bio. The following antibodies were used: Ki67 (ab16667) (purchased from Abcam), EGFR (#4267), and p‐EGFR (#3777) (purchased from Cell Signaling Technologies).
We employed the multiplicative quick score method (MQS) to assess the protein expression. This system accounts for both the intensity and the extent of cell staining. In brief, the proportion of positive cells was estimated and given a percentage score on a scale from 1 to 6 (1 = 1%‐4%; 2 = 5%‐19%; 3 = 20%‐39%; 4 = 40%‐59%; 5 = 60%‐79%; and 6 = 80%‐100%). The average intensity of the positively staining cells was given an intensity score from 0 to 3 (0 = no staining; 1 = weak, 2 = intermediate, and 3 = strong staining). The IHC score was then calculated by multiplying the percentage score by the intensity score to yield a minimum value of 0 and a maximum value of 18.
2.10. Statistical analysis
All data were presented as mean ± standard deviation (SD) or mean ± standard error of mean (SEM). Analysis of two samples was performed with unpaired two‐tailed Student's t test for equal variance. The specific details about statistical methods were introduced in respective figure legends.
3. RESULTS
3.1. Structure and kinase inhibitory activity of compound LS‐106
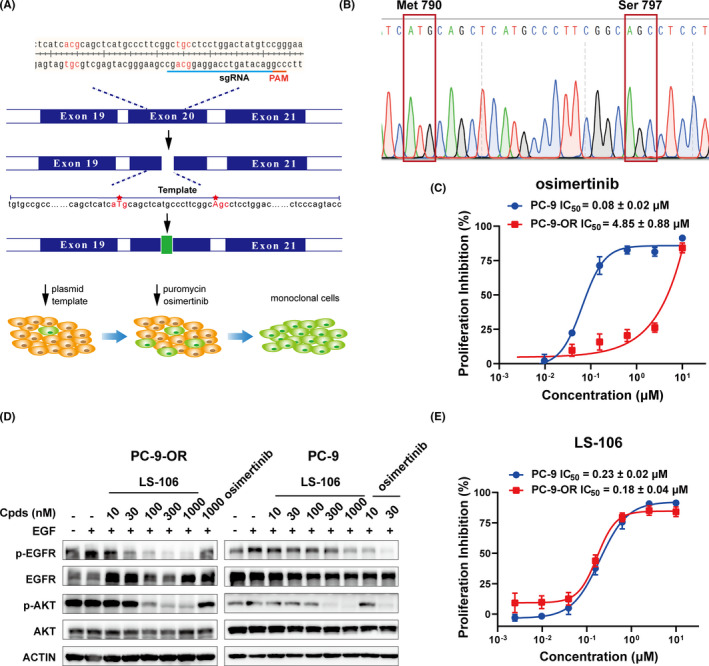
Using a structure‐based approach, we rationally designed and developed a series of novel molecules to target C797S‐mutant EGFR. Among them, LS‐106 was identified as a representative one (Figure 1A). It potently and dose‐dependently inhibited the kinase activity of EGFR19del/T790M/C797S, EGFRL858R/T790M/C797S, EGFRL858R/T790M, and EGFR19del/T790M with IC50 values of 2.4 ± 0.3 nmol/L, 3.1 ± 0.3 nmol/L, 7.3 ± 0.5 nmol/L, and 74.1 ± 15.6 nmol/L, respectively, and exhibited much weaker effect against EGFRwt (IC50 = 151.5 ± 26.2 nmol/L) and EGFR19del (IC50 = 402.9 ± 28.1 nmol/L) (Figure 1B). As expected, osimertinib possessed potent activity on EGFRL858R/T790M, EGFR19del/T790M, and EGFR19del (IC50S between 2.5 nmol/L to 11.1 nmol/L), while it showed much weaker activity against EGFR19del/T790M/C797S and EGFRL858R/T790M/C797S (IC50S between 179.1 nmol/L and 197.0 nmol/L) (Table 1).
FIGURE 1.
Structure and kinase inhibitory activity of LS‐106. A, Chemical structure of compound LS‐106. B, The kinase inhibitory activity of LS‐106 against EGFR19del/T790M/C797S, EGFRL858R/T790M/C797S, EGFR19del/T790M, EGFRL858R/T790M, EGFR19del, and EGFRwt. Data are shown as mean ± SD. C, The docking structure of LS‐106 with EGFRL858R/T790M/C797S (protein from PDB ID: 6LUD). The EGFR kinase is shown in grey stick and ribbon representation. LS‐106 is shown in green and blue stick. Hydrogen bonds are indicated by yellow hatched lines to key amino acids
TABLE 1.
The kinase inhibitory activity of LS‐106 against different EGFR mutations (IC50s, nmol/L)
| Compounds | EGFR19del/T790M/C797S | EGFRL858R/T790M/C797S | EGFRL858R/T790M | EGFR19del/T790M | EGFR19del | EGFRwt |
|---|---|---|---|---|---|---|
| LS‐106 | 2.4 ± 0.3 | 3.1 ± 0.3 | 7.3 ± 0.5 | 74.1 ± 15.6 | 402.9 ± 28.1 | 151.5 ± 26.2 |
| Osimertinib | 179.1 ± 65.6 | 197.0 ± 59.1 | 2.5 ± 0.5 | 3.9 ± 0.8 | 11.1 ± 0.7 | 196.5 ± 129.5 |
To investigate the potential binding mode of compound LS‐106 with triple‐mutant EGFR, molecular docking was performed with EGFRL858R/T790M/C797S protein derived from PDB code 6LUD. 31 According to the docking results, LS‐106 bound to EGFRL858R/T790M/C797S protein by hydrogen and hydrophobic bond rather than covalent binding, indicating that this compound might be a reversible inhibitor (Figure 1C). Classical bidentate hydrogen bonding network was formed between the nitrogen atoms of 2‐aminopyrimidine core and Met793 at the hinge region. Additionally, the phosphine oxide moiety acted as a hydrogen bond acceptor and fostered a strong hydrogen bond interaction with Lys745. Besides, 5‐chloride on the pyrimidine ring of LS‐106 was directed toward the “gatekeeper” residue Met790 and formed a hydrophobic interaction, which explained why this molecule has good inhibitory activity against EGFR containing T790M mutation.
Furthermore, we examined the inhibitory activities of LS‐106 against other 23 tyrosine kinases (Table S1). We found that LS‐106 not only strongly inhibited the EGFR‐C797S–mutant kinase but also inhibited some other kinases, such as RET, ACK1, and PDGFR‐β. These results suggested that LS‐106 was a multikinase inhibitor.
3.2. LS‐106 inhibits cell growth of EGFR‐C797S–mutant BaF3 cells by suppressing the activation of triple‐mutant EGFR
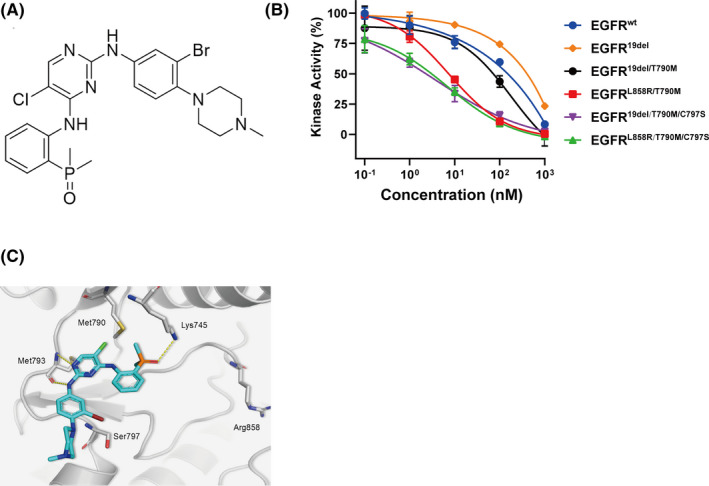
As no commercial tumor cell harboring EGFRC797S mutation was available, we constructed BaF3 cells exogenously expressing EGFR19del/T790M/C797S, EGFRL858R/T790M/C797S, or EGFR19del/T790M, respectively. These cells were incubated with increasing concentration of LS‐106 or osimertinib for 72 hours, and then cell growth was determined using CCK8 assay. Consistent with kinase assays results, although osimertinib exhibited strong antiproliferative effect in BaF3‐EGFR19del/T790M cells (IC50 = 0.04 μmol/L), it showed little antigrowth activity in BaF3‐EGFR19del/T790M/C797S (IC50 = 3.38 μmol/L) and BaF3‐EGFRL858R/T790M/C797S cells (IC50 = 4.14 μM) (Figure 2A). In contrast, LS‐106 not only inhibited the proliferation of BaF3‐EGFR19del/T790M cells (IC50 = 0.09 μmol/L) but also showed over 30‐fold stronger antigrowth activity than osimertinib in BaF3‐EGFR19del/T790M/C797S (IC50 = 0.09 μmol/L) and BaF3‐EGFRL858R/T790M/C797S (IC50 = 0.12 μmol/L) cells, respectively (Figure 2A). In addition, LS‐106 potently and dose‐dependently blocked EGFR phosphorylation both in BaF3‐EGFR19del/T790M/C797S and BaF3‐EGFRL858R/T790M/C797S cells (Figure 2B), and its blocking effect was more potent than that of osimertinib. Collectively, these results demonstrated that LS‐106 could effectively suppress the cellular phosphorylation of triple‐mutant EGFR and thus inhibited the proliferation of these constructed BaF3 cells, which were insensitive to osimertinib.
FIGURE 2.
Inhibition activity of LS‐106 on BaF3 cells expressing different EGFR mutations. A, The proliferative inhibition of LS‐106 and osimertinib were measured in BaF3‐EGFR19del/T790M/C797S cells, BaF3‐EGFRL858R/T790M/C797S cells, and BaF3‐EGFR19del/T790M cells, respectively. B, The expression of phospho‐EGFR in BaF3‐EGFR19del/T790M/C797S and BaF3‐EGFRL858R/T790M/C797S cells treated with LS‐106 or osimertinib were analyzed by Western blotting. The gray value was quantified by Image J software. Data are shown as mean ± SEM and were analyzed using t test, *P < .05, **P < .01, ***P < .001
3.3. LS‐106 shows antitumor activity in NSCLC tumor cells harboring EGFR C797S triple mutations
Besides the BaF3 tool cells mentioned above, we also constructed tumor cells expressing C797S triple mutations by introducing both T790M and C797S mutations into PC‐9 cells harboring exon 19 deletion of EGFR, using CRISPR/Cas9 knock‐in technology (Figure 3A). A monoclonal was then selected and was named PC‐9‐OR. Gene sequencing results proved the EGFR mRNAs of PC‐9‐OR cells successfully carried T790M and C797S mutations (Figure 3B), and mass spectrometry analysis also confirmed the presence of T790M and C797S mutations (data not shown). We next wanted to identify whether the T790M and C797S mutations occurred in cis (on the same allele) or in trans (on a different allele) with 19del in PC‐9‐OR cells. To address this issue, we sequenced EGFR cDNA (including exon 19 and 20) in PC‐9‐OR cells or PC‐9 cells using TA cloning technology. Sequencing analysis revealed that the T790M and C797S mutations coexisted with 19del on 83% of alleles in PC‐9‐OR cells, and the others had EGFRwt or exon 19 deletion only (Table 2). These data demonstrated that most alleles of EGFR were in cis (19del, T790M, and C797S occurred in the same strand). No form in trans was observed based on these data. We then compared the antiproliferation activity of osimertinib against PC‐9‐OR cells and PC‐9 cells. As expected, the proliferation of PC‐9‐OR cells was poorly inhibited by osimertinib with an IC50 value of 4.85 μmol/L, while PC‐9 parental cells were highly sensitive to osimertinib (IC50 = 0.08 μmol/L) (Figure 3B). These results confirmed that we had successfully constructed a tumor cell line containing EGFR19del/T790M/C797S.
TABLE 2.
Quantification of allelic reads of PC‐9 and PC‐9‐OR
| Cell lines | WT | exon 19 del | Total | |
|---|---|---|---|---|
| T790 C797 | M790 S797 | |||
| PC‐9‐OR (n = 100) | 4% | 13% | 83% | 100% |
| PC‐9 (n = 100) | 5% | 95% | 0 | 100% |
Western blot results demonstrated that LS‐106 potently inhibited the activation of intracellular EGFR19del/T790M/C797S and the downstream signal molecule AKT in a dose‐dependent manner in PC‐9‐OR. The phosphorylation of EGFR19del/T790M/C797S was obviously inhibited by 30 nmol/L and almost completely inhibited by 300 nmol/L LS‐106 treatment (Figure 3D). At the same time, LS‐106 hardly inhibited the activation of EGFR19del in PC‐9 cells, while osimertinib could effectively inhibit the activation of EGFR19del at 10 or 30 nmol/L (Figure 3D). We noticed that there was a slight difference in the expression of total EGFR under drug treatment in PC‐9‐OR; this change might be related to drug action or caused by other potential reasons and needs further study.
We then examined the antiproliferative activity of LS‐106 against PC‐9‐OR cells using SRB assay. LS‐106 exhibited potent antiproliferative activity on PC‐9‐OR cells with IC50 values of 0.18 μmol/L (Figure 3E), which was more powerful than the antiproliferative activity of osimertinib (IC50 = 4.85 μmol/L) (Figure 3C). Meanwhile, LS‐106 showed a strong inhibitory activity against PC‐9 (IC50 = 0.23 μmol/L). We assumed that the considerable inhibitory activity of LS‐106 against PC‐9 cells might be caused by its off‐target effects (Table S1), and the inhibition on PC‐9‐OR could not exclude this effect either.
3.4. LS‐106 elicits significant cell apoptosis in EGFR–triple‐mutant cells
As cell apoptosis was considered a major biological effect induced by EGFR TKIs, we then investigated the effect of LS‐106 on cell apoptosis in both BaF3‐EGFR19del/T790M/C797S cells and PC‐9‐OR cells. In BaF3‐EGFR19del/T790M/C797S, compared with the 9% apoptosis ratio in untreated control cells, LS‐106 triggered significant cell apoptosis in a concentration‐dependent manner with apoptosis ratios of 65% and 77% at 100 nmol/L and 300 nmol/L, respectively (Figure 4A). Similarly, in PC‐9‐OR cells, after 48 hours treatment, LS‐106 triggered significant cell apoptosis in a concentration‐dependent manner with apoptosis ratios of 41% and 47% at 100 nmol/L and 300 nmol/L, respectively (Figure 4B), compared with 9% apoptosis in the control group. In contrast, at concentrations between 30 nmol/L and 300 nmol/L, osimertinib hardly caused any significant apoptosis in both BaF3‐EGFR19del/T790M/C797S and PC‐9‐OR cells. These data demonstrated that LS‐106 induced cell apoptosis in EGFR‐C797S–triple‐mutant cells.
FIGURE 4.
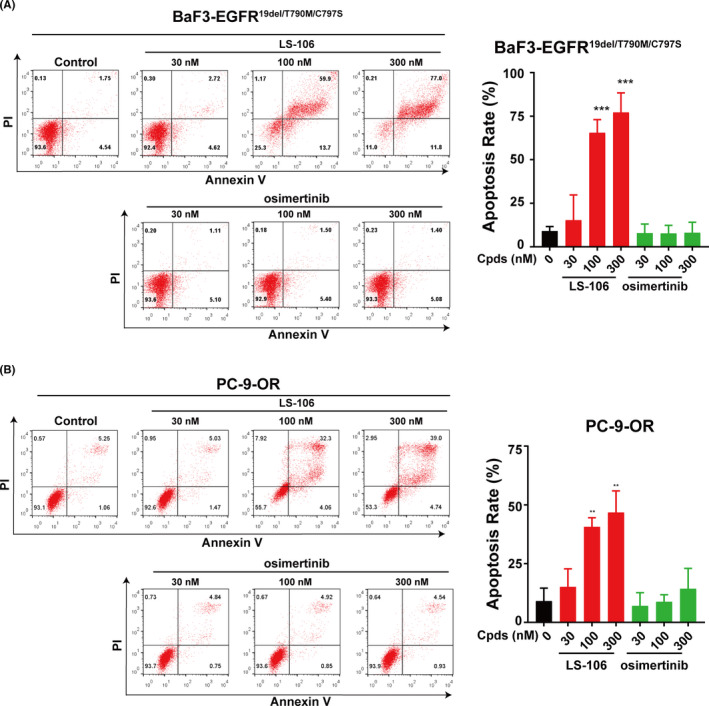
The apoptosis‐inducing effect of LS‐106 in EGFR–triple‐mutant cells. A, Cell apoptosis was measured in BaF3‐EGFR19del/T790M/C797S cells after incubation with LS‐106 or osimertinib for 48 h. B, Cell apoptosis was measured in PC‐9‐OR cells after incubation with LS‐106 or osimertinib for 48 h. Apoptosis rates were analyzed based on the data of three independent tests. Data are shown as mean ± SD and were analyzed using t test, *P < .05, **P < .01, ***P < .001
3.5. Oral administration of LS‐106 inhibits tumor progression in a PC‐9‐OR NSCLC xenograft model
To further assess the in vivo efficacy of LS‐106, we firstly evaluated the pharmacokinetic properties of LS‐106 on Sprague‐Dawley rats. LS‐106 exhibited a high AUClast of 3101 h/ng/mL upon oral administration to rats at a dose of 25 mg/kg (Table 3). After oral administration, the Cmax of LS‐106 reached 326.1 ng/mL (equivalent to 595 nmol/L), which far exceeded its effective concentration on target proteins. In addition, the half‐life (T 1/2 = 15.3 h) and mean residence time (MRTINF_obs = 22.8 hours) suggested that LS‐106 should be administered once a day in the subsequent experiment.
TABLE 3.
Pharmacokinetic parameters of LS‐106 after 25 mg/kg p.o. administration in male Sprague‐Dawley rats
| Animal no. | T 1/2 (h) | T max (h) | C max (ng/mL) | AUClast (h*ng/mL) | AUCINF_obs (h*ng/mL) | MRTINF_obs (h) |
|---|---|---|---|---|---|---|
| 1 | 26.6 | 2.0 | 210.0 | 2926.9 | 6745.6 | 40.1 |
| 2 | 7.1 | 0.3 | 372.0 | 3251.4 | 3652.0 | 10.5 |
| 3 | 12.1 | 0.5 | 396.4 | 3126.1 | 4342.0 | 18.0 |
| Mean | 15.3 | 0.9 | 326.1 | 3101.4 | 4913.2 | 22.8 |
| SD | 10.2 | 0.9 | 101.3 | 163.6 | 1624.0 | 15.4 |
Abbreviations: AUC, area under the curve; C max, maximum concentration; MRT, mean residence time, SD, standard deviation.
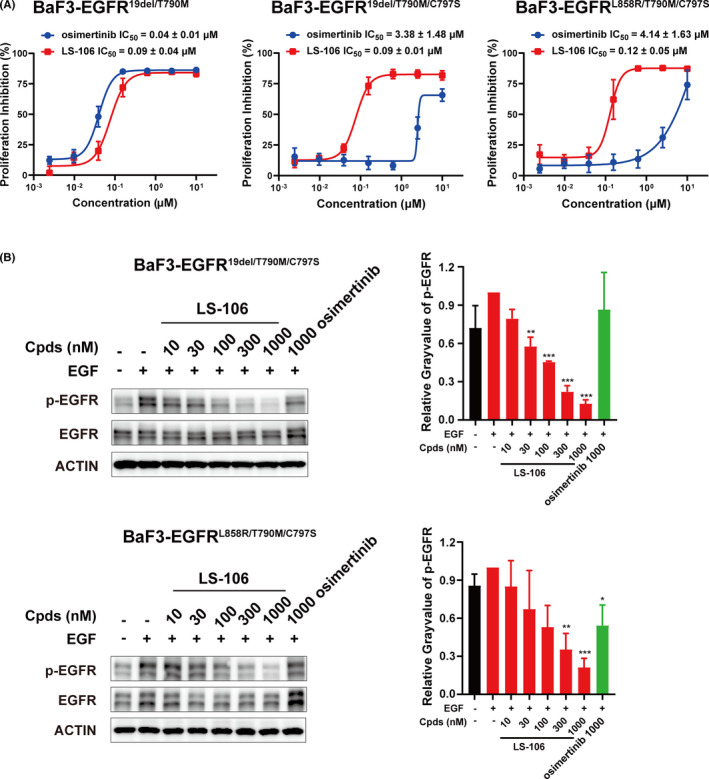
Then, a PC‐9‐OR–resistant lung cancer xenograft mouse model was established to assess the in vivo antitumor efficacy of LS‐106. Mice were orally administered with LS‐106 at doses of 30 mg/kg, 60 mg/kg, or vehicle control once daily for 14 days. At the study endpoint, LS‐106 showed potent antitumor effects with a TGI of 83.5% and 136.6% at doses of 30 and 60 mg/kg, respectively (Figure 5A). Meanwhile, we also observed that LS‐106 was not well tolerated, with 21% body weight loss observed after 14 days of treatment (Figure 5B). We assumed that this phenomenon might be caused, at least in part, by the off‐target toxicity of LS‐106. Then, the WB result showed that the phosphorylation of EGFR in the tumor tissue was markedly inhibited by the LS‐106 treatment (Figure 5C). Consistent with the WB result, IHC staining confirmed that the phosphorylation of EGFR was inhibited dramatically and the expression of total EGFR was not changed obviously (Figure 5D). Moreover, the expression of Ki67, a proliferation marker, was significantly decreased in the 30 mg/kg LS‐106 treatment group compared with the control group (Figure 5D), indicating that LS‐106 possessed potent antiproliferation activity in vivo. These data demonstrated that as a novel fourth‐generation EGFR inhibitor, LS‐106 possessed acceptable pharmacokinetic properties and exhibited potent antitumor activity in a PC‐9‐OR xenograft model.
FIGURE 5.
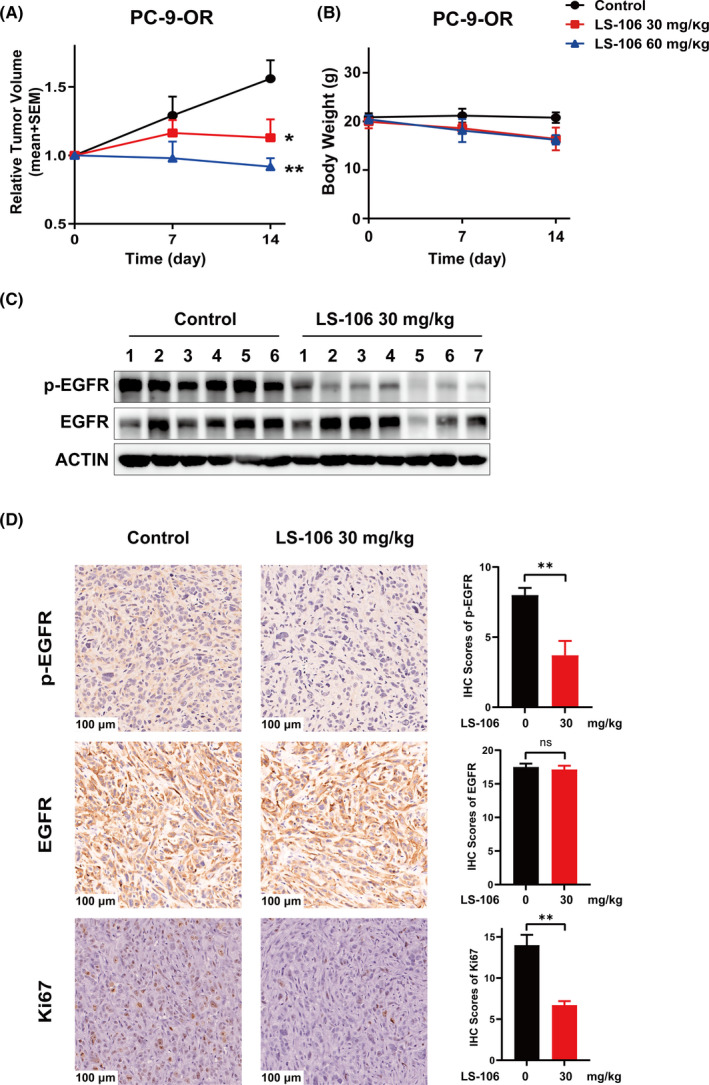
LS‐106 exhibits in vivo antitumor activity in a PC‐9‐OR tumor xenograft model. A, Tumor‐bearing mice were orally administered with vehicle or LS‐106 at doses of 30 mg/kg and 60 mg/kg once daily for 14 d. The relative tumor volumes (RTVs) are shown as mean ± SEM (*P < .05 vs control, Student's t test). B, The body weight of each mouse was monitored once a week. C, Inhibition of EGFR activation by LS‐106 in tumor tissue was examined using Western blot. D, The expression of p‐EGFR, EGFR, and Ki67 in tumor tissue was examined using immunohistochemistry (IHC). The IHC scores are shown as mean ± SEM and were analyzed using t test, *P < .05, **P < .01, ***P < .001
4. DISCUSSION
EGFR is a classic drug target for personal therapies in NSCLC. The representative third‐generation EGFR TKI osimertinib has been approved as a therapeutic agent for NSCLC patients with EGFR‐sensitive mutations or T790M mutation and has significantly improved the life quality of those patients. However, with the wide clinical use of osimertinib, acquired resistance inevitably developed. Resistance mechanisms to osimertinib can consist of EGFR modifications (including mutations and amplification), bypass pathway activation (such as MET or HER2 amplification, ACK1 activation, and oncogenic fusions), downstream pathway activation (such as RAS and PI3K pathway activation), epithelial‐to‐mesenchymal transition (EMT), histologic transformation from NSCLC to small cell lung cancer (SCLC), and cell‐cycle gene aberrations. 27 , 39 , 40 , 41 , 42 , 43 Among them, C797S mutation in EGFR is validated as an important reason mediating osimertinib resistance (10%‐26%), and the development of new fourth‐generation EGFR TKIs have attracted much attention. At present, the research of a fourth‐generation inhibitor is still at an early stage, and no such compound has been approved yet.
In this study, we rationally designed and synthesized novel compounds that target the EGFR C797S triple mutations and identified LS‐106 as a potent inhibitor based on in vitro and in vivo evaluation. LS‐106 potently inhibited the kinase activities of triple‐ or double‐mutant EGFR, while it exhibited much weaker inhibitory activity against EGFRwt. LS‐106 strongly inhibited EGFR activation and suppressed cell proliferation in BaF3‐EGFR19del/T790M/C797S and BaF3‐EGFRL858R/T790M/C797S cells. Of note, because of the difficulty obtaining tumor cell lines derived from C797S‐mutation patients, most of the reported compounds were evaluated in the modified BaF3 or NIH‐3T3 models which lacked complicated signaling pathways and metabolic environment of lung cancer. To better mimic the complex genetic background of lung cancer cells, we knocked in the T790M/C797S mutations of EGFR into PC‐9 cells (EGFR19del) and obtained the tumor cell line PC‐9‐OR harboring EGFR19del/T790M/C797S. As expected, LS‐106 strongly inhibited the activation of EGFR19del/T790M/C797S and thus induced apoptosis and suppressed cell proliferation of PC‐9‐OR cells. Moreover, LS‐106 possessed good pharmacokinetic properties in animals, and demonstrated in vivo monodrug anticancer efficacy in a xenograft mouse model with EGFR19del/T790M/C797S mutation.
Of special note, LS‐106 showed effective inhibition against both EGFR19del/T790M/C797S and EGFRL858R/T790M/C797S, which makes it more competitive than previously reported fourth‐generation EGFR TKIs, such as EAI‐045 and JBJ‐04‐125‐02 that failed to display antitumor effects against EGFR19del/T790M/C797S. Moreover, the combination with an EGFR antibody cetuximab was required for EAI045 to demonstrate in vivo therapeutic efficacy because of asymmetric dimerization of the receptor. In contrast, our result proved that LS‐106 possessed potent in vivo antitumor activities as a monotherapy, showing that this compound has outstanding advantages. In addition, the weak inhibition of LS‐106 against EGFRwt may enable it to avoid potential side‐effects that related to EGFRwt, such as rashes or diarrhea. Meanwhile, we also observed that LS‐106 did not perform very well in terms of selectivity and inhibited some other kinases except EGFR triple mutations, and this might lead to off‐target effects in cell tests and poor tolerance in animal experiments, which did occur in our subsequent experiments. For example, LS‐106 had a strong antiproliferation activity on PC‐9 cells, and the animal body weight decreased by about 21% after 14 days of treatment. These results suggest that LS‐106 has potential toxicity, and the chemical structure needs to be further optimized.
In summary, we established a comprehensive evaluation platform of fourth‐generation EGFR TKIs and identified LS‐106 as a novel fourth‐generation EGFR TKI, which showed in vitro and in vivo antitumor potency in EGFR C797S malignant models and deserves further investigation.
DISCLOSURE
The authors declare that they have no conflict of interest.
Supporting information
Table S1
ACKNOWLEDGEMENTS
This research was supported by grants from the National Science & Technology Major Project “Key New Drug Creation and Manufacturing Program,” China (2019ZX09301157‐003 and ‐004 to KD and HX, respectively), the "Personalized Medicines‐Molecular Signature‐based Drug Discovery and Development," Strategic Priority Research Program of the Chinese Academy of Sciences (XDA12020112 to HX), and the National Natural Science Foundation of China (81903638 to TZ, 21807045 to SL, and 81922062 to XYL). We thank the Xingxu Huang Laboratory of ShanghaiTech University for kindly providing the plasmid pGL3‐U6‐sgRNA‐PGK‐puromycin as a gift.
Liu Y, Lai M, Li S, et al. LS‐106, a novel EGFR inhibitor targeting C797S, exhibits antitumor activities both in vitro and in vivo. Cancer Sci.2022;113:709–720. doi: 10.1111/cas.15229
Yingqiang Liu, Mengzhen Lai, Shan Li, and Yanan Wang contributed equally to this work.
Contributor Information
Ke Ding, Email: dingke@jnu.edu.cn.
Ker Yu, Email: keryu@fudan.edu.cn.
Hua Xie, Email: hxie@simm.ac.cn.
Jian Ding, Email: jding@simm.ac.cn.
REFERENCE
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394‐424. doi: 10.3322/caac.21492 [DOI] [PubMed] [Google Scholar]
- 2. Sung H, Ferlay J, Siegel RL, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71(3):209‐249. doi: 10.3322/caac.21660 [DOI] [PubMed] [Google Scholar]
- 3. Bade BC, Dela Cruz CS. Lung Cancer 2020: Epidemiology, Etiology, and Prevention. Clin Chest Med. 2020;41:1‐24. [DOI] [PubMed] [Google Scholar]
- 4. Herbst RS, Morgensztern D, Boshoff C. The biology and management of non‐small cell lung cancer. Nature. 2018;553:446‐454. [DOI] [PubMed] [Google Scholar]
- 5. Harrison PT, Vyse S, Huang PH. Rare epidermal growth factor receptor (EGFR) mutations in non‐small cell lung cancer. Semin Cancer Biol. 2020;61:167‐179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jonna S, Subramaniam DS. Molecular diagnostics and targeted therapies in non‐small cell lung cancer (NSCLC): an update. Discovery Med. 2019;27:167‐170. [PubMed] [Google Scholar]
- 7. Tan C‐S, Kumarakulasinghe NB, Huang Y‐Q, et al. Third generation EGFR TKIs: current data and future directions. Mol Cancer. 2018;17:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Remon J, Steuer CE, Ramalingam SS, Felip E. Osimertinib and other third‐generation EGFR TKI in EGFR‐mutant NSCLC patients. Annal Oncol. 2018;29:i20‐i27. [DOI] [PubMed] [Google Scholar]
- 9. Westover D, Zugazagoitia J, Cho BC, Lovly CM, Paz‐Ares L. Mechanisms of acquired resistance to first‐ and second‐generation EGFR tyrosine kinase inhibitors. Annal Oncol. 2018;29:i10‐i19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ciardiello F, Caputo R, Bianco R, et al. Antitumor effect and potentiation of cytotoxic drugs activity in human cancer cells by ZD‐1839 (Iressa), an epidermal growth factor receptor‐selective tyrosine kinase inhibitor. Clin Can Res. 2000;6:2053‐2063. [PubMed] [Google Scholar]
- 11. Sirotnak FM, Zakowski MF, Miller VA, Scher HI, Kris MG. Efficacy of cytotoxic agents against human tumor xenografts is markedly enhanced by coadministration of ZD1839 (Iressa), an inhibitor of EGFR tyrosine kinase. Clin Can Res. 2000;6:4885‐4892. [PubMed] [Google Scholar]
- 12. Pedersen MW, Pedersen N, Ottesen LH, Poulsen HS. Differential response to gefitinib of cells expressing normal EGFR and the mutant EGFRvIII. Br J Cancer. 2005;93:915‐923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yun CH, Mengwasser KE, Toms AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci USA. 2008;105:2070‐2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non‐small‐cell lung cancer to gefitinib. N Engl J Med. 2005;352:786‐792. [DOI] [PubMed] [Google Scholar]
- 15. Li D, Ambrogio L, Shimamura T, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27:4702‐4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Park K, Tan EH, O'Byrne K, et al. Afatinib versus gefitinib as first‐line treatment of patients with EGFR mutation‐positive non‐small‐cell lung cancer (LUX‐Lung 7): a phase 2B, open‐label, randomised controlled trial. Lancet Oncol. 2016;17:577‐589. [DOI] [PubMed] [Google Scholar]
- 17. Hayashi H, Iihara H, Hirose C, et al. Effects of pharmacokinetics‐related genetic polymorphisms on the side effect profile of afatinib in patients with non‐small cell lung cancer. Lung Cancer. 2019;134:1‐6. [DOI] [PubMed] [Google Scholar]
- 18. Yang Z, Hackshaw A, Feng Q, et al. Comparison of gefitinib, erlotinib and afatinib in non‐small cell lung cancer: A meta‐analysis. Int J Cancer. 2017;140:2805‐2819. [DOI] [PubMed] [Google Scholar]
- 19. Cross DA, Ashton SE, Ghiorghiu S, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M‐mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4:1046‐1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang T, Qu R, Chan S, et al. Discovery of a novel third‐generation EGFR inhibitor and identification of a potential combination strategy to overcome resistance. Mol Cancer. 2020;19:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nagasaka M, Zhu VW, Lim SM, Greco M, Wu F, Ignatius Ou SH. Beyond osimertinib: The development of 3(rd)‐generation EGFR Tyrosine Kinase Inhibitors. J Thoracic Oncol. 2020;16:740‐763. [DOI] [PubMed] [Google Scholar]
- 22. Wang H, Pan R, Zhang X, Si X, Wang M, Zhang L. Abivertinib in patients with T790M‐positive advanced NSCLC and its subsequent treatment with osimertinib. Thoracic Cancer. 2020;11:594‐602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jänne PA, Yang JC, Kim DW, et al. AZD9291 in EGFR inhibitor‐resistant non‐small‐cell lung cancer. N Engl J Med. 2015;372:1689‐1699. [DOI] [PubMed] [Google Scholar]
- 24. Yang JC, Camidge DR, Yang CT, et al. Safety, Efficacy, and Pharmacokinetics of Almonertinib (HS‐10296) in Pretreated Patients With EGFR‐Mutated Advanced NSCLC: A Multicenter, Open‐label, Phase 1 Trial. J Thoracic Oncol. 2020;15:1907‐1918. [DOI] [PubMed] [Google Scholar]
- 25. Shi Y, Zhang S, Hu X, et al. Safety, Clinical Activity, and Pharmacokinetics of Alflutinib (AST2818) in Patients With Advanced NSCLC With EGFR T790M Mutation. J Thoracic Oncol. 2020;15:1015‐1026. [DOI] [PubMed] [Google Scholar]
- 26. Wang S, Tsui ST, Liu C, Song Y, Liu D. EGFR C797S mutation mediates resistance to third‐generation inhibitors in T790M‐positive non‐small cell lung cancer. J Hematol Oncol. 2016;9:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Leonetti A, Sharma S, Minari R, Perego P, Giovannetti E, Tiseo M. Resistance mechanisms to osimertinib in EGFR‐mutated non‐small cell lung cancer. Br J Cancer. 2019;121:725‐737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Oxnard GR, Hu Y, Mileham KF, et al. Assessment of resistance mechanisms and clinical implications in patients with EGFR T790M‐positive lung cancer and acquired resistance to osimertinib. JAMA Oncology. 2018;4:1527‐1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Piotrowska Z, Isozaki H, Lennerz JK, et al. Landscape of acquired resistance to osimertinib in EGFR‐mutant NSCLC and clinical validation of combined EGFR and RET inhibition with osimertinib and BLU‐667 for acquired RET fusion. Cancer Discov. 2018;8:1529‐1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mok TS, Wu YL, Ahn MJ, et al. Osimertinib or platinum‐pemetrexed in EGFR T790M‐positive lung cancer. N Engl J Med. 2017;376:629‐640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kashima K, Kawauchi H, Tanimura H, et al. CH7233163 overcomes osimertinib resistant EGFR‐Del19/T790M/C797S mutation. Mol Cancer Ther. 2020;19(11):2288‐2297. [DOI] [PubMed] [Google Scholar]
- 32. Jia Y, Yun CH, Park E, et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant‐selective allosteric inhibitors. Nature. 2016;534:129‐132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Starrett JH, Guernet AA, Cuomo ME, et al. Drug sensitivity and allele specificity of first‐line Osimertinib resistance EGFR mutations. Can Res. 2020;80:2017‐2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. To C, Jang J, Chen T, et al. Single and dual targeting of mutant EGFR with an allosteric inhibitor. Cancer Discov. 2019;9:926‐943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang Y, Yang N, Zhang Y, et al. Effective treatment of lung adenocarcinoma harboring EGFR‐activating mutation, T790M, and cis‐C797S triple mutations by Brigatinib and Cetuximab combination therapy. J Thoracic Oncol. 2020;15:1369‐1375. [DOI] [PubMed] [Google Scholar]
- 36. Liu X, Zhang X, Yang L, et al. Abstract 1320: Preclinical evaluation of TQB3804, a potent EGFR C797S inhibitor. Can Res. 2019;79:1320. [Google Scholar]
- 37. Lu X, Zhang T, Zhu SJ, et al. Discovery of JND3229 as a New EGFR(C797S) mutant inhibitor with in vivo Monodrug efficacy. ACS Med Chem Letters. 2018;9:1123‐1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Niederst MJ, Hu H, Mulvey HE, et al. The allelic context of the C797S mutation acquired upon treatment with third‐generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin Cancer Res. 2015;21(17):3924‐3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang Z, Yang JJ, Huang J, et al. Lung Adenocarcinoma Harboring EGFR T790M and In Trans C797S responds to combination therapy of first‐ and third‐generation EGFR TKIs and Shifts allelic configuration at resistance. J Thoracic Oncol. 2017;12:1723‐1727. [DOI] [PubMed] [Google Scholar]
- 40. Zhang Q, Zhang XC, Yang JJ, et al. EGFR L792H and G796R: Two Novel Mutations Mediating Resistance to the Third‐Generation EGFR Tyrosine Kinase Inhibitor Osimertinib. J Thoracic Oncol. 2018;13:1415‐1421. [DOI] [PubMed] [Google Scholar]
- 41. Nukaga S, Yasuda H, Tsuchihara K, et al. Amplification of EGFR Wild‐Type alleles in non‐small cell lung cancer cells confers acquired resistance to mutation‐selective EGFR tyrosine kinase inhibitors. Can Res. 2017;77:2078‐2089. [DOI] [PubMed] [Google Scholar]
- 42. Martinez‐Marti A, Felip E, Matito J, et al. Dual MET and ERBB inhibition overcomes intratumor plasticity in osimertinib‐resistant‐advanced non‐small‐cell lung cancer (NSCLC). Annal Oncol. 2017;28:2451‐2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nishiyama A, Takeuchi S, Adachi Y, et al. MET amplification results in heterogeneous responses to osimertinib in EGFR‐mutant lung cancer treated with erlotinib. Cancer Sci. 2020;111:3813‐3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1