

Abstract
Adult T‐cell leukemia/lymphoma (ATL) is a highly chemoresistant malignancy of peripheral T lymphocytes caused by human T‐cell leukemia virus type 1 infection, for which there is an urgent need for more effective therapeutic options. The molecular chaperone heat shock protein 90 (HSP90) plays a crucial role in nuclear factor‐κB (NF‐κB)‐mediated antiapoptosis in ATL cells, and HSP90 inhibitors are new candidate therapeutics for ATL. Accordingly, we investigated the anti‐ATL effects of a novel oral HSP90 inhibitor, TAS‐116 (pimitespib), and the mechanisms involved in ex vivo and in vivo preclinical models. TAS‐116 achieved IC50 values of less than 0.5 μmol/L in 10 ATL‐related cell lines and less than 1 μmol/L in primary peripheral blood cells of nine ATL patients; no toxicity was observed toward CD4+ lymphocytes from healthy donors, indicating the safety of this agent. Given orally, TAS‐116 also showed significant inhibitory effects against tumor cell growth in ATL cell‐xenografted mice. Furthermore, gene expression profiling of TAS‐116‐treated Tax‐positive or ‐negative cell lines and primary ATL cells using DNA microarray and multiple pathway analysis revealed the significant downregulation of the NF‐κB pathway in Tax‐positive cells and cell‐cycle arrest in Tax‐negative cells and primary ATL cells. TAS‐116 suppressed the activator protein‐1 and tumor necrosis factor pathways in all examined cells. These findings strongly indicate the efficacy of TAS‐116, regardless of the stage of ATL progression, and its potential application as a novel clinical anti‐ATL therapeutic agent.
Keywords: adult T‐cell leukemia/lymphoma, drug sensitivity, heat shock protein, HTLV‐1 infection, microarray analysis
A novel heat shock protein 90 inhibitor, TAS‐116, showed anti–adult T‐cell leukemia/lymphoma (ATL) effects in preclinical models and could be an effective therapeutic option for ATL. TAS‐116 has distinct as well as common therapeutic targets against Tax‐positive, Tax‐negative, and primary ATL cells.

Abbreviations
- AP‐1
activating protein‐1
- ATF3
activating transcription factor 3
- ATL
adult T‐cell leukemia/lymphoma
- CDKN2A
cyclin‐dependent kinase inhibitor 2A
- EGR
early growth response protein
- FDR
false discovery rate
- GEO
Gene Expression Omnibus
- GSEA
Gene Set Enrichment Analysis
- HBZ
human T‐cell leukemia virus type 1 bZIP factor
- HSP90
heat shock protein 90
- HTLV‐1
human T‐cell leukemia virus type 1
- IL‐2
interleukin‐2
- IPA
Ingenuity Pathway Analysis
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- NF‐κB
nuclear factor‐κB
- PARP
poly(ADP‐ribose) polymerase
- PDGFB
platelet‐derived growth factor B
- Rb
RB transcriptional corepressor 1
- Sp1
specificity protein 1
- TNF
tumor necrosis factor
1. INTRODUCTION
Human T‐cell leukemia virus type 1 infects CD4 lymphocytes mostly through breastfeeding in infancy or sexual intercourse in adults, or rarely, through the placenta. 1 , 2 , 3 The lifetime risk of development of ATL in HTLV‐1 carriers is 3%‐5%; however, approximately 5‐20 million individuals are infected with HTLV‐1 worldwide. 4 , 5 Among the four clinical subtypes of ATL, acute, lymphoma, and unfavorable chronic types are aggressive, and favorable chronic and smoldering types are indolent ATLs. 6 , 7 Although an international consensus meeting recommended chemotherapy as the first‐line treatment for aggressive ATL (with even better outcomes due to chemotherapy in Japan), the median overall survival is only approximately 1 year. 7 , 8 , 9 , 10 Other than chemotherapy, treatment options for patients with aggressive ATL include allogeneic hematopoietic stem cell transplantation and, more recently, anti‐C‐C motif chemokine receptor 4 Ab mogamulizumab, and the immunomodulatory agent lenalidomide. 10 , 11 However, no salvage therapy has been established for recurrent or resistant ATL and most patients still have a poor prognosis; thus, there is an urgent need for more effective options. 11
Importantly, the HTLV‐1 Tax oncoprotein is a major determinant of viral persistence and pathogenesis. It interacts with various cellular proteins, disrupts transcriptional activation and cell proliferation, and ultimately results in malignant transformation. 12 Human T‐cell leukemia virus type 1 activates the NF‐κB pathway through either Tax or NIK (NF‐κB‐inducing kinase) overexpression by targeting its suppressor micro‐RNA‐31. 13 In the early stage of ATL development, Tax physically interacts with NEMO, a regulatory subunit of NF‐κB to activate the IKK (IκB kinase) complex, and constitutively activates NF‐κB to immortalize HTLV‐1‐infected T cells. 14 However, Tax expression is frequently lost in ATL cases, probably because of accumulative genetic and epigenetic Tax modifications and Tax‐negative ATL cells utilize NF‐κB‐independent pathways such as ERK1/2 instead. 15
Another viral protein, HBZ, encoded by the reverse strand of HTLV‐1, is ubiquitously expressed in ATL cells and HTLV‐1 infected cells. Tax and HBZ exert opposite effects on most signaling pathways, but they cooperate to promote viral replication and proliferation in infected cells. 16 Particularly, HBZ promotes tumorigenic proliferation of HTLV‐1‐infected cells through the direct activation of the transcription factor JunD or the SMAD transcription complex and induces the expression of FOXP3 to evade the host immune surveillance machinery. 16 Overall, these two HTLV‐1 encoded transforming proteins enable the accumulation of multiple genetic or epigenetic modifications in HTLV‐1‐infected cells, and by extension their transformation into ATL cells. 15
The molecular chaperone HSP90 plays a crucial role in mediating the immune response and promoting cancer cell growth/survival by stabilizing multiple immune or cancer‐related kinases. In cancer cells, HSP90 inhibition induces the degradation of target proteins through the ubiquitin‐proteasome pathway and the downregulation of pathways crucial for tumor development. 17 , 18 Therefore, HSP90 inhibitors have been developed and are undergoing clinical trials. 18 , 19 , 20 , 21 However, the clinical use of HSP90 inhibitors has stalled, partly due to their association with adverse events such as hepatotoxicity and visual disturbance phenomenon, including blurred vision, flashes, delayed light/dark accommodation, or night blindness. 19 , 22 , 23 We have previously reported that the HSP90 inhibitors 17‐DMAG and NVP‐AUY922 exert anti‐ATL effects; 17‐DMAG induces Tax degradation and concomitant downregulation of NF‐κB signaling and viral replication, whereas NVP‐AUY922 acts through the inhibition of multiple cell cycle‐related kinases and by regulating the entry of cells into the sub‐G1 phase. These findings suggested that HSP90 inhibitors might provide therapeutic benefits in patients with ATL. 24 , 25 , 26 TAS‐116 (pimitespib) is a novel selective inhibitor of cytosolic HSP90. Given orally, it resulted in tumor shrinkage and depletion of multiple HSP90 target proteins in human tumor xenograft mouse models. 27 , 28 Furthermore, TAS‐116 has minimal hepatotoxicity and did not induce any detectable photoreceptor injury in a rat model; it is expected to show potent antitumor activity with minimal adverse effects. 27 , 28 A phase III trial evaluating the efficacy of TAS‐116 in comparison with placebo in patients with previously treated gastrointestinal stromal tumor showed significantly prolonged progression‐free survival. 29 , 30 , 31
In this study, we evaluated the effects of TAS‐116 against ATL cells both ex vivo and in vivo and elucidated the relevant pathways contributing to its cell growth inhibition effect.
2. MATERIALS AND METHODS
2.1. Ethics statement
This study was carried out in strict accordance with the Guidelines for Proper Conduct of Animal Experiments, Science Council of Japan. 32 All animal procedures were approved by the Animal Care Committee of the University of Miyazaki and undertaken in accordance with the Regulations for Animal Experiments (approval ID: 2013‐526‐4) and ARRIVE guidelines. All experiments using patient samples were carried out after obtaining approval from the Ethics Committee of the Nagasaki University Hospital (approval nos. 13102878 and 15122105).
2.2. Reagents, cells, and cell culture conditions
TAS‐116 (Taiho Pharmaceutical), 17‐DMAG (Selleck Chemicals), and NVP‐AUY922 (Selleck Chemicals) were dissolved in DMSO and added to the culture medium (1:20 ratio) to adjust for each concentration. The ATL‐related cell lines (Tax‐positive: HuT‐102, OATL4, and KOB; Tax‐negative: ED‐40515, Su9T01, ST1, KK1, SO4, LMY1, and LMY2) and non‐ATL leukemia cell lines (Jurkat, MOLT‐4, and HuT‐78) were maintained in RPMI‐1640 medium supplemented with 10% FCS. 1 , 24 , 33 Interleukin‐2 (10 ng/mL) was added in the cultures of IL‐2‐dependent ATL cell lines (KOB, KK1, SO4, LMY1, and LMY2). Peripheral blood samples were collected from nine patients diagnosed with aggressive ATL according to Shimoyama’s diagnostic criteria. 6 Adult T‐cell leukemia/lymphoma diagnosis was confirmed based on the detection of the monoclonal integration of HTLV‐1 proviral DNA by southern blotting. 34 Peripheral blood mononuclear cells were obtained from ATL patients and healthy adult volunteers and isolated using density‐gradient centrifugation on a Lympho‐prep (Axis Shield). CD4+ lymphocytes from healthy donors were purified from the PBMCs using the magnetic bead method (CD4+ T Cell Isolation Kit; Miltenyi Biotec). The PBMCs were cultured in RPMI‐1640 medium supplemented with 10% FCS and 10 ng/mL IL‐2.
2.3. Cell viability assay
Each cell line was treated with 0‐5 μmol/L TAS‐116 for 72 hours at 37℃. Cell viability was evaluated using a Cell Titer‐Glo Assay Kit (Promega). The clinical samples were subjected to the MTS cell viability assay using CellTiter 96 AQueous One Solution Cell Proliferation Assay Kit (Promega).
2.4. Flow cytometric analysis
After treatment with or without TAS‐116, cells were harvested and analyzed using a Cell Cycle Test kit (BD Biosciences), and the percentage of cells in each cell‐cycle phase (G0/G1, S, G2/M) was determined. Apoptosis induction was determined using annexin V/propidium iodide staining (Bender Medsystems). Assays were carried out on a FACSCalibur Flow Cytometer and analyzed using CellQuest software (BD Biosciences) or on a FACSCanto II Flow Cytometer with FACSDiva software (BD Biosciences). 25
2.5. Western blot analysis
OATL4 or ED‐40515 (1 × 106 cells/mL) treated with or without TAS‐116 (3 µmol/L) were lysed using the cell lysis buffer. Proteins present in the lysate were resolved by SDS‐PAGE followed by electroblotting onto PVDF membranes (Bio‐Rad Laboratories) and probed overnight at 4°C with specific Abs against proteins. The following Abs were used in this study: Hsp90 (Enzo), IκB‐α, phospho‐ERK1/2, ERK1/2, PARP, c‐MYC (Cell Signaling Technology), Tax, 24 and tubulin (Sigma). Bands corresponding to the proteins of interest were detected using HRP‐conjugated Abs and visualized using the ECL Prime Western Blotting Detection Reagent (GE Healthcare) in accordance with the manufacturer’s instructions.
2.6. Gene expression analysis using microarray technology
2.6.1. HuT‐102, OATL4, ED‐40515, and Su9T01 cells
Four ATL‐related cell lines were treated with DMSO (control) or with 1 μmol/L TAS‐116 for 16 hours in triplicate. Total RNA was isolated using an RNeasy mini kit (Qiagen) and subjected to DNase treatment (RNase‐Free DNase Set; Qiagen). A portion of the total RNA was reverse transcribed into cDNA. After the evaluation of RNA quality using an Agilent 2100 Bioanalyzer, each RNA was labeled using a Low Input Quick Amp Labeling Kit (one color method; Agilent) and hybridized using the SurePrint G3 Human GE microarray 8 × 60K version 2.0 (Agilent) for 17 hours at 65℃. Slides were scanned on the Agilent SureScan Microarray Scanner (G2600D) immediately after washing using one color scan setting for 8 × 60K array slides and the scanned images were analyzed using the Feature Extraction Software 11.5.1.1 (Agilent).
For the KEGG pathway enrichment analysis, data from each array (cells treated with DMSO or TAS‐116 in triplicate) were median‐normalized per chip. The data were then filtered based on the signal intensity and flagged values. Genes with |fold change| > 2 after TAS‐116 treatment of each cell line were identified using Student’s t test with the Benjamini and Hochberg multiple testing correction to restrict the FDR. Genes with corrected P values less than .05 were considered to be differentially expressed. Commonly upregulated or downregulated genes in ATL cells with (HuT‐102 and OATL4) or without (ED‐40515 and Su9T01) Tax expression were subjected to KEGG pathway analysis using DAVID 35 to investigate the key pathways dysregulated by TAS‐116. The KEGG terms with FDR less than .05 were considered significant.
For heatmap and upstream regulator analysis, commonly upregulated or downregulated (fold change > 1.5 in all TAS‐116‐treated samples relative to the average of DMSO‐treated samples) genes following TAS‐116 treatment of ATL cells with or without Tax expression were subjected to clustering, and the IPA (Qiagen) was carried out. 36 , 37 To measure functional activation in upstream regulator analysis, activation z scores were calculated. Positive and negative z scores indicate activation and inhibition, respectively. |Scores| > 2 were considered significant. Microarray data are available in the GEO database. The GEO accession numbers for HuT‐102, OATL4, ED‐40515, and Su9T01 are GSE168554, GSE168556, GSE168552, and GSE168558, respectively.
2.6.2. LMY1 and KK1 cells
For GSEA, 38 , 39 experiments and data analysis of the expression profiles of LYM1 and KK1 cells treated with or without TAS116 were carried out by the Dragon Genomics Center (Takara Bio) using the SurePrint G3 Human GE microarray 8 × 60K version 2.0 (Agilent) as described above. Expression profiles of LYM1 and KK1 cells were subjected to GSEA with Hallmark gene sets (version 6.2) from the Molecular Signatures Database. Gene sets with FDR less than .05 were considered significant and ranked according to the normalized enrichment score. The microarray data are available in the GEO database (accession no. GSE168555).
2.6.3. Adult T‐cell leukemia/lymphoma cases
The expression profiles of three primary ATL cells treated with or without TAS‐116 (1 μmol/L for 24 hours) were evaluated on an Agilent DNA microarray system (SurePrint G3 Human GE microarray 8 × 60K version 3.0) as described above. For the KEGG pathway enrichment analysis, data from each array were median‐normalized per chip. The data were then filtered based on the signal intensity and flagged values. Genes with |fold change| > 2 following TAS‐116 treatment were selected, and the upregulated and downregulated pathways common to all three cells were identified. The frequencies of the altered genes in these pathways were compared to the overall gene frequency in each pathway, and differences with P < .01 were considered significant. Microarray data are available on GEO (accession number GSE168557).
2.7. Adult T‐cell leukemia/lymphoma xenograft mouse experiments
Female CB17− SCID mice (body weight, 20 g; 20 per cell line) (CLEA Japan) were subcutaneously injected in the right flank with 5 × 106 HuT‐102 or Su9T01 cells resuspended in 100 μL normal saline containing 100 μL Matrigel (BD Biosciences). When the tumor volume reached 100 mm3, mice were randomized into the control or TAS‐116 groups (n ≥ 4 each), wherein each mouse was orally administered daily for 28 days following inoculation with 0.5% hydroxypropyl methylcellulose (Shin‐Etsu Chemical) or TAS‐116 (10 or 14 mg/kg), respectively. Tumor diameters were measured using vernier calipers twice weekly.
2.8. Statistical analysis
Data are expressed as the mean ± SEM. To compare between the control and TAS‐116 treated groups, data were analyzed using one‐way ANOVA followed by Dunnett’s test on GraphPad Prism 8.0. Results were considered significant when P < .05.
3. RESULTS
3.1. TAS‐116 inhibited the growth of ATL‐related cell lines and primary ATL cells
We first examined the growth inhibitory effects of TAS‐116 on ATL‐related and non‐ATL leukemia cell lines. TAS‐116 (0.5 μmol/L) inhibited the growth of approximately 60% of the ATL‐related cells and 40% of the non‐ATL leukemia cells (Figure 1A). Expression of HTLV‐1 Tax and the estimated IC50 values of TAS‐116 are summarized in Table S1. The IC50 values against ATL‐related cell lines were below 0.5 μmol/L with or without HTLV‐1 Tax, and these values were significantly lower than those against other leukemia cell lines. Similarly, TAS‐116 inhibited the proliferation of all primary ATL cells examined, with IC50 values ranging from 0.2 to 0.5 μmol/L. By contrast, TAS‐116 did not show substantial toxicity toward CD4+ lymphocytes derived from healthy donors (Figure 1B). Interestingly, TAS‐116 showed modest cytotoxicity compared to the HSP90 inhibitors we examined previously, although there were no significant differences in binding mechanism or target sites (Figure S1, Table S2).
FIGURE 1.
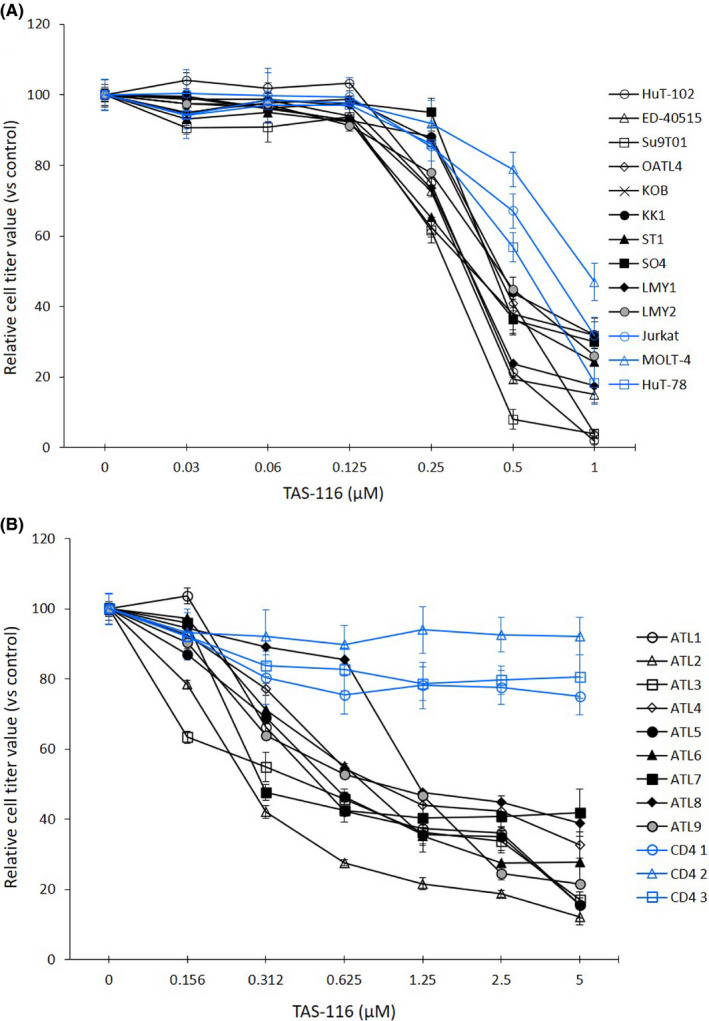
Growth inhibition of adult T‐cell leukemia/lymphoma (ATL)‐related cell lines and primary ATL cells after TAS‐116 treatment. A, ATL‐related cell lines (black), non‐ATL leukemia cell lines (blue) (2‐5 × 105 cells/mL). B, PBMCs from ATL patients (black), and CD4+ lymphocytes from healthy donors (blue) (1 × 106 cells/mL) were treated with DMSO (control) or various concentrations of TAS‐116 for 72 h and cell viability was evaluated. All experiments were carried out in triplicate, and data are expressed as the mean ± SD
3.2. TAS‐116 suppresses multiple cell growth‐promoting pathways
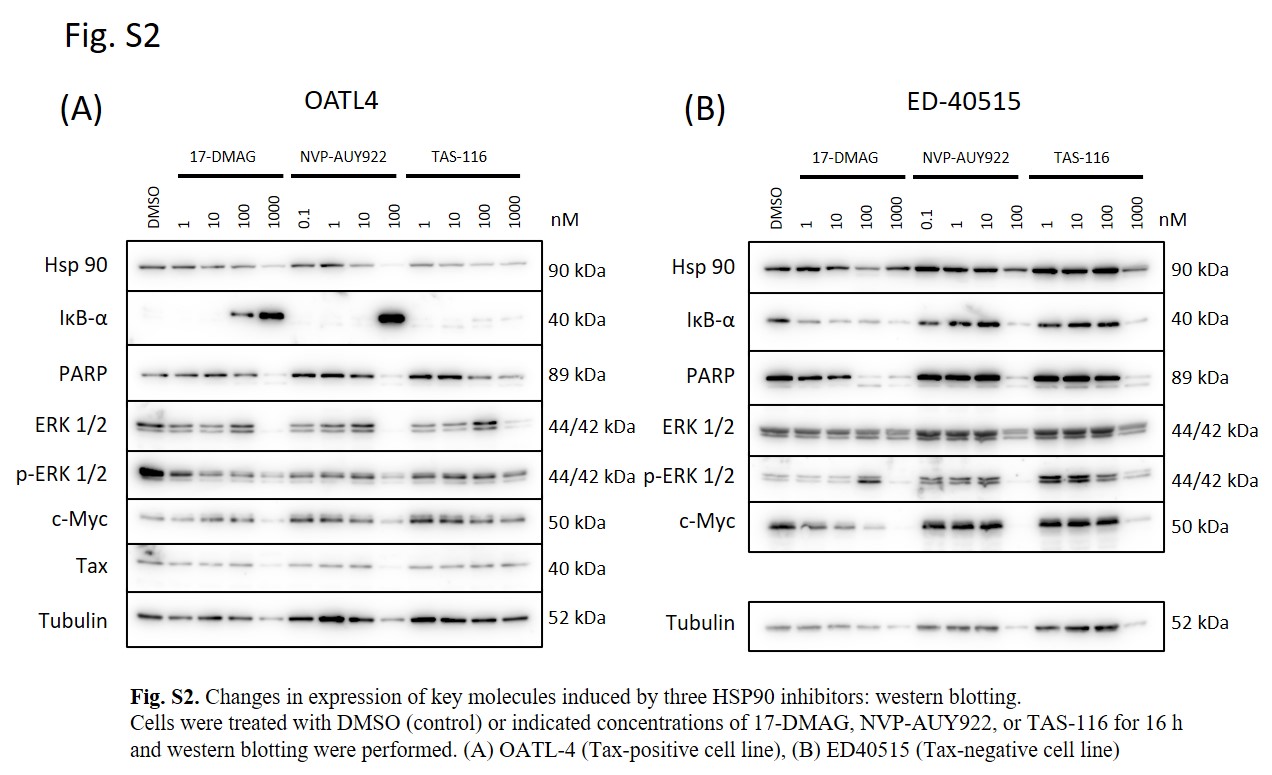
Next, we evaluated the effects of TAS‐116 on Tax degradation and NF‐κB downregulation. TAS‐116‐treated cells showed Tax downregulation until its complete elimination 60 hours after treatment (Figure 2A). By contrast, IκB‐α gradually accumulated up to 48 hours after treatment. TAS‐116 treatment completely inhibited Erk‐1/2 phosphorylation in Tax‐positive as well as ‐negative cell lines (Figure 2B). Comparing TAS‐116 with 17‐DMAG and NVP‐AUY922, IκB‐α accumulation by NVP‐AUY922 was prominent in Tax‐positive cells, whereas suppression of PARP and c‐MYC by 17‐DMAG was prominent in Tax‐negative cells (Figure S2). These results suggest that HSP90 inhibitors have NF‐κB inhibitory and apoptosis‐promoting effects, with the former being more prominent in Tax‐positive cells and the latter in Tax‐negative cells. Among three HSP90 inhibitors, these effects were not potent in TAS‐116.
FIGURE 2.
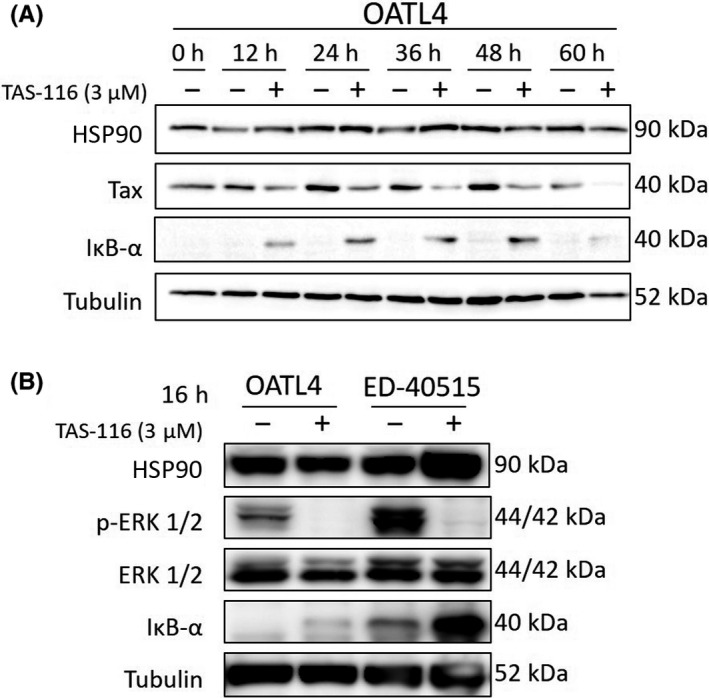
Time‐course analysis of Tax degradation or ERK inhibition after TAS‐116 treatment. Cells (1 × 106/mL) were treated with DMSO or TAS‐116 at various concentrations. A, Western blot analysis was carried out on cells collected every 12 h for a total of 60 h. B, After 16 h, cells were harvested, and western blotting was carried out to compare the Tax levels in Tax‐positive (OATL4) and Tax‐negative cells (ED‐40515). Tubulin was used as the internal control. HSP90, heat shock protein 90
3.3. Inhibition of growth‐promoting pathways in ATL cells following TAS‐116 treatment
Next, we evaluated the TAS‐116 mediated deregulation of cell‐cycle in four ATL cells using flow cytometry (Figure 3). Although dose‐dependent effects were noted, TAS‐116 generally increased the number of cells in the G0/G1 phase and decreased the number of cells in the S and G2/M phases, regardless of Tax expression. Similar changes were detected in KOB, LMY1, ST1, and KK1 cells (Figure S3A,B). Annexin V/propidium iodide staining revealed that approximately 50% of LMY1 cells, 30% of KOB and ST1, and 10% of KK1 cells underwent apoptosis following TAS‐116 treatment (Figure S3C).
FIGURE 3.
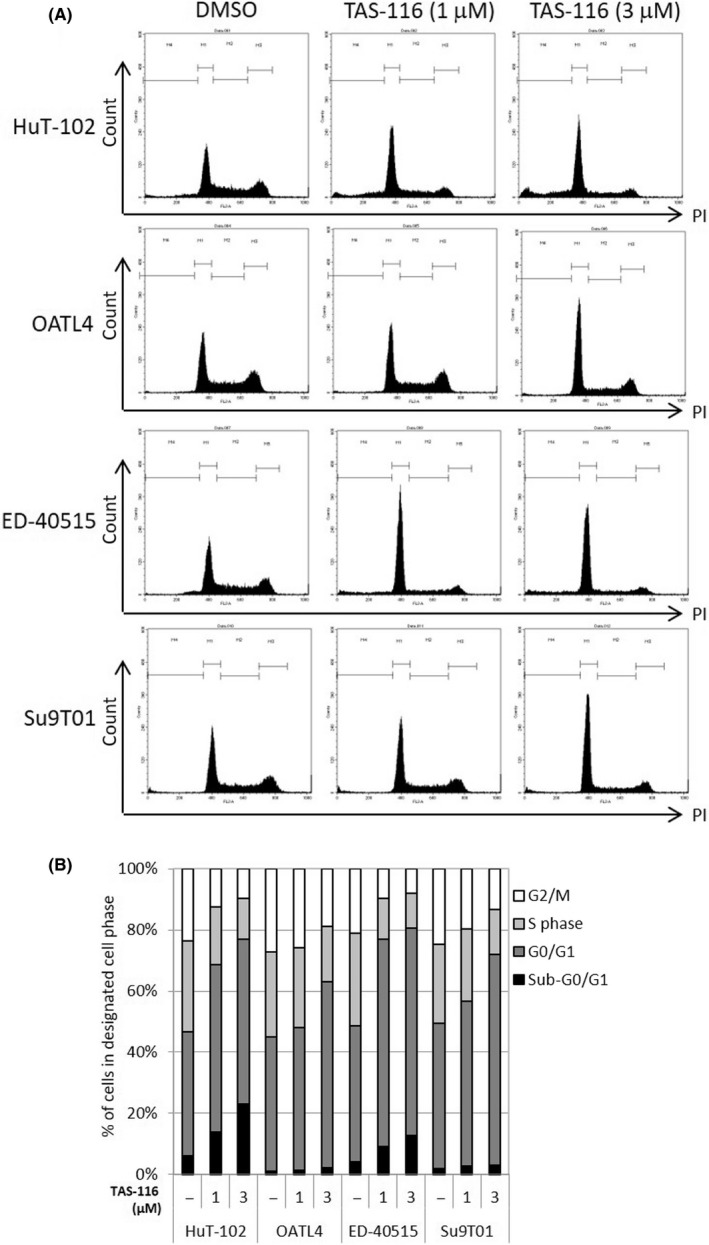
Cell‐cycle analysis of TAS‐116‐treated adult T‐cell leukemia/lymphoma‐related cell lines. A, B, Tax‐positive (HuT‐102 and OATL4; 1 × 106/mL) and Tax‐negative cells (ED‐40515 and Su9T01; 1 × 106/mL) were treated with DMSO or various concentrations of TAS‐116. After 48 h, cell‐cycle analysis was carried out. The percentage of cells arrested in the sub‐G0/G1, G0/G1, S, and G2/M phases was evaluated. The number of cells arrested in the G0/G1 phase and in the G1 phase increased after TAS‐116 treatment. –, untreated
3.4. Pathway analysis of TAS‐116‐induced cell growth inhibition
3.4.1. Tax‐positive cell lines
After DNA microarray analysis of four ATL‐related cell lines (Figure S4), IPA (Figure 4A) and DAVID (KEGG pathway) analyses (Figures 4B and S5A) were carried out. Prominent upstream regulators that showed increased activity included inhibitors for kinases targeting MEK, MAPK/ERK, and PI3K. Furthermore, significant downregulation of NF‐κB‐related factors was observed in Tax‐positive cell lines (Figure 4A). Figure 4B shows the top 10 downregulated KEGG pathways in these cells. “TNF signaling pathway” and “NF‐κB signaling pathway” were also identified. Because Tax is necessary for viral genome amplification, the “HTLV‐1 infection” pathway was also significantly downregulated. Table S3 summarizes the individual genes in the top three downregulated pathways common in Tax‐positive cell lines. No significantly upregulated pathways were identified in the KEGG pathway analysis.
FIGURE 4.
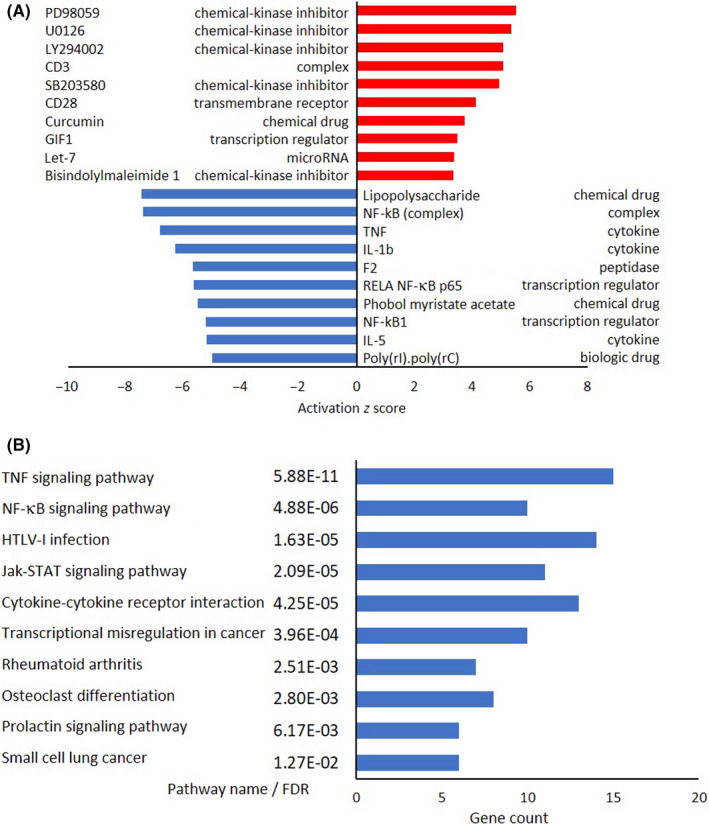
Microarray and pathway analyses of Tax‐positive adult T‐cell leukemia/lymphoma cell lines. A, Ingenuity Pathway Analysis of Tax‐positive cells (HuT‐102 and OATL4). Top 10 activated upstream regulators in Tax‐positive cells and their functions are shown according to activation z score. Red bars, increased; blue bars, decreased. B, Kyoto Encyclopedia of Genes and Genomes pathway analysis of Tax‐positive cells. Top 10 downregulated pathways in Tax‐positive cells, along with their gene count, are shown according to their false discovery rate (FDR). Blue bars, downregulated. GIF1, Growth‐regulating factor‐interacting factor 1; HTLV‐1, human T‐cell leukemia virus type 1; IL, interleukin; NF‐κB, nuclear factor‐κB; TNF, tumor necrosis factor
3.4.2. Tax‐negative cell lines
Similarly, the typical MEK pathway inhibitor PD98059 indicated increased activity in IPA (Figure 5A). We also detected activation of upstream regulators linked to cell‐cycle arrest, such as CDKN2A and Rb, and this result validated the findings of the cell‐cycle analysis (Figure 3). Thus, the expression of upstream regulators, such as MYC, E2F, and cyclin D1 reduced with CDKN2A and Rb activation. The KEGG pathway analysis revealed that five pathways were significantly downregulated in Tax‐negative cell lines (Figure 5B); of these, two were related to cell growth regulation‐associated processes such as “cell cycle” and “DNA replication.” The genes involved in these pathways (Table S4) did not include MYC or E2F, instead blockade of PCNA or CDK2‐cyclin E2 pathways was suggested to be responsible for cell cycle arrest. The top 10 significantly downregulated gene sets (FDR < 0.05) in the GSEA are shown in Figures 5C,D and S6. Of these, “TNFα signaling via NF‐κB”, “MYC targets V2”, and “IL‐2 STAT5 signaling” were common for both KK1 and LMY1 cell lines. Genes with the highest enrichment (leading‐edge subset) are listed in Table S5. These gene sets were in agreement with the findings of IPA (Figure 5A), and the downregulation of MYC was commonly observed in both analyses.
FIGURE 5.
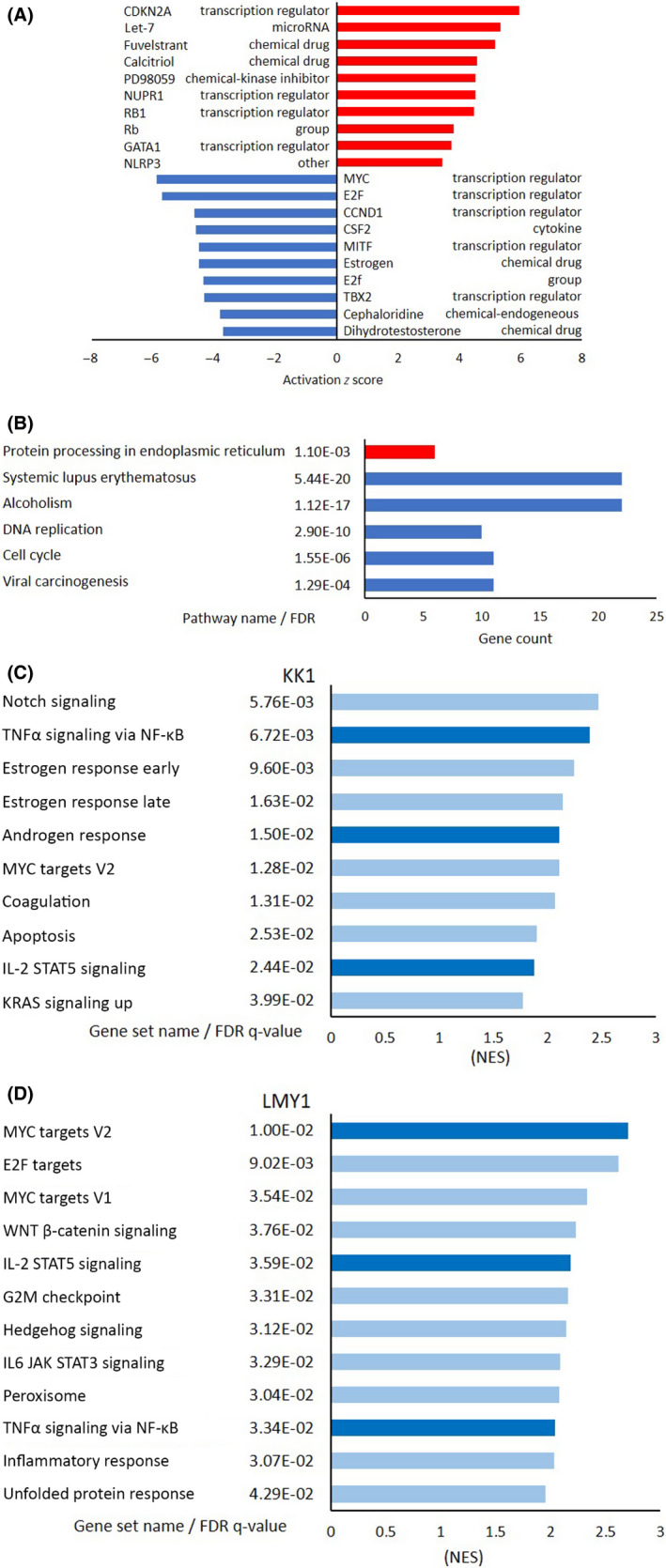
Microarray and pathway analyses of Tax‐negative adult T‐cell leukemia/lymphoma cell lines. A, Ingenuity Pathway Analysis of Tax‐negative cells (ED‐40515 and Su9T01). Top 10 activated upstream regulators in Tax‐negative cells and their functions are shown according to activation z score. Red bars, increased; blue bars, decreased. B, Kyoto Encyclopedia of Genes and Genomes pathway analysis of Tax‐negative cells. Top 10 common pathways with low false discovery rate (FDR) values (<0.01) in Tax‐negative cells, along with their gene count, according to their FDR. Red bars, upregulated; blue bars, downregulated. C, D, Gene Set Enrichment Analysis of Tax‐negative KK1 and LMY1 cells. Top 10 downregulated gene sets, along with their FDR values, are shown according to the normalized enrichment score. Light blue bars, downregulated; dark blue bars, commonly downregulated gene sets in KK1 and LMY1. CCND1, Cyclin D1; CDKN2A, cyclin‐dependent kinase inhibitor 2A; CSF2, Colony‐stimulating factor 2; GATA1, GATA‐binding factor 1; IL, interleukin; NLRP3, Nod‐like receptor family pyrin domain containing 3; NUPR1, Nuclear Protein 1; Rb, RB transcriptional corepressor 1; TBX2, T‐box transcription factor 2; TNFα, tumor necrosis factor α
3.4.3. Primary ATL cells
The top 10 downregulated KEGG pathways common in three primary ATL cells are displayed in Figure 6, and the details are listed in Table 1. The most significantly downregulated pathway was “Hepatitis B.” However, genes that were downregulated in this pathway, such as EGR, FOS, TNF, E2Fs, and MYC overlapped with those in the “HTLV‐1 infection” pathway, which was the third most significantly downregulated pathway. Interestingly, the other top 10 pathways always contained one or more of these genes. Among these, FOS, a subunit of the transcription factor AP‐1, was present in six of the top 10 pathways in Table 1 and in top three downregulated pathways in Table S3. Furthermore, other AP‐1 subunits, such as JUN, were included in Table S4, and FOSL1 and ATF3were included in Table S5. MYC, E2F1, and E2F2 were downregulated in Tax‐negative cell lines (Figure 5A and Table S5), whereas EGR1 and EGR2 were downregulated in Tax‐positive cell lines (Table S3).
FIGURE 6.
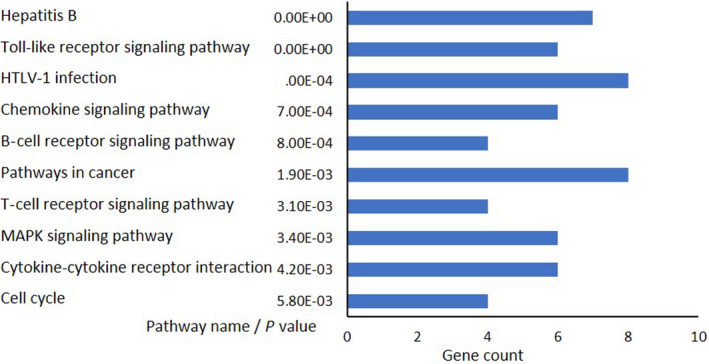
Microarray and pathway analyses of three primary adult T‐cell leukemia/lymphoma cells. Kyoto Encyclopedia of Genes and Genomes pathway analysis. Top 10 downregulated pathways, along with their gene count, are shown according to the P value. Blue bars, downregulated. HTLV‐1, human T‐cell leukemia virus type 1
TABLE 1.
Top 10 downregulated pathways according to the Kyoto Encyclopedia of Genes and Genomes and genes common in three primary adult T‐cell leukemia/lymphoma cells
| Hepatitis B | Toll‐like receptor signaling pathway | ||
| EGR2 | Early growth response 2 | CCL3L3 | Chemokine (C‐C motif) ligand 3‐like 3 |
| FOS | Fos proto‐oncogene, AP‐1 transcription factor subunit | TNF | Tumor necrosis factor |
| TNF | Tumor necrosis factor | CCL3 | Chemokine (C‐C motif) ligand 3 |
| STAT1 | Signal transducer and activator of transcription 1 | CCL4L2 | Chemokine (C‐C motif) ligand 4‐like 2 |
| E2F1 | E2F transcription factor 1 | FOS | Fos proto‐oncogene, AP‐1 transcription factor subunit |
| MYC | MYC proto‐oncogene, bHLH transcription factor | STAT1 | Signal transducer and activator of transcription 1 |
| E2F2 | E2F transcription factor 2 | ||
| Chemokine signaling | |||
| HTLV‐1 infection | CCL2 | Chemokine (C‐C motif) ligand 2 | |
| EGR1 | Early growth response 1 | CCL3L3 | Chemokine (C‐C motif) ligand 3‐like 3 |
| EGR2 | Early growth response 2 | CCL3 | Chemokine (C‐C motif) ligand 3 |
| FOS | Fos proto‐oncogene, AP‐1 transcription factor subunit | CCL4L2 | Chemokine (C‐C motif) ligand 4‐like 2 |
| TNF | Tumor necrosis factor | CXCR3 | Chemokine (C‐X‐C motif) receptor 3 |
| E2F1 | E2F transcription factor 1 | NCF1 | Neutrophil cytosolic factor 1 |
| MYC | MYC proto‐oncogene, bHLH transcription factor | ||
| LTA | Lymphotoxin α | Pathways in cancer | |
| E2F2 | E2F transcription factor 2 | FOS | Fos proto‐oncogene, AP‐1 transcription factor subunit |
| STAT1 | Signal transducer and activator of transcription 1 | ||
| T‐cell receptor signaling pathway | E2F1 | E2F transcription factor 1 | |
| FOS | Fos proto‐oncogene, AP‐1 transcription factor subunit | MYC | MYC proto‐oncogene, bHLH transcription factor |
| TNF | Tumor necrosis factor | E2F2 | E2F transcription factor 2 |
| PTPN6 | Protein tyrosine phosphatase, non‐receptor type 6 | NFKBIA | NF‐κB inhibitor α |
| NFKBIA | NF‐κB inhibitor α | ARNT2 | Aryl‐hydrocarbon receptor nuclear translocator 2 |
| CDC42 | Cell division cycle 42 | ||
| Cytokine‐cytokine receptor interaction | |||
| CCL2 | Chemokine (C‐C motif) ligand 2 | MAPK signaling pathway | |
| CCL3L3 | Chemokine (C‐C motif) ligand 3‐like 3 | FOS | Fos proto‐oncogene, AP‐1 transcription factor subunit |
| CCL3 | Chemokine (C‐C motif) ligand 3 | TNF | Tumor necrosis factor |
| TNF | Tumor necrosis factor | DUSP6 | Dual specificity phosphatase 6 |
| CCL4L2 | Chemokine (C‐C motif) ligand 4‐like 2 | MYC | MYC proto‐oncogene, bHLH transcription factor |
| CXCR3 | Chemokine (C‐X‐C motif) receptor 3 | CD14 | CD14 molecule, transcript variant 3 |
| DUSP4 | Dual specificity phosphatase 4 | ||
| Cell cycle | |||
| CDC45 | Cell division cycle 45 | ||
| E2F1 | E2F transcription factor 1 | ||
| MYC | MYC proto‐oncogene, bHLH transcription factor | ||
| E2F2 | E2F transcription factor 2 | ||
Abbreviations: AP‐1, activating protein‐1; HTLV‐1, human T‐cell leukemia virus type 1.
3.5. TAS‐116, given orally, suppresses the growth of xenografted ATL cells in mice
Finally, we evaluated the growth inhibitory effect of TAS‐116 in an ATL cell line (HuT‐102 [Tax‐positive] or Su9T01 [Tax negative])‐xenografted mouse model (Figure 7). The mean tumor volumes of both HuT‐102 and Su9T01 cells in the TAS‐116‐treated groups decreased after 3 days of treatment and remained significantly lower than those of the control group throughout the experiment. Furthermore, in two of the five HuT‐102‐transplanted mice treated with 14 mg/kg TAS‐116, tumors were completely eliminated after day 10.
FIGURE 7.
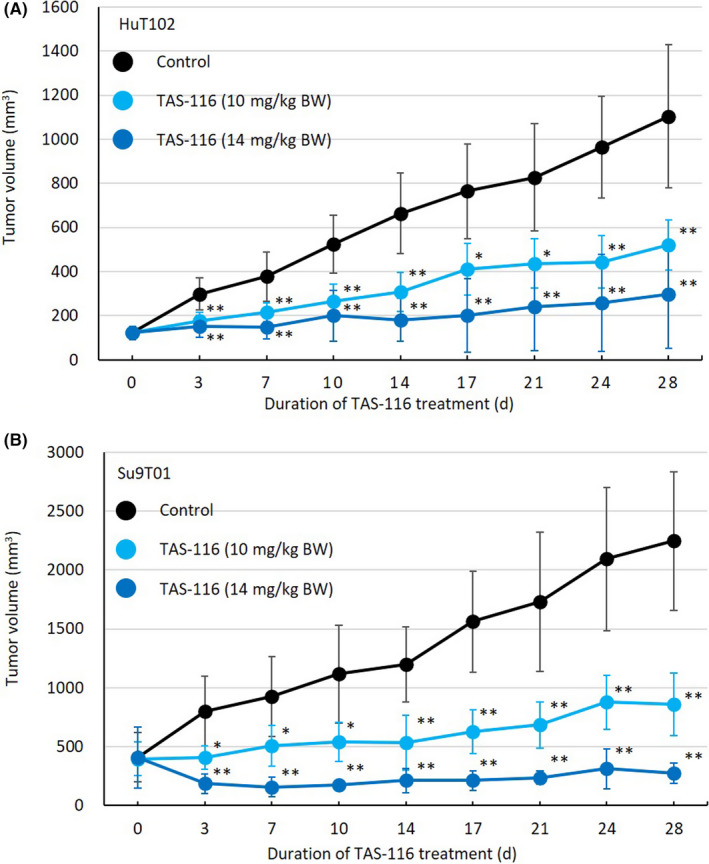
TAS‐116 reduces tumor volume in SCID mice. HuT‐102 or Su9T01 cells (5 × 106 cells/mouse) were injected subcutaneously into SCID mice. When the tumor volume reached approximately 100 mm3, mice were divided into three groups based on treatment (control, 10 mg/kg TAS‐116, or 14 mg/kg TAS‐116). Mice were given the treatment orally for 28 d from the day of grouping, and time‐course changes in tumor volume were monitored on the indicated days. Data are expressed as the mean ± SD of five (HuT‐102) or four (Su9T01) mice. *P < .05, **P < .01 (analyzed using ANOVA followed by Dunnett’s test). BW, body weight
4. DISCUSSION
In this study, we found that TAS‐116 effectively suppressed the growth of ATL‐related cell lines and primary ATL cells ex vivo and tumors implanted in mice in vivo. The mechanism by which TAS‐116 inhibited cell growth mainly involved cell‐cycle arrest at the G1 phase and apoptosis, consistent with previous studies. 24 , 25 We distinguished between Tax‐positive and Tax‐negative cell lines and divided them into three groups, including patient‐derived ATL cells; a few reports have made this distinction. Tax‐positive cell lines are helping to elucidate the pathogenesis of ATL, especially the behavior of Tax. 1 , 12 However, many primary ATL cells lose Tax expression, although the mechanism is not fully understood. Tax‐negative cell lines derived from ATL patients have not been widely used compared to Tax‐positive cell lines, partly because they are difficult to establish. To better understand how multiple cells were affected by TAS‐116 treatment, we focused on microarray analysis and on three different pathway analysis methods—IPA, DAVID (KEGG pathway), and GSEA—to identify commonalities and differences among each cell group. 37 , 40 However, we were not able to undertake the pathway analysis of primary ATL cells treated by TAS‐116 in vivo, which is a limitation of this study.
Our findings can be summarized in three main points. First, AP‐1 and TNF were commonly downregulated by TAS‐116 in all three cell groups. As a dimeric transcription factor composed of three Jun variants and/or Fos, AP‐1 is a key regulator linked to neoplastic transformation, including that in ATL. 41 , 42 Although Tax is known to interact with AP‐1 and contributes to the development of early stage ATL, many ATL cells do not express Tax. With respect to this, a previous report suggested that AP‐1 is activated in ATL cells through JunD in a Tax‐independent manner. 43 Our results indicate that TAS‐116 targets AP‐1 subunits, such as Fos in Tax‐positive cell lines and primary ATL cells and JUN, FOSL1, and ATF3 in Tax‐negative cell lines. Although our results suggest that the degradation of Tax by TAS‐116 contributes to the downregulation of Fos, TAS‐116 downregulates the expression of Fos in a Tax‐independent manner in primary ATL cells. These results suggest that TAS‐116 might show efficacy in various stages of ATL development. Tumor necrosis factor indirectly interacts with Tax and plays a central role in ATL pathogenesis. 44 It is interesting to note that TNF downregulation enables growth inhibition of ATL cells, in which Tax is not functional, and agents with anti‐TNF activity that exert anti‐ATL effects, such as thalidomide and its derivatives, are well known. Lenalidomide, an effective inhibitor of inflammation and TNF activity, is already a treatment option for ATL patients. 11 , 45 Considering the effects of lenalidomide in clinical settings, the TNF‐inhibitory activity of TAS‐116 makes it a potential therapeutic candidate for ATL. 11 , 46
Second, among the genes that are commonly downregulated in both primary ATL cells and Tax‐positive cell lines, EGRs of the “HTLV‐1 infection” pathway are prominent. Tax has been reported to form complexes with Sp1/Egr1 and Sp1/Ets1 and contributes to the transcriptional activation of the PDGFB proto‐oncogene. 47 Although there is no clear evidence indicating that EGR or PDGFB downregulation is effective at regulating the growth of ATL cells, inhibition of EGRs resulted in inhibition of PDGFB activity and could induce TAS‐116‐mediated anti‐ATL effects. 47 , 48 By contrast, the NF‐κB signaling pathway was downregulated to the second highest degree in Tax‐positive cell lines but not in primary ATL cells.
The third point is that genes significantly downregulated in both primary ATL cells and Tax‐negative cell lines include MYC and E2Fs, which are related to the “cell cycle.” Considering that TAS‐116 significantly upregulated the expression of Rb and CDKN2A in these cells, and did not damage normal CD4 lymphocytes, it can be considered to be an ideal antitumor agent that functions by inducing cell‐cycle arrest or apoptosis. 49 , 50
TAS‐116 exhibited NF‐κB inhibitor activities in cells with Tax expression, while showing anticancer properties by inducing cell‐cycle arrest and apoptosis in cells that did not express Tax. Consequently, TAS‐116 was pharmacologically active against cells in the early stage (immortalization by Tax) and late stage (frequent loss of Tax expression) of ATL development. We undertook several different pathway analyses using three different types of ATL cells, and the phenomena commonly observed in these analyses were confirmed. Our results strongly suggest that TAS‐116 is a promising therapeutic option for patients at any stage of ATL.
Compared with other HSP90 inhibitors examined in our previous studies, the cytotoxicity of TAS‐116 was modest. NF‐κB and c‐MYC inhibitory effects were observed dominant in Tax‐positive and Tax‐negative cells, respectively, both were modest in TAS‐116. Therefore, the efficacy of TAS‐116 in clinical trials will be more important. In a comparable clinical trial of TAS‐116 and NVP‐AUY922 for gastrointestinal stromal tumor, progression‐free survival and overall survival were slightly better with TAS‐116. 29 , 51 Both have grade 3 or higher toxicities (at clinically recommended doses), such as anemia and diarrhea, but there is no evidence of ocular toxicity with TAS‐116, which could be an advantage.
DISCLOSURE
The authors declares no competing financial interests.
Supporting information
Fig S1
{kind=link}
Fig S2
{kind=link}
Fig S3
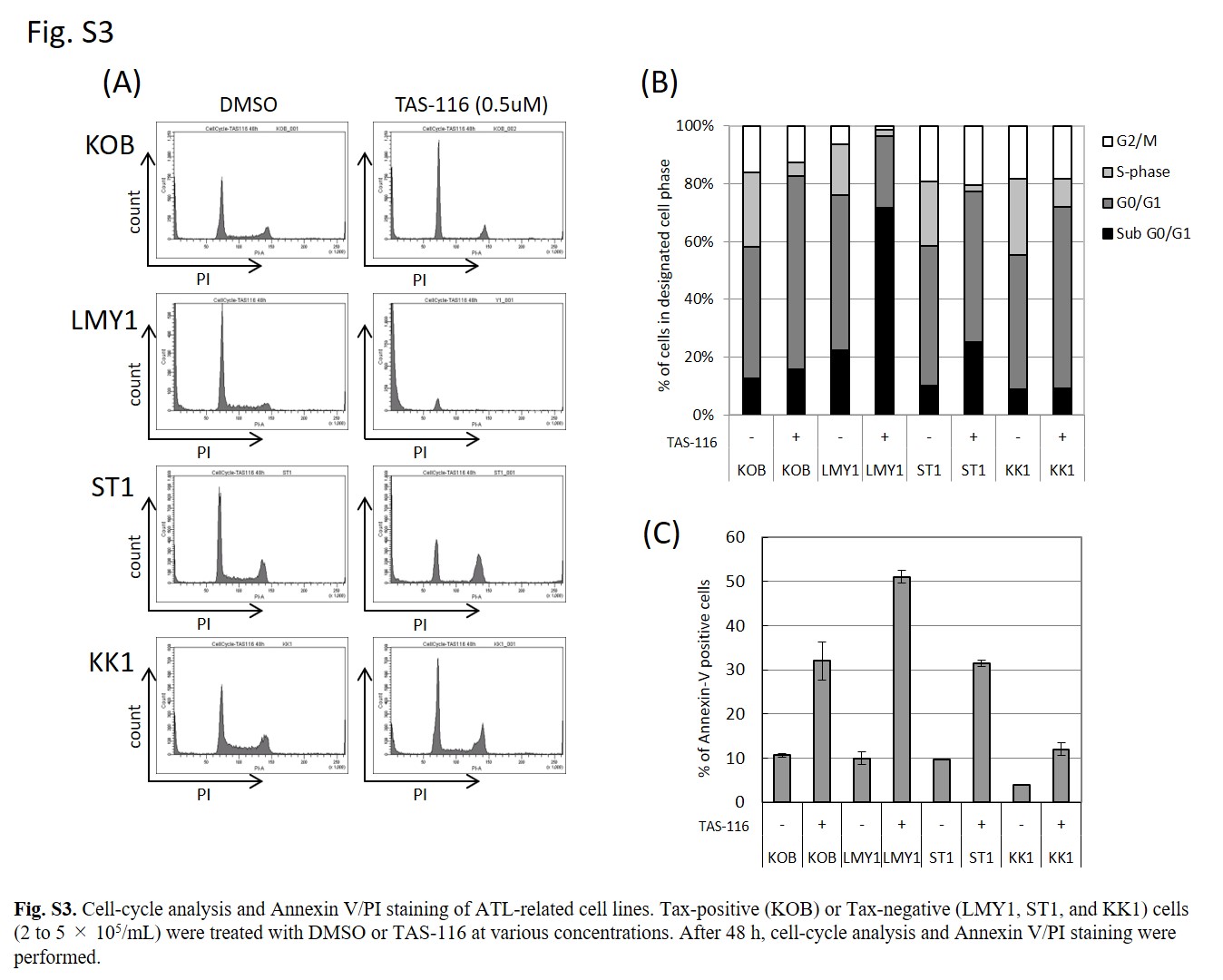{kind=link}
Fig S4
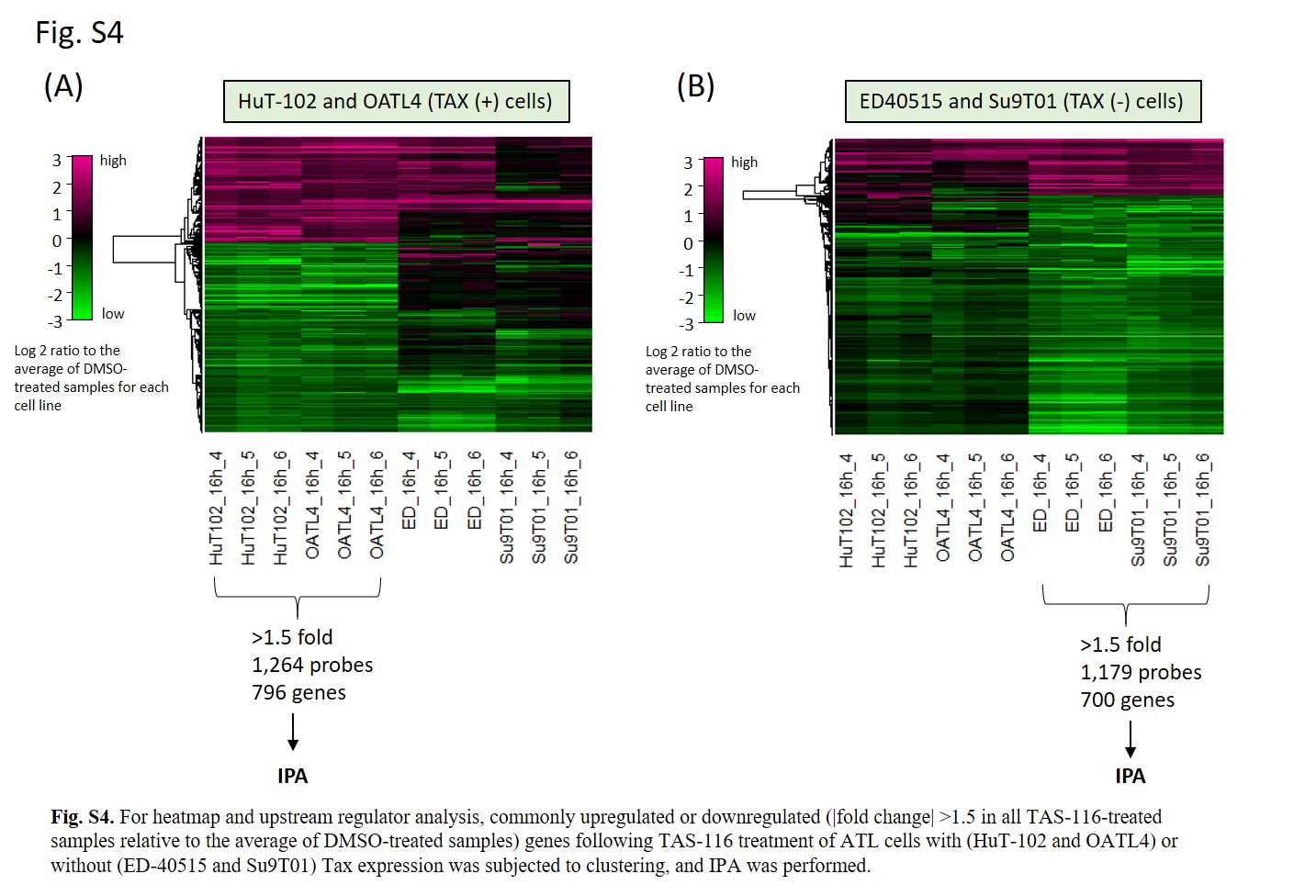{kind=link}
Fig S5
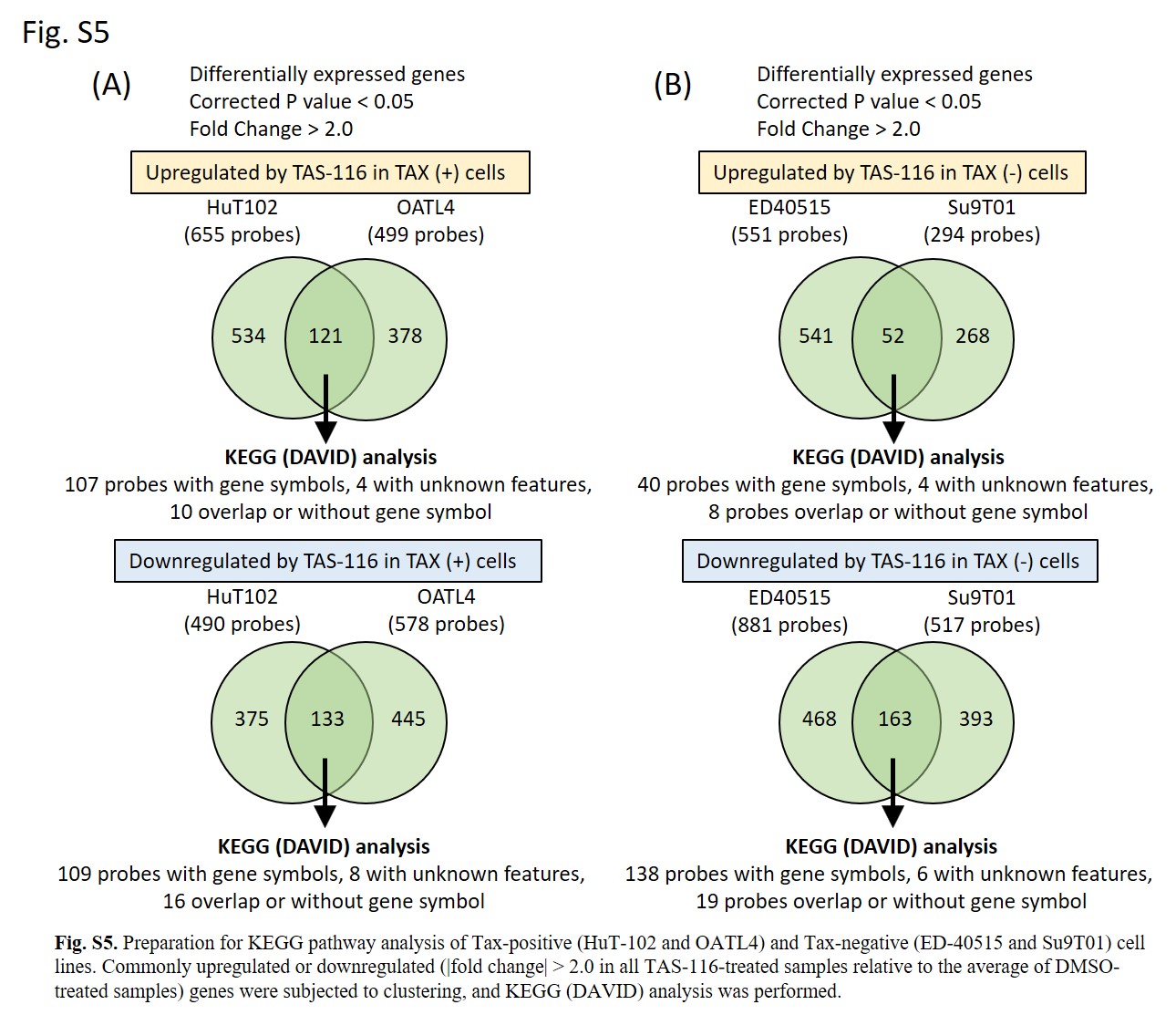{kind=link}
Fig S6
{kind=link}
Table S1
Table S2
Table S3
Table S4
Table S5
ACKNOWLEDGMENTS
This study was supported in part by research funding from the Japan Society for the Promotion of Science (grant numbers 23590641, 15K08647, and 18H02733) and a Research Program on Emerging and Re‐emerging Infectious Diseases grant from the Japan Agency for Medical Research and Development, AMED (20fk0108124h2001). We thank Ai Nishida for conducting cell culture experiments and the sample preparation. We would like to thank Editage for their assistance with English language editing.
Ikebe E, Shimosaki S, Hasegawa H, et al. TAS‐116 (pimitespib), a heat shock protein 90 inhibitor, shows efficacy in preclinical models of adult T‐cell leukemia. Cancer Sci.2022;113:684–696. doi: 10.1111/cas.15204
Emi Ikebe and Shunsuke Shimosaki contributed equally to this study.
Funding information
Japan Society for the Promotion of Science, Grant/Award Number: 23590641, 15K08647, 18H02733; Japan Agency for Medical Research and Development, Grant/Award Number: 20fk0108124h2001
Contributor Information
Hiroo Hasegawa, Email: hhase@nagasaki-u.ac.jp.
Hidekatsu Iha, Email: hiha@oita-u.ac.jp.
REFERENCES
- 1. Poiesz BJ, Ruscetti FW, Gazdar AF, Bunn PA, Minna JD, Gallo RC. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T‐cell lymphoma. Proc Natl Acad Sci U S A. 1980;77(12):7415‐7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Moriuchi H, Masuzaki H, Doi H, Katamine S. Mother‐to‐child transmission of human T‐cell lymphotropic virus type 1. Pediatr Infect Dis J. 2013;32(2):175‐177. [DOI] [PubMed] [Google Scholar]
- 3. Tezuka K, Fuchi N, Okuma K, et al. HTLV‐1 targets human placental trophoblasts in seropositive pregnant women. J Clin Invest. 2020;130(11):6171‐6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gessain A, Cassar O. Epidemiological aspects and world distribution of HTLV‐1 infection. Front Microbiol. 2012;3:388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Iwanaga M, Watanabe T, Yamaguchi K. Adult T‐cell leukemia: a review of epidemiological evidence. Front Microbiol. 2012;3:322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shimoyama M. Diagnostic criteria and classification of clinical subtypes of adult T‐cell leukaemia‐lymphoma. A report from the Lymphoma Study Group (1984‐87). Br J Haematol. 1991;79(3):428‐437. [DOI] [PubMed] [Google Scholar]
- 7. Tsukasaki K, Hermine O, Bazarbachi A, et al. Definition, prognostic factors, treatment, and response criteria of adult T‐cell leukemia‐lymphoma: a proposal from an international consensus meeting. J Clin Oncol. 2009;27(3):453‐459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yamada Y, Tomonaga M, Fukuda H, et al. A new G‐CSF‐supported combination chemotherapy, LSG15, for adult T‐cell leukaemia‐lymphoma: Japan Clinical Oncology Group Study 9303. Br J Haematol. 2001;113(2):375‐382. [DOI] [PubMed] [Google Scholar]
- 9. Tsukasaki K, Utsunomiya A, Fukuda H, et al. VCAP‐AMP‐VECP compared with biweekly CHOP for adult T‐cell leukemia‐lymphoma: Japan Clinical Oncology Group Study JCOG9801. J Clin Oncol. 2007;25(34):5458‐5464. [DOI] [PubMed] [Google Scholar]
- 10. Ishitsuka K, Tamura K. Human T‐cell leukaemia virus type I and adult T‐cell leukaemia‐lymphoma. Lancet Oncol. 2014;15(11):e517‐526. [DOI] [PubMed] [Google Scholar]
- 11. El Hajj H, Tsukasaki K, Cheminant M, Bazarbachi A, Watanabe T, Hermine O. Novel treatments of adult T cell leukemia lymphoma. Front Microbiol. 2020;11:1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boxus M, Twizere JC, Legros S, Dewulf JF, Kettmann R, Willems L. The HTLV‐1 Tax interactome. Retrovirology. 2008;5:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yamagishi M, Nakano K, Miyake A, et al. Polycomb‐mediated loss of miR‐31 activates NIK‐dependent NF‐kappaB pathway in adult T cell leukemia and other cancers. Cancer Cell. 2012;21(1):121‐135. [DOI] [PubMed] [Google Scholar]
- 14. Yamaoka S, Courtois G, Bessia C, et al. Complementation cloning of NEMO, a component of the IkappaB kinase complex essential for NF‐kappaB activation. Cell. 1998;93(7):1231‐1240. [DOI] [PubMed] [Google Scholar]
- 15. Kataoka K, Nagata Y, Kitanaka A, et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet. 2015;47(11):1304‐1315. [DOI] [PubMed] [Google Scholar]
- 16. Matsuoka M, Yasunaga J. Human T‐cell leukemia virus type 1: replication, proliferation and propagation by Tax and HTLV‐1 bZIP factor. Curr Opin Virol. 2013;3(6):684‐691. [DOI] [PubMed] [Google Scholar]
- 17. Miyata Y, Nakamoto H, Neckers L. The therapeutic target Hsp90 and cancer hallmarks. Curr Pharm Des. 2013;19(3):347‐365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li L, Wang L, You QD, Xu XL. Heat Shock Protein 90 inhibitors: an update on achievements, challenges, and future directions. J Med Chem. 2020;63(5):1798‐1822. [DOI] [PubMed] [Google Scholar]
- 19. Sanchez J, Carter TR, Cohen MS, Blagg BSJ. Old and new approaches to target the Hsp90 chaperone. Curr Cancer Drug Targets. 2020;20(4):253‐270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stingl L, Stuhmer T, Chatterjee M, Jensen MR, Flentje M, Djuzenova CS. Novel HSP90 inhibitors, NVP‐AUY922 and NVP‐BEP800, radiosensitise tumour cells through cell‐cycle impairment, increased DNA damage and repair protraction. Br J Cancer. 2010;102(11):1578‐1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee SL, Dempsey‐Hibbert NC, Vimalachandran D, Wardle TD, Sutton PA, Williams JH. Re‐examining HSPC1 inhibitors. Cell Stress Chaperones. 2017;22(2):293‐306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zagouri F, Sergentanis TN, Chrysikos D, Papadimitriou CA, Dimopoulos MA, Psaltopoulou T. Hsp90 inhibitors in breast cancer: a systematic review. Breast. 2013;22(5):569‐578. [DOI] [PubMed] [Google Scholar]
- 23. Zhou D, Liu Y, Ye J, et al. A rat retinal damage model predicts for potential clinical visual disturbances induced by Hsp90 inhibitors. Toxicol Appl Pharmacol. 2013;273(2):401‐409. [DOI] [PubMed] [Google Scholar]
- 24. Ikebe E, Kawaguchi A, Tezuka K, et al. Oral administration of an HSP90 inhibitor, 17‐DMAG, intervenes tumor‐cell infiltration into multiple organs and improves survival period for ATL model mice. Blood Cancer J. 2013;3:e132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Taniguchi H, Hasegawa H, Sasaki D, et al. Heat shock protein 90 inhibitor NVP‐AUY922 exerts potent activity against adult T‐cell leukemia‐lymphoma cells. Cancer Sci. 2014;105(12):1601‐1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gao L, Harhaj EW. HSP90 protects the human T‐cell leukemia virus type 1 (HTLV‐1) tax oncoprotein from proteasomal degradation to support NF‐kappaB activation and HTLV‐1 replication. J Virol. 2013;87(24):13640‐13654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ohkubo S, Kodama Y, Muraoka H, et al. TAS‐116, a highly selective inhibitor of heat shock protein 90alpha and beta, demonstrates potent antitumor activity and minimal ocular toxicity in preclinical models. Mol Cancer Ther. 2015;14(1):14‐22. [DOI] [PubMed] [Google Scholar]
- 28. Uno T, Kawai Y, Yamashita S, et al. Discovery of 3‐Ethyl‐4‐(3‐isopropyl‐4‐(4‐(1‐methyl‐1 H‐pyrazol‐4‐yl)‐1 H‐imidazol‐1‐yl)‐1 H‐pyrazolo[3,4‐ b]pyridin‐1‐yl)benzamide (TAS‐116) as a potent, selective, and orally available HSP90 inhibitor. J Med Chem. 2019;62(2):531‐551. [DOI] [PubMed] [Google Scholar]
- 29. Doi T, Kurokawa Y, Sawaki A, et al. Efficacy and safety of TAS‐116, an oral inhibitor of heat shock protein 90, in patients with metastatic or unresectable gastrointestinal stromal tumour refractory to imatinib, sunitinib and regorafenib: a phase II, single‐arm trial. Eur J Cancer. 2019;121:29‐39. [DOI] [PubMed] [Google Scholar]
- 30. Shimomura A, Yamamoto N, Kondo S, et al. First‐in‐Human Phase I Study of an Oral HSP90 inhibitor, TAS‐116, in patients with advanced solid tumors. Mol Cancer Ther. 2019;18(3):531‐540. [DOI] [PubMed] [Google Scholar]
- 31. Taiho Pharmaceutical's TAS‐116, an oral Hsp90 inhibitor, shows significantly prolonged progression‐free survival in patients with GIST in phase‐III clinical trial. Accessed May 25, 2021. https://www.taiho.co.jp/en/release/2021/20210222.html
- 32. Science Council of Japan . Guidelines for Proper Conduct of Animal Experiments. Accessed May 25, 2021. http://www.scj.go.jp/ja/info/kohyo/pdf/kohyo‐20‐k16‐2e.pdf
- 33. Hasegawa H, Yamada Y, Harasawa H, et al. Sensitivity of adult T‐cell leukaemia lymphoma cells to tumour necrosis factor‐related apoptosis‐inducing ligand. Br J Haematol. 2005;128(2):253‐265. [DOI] [PubMed] [Google Scholar]
- 34. Kamihira S, Sugahara K, Tsuruda K, et al. Proviral status of HTLV‐1 integrated into the host genomic DNA of adult T‐cell leukemia cells. Clin Lab Haematol. 2005;27(4):235‐241. [DOI] [PubMed] [Google Scholar]
- 35. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44‐57. [DOI] [PubMed] [Google Scholar]
- 36. Calvano SE, Xiao W, Richards DR, et al. A network‐based analysis of systemic inflammation in humans. Nature. 2005;437(7061):1032‐1037. [DOI] [PubMed] [Google Scholar]
- 37. Thomas S, Bonchev D. A survey of current software for network analysis in molecular biology. Hum Genomics. 2010;4(5):353‐360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mootha VK, Lindgren CM, Eriksson KF, et al. PGC‐1alpha‐responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267‐273. [DOI] [PubMed] [Google Scholar]
- 39. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545‐15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cirillo E, Parnell LD, Evelo CT. A review of pathway‐based analysis tools that visualize genetic variants. Front Genet. 2017;8:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gazon H, Barbeau B, Mesnard JM, Peloponese JM Jr. Hijacking of the AP‐1 signaling pathway during development of ATL. Front Microbiol. 2017;8:2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hess J, Angel P, Schorpp‐Kistner M. AP‐1 subunits: quarrel and harmony among siblings. J Cell Sci. 2004;117(Pt 25):5965‐5973. [DOI] [PubMed] [Google Scholar]
- 43. Mori N, Fujii M, Iwai K, et al. Constitutive activation of transcription factor AP‐1 in primary adult T‐cell leukemia cells. Blood. 2000;95(12):3915‐3921. [PubMed] [Google Scholar]
- 44. Twizere JC, Kruys V, Lefebvre L, et al. Interaction of retroviral Tax oncoproteins with tristetraprolin and regulation of tumor necrosis factor‐alpha expression. J Natl Cancer Inst. 2003;95(24):1846‐1859. [DOI] [PubMed] [Google Scholar]
- 45. Ishida T, Fujiwara H, Nosaka K, et al. Multicenter Phase II Study of Lenalidomide in relapsed or recurrent adult T‐Cell leukemia/lymphoma: ATLL‐002. J Clin Oncol. 2016;34(34):4086‐4093. [DOI] [PubMed] [Google Scholar]
- 46. Sakamoto H, Itonaga H, Sawayama Y, et al. Treatment with mogamulizumab or lenalidomide for relapsed adult T‐cell leukemia/lymphoma after allogeneic hematopoietic stem cell transplantation: the Nagasaki transplant group experience. Hematol Oncol. 2020;38(2):162‐170. [DOI] [PubMed] [Google Scholar]
- 47. Trejo SR, Fahl WE, Ratner L. The tax protein of human T‐cell leukemia virus type 1 mediates the transactivation of the c‐sis/platelet‐derived growth factor‐B promoter through interactions with the zinc finger transcription factors Sp1 and NGFI‐A/Egr‐1. J Biol Chem. 1997;272(43):27411‐27421. [DOI] [PubMed] [Google Scholar]
- 48. Fujii M, Niki T, Mori T, et al. HTLV‐1 Tax induces expression of various immediate early serum responsive genes. Oncogene. 1991;6(6):1023‐1029. [PubMed] [Google Scholar]
- 49. Dang CV. MYC on the path to cancer. Cell. 2012;149(1):22‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. McKeown MR, Bradner JE. Therapeutic strategies to inhibit MYC. Cold Spring Harb Perspect Med. 2014;4(10):a014266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bendell JC, Bauer TM, Lamar R, et al. A Phase 2 Study of the Hsp90 Inhibitor AUY922 as treatment for patients with refractory gastrointestinal stromal tumors. Cancer Invest. 2016;34(6):265‐270. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Fig S6
Table S1
Table S2
Table S3
Table S4
Table S5