

Abstract
Poly(lactic acid), PLA, which holds great promise as a biodegradable substitute of fossil resource-derived polyolefins, is industrially produced by the ring-opening polymerization of lactide using a potentially harmful tin catalyst. Based on mechanistic insights into the reaction of N-heterocyclic carbene (NHC) iron complexes with carbonyl substrates, we surmised and demonstrate here that such complexes are excellent catalysts for the bulk polymerization of lactide. We show that an iron complex with a triazolylidene NHC ligand is active at lactide/catalyst ratios of up to 10 000 : 1, produces polylactide with relatively high number-average molecular weights (up to 50 kg mol−1) and relatively narrow dispersity (Đ ∼ 1.6), and features an apparent polymerization rate constant kapp of up to 8.5 × 10−3 s−1, which is more than an order of magnitude higher than that of the industrially used tin catalyst. Kinetic studies and end-group analyses support that the catalytically active species is well defined and that the polymerization proceeds via a coordination–insertion mechanism. The robustness of the catalyst allows technical grade lactide to be polymerized, thus offering ample potential for application on larger scale in an industrially relevant setting.
Iron(ii) complexes containing a mesoionic triazolylidene ligand are highly efficient catalyst precursors for the ring opening polymerization of lactide to poly(lactic acid), surpassing other iron complexes and also industrially utilized Sn(oct)2.
Introduction
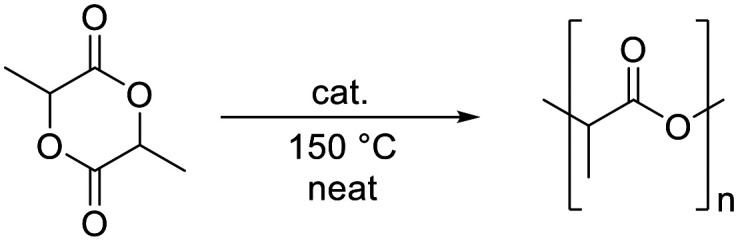
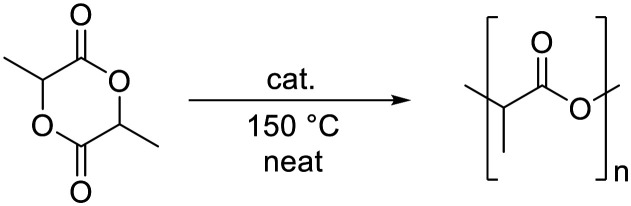
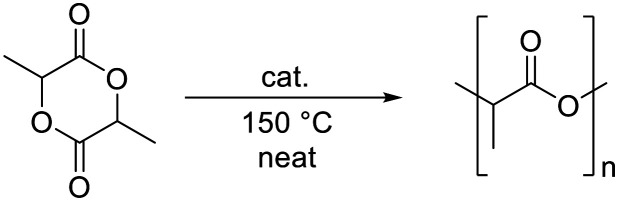
Despite the many benefits of polyolefins, these materials have become of increasing concern, especially because of significant micro- and macro-plastic pollution of oceans, arctic and high altitude areas,1,2 and also because of the limited availability of fossil resources, from which the corresponding monomers are produced.3 To address these issues, attempts are made to replace petrochemical-based, non-degradable polymers with biodegradable materials derived from renewable resources.4 At present, the most popular bio-based polymer is poly(lactic acid) (PLA), which is a non-toxic5,6 thermoplastic made from renewable resources, e.g. fermented corn starch.7 PLA is biodegradable and recyclable, and the possibility to vary the stereochemistry of the monomer allows access to different grades that cover a broad range of attractive properties.8 The synthesis of PLA can be accomplished via the polycondensation of lactic acid (Scheme 1, route A), but since this approach involves the formation of water as a by-product, which is difficult to remove, it is challenging to prepare high-molecular-weight polymers using this route. This problem can be overcome by employing the ring-opening polymerization (ROP) of lactide (LA) (Scheme 1, route B) in the melt.4
Scheme 1. Synthetic routes to poly(lactic acid) (PLA) include the direct condensation of lactic acid (route A) and the catalytic ring-opening polymerization (ROP) of lactide (LA, route B). Examples of previously employed iron catalysts for the ROP of lactide include complexes I–III (TFA = trifluoroacetate, NHC = N-heterocyclic carbene).

Currently, tin(ii)octanoate (Sn(Oct2)) is the prevailing catalyst used for the industrial production of PLA by lactide ROP.9 However, Sn(Oct2) is cytotoxic and requires a high purity of the monomer, since water, lactic acid, and other impurities reduce the control of the polymerization considerably.7 In the search for environmentally friendly, non-toxic catalysts for the ROP of LA, complexes based on Earth-abundant iron have shown some promise and are attracting increasing interest. Pioneering work in the late 1990's revealed catalytic activity of simple iron salts such as halides, acetates, or alkoxides, yet these systems require unfavorably low monomer (LA) to initiator (I) ratios, elevated temperatures of around 180 °C, and/or very long reaction times (>100 h) to achieve high monomer conversions.10–13 Effective polymerizations with higher [LA]/[cat] ratios have been reported for iron alkoxide catalysts in toluene,14,15 as well as for melt polymerization at 130 °C, leading to a monomer conversion of ca. 90% within 24 h (I, Scheme 1).16 Initial attempts to enhance the catalytic performance of iron complexes by introducing salen-type or other multidentate N,N or N,O-based ligands were met with little success; ROP with these catalysts generally required very low monomer to initiator ratios ([LA]/[cat] 100 : 1) and in many cases long reaction times.17–28 The Fe(N)4 system (II, Scheme 1) represents a notable exception, as this catalyst functions efficiently, even when used in low concentration.23 Recently, Thomas and co-workers developed a catalyst system that is based on an aminophenolate ligand and enables the stereoselective polymerization of LA at a [LA]/[cat] ratio of up to 800 : 1.29 The best performing iron catalyst to date was reported by Herres-Pawlis et al. in 2019 and is comprised of a guanidine-derived N,O-bidentate chelate. This complex (III, Scheme 1) shows a catalytic activity that is better than the industrially used Sn(Oct)2 and surpasses the activity of previously used iron catalysts by far.30
We reasoned that the use of N-heterocyclic carbenes (NHC)31–35 as spectator ligands might further enhance the robustness of the catalytically active species and hence improve the LA polymerization performance. Moreover, piano-stool iron NHC complexes36,37 have shown excellent catalytic activity in the conversion of carbonyl groups, e.g. in hydrosilylation reactions.38–40 Mechanistic studies indicated that the catalyst activation pathway of these complexes involves the initial interaction of the metal center with the carbonyl group, likely through CO dissociation.41 We suspected that a similar interaction of the iron center with the carbonyl group of LA might lead to substrate activation and cause ring-opening and polymerization. A classic coordination–insertion mechanism may then be accessible due to the presence of iodide or fortuitous water at the initiation stage, and subsequently the alkoxide of the growing polymer chain during propagation to promote the ring-opening of the coordinated LA.42,43 Here we demonstrate that iron complexes IV containing either an imidazolylidene or a triazolylidene44–46 as NHC unit constitute excellent catalyst precursors for LA polymerization with activities that exceed one of the industrially used Sn(Oct)2 and also considerably surpass the best iron catalysts reported so far.
Results and discussion
Complexes 1–4 (Fig. 1) were synthesized according to reported procedures.39,41,47 Complex 1 was analyzed by thermogravimetric analysis (TGA) which reveals that the complex is stable up to 250 °C (Fig. S1†), i.e., well above the typical temperature range for LA melt polymerization, which is generally performed between 180 and 200 °C.48 The initial, very moderate mass loss observed in the TGA trace is most likely caused by the evaporation of residual solvent trapped in the crystals.
Fig. 1. NHC iron complexes 1–4 used for the polymerization of lactide as well as carbene precursors trz-H and trz-CO2.

The catalytic activity of complex 1 for the ROP of rac-LA was initially tested by attempting the polymerization of technical grade monomer with 1,2-dichloroethane as solvent at a reaction temperature of 60 °C, and a 250 : 1 molar ratio of [LA]/[cat] (Table 1, entry 1). However, 1H NMR spectroscopic monitoring of the diagnostic resonances for the methine and CH3 protons, which appear at δH = 5.1 and 1.6, respectively for LA, and downfield shifted at δH = 5.2 and 1.7, respectively, for PLA, revealed that no polymerization took place under these conditions (Fig. S4†). When the solvent was removed and the temperature was increased to melt the monomer, polymerization took place (entries 2, 3), even though the [LA]/[cat] ratio was increased twenty-fold to 5000 : 1. The reaction proceeded much faster at 150 °C, where a conversion of 82% was observed after 2 h, than at 130 °C, where the conversion remained modest (23%), even after 24 h.
Key data for the polymerization of rac-LA with complex 1 and reference compoundsa.
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Pre-catalyst | [LA]/[cat] | m(LA) (mg) | cat (μmol) | Time (h) | Conversionb (%) | M n c (kg mol−1) | M w c (kg mol−1) | Đ c |
| 1d | 1 | 250 : 1 | 220 | 6.1 | 2 | 0 | n.d. | n.d. | n.d. |
| 2e | 1 | 5000 : 1 | 1600 | 2.2 | 24 | 23 | 24 | 42 | 1.8 |
| 3 | 1 | 5000 : 1 | 1600 | 2.2 | 2 | 82 | 48 | 89 | 1.9 |
| 4 | 1 | 5000 : 1 | 4800 | 6.6 | 2 | 57 | 38 | 63 | 1.6 |
| 5 | 1 | 1000 : 1 | 1600 | 11.1 | 2 | 91 | 21 | 53 | 2.5 |
| 6 | [FeCp(CO)2I] | 1000 : 1 | 1600 | 11.1 | 24 | 86f | 7.0 | 22 | 3.1 |
| 7 | trz-CO2 | 1000 : 1 | 1600 | 11.1 | 24 | 68g | 1.0 | 2.0 | 2.2 |
| 8 | trz-H | 1000 : 1 | 1600 | 11.1 | 24 | 3 | n.d. | n.d. | n.d. |
| 9 | — | — | 1600 | — | 24 | 2 | n.d. | n.d. | n.d. |
General conditions: bulk polymerization of rac-LA at 150 °C, unless noted otherwise; n.d. = not determined.
Determined by 1H NMR spectroscopy.
Determined by size-exclusion chromatography and corrected by the Mark–Houwink–Sakurada correction factor for PLA.
The polymerization was carried out in 1,2-dichloroethane (2.5 mL) at 60 °C.
The polymerization was carried out in bulk at 130 °C.
29% conversion after 2 h.
29% conversion after 2 h.
The observed activities, appreciable number-average molecular weights (Mn = 24 and 48 kg mol−1), and moderate dispersities (Đ = 1.8–1.9) established by size-exclusion chromatography (SEC) suggest that the catalytically active site is quite robust towards impurities that may be present in the technical grade LA, both at initiation and propagation stages. A small scale-up of the reaction (entry 4) led to a lower yield and a slightly reduced Mn under the same conditions, presumably due to less efficient stirring. When the [LA]/[cat] ratio was reduced to 1000 : 1 (entry 5), the rate of conversion was increased and Mn was reduced in comparison to the reaction carried out with the higher [LA]/[cat] ratio (entry 3), suggesting that (a fragment of) the iron complex may also act as initiator.
In order to investigate the function of the catalyst in more detail, control experiments with a carbene-free iron complex ([FeCp(CO)2I]) as well as with the carbene ligand (without the metal center) were performed. When the melt-polymerization was carried out with the carbene-free iron complex [FeCp(CO)2I] at 150 °C, the rate of polymerization was low, with only 29% of monomer conversion within 2 h (86% after 24 h, entry 6) compared to >90% conversion with complex 1 under otherwise identical conditions (entry 5). Moreover, the molecular weight was significantly reduced (Mn = 7 vs. 21 kg mol−1) and the dispersity higher (Đ = 3.1 vs. 2.5 with complex 1), suggesting a prominent role of the NHC ligand in keeping a well-defined and active catalytic site. Since it is known that free carbenes can initiate the polymerization of LA,49 the CO2-protected triazolylidene, trz-CO2 (Fig. 1),41 was evaluated as catalyst to probe whether complex 1 acts as a carbene releasing agent. Under the same conditions as applied for 1, trz-CO2 displayed some activity, but a reaction time of 24 h was required to reach a modest 68% conversion (entry 7). These results suggest that the catalytically active species in complex 1 is indeed an NHC–iron complex, rather than just the free carbene or the carbene-free iron center. Hardly any polymerization took place when the free triazolium salt trz-H (ref. 39) was used (entry 8), and no polymerization occurred in the absence of any catalyst (entry 9).
Further insights into the polymerization process were obtained by monitoring the Mn and the Đ of the PLA produced by the bulk polymerization of rac-LA at 150 °C catalyzed by complex 1 as a function of reaction time (Fig. 2a, Tables S1 and S2†) and monomer conversion (Fig. 2b). The Mn values saturate after an initial increase in the first minutes and scale linearly with the monomer conversion, up to a conversion of ca. 70%. Up to this point, the dispersity remains relatively low for melt conditions (Đ = 1.6).48 At higher conversion the dispersity increases and the Mn does not further grow, suggesting that the reaction kinetics change, perhaps due to the high viscosity and inhibited stirring, which favors side reactions such as chain transfer, termination and transesterifications. Similar effects have been noted when using Sn(Oct)2 as catalyst.50 The dispersity is comparable for reactions carried out with [LA]/[cat] ratios of 1000 : 1 and 5000 : 1 until about 50% conversion, while the molecular weights achieved with the higher [LA]/[cat] ratio are consistently higher by a factor of roughly two. The observed molecular weights correspond to only ca. 40% of the theoretical values for the reactions carried out with a [LA]/[cat] ratio of 1000 : 1 and only about 20% of the theoretical values for the reactions carried out with a higher [LA]/[cat] ratio of 5000 : 1 (Tables S1 and S2†). This discrepancy reflects that the polymerization is clearly not living, and suggests that either early termination is at play, possibly due to reactions with impurities in the technical grade monomer, or that the molecular weight is governed, at least to some extent, by the concentration of initiating species that are independent of the catalyst.
Fig. 2. Plots of the number-average molecular weight (Mn, black) and the dispersity (Đ, red) of PLA prepared by the bulk polymerization of rac-LA at 150 °C catalyzed by complex 1vs. a) time and b) conversion. Filled squares/circles are for [LA]/[1] = 1000 : 1 (m(LA) = 1.6 g) and open squares/circles are for [LA]/[1] = 5000 : 1 (m(LA) = 4.8 g).

To investigate how impurities in the technical grade LA impact the polymerization and gain further insights regarding the nature of the initiator, recrystallized LA was used for ROP with and without BnOH that was added as an auxiliary initiator (Tables S3 and S4†).30 These experiments were carried out with [LA]/[1] = 1000. Intriguingly, the polymerization of recrystallized LA in the absence of BnOH was slower than that of the technical grade monomer, while the addition of 1 eq of BnOH with respect to iron brings the conversion rate to a similar level (Fig. 3). These data allow the conclusion that iron complex 1, BnOH, and impurities in the technical grade monomer all serve as effective initiators for the iron-catalyzed LA polymerization. This is further supported by molecular weight data. When recrystallized LA was polymerized without BnOH, the Mn was close to the theoretical value calculated from [LA]/[1] up to a conversion of about 50% (Table S3†), consistent with initiation by the catalyst. As discussed above, under otherwise identical conditions, the polymerization of technical grade LA afforded polymers with an Mn of slightly less than half the theoretical value (Table S1†), suggesting that additional initiating species are in play. The deliberate addition of 1 eq BnOH as initiator afforded polymers whose Mn was in between those of the other series (technical grade or recrystallized LA), supporting the conclusion that BnOH and 1 initiate the reaction (Table S4†). Under all conditions, the polydispersity is generally higher than 1.5, except at short reaction times. At high monomer conversion the molecular weights drop and the distributions broaden, suggesting that depolymerization involving a back-biting mechanism and trans-esterification reactions become important.
Fig. 3. Plot of the monomer conversion in the melt-polymerization of LA (1.6 g, 11.1 mmol) catalyzed by complex 1 (11.1 μmol) vs. time. Conditions: 150 °C, 500 rpm using technical grade rac-LA (black squares), recrystallized rac-LA (red triangles), and recrystallized rac-LA with 1 eq BnOH as initiator (green circles).

In order to produce low-molecular weight polymers whose end groups can be analyzed by matrix-assisted laser desorption ionization time-of-flight (MALDI-ToF) spectrometry, crystallized rac-LA was polymerized in the melt at 150 °C with 1 as a catalyst and a [LA]/[cat] ratio of 50 : 1. In the absence of any auxiliary initiator, OH end-groups were identified by a signal at 903.10 Da (calcd. for (LA)6–OH–Na+ 904.25 Da), suggesting the presence of residual water as initiator (Fig. S5†). Notably, signals that can be attributed to cyclic oligomers (LA)n were also identified, e.g. at 887.78 Da, (calcd. for (LA)5Na+ 887.24 Da). These oligomers were present in the MALDI-ToF spectrum of a polymerization experiment with a higher monomer/catalyst ratio of 1000 : 1 at 1 min (Fig. S6†) and became dominant after prolonged reaction times (Fig. S7†). When BnOH was used as initiator at a 50 : 1 : 1 [LA]/[cat]/[I] ratio, BnO end-groups were indicated for most signals, e.g., 995.51 Da for H–(LA)6–OBn–Na+ (calcd. 995.84 Da; Fig. S8†). While no major signals indicative of macrocyclic products were detectable, the MALDI-data reveal signal separations for the oligomers of 72 Da, viz. one lactide monomer, hinting to transesterification reactions.20,27,51,52 Such post-polymerization modifications may be rationalized by the high activity of the catalyst as well as the elevated reaction temperatures.53
The MALDI-ToF spectra also reveal signals that can be attributed to small oligomers containing complex 1 without CO and I (666.55 Da, calculated for H–(LA)2–Fe(Cp)(trz) 666.25 Da) and without CO, I and Cp ligands (e.g. 890.07 Da, calcd. for H–(LA)4–Fe(trz) 890.30 Da; Fig. S9 and S10†). These data indicate that complex 1 or water can indeed initiate the polymerization, although there is no support for iodide as initiator. Notably, also signals attributed to (LA)2–trz-Na+ were detected (568.17 Da, calcd. 568.26 Da, Fig. S10†), which may indicate the contribution of free carbene as initiating species.54
A selected polymer sample produced by catalysis with complex 1 according to the conditions of entry 3, Table 1 was analyzed by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). The thermal decomposition of the polymer starts at 250 °C, well within the expected 230–260 °C range (Fig. S11†), while the DSC trace exhibits the typical features of amorphous poly(rac-lactide) with a glass transition temperature of around 52–55 °C and no melting or crystallizing events (Fig. S12†).55 Moreover, the polymer films do not show any significant coloration due to residual iron complex (Fig. S14†).
Because of the high activity and robustness of the polymerization catalyst derived from complex 1 with technical grade LA, related complexes 2–4 with different ligand architectures were investigated under comparable conditions (Fig. 1). Complexes 2–4 all feature the same iPr wingtip substituents but differ either in the heterocyclic carbene ligand (triazolylidene vs. imidazolylidene in 2vs.3), or in the arene ligand (Cp vs. Cp* in 3vs.4). The pertinent time-conversion profile indicates that the initial polymerization rates are lower for complex 2 than the parent complex 1 containing an aryl wingtip group, while the two imidazolylidene-based complexes 3 and 4 show similar initial activity (Fig. 4a). Notably, all three complexes 2–4 reach lower final conversions than 1 (62 ± 5% vs. 82%; Table 2). These different activities are also reflected in the apparent polymerization rate constants kapp, which were determined by linear regression of the polymerization data in a semilogarithmic plot (Fig. 4b, Tables S5–S7†).56 Complexes 1, 3, and 4 show all similar high rates with kapp as high as 8.5 × 10−4 s−1 for 1 (Table 2, entry 1). In comparison, exchanging the (Mes)(Bu) wingtip pattern in 1 for two iPr groups in complex 2 reduces kapp to 2.5 × 10−4 s−1 (entry 2). Introducing an imidazolylidene recovered activity (kapp = 7.3 × 10−4 s−1, entry 3), while the substitution of the Cp ligand with Cp* had no significant effect (kapp = 8.0 ± 0.6 × 10−4 s−1, entry 4). Notably, the rate differences observed as a function of ligand architecture reinforces the conclusion that a NHC iron species serves as initiator and polymerization catalyst.
Fig. 4. a) Plot of the monomer conversion during the bulk polymerization of technical grade rac-LA (1.6 g, 11.1 mmol) catalyzed by complexes 1–4 (2.2 μmol) vs. time. Conditions: 150 °C, 500 rpm, [LA]/[cat] = 5000 : 1. b) Plots of the ln([LA]0/[LA]t) values measured for the reactions reported in a) vs. time. The slope of the linear regressions yields kapp of complexes 1–4, (R2 > 0.98).

Key data for polymerization of rac-LA with different iron catalystsa.
| ||||
|---|---|---|---|---|
| Entry | Catalyst | k app (10−4 s−1) | Conversionb (%) | Conditions |
| 1 | 1 | 8.5 ± 0.6 | 82 | A, 2 h |
| 2 | 2 | 2.5 ± 0.2 | 57 | A, 2 h |
| 3 | 3 | 7.3 ± 0.3 | 63 | A, 2 h |
| 4 | 4 | 8.0 ± 0.6 | 67 | A, 2 h |
| 5 | II | 0.13 | 75 | A, 30 hc |
| 6 | 1 | 85.3 ± 3.3 | 82 | B, 4 min |
| 7 | III | 43.5 ± 3.5 | 68 | B, 1.6 mind |
Conditions A: rac-LA (1.6 g, 11.1 mmol), catalyst (2.2 μmol), [LA]/[cat] = 5000 : 1, 150 °C, 500 rpm, neat, time as indicated; conditions B: as A, except [LA]/[cat] ratio 500 : 1, 4 min, 260 rpm.
Determined by 1H NMR spectroscopy and average of two independent runs.
Data from ref. 23.
Data from ref. 30.
In order to benchmark the catalytic activity of complex 1 against state-of-the-art catalysts (cf.Scheme 1), polymerizations were performed neat at 150 °C, yet at a lower [LA]/[cat] ratio of 500 : 1. Under these conditions, the apparent polymerization constant kapp for complex 1 is one order of magnitude higher than under more dilute conditions (kapp = 85 × 10−4 s−1; Table 2, entry 6, see also Fig. S15, Table S8†). This activity is twice as high as that of complex III, which has been recognized as the fastest and most robust iron catalyst for the LA polymerization to date (kapp = 44 × 10−4 s−1, entry 7).30 The other iron complex reported to be highly active, complex II, displays an almost 100 times lower activity than 1 (kapp = 0.13 × 10−4 s−1, entry 5 vs. entry 1).23 These data underpin the considerable potential of complex 1 in LA polymerization.
Kinetic studies were performed with complex 1 by carrying out the polymerization or rac-LA at constant monomer concentration and varying catalyst loadings (Table 3, Fig. 5a). Under these conditions, the polymerization is clearly not controlled and the dispersity rather high. The apparent polymerization rate constant, kapp (Fig. 5b, Table S9†) displayed values ranging from 53.9 to 0.9 10−4 s−1 for [rac-LA]/[cat] ratios between 1000 : 1 and 10 000 : 1. The rate constant of propagation kp was obtained by plotting kappvs. the catalyst concentration (Fig. 5c, Table S9†).30 The linear fit suggests that the polymerization is first-order with respect to complex 1, in agreement with a molecularly well-defined active species that operates according to a coordination–insertion mechanism.42,57 An activated monomer mechanism would principally be conceivable as well, although the coordination insertion mechanism seems more likely based on the high monomer : initiator ratio, the occurrence of side reactions, the absence of a strong protic acid, and the insertion of single lactide units as shown with the 72 amu difference of polymers in MALDI-ToF spectrometry.58 Such a mechanism is also supported by the MALDI-ToF results, which reveal the iron NHC species as end group (Fig. S9 and S10†). However, the observed molecular weights are much lower than the ones calculated on the assumption that the iron complex serves as precursor for both initiation and polymerization (Table 3), which is in agreement with initiation by residual water or other impurities in the technical grade LA (see above). The propagation rate constant for the polymerization of LA with complex 1, kp = 0.74 (±0.04) M−1 s−1, exceeds that of the fastest iron catalyst known to date (viz.III, kp = 0.55 (±0.01) M−1 s−1) under identical reaction conditions.30 Importantly, this activity is also almost one order of magnitude higher than that of the industrially used Sn(Oct)2 catalyst (kp = 0.084 (±0.002) M−1 s−1). Moreover, the absence of induction period and the high conversions (25–70%) within the first 4 minutes of the reaction suggests that both initiation and propagation are fast.
Polymerization details at different [LA]/[cat] ratios with catalyst 1a.
| |||||||
|---|---|---|---|---|---|---|---|
| Entry | [LA]/[cat] | Conversion/yieldb (%) | k app (10−4 s−1) | theor Mnc (kg mol−1) | obs Mnd (kg mol−1) | M w d (kg mol−1) | Đ |
| 1 | 1000 : 1 | 91/69 | 53.9 ± 4.1 | 130 | 21 | 53 | 2.5 |
| 2 | 1500 : 1 | 86/80 | 33.0 ± 0.4 | 190 | 26 | 59 | 2.3 |
| 3 | 2775 : 1 | 74/64 | 12.3 ± 0.4 | 300 | 30 | 60 | 2.0 |
| 4 | 5000 : 1 | 82/67 | 8.5 ± 0.6 | 590 | 48 | 89 | 1.9 |
| 5 | 10 000 : 1 | 42/n.d. | 0.9 ± 0.1 | 600 | 17 | 49 | 2.8 |
General conditions: bulk polymerization of rac-LA (1.6 g, 11.1 mmol), cat (1.1–11.1 μmol), at 150 °C, 2 h.
The conversion was determined by 1H NMR spectroscopy, isolated yields gravimetrically after precipitation with ethanol; all values are averages of two independent polymerizations.
The theoretical number-average molecular weight was calculated from the monomer to catalyst ratio and the conversion of the reaction on the assumption that the catalyst initiates the polymerization.
Determined by size-exclusion chromatography and corrected by the Mark–Houwink–Sakurada correction factor for PLA.
Fig. 5. a) Plot of the monomer conversion during the bulk polymerization of rac-LA (1.6 g, 11.1 mmol) catalyzed by complex 1 (1.1–11.1 μmol) vs. time. Conditions: 150 °C, 500 rpm. b) Plots of the ln([LA]0/[LA]t) values measured for the reactions reported in a) vs. time. The slope of the linear regressions yields kapp of complex 1 for different [LA]/[cat] ratios (all R2 > 0.98). c) Plot of kappvs. the catalyst concentration. The slope of the linear fit to the data represents the propagation rate constant kp of the polymerization of LA with complex 1 (R2 = 0.986).

Conclusions
This study provides first evidence for catalytic activity of iron NHC complexes in lactide polymerization. The robustness of the catalysts enables the polymerization of technical grade rac-LA with all residual impurities, thus offering opportunities for utilizing commercially available rather than recrystallized lactide. The complexes are extraordinarily active and outperform other iron catalysts, featuring polymerization rates that are more than one order of magnitude higher than the industrially used Sn(Oct)2, thus offering a sustainable solution for LA polymerization. Conversions in the bulk reach 90% within hours and the PLA produced is characterized by high molar masses, with number-average molecular weights of up to 50 kg mol−1 and dispersity competitive to polymers from industrially used catalysts (Đ < 2). We show that the polymerization can be initiated by catalyst, impurities, and auxiliary initiator. Kinetic analyses support a well-defined molecular species as catalytically active entity, which offers opportunities for replacing currently implemented environmentally unfriendly catalysts such as tin(ii)octanoate with an environmentally more friendly, solvent-free process based on Earth-abundant iron for the industrial fabrication of biodegradable polylactide.
Experimental
1.1. General
ROP of rac-LA was carried out under an inert nitrogen atmosphere using standard Schlenk techniques. The rac-LA (Sigma-Aldrich, >96% purity by GC) was used without further purification (referred to as ‘technical grade LA’) or after recrystallization from hot toluene (‘recrystallized LA’) and stored at −35 °C in a nitrogen-filled glovebox. The synthesis of the triazolium carboxylate trz-CO2 (ref. 41) and complexes 1,392,413,47 and 4 (ref. 41) have been reported elsewhere. All other reagents were commercially available and used without further purification.
1.2. Physical methods
NMR spectra were measured at 25 °C on Bruker spectrometers operating at 300 or 400 MHz (1H NMR) and 75 or 101 MHz (13C{1H} NMR), respectively. Chemical shifts (δ in ppm, coupling constants J in Hz) were referenced to residual solvent resonances downfield to SiMe4. Assignments were made based on homo- and heteronuclear shift correlation spectroscopy. Elemental analyses and ESI and MALDI-ToF mass spectra were performed by the Mass Spectrometry Group at Universität Bern. UV irradiation was carried out using a UVP Blak-Ray B-100AP lamp.
1.3. Polymerization in Schlenk tubes
For a polymerization with a [LA]/[cat] ratio of 1000 : 1, a Schlenk tube equipped with a stir bar was filled with technical grade rac-lactide (1600 mg, 11.1 mmol) and the catalyst (0.011 mmol) in a glove box. The tube was closed with a septum and removed from the glovebox. The Schlenk tube was placed into a pre-heated oil bath at 150 °C. The reaction time was set to 0 when the reaction mixture was completely molten. A stirring speed of 500 rpm was maintained throughout the polymerization. For kinetic measurements, samples were withdrawn with a pipette under nitrogen and the conversion was determined by 1H NMR spectroscopy in CDCl3. The [LA]t/[LA]0 ratio was calculated by integration of the resonances corresponding to the H-atom for the polymer (δH = 5.24–5.09) and unreacted monomer (δH = 4.98–5.09). Apparent rate constants kapp were determined from the slopes of the ln([LA]t/[LA]0) vs. time plots. The rate constant of propagation kp was obtained from linear regression of kappvs. the catalyst concentration. After 2 h of reaction, the Schlenk tube was removed from the oil bath and the reaction mixture was dissolved in CH2Cl2. The dissolved polymer was twice precipitated from ethanol at room temperature, dried under vacuum, and characterized by gel permeation chromatography.
1.4. Size exclusion chromatography (SEC) and matrix-assisted laser desorption ionization time-of-flight (MALDI-ToF) analysis
Size exclusion chromatography (SEC) experiments were performed on an Agilent 1200 series HPLC system equipped with an Agilent PLgel mixed guard column (particle size = 5 μm) and two Agilent PLgel mixed-D columns (ID = 7.5 mm, L = 300 mm, particle size = 5 μm). Signals were recorded by a UV detector (Agilent 1200 series), an Optilab REX interferometric refractometer, and a miniDawn TREOS light scattering detector (Wyatt Technology Corp.). Samples were run using THF as the eluent at 30 °C and a flow rate of 1.0 mL min−1. Data analysis was carried out on Astra software (Wyatt Technology Corp.) and molecular weights were determined based on narrow molecular weight polystyrene standards calibration (from 540 to 2 210 000 g mol−1) and scaled by applying the Mark–Houwink–Sakurada correction factor59 deduced for PLA.60
The end group analysis was performed by MALDI-ToF on a Bruker Autoflex III Smartbeam equipped with a 337 nm Smartbeam laser in linear mode. The instrument was calibrated with the MSCAL1 calibration kit from Sigma Aldrich. Data were acquired by the FlexControl software and evaluated by the FlexAnalysis software (Bruker). A THF solution of trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) (10 mg mL−1) was used as matrix. The sample was dissolved in THF (100 μL, approx. 1 mg mL−1), of a solution of NaI (5 μL solution in iPrOH/H2O 1 : 1 at 1 mg ml−1) was added. A 0.5 μL sample solution was mixed with 0.5 μL of the matrix solution directly on the target spot. The spotted sample target was left to dry at room temperature. Spectra were acquired using 30% laser power, adding up 4–5 × 200 shots. The laser repetition rate was 100 Hz.
1.5. DSC and TGA measurements
Differential scanning calorimetry (DSC) was conducted on a Mettler Toledo DSC 2 STAR system. Samples were loaded in alumina crucibles Mettler Toledo part No. ME-26763 and measured under nitrogen up to 200 °C with a heating and cooling rate of 10 °C min−1. Thermogravimetric analysis (TGA) of the polymers was conducted on a Mettler Toledo TGA/DSC 1 STAR system under nitrogen between 25 and 550 °C with a heating rate of 10 °C min−1. Complex 1 was measured in a temperature range of 25–150 °C with a heating rate of 5 °C min−1 followed by 1 h at 150 °C.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
The authors thank Dr. Sètuhn Jimaja for assistance with SEC experiments and the European Research Council (ERC 615653), the Swiss National Science Foundation (Grants 200020_182633 and 200020_172619), and the Adolphe Merkle Foundation for generous financial support. The authors further acknowledge the Mark–Houwink–Sakaruda Correction developed by The Konkolewicz Group provided by the Macromolecular Alliance for Community Resources and Outreach.
Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy02143e
Notes and references
- Allen S. Allen D. Phoenix V. R. Le Roux G. Durántez Jiménez P. Simonneau A. Binet S. Galop D. Nat. Geosci. 2019;12:339–344. doi: 10.1038/s41561-019-0335-5. [DOI] [Google Scholar]
- Peeken I. Primpke S. Beyer B. Gütermann J. Katlein C. Krumpen T. Bergmann M. Hehemann L. Gerdts G. Nat. Commun. 2018;9:1505. doi: 10.1038/s41467-018-03825-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross G., Ross S. and Tighe B. J., Bioplastics: New Routes, New Products, in Brydson's Plastics Materials, Elsevier Ltd, 8th edn, 2016, pp. 631–652 [Google Scholar]
- Henton D. E. Gruber P. Lunt J. Randall J. Adv. Mater. 2000;12:527–577. doi: 10.1002/(SICI)1521-4095(200004)12:7<527::AID-ADMA527>3.0.CO;2-8. [DOI] [Google Scholar]
- Hébert C. D. Giles H. D. Heath J. E. Hogan D. B. Modderman J. P. Conn R. E. Food Chem. Toxicol. 1999;37:335–342. doi: 10.1016/S0278-6915(99)00014-9. [DOI] [PubMed] [Google Scholar]
- Conn R. E. Kolstad J. J. Borzelleca J. F. Dixler D. S. Filer L. J. Ladu B. N. Pariza M. W. Food Chem. Toxicol. 1995;33:273–283. doi: 10.1016/0278-6915(94)00145-E. [DOI] [PubMed] [Google Scholar]
- Rafael Auras H. T., Lim L.-T. and Selke S. E. M., Poly(Lactic Acid): Synthesis, Structures, Properties, Processing, and Applications, Wiley, Hoboken, 2011 [Google Scholar]
- Siracusa V. Rocculi P. Romani S. Rosa M. D. Trends Food Sci. Technol. 2008;19:634–643. doi: 10.1016/j.tifs.2008.07.003. [DOI] [Google Scholar]
- Mehta R. Kumar V. Bhunia H. Upadhyay S. N. J. Macromol. Sci., Polym. Rev. 2005;45:325–349. doi: 10.1080/15321790500304148. [DOI] [Google Scholar]
- Kricheldorf H. R. Serra A. Polym. Bull. 1985;14:497–502. doi: 10.1007/BF00271606. [DOI] [Google Scholar]
- Södergård A. Stolt M. Macromol. Symp. 1998;130:393–402. doi: 10.1002/masy.19981300133. [DOI] [Google Scholar]
- Stolt M. Södergård A. Macromolecules. 1999;32:6412–6417. doi: 10.1021/ma9902753. [DOI] [Google Scholar]
- Gibson V. C. Marshall E. L. Navarro-Llobet D. White A. J. P. Williams D. J. J. Chem. Soc., Dalton Trans. 2002:4321–4322. doi: 10.1039/B209703F. [DOI] [Google Scholar]
- O'Keefe B. J. O. Monnier S. M. Hillmyer M. A. Tolman W. B. J. Am. Chem. Soc. 2001;123:339–340. doi: 10.1021/ja003537l. [DOI] [PubMed] [Google Scholar]
- O'Keefe B. J. Breyfogle L. E. Hillmyer M. A. Tolman W. B. J. Am. Chem. Soc. 2002;124:4384–4393. doi: 10.1021/ja012689t. [DOI] [PubMed] [Google Scholar]
- Wang X. Liao K. Quan D. Wu Q. Macromolecules. 2005;38:4611–4617. doi: 10.1021/ma047545o. [DOI] [Google Scholar]
- Kricheldorf H. Boettcher C. Makromol. Chem. 1993;194:463–473. doi: 10.1002/macp.1993.021940209. [DOI] [Google Scholar]
- Idage B. B. Idage S. B. Kasegaonkar A. S. Jadhav R. V. Mater. Sci. Eng., B. 2010;168:193–198. doi: 10.1016/j.mseb.2009.10.037. [DOI] [Google Scholar]
- Fazekas E. Nichol G. S. Garden J. A. Shaver M. P. ACS Omega. 2018;3:16945–16953. doi: 10.1021/acsomega.8b02432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart J. A. McKeown P. Driscoll O. J. Mahon M. F. Ward B. D. Jones M. D. Macromolecules. 2019;52:5977–5984. doi: 10.1021/acs.macromol.9b01205. [DOI] [Google Scholar]
- Kang Y. Y. Park H. R. Lee M. H. An J. Kim Y. Lee J. Polyhedron. 2015;95:24–29. doi: 10.1016/j.poly.2015.04.006. [DOI] [Google Scholar]
- Silvino A. C. Rodrigues A. L. C. Resende J. A. L. C. Inorg. Chem. Commun. 2015;55:39–42. doi: 10.1016/j.inoche.2015.03.006. [DOI] [Google Scholar]
- Herber U. Hegner K. Wolters D. Siris R. Wrobel K. Hoffmann A. Lochenie C. Weber B. Kuckling D. Herres-Pawlis S. Eur. J. Inorg. Chem. 2017:1341–1354. doi: 10.1002/ejic.201601345. [DOI] [Google Scholar]
- Delle Chiaie K. R. Biernesser A. B. Ortuño M. A. Dereli B. Iovan D. A. Wilding M. J. T. Li B. Cramer C. J. Byers J. A. Dalton Trans. 2017;46:12971–12980. doi: 10.1039/C7DT03067C. [DOI] [PubMed] [Google Scholar]
- Duan R. Hu C. Li X. Pang X. Sun Z. Chen X. Wang X. Macromolecules. 2017;50:9188–9195. doi: 10.1021/acs.macromol.7b01766. [DOI] [Google Scholar]
- Liang Y. Duan R. L. Hu C. Y. Li L. L. Pang X. Zhang W. X. Chen X. S. Chin. J. Polym. Sci. 2018;36:185–189. doi: 10.1007/s10118-018-2068-0. [DOI] [Google Scholar]
- Driscoll O. J. Leung C. K. C. Mahon M. F. McKeown P. Jones M. D. Eur. J. Inorg. Chem. 2018:5129–5135. doi: 10.1002/ejic.201801239. [DOI] [Google Scholar]
- Driscoll O. J. Hafford-Tear C. H. McKeown P. Stewart J. A. Kociok-Köhn G. Mahon M. F. Jones M. D. Dalton Trans. 2019;48:15049–15058. doi: 10.1039/C9DT03327K. [DOI] [PubMed] [Google Scholar]
- Marin P. Tschan M. J. L. Isnard F. Robert C. Haquette P. Trivelli X. Chamoreau L. M. Guérineau V. del Rosal I. Maron L. Venditto V. Thomas C. M. Angew. Chem., Int. Ed. 2019;58:12585–12589. doi: 10.1002/anie.201903224. [DOI] [PubMed] [Google Scholar]
- Herres-Pawlis S. Rittinghaus R. D. Schäfer P. M. Albrecht P. Conrads C. Hoffmann A. Ksiazkiewicz A. Bienemann O. Pich A. ChemSusChem. 2019;12:2161–2165. doi: 10.1002/cssc.201900481. [DOI] [PubMed] [Google Scholar]
- Cazin C. S. J., N-Heterocyclic Carbenes in Transition Metal Catalysis and Organocatalysis, Springer, London, 2011 [Google Scholar]
- Díez-González S. Marion N. Nolan S. P. Chem. Rev. 2009;109:3612–3676. doi: 10.1021/cr900074m. [DOI] [PubMed] [Google Scholar]
- Hahn F. E. Jahnke M. C. Angew. Chem., Int. Ed. 2008;47:3122–3172. doi: 10.1002/anie.200703883. [DOI] [PubMed] [Google Scholar]
- Bourissou D. Guerret O. Gabbaï F. P. Bertrand G. Chem. Rev. 2000;100:39–91. doi: 10.1021/cr940472u. [DOI] [PubMed] [Google Scholar]
- Arduengo A. Bertrand G. Chem. Rev. 2009;109:3209–3210. doi: 10.1021/cr900241h. [DOI] [PubMed] [Google Scholar]
- Johnson C. Albrecht M. Coord. Chem. Rev. 2017;352:1–14. doi: 10.1016/j.ccr.2017.08.027. [DOI] [Google Scholar]
- Riener K. Haslinger S. Raba A. Högerl M. P. Cokoja M. Herrmann W. A. Kühn F. E. Chem. Rev. 2014;114:5215–5272. doi: 10.1021/cr4006439. [DOI] [PubMed] [Google Scholar]
- Jiang F. Bézier D. Sortais J. B. Darcel C. Adv. Synth. Catal. 2011;353:239–244. doi: 10.1002/adsc.201000781. [DOI] [Google Scholar]
- Johnson C. Albrecht M. Organometallics. 2017;36:2902–2913. doi: 10.1021/acs.organomet.7b00349. [DOI] [Google Scholar]
- Kandepi V. V. K. M. Cardoso J. M. S. Peris E. Royo B. Organometallics. 2010;29:2777–2782. doi: 10.1021/om100246j. [DOI] [Google Scholar]
- Nylund P. V. S. Segaud N. Albrecht M. Organometallics. 2021;40:1538–1550. doi: 10.1021/acs.organomet.1c00200. [DOI] [Google Scholar]
- Champouret Y. Hashmi O. H. Visseaux M. Coord. Chem. Rev. 2019;390:127–170. doi: 10.1016/j.ccr.2019.03.015. [DOI] [Google Scholar]
- Stanford M. J. Dove A. P. Chem. Soc. Rev. 2010;39:486–494. doi: 10.1039/B815104K. [DOI] [PubMed] [Google Scholar]
- Donnelly K. F. Petronilho A. Albrecht M. Chem. Commun. 2013;49:1145–1159. doi: 10.1039/C2CC37881G. [DOI] [PubMed] [Google Scholar]
- Vivancos Á. Segarra C. Albrecht M. Chem. Rev. 2018;118:9493–9686. doi: 10.1021/acs.chemrev.8b00148. [DOI] [PubMed] [Google Scholar]
- Schweinfurth D. Hettmanczyk L. Suntrup L. Sarkar B. Z. Anorg. Allg. Chem. 2017;643:554–584. doi: 10.1002/zaac.201700030. [DOI] [Google Scholar]
- Mercs L. Labat G. Neels A. Ehlers A. Albrecht M. Organometallics. 2006;25:5648–5656. doi: 10.1021/om060637c. [DOI] [Google Scholar]
- Schäfer P. M. Dankhoff K. Rothemund M. Ksiazkiewicz A. N. Pich A. Schobert R. Weber B. Herres-Pawlis S. ChemistryOpen. 2019;8:1020–1026. doi: 10.1002/open.201900199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor E. F. Nyce G. W. Myers M. Möck A. Hedrick J. L. J. Am. Chem. Soc. 2002;124:914–915. doi: 10.1021/ja0173324. [DOI] [PubMed] [Google Scholar]
- Yu Y. Storti G. Morbidelli M. Ind. Eng. Chem. Res. 2011;50:7927–7940. doi: 10.1021/ie200117n. [DOI] [Google Scholar]
- Ghosh S. Wölper C. Tjaberings A. Gröschel A. H. Schulz S. Dalton Trans. 2020;49:375–387. doi: 10.1039/C9DT04359D. [DOI] [PubMed] [Google Scholar]
- Culkin D. A. Jeong W. Csihony S. Gomez E. D. Balsara N. P. Hedrick J. L. Waymouth R. M. Angew. Chem., Int. Ed. 2007;46:2627–2630. doi: 10.1002/anie.200604740. [DOI] [PubMed] [Google Scholar]
- O'Keefe B. J. Hillmyer M. A. Tolman W. B. J. Chem. Soc., Dalton Trans. 2001:2215–2224. doi: 10.1039/B104197P. [DOI] [Google Scholar]
- Brulé E. Guérineau V. Vermaut P. Prima F. Balogh J. Maron L. Slawin A. M. Z. Nolan S. P. Thomas C. M. Polym. Chem. 2013;4:2414–2423. doi: 10.1039/C3PY00108C. [DOI] [Google Scholar]
- Lee T. S. and Tu B. S. T., Polylactic Acid: A Practical Guide for the Processing, Manufacturing, and Applications of PLA, Elsevier William Andrew Applied Science Publishers, 2019 [Google Scholar]
- Witzke D. R., Introduction of Properties, Engineering, and Prospects of Polylactide Polymers, Ph.D thesis, Michigan State University, 1997 [Google Scholar]
- Hermann A. Hill S. Metz A. Heck J. Hoffmann A. Hartmann L. Herres-Pawlis S. Angew. Chem., Int. Ed. 2020;59:21778–21784. doi: 10.1002/anie.202008473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basko M. Pure Appl. Chem. 2012;84:2081–2088. [Google Scholar]
- Eda G. Liu J. Shivkumar S. Eur. Polym. J. 2007;43:1154–1167. doi: 10.1016/j.eurpolymj.2007.01.003. [DOI] [Google Scholar]
- van Dijk J. A. P. P. Smit J. A. M. Kohn F. E. Feijen J. J. Polym. Sci. 1983;21:197–208. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.