

Abstract
There has been accumulating evidence that RNA splicing is frequently dysregulated in a variety of cancers and that hotspot mutations affecting key splicing factors, SF3B1, SRSF2 and U2AF1, are commonly enriched across cancers, strongly suggesting that aberrant RNA splicing is a new class of hallmark that contributes to the initiation and/or maintenance of cancers. In parallel, some studies have demonstrated that cancer cells with global splicing alterations are dependent on the transcriptional products derived from wild‐type spliceosome for their survival, which potentially creates a therapeutic vulnerability in cancers with a mutant spliceosome. It has been c. 10 y since the frequent mutations affecting splicing factors were reported in cancers. Based on these surprising findings, there has been a growing interest in targeting altered splicing in the treatment of cancers, which has promoted a wide variety of investigations including genetic, molecular and biological studies addressing how altered splicing promotes oncogenesis and how cancers bearing alterations in splicing can be targeted therapeutically. In this mini‐review we present a concise trajectory of what has been elucidated regarding the pathogenesis of cancers with aberrant splicing, as well as the development of therapeutic strategies to target global splicing alterations in cancers.
Keywords: antisense oligonucleotide, cancer, RNA binding protein, SF3b, splicing factor
Transcription and pre‐mRNA splicing are key steps in the control of gene expression in eukaryotic cells and mutations in genes regulating each of these processes are common in cancers. By reviewing the recent advances in this field, we described the pathogenic mechanisms in which the hotspot mutations in genes encoding splicing factors drive oncogenesis, and therapeutic strategies for targeting global alterations in splicing, including the use of splicing modulators, inhibition of splicing regulatory proteins, emerging technologies using antisense oligonucleotides and a potential tactic to improve the response to checkpoint blockade.
Abbreviations
- ASO
antisense oligonucleotide
- BPS
branchpoint sequence
- CLL
chronic lymphocytic leukemia
- CMML
chronic myelomonocytic leukemia
- ESE
exonic splicing enhancer
- ESS
exonic splicing silencer
- hnRNP
heterogeneous nuclear ribonucleoprotein
- ISE
intronic splicing enhancer
- ISS
intronic splicing silencer
- MDS
myelodysplastic syndrome
- MDS‐RS
MDS with ring sideroblasts
- PDX
patient‐derived xenograft
- RBP
RNA binding protein
- SF
splicing factor
- snRNP
small nuclear ribonucleoprotein particle
- SR
serine and arginine
- ss
splice site
- SSO
splicing switching oligonucleotide
- TCGA
The Cancer Genome Atlas
- UVM
uveal melanoma
1. INTRODUCTION
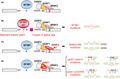
RNA splicing is the ingenious process that produces mature mRNA transcripts by removing introns from pre‐mRNA. A growing body of evidence has underlined the involvement of recurrent point mutations in the major RNA splicing factors SF3B1, SRSF2, and U2AF1 in the pathogenesis of cancers. These mutations in genes encoding SF mutations are frequently identified in a variety of hematopoietic malignancies 1 , 2 , 3 as well as in solid tumors such as breast cancers, lung cancers, and pancreatic cancers, 4 , 5 , 6 , 7 , 8 strongly suggesting that SF mutations plays an essential role in cancer pathogenesis (Figure 1A‐C). In this mini‐review we will summarize how SF mutations cause global alterations in splicing, how they contribute to cancer pathogenesis, and whether they can be therapeutically targeted.
FIGURE 1.
Mutational hotspots in genes encoding the major splicing factors. Frequent somatic mutations in SF3B1 (A), SRSF2 (B) and U2AF1 (C). The numbers in the center of the flame icon represent the percentage of samples with indicated mutations within “hotspot” mutations that were previously demonstrated to be pathogenic. These pan‐cancer samples with spliceosome mutations were collected from Columbia University Medical Center, Memorial Sloan Kettering Cancer Center, and public portal resources including TCGA, the ICGC database of Genotypes and Phenotypes (dbGaP), and the Gene Expression Omnibus (GEO), as previously described. 20 RRM, RNA recognition motif; RS domain, arginine/serine‐rich domain; ZnF, zinc finger; UHM, U2AF homology motif
2. THE REGULATORY MACHINERY OF RNA SPLICING
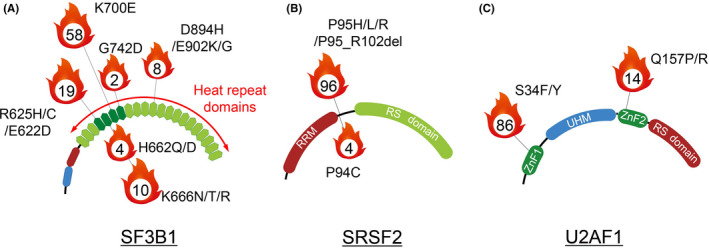
RNA splicing is essential for processing pre‐mRNA in >90% of human protein‐coding genes that contain more than 1 exon. The primary function of splicing is to remove non‐coding introns and essential splicing sequences include the 5′ss, the BPS and a 3′ss (Figure 2A). 9 , 10 Upon recognition of these sequences, the spliceosome catalyzes the splicing reaction. The core enzymatic machinery of splicing is accomplished by the major spliceosome that consists of 5 snRNP particles, U1, U2, U4, U5, and U6. The excision of >99% of human introns is carried out by this core enzymatic machinery. Each constituent snRNP contains a Sm or Sm‐like protein complex that is essential for the formation of the mature snRNP complex and proteins specific to each snRNP. 11 , 12
FIGURE 2.
Splicing catalysis, the spliceosome assembly pathway, and mechanisms of splice site selection. A, Diagram of an intron, 2 flanking exons, U1 and U2 snRNP, associated RNA binding proteins, and U2 snRNA, and the sequential reactions involved in removal of an intron. The U2 snRNP complex and U2AF complex recognize the BPS and 3′ss, respectively, which is an essential step for removing introns from pre‐mRNA. Accessory splicing regulatory proteins are also involved in splicing. B, Classification of alternative RNA splicing
In addition to core splicing sequences recognized by the spliceosome, both cis‐ and trans‐acting factors regulate the splicing pathways. The cis‐acting factors are splicing regulatory elements including the ESEs and ESS, and the ISEs and ISS (Figure 2A). 9 Trans‐acting RBPs recognize these sequences and recruit or repress the core splicing enzymatic machinery to regulate splicing. RBPs include SR‐rich proteins that generally bind to ESEs to promote the splicing, and the hnRNP family that recognize ESS and ISS and inhibit splicing. 9 Transcripts from almost all human multi‐exon genes are alternatively spliced, where a particular 5′ss can be joined to different 3′ss (or vice versa), frequently in a regulated fashion. 13 Alternative splicing events are further classified based on how they affect the exonic structure of the mature mRNA (Figure 2B).
3. MUTATIONS AFFECTING RNA SPLICING FACTORS IN CANCERS
3.1. Mutations in SF3B1
SF3B1 is the most commonly mutated splicing factor across cancers. Particularly, mutations in SF3B1 have an enrichment in several cancer types including MDS, CLL and UVM. Interestingly, SF3B1 mutations have specific values as biomarkers in certain types of cancer. For example, SF3B1 mutations are identified in >90% of patients with MDS‐RS. This form of MDS is characterized by anemia, iron‐laden mitochondria surrounding the nuclei of erythroid precursors and a favorable prognosis. 2 , 3 , 14 Because SF3B1 mutations have >97% positive predictive value for patients suspected to have MDS‐RS, 15 mutations in SF3B1 are currently part of the diagnostic criteria for MDS‐RS. Mutations in SF3B1 are generally early events in the disease progression of myeloid malignancies, whereas SF3B1 mutations in CLL are most commonly subclonal and enriched in more advanced and aggressive disease. 16 , 17
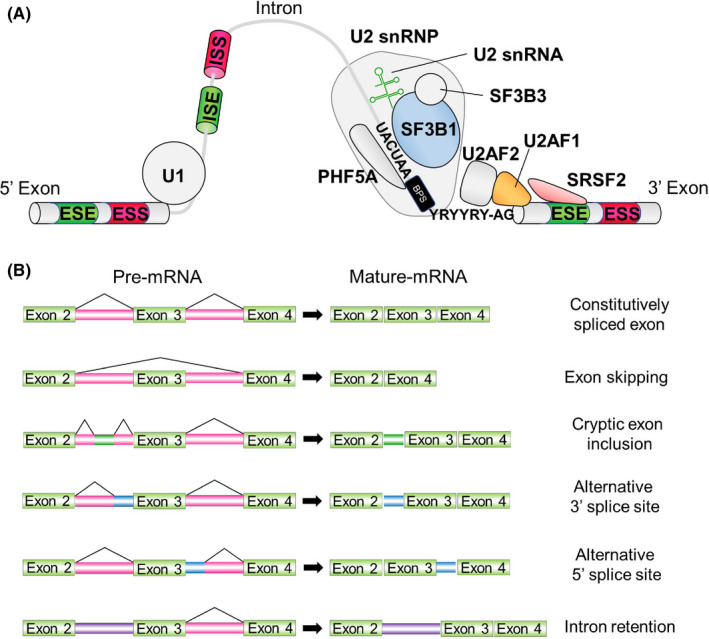
SF3B1 is a member of the U2 snRNP complex that physically associates with PHF5A, SF3B3 and the U2 snRNA (Figure 2A). The U2 snRNP complex is important in recognizing the BPS within the intron. In harmony with this, splicing analysis based on RNA sequence data from cancer cells and mouse models with mutations in SF3B1 revealed that SF3B1 mutant cells exhibit usage of an aberrant intron‐proximal 3′ss compared with canonical splicing (Figure 3A,B). 18 , 19 , 20 , 21 Mutations in SF3B1 are enriched in the 4th to 7th HEAT repeat domains, suggesting that mutant SF3B1 has altered conformation of the HEAT repeat domains. In fact, recent crystal structure analysis of the mutant SF3B1 protein clarified that SF3B1 mutations alter the conformation of the HEAT repeat domains that affects the interactions with U2AF2 or the SF3b complex. 22
FIGURE 3.
Functional consequences of SF3B1, SRSF2, and U2AF1 mutations in RNA splicing. A, Description of canonical splicing. B, Mutations in SF3B1 (marked in red) result in enhanced usage of an aberrant branchpoint to generate an alternative 3′ss. C, Mutations in SRSF2 are clustered at the proline 95 residue and alter the preference of ESE motif recognition. D, U2AF1 mutations alter the 3′ss consensus sequences. U2AF1 S34F/Y mutations favor inclusion of cassette exons with a C‐nucleotide at the “−3” position, whereas U2AF1 Q157P/R mutations favor inclusion of cassette exons with a G‐nucleotides at the “+1” position
SF3B1 is affected by some distinct hotspot mutations, and our recent pan‐cancer splicing analysis unveiled differential splicing events based on lineage as well as SF3B1 mutant allele. 20 The most common mutation in SF3B1 is the SF3B1 K700E mutation, which is found in myeloid malignancies, CLL and many solid tumors including breast cancers, pancreatic ductal adenocarcinomas, and others. In addition, there are several hotspot mutations in SF3B1 including SF3B1 R625 mutations (enriched in melanomas) and SF3B1 E902 mutations (enriched in bladder carcinomas). The biological and molecular bases for the lineage specificity of these hotspots still remain to be clarified. A recent paper has revealed that SF3B1K700E loses physical interaction with a poorly studied spliceosome protein, SUGP1, during BPS recognition. Loss of the interaction leads to aberrant usage of upstream BPSs that was partly rescued upon SUGP1 restoration. 23 This report also showed that the loss of interaction between mutant SF3B1 and SUGP1 is commonly observed among disease‐associated mutant SF3B1 proteins other than K700E, suggesting that the loss of interaction is one of the key mechanisms that promote aberrant usage of 3′ss by mutant SF3B1.
3.2. Mutations in SRSF2 and U2AF1
In addition to mutations in SF3B1, extensive studies have revealed more about how hotspot mutations in SRSF2 and U2AF1 impact splicing and disease development.
SRSF2 is an auxiliary splicing factor that binds ESEs to recruit the core spliceosome to promote splicing. Whereas wild‐type SRSF2 physically binds CCNG and GGNG sequences equally, 24 hotspot mutations in SRSF2 affecting proline 95 alter this preference. 19 , 25 , 26 , 27 As a result, mutant SRSF2 promotes splicing of exons with C‐rich sequences over G‐rich sequences (Figure 3C). Although the frequency of mutations in SRSF2 was originally estimated as low (<2%) in AML, 28 our group reanalyzed TCGA dataset and identified that 95% (18/19) of patients with SRSF2 mutations were missed in the previous TCGA publication, 26 which makes SRSF2 one of the most frequently mutated genes in AML. This was probably due to the markedly GC‐rich sequence around the SRSF2 mutational hotspot, leading to low coverage around this region in the next‐generation sequencing result. Interestingly, genomic analysis of the TCGA AML cohort using refined SRSF2 genotyping revealed the frequent and significant co‐occurrence in mutations affecting SRSF2 and IDH2 in AML. Further functional and biological studies clarified that aberrant RNA splicing and mutant IDH2‐mediated DNA hypermethylation closely cooperate with each other to drive leukemogenesis. One of the most robust splicing changes in IDH2/SRSF2 double‐mutant AML is the combined intron retention and exon skipping events in Integrator 3 (INTS3), whose loss results in dysregulated gene expression programs associated with hematopoietic cell differentiation and multiple signaling pathways, and blockade of myeloid differentiation leading to the development of myelodysplastic/myeloproliferative neoplasms in vivo. 26 Mutant SRSF2 also generates an EZH2 isoform that undergoes nonsense‐mediated decay, leading to loss of a key hematopoietic regulator. 25
U2AF1 is part of a heterodimeric U2AF complex with its partner, U2AF2, that recruits the U2 snRNP to the BPS (Figure 2A). U2AF1 associates with the AG dinucleotide at the 3′ss, whereas U2AF2 binds the polypyrimidine tract. Hotspot mutations in U2AF1 mainly affect the S34 or Q157 residue, each of which is located in 1 of its 2 zinc fingers. Similar to the hotspot mutations in SF3B1, U2AF1 mutations are linked to specific lineages of cancer. One of the examples is lung adenocarcinoma, in which U2AF1 S34 mutations are recurrent whereas U2AF1 Q157 mutations are absent. 29 Molecularly, U2AF1 S34 mutations promote inclusion of exons whose 3′ss is C‐rich, whereas U2AF1 Q157 mutations enhance inclusion of exons whose 3′ss is G‐rich (Figure 3D). 30
3.3. Mutations in other splicing factors
Mutations in ZRSR2 are identified in c. 10% of MDS patients. These mutations are located sporadically across the coding regions, suggesting that these mutations are loss‐of‐function mutations distinct from the change‐of‐function mutations in SF3B1, SRSF2, and U2FA1. In addition, a recent study investigating mutations in 404 SFs from 33 cancer types in TCGA identified potential driver mutations in 119 SFs. 31 Functional roles of these mutations in cancers remain to be addressed.
4. STRATEGIES FOR TARGETING SPLICING ALTERATIONS IN CANCERS
4.1. Inhibition of SF3b binding
Several natural products from bacterial species and their analogs that bind the SF3b complex have been discovered. These compounds, including E7107 (an analog of pladienolide B), 32 spliceostatin A, 33 and the sudemycins 34 inhibit the binding of the branchpoint binding region of U2 snRNP to the BPS and block the essential conformational change in U2 snRNP (Figure 4A). 35 , 36 , 37 , 38 H3 Biomedicine Inc and our group developed an orally bioavailable, clinical‐grade analog of E7107, H3B‐8800, and demonstrated that H3B‐8800 modulates splicing of wild‐type and mutant spliceosomes in vitro and preferentially kills spliceosome‐mutant cells. 39 Sequentially, we added studies for the evaluation of spliceosome inhibitor in isogeneic murine MDS/AML models and, more importantly, in preclinical settings using PDX models with mutant SF3B1 expression. In mice engrafted with SF3B1 K700E PDX, 10 d of H3B‐8800 treatment significantly reduced leukemic burden relative to that in vehicle‐treated mice. Given that mutations in SRSF2 are prevalent (46.9%) in CMML, 40 we have also tackled the generation of the first CMML PDX models that are robust enough to perform preclinical tests. Combined use of CD34‐enriched leukemia cells, intrafemoral injection, and NSGS mice as recipients enabled us to generate robust CMML PDX models with or without mutations in SRSF2. 41 Notably, the results that AML cells with mutations in spliceosome are more sensitive to H3B‐8800 were recapitulated in the CMML PDX models with or without SRSF2 mutations. 39
FIGURE 4.
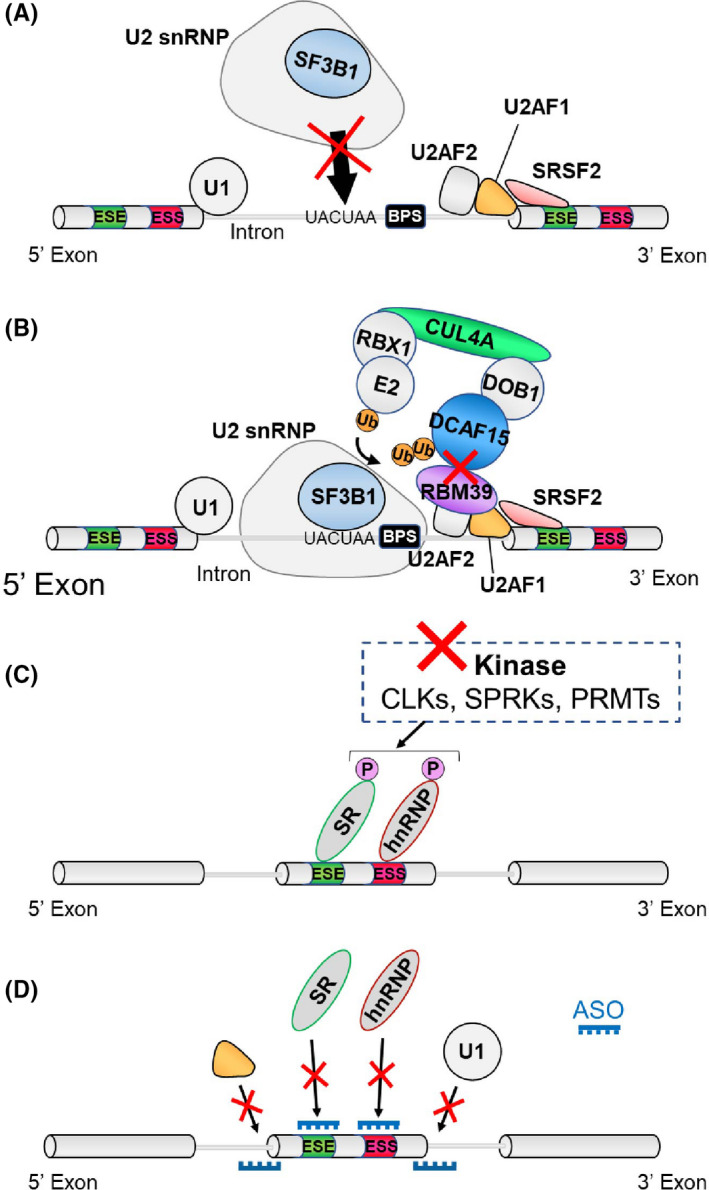
Strategies for targeting splicing alterations in cancers. A, The U2 snRNP inhibitor disrupts U2 snRNP’s ability to recognize the branchpoint region of the intron. B, Sulfonamide compounds physically link RBM39 to the CUL4‐DCAF15 ubiquitin ligase, resulting in ubiquitination of RBM39 and subsequent proteasomal degradation. C, CLK, SRPK, and PRMT inhibitors are promising because the function, cellular localization, and assembly of a variety of splicing proteins depend on post–translational modifications. D, Antisense oligonucleotides modify splicing of specific transcripts by blocking the RNA‐RNA base pairing or protein‐RNA binding interactions
These studies suggested that survival of spliceosome‐mutant cells may depend on the activity of the residual wild‐type allele and that cells heterozygous for spliceosomal mutations may therefore have increased sensitivity to spliceosomal inhibition. 39 Following this preclinical study, clinical trials to evaluate the therapeutic potential of SF3b inhibitors have been initiated. Very recently, the results from the Phase I First‐in‐Human dose escalation study of H3B‐8800 was reported (Table 1). 42 According to this study, H3B‐8800 treatment was associated with mostly low‐grade treatment‐related adverse events and induced red blood cell transfusion independence in some patients, especially in MDS patients with SF3B1 mutations and patients with high pre‐treatment splicing alterations in TMEM14C (a target of SF3B1 splicing encoding a mitochondrial porphyrin transporter). Future efforts, including the exploration of other dosing schedules will be needed to understand the safety and potential therapeutic efficacy of H3B‐8800 and other SF3b inhibitors.
TABLE 1.
Summary of agents targeting splicing
| Target | Molecular target | Agent | Clinical trial |
|---|---|---|---|
| SF3B1 inhibitor | SF3b | H3B‐8800 | NCT02841540 |
| Sulfonamide‐related agents | RBM39 |
E7820 Indisulam/E7070, chloroquinoxaline |
|
| PRMT inhibitor | PRMT5/MEP50 | GSK3326596 | |
| PRMT5 | PF06939999 | NCT03854227 | |
| JNJ‐64619178 | NCT03573310 | ||
| Type I PRMT | GSK3368715 | NCT03666988 | |
| Kinase inhibitor | CLK | CTX‐712 |
JapicCTI‐184188 |
| SRPK | SRPIN340 |
4.2. Sulfonamides and RBM39
Clinical trials of anticancer agents, sulfonamides, have shown only limited efficacy in cancer patients. 43 , 44 However, these trials were performed before the discovery of frequent SF mutations in cancers. Two studies have identified that RBM39, an RBP that plays a key role in regulating splicing, is the molecular target of sulfonamides including E7820, indisulam, and chloroquinoxaline. 45 , 46 Consonant with this, depletion of RBM39 resulted in global splicing changes such as increased exon skipping and intron retention. Sulfonamides function as molecular glue that connects RBM39 to the CUL4‐DCAF15 ubiquitin complex and induces ubiquitination‐mediated proteasomal degradation of RBM39 protein (Figure 4B). Following these observations, our group confirmed that leukemia cells with SF mutations are preferentially sensitive to pharmacological inhibition of RBM39. 47 These results may motivate further clinical trials to address whether anticancer sulfonamides preferentially kill cancer cells with SF mutations.
4.3. Spliceosome inhibition in MYC oncogene‐dependent cancers
Some reports have demonstrated that cancer cells with activated MYC are also preferentially sensitive to pharmacological spliceosome inhibition. For instance, multiple splicing regulatory proteins, such as hnRNP A1 and hnRNP A2, are transcriptionally regulated by MYC and promote tumorigenesis. 48 In addition, an analysis of the MYC transcriptional targets in lymphomas revealed that MYC directly upregulates some snRNP genes and snRNP assembly genes including PRMT5 (an arginine methyltransferase that methylates the Sm proteins of U2 snRNP). 49 Depletion of Prmt5 in MYC‐driven lymphoma cells led to global splicing changes and abrogated lymphomagenesis. Given the recent development of pharmacological inhibitors of PRMT5, 50 these data seem encouraging because activated MYC may create a therapeutic vulnerability to pharmacological inhibition of spliceosome and PRMT5 inhibitors through its effects on global splicing and the dependency on PRMT5.
Related to the above, our comprehensive splicing and transcriptomic analysis across cancers identified that SF3B1 K700E mutations induce a robust splicing change in PPP2R5A in CLL, which encodes one of the B‐subunits of the PP2A serine/threonine phosphatase complex. SF3B1 mutations promote decay of the PPP2R5A transcripts and increase MYC phosphorylation at serine 62, which in turn enhances MYC protein stability. Considering that activated MYC may sensitize cells to pharmacological inhibition of spliceosome, combined treatment with a spliceosome inhibitor with a PP2A activating agent would be a promising therapeutic option for CLL patients bearing SF3B1 K700E mutations.
4.4. Modulating splicing regulatory proteins
SR‐rich proteins belong to a family of RBPs that regulate splicing by recognizing splicing enhancer motifs. Post–translational modifications of SR proteins and other SFs regulate the formation of spliceosome and splicing catalysis (Figure 4C). For example, phosphorylation of SR proteins controls the nuclear shuttling of SR proteins. 51 , 52 , 53 Based on these observations, various kinase inhibitors and methyltransferase inhibitors that block CDC‐like kinases (CLKs), SR protein kinases (SRPKs), and PRMTs have been evaluated as splicing modulators. Among such newly developed agents is an orally available CLK inhibitor called CTX‐712, which inhibits phosphorylation of multiple SR proteins including SRSF3, SRSF4, SRRSF5, and SRSF6. In a preclinical study using 5 AML PDX models, leukemia cells were completely abrogated in 2 cases, including 1 SRSF2 mutant model. 54 Another class of compound to modulate splicing is the PRMT inhibitors. As mentioned above, genetic ablation or chemical inhibition of PRMT5 (which catalyzes symmetric arginine demethylation) leads to splicing inhibition and anticancer effects across cancer types. 49 , 55 Considering these observations, it would be important to determine whether inhibition of type I PRMT enzymes (which catalyze asymmetric arginine dimethylation) causes splicing alterations similar to PRMT5 inhibition. Of note, some of the PRMT5 inhibitors (such as GSK3326595, PF06939999 and JNJ‐64619178) and at least 1 type I PRMT inhibitor (GSK3368715) are in clinical trials for patients with relapsed/refractory malignancies (Table 1). Interestingly, a recent study has shown that the antitumor effects of the type I inhibitor synergizes with PRMT5 inhibition, 56 which is consistent with the notion that each has distinct substrates within the spliceosome. These studies will provide an opportunity to assess the safety and efficacy of PRMT inhibition in vivo.
4.5. Oligonucleotide‐based approach
Another approach to therapeutically target aberrant splicing in cancers is to design and use specific ASOs (or antisense SSOs) that bind complementarily to RNA through base pairing (Figure 4D). This class of therapies aims to target the RNA for degradation or to be used to affect splicing via hybridization to RNA, and has been recently approved in the United States for the treatment of neurodegenerative diseases such as spinal muscular atrophy 57 and Duchenne muscular dystrophy. 58
Although these approaches have not yet been applied successfully to cancers, there have been some promising experimental results for modulating STAT3 or Bcl‐x splicing in which switching in the usage of a specific exon by ASO treatment resulted in different or opposite functions from the original transcripts. For example, an ASO targeting the splicing enhancer of STAT3 successfully modified alternative splicing of STAT3 to enforce exon 23 skipping. 59 This caused a shift from the α‐isoform to the β‐isoform of STAT3, consequently increasing cell death and tumor regression. Similarly, an ASO targeting the alternative splice site of Bcl‐x caused exon 6 skipping, resulting in the expression of proapoptotic isoform Bcl‐xS that suppressed tumor load in preclinical models in vivo. 60 Furthermore, it has been revealed that diverse SF3B1 mutations converged on repression of BRD9, which is a core component of the non‐canonical BAF chromatin‐remodeling complex, and that treatment with ASO targeting cryptic exon inclusion of BRD9 increased the level of BRD9 protein and suppressed tumor growth. 61
Technologically, synthetic oligonucleotides composed of subunits with a morpholine ring (termed morpholino) were developed to improve the stability of ASOs. 62 The morpholino lacks the negatively charged backbone of traditional ASOs that may nonspecifically bind to other components of the cell and therefore may reduce the toxicity of ASOs. This technology is also suitable for targeting splicing because it is not recognized by RNase H and, therefore, does not cause direct degradation of the targeted pre‐mRNA.
Despite these technological advances, the utility of ASOs in the treatment of cancers remains challenging due to the lack of sufficient understanding of which mis‐splicing events are key to initiating and/or maintaining specific types of cancer, and to the lack of technological progress to efficiently deliver ASOs to the cancer cells in vivo.
5. CONCLUSION
Although our understanding of the genomics, molecular biology, and therapeutic implications of altered RNA splicing in cancer has been greatly improved since the frequent SF mutations in cancers were identified in 2011, standard treatment strategies targeting cancers bearing splicing alterations have not yet been established. In addition, the full contribution of aberrant RNA splicing to cancer pathogenesis has not been elucidated. The major direction of the field so far is the use of splicing modulators aimed at synthetic lethality in cancers with SF mutations. The tactics to inhibit regulatory proteins, such as CLKs, SRPKs and PRMTs, and RBPs including RBM39, are also being explored as therapeutic avenues. Although emerging technologies and the rapid development of treatment strategies using ASOs still have many challenges to be resolved before clinical use, these therapeutic strategies will expand the treatment options for cancers with aberrant splicing. Interestingly, the recent pan‐cancer TCGA splicing analysis suggested that cancer‐specific changes to RNA splicing may be an additional source of neoepitopes. 63 In fact, an elegant study has shown that the pharmacologic perturbation of splicing suppresses tumor growth in vivo, depending on host T cells and tumoral MHC I‐presented peptides. 64 The authors provided evidence that pharmacological modulation of global splicing enhances the effects of immune checkpoint blockade, providing a way to improve response to checkpoint blockade. We are just beginning to understand how neoepitopes generated by dysregulated alternative splicing through SF mutations and pharmacological perturbation overcomes immune tolerance to elicit an anticancer response. In summary, there still remains many unsolved problems in terms of the pathogenesis of spliceosome‐mutant cancers and the development of therapeutic avenues for cancers with aberrant splicing. In parallel, the ongoing efforts to modulate splicing will hopefully address the molecular and clinical questions of whether pharmacological intervention in global splicing is efficacious and safe in patients with cancers.
DISCLOSURE
The authors have no conflict of interest.
ACKNOWLEDGMENTS
AY acknowledges support from the Japan Society for the Promotion of Science (JSPS) Home‐Returning Researcher Development Research (grant number 19K24691), Grant‐in‐Aid for Scientific Research (A) (grant number 21H04828), AMED (Japan Agency for Medical Research and Development) (grant number JP20jm0210085h0002), National Cancer Center Research and Development Funds (2020‐A‐2), the Uehara Memorial Foundation, Kato Memorial Bioscience Foundation, the Tokyo Biochemical Research Foundation, the NOVARTIS Foundation (Japan) for the Promotion of Science, Japanese Foundation for Multidisciplinary Treatment of Cancer, the Japanese Society of Hematology, Brain Science Foundation, The Japan Neurosurgical Society, Narishige Neuroscience Research Foundation and Leukemia & Lymphoma Society CDP Achievement Award.
Yamauchi H, Nishimura K, Yoshimi A. Aberrant RNA splicing and therapeutic opportunities in cancers. Cancer Sci.2022;113:373–381. doi: 10.1111/cas.15213
REFERENCES
- 1. Graubert TA, Shen D, Ding LI, et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet. 2011;44:53‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Papaemmanuil E, Cazzola M, Boultwood J, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365:1384‐1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yoshida K, Sanada M, Shiraishi Y, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64‐69. [DOI] [PubMed] [Google Scholar]
- 4. Bailey P, Chang DK, Nones K, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47‐52. [DOI] [PubMed] [Google Scholar]
- 5. Imielinski M, Berger A, Hammerman P, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107‐1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Martin M, Maßhöfer L, Temming P, et al. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat Genet. 2013;45:933‐936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nik‐Zainal S, Davies H, Staaf J, et al. Landscape of somatic mutations in 560 breast cancer whole‐genome sequences. Nature. 2016;534:47‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Seiler M, Peng S, Agrawal AA, et al. Somatic mutational landscape of splicing factor genes and their functional consequences across 33 cancer types. Cell Rep. 2018;23:282‐296 e284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang Z, Burge CB. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA. 2008;14:802‐813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yoshimi A, Abdel‐Wahab O. Molecular pathways: understanding and targeting mutant spliceosomal proteins. Clin Cancer Res. 2017;23:336‐341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wahl MC, Will CL, Lührmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136:701‐718. [DOI] [PubMed] [Google Scholar]
- 12. Matera AG, Wang Z. A day in the life of the spliceosome. Nat Rev Mol Cell Biol. 2014;15:108‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Scamborova P, Wong A, Steitz JA. An intronic enhancer regulates splicing of the twintron of Drosophila melanogaster prospero pre‐mRNA by two different spliceosomes. Mol Cell Biol. 2004;24:1855‐1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yoshimi A, Abdel‐Wahab O. Splicing factor mutations in MDS RARS and MDS/MPN‐RS‐T. Int J Hematol. 2017;105:720‐731. [DOI] [PubMed] [Google Scholar]
- 15. Malcovati L, Karimi M, Papaemmanuil E, et al. SF3B1 mutation identifies a distinct subset of myelodysplastic syndrome with ring sideroblasts. Blood. 2015;126:233‐241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang L, Lawrence MS, Wan Y, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med. 2011;365:2497‐2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Quesada V, Conde L, Villamor N, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2011;44:47‐52. [DOI] [PubMed] [Google Scholar]
- 18. Darman R, Seiler M, Agrawal A, et al. Cancer‐associated SF3B1 hotspot mutations induce cryptic 3' splice site selection through use of a different branch point. Cell Rep. 2015;13:1033‐1045. [DOI] [PubMed] [Google Scholar]
- 19. DeBoever C, Ghia EM, Shepard PJ, et al. Transcriptome sequencing reveals potential mechanism of cryptic 3' splice site selection in SF3B1‐mutated cancers. PLoS Comput Biol. 2015;11:e1004105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu Z, Yoshimi A, Wang J, et al. Mutations in the RNA splicing factor SF3B1 promote tumorigenesis through MYC stabilization. Cancer Discov. 2020;10:806‐821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Obeng E, Chappell R, Seiler M, et al. Physiologic expression of Sf3b1(K700E) causes impaired erythropoiesis, aberrant splicing, and sensitivity to therapeutic spliceosome modulation. Cancer Cell. 2016;30:404‐417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cretu C, Schmitzová J, Ponce‐Salvatierra A, et al. Molecular architecture of SF3b and structural consequences of its cancer‐related mutations. Mol Cell. 2016;64:307‐319. [DOI] [PubMed] [Google Scholar]
- 23. Zhang J, Ali AM, Lieu YK, et al. Disease‐causing mutations in SF3B1 alter splicing by disrupting interaction with SUGP1. Mol Cell. 2019;76:82‐95 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Daubner GM, Clery A, Jayne S, Stevenin J, Allain FH. A syn‐anti conformational difference allows SRSF2 to recognize guanines and cytosines equally well. EMBO J. 2012;31:162‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim E, Ilagan J, Liang Y, et al. SRSF2 mutations contribute to myelodysplasia by mutant‐specific effects on exon recognition. Cancer Cell. 2015;27:617‐630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yoshimi A, Lin K‐T, Wiseman DH, et al. Coordinated alterations in RNA splicing and epigenetic regulation drive leukaemogenesis. Nature. 2019;574:273‐277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang J, Lieu YK, Ali AM, et al. Disease‐associated mutation in SRSF2 misregulates splicing by altering RNA‐binding affinities. Proc Natl Acad Sci USA. 2015;112:E4726‐4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cancer Genome Atlas Research N , Ley TJ, Miller C, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059‐2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brooks AN, Choi PS, de Waal L, et al. A pan‐cancer analysis of transcriptome changes associated with somatic mutations in U2AF1 reveals commonly altered splicing events. PLoS One. 2014;9:e87361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ilagan JO, Ramakrishnan A, Hayes B, et al. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res. 2015;25:14‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Seiler M, Peng S, Agrawal AA, et al. Somatic mutational landscape of splicing factor genes and their functional consequences across 33 cancer types. Cell Rep. 2018;23:282‐296 e284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kotake Y, Sagane K, Owa T, et al. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat Chem Biol. 2007;3:570‐575. [DOI] [PubMed] [Google Scholar]
- 33. Kaida D, Motoyoshi H, Tashiro E, et al. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre‐mRNA. Nat Chem Biol. 2007;3:576‐583. [DOI] [PubMed] [Google Scholar]
- 34. Fan L, Lagisetti C, Edwards CC, Webb TR, Potter PM. Sudemycins, novel small molecule analogues of FR901464, induce alternative gene splicing. ACS Chem Biol. 2011;6:582‐589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bonnal S, Vigevani L, Valcarcel J. The spliceosome as a target of novel antitumour drugs. Nat Rev Drug Discov. 2012;11:847‐859. [DOI] [PubMed] [Google Scholar]
- 36. Effenberger KA, Urabe VK, Jurica MS. Modulating splicing with small molecular inhibitors of the spliceosome. Wiley Interdisciplinary Rev RNA. 2017;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Folco EG, Coil KE, Reed R. The anti‐tumor drug E7107 reveals an essential role for SF3b in remodeling U2 snRNP to expose the branch point‐binding region. Genes Dev. 2011;25:440‐444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee S‐W, Dvinge H, Kim E, et al. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat Med. 2016;22:672‐678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Seiler M, Yoshimi A, Darman R, et al. H3B–8800, an orally available small‐molecule splicing modulator, induces lethality in spliceosome‐mutant cancers. Nat Med. 2018;24:497‐504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Meggendorfer M, Roller A, Haferlach T, et al. SRSF2 mutations in 275 cases with chronic myelomonocytic leukemia (CMML). Blood. 2012;120:3080‐3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yoshimi A, Balasis ME, Vedder A, et al. Robust patient‐derived xenografts of MDS/MPN overlap syndromes capture the unique characteristics of CMML and JMML. Blood. 2017;130:397‐407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Steensma DP, Wermke M, Klimek VM, et al. Phase I first‐in‐human dose escalation study of the oral SF3B1 modulator H3B–8800 in myeloid neoplasms. Leukemia. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Haddad RI, Weinstein LJ, Wieczorek TJ, et al. A phase II clinical and pharmacodynamic study of E7070 in patients with metastatic, recurrent, or refractory squamous cell carcinoma of the head and neck: modulation of retinoblastoma protein phosphorylation by a novel chloroindolyl sulfonamide cell cycle inhibitor. Clin Cancer Res. 2004;10:4680‐4687. [DOI] [PubMed] [Google Scholar]
- 44. Rigas JR, Tong WP, Kris MG, Orazem JP, Young CW, Warrell RP Jr. Phase I clinical and pharmacological study of chloroquinoxaline sulfonamide. Cancer Res. 1992;52:6619‐6623. [PubMed] [Google Scholar]
- 45. Han T, Goralski M, Gaskill N, et al. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science. 2017;356. [DOI] [PubMed] [Google Scholar]
- 46. Uehara T, Minoshima Y, Sagane K, et al. Selective degradation of splicing factor CAPERalpha by anticancer sulfonamides. Nat Chem Biol. 2017;13:675‐680. [DOI] [PubMed] [Google Scholar]
- 47. Wang E, Lu SX, Pastore A, et al. Targeting an RNA‐binding protein network in acute myeloid leukemia. Cancer Cell. 2019;35:369‐384 e367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. David CJ, Chen M, Assanah M, Canoll P, Manley JL. HnRNP proteins controlled by c‐Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2010;463:364‐368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Koh CM, Bezzi M, Low DHP, et al. MYC regulates the core pre‐mRNA splicing machinery as an essential step in lymphomagenesis. Nature. 2015;523:96‐100. [DOI] [PubMed] [Google Scholar]
- 50. Chan‐Penebre E, Kuplast KG, Majer CR, et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat Chem Biol. 2015;11:432‐437. [DOI] [PubMed] [Google Scholar]
- 51. Cho S, Hoang A, Sinha R, et al. Interaction between the RNA binding domains of Ser‐Arg splicing factor 1 and U1–70K snRNP protein determines early spliceosome assembly. Proc Natl Acad Sci USA. 2011;108:8233‐8238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tacke R, Chen Y, Manley JL. Sequence‐specific RNA binding by an SR protein requires RS domain phosphorylation: creation of an SRp40‐specific splicing enhancer. Proc Natl Acad Sci USA. 1997;94:1148‐1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Xiao SH, Manley JL. Phosphorylation‐dephosphorylation differentially affects activities of splicing factor ASF/SF2. EMBO J. 1998;17:6359‐6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yoda A, Morishita DM, Satoh A, et al. CTX‐712, a novel Clk inhibitor targeting myeloid neoplasms with SRSF2 mutation. Blood J. 2019;134(Suppl_1):404. [Google Scholar]
- 55. Banasavadi‐Siddegowda YK, Welker AM, An M, et al. PRMT5 as a druggable target for glioblastoma therapy. Neuro‐Oncology. 2018;20:753‐763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fedoriw A, Rajapurkar SR, O'Brien S, et al. Anti‐tumor activity of the type I PRMT inhibitor, GSK3368715, synergizes with PRMT5 inhibition through MTAP loss. Cancer Cell. 2019;36:100‐114 e25. [DOI] [PubMed] [Google Scholar]
- 57. Touznik A, Maruyama R, Hosoki K, Echigoya Y, Yokota T. LNA/DNA mixmer‐based antisense oligonucleotides correct alternative splicing of the SMN2 gene and restore SMN protein expression in type 1 SMA fibroblasts. Sci Rep. 2017;7:3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cirak S, Arechavala‐Gomeza V, Guglieri M, et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open‐label, phase 2, dose‐escalation study. Lancet. 2011;378:595‐605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zammarchi F, de Stanchina E, Bournazou E, et al. Antitumorigenic potential of STAT3 alternative splicing modulation. Proc Natl Acad Sci USA. 2011;108:17779‐17784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bauman JA, Li SD, Yang A, Huang L, Kole R. Anti‐tumor activity of splice‐switching oligonucleotides. Nucleic Acids Res. 2010;38:8348‐8356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Inoue D, Chew G‐L, Liu BO, et al. Spliceosomal disruption of the non‐canonical BAF complex in cancer. Nature. 2019;574:432‐436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Corey DR, Abrams JM. Morpholino antisense oligonucleotides: tools for investigating vertebrate development. Genome Biol. 2001;2:REVIEWS1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kahles A, Lehmann KV, Toussaint NC, et al. Comprehensive analysis of alternative splicing across tumors from 8,705 patients. Cancer Cell. 2018;34:211‐224 e216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lu SX, De Neef E, Thomas JD, et al. Pharmacologic modulation of RNA splicing enhances anti‐tumor immunity. Cell. 2021;184:4032‐4047 e4031. [DOI] [PMC free article] [PubMed] [Google Scholar]