

Summary
Because epigenetics is a critical component for gene expression, the hypothesis was tested that DNA methylation alterations are dynamic and continually change throughout gametogenesis to generate the mature sperm. Developmental alterations and stage-specific DNA methylation during gametogenesis from primordial germ cells (PGCs) to mature sperm are investigated. Individual developmental stage germ cells were isolated and analyzed for differential DNA methylation regions (DMRs). The number of DMRs was highest in the first three comparisons with mature PGCs, prospermatogonia, and spermatogonia. The most statistically significant DMRs were present at all stages of development and had variations involving both increases or decreases in DNA methylation. DMR-associated genes were identified and correlated with gene functional categories, pathways, and cellular processes. Observations identified a dynamic cascade of epigenetic changes during development that is dramatic during the early developmental stages. Complex epigenetic alterations are required to regulate genome biology and gene expression during gametogenesis.
Subject areas: Epigenetics, Stem cells research, Developmental biology
Graphical abstract
Highlights
-
•
A dynamic cascade of epigenetic change throughout gametogenesis from PGC to sperm
-
•
Most dramatic epigenetic alterations in PGC and spermatogenic stem cell stages
-
•
Different DNA methylation regions between and within stages were identified
-
•
Complex epigenetic alterations required for gene expression during gametogenesis
Epigenetics; Stem cells research; Developmental biology
Introduction
In contrast to somatic cells, germ cells go through a series of mitotic stages and then meiosis to generate the differentiated sperm or egg. During fertilization, the two mature gametes come together to produce the zygote, which will give rise to the embryo totipotent cells that will generate all somatic cells and tissues in the individual, as well as the gametes for the next generation. In mammals, the precursor primordial germ cells (PGCs) arise in the equipotent epiblast in response to bone morphogenic protein (BMP) signals (Coucouvanis and Martin, 1999; de Sousa Lopes et al., 2004; Lawson et al., 1999). The PGCs will develop and migrate to the genital ridge to colonize the undifferentiated gonad (Seisenberger et al., 2012). Upon gonadal sex determination, depending on the chromosomal sex, the mature PGCs will differentiate into the male or female germline lineage (Jost et al., 1973). In males, testis morphogenesis is initiated when the testis determining factor, SRY, is first expressed in the XY genital ridge Sertoli cell precursors (Gubbay et al., 1990; Hawkins et al., 1992; Koopman et al., 1991; Lovell-Badge and Robertson, 1990) and then expands toward the gonadal poles (Albrecht and Eicher, 2001; Bullejos and Koopman, 2005). In the outbred rat model gonad, at embryonic day 16 (E16), the early testis germ cells will develop into prospermatogonia during the male gonadal sex determination period (McCarrey, 2012; McCarrey, 2013). The germline then develops into spermatogonia by postnatal day 10 (P10) in the rat. Once a rat reaches puberty, the initial wave of spermatogenesis starts in the testes where the spermatocytes (including the meiotic stage of pachytene spermatocytes) develop. Following meiosis, the round haploid spermatids differentiate, and once they reach the mature spermatozoa stage, the cells are released into the lumen of the seminiferous tubules (Dym and Fawcett, 1971). The spermatozoa differentiate further after entering the initial caput stage of the epididymis and then mature during epididymal transit. Upon entering the cauda, which is the final stage of the epididymis, the sperm have gained the potential for motility and fertilization capacity (Orgebin-Crist et al., 1976; Cornwall, 2009). The mature caudal epididymal sperm are then stored in the vas deferens before ejaculation or degradation.
The migration of the primordial germ cells (PGCs) into the fetal gonad prior to gonadal sex determination precedes the differentiation of the PGC into prospermatogonia. In the testis, the expression of SRY in the supportive fetal somatic cells specifies these cells to differentiate into SOX9+ Sertoli cells. These cells will in turn proliferate in response to FGF9 (Chaboissier et al., 2004; Kim et al., 2006; Palmer and Burgoyne, 1991; Schmahl et al., 2004; Willerton et al., 2004) and TGFα (Levine et al., 2000). SOX9 and SRY regulate a significant number of shared and distinct downstream gene targets (Li et al., 2014; Bhandari et al., 2012), a finding consistent with studies demonstrating that ectopic expression of SOX9 is necessary and sufficient for testis differentiation (Chaboissier et al., 2004; Vidal et al., 2001). In the absence of SRY, FGF9, or SOX9, pregranulosa cells are specified and ovarian development initiates in XY males (Capel, 2006; Chaboissier et al., 2004; Colvin et al., 2001; Jeays-Ward et al., 2004; Kent et al., 1996; Koopman, 1999; Koopman et al., 1991). When somatic cells commit to testis or ovary lineages, the germ cells also undergo sex differentiation in the fetal gonad and become developmentally restricted (Adams and McLaren, 2002). In the male gonad, the prospermatogonia undergo a cell-cycle arrest at G0 and remain quiescent until after birth in the rodent (Western et al., 2008). DAZL (deleted in azoospermia-like) is a key factor enabling PGCs to respond to cues from the somatic environment and commit to gametogenesis. This RNA binding protein is required for meiotic chromosome condensation and meiotic prophase protein expression (Lin and Page, 2005). Dazl mRNA is detected in both male and female germ cells during fetal gonadal development (Seligman and Page, 1998). Dazl-deficient mice are infertile due to germ cell differentiation defects (Lin and Page, 2005). Stimulation with retinoic acid gene 8 (STRA8) and meiotic recombination protein 8 (REC8) are also required for meiotic DNA replication, as well as the subsequent processes of meiotic prophase, chromosome synapsis, and chromosome segregation (Koubova et al., 2006). Several of these genes were used in the current study as markers of the various gametogenesis stages.
Sperm production is a continuous developmental process throughout a male’s lifespan; this relies on a constant supply of an adult stem cell population with long-term renewal potential, the spermatogonial stem cells (SSCs). They are located along the basement membrane of the basal surface of the seminiferous tubules in the mature testes. The proliferation of these spermatogonial cells and their differentiation into mature spermatozoa is a highly regulated developmental process, and alterations in spermatogenesis can result in infertility. Although some genes (e.g., ZBTB16, SALL4, LIN28, CDH1 and FOXO1) have been shown to be markers for the undifferentiated spermatogonia (Buaas et al., 2004; Costoya et al., 2004; Eildermann et al., 2012; Goertz et al., 2011; Hobbs et al., 2012; Tokuda et al., 2007), others are specific to a subset of undifferentiated spermatogonia (e.g., ID4, PAX7, BMI1) that are specific to type A spermatogonia (Aloisio et al., 2014; Komai et al., 2014; Oatley et al., 2011) or genes (e.g., GFRa1 and NANOS2) specific to type A aligned spermatogonia (Meng et al., 2000; Suzuki et al., 2009; van Bragt et al., 2008). The regulation of this gene expression specificity to spermatogenic stages for the most part remains to be evaluated. The current study investigates the potential role of epigenetics during the developmental process and correlates the DNA methylation alterations observed with some of the above genes.
The first wave of epigenetic reprogramming occurs in the embryo, where the partial epigenetic erasure of DNA methylation is incomplete and maternal and paternal imprinted gene DNA methylation are retained (i.e., protected) for all subsequent somatic lineages. The second wave of partial DNA methylation erasure and reprogramming is more comprehensive and occurs upon subsequent development of the mature PGCs, which are the stem cells for the germline. Despite these two partial epigenetic DNA methylation erasures, epigenetic information can be passed from one generation to the next transgenerationally (Anway et al., 2005; Nilsson et al., 2018). During the first wave of embryonic epigenetic reprogramming, a repression of de novo DNA methyltransferases DNMT3a/b occurs, leading to partial DNA methylation erasure (Kagiwada et al., 2013; Kurimoto et al., 2008; Seisenberger et al., 2012; Seki et al., 2007; Shirane et al., 2016). During this developmental window, the process appears to be active (paternal genome) or passive (maternal genome) DNA demethylation erasure. During the second wave of partial PGC demethylation, the AID, APOBEC, or the TET family proteins, which are all involved in DNA methylation erasure, are either not transcribed in early PGCs or their enzymatic activity is not essential for the decrease in DNA methylation levels observed (Kagiwada et al., 2013; Popp et al., 2010; Vincent et al., 2013). However, DNMT1 (DNA methyltransferase 1) is maintained to prevent the dilution of DNA methylation at imprinted loci and some meiotic gene promoters (Hargan-Calvopina et al., 2016). The loss of Dnmt1 in PGCs results in female infertility due to premature meiotic entry and infertility in males, which is due to precocious differentiation of prospermatogonia (Hargan-Calvopina et al., 2016). During the second wave of epigenetic reprogramming, DNA methylation is at its lowest from TET1 and TET2 active removal of DNA methylation (Vincent et al., 2013; Hackett et al., 2013; Yamaguchi et al., 2013a). This DNA methylation erasure window generates the mature pluripotent PGCs that are within the indifferent early fetal gonad and provides the germline stem cells with the highest level of genome hypomethylation (Kobayashi et al., 2013; Yamaguchi et al., 2013b). Proper regulation of epigenetic processes is crucial throughout subsequent spermatogenesis to ensure normal embryonic development and sperm function. The sperm epigenome plays an essential role in establishing epigenetic marks in the early embryo following fertilization (Ben Maamar et al., 2021).
Although specific stages of PGC development have been extensively analyzed (Morgan et al., 2005; Hill et al., 2018; Molaro et al., 2014; Seisenberger et al., 2013), there is negligible comprehensive genome-wide epigenetic information on the developmental DNA methylation patterns from the mature PGCs through adult spermatogenesis. Previous studies in the mouse have extensively studied alterations in DNA methylation during PGC maturation (Morgan et al., 2005; Hill et al., 2018; Molaro et al., 2014; Seisenberger et al., 2013). Spermatogonia development and commitment to spermatocytes have also been studied with regard to DNA methylation studies (Liu et al., 2019; Fend-Guella et al., 2019; Gaysinskaya et al., 2018; Kubo et al., 2015). Fewer studies at the other stages of development such as during meiosis and haploid spermatid cell development have been reported (Mallol et al., 2019; Schutte et al., 2013). Although discussed in reviews on methylation dynamics during gametogenesis (Morgan et al., 2005; Seisenberger et al., 2013), few studies have examined DNA methylation from mature PGC through sperm development. Similarly, analysis of specific gene expression and/or transcription analysis have been performed at specific stages of development, but not concurrently for all stages (Carrell et al., 2016). Other epigenetic processes such as noncoding RNAs (e.g., piRNAs) have also been examined in isolated stages of gametogenesis (Luk et al., 2014; Kotaja, 2014; Bao et al., 2014; Yadav and Kotaja, 2014). Therefore, a better understanding of the normal developmental DNA methylation patterns is needed throughout gametogenesis.
Previous studies have shown that an exposure of a gestating female to environmental toxicants, such as the pesticide DDT (dichlorodiphenyltrichloroethane) or the fungicide vinclozolin, during the fetal gonadal sex determination developmental window promotes an altered developmental programming of the male germline epigenome (Anway et al., 2005; Skinner et al., 2013b, 2018, 2019; Nilsson et al., 2018; Ben Maamar et al., 2019). Stress, high-fat diets, caloric restriction, and a variety of toxicants have also been linked to the epigenetic transgenerational inheritance phenomenon (Nilsson et al., 2018; Skinner, 2014; Soubry, 2015; Vaiserman et al., 2017). These studies show changes in epigenetic DNA methylation programming. Epigenetic transgenerational inheritance refers to the transmission of epigenetic information to the next generation through the germline in the absence of direct exposure. The main epigenetic processes previously studied are DNA methylation, noncoding RNAs, chromatin structure, histone modifications, and RNA methylation (Jirtle and Skinner, 2007). These epigenetic changes can have short- or long-term effects on genome activity and become transgenerational when the germline is involved (Anway et al., 2005).
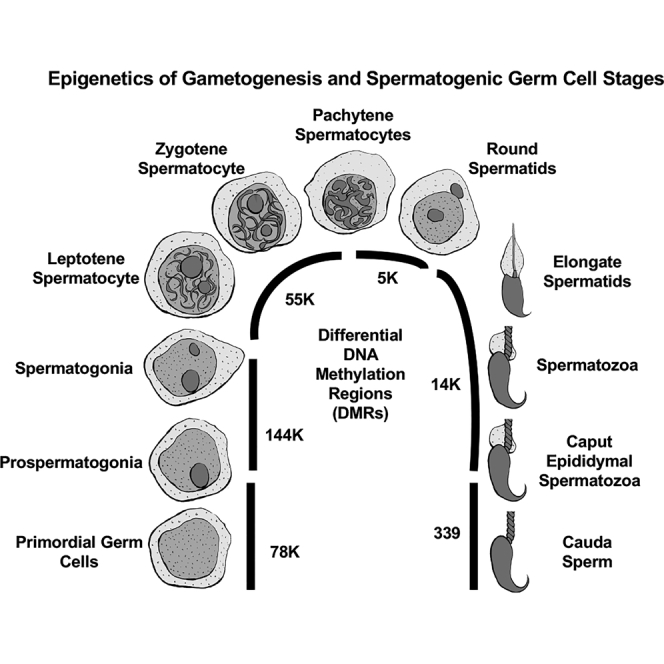
In the current study, the outbred rat model is used to assess normal development patterns of DNA methylation from the mature PGCs to sperm. An outbred rat model was used to avoid the issues of inbreeding depression of epigenetics known to occur in most mouse models and other inbred models (Vergeer et al., 2012; Pennisi, 2011; Guerrero-Bosagna et al., 2012). The control generation animals from previous toxicant exposure studies (Skinner et al., 2019; Ben Maamar et al., 2019) were used for the current study to establish the baseline normal control epigenetic developmental programming during spermatogenesis of mature PGCs to sperm. The rat male germline developmental stages examined include embryonic day 13 (E13) mature primordial germ cells (PGCs), embryonic day 16 (E16) prospermatogonia, postnatal day 10 (P10) spermatogonia, adult pachytene spermatocytes, round spermatids, caput epididymal spermatozoa, and caudal epididymal sperm (Figure 1). The current study was designed to investigate the developmental programming and origins of differential DNA methylation regions (DMRs) between developmental stages during normal control development without any environmental exposure. The hypothesis tested is that a dramatic cascade of epigenetic changes is initiated at the early stages of male germline development to then form the final DNA methylation patterns in the mature sperm.
Figure 1.

Developmental stage comparisons
Black line indicates stages compared.
(A) Spermatogenesis and spermatogenic germ cell stages.
(B) Differential DNA methylation during gametogenesis. Total number DMR at p < 1e-04 for each stage comparison, indicated in horizontal bar.
Results
Gametogenesis stage germ cell isolations
Seven different germ cell stages from male outbred rats were used in this experimental design for epigenetic analysis. Gestating female rats at 90 days of age were used to isolate the developing fetus at gestational day 13 or 16. At the embryonic day 13 (E13) stage of rat fetal development, the mature PGCs have migrated to colonize the indifferent gonad prior to gonadal sex determination. At embryonic day 16 (E16), the early male gonad (testis) contains prospermatogonia (McCarrey, 2012; McCarrey, 2013; Jirtle and Skinner, 2007; Harikae et al., 2013). At the postnatal day 10 (P10), the testis contains predominantly spermatogonia and at 10 months of development, the adult pachytene spermatocytes, round spermatids, caput spermatozoa, and cauda epididymal sperm are all present. The isolation of PGCs, prospermatogonia, spermatogonia, pachytene spermatocytes, and round spermatid cell types was done using a gravity sedimentation StaPut protocol, and cell purity assessed (McCarrey, 2012; McCarrey, 2013; Skinner et al., 2019; Ben Maamar et al., 2019), as described in the Methods. The epididymal caput spermatozoa and cauda sperm were isolated from the appropriate section/stage of the epididymis from the same animals, as described in the Methods (Skinner et al., 2019; Ben Maamar et al., 2019). Different sample pools, each with different individual sets of animals from different litters, were obtained with approximately 20 different males' gonads per pool for the E13 stage, 5–6 males' gonads per pool for the E16 stage, 6–7 males' gonads per pool for the P10 stage, and three adult males' gonads per pool for the adult stages. Therefore, 3–20 different males' gonads, depending on the developmental stage, were used in the analysis of each of the different sample pools studied. Three pools were collected for each of the developmental stages, except for E13 stage, which was limited to two pools. The purity of the cell populations has previously been shown to be PGCs (80%–85%), prospermatogonia (80%–85%), spermatogonia (80%–85%), pachytene spermatocytes (85%–90%), round spermatids (85%–90%), caput spermatozoa (100%), and cauda sperm (100%) (McCarrey, 2012; McCarrey, 2013; Ben Maamar et al., 2019; Skinner et al., 2019). Recent studies using more advanced analyses have also confirmed these previous purity assessments in the spermatogenic cell stages (Mutoji et al., 2016; Lesch et al., 2013). The genomic DNA was isolated from each of the purified developmental stage (Figure 1) pools and processed for subsequent epigenetic analysis.
Gametogenesis stage germ cell DNA methylation analysis
A methylated DNA immunoprecipitation (MeDIP) procedure followed by next-generation sequencing for a MeDIP-Seq protocol was performed to analyze the differential DNA methylation regions (DMRs), as described in the Methods (Beck et al., 2017, 2021). The MeDIP-Seq procedure was selected due to its efficiency of assessing low-density CpG regions that constitute >90% of the genome, so provides a more genome-wide analysis than other procedures such as bisulfite sequencing due to CpG density bias (Beck et al., 2021). The E13 PGCs were compared with the E16 prospermatogonia, the prospermatogonia with the P10 spermatogonia, the spermatogonia with the adult pachytene spermatocytes, the pachytene spermatocytes with the adult round spermatids, the round spermatids with the adult caput epididymal spermatozoa, and the caput epididymal spermatozoa with the adult caudal epididymal sperm (Figure 1) in order to identify the differential DNA methylation regions (DMRs) that changed between each developmental stage of the male germline (Figure 2). The sequencing alignment was 90%–95% for all analyses, and a minimum of 40 reads was required for a DMR. A DMR EdgeR p value was used to identify statistically significant DMRs from each comparison (Figure 2), which correlated with a multiple testing correction FDR (false discovery rate) value of <0.05 for all comparisons, except the caput versus cauda comparison at <0.1. The All Windows is all DMRs with at least one 1000 bp region (window) with statistical significance and the Multiple Windows being ≥2 nearby significant 1000 bp windows. The number of DMRs with multiple windows is also presented for the bolded p values in Figure 2. Developmental stage comparisons showed approximately 336–144,046 DMRs at p < 1e-04 (Figures 1B and 2). The epigenetic alterations were highest between the E16 prospermatogonia and the P10 spermatogonia, which had more than 38,000 DMRs at p < 1e-07 (Figure 2). The top 1000 statistically significant DMRs were further analyzed for general genomic characteristics. The number of DMRs with an increase versus decrease in DNA methylation is presented in Figures 2C and 2G, with between 40% and 60% of the DMRs having an increase in DNA methylation. Interestingly, 75% of the prospermatogonia versus the spermatogonia comparison DMRs showed a decrease in DNA methylation, and 25% of the DMRs showed an increase in DNA methylation, whereas the spermatogonia versus pachytene spermatocyte DMRs showed 25% with a decrease in methylation and 75% of the DMRs with an increase in DNA methylation (Figure 2G and Tables S1–S6). These individual stage comparisons were investigated such that the comparisons of all can provide insight into the dynamic DNA methylation changes throughout development.
Figure 2.

DMR identification
The number of DMRs found using different p value cutoff thresholds. The All Window column shows all DMR sites. The Multiple Window column shows the number of DMRs with ≥2 significant nearby windows at a p value threshold indicated in the bolded line.
(A) E13 primordial germ cells versus E16 prospermatogonia.
(B) E16 prospermatogonia versus P10 spermatogonia.
(C) P10 spermatogonia versus pachytene spermatocytes.
(D) Pachytene spermatocytes versus round spermatids.
(E) Round spermatids versus caput epididymal spermatozoa.
(F) Caput epididymal spermatozoa versus cauda sperm.
(G) Developmental DMR numbers with an increase or decrease in methylation alteration (top 1000 DMR) from the maximum log-fold-change (maxLFC). Light shading, positive increase in methylation and dark shading, negative decrease in DNA methylation.
The chromosomal locations of the DMRs detected from each developmental stage comparison are presented in Figure 3. The top 1000 statistically significant DMRs were used, due to high DMR numbers in the initial stages. The locations of DMRs are designated by the red arrowheads, and the black boxes indicate clusters of DMRs. In most stages, all the chromosomes, except in some comparisons the Y or mitochondrialtDNA (MT), were found to contain DMRs. Interestingly, some of the DMR clusters were in common regions on the same chromosome during different developmental stage comparisons (Figure 3). The genomic features of the DMRs detected at each developmental stage comparison were investigated. The CpG density of the DMRs at all stages was 1–5 CpG per 100 bp with 1 or 2 CpG being prevalent (Figure S1). Similar observations were observed with DMRs from p < 1e-05 in comparison to the top 1000, Figure S2. This represents a low-density CpG desert (Skinner and Guerrero-Bosagna, 2014), which has been observed with previous transgenerational DMRs. The lengths of the DMRs detected in all the developmental stage comparisons for the top 1000 were between 1 and 10 kb, with 1 or 2 kb length being predominant (Figure S3). Similar observations were made with DMRs from p < 1e-05 in comparison with the top 1000 (Figure S4). In considering the genomic features of the DMRs generated from each comparison, a principal component analysis (PCA) was performed with each comparison to show distinct principal component separations in the pools for each stage (Figure S5). Therefore, each individual stage comparison DMRs during development are distinct in the PCA.
Figure 3.

DMR chromosomal locations
The DMR locations on the individual chromosomes. All DMRs at p value threshold for top 1000 DMR presented. Chromosome number and length (megabase) presented. The red arrowhead identifies a DMR site and black box a cluster of DMRs.
(A) E13 primordial germ cells versus E16 prospermatogonia.
(B) E16 prospermatogonia versus P10 spermatogonia.
(C) P10 spermatogonia versus pachytene spermatocytes.
(D) Pachytene spermatocytes versus round spermatids.
(E) Round spermatids versus caput epididymal spermatozoa.
(F) Caput epididymal spermatozoa versus cauda sperm.
DNA methylation alterations during gametogenesis
Specific DNA methylation alterations of the DMRs for each stage of development were investigated to identify the patterns of change developmentally. The top 100 statistically significant DMRs from each developmental stage comparisons were studied to assess the percent mean read depth for each stage comparison DMR data. The percent mean read depth values for each DMR were separated into an increase or decrease in DNA methylation, for instance, caput greater than cauda in the caput versus cauda data (represents a decrease in DNA methylation) and cauda greater than caput in caput versus cauda data (represents an increase in DNA methylation), which are presented as a percent scaled mean read depth (Figure 4). The 100 most significant caput versus cauda DMRs included 34 DMRs with decreased DNA methylation (caput > cauda) and 66 DMRs with increased DNA methylation (cauda > caput) (Figures 4K and 4L). For this developmental stage comparison, the methylation alterations generally became more variable with no distinct patterns at earlier stages. The round spermatids versus caput included 81 DMRs with decreased DNA methylation (round spermatid > caput) and 19 DMRs with increased DNA methylation (round spermatid > caput) (Figures 4I and 4J). The pachytene spermatocyte versus round spermatid included 44 DMRs with decreased DNA methylation (pachytene spermatocyte > round spermatid) and 56 DMRs with increased DNA methylation (pachytene spermatocyte < round spermatid) (Figures 4G and 4H). There was a dramatic alteration in DNA methylation between the pachytene spermatocyte and round spermatid stages and between the P10 spermatogonia and adult pachytene spermatocyte stages. The P10 spermatogonia versus pachytene spermatocytes included 74 DMRs with decreased DNA methylation (P10 spermatogonia > pachytene spermatocytes) and 26 DMRs with increased DNA methylation (P10 spermatogonia < pachytene spermatocytes) (Figures 4E and 4F). At this developmental stage comparison, a dramatic alteration in DNA methylation was also observed between the P10 spermatogonia and the pachytene spermatocytes. The E16 prospermatogonia versus P10 spermatogonia included 82 DMRs with decreased DNA methylation (E16 prospermatogonia > P10 spermatogonia) and 18 DMRs with increased DNA methylation (E16 prospermatogonia < P10 spermatogonia) (Figures 4C and 4D). Therefore, the E16 prospermatogonia and PGCs or P10 spermatogonia stage DMRs appear to have consistent patterns of DNA hyper- or hypomethylation. The PGC versus E16 prospermatogonia included 63 DMRs with decreased DNA methylation (PGC > E16 prospermatogonia) and 37 DMRs with increased DNA methylation (PGC < E16 prospermatogonia) (Figures 4A and 4B).
Figure 4.

DMR developmental time course
The scaled mean RPKM read depth is shown for the top 100 statistically significant DMRs for each developmental stage comparison.
(A) PGC greater than E16 (∗ = p < 1e-05) n = 72.
(B) E16 greater than PGC (∗ = p < 1e-05) n = 28.
(C) E16 greater than P10 (∗ = p < 1e-05) n = 15.
(D) P10 greater than E16 (∗ = p < 1e-05) n = 85.
(E) P10 greater than pachy (∗ = p < 1e-05) n = 81.
(F) Pachy greater than P10 (∗ = p < 1e-05) n = 19.
(G) Pachy greater than RS (∗ = p < 1e-05) n = 41.
(H) RS greater than pachy (∗ = p < 1e-05) n = 59.
(I) RS greater than caput (∗ = p < 1e-05) n = 64.
(J) Caput greater than RS (∗ = p < 1e-05) n = 36.
(K) Caput greater than cauda (∗ = p < 1e-05) n = 35.
(L) Cauda greater than caput (∗ = p < 1e-05) n = 65.
Following the analysis of individual stage comparison DMR developmental patterns (Figure 4), the next analysis determined the potential overlap and specificity of DMRs for each comparison. A Venn diagram of the PGCs and spermatogonia comparison demonstrated negligible overlap, with the highest being 120 DMRs between the PGCs versus prospermatogonia comparison and the prospermatogonia versus spermatogonia comparison (Figure 5A). In addition, a comparison of the spermatogonia stages and the sperm demonstrates negligible overlap, with the highest being 62 DMRs common to both the round spermatid versus spermatozoa and spermatogonia versus pachytene spermatocytes comparisons (Figure 5B). Therefore, the specific DMRs detected at each developmental stage comparison were primarily unique at the highest statistical threshold cutoff (i.e., top 1000). However, some DMRs were present throughout development (Figure 4). An extended overlap involving a more stringent top 1000 threshold DMRs with the other stages at a lower stringency threshold (p< 0.001) demonstrated that several comparisons have high overlap (Figure 5C). The major overlaps were observed between the PGCs, prospermatogonia, and spermatogonia, with the PGCs versus E16 prospermatogonia having an 82.4% overlap with the E16 prospermatogonia versus P10 spermatogonia. Interestingly, DMRs from the comparison of E16 prospermatogonia and spermatogonia had a 94.8% overlap with the DMRs from the PGCs and E16 prospermatogonia comparison. There was also a 71.1% overlap with the E16 prospermatogonia versus spermatogonia comparison DMRs with the spermatogonia versus pachytene spermatocytes comparison. A 51.5% overlap was observed between the pachytene versus round spermatids and spermatogonia versus pachytene. Interestingly, there was a 53.1% overlap with the round spermatid versus caput spermatozoa comparison with the spermatogonia versus pachytene comparison and 55.1% with the prospermatogonia versus spermatogonia comparison (Figure 5C). The other comparisons were lower and ranged from 0.1% to 43%. Therefore, the specific DMRs are present at many stages when a reduced stringency is considered. This analysis examined the alterations in DNA methylation between two adjacent stages of development, and a complex developmental cascade of DMR development is observed.
Figure 5.

Developmental DMR overlaps
(A) Venn diagram DMR overlaps between PGC stage and spermatogenic stages (top 1000 DMR).
(B) DMR overlaps between sperm stage and spermatogenic stages (top 1000 DMR).
(C) Extended overlap DMRs for all stage comparisons (top 1000 DMR versus p < 0.001).
An alternative type of analysis from the DMRs between stages performed involved DNA methylation site read depth comparisons. The MeDIP-Seq for methylated site read depth was used to compare the level of methylation for each stage, as described in the Methods. This type of analysis is not dependent on alterations in DNA methylation between stages for DMRs, but general changes in DNA methylation compared with a specific stage (e.g., cauda sperm). The DNA methylation for read depth was normalized for library size and sequence efficiency then reported as RPKM (reads per kilobase per million reads) where RPKM of 20 is approximately 150 raw reads (Figure 6). The top 100 DNA methylation sites in cauda sperm were compared with all the other stages and presented in Figure 6A. All stages had similar sites as the top 100 cauda sperm that were more variable with the stem cell stages. A combination of all other stages of top 100 DNA methylation sites with a reduction in cauda sperm identified the largest reduction in read depth in cauda sperm, Figure 6B. A comparison of each individual stage top 100 DNA methylation sites with all the other stages is presented in Figure S6. In all cases DNA methylation was detected at all stages with often similar profile of change. A pairwise comparison of all the different stages of top 100 DNA methylation sites is presented in Figure 6C. A high level of overlap was observed among the top 100 sites. Therefore, a similar analysis of the top 1000 DNA methylation sites between the different developmental stages was performed (Figure 6D). Both top 100 and 1000 DNA methylation comparisons demonstrate common sites between the stages, with the PGC having approximately 50% of the sites common with the cauda sperm. The other developmental stage sites demonstrate the PGC, prospermatogonia, and spermatogonia being more similar, the pachytene and round spermatid being similar, and caput and cauda sperm similar (Figures 6C and 6D). Therefore, the observations with the DNA methylation sites between developmental stages demonstrated similar observations as observed with the DMR analysis. The concept that DNA methylation is simply increased from PGC to cauda sperm is not supported, but rather is dynamic and complex.
Figure 6.

Gametogenesis stage methylation sites read depth RPKM comparison with other stages
(A) Cauda sperm top 100 methylation sites compared with other stages.
(B) Other stage top 100 methylation sites compared with cauda sperm.
(C) Pairwise comparison top 100 stage-specific methylation sites.
(D) Pairwise comparison top 1000 stage-specific methylation sites.
Differential DNA methylation region gene associations
The DMRs from each stage comparison are listed for the top 1000 DMRs in Tables S1–S6. The genomic locations and features are listed, as well as the associated genes within 10 kb to include potential promoters. The gene associations for all DMRs for the top 1000 for each stage were identified and functionally categorized. The gene functional categories are presented in Figure 7A for DMRs derived from each stage comparison. The signaling, transcription, metabolism, receptor, and cytoskeleton categories were the most predominant for all comparisons. The top five categories in terms of number of DMR-associated genes were selected to examine the dynamics of these gene categories during gametogenesis (Figure 7B). In general, the numbers of DMR-associated genes in a particular category were seen throughout gametogenesis or increase in the later stages of development. Therefore, similar DMR-associated gene categories are observed between the stages, and the specific category genes were considered throughout gametogenesis.
Figure 7.

The DMR-associated gene functional categories
(A) The top 1000 statistically significant DMRs are presented for each developmental stage comparison, indicated in the inset, with number of DMRs for each category.
(B) The top categories are individually presented for DMR numbers versus the different developmental stages.
The DMR-associated genes at each stage were also used to identify predominant pathways at each stage comparison with a KEGG pathway analysis. The predominant DMR-associated gene pathways identified were metabolism, olfactory transduction, pathways in cancer, MAPK, and PI3K-Akt signaling (Figure S7). Further analysis of DMR-associated genes with cellular components and processes for each stage comparison identified common extracellular, membrane, organismal, and nuclear associations (Figure 8). Observations demonstrate all developmental comparisons were represented in most of the cellular components, with Golgi and mitochondrial processes most represented in the prospermatogonia versus spermatogonia comparison.
Figure 8.

DMR-associated genes overrepresented in cellular and biological processes pathways as determined with Pathway Studio software (Elsevier Inc.)
The DMR-associated genes present in the top 5–10 pathways having the lowest p values (<1e-03) for overrepresentation for each comparison are shown. Index for colors designating different developmental comparisons presented.
An additional analysis investigated the utility of the dataset generated to examine specific genes known to be expressed in specific stages of development. The genes for PGCs were Tet and Dazl (Tang et al., 2015), for spermatogonia Id4 (Lord and Oatley, 2017), spermatocytes Stra8 (Anderson et al., 2008), and for spermatids protamines Prm1 and Prm2 (Steger, 1999). The developmental pattern of the associated DNA methylation at the gene locations is presented in Figure 9. Observations indicate that methylation changes are associated with many of the developmental genes chosen and that DNA methylation, assessed with read depth, had a tendency to increase with Stra8, Prm1, and Prm2 and decrease with Dazl and Id4, during development of the more advanced stage of gametogenesis (Figure 9). The Tet1 and Tet2 sites show more consistency across developmental stages. This helps demonstrate the future use of the dataset to potentially identify candidate DMR regulatory sites.
Figure 9.

Methylation at the sites of genes known to be expressed at the different developmental stages
The PGC-associated Tet and Dazl, spermatogonia-associated Id4, prospermatogonia-associated Stra8, and spermatid-associated protamines Prm1 and Prm2 are presented. The relative level of DNA methylation of the chromosomal sites of each developmental stage-specific gene. The color insert identifies the specific genes. The relative DNA methylation was determined as mean RPKM read depth.
Gametogenesis DNA methylation and weighted gene co-expression network analysis
The analyses presented earlier all focused on the analysis of DNA methylation alterations between adjacent stages of development to identify DMRs. An alternate consideration is to examine the DNA methylation at specific stages, which allows stage-specific DNA methylation patterns to be identified. One approach has been previously developed for gene expression during development and is termed Weighted Gene Co-Expression Network Analysis (WGCNA) (Zhao et al., 2010; Deyssenroth et al., 2017). A previous analysis of ovarian developmental biology and follicle development to identify gene networks involved in primordial follicle development used this WCGNA analysis (Nilsson et al., 2010). The MeDIP-Sequencing data was all used in a WGCNA analysis to identify DNA methylation modules and correlate with the different stages of germ cell development, as described in the Methods. This analysis generated 18 different modules and correlations, as shown in Figure 10. The correlation coefficient and p value statistics for each module and correlated developmental stage are presented. The number of DNA methylation sites for each module are presented in Figure 10B. The dendrograms used in generating the modules are presented in Figure S8. The DNA methylation sites within the various modules' chromosomal locations are presented in Figure S9 for all modules except the Blue and Turquoise due to the high number of DNA methylation sites in those modules (Figure 10B). The WGCNA DNA methylation module sites are found on all chromosomes and presented throughout the genome (Figure S9). The majority of modules correlated with developmental sites were not statistically significant, p> 0.05, but selected modules were statistically significant and associated with specific developmental stages (Figure 10A). The statistically significant correlations, p< 0.001, were identified and presented in Figure 11A. Those significant and correlated are shown in the black boxed module correlations. Seven modules had statistical significance for minimally one stage, but some with multiple stages. The PGC had module association with the Magenta and Midnight Blue modules; the prospermatogonia with the Red, Blue, and Turquoise modules; and the spermatogonia with the Tan and Cyan modules. The pachytene spermatocytes had significance with several modules in Figure 10, but none in the modules in Figure 11. The round spermatids had no significant correlations (Figures 10 and 11). The caput spermatozoa and cauda sperm had reduced significance of p = 0.02 and p = 0.04, respectively, with the Blue module (Figure 11). Therefore, a number of specific modules identified had significant correlations with specific stages of development. The round spermatids had no significant correlations, but the caput spermatozoa and cauda sperm had correlations at a lower significant threshold (Figure 11A).
Figure 10.

Weighted gene co-expression network analysis (WGCNA)
(A) WGCNA DNA methylation module and developmental stage relationships. Modules with different colors are correlated with stages PGC, prospermatogonia, spermatozoa, pachytene spermatocytes, round spermatids, caput spermatozoa, and cauda sperm. The colored correlation coefficient reflects the modules with the correlation coefficient number and statistical p value in brackets for each module stage comparison.
(B) The number of DNA methylation sites in each module is listed under each module color.
Figure 11.

WGCNA module and DNA methylation
(A) Module with DNA methylation relationships with correlations p < 0.001. The module colors relationships with developmental stage PGC, prospermatogonia, spermatogonia, pachytene spermatocytes, spermatozoa, and sperm. The correlation coefficient range and statistical significance module stage correlations with the correlation coefficient for each individual and the p value significance in brackets. The most significant for each developmental stage are identified with a black box.
(B) The DMR numbers for specifically developmental stage comparisons that overlap with the module DNA methylation sites are presented. These statistical significance module overlaps are underlined. All the other module DMR overlaps are presented in Figure S9.
The final analysis used the WGCNA-identified developmental stage DNA methylation module sites and examined potential overlaps with the DMRs identified between developmental stages (Figure S10). All the modules' identified DNA methylation sites were compared with the DMR sites between developmental stages to identify overlapping DMRs within the stage-specific modules. The number of DMR (p < 1e-05) sites present within the various modules for each developmental stage comparison is presented (Figure S10). Those WGCNA DNA methylation modules with statistically significant correlations with developmental stages had overlap with DMRs from various stage comparisons and are presented in Figure 11B. Therefore, DMR sites identified between developmental stage comparisons were also observed within the DNA methylation modules identified with WGCNA and were statistically correlated with specific stages of development. Those DMR correlations are identified in Figure 11B with the individual DNA methylation sites.
As an example of a network analysis of these observations, the Red module’s prospermatogonia-stage-specific DNA methylation site overlap with 451 DMRs was analyzed. These 451 DMRs with overlap to the Red module DNA methylation sites are presented in Table S7. Those DMRs with annotated genes (74 total) were used in a network analysis, but no integrated gene network was identified. The cellular gene locations and processes are shown in Figure 12. These Red module DNA methylation sites are prospermatogonia stage specific and altered as a DMR with adjacent stages of development.
Figure 12.

WGCNA Red module DNA methylation site overlap with DMRs for prospermatogonia stage
DMR-associated genes are shown with their associations to cellular processes and localization, as determined with Pathway Studio.
Discussion
The hypothesis tested in the current study is that a dramatic cascade of epigenetic changes is initiated at the early stages of male germline development to form the final DNA methylation pattern in the mature sperm. The study is designed to identify the developmental origins of differential DNA methylation in the germline DMRs between major developmental stages, as well as DNA methylation within specific stages of development. The primordial germ cells (PGCs) migrate between embryonic day E8 and E14 in the outbred rat in order to colonize the undifferentiated gonad. The outbred rat model was used to avoid inbreeding depression of epigenetics. Seven different stages of male germ cell development were assessed: embryonic day E13 PGCs, E16 prospermatogonia, postnatal day P10 spermatogonia, adult pachytene spermatocytes, round spermatids, caput epididymal spermatozoa, and cauda mature sperm (Figure 1). These stages were considered in part due to the functional role of the cellular stages during development, which include the primordial germ cells as the germline stem cells in the undifferentiated gonad, the prospermatogonia precursor cell population in the fetal testis, the testicular spermatogonia adult stem cell population, the meiotic pachytene spermatocyte cell population, the postmeiotic spermatid population, the epididymal caput spermatozoa, and the mature cauda sperm. To isolate and purify the caput spermatozoa and cauda sperm from any somatic cell contamination, the samples were sonicated and washed as described in the Methods. For the four other cell stages, a gravity sedimentation on a StaPut apparatus procedure was used to isolate the specific cell populations, as previously described (Hermann et al., 2015; Skinner and Guerrero-Bosagna, 2014). Although the developmental stages generally have an 85%–95% purity and the sperm 100% purity, the potential cell contamination is a limitation to consider in the data interpretation.
The developmental stages were compared with each other as follows: the E13 PGCs were compared with the E16 prospermatogonia; the E16 prospermatogonia with the P10 spermatogonia; the P10 spermatogonia with the adult pachytene spermatocytes; the adult pachytene spermatocytes with the round spermatids; the round spermatids with the caput epididymal spermatozoa; and the caput epididymal spermatozoa with the cauda mature sperm. The current study initially focused on the developmental stage comparisons to assess the majority of the epigenome changes throughout male germline development. Few studies have concurrently examined on a genome-wide level all developmental stages, but instead focused on PGC or spermatogonia stages (Morgan et al., 2005; Hill et al., 2018; Molaro et al., 2014; Seisenberger et al., 2013; Liu et al., 2019; Fend-Guella et al., 2019; Gaysinskaya et al., 2018; Kubo et al., 2015; Mallol et al., 2019; Schutte et al., 2013). All developmental stage comparison DMR sets were distinct from each other at a high statistical threshold but had high overlap at a lower statistical threshold. Observations imply a cascade of epigenetic changes occurs during spermatogenesis to program the sperm epigenome.
The different developmental stage comparisons were examined in regard to DNA methylation alterations to identify differential DNA methylation regions (DMRs). High numbers of DMRs with unique patterns of an increase or decrease in DNA methylation were observed between E13 PGCs and E16 prospermatogonia, between prospermatogonia and P10 spermatogonia stages, or between spermatogonia and pachytene spermatocytes (Figure 2). The caput spermatozoa versus the cauda epididymal sperm comparison presented the lowest number of DMRs, which suggests common DNA methylation between the two stages. In contrast, round spermatids, caput epididymal spermatozoa, and caudal sperm showed less variable alterations and appear more conserved with similar alterations in DNA methylation (Figure 2). At the beginning of spermatogenesis, a dramatic cascade of epigenetic programming and DNA methylation occurs in the spermatogenic cell populations, but this is reduced during epididymal maturation for the spermatozoa. During this epididymal maturation, the developing sperm undergo structural and molecular alterations, which also involve sperm DMR programing (Boskovic and Rando, 2018; Ariel et al., 1994), but less extensively than in the earlier pluripotent PGC and adult spermatozoa stem cell stages of development.
The dynamic genome-wide DNA methylation reprogramming in the PGCs and the preimplantation embryo has previously been described (Seisenberger et al., 2012). During migration and the colonization of the fetal gonad, an almost complete DNA methylation erasure occurs (Seisenberger et al., 2012). Following gonadal sex determination, this process enables the germline pluripotent PGC stem cell population to generate the male or female germline (Seisenberger et al., 2012; Tang et al., 2016). In the adult testis, the spermatogonial stem cell population goes through a stable epigenetic cascade of events during spermatogenesis. Numerous studies have shown that environmental toxicants can promote epigenetic DNA methylation alterations in the PGCs and in the subsequent germline, which induces distinct alterations in the DMRs found in the cauda epididymal sperm (Skinner et al., 2013a, 2019; Ben Maamar et al., 2019). Although previous studies have used an environmental exposure compared with control to assess the DMRs at each stage of germline development (Ben Maamar et al., 2019; Skinner et al., 2019), the objective of the current study was to develop a better perspective of the DNA methylation changes throughout normal development between the stages.
The level of DNA methylation change in the first three stage comparisons between the PGCs, prospermatogonia, spermatogonia, and pachytene spermatocytes was orders of magnitude higher than that observed in the later stages of round spermatids, caput spermatozoa, and cauda sperm (Figure 1). Interestingly, each stage comparison at a stringent threshold had a primarily unique set of DMRs at a higher statistical threshold (Figure 5A). Because the level of genome-wide developmental change between the initial stages is biologically more dramatic involving stem cell differentiation to subsequent stages, it is anticipated a greater degree of DNA methylation change occurs and correlates with greater gene expression changes. Therefore, a dynamic cascade of genome-wide DNA methylation change is observed throughout the PGC to sperm development, which is characterized in the current study. Similar observations were obtained with analysis of DNA methylation site read depths, in contrast to DMRs (Figure 6). The top DNA methylation sites were present at all stages and often declined from PGC to mature sperm (Figures 6 and S6). Therefore, the concept that DNA methylation simply increases between PGC to sperm is not accurate; this was further studied by examining stage-specific DMR DNA methylation and associated genes.
The DMR genomic features were consistent among the developmental stage comparisons. Primarily low-density CpG deserts (Skinner and Guerrero-Bosagna, 2014) were observed with a predominance of 1–2 CpG per 100 bp in the DMRs (Figure S1) and a size of 1–5 kb (Figure S3). The majority of most genomes, including mouse, rat, and human, have greater than 90% of the genome with a CpG density of 1 or 2 CpG per 100 bp, whereas higher CpG density islands constitute less than 5% of the genome (Beck et al., 2021). The MeDIP-Seq procedure used in the current study is optimal for analysis of lower density CpG analysis, which constitute >90% of the genome (Nair et al., 2011; Beck et al., 2021), whereas bisulfite analysis or methyl-CpG-binding protein MBP analysis is optimal for higher density CpG analysis (Nair et al., 2011; Pomraning et al., 2009; Beck et al., 2021). The sequence alignment issues associated with bisulfite also bias the procedure to higher density CpG (Pomraning et al., 2009). Therefore, the current study used MeDIP-Seq analysis to capture a more representative majority of the genome that was not biased toward CpG islands, which constitute less than 5% of the genome. This is a limitation that needs to be considered in data interpretation.
The DMRs identified were predominantly intergenic, with less than half of the DMRs having gene associations for all developmental stage comparisons (Tables S1–S6). This is expected, as only approximately 5%–8% of the mammalian genome contains genes. Approximately half of the DMRs had an increase in DNA methylation and half a decrease in DNA methylation at each stage of development. The exception was at the prospermatogonia versus spermatogonia comparison that had 75% of the DMR with a decrease in DNA methylation and the spermatogonia versus pachytene spermatocyte comparison that had a 75% increase in DNA methylation (Figure 2 and Tables S1–S6). These stage comparisons also had the highest numbers of DMRs identified. These changes in DNA methylation may be associated with similar DMR sites, although the higher statistical threshold comparison suggested the DMR sites were unique to each developmental stage comparison (Figures 5A and 5B). However, the extended overlap low statistical threshold comparisons did show a significant overlap between the PGCs, prospermatogonia, and spermatogonia stages, as well as a 71% overlap among the prospermatogonia, spermatogonia, and pachytene spermatocyte stages (Figure 5C). Therefore, variable modifications of the specific DMR throughout development appears to occur; this is supported by the time course data shown in Figure 4, which indicates individual DMR can be present throughout development at each stage with different levels of DNA methylation. Dynamic DNA methylation changes at similar sites can occur, as well as alterations at unique sites from the PGCs to later stages of sperm development exists. Interestingly, the PGC did not have a low methylation level in considering DMRs and highest methylation site read depth (Figure 6), so simple increases in DNA methylation levels were not observed.
Although the majority of DMRs do not show gene associations and appear intergenic (Figures S1–S6), some did have gene associations within 10 kb so the promoter could be considered. The intergenic DMRs have been shown previously to have critical roles as enhancers, in chromatin structure, and ncRNA expression to regulate gene expression at a distance. The initial examples of this were imprinted genes that, through a DNA methylation site, regulate a long ncRNA to influence distal gene expression megabases away from genes (Barlow and Bartolomei, 2014; Sanli and Feil, 2015). These types of distal regulatory sites are considered epigenetic control regions (ECR) that can influence multiple gene expression events from intergenic regions (Haque et al., 2016). However, the large number of DMRs associated with genes within 10 kb may act in a more traditional manner at promoters. The current study identified these DMR-associated genes and found a number of gene categories in common between the different developmental stage comparisons (Figure 7). The dynamics of changes in the DMR-associated gene categories were found to be similar during gametogenesis (Figure 7B). The specific DMR-associated genes at each individual stage comparison were used to identify potential regulation of cellular and biological processes (Figure 8). Interestingly, similar cellular processes appear regulated within the different developmental stage comparisons. Although the DMR-associated genes are unique, similar gene processes are associated. Therefore, the DNA methylation changes during PGC to sperm development will have a critical role in the regulation of gene expression to help establish the unique cellular functions and processes required during gametogenesis and spermatogenesis.
A validation analysis of selected individual genes that have been shown to have functions and gene expression at these developmental stages were examined for DNA methylation level dynamics of the developmental gene sites. The genes selected were Tet for PGCs, Dazl for PCCs, ID4 for spermatogonia, Stra8 for spermatocytes, and protamines for round spermatids. The methylation dynamics during the developmental processes for each of the development-associated genes are presented in Figure 9. Observations indicate the relative methylation changes for all the DMR associated with these genes during gametogenesis. Generally, an increase in methylation is observed for most of the DMR-associated genes during the progression of development, but the Id4 and Dazl had a decrease in methylation in their associated DMRs. This demonstrates the utility of the dataset developed to investigate individual gene associated DNA methylation alterations.
An analysis of the DNA methylation at individual stages was used to compare with the alterations in DNA methylation between stages for DMR analysis. This stage-specific analysis used Weighted Gene Co-Expression Network Analysis (WGCNA) (Zhao et al., 2010; Deyssenroth et al., 2017; Nilsson et al., 2010). The MeDIP-Seq data from all the stages were combined and used to identify DNA methylation modules and correlate with specific developmental stages (Figure 10). A number of the DNA methylation modules were found to statistically correlate with specific developmental stages. A comparison of the DMRs identified between stages of development was made with the stage-specific DNA methylation modules and demonstrated a good overlap (Figures 11 and S10). Therefore, the correlation of the DMRs with the significant stage-specific module DNA methylation helps validate both distinct analyses and identifies potentially critical DNA methylation sites. Further analysis of those altered DNA methylation sites is anticipated to be critical to understand germline development and spermatogenesis. As an example, the Red module DNA methylation sites that overlapped with DMRs in the comparison of prospermatogonia identified DNA methylation sites that alter at this stage of development. These DMR-associated genes are localized with cellular function and processes (Figure 12). Therefore, the observations can be further investigated to provide insights into the molecular control of gametogenesis.
The current study was designed to examine the DNA methylation alteration from the mature fetal primordial germ cell (PGC) through development stages to the mature differentiated sperm. This is one of the initial studies to examine the concurrent genome-wide developmental changes in epigenetics between each developmental stage, from the fetal germ cell stage, to the adult stem cell stage, to the adult differentiated germ cell. Previously our laboratory has examined the impacts of environmental exposures (i.e., pesticide DDT and fungicide vinclozolin) on the DNA methylation alterations at each of the stages of PGC to sperm development (Ben Maamar et al., 2019; Skinner et al., 2019). Clearly, a dynamic cascade of epigenetic DNA methylation alterations is observed throughout the gametogenesis process. The initial fetal germ cell transition to adult stem cells had the most dramatic numbers of DMRs, which far exceed the number of genes in the genome by an order of magnitude. The later stages of development to obtain the mature sperm showed lower numbers and less variability between developmental stages. A similar ratio of an increase or decrease in DNA methylation was observed, and the genomic features were similar for the DMRs at all stages of development. The development cascade of individual DMRs throughout gametogenesis supports the concept that a dynamic cascade of epigenetic changes occurs during each developmental stage. The highest overlap was observed during the fetal stem cell stage transition to the adult stem cell transition (i.e. primordial germ cell, prospermatogonia, and spermatogonia). This period showed the highest number of DMRs and greatest overlap at specific DMR sites. The DMR-associated genes had common gene categories and cell processes at all stages of development. The study provides insights into the developmental biology of an epigenetic process and suggests a dynamic and complex process to regulate genome biology and gene expression during gametogenesis.
Previous studies have demonstrated the ability of environmental exposures to modify germ cell epigenetics that can have generational impacts on physiological traits and disease (Nilsson et al., 2018). Specific studies have demonstrated major impacts on the early stem cell stages during gametogenesis by toxicants such as DDT or vinclozolin (Ben Maamar et al., 2019; Skinner et al., 2019). The more significant actions of the environmental exposure on the PGC and spermatogonia are supported by the current study due to the high number of epigenetic alterations at these stages of development and potential implications on later stages of development (e.g., sperm). The current study provides a molecular basis to understand these environmental exposures and impacts on gametogenesis and male reproduction. In addition, the study helps better understand the DNA methylation alterations during normal control development and helps elucidate the potential role of epigenetics during gametogenesis.
Limitations of the study
The current study examined epigenetic alterations at various stages of gametogenesis from PGC to sperm. The purity of the cell populations is important to consider for data interpretations; this varies from 80% to 85% in the early stages to 100% for the caput and cauda sperm stages. Contaminating cells will be somatic cells. This limitation needs to be considered in data interpretation. The DNA methylation procedure used involved a methylated DNA immunoprecipitation (MeDIP) followed by DNA sequencing. This procedure assesses greater than 95% of the genome DNA methylation (Beck et al., 2021) but is biased to lower density CpG density. This MeDIP-Seq procedure will be less efficient at assessing high-density CpG islands, which constitutes less than 5% of the genome, so needs to be considered in data interpretation.
Data archiving
All molecular data have been deposited into the public database at NCBI (GEO # GSE117995 and GSE151458), and R code computational tools are available at GitHub (https://github.com/skinnerlab/MeDIP-seq) and www.skinner.wsu.edu.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| 5-mC monoclonal antibody cl.b Classic | Diagenode | C15200006-500 |
| Magnetic beads (Dynabeads M−280 Sheep anti-Mouse IgG; 11201D) | Life technologies | 11202D |
| Biological samples | ||
| Female and male rats of an outbred strain | Harlan/Envigo (Indianapolis, IN) | Hsd:Sprague Dawley SD |
| Aspen Sani chips | Harlan | |
| 8640 Teklad 22/5 Rodent Diet | Harlan | 8640 |
| Chemicals, peptides, and recombinant proteins | ||
| Protein precipitation solution | Promega | A7953 |
| Bovine serum albumen (BSA) | Sigma | A2153 |
| Collagenase (Sigma C1639) | Sigma | C1639 |
| F-12 culture medium | Gibco-Life Technologies, USA. | Ref 11765-054 |
| Glycoblue | Life Technologies | AM 9516 |
| Proteinase K | Bio Basic | PB0451 |
| Buffered Phenol-Chloroform-Isoamylalcohol solution | Fisher Scientific | 327111000 |
| Dimethyl sulfoxide (DMSO) | J. T. Baker | 9224-01 |
| Oligonucleotides | ||
| NEBNext Multiplex Oligos for Illumina | NEB, San Diego, CA | E7335L |
| NEBNext Multiplex Oligos for Illumina | NEB, San Diego, CA | E7500L |
| Critical commercial kits | ||
| Qubit ssDNA kit (Molecular Probes Q10212) | Life Technologies | Q10212 |
| NEBNext Ultra II RNA Library Prep Kit for Illumina | NEB, San Diego, CA | E7770L |
| Deposited data | ||
| The public database at NCBI GEO |
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE117995 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE151458 https://www.ncbi.nlm.nih.gov/geo/ |
GEO # GSE117995 |
| The public database at NCBI GEO |
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE117995 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE151458 https://www.ncbi.nlm.nih.gov/geo/ |
GEO # GSE151458 |
| The public database at NCBI GEO |
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE117995 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE151458 https://www.ncbi.nlm.nih.gov/geo/ |
GEO # GSE121585 |
| Software and algorithms | ||
| FastQC program | http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ | |
| Trimmomatic | http://www.usadellab.org/cms/?page=trimmomatic | PMID: 24695404 |
| Bowtie2 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml | PMID: 22388286 |
| MEDIPS | https://bioconductor.org/packages/release/bioc/html/MEDIPS.html | PMID: 24227674 |
| BiomaRt R package | https://bioconductor.org/packages/release/bioc/html/biomaRt.html | PMID: 19617889 PMID: 16082012 |
| Ensembl database | https://www.ensembl.org/index.html | PMID: 31691826 |
| KEGG pathway search | https://www.genome.jp/kegg/mapper/search.html | PMID: 31423653 |
| DAVID | https://david.ncifcrf.gov/home.jsp | PMID: 19131956 PMID: 19033363 |
| WGCNA R package | https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA/ | PMID: 19114008 |
| Pathway Studio | https://www.pathwaystudio.com | |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Michael K. Skinner (skinner@wsu.edu).
Materials availability
This study did not generate new unique reagents and there are restrictions to availability.
Experimental model and subject details/animal studies and breeding
Female and male rats of an outbred strain Hsd:Sprague Dawley SD obtained from Harlan/Envigo (Indianapolis, IN) at about 70 to 100 days of age were maintained in cages with dimensions of 10 ¾″ W×19 ¼″ D×10 ¾″ H, 143 square inch floor space, containing Aspen Sani chips (pinewood shavings from Harlan) as bedding, and a 14 h light: 10 h dark regimen, at a temperature of 70 F and humidity of 25% to 35%. The mean light intensity in the animal rooms ranged from 22 to 26 ft-candles. Rats were fed ad lib with standard rat diet (8640 Teklad 22/5 Rodent Diet; Harlan) and ad lib tap water for drinking. To obtain time-pregnant females, the female rats in proestrus were pair-mated with male rats. The sperm-positive (i.e., sperm plug present) (day 0) rats were monitored for diestrus and body weight. The gestating female rats were designated as the F0 generation. On days 8 through 14 of gestation (Manikkam et al., 2014), the F0 generation females received daily intraperitoneal injections of dimethyl sulfoxide (DMSO, 1 mL/kg body weight/day). This lineage of rats served as the vehicle controls for other studies in which treatment compounds were dissolved in DMSO (Ben Maamar et al., 2019; Skinner et al., 2019). Only the F0 generation of pregnant females received DMSO treatment.
The offspring of the F0 generation rats were the F1 generation. Unrelated F1 generation rats were bred to produce the F2 generation, and similarly F2 generation rats were bred to produce the F3 generation. No sibling or cousin breedings were performed. Testes from F3 generation rats were collected at embryonic day 13 (E13), E16, and at 10 days of age (P10), as described below. Epididymal sperm was collected from adult males at 10-11 months of age as described below. All experimental protocols for the procedures with rats were pre-approved by the Washington State University Animal Care and Use Committee (IACUC approval # 6252).
Epididymal caput spermatozoa and cauda sperm collections and DNA isolation
The epididymis was dissected free of connective tissue, a small cut made to the caput or cauda and tissue placed separately in 5 mL of 1X PBS solution for up to 2 hours at 4°C. The epididymal tissue was minced and the released caput spermatozoa or cauda sperm was centrifuged at 6,000 x g, then the supernatant was removed, and the pellets were resuspended in NIM buffer, to be stored at −80°C until further use. One hundred μl of spermatozoa or sperm suspension was sonicated to destroy contaminating somatic cells and tissue, spun down at 6,000 x g, the sperm pellet was washed with 1X PBS once, and then combined with 820 μL DNA extraction buffer and 80 μL 0.1M DTT. The samples were incubated at 65°C for 15 minutes. Following this incubation 80 μL proteinase K (20 mg/mL) was added and the sample was incubated at 55°C for at least 2 hours under constant rotation. Then 300 μL of protein precipitation solution (Promega, A7953) was added, the sample was mixed thoroughly and then it was incubated for 15 min on ice. The sample was centrifuged at 12,500 x g for 30 minutes at 4°C. One mL of the supernatant was transferred to a 2 mL tube and 2 μL of Glycoblue and 1 mL of cold 100% isopropanol were added. The sample was mixed well by inverting the tube several times, then it was left at −20°C for at least one hour. After precipitation the sample was centrifuged at 12,500 x g for 20 min at 4°C. The supernatant was taken off and discarded without disturbing the (blue) pellet. The pellet was washed with 70% cold ethanol by adding 500 μL of 70% ethanol to the pellet and returning the tube to the freezer for 20 minutes. After the incubation the tube was centrifuged for 10 min at 4°C at 12,500 x g and the supernatant discarded. The tube was spun again briefly to collect residual ethanol to bottom of tube and then as much liquid as possible was removed with a gel loading tip. Pellet was air-dried at room temperature until it looked dry (about 5 minutes). Pellets were then resuspended in 100 μL of nuclease free water.
Developing germ cell stage isolation and DNA preparation
Pregnant rats were euthanized at embryonic day 13 (E13) or 16 (E16) of gestation, and fetal gonads were collected for germ cell preparations. Sex was determined on the basis of gonadal morphology for E16 testis cords. Germ cells were isolated exclusively from males. Sex was determined with an SRY PCR analysis for E13 rats due to the lack of defined morphology, as previously described (McCarrey et al., 1992; Kafri et al., 1992; Levine et al., 2000).
Purified populations of male PGCs (at E13) and type T1 prospermatogonia (at E16) were prepared using a mini StaPut gradient method as previously described (McCarrey et al., 1992; Kafri et al., 1992). Briefly, fetal testes were pooled and dissociated by incubation in 0.25% trypsin-EDTA (Sigma) with vigorous pipetting using a 1000 microliter pipette tip, and the resulting cell solution was filtered through 100 micron nylon mesh to yield a single cell suspension. This cell suspension was then loaded onto a 50 mL 2–4% bovine serum albumen (BSA) gradient prepared in KREBS buffer, and the cells were allowed to sediment at unit gravity at 4°C for two hours, as previously described (McCarrey et al., 1992; Kafri et al., 1992). The gradient was then fractionated and aliquots of the fractions were examined under phase optics to identify those enriched for the appropriate cell types on the basis of morphological characteristics. The enriched fractions were pooled to yield the final sample which was >85% pure for the desired male germ cell type in each case.
A similar mini StaPut gradient method (McCarrey et al., 1992; Kafri et al., 1992) was used to isolate spermatogonia from testes of 10-day old rats, with the addition of incubation of the testes in 0.5 mg/mL collagenase (Sigma C1639) at 33°C for 20 min with agitation to dissociate the seminiferous tubules. Three pools of spermatogonia from 10-day-old rats were prepared for each treatment group, with each pool derived from testes of six to seven rats from different litters.
To isolate pachytene spermatocytes and round spermatids, testes were collected from 10 to 11 month old rats suspended in F-12 culture medium (Gibco-Life Technologies, USA. Ref 11,765-054) and shipped overnight on ice to Dr. John McCarrey. A StaPut gradient method was used to isolate the developing germ cell stages as previously described (McCarrey et al., 1992; Kafri et al., 1992). Three pools of cells of each cell type were prepared for each treatment group, with each pool derived from testes of three rats from different litters. DNA was isolated from prospermatogonia, spermatogonia, pachytene spermatocytes and round spermatids using the same procedure as was used for sperm, with the omission of sonication and DTT treatments.
Methylated DNA immunoprecipitation MeDIP
Methylated DNA Immunoprecipitation (MeDIP) with genomic DNA was performed as follows: rat sperm DNA pools were generated using the appropriate amount of genomic DNA from each individual for 2 pools in the PGC stage and 3 pools in each of the subsequent stages. Genomic DNA pools were sonicated using the Covaris M220 the following way: the pooled genomic DNA was diluted to 130 μL with 1X TE buffer into the appropriate Covaris tube. Covaris was set to 300 bp program and the program was run for each tube in the experiment. 10 μL of each sonicated DNA was run on 1.5% agarose gel to verify fragment size. The sonicated DNA was transferred from the Covaris tube to a 1.7 mL microfuge tube and the volume measured. The sonicated DNA was then diluted with 1X TE buffer (10mM Tris HCl, pH7.5; 1mM EDTA) to 400 μL, heat-denatured for 10 min at 95°C, then immediately cooled on ice for 10 min. Then 100 μL of 5X IP buffer and 5 μg of antibody (monoclonal mouse anti 5-methyl cytidine; Diagenode #C15200006) were added to the denatured sonicated DNA. The DNA-antibody mixture was incubated overnight on a rotator at 4°C.
The following day magnetic beads (Dynabeads M−280 Sheep anti-Mouse IgG; 11201D) were pre-washed as follows: The beads were resuspended in the vial, then the appropriate volume (50 μL per sample) was transferred to a microfuge tube. The same volume of Washing Buffer (at least 1 mL PBS with 0.1% BSA and 2mM EDTA) was added and the bead sample was resuspended. Tube was then placed into a magnetic rack for 1–2 minutes and the supernatant discarded. The tube was removed from the magnetic rack and the beads washed once. The washed beads were resuspended in the same volume of IP buffer (50 mM sodium phosphate ph7.0, 700 mM NaCl, 0.25% Triton X-100) as the initial volume of beads. 50μL of beads were added to the 500μL of DNA-antibody mixture from the overnight incubation, then incubated for 2h on a rotator at 4°C.
After the incubation the bead-antibody-DNA complex was washed three times with 1X IP buffer as follows: The tube was placed into a magnetic rack for 1–2 minutes and the supernatant discarded, then washed with IP buffer 3 times. The washed bead-DNA solution is then resuspended in 250 μL digestion buffer with 3.5 μL Proteinase K (20 mg/ml). The sample was then incubated for 2–3 hours on a rotator at 55°C and then 250 μL of buffered Phenol-Chloroform-Isoamylalcohol solution was added to the supernatant and the tube vortexed for 30 sec then centrifuged at 12,500 x g for 5 min at room temperature. The aqueous supernatant was carefully removed and transferred to a fresh microfuge tube. Then 250 μL chloroform were added to the supernatant from the previous step, vortexed for 30 sec and centrifuged at 12,500 x g for 5 min at room temperature. The aqueous supernatant was removed and transferred to a fresh microfuge tube. To the supernatant 2μL of Glycoblue (20 mg/ml), 20μL of 5M NaCl and 500μL ethanol were added and mixed well, then precipitated in −20°C freezer for 1 hour to overnight.
The precipitate was centrifuged at 12,500 x g for 20 min at 4°C and the supernatant removed, while not disturbing the pellet. The pellet was washed with 500μL cold 70% ethanol in −20°C freezer for 15 min then centrifuged again at 12,500 x g for 5 min at 4°C and the supernatant discarded. The tube was spun again briefly to collect residual ethanol to bottom of tube and as much liquid as possible was removed with gel loading tip. Pellet was air-dried at RT until it looked dry (about 5 minutes) then resuspended in 20μL H2O or TE. DNA concentration was measured in Qubit (Life Technologies) with ssDNA kit (Molecular Probes Q10212).
MeDIP-seq analysis
The MeDIP pools were used to create libraries for next generation sequencing (NGS) using the NEBNext Ultra II RNA Library Prep Kit for Illumina (NEB, San Diego, CA) starting at step 1.4 of the manufacturer’s protocol to generate double-stranded DNA. After this step the manufacturer’s protocol was followed. Each pool received a separate index primer. NGS was performed at the WSU Spokane Genomics Core laboratory using the Illumina HiSeq 2500 with a PE50 application, with a read size of approximately 50 bp and approximately 30 million reads per pool. Five to six libraries were run in one lane.
Quantification and statistical analysis
For the DMR analyses, the basic read quality was verified using summaries produced by the FastQC program http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. The raw reads were trimmed and filtered using Trimmomatic (Bolger et al., 2014). The reads for each MeDIP sample were mapped to the Rnor 6.0 rat genome using Bowtie2 (Langmead and Salzberg, 2012) with default parameter options. The sequence alignment was generally 90–95% with three samples at 88%. The mapped read files were then converted to sorted BAM files using SAMtools (Li et al., 2009). Genomic windows with a minimum read depth of 40 reads were analyzed for differential methylation. To identify DMRs, the reference genome was broken into 1000 bp windows. The MEDIPS (Lienhard et al., 2014) and edgeR (Robinson et al., 2010) R packages were used to calculate differential coverage between control and exposure sample groups. The edgeR p value was used to determine the relative difference between the two groups for each genomic window. Windows with an edgeR p value less than 1e-5 were considered DMRs. Multiple testing correction using FDR was performed. All DMR at an edgeR p < 1e-5 also met an FDR p < 0.05 except for the caput vs cauda comparison. The DMR edges were extended until no genomic window with a p value < 0.1 remained within 1000 bp of the DMR. CpG density and other information was then calculated for the DMR based on the reference genome. Due to high numbers of DMRs, 1000 of the most significant DMR were selected for further examination and annotation. DMRs were annotated using the biomaRt R package (Durinck et al., 2005, 2009) to access the Ensembl database (Howe et al., 2020). The genes that fell within 10kbp of the DMR edges were then input into the KEGG pathway search (Kanehisa and Goto, 2000) to identify associated pathways. The associated genes were then sorted into functional groups by consulting information provided by the DAVID (Huang da et al., 2009b, Huang da et al., 2009a), and Panther (Mi et al., 2017) databases incorporated into an internal curated database (www.skinner.wsu.edu under genomic data).
For Figure 4, the top 100 most significant DMRs for each analysis were selected. The DMR were split into two groups of DMR based on whether the read depth at the DMR site was elevated in the first set of samples or the second. The average RPKM read depth was then calculated for each DMR site for every analysis. This value was then scaled such that the analysis with the lowest read depth is 0 and the analysis with the highest read depth is 1.
The Venn diagrams occasionally resulted in two DMR in one analysis overlapping with a single DMR in a second analysis. When this occurred, the average number of DMR in the overlap was calculated. The resulting decimal number was then truncated to obtain an integer overlap. For the expanded overlaps, a DMR in one analysis was considered present in a second analysis if it had a p value less than 1e-3 in the second analysis.
All molecular data has been deposited into the public database at NCBI (GEO # GSE117995 & GSE151458).
Weighted gene co-expression network analysis (WGCNA)
The weighted correlation network analysis (WGCNA) (Zhang and Horvath, 2005) was performed using the WGCNA R package (Langfelder and Horvath, 2008). All MeDIP-Seq genomic windows were ranked by the mean RPKM read depth across all samples. The top 100,000 sites were chosen for inclusion in the analysis. The size of this subset was chosen to allow for reasonable computational requirements. WGCNA is a parameter rich analysis and only limited exploration of parameter variations was performed. Modules were calculated using the blockwiseModules function with the following parameters: maxBlockSize = 15000, power = 9, TOMType = "unsigned", minModuleSize = 30, reassignThreshold = 0, and mergeCutHeight = 0.25. The Pearson correlation was calculated for each development stage and module. The p value for each correlation was calculated using the corPvalueStudent function. Sites within each module were annotated using the same methods as the DMRs. For the module sites, associated genes were required to be overlapping with the site, in contrast to the DMRs, which only required the gene to be within 10kb of the DMR edge.
Acknowledgments
We acknowledge Dr. Ingrid Sadler-Riggleman, Ms. Michelle Pappalardo, and Mr. Ryan Thompson for technical assistance. We acknowledge Ms. Amanda Quilty for editing and Ms. Heather Johnson for assistance in preparation of the manuscript. We thank the Genomics Core laboratory at WSU Spokane for sequencing data. This study was supported by John Templeton Foundation (50183 and 61174) (https://templeton.org/) grants to MKS and NIH (ES012974) (https://www.nih.gov/) grant to MKS. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions
MBM, molecular analysis, data analysis, wrote manuscript. DB, bioinformatic analysis, data analysis, edited manuscript. EN, animal studies, cell isolations, data analysis, edited manuscript. JRM, cell isolations, data analysis, edited manuscript. MKS, conceived, oversight, obtained funding, edited manuscript.
Declaration of interests
The authors declare no competing interests.
Published: February 18, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.103786.
Supplemental information
Data and code availability
-
•
Data availability: All molecular data has been deposited into the public database at NCBI (GEO # GSE117995 & GSE151458).
-
•
Code availability: R code computational tools are available at GitHub (https://github.com/skinnerlab/MeDIP-seq) and www.skinner.wsu.edu.
References
- Adams I.R., McLaren A. Sexually dimorphic development of mouse primordial germ cells: switching from oogenesis to spermatogenesis. Development. 2002;129:1155–1164. doi: 10.1242/dev.129.5.1155. [DOI] [PubMed] [Google Scholar]
- Albrecht K.H., Eicher E.M. Evidence that Sry is expressed in pre-Sertoli cells and Sertoli and granulosa cells have a common precursor. Dev. Biol. 2001;240:92–107. doi: 10.1006/dbio.2001.0438. [DOI] [PubMed] [Google Scholar]
- Aloisio G.M., Nakada Y., Saatcioglu H.D., Pena C.G., Baker M.D., Tarnawa E.D., Mukherjee J., Manjunath H., Bugde A., Sengupta A.L., et al. PAX7 expression defines germline stem cells in the adult testis. J. Clin. Invest. 2014;124:3929–3944. doi: 10.1172/JCI75943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson E.L., Baltus A.E., Roepers-Gajadien H.L., Hassold T.J., de Rooij D.G., Van Pelt A.M., Page D.C. Stra8 and its inducer, retinoic acid, regulate meiotic initiation in both spermatogenesis and oogenesis in mice. Proc. Natl. Acad. Sci. U S A. 2008;105:14976–14980. doi: 10.1073/pnas.0807297105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anway M.D., Cupp A.S., Uzumcu M., Skinner M.K. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science. 2005;308:1466–1469. doi: 10.1126/science.1108190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariel M., Cedar H., McCarrey J. Developmental changes in methylation of spermatogenesis-specific genes include reprogramming in the epididymis. Nat. Genet. 1994;7:59–63. doi: 10.1038/ng0594-59. [DOI] [PubMed] [Google Scholar]
- Bao J., Zhang Y., Schuster A.S., Ortogero N., Nilsson E.E., Skinner M.K., Yan W. Conditional inactivation of Miwi2 reveals that MIWI2 is only essential for prospermatogonial development in mice. Cell Death Differ. 2014;21:783–796. doi: 10.1038/cdd.2014.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow D.P., Bartolomei M.S. Genomic imprinting in mammals. Cold Spring Harb. Perspect. Biol. 2014;6:1–21. doi: 10.1101/cshperspect.a018382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck D., Ben Maamar M., Skinner M.K. Genome-wide CpG density and DNA methylation analysis method (MeDIP, RRBS, and WGBS) comparisons. Epigenetics. 2021:1–13. doi: 10.1080/15592294.2021.1924970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck D., Sadler-Riggleman I., Skinner M.K. Generational comparisons (F1 versus F3) of vinclozolin induced epigenetic transgenerational inheritance of sperm differential DNA methylation regions (epimutations) using MeDIP-Seq. Environ. Epigenet. 2017;3:dvx016. doi: 10.1093/eep/dvx016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Maamar M., Nilsson E., Sadler-Riggleman I., Beck D., McCarrey J.R., Skinner M.K. Developmental origins of transgenerational sperm DNA methylation epimutations following ancestral DDT exposure. Dev. Biol. 2019;445:280–293. doi: 10.1016/j.ydbio.2018.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Maamar M., Nilsson E.E., Skinner M.K. Epigenetic transgenerational inheritance, gametogenesis and germline development. Biol. Reprod. 2021;105:570–592. doi: 10.1093/biolre/ioab085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhandari R., Haque Md. M., Skinner M. Global genome analysis of the downstream binding targets of testis determining factor SRY AND SOX9. PLoS One. 2012;7:e43380. doi: 10.1371/journal.pone.0043380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boskovic A., Rando O.J. Transgenerational epigenetic inheritance. Annu. Rev. Genet. 2018;52:21–41. doi: 10.1146/annurev-genet-120417-031404. [DOI] [PubMed] [Google Scholar]
- Buaas F.W., Kirsh A.L., Sharma M., Mclean D.J., Morris J.L., Griswold M.D., de Rooij D.G., Braun R.E. Plzf is required in adult male germ cells for stem cell self-renewal. Nat. Genet. 2004;36:647–652. doi: 10.1038/ng1366. [DOI] [PubMed] [Google Scholar]
- Bullejos M., Koopman P. Delayed Sry and Sox9 expression in developing mouse gonads underlies B6-Y(DOM) sex reversal. Dev. Biol. 2005;278:473–481. doi: 10.1016/j.ydbio.2004.11.030. [DOI] [PubMed] [Google Scholar]
- Capel B. R-spondin1 tips the balance in sex determination. Nat. Genet. 2006;38:1233–1234. doi: 10.1038/ng1106-1233. [DOI] [PubMed] [Google Scholar]
- Carrell D.T., Aston K.I., Oliva R., Emery B.R., De Jonge C.J. The "omics" of human male infertility: integrating big data in a systems biology approach. Cell Tissue Res. 2016;363:295–312. doi: 10.1007/s00441-015-2320-7. [DOI] [PubMed] [Google Scholar]
- Chaboissier M.C., Kobayashi A., Vidal V.I., Lutzkendorf S., van de Kant H.J., Wegner M., de Rooij D.G., Behringer R.R., Schedl A. Functional analysis of Sox8 and Sox9 during sex determination in the mouse. Development. 2004;131:1891–1901. doi: 10.1242/dev.01087. [DOI] [PubMed] [Google Scholar]
- Colvin J.S., Green R.P., Schmahl J., Capel B., Ornitz D.M. Male-to-female sex reversal in mice lacking fibroblast growth factor 9. Cell. 2001;104:875–889. doi: 10.1016/s0092-8674(01)00284-7. [DOI] [PubMed] [Google Scholar]
- Cornwall G.A. New insights into epididymal biology and function. Hum. Reprod. Update. 2009;15:213–227. doi: 10.1093/humupd/dmn055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costoya J.A., Hobbs R.M., Barna M., Cattoretti G., Manova K., Sukhwani M., Orwig K.E., Wolgemuth D.J., Pandolfi P.P. Essential role of Plzf in maintenance of spermatogonial stem cells. Nat. Genet. 2004;36:653–659. doi: 10.1038/ng1367. [DOI] [PubMed] [Google Scholar]
- Coucouvanis E., Martin G.R. BMP signaling plays a role in visceral endoderm differentiation and cavitation in the early mouse embryo. Development. 1999;126:535–546. doi: 10.1242/dev.126.3.535. [DOI] [PubMed] [Google Scholar]
- de Sousa Lopes S.M., Roelen B.A., Monteiro R.M., Emmens R., Lin H.Y., Li E., Lawson K.A., Mummery C.L. BMP signaling mediated by ALK2 in the visceral endoderm is necessary for the generation of primordial germ cells in the mouse embryo. Genes Dev. 2004;18:1838–1849. doi: 10.1101/gad.294004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deyssenroth M.A., Peng S., Hao K., Lambertini L., Marsit C.J., Chen J. Whole-transcriptome analysis delineates the human placenta gene network and its associations with fetal growth. BMC Genomics. 2017;18:520. doi: 10.1186/s12864-017-3878-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S., Moreau Y., Kasprzyk A., Davis S., De Moor B., Brazma A., Huber W. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics. 2005;21:3439–3440. doi: 10.1093/bioinformatics/bti525. [DOI] [PubMed] [Google Scholar]
- Durinck S., Spellman P.T., Birney E., Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009;4:1184–1191. doi: 10.1038/nprot.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dym M., Fawcett D.W. Further observations on the numbers of spermatogonia, spermatocytes, and spermatids connected by intercellular bridges in the mammalian testis. Biol. Reprod. 1971;4:195–215. doi: 10.1093/biolreprod/4.2.195. [DOI] [PubMed] [Google Scholar]
- Eildermann K., Aeckerle N., Debowski K., Godmann M., Christiansen H., Heistermann M., Schweyer S., Bergmann M., Kliesch S., Gromoll J., et al. Developmental expression of the pluripotency factor sal-like protein 4 in the monkey, human and mouse testis: restriction to premeiotic germ cells. Cells Tissues Organs. 2012;196:206–220. doi: 10.1159/000335031. [DOI] [PubMed] [Google Scholar]
- Fend-Guella D.L., Von Kopylow K., Spiess A.N., Schulze W., Salzbrunn A., Diederich S., El Hajj N., Haaf T., Zechner U., Linke M. The DNA methylation profile of human spermatogonia at single-cell- and single-allele-resolution refutes its role in spermatogonial stem cell function and germ cell differentiation. Mol. Hum. Reprod. 2019;25:283–294. doi: 10.1093/molehr/gaz017. [DOI] [PubMed] [Google Scholar]
- Gaysinskaya V., Miller B.F., de Luca C., van der Heijden G.W., Hansen K.D., Bortvin A. Transient reduction of DNA methylation at the onset of meiosis in male mice. Epigenetics Chromatin. 2018;11:15. doi: 10.1186/s13072-018-0186-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goertz M.J., Wu Z., Gallardo T.D., Hamra F.K., Castrillon D.H. Foxo1 is required in mouse spermatogonial stem cells for their maintenance and the initiation of spermatogenesis. J. Clin. Invest. 2011;121:3456–3466. doi: 10.1172/JCI57984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubbay J., Collignon J., Koopman P., Capel B., Economou A., Munsterberg A., Vivian N., Goodfellow P., Lovell-Badge R. A gene mapping to the sex-determining region of the mouse Y chromosome is a member of a novel family of embryonically expressed genes. Nature. 1990;346:245–250. doi: 10.1038/346245a0. [DOI] [PubMed] [Google Scholar]
- Guerrero-Bosagna C., Covert T., Haque M.M., Settles M., Nilsson E.E., Anway M.D., Skinner M.K. Epigenetic transgenerational inheritance of vinclozolin induced mouse adult onset disease and associated sperm epigenome biomarkers. Reprod. Toxicol. 2012;34:694–707. doi: 10.1016/j.reprotox.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett J.A., Sengupta R., Zylicz J.J., Murakami K., Lee C., Down T.A., Surani M.A. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science. 2013;339:448–452. doi: 10.1126/science.1229277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque M.M., Nilsson E.E., Holder L.B., Skinner M.K. Genomic Clustering of differential DNA methylated regions (epimutations) associated with the epigenetic transgenerational inheritance of disease and phenotypic variation. BMC Genomics. 2016;17:1–13. doi: 10.1186/s12864-016-2748-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargan-Calvopina J., Taylor S., Cook H., Hu Z., Lee S.A., Yen M.R., Chiang Y.S., Chen P.Y., Clark A.T. Stage-specific demethylation in primordial germ cells safeguards against precocious differentiation. Dev. Cell. 2016;39:75–86. doi: 10.1016/j.devcel.2016.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harikae K., Miura K., Kanai Y. Early gonadogenesis in mammals: significance of long and narrow gonadal structure. Dev. Dyn. 2013;242:330–338. doi: 10.1002/dvdy.23872. [DOI] [PubMed] [Google Scholar]
- Hawkins J.R., Taylor A., Berta P., Levilliers J., Van der Auwera B., Goodfellow P.N. Mutational analysis of SRY: nonsense and missense mutations in XY sex reversal. Hum. Genet. 1992;88:471–474. doi: 10.1007/BF00215684. [DOI] [PubMed] [Google Scholar]
- Hermann B.P., Mutoji K.N., Velte E.K., Ko D., Oatley J.M., Geyer C.B., Mccarrey J.R. Transcriptional and translational heterogeneity among neonatal mouse spermatogonia. Biol. Reprod. 2015;92:54. doi: 10.1095/biolreprod.114.125757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill P.W.S., Leitch H.G., Requena C.E., Sun Z., Amouroux R., Roman-Trufero M., Borkowska M., Terragni J., Vaisvila R., Linnett S., et al. Epigenetic reprogramming enables the transition from primordial germ cell to gonocyte. Nature. 2018;555:392–396. doi: 10.1038/nature25964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs R.M., Fagoonee S., Papa A., Webster K., Altruda F., Nishinakamura R., Chai L., Pandolfi P.P. Functional antagonism between Sall4 and Plzf defines germline progenitors. Cell Stem Cell. 2012;10:284–298. doi: 10.1016/j.stem.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe K.L., Contreras-Moreira B., De Silva N., Maslen G., Akanni W., Allen J., Alvarez-Jarreta J., Barba M., Bolser D.M., Cambell L., et al. Ensembl Genomes 2020-enabling non-vertebrate genomic research. Nucleic Acids Res. 2020;48:D689–D695. doi: 10.1093/nar/gkz890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W., Sherman B.T., Lempicki R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Jeays-Ward K., Dandonneau M., Swain A. Wnt4 is required for proper male as well as female sexual development. Dev. Biol. 2004;276:431–440. doi: 10.1016/j.ydbio.2004.08.049. [DOI] [PubMed] [Google Scholar]
- Jirtle R.L., Skinner M.K. Environmental epigenomics and disease susceptibility. Nat. Rev. Genet. 2007;8:253–262. doi: 10.1038/nrg2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost A., Vigier B., Prepin J., Perchellet J.P. Studies on sex differentiation in mammals. Recent Prog. Horm. Res. 1973;29:1–41. doi: 10.1016/b978-0-12-571129-6.50004-x. [DOI] [PubMed] [Google Scholar]
- Kafri T., Ariel M., Brandeis M., Shemer R., Urven L., Mccarrey J., Cedar H., Razin A. Developmental pattern of gene-specific DNA methylation in the mouse embryo and germ line. Genes Dev. 1992;6:705–714. doi: 10.1101/gad.6.5.705. [DOI] [PubMed] [Google Scholar]
- Kagiwada S., Kurimoto K., Hirota T., Yamaji M., Saitou M. Replication-coupled passive DNA demethylation for the erasure of genome imprints in mice. EMBO J. 2013;32:340–353. doi: 10.1038/emboj.2012.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M., Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent J., Wheatley S.C., Andrews J.E., Sinclair A.H., Koopman P. A male-specific role for SOX9 in vertebrate sex determination. Development. 1996;122:2813–2822. doi: 10.1242/dev.122.9.2813. [DOI] [PubMed] [Google Scholar]
- Kim Y., Kobayashi A., Sekido R., Dinapoli L., Brennan J., Chaboissier M.C., Poulat F., Behringer R.R., Lovell-Badge R., Capel B. Fgf9 and Wnt4 act as antagonistic signals to regulate mammalian sex determination. PLoS Biol. 2006;4:e187. doi: 10.1371/journal.pbio.0040187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H., Sakurai T., Miura F., Imai M., Mochiduki K., Yanagisawa E., Sakashita A., Wakai T., Suzuki Y., Ito T., et al. High-resolution DNA methylome analysis of primordial germ cells identifies gender-specific reprogramming in mice. Genome Res. 2013;23:616–627. doi: 10.1101/gr.148023.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komai Y., Tanaka T., Tokuyama Y., Yanai H., Ohe S., Omachi T., Atsumi N., Yoshida N., Kumano K., Hisha H., et al. Bmi1 expression in long-term germ stem cells. Sci. Rep. 2014;4:6175. doi: 10.1038/srep06175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman P. Sry and Sox9: mammalian testis-determining genes. Cell Mol. Life Sci. 1999;55:839–856. doi: 10.1007/PL00013200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman P., Gubbay J., Vivian N., Goodfellow P., Lovell-Badge R. Male development of chromosomally female mice transgenic for Sry. Nature. 1991;351:117–121. doi: 10.1038/351117a0. [DOI] [PubMed] [Google Scholar]
- Kotaja N. MicroRNAs and spermatogenesis. Fertil. Steril. 2014;101:1552–1562. doi: 10.1016/j.fertnstert.2014.04.025. [DOI] [PubMed] [Google Scholar]
- Koubova J., Menke D.B., Zhou Q., Capel B., Griswold M.D., Page D.C. Retinoic acid regulates sex-specific timing of meiotic initiation in mice. Proc. Natl. Acad. Sci. U S A. 2006;103:2474–2479. doi: 10.1073/pnas.0510813103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo N., Toh H., Shirane K., Shirakawa T., Kobayashi H., Sato T., Sone H., Sato Y., Tomizawa S., Tsurusaki Y., et al. DNA methylation and gene expression dynamics during spermatogonial stem cell differentiation in the early postnatal mouse testis. BMC Genomics. 2015;16:624. doi: 10.1186/s12864-015-1833-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurimoto K., Yamaji M., Seki Y., Saitou M. Specification of the germ cell lineage in mice: a process orchestrated by the PR-domain proteins, Blimp1 and Prdm14. Cell Cycle. 2008;7:3514–3518. doi: 10.4161/cc.7.22.6979. [DOI] [PubMed] [Google Scholar]
- Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson K.A., Dunn N.R., Roelen B.A., Zeinstra L.M., Davis A.M., Wright C.V., Korving J.P., Hogan B.L. Bmp4 is required for the generation of primordial germ cells in the mouse embryo. Genes Dev. 1999;13:424–436. doi: 10.1101/gad.13.4.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesch B.J., Dokshin G.A., Young R.A., Mccarrey J.R., Page D.C. A set of genes critical to development is epigenetically poised in mouse germ cells from fetal stages through completion of meiosis. Proc. Natl. Acad. Sci. U S A. 2013;110:16061–16066. doi: 10.1073/pnas.1315204110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine E., Cupp A.S., Miyashiro L., Skinner M.K. Role of transforming growth factor-alpha and the epidermal growth factor receptor in embryonic rat testis development. Biol. Reprod. 2000;62:477–490. doi: 10.1095/biolreprod62.3.477. [DOI] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., Genome Project Data Processing, Subgroup The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Zheng M., Lau Y.F. The sex-determining factors SRY and SOX9 regulate similar target genes and promote testis cord formation during testicular differentiation. Cell Rep. 2014;8:723–733. doi: 10.1016/j.celrep.2014.06.055. [DOI] [PubMed] [Google Scholar]
- Lienhard M., Grimm C., Morkel M., Herwig R., Chavez L. MEDIPS: genome-wide differential coverage analysis of sequencing data derived from DNA enrichment experiments. Bioinformatics. 2014;30:284–286. doi: 10.1093/bioinformatics/btt650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y., Page D.C. Dazl deficiency leads to embryonic arrest of germ cell development in XY C57BL/6 mice. Dev. Biol. 2005;288:309–316. doi: 10.1016/j.ydbio.2005.06.032. [DOI] [PubMed] [Google Scholar]
- Liu Y., Zhang Y., Yin J., Gao Y., Li Y., Bai D., He W., Li X., Zhang P., Li R., et al. Distinct H3K9me3 and DNA methylation modifications during mouse spermatogenesis. J. Biol. Chem. 2019;294:18714–18725. doi: 10.1074/jbc.RA119.010496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord T., Oatley J.M. A revised Asingle model to explain stem cell dynamics in the mouse male germline. Reproduction. 2017;154:R55–R64. doi: 10.1530/REP-17-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell-Badge R., Robertson E. XY female mice resulting from a heritable mutation in the primary testis-determining gene, Tdy. Development. 1990;109:635–646. doi: 10.1242/dev.109.3.635. [DOI] [PubMed] [Google Scholar]
- Luk A.C., Chan W.Y., Rennert O.M., Lee T.L. Long noncoding RNAs in spermatogenesis: insights from recent high-throughput transcriptome studies. Reproduction. 2014;147:R131–R141. doi: 10.1530/REP-13-0594. [DOI] [PubMed] [Google Scholar]
- Mallol A., Guirola M., Payer B. PRDM14 controls X-chromosomal and global epigenetic reprogramming of H3K27me3 in migrating mouse primordial germ cells. Epigenet. Chromatin. 2019;12:38. doi: 10.1186/s13072-019-0284-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manikkam M., Haque M.M., Guerrero-Bosagna C., Nilsson E., Skinner M.K. Pesticide methoxychlor promotes the epigenetic transgenerational inheritance of adult onset disease through the female germline. PLoS ONE. 2014;9:e102091. doi: 10.1371/journal.pone.0102091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarrey J.R. The epigenome as a target for heritable environmental disruptions of cellular function. Mol. Cell. Endocrinol. 2012;354:9–15. doi: 10.1016/j.mce.2011.09.014. [DOI] [PubMed] [Google Scholar]
- McCarrey J.R. Toward a more precise and informative nomenclature describing fetal and neonatal male germ cells in rodents. Biol. Reprod. 2013;89:47. doi: 10.1095/biolreprod.113.110502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarrey J.R., Berg W.M., Paragioudakis S.J., Zhang P.L., Dilworth D.D., Arnold B.L., Rossi J.J. Differential transcription of Pgk genes during spermatogenesis in the mouse. Dev. Biol. 1992;154:160–168. doi: 10.1016/0012-1606(92)90056-m. [DOI] [PubMed] [Google Scholar]
- Meng X., Lindahl M., Hyvonen M.E., Parvinen M., de Rooij D.G., Hess M.W., Raatikainen-Ahokas A., Sainio K., Rauvala H., Lakso M., et al. Regulation of cell fate decision of undifferentiated spermatogonia by GDNF. Science. 2000;287:1489–1493. doi: 10.1126/science.287.5457.1489. [DOI] [PubMed] [Google Scholar]
- Mi H., Huang X., Muruganujan A., Tang H., Mills C., Kang D., Thomas P.D. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017;45:D183–D189. doi: 10.1093/nar/gkw1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molaro A., Falciatori I., Hodges E., Aravin A.A., Marran K., Rafii S., Mccombie W.R., Smith A.D., Hannon G.J. Two waves of de novo methylation during mouse germ cell development. Genes Dev. 2014;28:1544–1549. doi: 10.1101/gad.244350.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan H.D., Santos F., Green K., Dean W., Reik W. Epigenetic reprogramming in mammals. Hum. Mol. Genet. 2005;14:R47–R58. doi: 10.1093/hmg/ddi114. [DOI] [PubMed] [Google Scholar]
- Mutoji K., Singh A., Nguyen T., Gildersleeve H., Kaucher A.V., Oatley M.J., Oatley J.M., Velte E.K., Geyer C.B., Cheng K., et al. TSPAN8 expression distinguishes spermatogonial stem cells in the prepubertal mouse testis. Biol. Reprod. 2016;95:117. doi: 10.1095/biolreprod.116.144220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair S.S., Coolen M.W., Stirzaker C., Song J.Z., Statham A.L., Strbenac D., Robinson M.D., Clark S.J. Comparison of methyl-DNA immunoprecipitation (MeDIP) and methyl-CpG binding domain (MBD) protein capture for genome-wide DNA methylation analysis reveal CpG sequence coverage bias. Epigenetics. 2011;6:34–44. doi: 10.4161/epi.6.1.13313. [DOI] [PubMed] [Google Scholar]
- Nilsson E., Sadler-Riggleman I., Skinner M.K. Environmentally induced epigenetic transgenerational inheritance of disease. Environ. Epigenetics. 2018;4:1–13. doi: 10.1093/eep/dvy016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson E.E., Savenkova M.I., Schindler R., Zhang B., Schadt E.E., Skinner M.K. Gene bionetwork analysis of ovarian primordial follicle development. PLoS ONE. 2010;5:e11637. doi: 10.1371/journal.pone.0011637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oatley M.J., Kaucher A.V., Racicot K.E., Oatley J.M. Inhibitor of DNA binding 4 is expressed selectively by single spermatogonia in the male germline and regulates the self-renewal of spermatogonial stem cells in mice. Biol. Reprod. 2011;85:347–356. doi: 10.1095/biolreprod.111.091330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orgebin-Crist M.C., Danzo B.J., Cooper T.G. Re-examination of the dependence of epididymal sperm viability on the epididymal environment. J. Reprod. Fertil. 1976:115–128. [PubMed] [Google Scholar]
- Palmer S.J., Burgoyne P.S. In situ analysis of fetal, prepuberal and adult XX----XY chimaeric mouse testes: sertoli cells are predominantly, but not exclusively, XY. Development. 1991;112:265–268. doi: 10.1242/dev.112.1.265. [DOI] [PubMed] [Google Scholar]
- Pennisi E. European society for evolutionary biology meeting. epigenetics linked to inbreeding depression. Science. 2011;333:1563. doi: 10.1126/science.333.6049.1563. [DOI] [PubMed] [Google Scholar]
- Pomraning K.R., Smith K.M., Freitag M. Genome-wide high throughput analysis of DNA methylation in eukaryotes. Methods. 2009;47:142–150. doi: 10.1016/j.ymeth.2008.09.022. [DOI] [PubMed] [Google Scholar]
- Popp C., Dean W., Feng S., Cokus S.J., Andrews S., Pellegrini M., Jacobsen S.E., Reik W. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature. 2010;463:1101–1105. doi: 10.1038/nature08829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.D., Mccarthy D.J., Smyth G.K. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanli I., Feil R. Chromatin mechanisms in the developmental control of imprinted gene expression. Int. J. Biochem. Cell Biol. 2015;67:139–147. doi: 10.1016/j.biocel.2015.04.004. [DOI] [PubMed] [Google Scholar]
- Schmahl J., Kim Y., Colvin J.S., Ornitz D.M., Capel B. Fgf9 induces proliferation and nuclear localization of FGFR2 in Sertoli precursors during male sex determination. Development. 2004;131:3627–3636. doi: 10.1242/dev.01239. [DOI] [PubMed] [Google Scholar]
- Schutte B., El Hajj N., Kuhtz J., Nanda I., Gromoll J., Hahn T., Dittrich M., Schorsch M., Muller T., Haaf T. Broad DNA methylation changes of spermatogenesis, inflammation and immune response-related genes in a subgroup of sperm samples for assisted reproduction. Andrology. 2013;1:822–829. doi: 10.1111/j.2047-2927.2013.00122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seisenberger S., Andrews S., Krueger F., Arand J., Walter J., Santos F., Popp C., Thienpont B., Dean W., Reik W. The dynamics of genome-wide DNA methylation reprogramming in mouse primordial germ cells. Mol. Cell. 2012;48:849–862. doi: 10.1016/j.molcel.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seisenberger S., Peat J.R., Reik W. Conceptual links between DNA methylation reprogramming in the early embryo and primordial germ cells. Curr. Opin. Cell Biol. 2013;25:281–288. doi: 10.1016/j.ceb.2013.02.013. [DOI] [PubMed] [Google Scholar]
- Seki Y., Yamaji M., Yabuta Y., Sano M., Shigeta M., Matsui Y., Saga Y., Tachibana M., Shinkai Y., Saitou M. Cellular dynamics associated with the genome-wide epigenetic reprogramming in migrating primordial germ cells in mice. Development. 2007;134:2627–2638. doi: 10.1242/dev.005611. [DOI] [PubMed] [Google Scholar]
- Seligman J., Page D.C. The Dazh gene is expressed in male and female embryonic gonads before germ cell sex differentiation. Biochem. Biophys. Res. Commun. 1998;245:878–882. doi: 10.1006/bbrc.1998.8530. [DOI] [PubMed] [Google Scholar]
- Shirane K., Kurimoto K., Yabuta Y., Yamaji M., Satoh J., Ito S., Watanabe A., Hayashi K., Saitou M., Sasaki H. Global landscape and regulatory principles of DNA methylation reprogramming for germ cell specification by mouse pluripotent stem cells. Dev. Cell. 2016;39:87–103. doi: 10.1016/j.devcel.2016.08.008. [DOI] [PubMed] [Google Scholar]
- Skinner M., Guerrero-Bosagna C., Haque M.M., Nilsson E., Bhandari R., Mccarrey J. Environmentally induced transgenerational epigenetic reprogramming of primordial germ cells and subsequent germline. PLoS ONE. 2013;8:1–15. doi: 10.1371/journal.pone.0066318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner M.K. Endocrine disruptor induction of epigenetic transgenerational inheritance of disease. Mol. Cell. Endocrinol. 2014;398:4–12. doi: 10.1016/j.mce.2014.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner M.K., Ben Maamar M., Sadler-Riggleman I., Beck D., Nilsson E., Mcbirney M., Klukovich R., Xie Y., Tang C., Yan W. Alterations in sperm DNA methylation, non-coding RNA and histone retention associate with DDT-induced epigenetic transgenerational inheritance of disease. Epigenet. Chromatin. 2018;11:1–24. doi: 10.1186/s13072-018-0178-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner M.K., Guerrero-Bosagna C. Role of CpG deserts in the epigenetic transgenerational inheritance of differential DNA methylation regions. BMC Genomics. 2014;15:692. doi: 10.1186/1471-2164-15-692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner M.K., Manikkam M., Tracey R., Guerrero-Bosagna C., Haque M.M., Nilsson E. Ancestral dichlorodiphenyltrichloroethane (DDT) exposure promotes epigenetic transgenerational inheritance of obesity. BMC Med. 2013;11:1–16. doi: 10.1186/1741-7015-11-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner M.K., Nilsson E., Sadler-Riggleman I., Beck D., McCarrey J.R. Transgenerational sperm DNA methylation epimutation developmental origins following ancestral vinclozolin exposure. Epigenetics. 2019;14:721–739. doi: 10.1080/15592294.2019.1614417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubry A. Epigenetic inheritance and evolution: a paternal perspective on dietary influences. Prog. Biophys. Mol. Biol. 2015;118:79–85. doi: 10.1016/j.pbiomolbio.2015.02.008. [DOI] [PubMed] [Google Scholar]
- Steger K. Transcriptional and translational regulation of gene expression in haploid spermatids. Anat. Embryol. 1999;199:471–487. doi: 10.1007/s004290050245. [DOI] [PubMed] [Google Scholar]
- Suzuki H., Sada A., Yoshida S., Saga Y. The heterogeneity of spermatogonia is revealed by their topology and expression of marker proteins including the germ cell-specific proteins Nanos2 and Nanos3. Dev. Biol. 2009;336:222–231. doi: 10.1016/j.ydbio.2009.10.002. [DOI] [PubMed] [Google Scholar]
- Tang W.W., Dietmann S., Irie N., Leitch H.G., Floros V.I., Bradshaw C.R., Hackett J.A., Chinnery P.F., Surani M.A. A unique gene regulatory network resets the human germline epigenome for development. Cell. 2015;161:1453–1467. doi: 10.1016/j.cell.2015.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W.W., Kobayashi T., Irie N., Dietmann S., Surani M.A. Specification and epigenetic programming of the human germ line. Nat. Rev. Genet. 2016;17:585–600. doi: 10.1038/nrg.2016.88. [DOI] [PubMed] [Google Scholar]
- Tokuda M., Kadokawa Y., Kurahashi H., Marunouchi T. CDH1 is a specific marker for undifferentiated spermatogonia in mouse testes. Biol. Reprod. 2007;76:130–141. doi: 10.1095/biolreprod.106.053181. [DOI] [PubMed] [Google Scholar]
- Vaiserman A.M., Koliada A.K., Jirtle R.L. Non-genomic transmission of longevity between generations: potential mechanisms and evidence across species. Epigenet. Chromatin. 2017;10:38. doi: 10.1186/s13072-017-0145-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bragt M.P., Roepers-Gajadien H.L., Korver C.M., Bogerd J., Okuda A., Eggen B.J., de Rooij D.G., Van Pelt A.M. Expression of the pluripotency marker UTF1 is restricted to a subpopulation of early A spermatogonia in rat testis. Reproduction. 2008;136:33–40. doi: 10.1530/REP-07-0536. [DOI] [PubMed] [Google Scholar]
- Vergeer P., Wagemaker N.C., Ouborg N.J. Evidence for an epigenetic role in inbreeding depression. Biol. Lett. 2012;8:798–801. doi: 10.1098/rsbl.2012.0494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal V.P., Chaboissier M.C., de Rooij D.G., Schedl A. Sox9 induces testis development in XX transgenic mice. Nat. Genet. 2001;28:216–217. doi: 10.1038/90046. [DOI] [PubMed] [Google Scholar]
- Vincent J.J., Huang Y., Chen P.Y., Feng S., Calvopina J.H., Nee K., Lee S.A., Le T., Yoon A.J., Faull K., et al. Stage-specific roles for tet1 and tet2 in DNA demethylation in primordial germ cells. Cell Stem Cell. 2013;12:470–478. doi: 10.1016/j.stem.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Western P.S., Miles D.C., van den Bergen J.A., Burton M., Sinclair A.H. Dynamic regulation of mitotic arrest in fetal male germ cells. Stem Cells. 2008;26:339–347. doi: 10.1634/stemcells.2007-0622. [DOI] [PubMed] [Google Scholar]
- Willerton L., Smith R.A., Russell D., Mackay S. Effects of FGF9 on embryonic Sertoli cell proliferation and testicular cord formation in the mouse. Int. J. Dev. Biol. 2004;48:637–643. doi: 10.1387/ijdb.031778lw. [DOI] [PubMed] [Google Scholar]
- Yadav R.P., Kotaja N. Small RNAs in spermatogenesis. Mol. Cell. Endocrinol. 2014;382:498–508. doi: 10.1016/j.mce.2013.04.015. [DOI] [PubMed] [Google Scholar]
- Yamaguchi S., Hong K., Liu R., Inoue A., Shen L., Zhang K., Zhang Y. Dynamics of 5-methylcytosine and 5-hydroxymethylcytosine during germ cell reprogramming. Cell Res. 2013;23:329–339. doi: 10.1038/cr.2013.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi S., Shen L., Liu Y., Sendler D., Zhang Y. Role of Tet1 in erasure of genomic imprinting. Nature. 2013;504:460–464. doi: 10.1038/nature12805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B., Horvath S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005;4:17. doi: 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- Zhao W., Langfelder P., Fuller T., Dong J., LI A., Hovarth S. Weighted gene coexpression network analysis: state of the art. J. Biopharm. Stat. 2010;20:281–300. doi: 10.1080/10543400903572753. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All molecular data have been deposited into the public database at NCBI (GEO # GSE117995 and GSE151458), and R code computational tools are available at GitHub (https://github.com/skinnerlab/MeDIP-seq) and www.skinner.wsu.edu.
-
•
Data availability: All molecular data has been deposited into the public database at NCBI (GEO # GSE117995 & GSE151458).
-
•
Code availability: R code computational tools are available at GitHub (https://github.com/skinnerlab/MeDIP-seq) and www.skinner.wsu.edu.