

Abstract
We previously examined the utility of rituximab‐bendamustine (RB) in patients with follicular lymphoma (FL) exhibiting less than optimal responses to 2 cycles of the R‐CHOP chemotherapy regimen. The aim of this study was to identify molecular biomarkers that can predict prognosis in RB‐treated patients in the context of the prospective cohort. We first analyzed the mutational status of 410 genes in diagnostic tumor specimens by target capture and Sanger sequencing. CREBBP, KMT2D, MEF2B, BCL2, EZH2, and CARD11 were recurrently mutated as reported before, however none was predictive for progression‐free survival (PFS) in the RB‐treated patients (n = 34). A gene expression analysis by nCounter including 800 genes associated with carcinogenesis and/or the immune response showed that expression levels of CD8+ T‐cell markers and half of the genes regulating Th1 and Th2 responses were significantly lower in progression of disease within the 24‐mo (POD24) group (n = 8) than in the no POD24 group (n = 31). Collectively, we selected 10 genes (TBX21, CXCR3, CCR4, CD8A, CD8B, GZMM, FLT3LG, CD3E, EOMES, GZMK), and generated an immune infiltration score (IIS) for predicting PFS using principal component analysis, which dichotomized the RB‐treated patients into immune IIShigh (n = 19) and IISlow (n = 20) groups. The 3‐y PFS rate was significantly lower in the IISlow group than in the IIShigh group (50.0% [95% CI: 27.1‐69.2%] vs. 84.2% [95% CI: 58.7‐94.6%], P = .0237). Furthermore, the IIS was correlates with absolute lymphocyte counts at diagnosis (r = 0.460, P = .00355). These results suggest that the T‐cell‐associated immune markers could be useful to predict prognosis in RB‐treated FL patients. (UMIN:000 013 795, jRCT:051 180 181)
Keywords: bendamustine, follicular lymphoma, lymphopenia, POD24, tumor microenvironment
The expression levels of T‐cell‐associated immune markers were significantly lower in RB‐treated patients with POD24 than those without POD24. Furthermore, FL patients were classified into infiltration low and high scores based on the expression of T‐cell‐associated genes, which was useful to predict the prognosis of RB‐treated FL patients.

Abbreviations
- ALCs
absolute lymphocyte counts
- B
bendamustine
- CR
complete response
- Cru
complete response unconfirmed
- CTLs
cytotoxic T cells
- FDCs
follicular dendritic cells
- FFPE
formalin‐fixed paraffin‐embedded
- FL
follicular lymphoma
- FLIPI
Follicular lymphoma International Prognostic Index
- G
obinutuzumab
- GELF
Groupe d'Etude des Lymphomes Folliculaires
- GEP
gene expression profiling
- HR
hazard ratio
- IIS
immune infiltration score
- infilhigh
immune infiltrationhigh
- infillow
immune infiltrationlow
- OS
overall survival
- PFS
progression‐free survival
- POD24
progression of disease within 24 mo
- R
rituximab
- RB
rituximab‐bendamustine
- ROC
receiver operating characteristic
- SNVs
single nucleotide variants
- TAMs
tumor‐associated macrophages
- Tfh
T follicular helper
- TME
tumor microenvironment
- Treg
regulatory T
1. INTRODUCTION
Follicular lymphoma is the most common indolent subtype of B‐cell non‐Hodgkin lymphomas. 1 The majority of FL patients present with advanced stages, 1 and those with high tumor burden and/or disease‐related symptoms require immunochemotherapy.
To date, several chemotherapy regimens, including CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), CVP (cyclophosphamide, vincristine, and prednisone), and bendamustine (B) in combination with anti‐CD20 monoclonal antibodies such as rituximab (R) or obinutuzumab (G) 2 , 3 have been recommended as front‐line therapies in several guidelines. 4 , 5 , 6 , 7 , 8 Most of the FL patients receiving these front‐line immunochemotherapy regimens can obtain long‐term survival benefit. 9 However, c. 10%‐20% of the patients experience progression of disease within 24 mo of diagnosis or treatment initiation (which is called POD24), and these patients have a substantially increased risk of death within 5 y after diagnosis. 9 , 10 , 11 , 12
Given the heterogeneity of disease courses, several clinical risk stratification models, including FLIPI‐1 and FLIPI‐2, PRIMA‐PI, and FLEX were proposed to predict the clinical outcomes of FL patients such as PFS and OS. 13 , 14 , 15 , 16 However, most of these models have limited value to predict POD24. In addition, accumulated findings from genetic and GEP analyses showed that several molecular risk models mainly based on the intrinsic tumor properties, such as m7‐FLIPI, 17 POD24‐PI 18 and 23‐GEP score, 19 , 20 were useful tools to identify FL patients at high or low risk of progression after treatment with immunochemotherapy. In addition, a gene expression signature based on the extrinsic tumor environment, particularly focusing on the TME of FL, was established to predict POD24. 11 However, most of these models were mainly based on the data from the patients who received R‐CHOP or R‐CVP, and few reports are available to predict the prognosis of FL patients receiving B‐based regimens.
We previously conducted a multicenter prospective phase 2 (CONVERT) trial, and demonstrated the utility of RB in patients with advanced FL exhibiting less than optimal responses to the initial 2 cycles of R‐CHOP therapy. 21 In addition, we reported that peripheral lymphopenia (<869/μL) before treatment was an independent poor prognostic factor for RB‐treated FL patients in both our trial and validation cohorts. 21 The main object of this study was to identify molecular biomarker(s) that could predict outcomes of the RB‐treated FL patients in the context of the prospective cohort by genetic and GEP analyses.
2. MATERIALS AND METHODS
2.1. Study design and patients
The design and characteristics of the registered patients in the CONVERT trial have been described previously. 21 All patients initially received 2 cycles of standard R‐CHOP. Patients who achieved CR/CR unconfirmed (CRu) according to Cheson's morphological criteria 1999 22 (defined as an optimal response [OR] group) further received 4‐6 cycles of R‐CHOP treatment. The remaining patients who failed to achieve CR/CRu (defined as a non‐OR [nOR] group), subsequently received 6 cycles of RB (R 375 mg/m2 d1, B 90 mg/m2 d1‐2) every 28 d.
This study was designed and conducted according to the Declaration of Helsinki and was registered in UMIN (000 013 795) and jRCT (051 180 181). All patients provided written informed consent. The protocol, informed consent forms, and any amendments were approved by the institutional review boards of each hospital and certified review board.
2.2. Target capture sequencing assay
DNA was extracted from FFPE samples using a DNA storm kit (Cell Data Sciences, Inc). The targeted DNA library for panel sequencing that was comprised of c. 1.65 Mb coding regions of all exons in 409 genes (Table S1) was constructed using an Oncomine Tumor Mutation Load Assay (Thermo Fisher Scientific) according to the manufacturer's protocol. The purified libraries were pooled and then sequenced with an Ion Torrent S5 instrument and Ion 550 Chip Kit (Thermo Fisher Scientific). DNA sequencing data were accessed through the Torrent Suite v.5.10 program (Thermo Fisher Scientific). Reads were aligned against the hg19 human reference genome, and variants were analyzed with the use of Ion reporter v.5.10 (Thermo Fisher Scientific). Raw variant calls were manually checked using the Integrative Genomics Viewer (IGV; Broad Institute, MA). In this analysis, we defined the following criteria for the sequence quality: (1) the uniformity of amplicon coverage of >60%, and (2) the minimum quality criterion was 80% of target bases with ≥100× sequencing coverage. The mutations were filtered with the exclusion of: (1) Fisher exact test, P‐value >10‐4, (2) Phred QUAL score of <99, and (3) allele frequency in tumor <0.025, and missense SNVs with an allele frequency of 0.45‐0.55 in copy‐neutral regions, unless they were listed in the COSMIC database (v.70) or reported to be mutated in FL. Germline mutations were excluded using the NCBI dbSNP build 131 and UCSC genome browser database. 23 , 24
2.3. Sanger sequencing analysis
Exons 2 and 3 of MEF2B were analyzed using the PCR primers designed with Primer3 software. The MEF2B coding exons 2 and 3 were amplified using PCR. After visual confirmation of amplification, the PCR product was submitted for Sanger sequence analysis as previously described. 25 The sequences of the PCR primers used in this study are shown in Table S2.
2.4. The m7‐FLIPI score calculation
The m7‐FLIPI score was calculated using the calculator presented at the German Low‐Grade Lymphoma Study Group official internet site (www.glsg.de/m7‐flipi/). The same cut‐off level as used in the original publication was applied to determine high‐risk and low‐risk categories (m7‐FLIPI scores ≥0.8 and <0.8, respectively). 17
2.5. Gene expression profiling analysis
RNA was extracted from FFPE samples using an RNA storm kit (Cell Data Sciences, Inc). Details of the nCounter assay (NanoString Technologies, Seattle, WA, USA) have been reported previously. 24 , 26 In this study, a customized pan‐cancer immune‐profiling panel (NanoString Technologies), which consists of 770 genes related to cancer or immune cells and an additional 30 genes (Table S3), was used for nCounter‐based gene expression measurements. The data were analyzed using nSolver 4.0 software (NanoString) and JMP version 13.0 software (SAS Institute).
2.6. Immune infiltration score calculation
To generate an IIS for predicting PFS, we performed a principal component analysis, which used orthogonal rotation to extract the components whose eigenvalues were greater than 1.0 (SPSS statistics v.24.0). Subsequently, a multivariate Cox proportional hazards analysis for PFS was performed with the extracted components as explanatory variables. Finally, the IIS was calculated as the sum of the score matrix of each gene. Expression levels from selected genes were multiplied by z‐score. ROC analyses were performed to determine the optimal cut‐off values of the IIS to predict PFS by the Youden index calculation algorithm. The formula of the IIS was as followed: (CD8A × 0.161 + CD3E × 0.160 + CD8B × 0.157 + EOMES × 0.157 + GZMK × 0.153 + CXCR3 × 0.139 + GZMM × 0.132 + FLT3LG × 0.121 + TBX21 × 0.075 + CCR4 × 0.072).
2.7. Statistical analysis
The cut‐off date for this analysis was December 31, 2019. The survival analysis was performed using a Kaplan‐Meier method, which was evaluated by log‐rank test. Fisher exact test and Benjamini‐Hochberg correction for multiple testing were used for between‐category data comparisons. For the comparison of 2 continuous variables, data were tested by Wilcoxon rank sum test. Spearman correlation coefficients were estimated as continuous variables. Statistical analyses were performed with R 4.1.0 software (The R Foundation for Statistical Computing), and a P‐value < .05 was considered statistically significant.
3. RESULTS
3.1. Patient characteristics and clinical outcomes
In total, 56 patients initially received 2 cycles of R‐CHOP. Then, 13 patients (23.2%), who achieved CR/CRu (OR group), continued R‐CHOP for further 4‐6 cycles. The remaining 43 patients (76.8%), who were judged as nOR, received RB as the protocol treatment. The FFPE samples were available for extraction of DNA and/or RNA in 50/56 cases (89.3%, [OR n = 10, nOR n = 40]) for this study. The baseline characteristics of these 50 cases are shown in Table 1. The median treatment cycle of RB in the nOR group (n = 40) was 6 cycles (range, 2‐6), and 30/40 cases (75.0%) completed 6 cycles of RB. At a median follow‐up time of 47.4 mo (range: 3.20‐79.3), the PFS and OS rates at 3 y in all cases (n = 50) were 71.0% (95% CI: 56.0%‐81.7%) and 93.5% (95% CI: 81.2%‐97.9%), respectively (Figure S1A, B). The 3‐y PFS rates were 87.5% in the OR group (95% CI: 38.7%‐98.1%) and 67.5% in the nOR group (95% CI: 50.7%‐9.7%), respectively, with no statistically significant difference (P = .213).
TABLE 1.
Patient characteristics
| All (n = 50) | OR (N = 10) | nOR (N = 40) | P‐value | |
|---|---|---|---|---|
| Age (y) | ||||
| Median [range] | 62.5 [36.0, 78.0] | 64.0 [54.0, 78.0] | 65.0 [36.0, 76.0] | .622 |
| Sex n(%) | ||||
| Female | 29 (58.0) | 6 (60.0) | 23 (57.5) | 1 |
| Male | 31 (42.0) | 4 (40.0) | 17 (42.5) | |
| Grade n (%) | ||||
| 1 | 12 (24.0) | 1 (10.0) | 11 (29.7) | .532 |
| 2 | 23 (46.0) | 6 (60.0) | 17 (45.9) | |
| 3a | 12 (24.0) | 3 (30.0) | 9 (24.3) | |
| Stage n (%) | ||||
| Ⅱ | 3 (6.00) | 0 (00.0) | 3 (7.50) | .036 |
| Ⅲ | 17 (34.0) | 7 (70.0) | 10 (25.0) | |
| Ⅳ | 30 (60.0) | 3 (30.0) | 27 (67.5) | |
| B symptoms n (%) | 9 (18.0) | 3 (30.0) | 6 (18.2) | 1 |
| Bone marrow involvement n (%) | 27 (54.0) | 4 (30.8) | 23 (57.5) | .164 |
| Bulky disease >7 cm n (%) | 18 (36.0) | 0 (0.00) | 18 (45.0) | .009 |
| GELF criteria | ||||
| Low | 2 (4.00) | 1 (10.0) | 1 (2.50) | .363 |
| FLIPI n (%) | ||||
| Low | 3 (6.00) | 1 (10.0) | 2 (5.00) | .07 |
| Intermediate | 22 (44.0) | 7 (70.0) | 15 (37.5) | |
| High | 25 (50.0) | 2 (20.0) | 23 (57.5) | |
| FLIPI‐2 n (%) | ||||
| Low | 0 (0.00) | 0 (0.00) | 0 (0.00) | .308 |
| Intermediate | 27 (54.0) | 7 (70.0) | 20 (50.0) | |
| High | 23 (46.0) | 3 (30.0) | 20 (50.0) | |
We next investigated whether FLIPI could be applied in the nOR group (n = 40). The proportions of patients in the low‐, intermediate‐, and high‐risk groups by FLIPI were 5.0% (2/40) and 37.5% (15/40), and 57.5% (23/40), respectively (Table 1). The high‐risk group tended to have an inferior 3‐y PFS rate (60.9%, [95% CI: 38.3%‐77.4%]) compared with the low‐risk group (100% [95% CI: NA]) and the intermediate‐risk group (73.3%, [95% CI: 43.6%‐89.1%]), but without a statistically significant difference (P = .323) (Figure S1C).
3.2. Mutational landscape of the whole population
To investigate the mutational landscape in this cohort, we performed the target capture sequence using tumor samples from the above‐described 50 cases. Eight cases (OR n = 2, nOR n = 6) were excluded from this analysis because of their poor DNA qualities. Therefore, 42 cases (OR n = 8, nOR n = 34) were analyzed on the targeted panel for 409 genes, in which most of the recurrently mutated genes in FL were included except MEF2B. MEF2B encodes a transcriptional activator and is mutated in 10%‐20% of FL and its mutational status is applied in m7‐FLIPI. 27 Therefore, we analyzed its mutation status by Sanger sequencing independently. In this analysis, we focused on exon 2 and exon 3 of MEF2B, because most of MEF2B mutations in FL were scattered around these regions. 28 As a result, MEF2B mutations were identified in 13/42 cases (31.0%), all consisting of L67R in the exon 3, in which the mutated site was reported to play an important role in the interaction with EP300 proteins. 28 The double mutations with L67R and G2R in exon 2 were identified in 1 case (Figure 1A and Table S4).
FIGURE 1.
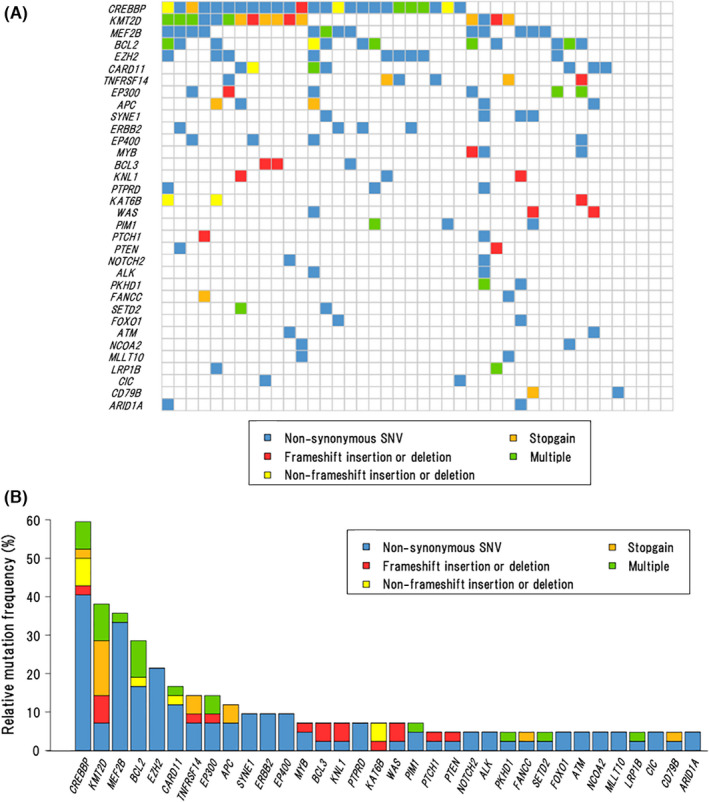
Landscape of somatic mutations. A, Co‐mutation plot showing the spectrum of recurrently mutated genes across all cases (n = 42). B, Frequencies and types of somatic mutations in 34 recurrently mutated genes (found in ≥2 cases) for all cases (n = 42)
When the results from target capture sequencing and Sanger sequencing were combined, a total of 34 genes was found to be recurrently mutated (in ≥2 cases), including 107 non‐synonymous SNVs, 19 multiple mutations, 16 frameshift insertions or deletions, 13 stop‐gain mutations, and 7 non‐frameshift insertions or deletions. Overall, 37/42 cases (88.1%) had at least 1 coding mutation within 410 genes with a median of 4 mutations per patient (range 0‐10) (Figure 1A), and the average depth of sequencing coverage in the 37 cases was 882× (103‐3999×). The most recurrently mutated genes were CREBBP (59.5%), KMT2D (38.1%), MEF2B (31.0%), BCL2 (28.6%), EZH2 (21.4%), CARD11 (16.7%), TNFRSF14, and EP300 (14.3% each), APC (11.9%), with the landscape closely resembling the previous reports 1 , 29 , 30 (Figure 1B). Positions and types of somatic mutations in these genes are shown in Figure S2 and Table S4.
3.3. Association of mutation profile with clinical outcomes in the nOR group
Among recurrently mutated genes, their mutation frequencies (in ≥10%) in the OR and nOR groups are shown in Table 2. Next, we analyzed whether mutation status influenced the outcomes in the nOR group (n = 34). As shown in Figure 2A, the mutation status of the specific gene did not influence PFS significantly in univariate analyses. We also evaluated the influence of gene mutations on POD24; however, there were no statistically significant differences between the 2 groups (Figure 2B). Interestingly, no POD24 event occurred in RB‐treated patients with CARD11 mutations in the nOR group. In addition, the cases with CARD11 mutations tended to have a better 3‐y PFS rate than unmutated cases but with no statistically significant difference (100% vs. 59.3%, P = .174) (Figure 2C).
TABLE 2.
Mutation frequencies of recurrently mutated genes in all cases, OR group, and nOR group
| Genes | Mutation presence | All (n = 42) | OR (N = 8) | nOR (N = 34) | P‐value |
|---|---|---|---|---|---|
| APC n (%) | Absence | 37 (88.1) | 7 (87.5) | 30 (88.2) | 1 |
| Presence | 5 (11.9) | 1 (12.5) | 4 (11.8) | ||
| ARID1A n (%) | Absence | 40 (95.2) | 8 (100.0) | 32 (94.1) | 1 |
| Presence | 2 (4.80) | 0 (0.00) | 2 (5.90) | ||
| BCL2 n (%) | Absence | 30 (71.4) | 7 (87.5) | 23 (67.6) | .402 |
| Presence | 12 (28.6) | 1 (12.5) | 11 (32.4) | ||
| CARD11 n (%) | Absence | 35 (83.3) | 8 (100.0) | 27 (79.4) | .312 |
| Presence | 7 (16.7) | 0 (0.00) | 7 (20.6) | ||
| CREBBP n (%) | Absence | 17 (40.5) | 5 (62.5) | 12 (35.3) | .235 |
| Presence | 25 (59.5) | 3 (37.5) | 22 (64.7) | ||
| EP300 n (%) | Absence | 36 (85.7) | 8 (100.0) | 28 (82.4) | .576 |
| Presence | 6 (14.3) | 0 (0.00) | 6 (17.6) | ||
| EP400 n (%) | Absence | 38 (90.5) | 8 (100.0) | 30 (88.2) | .572 |
| Presence | 4 (9.50) | 0 (0.00) | 4 (11.8) | ||
| ERBB2 n (%) | Absence | 38 (90.5) | 7 (87.5) | 31 (91.2) | 1 |
| Presence | 4 (9.50) | 1 (12.5) | 3 (8.80) | ||
| EZH2 n (%) | Absence | 33 (78.6) | 8 (100.0) | 25 (73.5) | .168 |
| Presence | 9 (21.4) | 0 (0.00) | 9 (26.5) | ||
| FOXO1 n (%) | Absence | 40 (95.2) | 8 (100.0) | 32 (94.1) | 1 |
| Presence | 2 (4.80) | 0 (0.00) | 2 (5.9) | ||
| KMT2D n (%) | Absence | 26 (61.9) | 7 (87.5) | 19 (55.9) | .127 |
| Presence | 16 (38.1) | 1 (12.5) | 15 (44.1) | ||
| MEF2B n (%) | Absence | 29 (69.0) | 5 (62.5) | 24 (70.6) | .686 |
| Presence | 13 (31.0) | 3 (37.5) | 10 (29.4) | ||
| SYNE1 n (%) | Absence | 38 (90.5) | 7 (87.5) | 31 (91.2) | 1 |
| Presence | 4 (9.50) | 1 (12.5) | 3 (8.80) | ||
| TNFRSF14 n (%) | Absence | 36 (85.7) | 7 (87.5) | 29 (85.3) | 1 |
| Presence | 6 (14.3) | 1 (12.5) | 5 (14.7) | ||
| m7FLIPI n (%) | Low | 32 (76.2) | 8 (100.0) | 24 (70.6) | .165 |
| High | 10 (23.8) | 0 (0.00) | 10 (29.4) |
FIGURE 2.
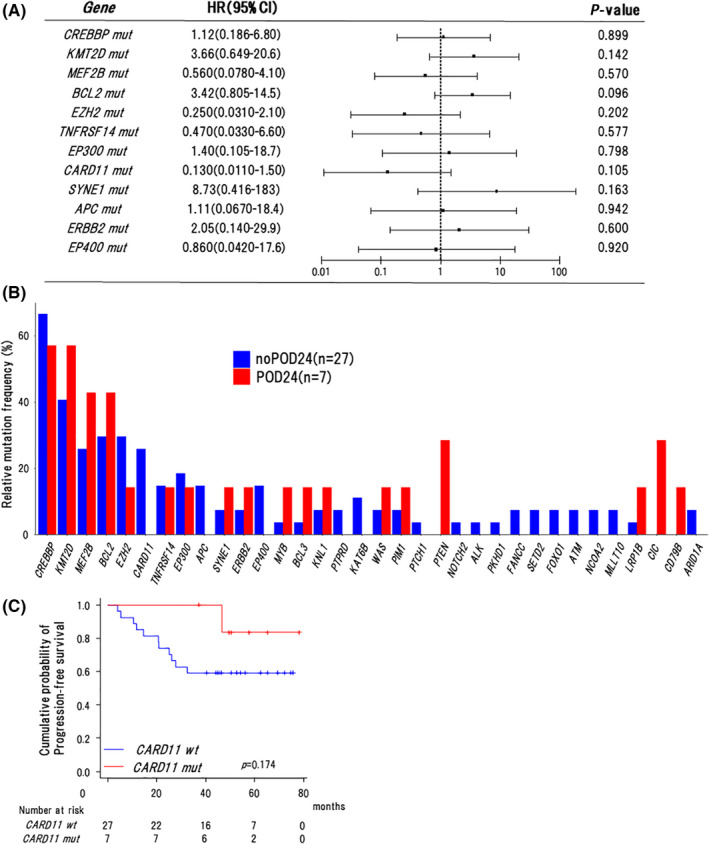
Association of mutation profile with clinical outcomes in the nOR group. A, Forest plot for progression‐free survival in univariate analyses. HR, hazard ratio. Mut, mutations. B, Frequencies of mutations in 34 recurrently mutated genes in cases with or without POD24 in the nOR group (n = 34). C, Kaplan‐Meier curves for progression‐free survival of cases with or without CARD11 mutations in the nOR group (n = 34). Log‐rank P‐values are shown in the graphs. Wt, wild type. Mut, mutations
3.4. Association of m7‐FLIPI with clinical outcomes
We next investigated whether m7‐FLIPI could be applied to this total cohort (n = 42). As expected, no cases (0/8) in the OR group were classified into the high‐risk category in the m7‐FLIPI compared with 29.4% (10/34) in the nOR group but without a significant difference (P = .165) (Table 2). The high‐risk group tended to have an inferior 3‐y PFS rate compared with the low‐risk group (76.8% [95% CI: 57.4%‐88.2%] vs. 60.0% [95% CI: 25.3%‐82.7%]) but without a significant difference (P = .104) (Figure 3A).
FIGURE 3.
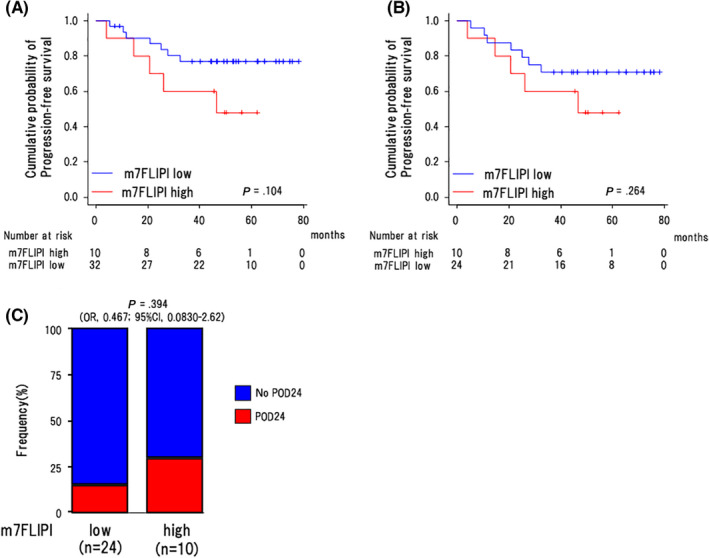
Association of m7‐FLIPI with clinical outcomes. Comparison of progression‐free survival between the cases in the low‐risk and high‐risk categories classified by m7‐FLIPI for (A) all cases (n = 42) and (B) nOR group (n = 34). Log‐rank P‐values are shown in the graphs. Int, intermediate. C, The proportions of the low‐risk and high‐risk categories classified by m7‐FLIPI in the cases with and without POD24 in the nOR group (n = 34). P‐values by Fisher exact test are shown in the graphs
Next, we focused on RB‐treated patients (n = 34) in the nOR group. In this group, the proportion of cases in the low‐ and high‐risk categories in m7‐FLIPI was 70.6% (24/34) and 29.4% (10/34), respectively (Table 2). At a median follow‐up time of 48.7 mo (4.07‐79.3), the 3‐y PFS rates were 70.8% in the low‐risk group (95% CI: 48.4%‐84.9%) and 60.0% in the high‐risk group (95% CI: 25.3%‐82.7%), without a significant difference (P = .264) (Figure 3B). Among 24 cases in the low‐risk group, 4 cases experienced POD24 (16.7%), while 3 from 10 cases in high‐risk group experienced POD24 (30%) without a significant difference (OR: 0.467, [95%CI: 0.0830‐2.62], P = .394) (Figure 3C).
3.5. Association of the 23‐gene expressions with clinical outcomes
We next analyzed gene expression levels using the FFPE samples from the above‐described 50 patients. Two cases (OR n = 1, nOR n = 1) were excluded from this analysis because of their poor RNA qualities. Therefore, 48 cases (OR n = 9, nOR n = 39) were analyzed by the nCounter system. Sample quality was assessed by mRNA levels of 40 housekeeping genes in each sample (Table S3). It has been previously reported that the GEP score based on the expression of 23 genes was associated with clinical outcomes of FL cases, 19 which was confirmed by the following study. 20 Furthermore, the 23 genes could identify 2 main clusters characterized by high or low expression associated with favorable or poor outcomes. 20 So, we subjected our 48 cases to hierarchical clustering using these 23 genes. Consistent with the previous reports, 19 , 20 we could identify 2 main clusters (cluster 1 and cluster 2) (Figure 4A), however, at a median follow‐up time of 47.4 mo (3.2‐79.3), 3‐y PFS rates did not differ significantly between the 2 clusters (74.7% [95% CI: 55.6%‐86.5%] vs. 60.0% [95% CI: 31.8%‐79.7%], P = .436) (Figure 4B).
FIGURE 4.
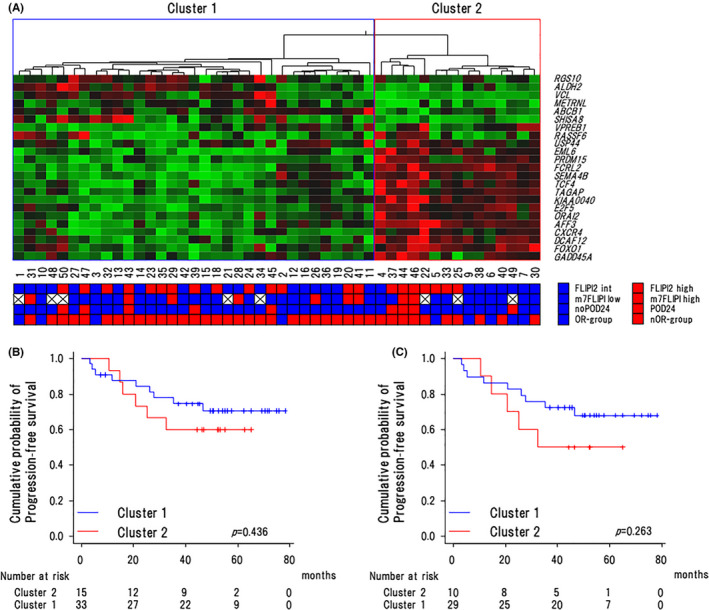
Association of the 23‐gene expression levels with clinical outcomes. A, Heat map and unsupervised hierarchical clustering showing the genes along rows and cases along columns in all cases. Green denotes low and red indicates high gene expression. Clusters 1 and 2 were characterized by high expression of genes associated with favorable and poor outcome, respectively. B, Kaplan‐Meier curves for progression‐free survival of cases with clusters 1 and 2 in all cases (n = 48). C, the nOR group (n = 39). Log‐rank P‐values are shown in the graphs
Next, we compared 3‐y PFS rates between clusters 1 and 2 in the RB‐treated 39 patients. At a median follow‐up time of 46.1 mo (3.2‐79.3), the patients in cluster 2 tended to have an inferior 3‐y PFS rate compared with those in cluster 1 (50.0% [95% CI: 18.4%‐75.3%] vs. 72.4% [95% CI: 52.3%‐85.1%]) but without a significant difference (P = .263) (Figure 4C). We further compared the expression of 23 genes between 8 patients with POD24 and 31 patients without POD24. Although these genes were reported to portend good or poor risk, the expression levels of these genes were almost equivalent in the 2 groups (Figure S3).
3.6. Association of the T‐cell‐associated gene expression with clinical outcomes
In this trial, we previously reported that a low ALCs was an independent poor prognostic factor for RB‐treated patients. 21 Because lymphopenia was considered to reflect an immune‐suppressive state, we applied a customized pan‐cancer immune‐profiling panel, which consisted of 800 genes related to cancer development and/or immune reactions, to the RB‐treated case in the nOR group (n = 39). As a result, a total of 33 genes, including 2 upregulated (CD79a and POU2F2) and 31 downregulated genes (each with log2 fold change > |0.5|, and corrected [−log10 P] > 1.5) were differentially expressed in the POD24 group (n = 8) compared with the no POD24 group (n = 31). Of interest, the top genes downregulated in the POD24 group were markedly enriched with T‐cell‐associated genes (Figure 5A).
FIGURE 5.
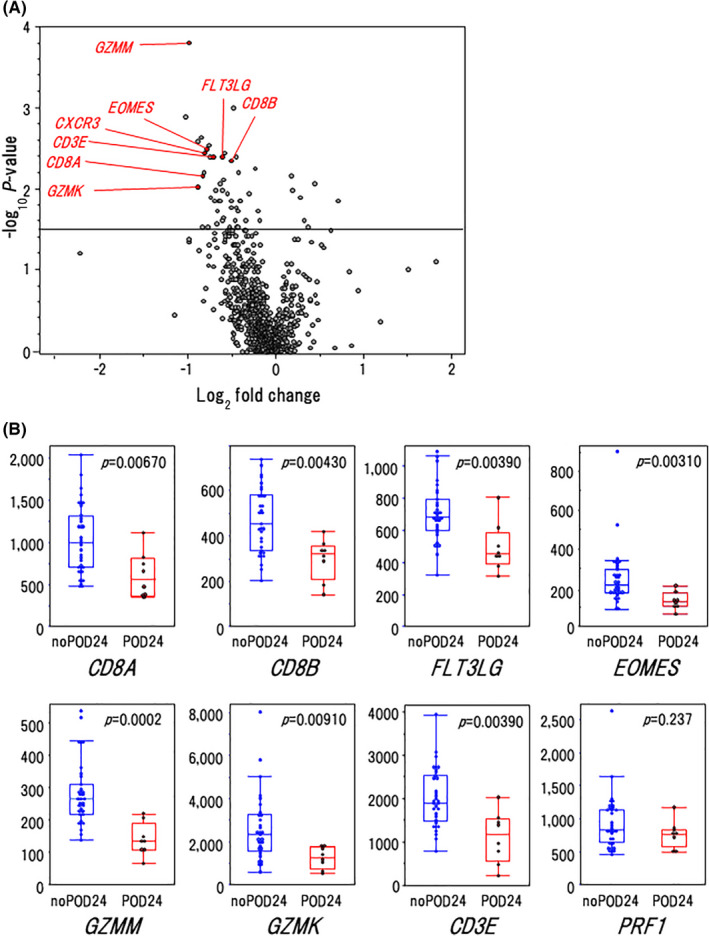
Association of T‐cell‐associated gene expression with clinical outcomes. A, Volcano plot indicating differentially expressed genes in cases with or without POD24 in the nOR group (n = 39). B, Gene expression levels of the CD8+ T‐cell markers in the cases with POD24 and without POD24. P‐values by Fisher exact test are shown in the graphs. In the box plots, the center line and lower and upper hinges correspond to the median, and the first and third quartiles (25 and 75 percentiles), respectively. The upper and lower whiskers extend from the upper and lower hinges to the largest or smallest values no further than a 1.5× inter‐quartile range from the hinges
Therefore, we further compared the expression levels of molecules specific for various types of T cells (Th1, Th2, and Th17, Tfh, Treg, natural killer (NK), and CD8+ T cells) and of immune checkpoint markers between the 2 groups. Interestingly, the expression levels of CD8+ T‐cell marker genes (CD8A, CD8B, FLT3LG, EOMES, GZMM, GZMK) except for PRF1 were significantly lower in the POD24 group compared with the no POD24 group (Figure 5B). Furthermore, about half of the genes related to Th1 and Th2 responses in T cells were downregulated in the POD24 group compared with those in the no POD24 group (Figure S4). Among them, expression levels of several genes that had been reported to regulate differentiation (EOMES, 31 , 32 TBX‐21 33 ) or migration (CXCR3 34 , 35 ) of CD8+ T cells were significantly lower in the POD24 group. Unexpectedly, CCR4, known to be mainly expressed on Treg cells, was downregulated in the POD24 group. In contrast, NK cell markers and immune checkpoint markers were almost equivalently expressed in the 2 groups (Figures 5B and S4). So, we speculated that the prognosis of the FL patients might be influenced by CD8+ T‐cell infiltration.
3.7. Association of the immune infiltration score with clinical outcomes
Next, we selected 10 genes (TBX21, CXCR3, CCR4, CD8A, CD8B, GZMM, FLT3LG, CD3E, EOMES, GZMK) by manual curation that are involved in differentiation and/or migration of CD8+ T cells and performed unsupervised hierarchical clustering. As a result, we could dichotomize 39 RB‐treated cases into immune infiltrationhigh (infilhigh) and infiltrationlow (infillow) clusters (Figure 6A). Of note, POD24 was observed in 8/20 (40%) cases in the infillow cluster, and no POD24 event occurred in the infilhigh cluster with significant difference (P = .0084) (Figure 6B).
FIGURE 6.
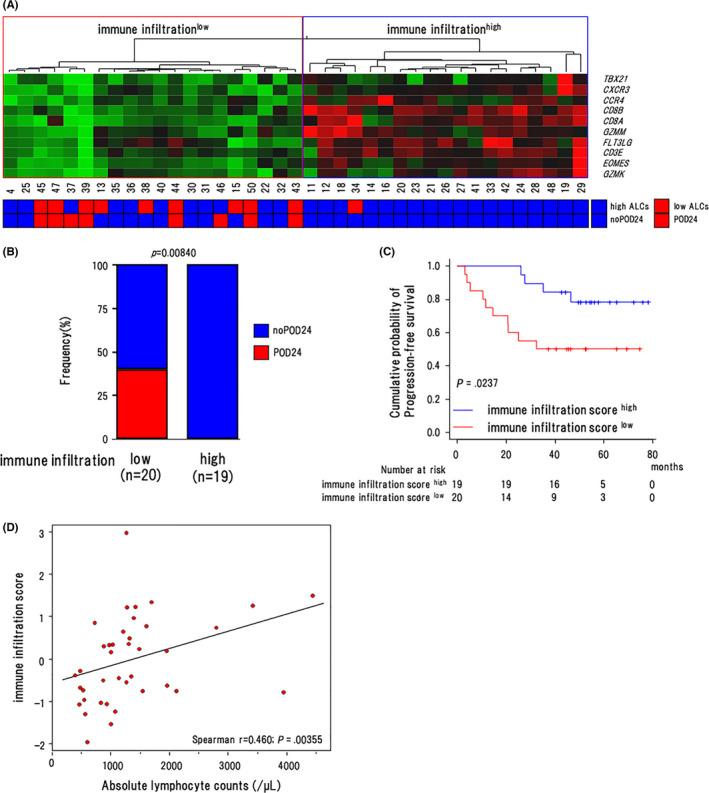
Association of the immune infiltration score with clinical outcomes. A, Heat map and unsupervised hierarchical clustering showing the genes along rows and cases along columns in the nOR group. Green denotes low and red indicates high gene expression. The regions indicate the low (shown in red) and high immune infiltration cluster (shown in blue). B, The proportion of cases with POD24 in the infillow and infilhigh clusters in the nOR group. P‐values by Fisher exact test are shown in the graphs (n = 39). C, Progression‐free survival of cases in the IISlow (n = 20) and IIShigh (n = 19) groups in the nOR group (n = 39). Log‐rank P‐values are shown in the graphs. D, Scattergram shows the correlation between ALCs (horizontal axis) and IIS (vertical axis). Spearman's correlation coefficients are estimated as continuous variables
Next, we attempted to generate an IIS using these 10 genes. For this purpose, we conducted a principal component analysis on 39 cases in the nOR group and extracted 3 principal components (Table S5). Eventually, principal component 1 had a prognostic value for PFS (HR 0.382, 95% CI: 0.170‐0.856, P = .0194; Table S6). So, we calculated the IIS using this component. The IIS ranged from −1.986 to 2.937, and a score of −0.197 was determined as the optimum threshold to separate these cases into IISlow (n = 20) and IIShigh (n = 19) groups from ROC analysis (specificity = 0.624, sensitivity = 0.786, AUC = 0.719; Figure S5). The 3‐y PFS rate was significantly lower in the IISlow group compared with in the IIShigh group (50.0% [95% CI: 27.1%‐69.2%] vs. 84.2% [95% CI: 58.7%‐94.6%], P = .0237) (Figure 6C). We also examined the relationship between the IIS and ALCs in these 39 patients and found that the IIS was correlated with ALCs (r = 0.460, P = .00355; Figure 6D).
3.8. Association of the T‐cell‐associated gene expression with their mutation profile
We next analyzed whether gene mutations influenced the expression of T‐cell‐associated genes in 40 patients. However, the frequencies of mutations were roughly the same between infillow and infilhigh clusters in most genes (Figure 7A). For example, the mutation frequencies in CREBBP, 36 EZH2, 37 and TNFRSF14, 38 which are known to regulate the tumor microenvironment, did not differ significantly between the 2 clusters.
FIGURE 7.
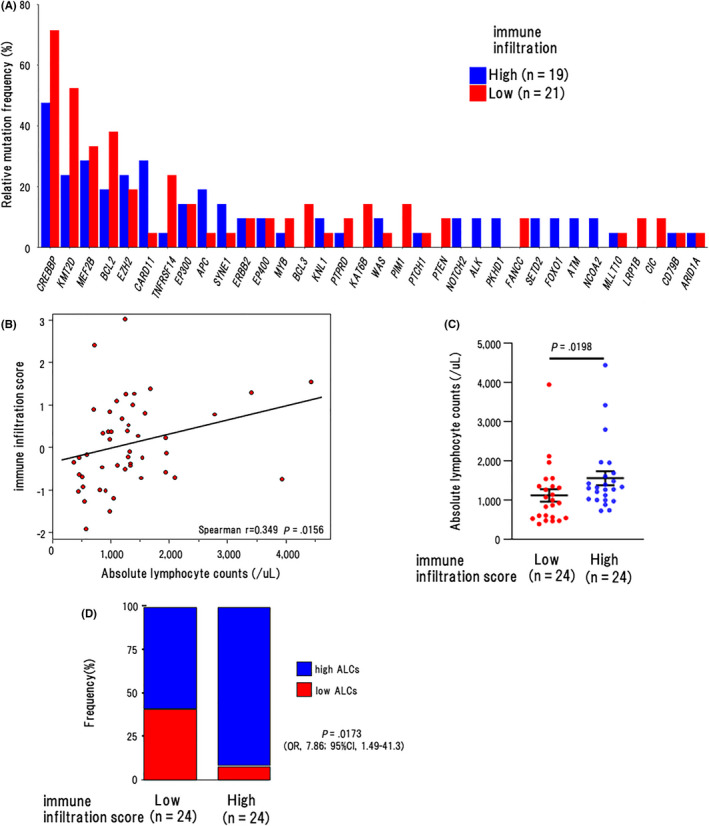
Association of the T‐cell‐associated gene expression with their mutation profile. A, Mutation frequencies of recurrently mutated genes by infillow and infilhigh clusters in all cases (n = 40). B, Scattergram shows the correlation between ALCs (horizontal axis) and IIS (vertical axis). Spearman correlation coefficients were estimated as continuous variables. C, The number of ALCs in the IISlow (n = 24) and IIShigh (n = 24) groups. P‐values using Wilcoxon rank sum test are shown in the graphs. The graph shows the mean and SEM of data from the cases in the respective groups. D, The proportion of cases with low ALCs (< 869/μL) in the IISlow (n = 24) and IIShigh (n = 24) groups. P‐values using Fisher exact test are shown in the graphs
Finally, we investigated whether the IIS was correlated with ALCs using all cases (n = 48) and found that the IIS was correlated with ALCs also in this population (r = 0.349, P = .0156; Figure 7B). Of note, the mean number of ALCs was significantly lower in the IISlow group (1120/μL) than in the IIShigh group (1560/μL) (P = .0198) (Figure 7C). Furthermore, the proportion of cases with low ALCs (<869/μL) was higher in the IISlow group than in the IIShigh group (41.7% vs.8.33%, OR: 7.86 [95%CI, 1.49‐41.3], P = .0173) (Figure 7D).
4. DISCUSSION
In this study, we attempted to identify biomarker(s) that can predict clinical outcomes of RB‐treated FL patients. At first, we examined gene mutation profiles in our patients. As a result, we found that types of mutated genes and their frequencies were roughly the same as previously reported, 1 , 29 , 30 and we found that none of those mutations influenced PFS in univariate analyses. Among them, mutations of CARD11, which functions as a positive regulator of the NF‐κB pathway in normal B and T lymphocytes, were detected in 16.7% of the cases. A CARD11 mutation was more frequently observed in transformed FL than in untransformed FL 29 , 39 and counted as a poor prognostic factor in m7‐FLIPI. 17 In accordance with these results, all patients harboring CARD11 mutations did not show optimal responses to the initial 2 cycles of R‐CHOP and were classified into the nOR group. However, all these patients achieved and maintained CR at least for 3 y by the early conversion to RB treatment. These results raised the possibility that the negative impact of CARD11 mutations observed under R‐CHOP treatment might be canceled by RB treatment.
We also assessed whether m7‐FLIPI is useful to predict prognosis of our RB‐treated FL patients. Although the 3‐y PFS rate tended to be higher in the m7‐FLIPI low group than in the m7‐FLIPI high group, this difference was not significant. Also, whereas m7‐FLIPI was reported to be able to predict POD24 in R‐CHOP‐treated FL patients, POD24 was observed regardless of m7‐FLIPI risk group in our BR‐treated cases. It must be noted that the results reported in this study may lack statistical power due to the small number of cases. In accordance with our results, however, m7‐FLIPI had no prognostic value in RB‐treated or GB (G + B)‐treated FL patients, as reported in a randomized phase III (GALLIUM) trial. 3 , 40 Together, these results suggested that m7‐FLIPI may not be an accurate prognostic factor in FL patients treated with B‐based regimens.
A GEP score based on the expression of 23 genes was initially established to predict clinical outcomes in FL patients, 19 and based on the 23‐gene expression panel, it was possible to identify 2 main clusters associated with favorable or poor outcome in the subsequent study. 20 However, these studies included only a few patients treated with RB. Subsequently, this model was shown to have no prognostic value in the RB‐treated or GB‐treated cohort in the GALLIUM study. 41 In accordance with this result, we found that the 3‐y PFS rates did not differ between the patients in cluster 1 and cluster 2. However, cluster 2 patients tended to have an inferior 3‐y PFS rate compared with cluster 1 patients. Considering the small number of patients in this study, further studies are needed to draw a definite conclusion as to the efficacy of this model in predicting the prognosis of B‐treated FL cases.
Several lines of evidence indicated that FL cells depend on the interactions with non‐malignant cells that constitute the TME for their growth and survival. 1 , 42 Particularly, because of the indolent feature of FL cells, the prognosis of FL is substantially affected by the TME, which consists of various types of T lymphocytes (CTLs, Tregs, and so on), B lymphocytes, and TAMs. These cells act as positive or negative regulators of the immune response in FL, thereby influencing the clinical outcomes in this disease. At first, Dave and colleagues showed that FL patients with high levels of immune infiltration in tumoral tissues had a better prognosis compared with those with low immune infiltration. 43 However, this study was conducted before the R era, including some patients who initially received a watch‐and‐wait approach. Another study showed that a cytotoxic effector T‐cell signature based on a signature containing 6 genes (GZMA, GZMB, PRF1, IFN‐γ, EOMES, and CD8A) could be a prognostic factor, in which the “inflamed” subset (effector T‐cellhigh) showed better PFS than the “uninflamed” subset (effector T‐celllow). 44 More recently, Tobin and co‐workers analyzed the immune infiltration profile in FL cases with or without POD24, and observed that a low expression of immune effectors (TNFα, CD4), checkpoints (programmed death‐ligand 2 [PD‐L2]), or macrophage markers (CD68), were associated with POD24. Among them, PD‐L2 was the most sensitive marker in identifying patients with a poor prognosis. 11 However, most of the patients enrolled in these studies received R‐CHOP/R‐CVP.
To elucidate the roles of the immune microenvironment in RB‐treated FL patients, we applied a customized pan‐cancer immune‐profiling panel consisting of 800 genes to the RB‐treated cases in the nOR group. As a result, we found that CD8+ T‐cell markers such as CD8A, CD8B, FLT3LG, GZMM, and GZMK, were downregulated in the POD24 group, while markers for NK/T cells and immune checkpoint molecules were almost equivalently expressed in cases with or without POD24. Among the genes downregulated in the POD24 group, GZMM and GZMK, which are commonly expressed in CTLs and NK cells, have pro‐apoptotic activities on tumor cells. 45 A low expression of GZMM and GZMK in tumor tissues has been associated with an unfavorable prognosis in cutaneous melanoma, 46 and GZMK was also found to be downregulated in the process of FL transformation. 47 Together, these results suggested that anti‐FL immune responses may be weakened in RB‐treated FL patients undergoing POD24. However, future studies using high‐throughput technologies, such as single‐cell transcriptome sequencing, multicolor immunohistochemistry, and imaging mass cytometry are needed to determine which T‐cell subpopulations indeed express each cytotoxic molecule.
In the presented study, some genes related to differentiation (EOMES, 31 , 32 TBX‐21 33 ) or migration (CXCR3 34 , 35 ) of CD8+ T cells were also significantly downregulated in the POD24 group. Unexpectedly, CCR4 was also downregulated in the POD24 group. This result seems to be somewhat unreasonable because targeting therapies against CCR4 were expected to have great potential in cancer immunotherapies through depletion of Tregs. 48 However, using single‐cell transcriptome sequencing, a recent study has reported that the expression of CCR4 was positively correlated with CTLs signature and a good prognosis in patients with nasopharyngeal carcinoma. 49 Collectively, we further classified RB‐treated cases into infilhigh and infillow clusters using the 10 manually selected genes. POD24 was observed in 8/20 (40%) cases in the infillow cluster, while none of infilhigh cases underwent POD24 (P = .0084). Furthermore, we generated the IIS using principal component analysis based on the 10 genes. The 3‐y PFS rate was significantly lower in the IISlow group than in the IIShigh group (P = .0237). These results suggested that our model would be useful for predicting clinical outcomes including POD24 in RB‐treated FL patients. Although it has already been shown that an increased number of CD8+ T cells in neoplastic follicles of FL was associated with a favorable prognosis, 50 CD8+ T cells present in neoplastic follicles are functionally heterogeneous and include some exhausted T cells with inhibitory receptors (ie, PD‐1, LAG3). 51 So, to identify truly functional CTLs against FL cells among CD8+ T cells by immunohistochemistry in daily practice, we should identify several surface markers and utilize them in combination. Furthermore, it should be noted that our RB‐treated patients constituted a unique population, namely those who received RB after 2 cycles of R‐CHOP. Therefore, future studies are required to determine whether our results are applicable to de novo FL patients treated with a B‐based regimen as the 1st‐line treatment using larger prospective cohorts.
To clarify molecular mechanisms underlying the difference in infillow and infilhigh immune profiles, we compared the frequency of mutations in both clusters. However, the frequencies of mutations were roughly the same between infillow and infilhigh clusters in most genes, especially those that regulate the TME such as CREBBP, EZH2, and TNFRSF14. These results raised the possibility that epigenetic regulation might be different between the 2 clusters. Of importance, the only significant clinical parameter related to the IISlow group was lymphopenia, which we had previously identified as a poor prognostic marker for RB‐treated patients. 21 These results suggested that lymphopenia at diagnosis may reflect reduced T‐cell‐mediated immune reactions in the TME and that it may be possible to substitute this for the immune profiling utilized in this study.
In conclusion, we here found that the expression of CD8+ T‐cell markers was significantly lower in RB‐treated patients with POD24 than those without POD24. Furthermore, FL cases were classified into IISlow and IIShigh groups based on the expression of T‐cell‐associated genes, which was useful to predict the prognosis of RB‐treated FL patients.
CONFLICT OF INTEREST
IM has received research funding from Chugai and Eizai Pharmaceuticals. SR has received payment for lectures from Chugai Pharmaceuticals. No other potential conflicts of interest were reported.
Supporting information
Appendix S1
ACKNOWLEDGMENTS
The authors thank the patients who participated in the study, their supporters, the investigators, and clinical research staff from the study centers.
Rai S, Inoue H, Sakai K, et al. Decreased expression of T‐cell‐associated immune markers predicts poor prognosis in patients with follicular lymphoma. Cancer Sci.2022;113:660–673. doi: 10.1111/cas.15224
REFERENCES
- 1. Carbone A, Roulland S, Gloghini A, et al. Follicular lymphoma. Nat Rev Dis Primers. 2019;5:83. [DOI] [PubMed] [Google Scholar]
- 2. Hiddemann W, Barbui AM, Canales MA, et al. Immunochemotherapy With Obinutuzumab or Rituximab for Previously Untreated Follicular Lymphoma in the GALLIUM Study: Influence of Chemotherapy on Efficacy and Safety. J Clin Oncol. 2018;36:2395‐2404. [DOI] [PubMed] [Google Scholar]
- 3. Marcus R, Seymour JF, Hiddemann W. Obinutuzumab Treatment of Follicular Lymphoma. N Engl J Med. 2017;377:2605‐2606. [DOI] [PubMed] [Google Scholar]
- 4. Federico M, Luminari S, Dondi A, et al. R‐CVP versus R‐CHOP versus R‐FM for the initial treatment of patients with advanced‐stage follicular lymphoma: results of the FOLL05 trial conducted by the Fondazione Italiana Linfomi. J Clin Oncol. 2013;31:1506‐1513. [DOI] [PubMed] [Google Scholar]
- 5. Luminari S, Ferrari A, Manni M, et al. Long‐Term Results of the FOLL05 Trial Comparing R‐CVP Versus R‐CHOP Versus R‐FM for the Initial Treatment of Patients With Advanced‐Stage Symptomatic Follicular Lymphoma. J Clin Oncol. 2018;36:689‐696. [DOI] [PubMed] [Google Scholar]
- 6. Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first‐line treatment for patients with indolent and mantle‐cell lymphomas: an open‐label, multicentre, randomised, phase 3 non‐inferiority trial. Lancet. 2013;381:1203‐1210. [DOI] [PubMed] [Google Scholar]
- 7. Flinn IW, van der Jagt R, Kahl BS, et al. Randomized trial of bendamustine‐rituximab or R‐CHOP/R‐CVP in first‐line treatment of indolent NHL or MCL: the BRIGHT study. Blood. 2014;123:2944‐2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Flinn IW, van der Jagt R, Kahl B, et al. First‐Line Treatment of Patients With Indolent Non‐Hodgkin Lymphoma or Mantle‐Cell Lymphoma With Bendamustine Plus Rituximab Versus R‐CHOP or R‐CVP: Results of the BRIGHT 5‐Year Follow‐Up Study. J Clin Oncol. 2019;37:984‐991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sarkozy C, Maurer MJ, Link BK, et al. Cause of death in follicular lymphoma in the first decade of the rituximab era: A pooled analysis of French and US Cohorts. J Clin Oncol. 2019;37:144‐152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Casulo C, Byrtek M, Dawson KL, et al. Early Relapse of Follicular Lymphoma After Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone Defines Patients at High Risk for Death: An Analysis From the National LymphoCare Study. J Clin Oncol. 2015;33:2516‐2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tobin JWD, Keane C, Gunawardana J, et al. Progression of Disease Within 24 Months in Follicular Lymphoma Is Associated With Reduced Intratumoral Immune Infiltration. J Clin Oncol. 2019;37:3300‐3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Seymour JF, Marcus R, Davies A, et al. Association of early disease progression and very poor survival in the GALLIUM study in follicular lymphoma: benefit of obinutuzumab in reducing the rate of early progression. Haematologica. 2019;104:1202‐1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Solal‐Céligny P, Roy P, Colombat P, et al. Follicular lymphoma international prognostic index. Blood. 2004;104:1258‐1265. [DOI] [PubMed] [Google Scholar]
- 14. Federico M, Bellei M, Marcheselli L, et al. Follicular lymphoma international prognostic index 2: a new prognostic index for follicular lymphoma developed by the international follicular lymphoma prognostic factor project. J Clin Oncol. 2009;27:4555‐4562. [DOI] [PubMed] [Google Scholar]
- 15. Bachy E, Maurer MJ, Habermann TM, et al. A simplified scoring system in de novo follicular lymphoma treated initially with immunochemotherapy. Blood. 2018;132:49‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mir F, Mattiello F, Grigg A, et al. Follicular Lymphoma Evaluation Index (FLEX): A new clinical prognostic model that is superior to existing risk scores for predicting progression‐free survival and early treatment failure after frontline immunochemotherapy. Am J Hematol. 2020;95:1503‐1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pastore A, Jurinovic V, Kridel R, et al. Integration of gene mutations in risk prognostication for patients receiving first‐line immunochemotherapy for follicular lymphoma: a retrospective analysis of a prospective clinical trial and validation in a population‐based registry. Lancet Oncol. 2015;16:1111‐1122. [DOI] [PubMed] [Google Scholar]
- 18. Jurinovic V, Kridel R, Staiger AM, et al. Clinicogenetic risk models predict early progression of follicular lymphoma after first‐line immunochemotherapy. Blood. 2016;128:1112‐1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huet S, Tesson B, Jais JP, et al. A gene‐expression profiling score for prediction of outcome in patients with follicular lymphoma: a retrospective training and validation analysis in three international cohorts. Lancet Oncol. 2018;19:549‐561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Silva A, Bassim S, Sarkozy C, et al. Convergence of risk prediction models in follicular lymphoma. Haematologica. 2019;104:e252‐e255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rai S, Inoue H, Hanamoto H, et al. Low absolute lymphocyte count is a poor prognostic factor for untreated advanced follicular lymphoma treated with rituximab plus bendamustine: results of the prospective phase 2 CONVERT trial. Int J Hematol. 2021;114(2):205‐216. [DOI] [PubMed] [Google Scholar]
- 22. Cheson BD, Horning SJ, Coiffier B, et al. Report of an international workshop to standardize response criteria for non‐Hodgkin's lymphomas. NCI Sponsored International Working Group. J Clin Oncol. 1999;17:1244. [DOI] [PubMed] [Google Scholar]
- 23. Sakai K, Tsuboi M, Kenmotsu H, et al. Tumor mutation burden as a biomarker for lung cancer patients treated with pemetrexed and cisplatin (the JIPANG‐TR). Cancer Sci. 2021;112:388‐396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Watatani Y, Sato Y, Miyoshi H, et al. Molecular heterogeneity in peripheral T‐cell lymphoma, not otherwise specified revealed by comprehensive genetic profiling. Leukemia. 2019;33:2867‐2883. [DOI] [PubMed] [Google Scholar]
- 25. Rai S, Tanaka H, Suzuki M, et al. Chlorpromazine eliminates acute myeloid leukemia cells by perturbing subcellular localization of FLT3‐ITD and KIT‐D816V. Nat Commun. 2020;11:4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sugio T, Miyawaki K, Kato K, et al. Microenvironmental immune cell signatures dictate clinical outcomes for PTCL‐NOS. Blood Adv. 2018;2:2242‐2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huet S, Sujobert P, Salles G. From genetics to the clinic: a translational perspective on follicular lymphoma. Nat Rev Cancer. 2018;18:224‐239. [DOI] [PubMed] [Google Scholar]
- 28. Morin RD, Mendez‐Lago M, Mungall AJ, et al. Frequent mutation of histone‐modifying genes in non‐Hodgkin lymphoma. Nature. 2011;476:298‐303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bouska A, Zhang W, Gong Q, et al. Combined copy number and mutation analysis identifies oncogenic pathways associated with transformation of follicular lymphoma. Leukemia. 2017;31:83‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Okosun J, Bödör C, Wang J, et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat Genet. 2014;46:176‐181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pearce EL, Mullen AC, Martins GA, et al. Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science. 2003;302:1041‐1043. [DOI] [PubMed] [Google Scholar]
- 32. Takemoto N, Intlekofer AM, Northrup JT, Wherry EJ, Reiner SL. Cutting Edge: IL‐12 inversely regulates T‐bet and eomesodermin expression during pathogen‐induced CD8+ T cell differentiation. J Immunol. 2006;177:7515‐7519. [DOI] [PubMed] [Google Scholar]
- 33. Joshi NS, Cui W, Chandele A, et al. Inflammation directs memory precursor and short‐lived effector CD8(+) T cell fates via the graded expression of T‐bet transcription factor. Immunity. 2007;27:281‐295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kohli K, Pillarisetty VG, Kim TS. Key chemokines direct migration of immune cells in solid tumors. Cancer Gene Ther. 2021. 10.1038/s41417-021-00303-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hensbergen PJ, Wijnands PG, Schreurs MW, Scheper RJ, Willemze R, Tensen CP. The CXCR3 targeting chemokine CXCL11 has potent antitumor activity in vivo involving attraction of CD8+ T lymphocytes but not inhibition of angiogenesis. J Immunother. 2005;28:343‐351. [DOI] [PubMed] [Google Scholar]
- 36. Andor N, Simonds EF, Czerwinski DK, et al. Single‐cell RNA‐Seq of follicular lymphoma reveals malignant B‐cell types and coexpression of T‐cell immune checkpoints. Blood. 2019;133:1119‐1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Béguelin W, Teater M, Meydan C, et al. Mutant EZH2 Induces a Pre‐malignant Lymphoma Niche by Reprogramming the Immune Response. Cancer Cell. 2020;37:655‐673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Boice M, Salloum D, Mourcin F, et al. Loss of the HVEM Tumor Suppressor in Lymphoma and Restoration by Modified CAR‐T Cells. Cell. 2016;167:405‐418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Devan J, Janikova A, Mraz M. New concepts in follicular lymphoma biology: From BCL2 to epigenetic regulators and non‐coding RNAs. Semin Oncol. 2018;45:291‐302. [DOI] [PubMed] [Google Scholar]
- 40. Jurinovic V, Passerini V, Oestergaard MZ, et al. Evaluation of the m7‐FLIPI in Patients with Follicular Lymphoma Treated within the Gallium Trial: EZH2 mutation Status May be a Predictive Marker for Differential Efficacy of Chemotherapy. Blood. 2019;134:122. [Google Scholar]
- 41. Bolen CR, Hiddemann W, Marcus R, et al. Treatment‐dependence of high‐risk gene expression signatures in de novo folicular lymphoma. International Conference on Malignant Lymphoma (ICML), June 18th‐22nd, 2019, Abstract 143. [Google Scholar]
- 42. Inoue H, Rai S, Tanaka H, et al. Tumour‐immune microenvironment in duodenal‐type follicular lymphoma. Br J Haematol. 2020;191(2):243‐252. [DOI] [PubMed] [Google Scholar]
- 43. Dave SS, Wright G, Tan B, et al. Prediction of survival in follicular lymphoma based on molecular features of tumor‐infiltrating immune cells. N Engl J Med. 2004;351:2159‐2169. [DOI] [PubMed] [Google Scholar]
- 44. Bolen CR, McCord R, Huet S, et al. Mutation load and an effector T‐cell gene signature may distinguish immunologically distinct and clinically relevant lymphoma subsets. Blood Adv. 2017;1:1884‐1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Voskoboinik I, Whisstock JC, Trapani JA. Perforin and granzymes: function, dysfunction and human pathology. Nat Rev Immunol. 2015;15:388‐400. [DOI] [PubMed] [Google Scholar]
- 46. Wu X, Wang X, Zhao Y, Li K, Yu B, Zhang J. Granzyme family acts as a predict biomarker in cutaneous melanoma and indicates more benefit from anti‐PD‐1 immunotherapy. Int J Med Sci. 2021;18:1657‐1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. de Vos S, Hofmann WK, Grogan TM, et al. Gene expression profile of serial samples of transformed B‐cell lymphomas. Lab Invest. 2003;83:271‐285. [DOI] [PubMed] [Google Scholar]
- 48. Kang S, Xie J, Ma S, Liao W, Zhang J, Luo R. Targeted knock down of CCL22 and CCL17 by siRNA during DC differentiation and maturation affects the recruitment of T subsets. Immunobiology. 2010;215:153‐162. [DOI] [PubMed] [Google Scholar]
- 49. Li H, Chen X, Zeng W, et al. Radiation‐enhanced expression of CCL22 in nasopharyngeal carcinoma is associated with CCR4. Int J Radiat Oncol Biol Phys. 2020;108:126‐139. [DOI] [PubMed] [Google Scholar]
- 50. Alvaro T, Lejeune M, Salvadó MT, et al. Immunohistochemical patterns of reactive microenvironment are associated with clinicobiologic behavior in follicular lymphoma patients. J Clin Oncol. 2006;24:5350‐5357. [DOI] [PubMed] [Google Scholar]
- 51. Kurachi M. CD8. Semin Immunopathol. 2019;41:327‐337. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1