

Abstract
The emergence of tyrosine kinase inhibitors as part of a front‐line treatment has greatly improved the clinical outcome of the patients with Ph+ acute lymphoblastic leukemia (ALL). However, a portion of them still become refractory to the therapy mainly through acquiring mutations in the BCR‐ABL1 gene, necessitating a novel strategy to treat tyrosine kinase inhibitor (TKI)‐resistant Ph+ ALL cases. In this report, we show evidence that RUNX1 transcription factor stringently controls the expression of BCR‐ABL1, which can strategically be targeted by our novel RUNX inhibitor, Chb‐M'. Through a series of in vitro experiments, we identified that RUNX1 binds to the promoter of BCR and directly transactivates BCR‐ABL1 expression in Ph+ ALL cell lines. These cells showed significantly reduced expression of BCR‐ABL1 with suppressed proliferation upon RUNX1 knockdown. Moreover, treatment with Chb‐M' consistently downregulated the expression of BCR‐ABL1 in these cells and this drug was highly effective even in an imatinib‐resistant Ph+ ALL cell line. In good agreement with these findings, forced expression of BCR‐ABL1 in these cells conferred relative resistance to Chb‐M'. In addition, in vivo experiments with the Ph+ ALL patient‐derived xenograft cells showed similar results. In summary, targeting RUNX1 therapeutically in Ph+ ALL cells may lead to overcoming TKI resistance through the transcriptional regulation of BCR‐ABL1. Chb‐M' could be a novel drug for patients with TKI‐resistant refractory Ph+ ALL.
Keywords: bcr‐abl; fusion proteins; gene expression regulation; leukemia; lymphoid; Philadelphia chromosome; RUNX1 protein, human
Targeting RUNX1 therapeutically in Ph+ALL cells may lead to overcoming TKI resistance through the transcriptional regulation of BCR‐ABL1. Chb‐M' could be a novel drug for patients with TKI‐resistant refractory Ph+ALL.

Abbreviations
- BCR‐ABL1
breakpoint cluster region‐Abelson 1
- Ph+ ALL
Philadelphia chromosome positive acute lymphoblastic leukemia
- RUNX1
Runt‐related transcription factor 1
- TKI
tyrosine kinase inhibitor
1. INTRODUCTION
Acute lymphoblastic leukemia (ALL) is an acute form of leukemia characterized by the emergence of highly proliferative immature white blood cells, known as lymphoblasts. Approximately 6000 new cases are reported yearly in the United States and ALL is the most frequently encountered malignancy in childhood. 1 , 2 , 3 ALL is one of the first cancers for which an effective chemotherapeutic treatment was developed and its cure is now a realistic goal and is achieved in more than 90% of affected children, 3 , 4 , 5 , 6 while only 20%‐40% of adults respond to and survive courses of intensified chemotherapies. 7 , 8 This difference is supposed to originate from the vulnerability of elderly patients who have weakened immune and circulatory organ systems. Philadelphia chromosome positive ALL (Ph+ ALL) marks a subset of leukemia with distinctive treatment strategy and outcomes due to the existence of the BCR‐ABL1 pathogenic fusion gene that is created by juxtaposing the ABL1 gene on chromosome 9 to part of the BCR gene on chromosome 22. 3 , 9 The emergence of imatinib mesylate, a tyrosine kinase inhibitor (TKI) that inhibits ABL1, KIT and PDGFR, entirely changed the game of anti‐leukemia strategy toward Ph+ ALL. 10 , 11 Adding imatinib to standard therapy improved the outcomes for adults with Ph+ ALL, at least in part, by facilitating allogeneic stem cell transplant. 12 However, a portion of adults steadily develop resistance to TKI therapy, mainly through acquiring point mutations in the kinase domain of BCR‐ABL1 in ALL cells. 13 These patients can be treated by the next generation of tyrosine kinase inhibitors such as nilotinib, dasatinib, or ponatinib. In particular, the third‐generation TKI, ponatinib, is a potent orally bioavailable pan BCR‐ABL1 inhibitor that inhibits both wild‐type and mutant BCR‐ABL1 kinase, including the “gatekeeper” T315I mutation, which is resistant to all other currently available TKIs. 14 , 15 However, because of the risk of cardiovascular side effects, the risk/benefit balance must be evaluated for each patient. 14 Therefore, a new treatment modality against TKI treatment‐resistant Ph+ ALL with no side effects is highly needed.
Runt‐related transcription factor 1 (RUNX1), also known as acute myeloid leukemia 1 protein (AML1), is an essential master transcription factor implicated in the differentiation and the maintenance of hematopoietic stem cells. 16 In ALL, a well known t(12;21)(p13.1;q22) translocation causes the fusion of the ETS variant 6 (ETV6) and RUNX1 genes (ETV6‐RUNX1, formerly TEL‐AML1). It is the most common translocation in childhood ALL, 17 suggesting a fundamental involvement of RUNX1 in the pathogenesis of a subset of ALL cases. Intriguingly, Yamamoto K et.al. 18 reported that the elevated expressions of wild‐type RUNX1 closely correlates with worse outcomes in chronic myeloid leukemia (CML) patients, another type of leukemia caused by the same chimeric protein BCR‐ABL1 as Ph+ ALL but with a different break point. The molecular mechanisms underlying the possible interaction of RUNX1 and BCR‐ABL1, however, have poorly been elucidated so far. We have previously reported the requirement of RUNX1 in the development and the maintenance of AML, 19 , 20 , 21 , 22 , 23 , 24 another form of acute leukemia originating in myeloid progenitor cells. In this report, we addressed the leukemogenic role of RUNX1 in Ph+ ALL and elaborated to elucidate the molecular mechanisms in the regulation of BCR‐ABL1 expression and in the proliferation of Ph+ leukemia cells.
2. MATERIALS AND METHODS
2.1. Cell lines and plasmids
SU‐Ph2 is an imatinib‐sensitive cell line established from a patient with Ph+ALL. SU/SR is an imatinib‐resistant subline of SU‐Ph2 obtained after long‐term exposure to imatinib until they finally acquired the T315I mutation in BCR‐ABL1 gene. These cells were kindly gifted from Dr. A. Kanamaru (Department of Internal Medicine, Kinki University School of Medicine, Osaka, Japan). ALL‐derived BALL‐1, KOCL‐45, SUP‐B15, SU‐Ph2 and SU/SR cells as well as CML‐derived MYL, BV173 and K562 cells were maintained in RPMI 1640 medium with 10% heat‐inactivated FBS and 1% penicillin‐streptomycin at 37°C in 5% CO2.
Human BCR‐ABL1 was a kind gift from Nora Heisterkamp (Addgene plasmid # 31 285). pENTR1A Dual Selection vector (Thermo Fisher Scientific), CSIV‐TRE‐RfA‐UbC‐KT and CSII‐EF‐MCS‐IRES‐hKO1 (RIKEN BRC) were used to construct expression vectors. All of the products were verified by DNA sequencing.
2.2. Dual luciferase reporter assay
HEK293T cells were seeded in 10 mL DMEM supplemented with 10% heat‐inactivated FBS and 1% PS 1 d before transfection. Cells were transfected with 10 µg of pGL4.20 harboring the BCR promoter and 1 µg pRL‐CMV with polyethylenimine (PEI; Sigma‐Aldrich). The BCR promoter region was amplified from the genomic DNA of SU/SR cells using specific primers (F 5′‐TTAGAGGGAGGCTAATCAGGG‐3′ and R 5′‐TCCTCGGACGCTAAGCTC‐3′). At 24 h after transfection, doxycycline was added at 3 µmol L−1 and incubated for another 24 h. The cells were then rinsed twice with PBS and lysed with 1× lysis buffer as supplied in the PicaGene® Dual Sea Pansy Luminescence kit (TOYO B‐net). The luciferase and Renilla luciferase activity were measured using ARVO X5 (PerkinElmer).
2.3. IC50 evaluation
For cell survival assay, 3 × 104 cells were seeded onto 96‐well flat plates. The indicated concentrations of PI polyamides or drugs were added to the culture medium and cells were incubated for 48 h. Cell viability was then assessed using the Cell Count Reagent SF (nacalai tesque, Inc) and the Infinite® 200 PRO multimode reader (TECAN). Percent inhibition curves were drawn and IC50 of the indicated compounds was calculated based on median‐effect method. 25
2.4. Statistics
Statistical significance of differences between control and experimental groups was assessed using a 2‐tailed unpaired Student t test and was declared if the P‐value was less than .05. Equality of variances in 2 populations was calculated using the F test. The results were represented as the average ± SD values obtained from 3 independent experiments.
2.5. Quantitative RT‐PCR
Quantitative RT‐PCR (qRT‐PCR) was conducted as previously described. 26 Briefly, total RNA was extracted from cultured cells using the RNeasy mini kit (Quiagen) and reverse transcribed using the ReverTra Ace® qPCR RT Master Mix (TOYOBO) to generate cDNA. qRT‐PCR was conducted on the StepOne™ real‐time PCR system (Applied Biosystems). Relative expression levels were calculated using the 2−ΔΔCt method. Primers used for qRT‐PCR are listed in Table S1.
2.6. ChIP‐PCR
ChIP assay was performed using SimpleChIP® Plus enzymatic Chromatin IP Kit (Cell Signaling Technology) according to the manufacturer's instructions. Chromatin preparation was processed for immunoprecipitation with anti‐RUNX1 antibody (ab23980, abcam) at 4°C overnight. Following ChIP, DNA was amplified with specific primers listed in Table S2 using Ex Taq ® polymerase (Takara Bio Inc). Obtained DNA was analyzed using agarose gel electrophoresis.
2.7. Immunoblotting
Cells were washed twice in ice‐cold PBS and lysed in lysis buffer as previously described. 21 Equal amounts of protein samples were loaded onto the gels for each target proteins, separated using SDS‐PAGE and electrotransferred onto 45‐µm pore size polyvinylidene difluoride membranes (Millipore, IPVH00010). Membranes were probed with the following primary antibodies: anti‐c‐abl (Cell Signaling Technology, 2862), anti‐RUNX1 (Santa Cruz Biotechnology, clone A‐2), anti‐GAPDH (Santa Cruz Biotechnology, clone 0411), anti‐phospho‐AKT(Ser473; Cell Signaling Technology, 9271), anti‐AKT (Cell Signaling Technology, 9272) and anti‐p53 (Santa Cruz Biotechnology, clone DO‐1) antibodies. For secondary antibodies, HRP‐conjugated anti‐rabbit IgG and anti‐mouse IgG (Cell Signaling Technology, 7074 and 7076) were used. Primary antibodies and secondary antibodies were diluted to 1:1000 and 1:5000. Blots were visualized using Chemi‐Lumi One Super (Nacalai Tesque) and the ChemiDoc XRS + Imager (Bio‐Rad Laboratories).
2.8. shRNA interference
shRNA targeting human RUNX1, BCR‐ABL1, and p53 were designed and sub‐cloned into pENTR4‐H1tetOx1, CS‐RfA‐ETV, CS‐RfA‐ETBsd vectors (RIKEN BRC). Non‐targeting control shRNA was designed against luciferase (sh_Luc). The target sequences were provided in Table S3.
2.9. Xenograft mouse model
NOD/Shi‐scid, IL‐2RγKO (NOG) mice were purchased from the Central Institute for Experimental Animals, Japan and were used as controls in all experiments. For leukemia cell lines mouse xenograft models, 2 × 106 cells/body of SU/SR cells with doxycycline‐inducible shRNA expression vector targeting Luciferase or RUNX1 were injected intravenously into NOG mice. At 7 d after transplantation, 1 mg/mL doxycycline (Sigma) and 30 mg/mL sucrose (Wako) were added to the drinking water and started to be given orally. Peripheral blood was then collected every week and chimerism was checked by a flow cytometer. For the patient‐derived xenograft (PDX) study, PDX cells were provided by Dr. Itaru Kato's group. Appropriate informed consent was obtained from this patient. At the age of 6, she was diagnosed with Ph1‐positive BCP‐ALL (minor BCR‐ABL1‐positive), and was in remission with multidrug chemotherapy including imatinib. At 1 y and 6 mo after the diagnosis, she had a isolated central nervous system (CNS) recurrence. She achieved remission again after switching to dasatinib, Hyper‐CVAD, and intensified intrathecal injections. Bone marrow transplantation was performed from an HLA‐matched relative donor, but she had the second relapse in the CNS. At the second CNS recurrence, the T315I mutation was tested and was negative. She became refractory to treatment and died 1 y and 4 mo after transplantation. The PDX cells used in this study were established using leukemia cells collected from cerebrospinal fluid at the time of the first relapse of the CNS alone. These PDX cells were intravenously transplanted into NOG mice. At 2 wk after transplantation, Chb‐M' (320 μg/kg body weight, twice per week) or DMSO (the equivalent amount, twice per week) administration was intravenously started, and oral administration of imatinib mesylate (Tokyo Chemical Industry Co., Ltd., 100 mg/kg body weight, daily) was started. Bone marrow was then collected every week and chimerism was checked using a flow cytometer and an anti‐human CD45 antibody and an anti‐mouse CD45 antibody (BD Biosciences). Overall survival was monitored until the mice succumbed to their disease. For the bone marrow of 1 representative of each group at day 36, H&E staining and immunohistochemical staining with anti‐human CD45 antibody (Thermo Fisher Scientific, MA5‐13197), anti‐Ki‐67 antibody (Agilent, M7240), anti‐RUNX1 antibody (Abcam, ab35962) and anti‐BCR (BCR‐ABL1 p190/p210) antibody (Santa Cruz Biotechnology, G6) were done.
2.10. Study approval
All animal studies were properly conducted according to the Regulations on Animal Experimentation at Kyoto University, based on International Guiding Principles for Biomedical Research Involving Animals. All procedures used in this study were approved by the Kyoto University Animal Experimentation Committee (Permit Number: Med Kyo 14 332). PDX analysis was approved by the Kyoto University Hospital Ethical Board (Approval number: G‐1030).
3. RESULTS
3.1. Knockdown of RUNX1 suppresses the proliferation of Ph+ ALL cell lines
To explore the role of RUNX1 in the maintenance of Ph+ ALL cells, we first modulated the expression of RUNX1 in human Ph+ ALL‐derived SU/SR cells with doxycycline‐inducible shRNA. SU/SR cells are genetically identical to SU‐Ph2 cells except for the T315I point mutation in the ABL1 protein, which confers major resistance to TKI treatment (Figure S1). 27 , 28 , 29 , 30 As shown in Figure 1A,B, silencing of RUNX1 significantly suppressed the cell growth of SU/SR imatinib‐resistant Ph+ ALL cells in vitro. Intriguingly, this RUNX1 inhibition‐mediated suppression of tumor growth was observed not only in Ph+ ALL‐derived SU/SR and SU/Ph2 cells, but also in CML‐derived MYL and K562 cells (Figure S2). As widely known, while BCR‐ABL1 p190 occurs in the majority of Ph+ ALL cases, BCR‐ABL1 p210 is the hallmark of CML, and both fusion genes are thought to be under the control of the BCR promoter. These results prompted us to further investigate the role of RUNX1 in BCR‐ABL1‐dependent hematologic malignancies.
FIGURE 1.
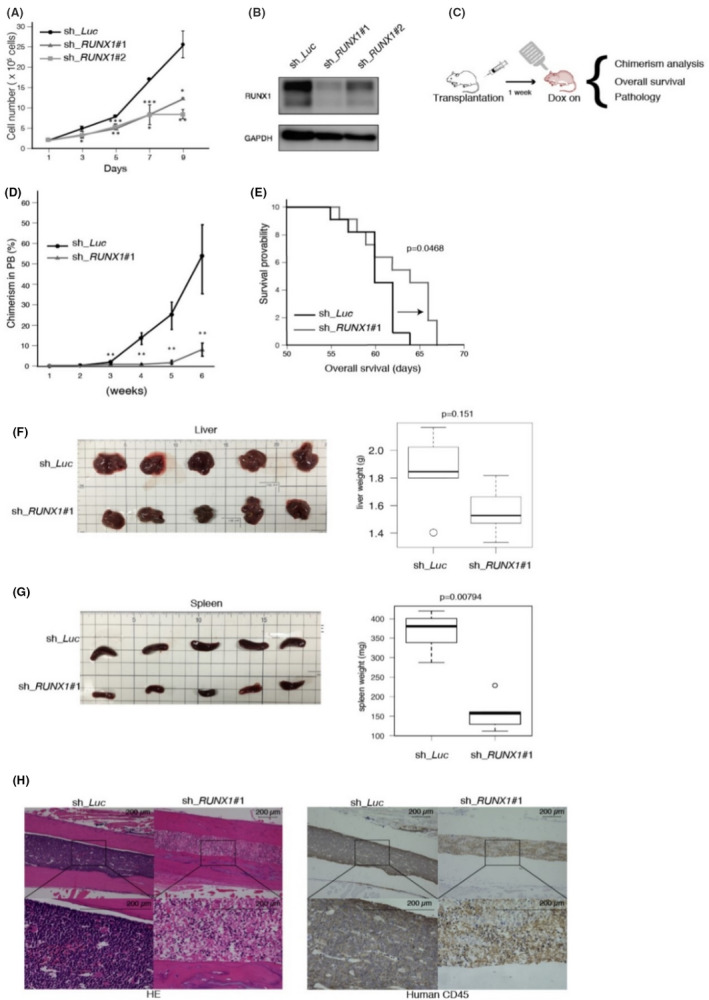
The expression of RUNX1 is required in the maintenance of Ph+ ALL cells. A, Cell growth curves of SU/SR cells transduced with shRNAs targeting RUNX1 (sh_RUNX1 #1 and sh_RUNX1 #2) or luciferase (sh_Luc). B, Immunoblot of RUNX1 and GAPDH in SU/SR cells transfected with sh_Luc, sh_RUNX1 #1 and sh_RUNX1 #2. Cells were treated with 3 µmol L−1 doxycycline for 24 h. C, Schema of xenotransplantation assay in NOG mice with SU/SR cells (sh_Luc or sh_RUNX1#1). D, Chimerism of transplanted leukemia cells in (C; n = 5). E, Overall survival of NOG mice in (C; n = 11). F, G, Organ images of the livers (F) and the spleens (G) with the weight boxplots at day 40 in (C; n = 5). H, Representative histology pictures of the bone marrow at day 40 in (C). H&E staining and immunohistochemical staining with anti‐human CD45 antibody were done for each slide (original magnification; ×10 (upper panels) and ×40 (lower panels), Scale bars; 200 μm). Mean ± SD. *P < .05, **P < .01, ***P < .001, using two‐tailed Student t test (A, D), log‐rank (E), Mann‐Whitney U test (F, G)
We next investigated the effect of RUNX1 inhibition in Ph+ ALL cells in vivo, and prepared a Ph+ ALL xenograft model. We transplanted SU/SR cells that had been stably transduced with lentivirus expressing control sh_Luc or sh_RUNX1 into immunodeficient NOG mice. At 7 d after the transplantation, doxycycline administration was started to induce in vivo RUNX1 knockdown (Figure 1C). Peripheral blood was collected every week to check the chimerism of transplanted ALL cells (Figure 1D). Overall survival periods were monitored until they succumbed to their disease. Thoroughly consistent with the results observed in the in vitro experiments, NOG mice transplanted with RUNX1‐silenced SU/SR cells exhibited prolonged survival with statistical significance (Figure 1E). These mice showed lessened tumor burdens in the spleen and the bone marrow relative to the control (Figure 1F‐H).
3.2. RUNX1 directly transactivates the expression of BCR‐ABL1
As we found that RUNX1 expression is a prerequisite for the proliferation of Ph+ ALL cell lines, we assumed that its expression might be elevated in Ph+ ALL patients. As shown in Figure 2A, analysis of a microarray dataset elucidated that the expression of RUNX1 indeed increased in the bone marrow cells and peripheral blood cells derived from Ph+ ALL patients relative to those from the healthy donors and non‐leukemic patients. In this data set (GSE13204), non‐leukemic patients included those with megaloblastic anemia, hemolysis, iron deficiency, or idiopathic thrombocytopenic purpura. As the expression of the oncogenic BCR‐ABL1 fusion gene is regulated under the BCR promoter, as we have mentioned, this finding led us to hypothesize that the expression of the BCR‐ABL1 fusion gene might be transcriptionally controlled by RUNX1.
FIGURE 2.

R Runt‐related transcription factor 1 (RUNX1) directly transactivates the expression of BCR‐ABL1. A, RUNX1 expression (probe ID:209360_s_at., GSE13204) in pediatric Ph+ ALL (mean = 0.5123, n = 122) and in the control samples (mean = 0.4278, n = 79). B, Relative mRNA expression of BCR‐ABL1 in SU/SR cells stably transduced with sh_Luc, sh_RUNX1 #1 or sh_RUNX1 #2. Cells were treated with 3 µmol L−1 doxycycline for 24 h. C, Immunoblot of RUNX1, BCR‐ABL1, phosphorylated‐AKT (p‐AKT), AKT and GAPDH in the same SU/SR cells as (B). Cells were treated with 3 µmol L−1 doxycycline for 24 h, then lysed for protein extraction. D, Luciferase reporter activity of BCR promoter in HEK293T cells upon knockdown (sh_RUNX1#1) or overexpression (RUNX1 O/E) of RUNX1 with immunoblot images of RUNX1 and GAPDH in the samples. E, Gel image of ChIP‐PCR in SU/SR cells with RUNX1 antibody. The binding of RUNX1 transcription factors to the BCR promoter was assessed with the primers amplifying the region including the RUNX1 consensus binding site (5′‐TGTGGT‐3′) located at 802 bp upstream of TSS (R1). RPL30 was used as RUNX1‐irrelevant negative control. Mean ± SD. *P < .05, ***P < .001, using Mann‐Whitney U test (A), two‐tailed Student t test (B, D)
To test our hypothesis, we first examined the expression of BCR‐ABL1 upon RUNX1 knockdown in SU/SR cells. As shown in Figure 2B,C, the expression of BCR‐ABL1 was significantly downregulated in RUNX1‐silenced SU/SR cells relative to the control both at mRNA and protein levels. In addition, the phosphorylation level of AKT, one of the most important downstream targets of BCR‐ABL1, was also significantly reduced upon knockdown of RUNX1 in SU/SR cells (Figure 2C). Of note, the growth rate of SU/SR cells was attenuated upon BCR‐ABL1 knockdown to the extent of RUNX1‐silencing, underpinning the importance of RUNX1 in the regulation of BCR‐ABL1 expression (Figure S3). To address whether RUNX1 directly transactivates BCR‐ABL1 expression, we next conducted luciferase reporter assays using the BCR promoter in HEK293T cells. We prepared HEK293T cells that were stably transduced with shRNAs targeting RUNX1 or lentivirus expressing RUNX1. These cells were transiently transfected with a vector harboring a luciferase reporter fused to the BCR promoter (located at −1000 to +200 bp relative to the transcription start site [TSS] of BCR gene), and the expression of shRNAs or RUNX1 was induced by doxycycline. As shown in Figure 2D, while inhibition of RUNX1 downregulated the activity of the BCR promoter, additional RUNX1 expression consistently upregulated its activity. Close inspection of the BCR promoter uncovered the RUNX1 consensus binding site of 5′‐TGTGGT‐3′ at 802 bp upstream of the TSS of BCR. ChIP experiments confirmed the actual binding of RUNX1 in this region (Figure 2E). These results collectively suggested that RUNX1 binds to the promoter of BCR‐ABL1 in Ph+ ALL cells and positively regulates it, which could potentially be targeted in anti‐leukemia therapy toward this cancer.
3.3. Novel RUNX inhibitor, Chb‐M’, induces Ph+ALL cell death BCR‐ABL1‐dependently
To further investigate the role of RUNX1 in Ph+ALL cells, we next pharmacologically inhibited RUNX1 by our novel RUNX inhibitor Chb‐M’ 21 and examined its anti‐leukemia effect on Ph+ ALL cells. Chb‐M’ is a pyrrole‐imidazole polyamide interlocked with a hairpin conjugated with alkylating reagent chlorambucil that specifically recognizes DNA sequences containing 5′‐TGTGGT‐3′, a canonical RUNX1 recognition site. To start with, we examined the specificity of the pyrrole‐imidazole polyamide to the 5′‐TGTGGT‐3′ region in the BCR promoter by ChIP assay. For this purpose, we prepared alkylating agent‐free Chb‐M' (Simple‐M') and tested whether the binding of RUNX1 to the 5′‐TGTGGT‐3′ site in the BCR promoter was competitively inhibited by adding Simple‐M'. As shown in Figure 3A, Simple‐M' apparently removed RUNX1 from the BCR promoter in our ChIP experiment dose dependently.
FIGURE 3.
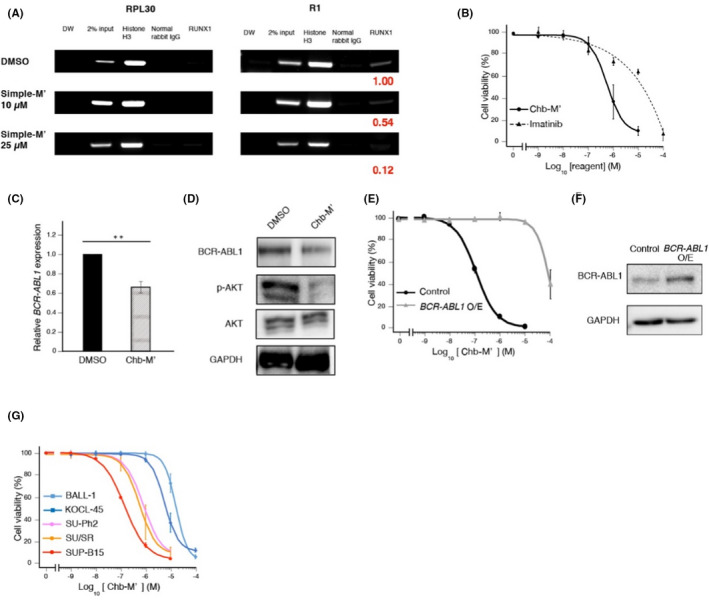
Anti‐leukemic efficacy of RUNX inhibitor Chb‐M' in Ph+ ALL cells. A, Gel image of ChIP‐PCR in SU/SR cells treated with DMSO or Simple‐M' (10, 25 µmol L−1) for 12 h in the same way as Figure 2E. Bands were quantified using Image Lab software (Bio‐Rad Laboratories) and normalized to that of the control. B, Dose‐response curves of SU/SR cells treated with the indicated doses of Chb‐M' (IC50: 658 nmol L−1) and imatinib (IC50: 18.2 µmol L−1) for 48 h. C, BCR‐ABL1 mRNA expression in SU/SR cells treated with DMSO or Chb‐M' (1 µmol L−1) for 9 h. D, Immunoblot of BCR‐ABL1, phosphorylated‐AKT (p‐AKT), AKT and GAPDH in SU/SR cells treated with DMSO or Chb‐M' (1 µmol L−1) for 24 h. E, Dose‐response curves of SU/SR cells stably transduced with control (IC50: 143 nmol L−1) or BCR‐ABL1 expressing vectors (IC50: 33.1 µmol L−1) for 48 h. F, Immunoblot of BCR‐ABL1 and GAPDH in (E). Cells were treated with 3 µmol L−1 doxycycline for 48 h. G, Dose‐response curves of ALL cell lines with BCR‐ABL1 (SU‐Ph2 [IC50: 849 nmol L−1], SU/SR (IC50: 658 nmol L−1) and SUP‐B15 [IC50: 167 nmol L−1]) and ALL cell lines without BCR‐ABL1 (BALL‐1 [IC50: 21.7 µmol L−1] and KOCL‐45 [IC50: 6.04 µmol L−1]) treated with the indicated doses of Chb‐M' for 48 h. Mean ±SD. **P < .01, using two‐tailed Student t test (C)
With respect to the antitumor effect on Ph+ ALL cells, Chb‐M' effectively controlled their proliferation in several Ph+ ALL cell lines that we tested in this study (Figure 3B, Figure S4A‐D). Furthermore, treatment with Chb‐M' downregulated the expression of BCR‐ABL1 both at mRNA and protein levels in these cells (Figures 3C,D and S4E‐H). Contrary to Figure 3A, Chb‐M' suppressed BCR‐ABL1 expression at lower concentration, suggesting that DNA alkylation by chlorambucil is important for transcriptional regulation, as described in our previous reports. 31 , 32 The phosphorylation of AKT was also consistently reduced in SU/SR cells upon Chb‐M' treatment (Figure 3D). These results were thoroughly consistent with those obtained in the RUNX1 knockdown experiments. Of note, additional BCR‐ABL1 expression in SU/SR cells and MYL conferred relative resistance to Chb‐M' treatment (Figures 3E,F and S5). Moreover, we found that Chb‐M' preferentially suppresses the growth of ALL cells with BCR‐ABL1 relative to those without it (Figure 3G). These results collectively suggested that the anti‐leukemia effect of Chb‐M' largely depended on this oncogenic fusion gene.
We have previously found and reported that the growth suppression induced by Chb‐M' is highly dependent on the p53 cell death pathway. 21 Therefore, we tested whether p53 significantly contributed to the Chb‐M'‐mediated growth suppression in SU/SR cells. For this purpose, we prepared p53‐knocked down SU/SR cells and challenged them with Chb‐M'. As shown in Figure S6A‐F, p53 knockdown indeed conferred relative resistance to Chb‐M' to a certain extent, suggesting a possible involvement of p53 in the Chb‐M'‐mediated tumor suppression in these cells, however, the growth of p53‐silenced SU/SR cells was still effectively controlled by Chb‐M' at submicromolar levels. Considering the significant resistance to Chb‐M' conferred by BCR‐ABL1 overexpression in these cells (Figure 3E), these results overall indicated that the growth suppression mediated by Chb‐M' was dependent on both functional p53 and BCR‐ABL1, however possibly more on BCR‐ABL1 in these Ph+ ALL cells.
3.4. Chb‐M' significantly suppresses the growth of Ph+ ALL PDX cells by downregulating BCR‐ABL1 expression in vivo
We investigated the effects of Chb‐M' on Ph+ ALL PDX cells in vivo. We transplanted Ph+ ALL PDX cells derived from the first relapse patient into NOG mice. At 2 wk after the transplantation, Chb‐M' administration was started to treat these mice. DMSO and imatinib mesylate were injected as controls (Figure 4A). Bone marrow was collected every week to check the chimerism of transplanted ALL cells. Chb‐M' significantly suppressed the cell growth of Ph+ ALL PDX cells in the bone marrow, compared with DMSO at week 5 (Figure 4B). NOG mice treated with Chb‐M' had significantly prolonged overall survival compared with mice treated with DMSO (Figure 4C), which is consistent with the results observed in our previous in vivo experiments with the SU/SR Ph+ ALL cell line. 21 The patient sample was negative for the T315I mutation, but imatinib did not prolong survival compared with controls in PDX experiments. To investigate the mechanism of imatinib resistance, we performed mutation analysis on the RNA‐seq data of the PDX cells, and the results are listed in Table S4, which showed no mutations in the ABL1 gene, including T315. The underlying mechanism of imatinib resistance in Ph+ leukemia patients, in addition to mutations in the kinase domain of ABL1, has recently been shown to be due to the genomic amplification of BCR‐ABL1 or the upregulation of the BCR‐ABL1 transcript level. 33 , 34 , 35 FISH of BCR‐ABL1 showed that most leukemic cells at the patient's initial diagnosis had 3 signals of BCR‐ABL1, indicating genomic amplification of BCR‐ABL1. In addition, the mRNA expression of RUNX1 and BCR‐ABL1 was increased in relapse‐derived PDX cells compared with that in primary‐derived PDX cells (Figure S7). This is consistent with the previous report that high expression of RUNX1 is associated with disease progression of CML. 18 From these results, the imatinib resistance in the PDX cells may be due to the increased expression of BCR‐ABL1 associated with increased copy number of BCR‐ABL1 and upregulation by RUNX1. As shown in the H&E staining and immunohistochemistry (human CD45 and Ki‐67) panels, Chb‐M' lessened the tumor burdens in the bone marrow relative to the controls. In addition, Chb‐M' suppressed RUNX1 and BCR‐ABL1 expression of leukemic cells as shown by immunohistochemistry (Figure 4D). Taken together, our RUNX inhibitor, Chb‐M', could be used as a novel drug for patients with TKI‐resistant refractory Ph+ ALL through the downregulation of BCR‐ABL1 (Figure 4E).
FIGURE 4.
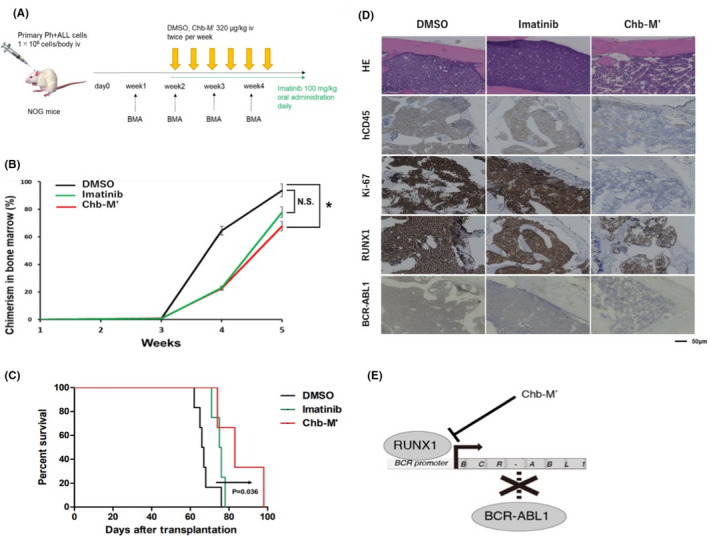
Chb‐M' significantly suppresses the growth of Ph+ ALL PDX cells by downregulating BCR‐ABL1 expression in vivo. A, Schema of transplantation assay in NOG mice with Ph+ ALL PDX cells. These mice were treated with DMSO, imatinib mesylate or Chb‐M'. B, Chimerism of transplanted leukemia cells in bone marrow (n = 7). C, Overall survival of NOG mice transplanted with Ph+ ALL PDX cells (n = 6). D, Representative histology pictures of bone marrow at day 36. H&E staining and immunohistochemical staining with anti‐human CD45 antibody, anti‐Ki‐67 antibody, anti‐RUNX1 antibody and anti‐BCR (BCR‐ABL1) antibody were done for each slide (original magnification; ×10, Scale bars; 50 μm). E, Graphical abstract of this study. RUNX1‐silencing inhibits the transactivation of BCR‐ABL1 expression and therefore attenuates the proliferation of BCR‐ABL1 fusion gene‐dependent leukemia cells. Our RUNX inhibitor, Chb‐M', could be potentially a novel drug for Ph+ ALL with TKI resistance through the downregulation of BCR‐ABL1. Mean ± SD. *P < .05, NS; not significant, using two‐tailed Student t test (B), log‐rank (C)
4. DISCUSSION
Runt‐related transcription factor 1 (RUNX1) forms a heterodimeric complex with core binding factor‐β (CBFβ) on DNA promoter regions and regulates the expression of diverse target genes that are essential for the survival of certain cancers. Yamamoto et al 18 have previously reported that functionally deregulated RUNX1 cooperates with BCR‐ABL1 and induces a blastic phase‐like phenotype of CML in mice. In this study, we found that RUNX1 directly targets BCR‐ABL1 in Ph+ ALL cells through regulating the BCR promoter. According to Shah et al 36 , a functional promoter of BCR is localized in a region 1000 bp upstream of the BCR exon 1 coding sequence, which includes the RUNX consensus binding sequence we identified in this study. In addition to this study, a few groups have previously studied and reported the functional regulation of the BCR promoter. For example, Sharma et al 37 have shown that MYC and MAX genes interact with the BCR promoter and regulate its transcription. To our knowledge, however, this is the first study that provides evidence for a possibility of pharmacological intervention in the transcriptional regulation of BCR‐ABL1 gene. As acquisition of point mutations in the BCR‐ABL1 gene is the major mechanism that hampers TKI‐mediated tumor suppression in Ph+ ALL patients, therapies that directly modulate the expression of BCR‐ABL1 can be a reasonable strategy to overcome the current clinical problems related to TKIs. Together with our previous finding that Chb‐M' is highly effective against T315I mutation positive Ph+ ALL cells even in vivo with minimal side effects, 21 our work not only unveiled the novel role of RUNX1 transcription factor in the transactivation of BCR‐ABL1 expression, but also potentially provides alternative choice for the patients with TKI treatment‐resistant Ph+ ALL. Moreover, our study provides pieces of evidence that not only Ph+ ALL cells but also CML cells might be efficiently controlled by RUNX1 inhibition.
Conversely, other RUNX inhibitors that stand on other mechanisms of action (ex. Ro5‐3335 38 ) should also be tested in these tumors to further validate our results. In addition, addressing the roles of other RUNX family members such as RUNX2 and RUNX3 will help elucidate how RUNX family transcription factors generally contribute to the pathogenesis of BCR‐ABL1 positive tumors including Ph+ ALL. Although the role of BCR itself has not been fully elucidated in tumorigenesis, we are assuming that the RUNX inhibition strategy can potentially be applied to cancers that are dependent on BCR, such as metastatic colorectal cancer. 39 The efficacy of available RUNX inhibitors should also be tested in these tumors in future studies. From mutations of PDX cells (Table S4), based on known driver genes in pediatric B‐cell precursor ALL, 40 we extracted the 2 driver genes, MSH6 and CREBBP. Of them, CREBBP mutations have been identified as a mechanism of resistance in ALL, 41 and somatic variants in epigenetic modifiers including CREBBP can predict failure of response to imatinib in chronic‐phase CML. 42 These suggest that imatinib resistance in PDX cells may be due to the CREBBP mutation in addition to the high expression of BCR‐ABL1.
In conclusion, we have discovered a vital role of the RUNX1 transcription factor in the regulation of BCR‐ABL1 expression and in the maintenance of Ph+ ALL cells not only in human leukemia cell lines but also in PDX cells. RUNX1 could be an ideal target in the treatment of Ph+ ALL, and future clinical trials with our novel RUNX inhibitor Chb‐M' in these patients are awaited.
DISCLOSURE
The authors have no conflict of interest.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)); 19am0101101j0003, Basic Science and Platform Technology Program for Innovative Biological Medicine from the Japan Agency for Medical Research and Development (AMED); 15am0301005h0002, Grants from International Joint Usage/Research Center, the Institute of Medical Science, the University of Tokyo and Kanazawa University and Grant‐in‐Aid for Scientific Research (KAKENHI); 17H03597 and 16K14632. We thank Dr. H. Miyoshi (RIKEN, BRC) for kindly providing the following vectors; CSIV‐TRE‐RfA‐UbC‐KT, pENTR4‐H1tetOx1, CS‐RfA‐ETV, and CS‐RfA‐ETBsd.
Masuda T, Maeda S, Shimada S, et al. RUNX1 transactivates BCR‐ABL1 expression in Philadelphia chromosome positive acute lymphoblastic leukemia. Cancer Sci.2022;113:529–539. doi: 10.1111/cas.15239
Shintaro Maeda, Sae Shimada, Naoya Sakuramoto and Ken Morita contributed equally to this work.
Funding information
Japan Society for the Promotion of Science, Grant/Award Number: 17H03597; Japan Agency for Medical Research and Development, Grant/Award Number: 15am0301005h0002 and 19am0101101j0003
Contributor Information
Hiroshi Sugiyama, Email: hs@kuchem.kyoto-u.ac.jp.
Yasuhiko Kamikubo, Email: kamikubo.yasuhiko.7u@kyoto-u.ac.jp.
REFERENCES
- 1. Malard F, Mohty M. Acute lymphoblastic leukaemia. Lancet. 2020;395(10230):1146‐1162. [DOI] [PubMed] [Google Scholar]
- 2. Pui CH, Yang JJ, Hunger SP, et al. Childhood acute lymphoblastic leukemia: progress through collaboration. J Clin Oncol. 2015;33(27):2938‐2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006;354(2):166‐178. [DOI] [PubMed] [Google Scholar]
- 4. Linabery AM, Ross JA. Trends in childhood cancer incidence in the U.S. (1992‐2004). Cancer. 2008;112(2):416‐432. [DOI] [PubMed] [Google Scholar]
- 5. Smith MA, Seibel NL, Altekruse SF, et al. Outcomes for children and adolescents with cancer: challenges for the twenty‐first century. J Clin Oncol. 2010;28(15):2625‐2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pui CH, Pei D, Campana D, et al. A revised definition for cure of childhood acute lymphoblastic leukemia. Leukemia. 2014;28(12):2336‐2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jabbour E, O'Brien S, Konopleva M, Kantarjian H. New insights into the pathophysiology and therapy of adult acute lymphoblastic leukemia. Cancer. 2015;121(15):2517‐2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sive JI, Buck G, Fielding A, et al. Outcomes in older adults with acute lymphoblastic leukaemia (ALL): results from the international MRC UKALL XII/ECOG2993 trial. Br J Haematol. 2012;157(4):463‐471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ottmann OG, Pfeifer H. Management of Philadelphia chromosome‐positive acute lymphoblastic leukemia (Ph+ ALL). Hematology Am Soc Hematol Educ Program. 2009;1:371‐381. [DOI] [PubMed] [Google Scholar]
- 10. Druker BJ, Sawyers CL, Kantarjian H, et al. Activity of a specific inhibitor of the BCR‐ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome [published correction appears in N Engl J Med 2001 Jul 19;345(3):232]. N Engl J Med. 2001;344(14):1038‐1042. [DOI] [PubMed] [Google Scholar]
- 11. Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347(7):472‐480. [DOI] [PubMed] [Google Scholar]
- 12. Fielding AK, Rowe JM, Buck G, et al. UKALLXII/ECOG2993: addition of imatinib to a standard treatment regimen enhances long‐term outcomes in Philadelphia positive acute lymphoblastic leukemia. Blood. 2014;123(6):843‐850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Soverini S, De Benedittis C, Papayannidis C, et al. Drug resistance and BCR‐ABL kinase domain mutations in Philadelphia chromosome‐positive acute lymphoblastic leukemia from the imatinib to the second‐generation tyrosine kinase inhibitor era: the main changes are in the type of mutations, but not in the frequency of mutation involvement. Cancer. 2014;120(7):1002‐1009. [DOI] [PubMed] [Google Scholar]
- 14. Luciano L, Annunziata M, Attolico I, et al. The multi‐tyrosine kinase inhibitor ponatinib for chronic myeloid leukemia: real‐world data. Eur J Haematol. 2020;105(1):3‐15. [DOI] [PubMed] [Google Scholar]
- 15. Pavlovsky C, Chan O, Talati C, Pinilla‐Ibarz J. Ponatinib in the treatment of chronic myeloid leukemia and Philadelphia chromosome positive acute lymphoblastic leukemia. Future Oncol. 2019;15(3):257‐269. [DOI] [PubMed] [Google Scholar]
- 16. Okuda T, van Deursen J, Hiebert SW, Grosveld G, Downing JR. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell. 1996;84(2):321‐330. [DOI] [PubMed] [Google Scholar]
- 17. Hein D, Borkhardt A, Fischer U. Insights into the prenatal origin of childhood acute lymphoblastic leukemia. Cancer Metastasis Rev. 2020;39(1):161‐171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yamamoto K, Tsuzuki S, Minami Y, et al. Functionally deregulated AML1/RUNX1 cooperates with BCR‐ABL to induce a blastic phase‐like phenotype of chronic myelogenous leukemia in mice. PLoS One. 2013;8(9):e74864. Published 2013 Sep 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kamikubo Y, Zhao L, Wunderlich M, et al. Accelerated leukemogenesis by truncated CBF beta‐SMMHC defective in high‐affinity binding with RUNX1. Cancer Cell. 2010;17(5):455‐468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hyde RK, Zhao L, Alemu L, Liu PP. Runx1 is required for hematopoietic defects and leukemogenesis in Cbfb‐MYH11 knock‐in mice. Leukemia. 2015;29(8):1771‐1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Morita K, Suzuki K, Maeda S, et al. Genetic regulation of the RUNX transcription factor family has antitumor effects. J Clin Invest. 2017;127(7):2815‐2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Morita K, Maeda S, Suzuki K, et al. Paradoxical enhancement of leukemogenesis in acute myeloid leukemia with moderately attenuated RUNX1 expressions. Blood Adv. 2017;1(18):1440‐1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Morita K, Noura M, Tokushige C, et al. Autonomous feedback loop of RUNX1‐p53‐CBFB in acute myeloid leukemia cells. Sci Rep. 2017;7(1):16604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Morita K, Tokushige C, Maeda S, et al. RUNX transcription factors potentially control E‐selectin expression in the bone marrow vascular niche in mice. Blood Adv. 2018;2(5):509‐515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chou TC, Talalay P. Quantitative analysis of dose‐effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27‐55. [DOI] [PubMed] [Google Scholar]
- 26. Morita K, Masamoto Y, Kataoka K, et al. BAALC potentiates oncogenic ERK pathway through interactions with MEKK1 and KLF4. Leukemia. 2015;29(11):2248‐2256. [DOI] [PubMed] [Google Scholar]
- 27. Weisberg E, Manley PW, Cowan‐Jacob SW, Hochhaus A, Griffin JD. Second generation inhibitors of BCR‐ABL for the treatment of imatinib‐resistant chronic myeloid leukaemia. Nat Rev Cancer. 2007;7(5):345‐356. [DOI] [PubMed] [Google Scholar]
- 28. Hirase C, Maeda Y, Takai S, Kanamaru A. Hypersensitivity of Ph‐positive lymphoid cell lines to rapamycin: possible clinical application of mTOR inhibitor. Leuk Res. 2009;33(3):450‐459. [DOI] [PubMed] [Google Scholar]
- 29. Redaelli S, Piazza R, Rostagno R, et al. Activity of bosutinib, dasatinib, and nilotinib against 18 imatinib‐resistant BCR/ABL mutants. J Clin Oncol. 2009;27(3):469‐471. [DOI] [PubMed] [Google Scholar]
- 30. Müller MC, Cortes JE, Kim DW, et al. Dasatinib treatment of chronic‐phase chronic myeloid leukemia: analysis of responses according to preexisting BCR‐ABL mutations. Blood. 2009;114(24):4944‐4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bando T, Sugiyama H. Synthesis and biological properties of sequence‐specific DNA‐alkylating pyrrole‐imidazole polyamides. Acc Chem Res. 2006;39(12):935‐944. [DOI] [PubMed] [Google Scholar]
- 32. Minoshima M, Bando T, Shinohara K, Sugiyama H. Molecular design of sequence specific DNA alkylating agents. Nucleic Acids Symp Ser (Oxf). 2009;53:69‐70. [DOI] [PubMed] [Google Scholar]
- 33. Ossard‐Receveur A, Bernheim A, Clausse B, et al. Duplication of the Ph‐chromosome as a possible mechanism of resistance to imatinib mesylate in patients with chronic myelogenous leukemia. Cancer Genet Cytogenet. 2005;163(2):189‐190. [DOI] [PubMed] [Google Scholar]
- 34. Melo JV, Chuah C. Resistance to imatinib mesylate in chronic myeloid leukaemia. Cancer Lett. 2007;249(2):121‐132. [DOI] [PubMed] [Google Scholar]
- 35. Otero L, Ornellas MH, Dobbin J, de Souza FT. Double Philadelphia‐chromosome: a resistance factor on the imatinib mesylate therapy for chronic myeloid leukemia. Int J Lab Hematol. 2008;30(4):346‐348. [DOI] [PubMed] [Google Scholar]
- 36. Shah NP, Witte ON, Denny CT. Characterization of the BCR promoter in Philadelphia chromosome‐positive and ‐negative cell lines. Mol Cell Biol. 1991;11(4):1854‐1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sharma N, Magistroni V, Piazza R, et al. BCR/ABL1 and BCR are under the transcriptional control of the MYC oncogene. Mol Cancer. 2015;14:132. Published 2015 Jul 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cunningham L, Finckbeiner S, Hyde RK, et al. Identification of benzodiazepine Ro5‐3335 as an inhibitor of CBF leukemia through quantitative high throughput screen against RUNX1‐CBFβ interaction. Proc Natl Acad Sci USA. 2012;109(36):14592‐14597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jeitany M, Leroy C, Tosti P, et al. Inhibition of DDR1‐BCR signalling by nilotinib as a new therapeutic strategy for metastatic colorectal cancer. EMBO Mol Med. 2018;10(4):e7918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ueno H, Yoshida K, Shiozawa Y, et al. Landscape of driver mutations and their clinical impacts in pediatric B‐cell precursor acute lymphoblastic leukemia. Blood Adv. 2020;4(20):5165‐5173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mullighan CG, Zhang J, Kasper LH, et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature. 2011;471(7337):235‐239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nteliopoulos G, Bazeos A, Claudiani S, et al. Somatic variants in epigenetic modifiers can predict failure of response to imatinib but not to second‐generation tyrosine kinase inhibitors. Haematologica. 2019;104(12):2400‐2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material