

Abstract
APOBEC3A (A3A) deaminates deoxycytidine in target motif TC in a single‐stranded DNA (we termed it as TC DNA), which mortally mutates viral pathogens and immunoglobulins, and leads to the diversification and lethality of cancers. The crystal structure of A3A‐DNA revealed a unique U‐shaped recognition mode of target base dC0. However, when TC DNA was titrated into 15N‐labeled A3A solution, we observed two sets of 1H‐15N cross‐peaks of A3A in HSQC spectra, and two sets of 1H‐1H cross‐peaks of DNA in two‐dimensional 13C,15N‐filtered TOCSY spectra, indicating two different kinds of conformers of either A3A or TC DNA existing in solution. Here, mainly by NMR, we demonstrated that one DNA conformer interacted with one A3A conformer, forming a specific complex A3AS‐DNAS in a way almost similar to that observed in the reported crystal A3A‐DNA structure, where dC0 inserted into zinc ion binding center. While the other DNA conformer bound with another A3A conformer, but dC0 did not extend into the zinc‐binding pocket, forming a nonspecific A3ANS‐DNANS complex. The NMR solution structure implied three sites Asn61, His182 and Arg189 were necessary to DNA recognition. These observations indicate a distinctive way from that reported in X‐ray crystal structure, suggesting an unexpected mode of deaminase APOBEC3A to identify target motif TC in DNA in solution.
Keywords: APOBEC3A, DNA, identification, nuclear magnetic resonance, structure
Short abstract
PDB Code(s): 7D3X, 3D3W, 5W2M, 5ZVA, 5ZVB, 5K83, 6K3J, 6K3K, 6bux, 5KEG, 5SWW and 5TD5
1. INTRODUCTION
APOBEC3 (A3), a family of activation‐induced cytidine deaminases, contains seven members APOBEC3A (A3A), APOBEC3B (A3B), APOBEC3C (A3C), APOBEC3DE (A3DE), APOBEC3F (A3F), APOBEC3G (A3G) and APOBEC3H (A3H). They deaminate deoxycytidines into uridines in single‐stranded DNA (ssDNA) during reverse transcription, which produces G to A hypermutations in a viral genome, generates defective proteins and proviruses, thus decreases the possibility of further viral replication. 1 , 2 , 3 , 4 A3s generally display differential substrate binding specificities (CC or TC), leading to altered frequencies of mutation of deoxycytidines. 1 , 2 , 3 , 5 , 6 , 7 , 8 Among them, A3DE, A3F, A3G and A3H restrict replication of HIV‐1 virus in strains lacking the virus infectivity factor (Vif) by deaminating cytidine in virus cDNA. 9 , 10 , 11 , 12 , 13 Vif facilitates polyubiquitination of A3 members, leading to proteasomal degradation. 14 , 15 , 16 , 17 , 18 , 19 Specifically, A3A suppresses HIV‐1 primary infection in macrophages, 20 , 21 as well as other viruses (including hepatitis B virus, human papillomavirus, Epstein–Barr virus, cytomegalovirus, and herpes simplex virus type 1) by inducing the lethal mutation in viral genomic DNA through cytidine deamination. 21 , 22 , 23 , 24
To study how A3s specifically recognize the target motifs TC or CC in DNA, many structures of A3‐DNA complexes had been reported. 25 , 26 The structure of human A3F‐CD2 (i.e., hA3F‐CD2) in complex with a 10‐dT ssDNA (PDB code 5W2M) 27 shows that DNA is far away from catalytic zinc‐binding motif, while the structures of chimeric hA3F‐CD2 (i.e., hA3Fc‐CD2) in complex with DNA strands (PDB codes 5ZVA and 5ZVB) demonstrate two DNA binding sites on hA3Fc‐CD2. 28 One is formed by Tyr333, Lys358 and Tyr359 in one monomer of hA3Fc‐CD2, the other is composed by conserved residues Trp277, Y307YFW310 near to Zn2+ binding region in another monomer hA3Fc‐CD2. The structure of inactive pseudo‐catalytic rhesus macaque A3G‐NTD (i.e., rA3G‐NTD) in complex with poly‐dT ssDNA (PDB code 5K83) 29 reveals that only one deoxythymidine is in a shallow cleft close to the pseudo‐catalytic zinc‐binding motif. The structures of A3G‐CD2 in complex with two deamination product DNA strands containing 5‐iodinated CUCI and CCUI motifs (PDB codes 6K3J and 6K3K) 30 imply the structural basis of the two DNA binding modes responsible for cytidine deamination in TCC and CCC motifs catalyzed by A3G, respectively. The 10‐site A3G‐CD2 variant (i.e., P200A, L234K, N236A, C243A, P247K, Q318K, F310K, C321A, Q322A, and C356A, termed as A3G‐CTD2) was reported to have much stronger binding affinity to DNA and higher catalytic efficiency than wild‐type A3G‐CD2. 31 The structure of the inactive form A3G‐CTD2 E259A variant (i.e., A3G‐CTD2*) complexed with ssDNA (PDB code 6bux) 31 demonstrates that only base dC0 in the target motif 5′‐C−1C0C1 inserts into the pocket of the zinc‐binding active site region. Residues Asp316 and Asp317 of A3G‐CTD2* form hydrogen‐bonds with base dC−1, accounting for their roles in identifying base dC−1. The structures of inactive variant A3A E72A or E72A/C171A 32 , 33 (Glu72 is conserved in A3 family, Figure S1, Supporting Information) and chimeric A3B variant E255A‐CD2 33 (i.e., A3Bctd‐QMloop3A3Aloop1 or A3B‐CTD*, in which loop 3 of A3B was replaced by loop 1 of A3A) in complexes with DNA (PDB codes 5KEG, 5SWW and 5TD5) also show that only cytidine in hotspot 5′‐TC inserts into zinc‐binding active site pocket. One molecular DNA binds to one molecular A3A or chimeric A3B‐CD2 in a U‐shaped mode, similar to that observed in A3G‐CTD2*‐DNA complex. These three structures obviously visualize the active sites poised for catalysis of A3A and A3B, and pinpoints the residues conferring the specificity of TC motif.
All those reported results indicated that A3 proteins (including A3A) identified the target base dC0 in one mode, where the base dC0 must insert into the active site of A3A. From this point, the specific identification of base dC0 is in a unique way. It is well known that, by nuclear magnetic resonance (NMR) technique, the dynamic process can be sometimes observed. Thus, we decided to investigate the interactions of A3A with DNA in solution by NMR, since the assignment of the atoms 13C, 15N and 1H of inactive A3A variant L63N/C64S/E72Q/C171Q (i.e., A3A4M in the current report) was accessible. 34 The residues L63, C64 and C171 are located in the surface of A3A, the mutation from L63 to N63 improves protein solubility, while the mutations C64S and C171Q are helpful to avoid A3A aggregation by forming disulfide bond between two A3A monomers, thus resolving ambiguous assignments of free A3A. 34 We made 15N‐labled NMR sample, and acquired 1H‐15N HSQC spectrum, which displayed resolved cross‐peaks (Figure S2), consistent with the previous observation. 34 However, during our NMR studies, we found that, upon TC DNA (with a sequence of 5′‐ATTTT−1C0A+1ATT‐3′, Table 1) being titrated into the solution of 15N labeled inactive A3A4M, A3A4M variant two sets of 1H‐15N cross‐peak signals belonging to A3A4M in two‐dimensional (2D) 1H‐15N HSQC spectrum (Figure 1a) were observed even at a molar ratio less than 1:1, and two sets of 1H‐1H correlation signals belonging to TC DNA were either found in 2D 13C,15N‐filtered 1H‐1H TOCSY (Figure 1b,c). After we finished assignment of the backbone atoms of A3A4M, we selected several cross‐peaks belonging to residues L12, V150, S187 and G198 in 1H‐15N HSQC spectra acquired at different molar ratio, and measured the intensity ratios of these cross‐peaks, the results were shown in Figure S3. From Figure S3, we can conclude that, after A3A4M was mixed with DNA at different molar ratios (TC DNA vs. A3A4M), the intensity ratio of the two complexes (specific vs. nonspecific binding states) is almost unchanged. Obviously, these observations indicated that A3A4M interacted with TC DNA in solution in a manner, not completely identical to those observed in the reported crystal structures of A3‐DNA complexes mentioned above. To account for these observations, we here conducted extensive NMR studies on the interactions between A3A4M and TC DNA.
TABLE 1.
5′‐FAM‐labeled DNA strands used in this manuscript
| ssDNA | K d (μM) | Sequence |
|---|---|---|
| TC | 0.086 ± 0.010 | 5′‐ATT TTC AAT T‐3′ |
| TTC‐1 | 5.683 ± 0.236 | 5′‐TTT CTT T‐3′ |
| TTC‐2 | 3.486 ± 0.172 | 5′‐TTT TCT TTT‐3′ |
| TTC‐3 | 2.624 ± 0.070 | 5′‐TTT TCT TTT TT‐3′ |
| TTC‐4 | 4.280 ± 0.273 | 5′‐TTT TCT TTT TTT T‐3′ |
| TTC‐5 | 3.852 ± 0.294 | 5′‐TTT TCT TTT TTT TTT‐3′ |
| TTC‐6 | 4.193 ± 0.306 | 5′‐TTT TCT TTT TTT TTT TT‐3′ |
| TTC‐7 | 3.610 ± 0.091 | 5′‐TTT TTT CTT TT‐3′ |
| TTC‐8 | 19.925 ± 2.091 | 5′‐TTT TTT TTC TTT T‐3′ |
| TTC‐9 | 10.490 ± 1.104 | 5′‐TTT TTT TTT TCT TTT‐3′ |
| TTC‐10 | 9.188 ± 0.846 | 5′‐TTT TTT TTT TTT CTT TT‐3′ |
| TTC‐11 | 39.184 ± 2.524 | 5′‐TTT TTT TTT TTT TTT TCT TTT‐3′ |
FIGURE 1.
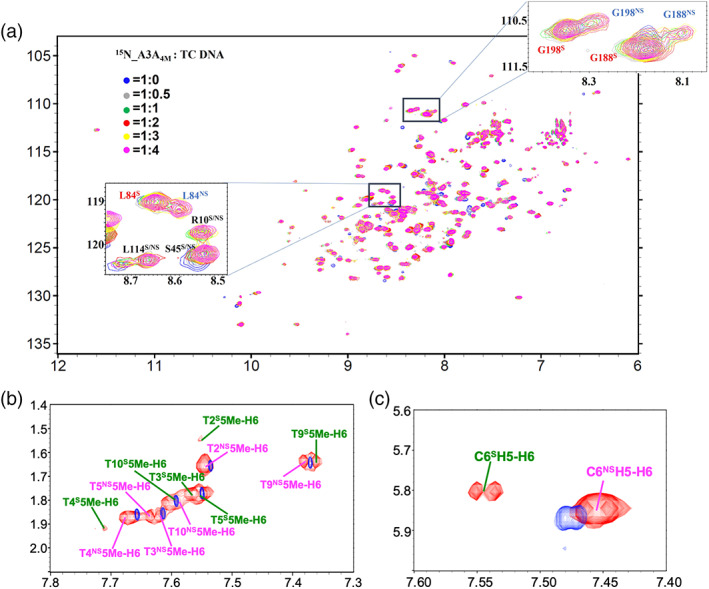
Two kinds of conformers of either A3A4M or TC DNA in solution upon their interaction with each other were observed by NMR technique. (a) 1H‐15N HSQC spectra superimposition at different molar ratios of A3A4M versus DNA. (b) 13C /15N filtered two‐dimensional (2D) 1H‐1H TOCSY spectrum was overlapped with the 2D TOCSY spectrum of free TC DNA. In (b, c), the 1H‐1H cross‐peaks of free TC DNA were displayed in blue. The assignment of the signals in (a–c) indicated the residues in A3AS or DNAS (in green) and A3ANS or DNANS (in pink)
2. RESULTS AND DISCUSSION
2.1. Two kinds of A3A4M‐DNA complexes observed in solution
To study how A3A interacted with TC DNA in solution, we first conducted extensive NMR studies on the interactions between A3A4M and TC DNA. We titrated TC DNA into 15N‐labeled A3A4M solution. In the absence of DNA, there is only one set of 1H‐15N cross peaks in the HSQC spectra, although some peaks have weak intensity due to the mixed forms of monomer and dimer A3A4M in solution, determined by running ultracentrifugation (Figure S4). However, the addition of TC DNA into A3A4M solution led to two sets of 1H‐15N cross peaks of A3A4M shown up in 1H‐15N HSQC spectra, indicating that two kinds of A3A4M conformers existing in solution. Moreover, this observation was independent on the molar ratio of A3A4M versus TC DNA. When the molar ratio was increased up to 1:2, the cross‐peaks did not shift any longer. So, to do NMR studies on the interactions between A3A4M and TC DNA, we made NMR samples of double isotope 13C, 15N labeled or triple isotope 13C, 15N, 2H labeled A3A4M in complex with TC DNA at a molar ratio 1:2 for further NMR experiments.
To clearly identify the signals belonging to the bound TC DNA, we acquired 2D 1H‐31P HETCOR spectrum, 13C/15N‐filtered 1H‐1H TOCSY and 1H‐1H NOESY spectra. Interestingly, in the 2D filtered TOCSY spectrum (Figure 1b,c), two sets of 1H‐1H cross‐peaks of DNA were also observed, indicating that two kinds of DNA conformers existing in solution, either. Compared the chemical shifts of A3A‐bound DNA with those of free DNA, we found that, in one A3A‐bound TC DNA conformer, which displayed more protons displaying chemical shift changes (Figure S5a), its protons demonstrated different intra‐DNA 1H‐1H NOE patterns from those observed in NOESY spectrum of free TC DNA. So, we tentatively named this kind of DNA conformation as A3A4M specifically bound conformer (i.e., DNAS). In contrast, in another A3A‐bound TC DNA conformer, which had fewer protons demonstrating chemical shifts changes (Figure S5b), its protons had intra‐DNA 1H‐1H NOE patterns almost similar to those observed in NOESY spectrum of free TC DNA. We thus termed this kind of TC DNA conformation as A3A4M nonspecifically bound conformer (i.e., DNANS). To assign NMR signals of A3A in different states, a series of 2D and three‐dimensional (3D) NMR experiments were then performed to assign NMR signals belonging to backbone and side‐chain atoms of A3A4M, and intramolecular NOEs in A3A4M (described in section 4.1). Compared to the free A3A4M, the chemical shifts changes of the backbone nitrogen atoms and amide protons of A3A4M (Figure 2) implied that the TC DNA binding regions in two kinds of A3A4M conformers were almost similar to each other, mainly locating in the loops 1, 3, 5 and 7. Subsequently, by analyzing 3D 13C‐F1 edited, 13C/15N‐F3 filtered NOESY spectra, we got intermolecular NOEs between A3A4M and TC DNA. We found that one A3A4M conformer individually displayed intermolecular NOEs with only one TC DNA conformer. We thus termed two different kinds of A3A4M conformers as DNAS bound A3A4M (i.e., A3AS) and DNANS bound A3A4M (i.e., A3ANS). To explore how DNAS and DNANS interacted with A3AS and A3ANS, respectively, we further decided to determine the solution structures of the A3ANS‐DNANS and A3AS‐DNAS complexes. So, we assigned all intramolecular NOEs in DNANS, DNAS, A3AS and A3ANS, intermolecular NOEs between A3AS and DNAS, and between A3ANS and DNANS.
FIGURE 2.
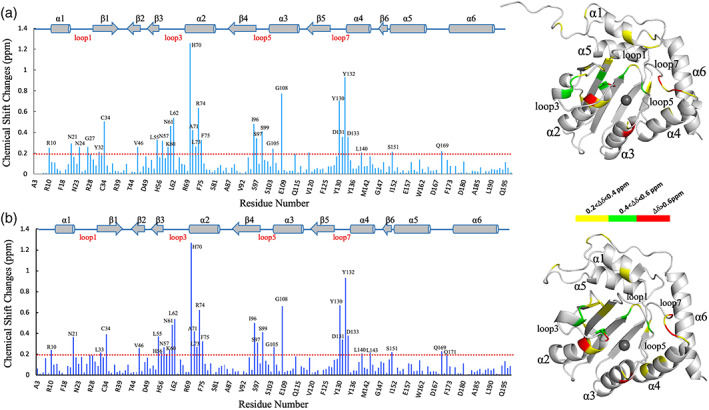
The chemical shift changes of backbone amide nitrogen and protons in two DNA‐bound states of A3A4M compared to free A3A4M. (a) DNA specifically bound A3A4M. (b) DNA nonspecifically bound A3A4M. In the left of (a, b), the secondary regions were labeled and drawn above the bar charts. The dashed lines indicated the lowest values, below which the chemical shift changes were not considered. In the right of (a, b), the residues in the surface of A3A4M structure with different scales (0.2–0.4 ppm, 0.4–0.6 ppm and >0.6 ppm) of the chemical shift changes were displayed in yellow, green and red, respectively
2.2. DNAS binds A3A4M in a manner almost similar to that observed in the reported crystal structures of A3A‐DNA or A3B‐DNA complexes
To calculate NMR structures of A3ANS‐DNANS and A3AS‐DNAS complexes, we generated 3,346 and 3,409 distance constraints derived from NOE intensities in total (including 50 and 95 intermolecular NOEs, which were listed in Tables S1 and S2), 184 phi and 185 psi dihedral angel constrains based on the chemical shifts of backbone atoms of A3A4M, 174 hydrogen‐bond constraints based on the secondary structure of A3A4M, as shown in Table S3. Constraints between the ligated residues (His70, Cys101 and Cys106) in the protein and the zinc ion were added using the procedure of Neuhaus et al as described previously. 35 , 36 The final two ensembles, each of which contained 20 structures of A3A4M‐DNA complexes with the lowest energies, were displayed in Figure 3a,b, with RMSD values of 0.55 ± 0.11 Å and 0.54 ± 0.07 Å for backbone atoms in the secondary structural regions, respectively, upon overlapping the backbone atoms at the secondary regions of A3A4M. The Ramachandran plot displayed 89.0 and 86.6% of the residues in the most‐favored regions, and no residues (0%) located in the disallowed regions, indicating that all structures were reasonable. On the whole, as shown in Figure 3c,d, the TC DNA mainly bound in a groove between loop 1 or 3 and loop 5 or 7, consistent with the NMR titration results, indicating the roles of these loops in the interactions between A3A4M and DNA. The global folding of A3A4M is almost identical in two bound states with an RMSD value of 1.27 Å upon 162 Cα atoms of the residues in the secondary structures. The loops 1, 3, 5 and 7 display plasticity to accommodate the conformational changes of the DNA upon its binding to A3A4M.
FIGURE 3.
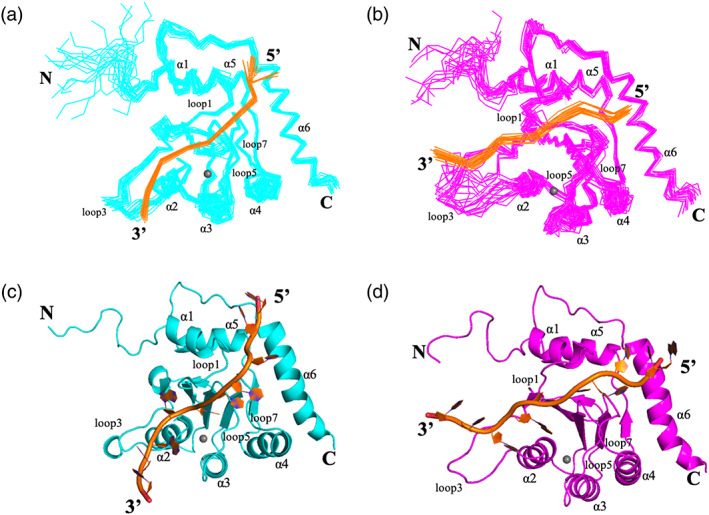
NMR structures of A3A‐TC DNA complexes. (a) The bundle of 20 structures of A3AS‐DNAS complex with the lowest energies. (b) The bundle of 20 structures of A3ANS‐DNANS complex with the lowest energies. (c) One structure of A3AS‐DNAS complex. (d) One structure of A3ANS‐DNANS complex. In (a, b) and (c, d), DNA (in orange) and A3A4M (in cyan or pink) were displayed in ribbon and cartoon modes, respectively. The N‐ and C‐termini, the loops 1, 3, 5 and 7, the helices α1, α2, α3, α4, α5 and α6 of A3A4M, and 5′‐ and 3′‐ ends of DNA were labeled, respectively. The grey balls represented zinc ions
To verify the effects of these loops on enhancing DNA binding, we varied the length of ssDNA (containing only one target motif TC) by cutting off or adding bases at either 5′‐ end or 3′‐ end, respectively, from TTC‐1 to TTC‐11 (Table 1 and Figure S6). Without adenine adjacent to TC motif at its 3′‐ end (i.e., TCA motif in TC DNA), all these DNA strands had dramatically decreased binding affinities to A3A4M, compared to TC DNA. Compared to TTC‐2 (containing 9 bases in total), A3A4M demonstrated no significant changes in binding affinities (all K D < 5 μM) to TTC‐3 (11 bases in total, adding 2 dT bases at 3′‐ end), TTC‐4 (13 bases in total, adding 4 dT bases at 3′‐ end), TTC‐5 (15 bases in total, adding 6 dT bases at 3′‐ end) and TTC‐6 (17 bases in total, adding 8 dT bases at 3′‐ end). Since 3′‐ end bases of DNA mainly interacted with loops 1, 3, 5 and 7 of A3A4m, as shown in Figure 3, this observation implied the indispensable roles of these loops in stabilizing the conformation of A3A‐DNA complex. In contrast, compared to TTC‐2, when the length of DNA was extended by adding different numbers of dT into 5′‐ end, the binding affinities (20 μM < K D < 40 μM) of TTC‐8 (13 bases in total, adding 4 dT bases at 5′‐ end), TTC‐9 (15 bases in total, adding 6 dT bases at 5′‐ end), TTC‐10 (17 bases in total, adding 8 dT bases at 5′‐ end) and TTC‐11 (21 bases in total, adding 12 bases at 5′‐ end) were remarkably impaired by 5–10 fold. This finding indicated that, the DNA strands, with larger number of flanking nucleotides at 5′‐ end than TTC‐2, obviously weakened the interactions between A3A4M and DNA, and the conformation of DNA became more flexible in A3A‐DNA complex. Meanwhile, TTC‐7 DNA (11 bases in total, adding 2 dT bases at 5′‐ end) displayed binding affinity to A3A4M stronger binding affinities than TTC‐1 DNA (7 bases in total) (Figure S6a), and in an almost similar order of magnitude to those of TTC‐2 DNA (9 bases in total) and TTC‐3 DNA (11 bases in total) (Figure S6b), further implying that DNA with a length of 9–11 bases was most suitable for A3A4M interaction, consistent with the previous report. 34
In A3AS‐DNAS complex structure, the zinc ion coordinated with residues His70, Cys101, Cys106, the whole DNA interacted with A3A4M also in a U‐shaped manner. The target base dC0 extended into the pocket of zinc ion binding site (Figure 4a,b), and had parallel or T‐shaped π–π stacking interactions with the aromatic side‐chains of the conserved residues His70 and Tyr130 (Figure 4a,c), respectively, similar to those observed in the reported structures of A3‐DNA complexes (Figure 4d–f). 31 , 32 , 33 The conformation of dC0 was stabilized by several hydrogen‐bonds between Ala71 backbone NH and dC0 O2 atoms, between the carbonyl oxygen atoms of Trp98 and Ser99 and NH2 of dC0, and between the hydroxyl group of the side‐chain of Thr31 and O3 atom of the sugar ring of dC0, which also addressed the specificity for dC0 over dT−1 (Figure 4a). The base dT−1 was identified by the conserved residue Asp131 in loop 7 through hydrogen‐bond between amide nitrogen of Asp131 and oxygen of dT−1 (Figure S7a,e). Identical to those observed in the crystal structures of A3A‐DNA complex, 32 , 33 A3Bctd*‐DNA complex 33 and A3G‐CTD2*‐DNA complex 31 (Figure S7b–d), the Asp131 side chain had a salt bridge to the side‐chain of the conserved residue Arg189 in helix α6, which might enhance all hydrogen‐bond interactions between loop 7 and base dT−1. Similarly, as shown in Figure S8, the conserved residue His 29 in loop 1 had π–π stacking interactions with dA+1, as reported in the crystal structures of A3A‐DNA, 32 A3Bctd*‐DNA 33 and A3G‐CTD2*‐DNA 31 complexes, which once again confirmed that residue His 29 might work as “latch” in the process of A3A recognizing the TCA motif in the substrate DNA.
FIGURE 4.
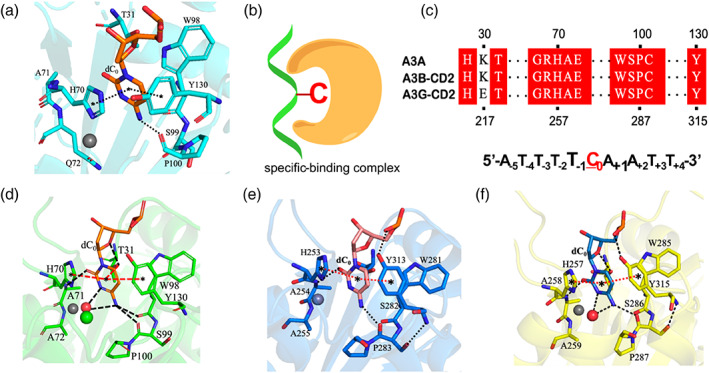
Structural analysis of A3AS‐DNAS complex. (a) The target dC0 identified by A3 members in structures of A3AS‐DNAS complex. (b) The specific binding complex displayed in a cartoon mode, where dC0 inserted into zinc‐ion binding center. (c) Conserved residues among A3A, A3B and A3G interact with dC0. (d) The target dC0 identified by A3 members in structures of A3A‐DNA complex (5KEG). (e) The target dC0 identified by A3 members in structures of A3Bctd*‐DNA complex (5TD5), and (f) A3G‐CTD2* complex (6BUX). The dashed lines with stars in both ends indicate stacking interactions. The dash lines without stars in both ends imply hydrogen‐bonds
2.3. A3ANS‐DNANS structure implies three new sites important to DNA binding
In A3ANS‐DNANS complex structure (Figure 5a), DNANS located in a region almost similar to that observed in the structure of A3AS‐DNAS complex. However, the target base dC0 did not extend into the zinc ion binding pocket. The averaged distances between zinc ion and N4 atom of dC0 in all final 20 structures were measured as 12.0 Å, which was much farther than that (5.0 Å) in all final 20 structures of A3AS‐DNAS complex. Therefore, we thought that this kind of interaction between A3ANS and DNANS was nonspecific, however, which was not single one case reported between DNA and A3 family members. For examples, as displayed in Figure S9, poly‐dT DNA interactions with rA3G‐CD1 only led to the conformational change of the loops and residues surrounding the Zn2+‐coordinated center. 29 The hA3F‐CD2 bound to poly‐dT DNA through a positively charged site distal to the active zinc ion binding center. 27 DNA (containing two TC motifs at both ends) interacted with hA3Fc‐CD2 dimer at two sites existing in the hA3Fc‐CD2 monomers. 28 In all these structures, as well as that of A3ANS‐DNANS complex, the target dC0 was not in the zinc ion binding pocket. However, obviously, DNA bound to different regions of A3 proteins, suggesting that nonspecific interactions between DNA and A3 members were in varied manners.
FIGURE 5.
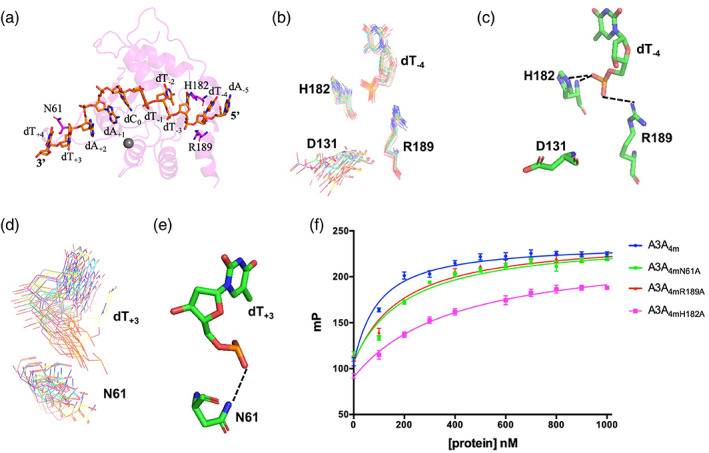
The NMR structure of A3ANS‐DNANS complex indicated three new sites important to DNA binding. (a) The positions of residues N61, H182 and R189. DNA was displayed in stick mode, A3A4M was shown in transparent, pink cartoon mode. (b, c) Residues H182 and R189 formed H‐bonds with DNA base dT−4. (d, e) Residue N61 formed H‐bond with dT+3 . In (b, d), the residues N61, D131, H182 and R189 in A3A4M and bases dT+3 and dT−4 in DNA were shown in line mode in all final 20 structures with the lowest energies, indicating the convergent orientations of their side‐chains. H182 located in regid α6 helix, while N61 was in flexible loop 3, which resulted in convergent conformation of H182, and shifted‐conformation of N61. In (c, e), residues N61, D131, H182 and R189 in A3A and bases dT−4 and dT+3 in DNA were shown in stick mode. The dashed lines represented H‐bonds. (f) The binding affinities of A3A4M and its variants to TC DNA were measured by fluorescent polarization assay
Structural analysis on A3ANS‐DNANS complex showed that the backbone phosphate ions of base dT−4 at 5′‐ end and of base dT+3 at 3′‐ end formed hydrogen‐bonds with the side‐chains of the residues Asn61, His182 and Arg189 (Figure 5b–e), respectively. This observation explained why the chemical shifts of backbone atoms of residues His182 and Arg189 did not change (Figure 2b) upon DNA nonspecifically interacting with A3A4M. At the same time, the side‐chain of Asp131 did not have a salt‐bridge with the side‐chain of residue Arg189, different from the observation in structure of A3AS‐DNAS complex (Figure S7a,b). To confirm whether these three H‐bonds stabilized the conformation of A3ANS‐DNANS complex, we replaced them into alanine, and measured the binding affinities of these A3A4M variants to TC DNA (Figure 5f). Their binding affinities were decreased by 2–4.5 folds, compared to A3A4M. Thus, A3ANS‐DNANS structure explored three key sites useful for A3A interaction with DNA.
3. CONCLUSIONS
In this report, we determined two solution structures of A3A4M in complexes with TC DNA in specific and nonspecific binding ways, respectively. The nonspecific binding mode of A3A4M to ssDNA is not unique. Previously, the catalytic domain of APOBEC3B (i.e., A3B‐CTD) was suggested to specifically identify target base dC0 by sliding along ssDNA (i.e., nonspecific interaction with ssDNA). 37 A3B slides only for a relatively short distance and tends to dissociate from the ssDNA before reaching the target sequence. The full‐length A3G (i.e., FL‐A3G, containing inactive A3G‐NTD and active A3G‐CTD domains) has higher deaminase activity than A3G‐CTD. FL‐A3G interacts with ssDNA in specific and nonspecific modes, in which A3G displayed elevated affinity for specific sequence ssDNA than for nonspecific sequence ssDNA. 38 A3G‐NTD interacts with ssDNA in a nonspecific mode, while A3G‐CTD interacts with ssDNA in a specific mode. Both domains of A3G contribute to the sequence specific binding of ssDNA. To identify target motif 5′‐CC‐3′, A3G first binds to ssDNA in a sequence nonspecific manner and slides along the ssDNA without directional preference. 39 Similar to A3B and A3G, before the target base dC0 in TC motif was specifically identified, A3A must slide along ssDNA through nonspecific interaction with ssDNA. Thus, the nonspecific interaction between A3A and ssDNA plays a role in guiding substrate DNA to be specifically recognized by the active center of A3A. This not only revealed that A3s had multiple nonspecific recognition manners for substrate ssDNA, but also provide implications of how A3s deaminate ssDNA in solution.
4. MATERIALS AND METHODS
4.1. Supplemental methods and materials
4.1.1. Expression and purification of A3A4M
The gene of A3A4M or its variants was cloned into the region between BamHI and XhoI cleavage sites of a recombined pGEX‐6p‐1 plasmid which contained an His6‐tag adjacent to the N‐terminal GST tag, and HRV 3c cleavage site. A3A4M and its variants were expressed in BL‐21(DE3) Escherichia coli cells. Cell cultures were grown to OD600 value equal to about 0.8 and induced with a final concentration of 0.4 mM IPTG for 20 hr at 16°C. Cells were re‐suspended in Ni2+‐binding buffer A (50 mM Tris–HCl pH 7.5, 300 mM NaCl, 50 μM ZnCl2 and 10 mM β‐mercaptoethanol [β‐ME]) with protease inhibitor (PMSF) and lysed at 15 kpsi using a hydraulic cell disruption system (Constant System JINBO Benchtop) (Guangzhou Juneng Biology and Technology Co., Ltd., Guangzhou, China). The lysate was centrifuged at 12,000 rpm and 4°C for 55 min to remove cellular debris prior to loading into a Ni‐NTA resin (GE Health). The column was washed with 10 column volumes buffer A (50 mM Tris–HCl pH 7.5, 300 mM NaCl, 50 μM ZnCl2 and 10 mM β‐ME) followed by 10 volumes buffer B (50 mM Tris–HCl pH 7.5, 300 mM NaCl, 20 mM imidazole, 50 μM ZnCl2 and 10 mM β‐ME). The bound protein was eluted with 10 column volumes of buffer C (50 mM Tris–HCl pH 7.5, 300 mM NaCl, 20 mM imidazole, 50 μM ZnCl2 and 10 mM β‐ME). The elute was then dialyzed with buffer D (25 mM Na2HPO4 pH 7.3, 200 mM NaCl, 50 μM ZnCl2 and 5 mM DTT). Fractions containing A3A4M were then concentrated and purified on a Superdex75 16/300 GL column (GE Health) previously equilibrated with buffer D (25 mM Na2HPO4 pH 7.3, 200 mM NaCl, 50 μM ZnCl2 and 5 mM DTT).
4.2. DNA synthesis and purification
All DNA strands with or without FAM label were commercially synthesized from Shanghai Biosune Biotech Co., Ltd., China at a PAGE grade. The molecular weight of all strands was confirmed by running PAGE gel and MALDI TOF mass spectroscopy.
4.3. Fluorescence polarization
To determine the binding affinities of A3A4M and its variants to different DNA strands, or 5′‐fluoresceinated DNA strands were commercially synthesized at a HPLC grade (Shanghai Sangon Biotech Co., Ltd., China) with the corresponding sequences. The fluorescence polarization (FP) was performed, where all proteins were diluted in buffer D (25 mM Na2HPO4 pH 7.3, 200 mM NaCl, 50 μM ZnCl2 and 5 mM DTT) for FP and incubated with a 10 nM 5′‐ end 6‐FAM‐labeled ssDNA at room temperature in a total reaction volume of 200 μL. FP assay was measured at 25°C using SpectraMax i3x Platform (Molecular Devices, Inc.) with 490 nm excitation and 535 nm emission wavelengths, respectively. The dissociation constants (K D) were determined by a nonlinear least‐squares analysis using the program Prism 5 (GraphPad, Inc.). Data shown are averaged values of three repeated measurements.
4.4. NMR data collection and spectra analysis
To correctly assign NMR signals of DNA used in the structural determination of A3A‐DNA complex, we first performed NMR experiments on DNA samples of TC DNA, CC DNA, TU DNA and TT DNA. All DNA samples were about 1 mM dissolved in buffer containing 25 mM Na2HPO4 pH 7.3, 200 mM NaCl, 50 μM ZnCl2, 5 mM DTT, 100% D2O. All NMR samples were placed in 5‐mm Shigemi NMR tubes. All NMR experiments were performed on a Varian Unity Inova 600 NMR spectrometer (with cryo‐probe) equipped with triple resonances and pulsed field gradients. Two‐dimensional (2D) 1H‐1H NOESY (with a mixing time of 250 ms), TOCSY (with a mixing time of 80 ms), and DQF‐COSY spectra were acquired at 20°C using a spectral width of 6,100 Hz in both dimensions. The acquisition data points were set to 2048 × 512 (complex points). The watergate sequence was used for water suppression. During data processing, the 45° or 60° shifted sine‐squared functions were applied to NOESY and TOCSY spectra. The fifth‐order polynomial functions were employed for the baseline corrections. The final spectral sizes are 2,048 × 1,024. The 31P NMR spectra were collected at about 1.5 mM concentration in D2O (25 mM Na2HPO4 pH 7.3, 200 mM NaCl, 50 μM ZnCl2 and 5 mM dithiothreitol‐d10 [d10‐DTT]) at 20°C at 600 MHz Varian spectrometer including the one dimensional proton‐decoupled phosphorus spectrum, and 2D heteronuclear 31P‐1H Correlation Spectroscopy (31P‐1H HETCOR). Assignments of the individual 31P resonance were accomplished by a combination of 2D 1H‐1H NOESY, COSY, TOCSY and 31P‐1H HETCOR spectra.
To determine NMR structure of A3A4M in complex with TC DNA, 0.25 mM uniformly 15N‐/13C double isotope labeled or 15N‐/13C‐/70% 2H triple labeled A3A4M plus 0.5 mM unlabeled TC DNA was prepared in NMR buffer (25 mM Na2HPO4 pH 7.3, 200 mM NaCl, 50 μM ZnCl2 and 5 mM d10‐DTT) containing 10% or 100% D2O. All NMR experiments were performed at 20°C on a Varian Unity Inova or Agilent 600 NMR spectrometer (with cryo‐probe) equipped with triple resonances and pulsed field gradients, or on Bruker Avance or Agilent III‐800MHz, 850MHz, 900MHz NMR spectrometers (with cryo‐probe) equipped with four channels and z‐axis pulsed‐field gradient.
The standard suite of experiments were acquired for assigning the 1H, 13C and 15N backbone and side‐chain chemical shifts of 13C and 15N double labeled A3A4M in complex with unlabeled DNA, and for the collection of NOE‐based distance restraints were measured, 40 , 41 including the 2D 13C‐edited HSQC in both aliphatic and aromatic regions, and 15N‐edited HSQC; the three‐dimensional (3D) HNCA, HNCO, HN(CO)CA, HNCACB, CBCA(CO)NH, 15N‐resolved HSQC‐TOCSY, HCCH‐TOCSY in both aliphatic and aromatic regions, 15N‐resolved HSQC‐NOESY, 13C‐resolved HSQC‐NOESY for both aliphatic and aromatic resonances, 2D (Hβ)Cβ(CγCδ)Hδ and (Hβ)Cβ(CγCδCε)Hε spectra for correlation of Cβ and Hδ or Hε in aromatic rings used in aromatic protons assignment. 42 To obtain NMR signals of bound TC DNA, 2D 13C/15N filtered 1H‐1H TOCSY, NOESY spectra and 2D 31P‐1H HETCOR spectra were collected. The intermolecular NOEs between 13C‐/15N‐labeled A3A4M and unlabeled TC DNA were obtained by analyzing 3D 13C‐F1 edited, 13C/15N‐F3 filtered NOESY spectra.
All NMR spectra were processed with the program NMRPipe 43 and analyzed with Sparky 3 software. 44 The 1H chemical shifts were referenced to 2, 2‐dimethylsilapentane‐5‐sulfonic acid (DSS), the 13C‐ and 15N‐resonances were indirectly referenced to DSS, and the 31P chemical shifts were referenced to an external standard of 85% H3PO4.
4.5. Analysis of ultracentrifugation experiments
Sedimentation velocity experiments were performed on a Beckman XL‐I analytical ultracentrifuge equipped with an eight‐cell rotor under 42,000 rpm at 20°C. The partial specific volume of different samples and the buffer density were calculated using the program SEDNTERP (http://www.rasmb.bbri.org/). The final sedimentation velocity data were analyzed and fitted to a continuous sedimentation coefficient distribution model using the program SEDFIT. The fitting results were further output to the Origin 9.0 software and aligned with each other.
4.6. NMR structure determination
The structural calculations of the A3AS‐DNAS and A3ANS‐DNANS complexes were carried out using a standard simulated annealing protocol implemented in the program XPLOR‐2.37 (NIH version). 45 3,409 and 3,346 distance constraints derived from NOE intensities were classified into 1.8–2.9 Å, 1.8–3.5 Å, 1.8–5.0 Å groups, corresponding to strong, medium and weak NOEs, respectively, while intermolecular distance restrains were sorted into weak (1.8–5.0 Å) and very weak (1.8–6.0 Å) groups. 184 phi and 185 psi dihedral angles were derived from the chemical shifts of the backbone atoms (HN, HA, CO, CA) by the program TALOS. 43 , 46 One hundred and seventy‐four hydrogen‐bond restraints for secondary structures were generated by analyzing secondary structural regions in the reported free NMR and X‐ray apo‐A3A structures 47 , 48 , 49 , 50 for the final structure calculation. Constraints between the protein ligands and the zinc ion were added using the procedure of Neuhaus et al as described previously. 35 , 36 A total of 10 iterations (50 structures in the initial 10 iterations) were performed. One hundred structures were computed in the last five iterations, 20 conformers with the lowest energy are used to represent the 3D structures. In the ensemble of the simulated annealing 20 structures, there was no distance constraint violation more than 0.3 Å and no torsion angle violation more than 5°. The final 20 structures of the complex A3A with different DNA with lowest energy were evaluated with the program PROCHECK‐NMR and PROCHECK. 51 All figures related to structures were generated using the program PyMOL.
AUTHOR CONTRIBUTIONS
Yaping Liu: Data curation (equal); formal analysis (equal); investigation (supporting); validation (equal). Wenxian Lan: Methodology (equal); software (equal); validation (equal). Chunxi Wang: Data curation (equal); software (equal); supervision (equal); validation (equal). Chunyang Cao: Conceptualization (equal); funding acquisition (lead); project administration (lead); supervision (lead).
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
Supporting information
Appendix S1: Supporting information
ACKNOWLEDGMENTS
This work was supported by National Program on Key Basic Research Project of China (2017YFE0108200 and 2016YFA0502302), by National Science Foundation of China (NSFC) under No. 91753119, 21977110, 21778065 and 21807105, by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB 20000000) and by Center for Excellence in Molecular Synthesis, CAS (FZHCZY020600). The authors sincerely thank facility team members in National Center of Protein Sciences Shanghai (NCPSS), in State Key Laboratory of Magnetic Resonance and Atomic Molecular Physics, Wuhan Institute of Physics and Mathematics, Chinese Academy of Sciences (CAS), the NMR center of Shanghai Institute of Materia Medica, CAS, in High Magnetic Field Laboratory (HMFL), CAS, for their great help with NMR spectra data acquirement. The atom coordinates of NMR structures were deposited into protein data bank (RCSB) with access codes 7D3X (A3AS‐DNAS) and 7D3W (A3ANS‐DNANS), the NMR chemical shifts of TC DNA strand and specifically and nonspecifically DNA‐bound A3A4M were deposited into BMRB with access numbers 36389 (A3AS‐DNAS) and 36388 (A3ANS‐DNANS).
Liu Y, Lan W, Wang C, Cao C. Two different kinds of interaction modes of deaminase APOBEC3A with single‐stranded DNA in solution detected by nuclear magnetic resonance. Protein Science. 2022;31:443–453. 10.1002/pro.4242
Yaping Liu and Wenxian Lan contributed equally to this study.
Funding information Center for Excellence in Molecular Synthesis, CAS, Grant/Award Number: FZHCZY020600; National Natural Science Foundation of China, Grant/Award Numbers: 21778065, 21807105, 21977110, 22174155, 22177127; National Program on Key Basic Research Project of China, Grant/Award Numbers: 2016YFA0502302, 2017YFE0108200; Strategic Priority Research Program of the Chinese Academy of Sciences, Grant/Award Number: XDB 20000000
REFERENCES
- 1. Yu Q, Konig R, Pillai S, et al. Single‐strand specificity of APOBEC3G accounts for minus‐strand deamination of the HIV genome. Nat Struct Mol Biol. 2004;11:435–442. [DOI] [PubMed] [Google Scholar]
- 2. Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003;424:99–103. [DOI] [PubMed] [Google Scholar]
- 3. Zhang H, Yang B, Pomerantz RJ, Zhang C, Arunachalam SC, Gao L. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV‐1 DNA. Nature. 2003;424:94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lada AG, Dhar A, Boissy RJ, et al. AID/APOBEC cytosine deaminase induces genome‐wide kataegis. Biol Direct. 2012;7:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chelico L, Sacho EJ, Erie DA, Goodman MF. A model for oligomeric regulation of APOBEC3G cytosine deaminase‐dependent restriction of HIV. J Biol Chem. 2008;283:13780–13791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chelico L, Pham P, Calabrese P, Goodman MF. APOBEC3G DNA deaminase acts processively 3′–>5′ on single‐stranded DNA. Nat Struct Mol Biol. 2006;13:392–399. [DOI] [PubMed] [Google Scholar]
- 7. Rausch JW, Chelico L, Goodman MF, Le Grice SF. Dissecting APOBEC3G substrate specificity by nucleoside analog interference. J Biol Chem. 2009;284:7047–7058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang H, Ito F, Wolfe AD, et al. Understanding the structural basis of HIV‐1 restriction by the full length double‐domain APOBEC3G. Nat Commun. 2020;11:632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV‐1 infection and is suppressed by the viral Vif protein. Nature. 2002;418:646–650. [DOI] [PubMed] [Google Scholar]
- 10. Olson ME, Harris RS, Harki DA. APOBEC enzymes as targets for virus and cancer therapy. Cell Chem Biol. 2018;25:36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Venkatesan S, Rosenthal R, Kanu N, et al. Perspective: APOBEC mutagenesis in drug resistance and immune escape in HIV and cancer evolution. Ann Oncol. 2018;29:563–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ran X, Ao Z, Yao X. Apobec3G‐based strategies to defeat HIV infection. Curr HIV Res. 2016;14:217–224. [DOI] [PubMed] [Google Scholar]
- 13. Goila‐Gaur R, Strebel K. HIV‐1 Vif, APOBEC, and intrinsic immunity. Retrovirology. 2008;5:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang WY, Du J, Evans SL, Yu YK, Yu XF. T‐cell differentiation factor CBF‐beta regulates HIV‐1 Vif‐mediated evasion of host restriction. Nature. 2012;481:376–379. [DOI] [PubMed] [Google Scholar]
- 15. Jäger S, Kim DY, Hultquist JF, et al. Vif hijacks CBF‐beta to degrade APOBEC3G and promote HIV‐1 infection. Nature. 2012;481:371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marin M, Rose KM, Kozak SL, Kabat D. HIV‐1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat Med. 2003;9:1398–1403. [DOI] [PubMed] [Google Scholar]
- 17. Sheehy AM, Gaddis NC, Malim MH. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV‐1 Vif. Nat Med. 2003;9:1404–1407. [DOI] [PubMed] [Google Scholar]
- 18. Yu X, Yu Y, Liu B, et al. Induction of APOBEC3G ubiquitination and degradation by an HIV‐1 Vif‐Cul5‐SCF complex. Science. 2003;302:1056–1060. [DOI] [PubMed] [Google Scholar]
- 19. Lecossier D, Bouchonnet F, Clavel F, Hance AJ. Hypermutation of HIV‐1 DNA in the absence of the Vif protein. Science. 2003;300:1112. [DOI] [PubMed] [Google Scholar]
- 20. Berger G, Durand S, Fargier G, et al. APOBEC3A is a specific inhibitor of the early phases of HIV‐1 infection in myeloid cells. PLoS Path. 2011;7:e1002221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Taura M, Song E, Ho YC, Iwasaki A. Apobec3A maintains HIV‐1 latency through recruitment of epigenetic silencing machinery to the long terminal repeat. Proc Natl Acad Sci U S A. 2019;116:2282–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Warren CJ, Xu T, Guo K, et al. APOBEC3A functions as a restriction factor of human papillomavirus. J Virol. 2015;89:688–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nakaya Y, Stavrou S, Blouch K, Tattersall P, Ross SR. In vivo examination of mouse APOBEC3‐ and human APOBEC3A‐ and APOBEC3G‐mediated restriction of parvovirus and herpesvirus infection in mouse models. J Virol. 2016;90:8005–8012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Suspene R, Aynaud MM, Koch S, et al. Genetic editing of herpes simplex virus 1 and Epstein‐Barr herpesvirus genomes by human APOBEC3 cytidine deaminases in culture and in vivo. J Virol. 2011;85:7594–7602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jin JY, Yan XY, Liu YP, Lan WX, Wang CX, Xu B, & Cao, CY . Recent advances in the structural studies on cytosine deaminase APOBEC3 family members and their nucleic acid complexes. Acta Chim Sinica. 2019;77:1089–1098. [Google Scholar]
- 26. Salter JD, Smith HC. Modeling the embrace of a mutator: APOBEC selection of nucleic acid ligands. Trends Biochem Sci. 2018;43:606–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fang Y, Xiao X, Li SX, Wolfe A, Chen XS. Molecular interactions of a DNA modifying enzyme APOBEC3F catalytic domain with a single‐stranded DNA. J Mol Biol. 2018;430:87–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cheng C, Zhang TL, Wang CX, Lan WX, Ding JP, Cao CY. Crystal structure of cytidine deaminase human APOBEC3F chimeric catalytic domain in complex with DNA. Chinese J Chem. 2018;36:1241–1248. [Google Scholar]
- 29. Xiao X, Li SX, Yang H, Chen XS. Crystal structures of APOBEC3G N‐domain alone and its complex with DNA. Nat Commun. 2016;7:12193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yan XX, Lan W, Wang CX, Cao C. Structural investigations on the interactions between cytidine deaminase human APOBEC3G and DNA. Chem Asian J. 2019;14:2235–2241. [DOI] [PubMed] [Google Scholar]
- 31. Maiti A, Myint W, Kanai T, et al. Crystal structure of the catalytic domain of HIV‐1 restriction factor APOBEC3G in complex with ssDNA. Nat Commun. 2018;9:2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kouno T, Silvas TV, Hilbert BJ, et al. Crystal structure of APOBEC3A bound to single‐stranded DNA reveals structural basis for cytidine deamination and specificity. Nat Commun. 2017;8:15024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shi K, Carpenter MA, Banerjee S, et al. Structural basis for targeted DNA cytosine deamination and mutagenesis by APOBEC3A and APOBEC3B. Nat Struct Mol Biol. 2017;24:131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Byeon IJ, Ahn J, Mitra M, et al. NMR structure of human restriction factor APOBEC3A reveals substrate binding and enzyme specificity. Nat Commun. 2013;4:1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Neuhaus D, Nakaseko Y, Schwabe JW, Klug A. Solution structures of two zinc‐finger domains from SWI5 obtained using two‐dimensional 1H nuclear magnetic resonance spectroscopy. A zinc‐finger structure with a third strand of beta‐sheet. J Mol Biol. 1992;228:637–651. [DOI] [PubMed] [Google Scholar]
- 36. Kwon K, Cao C, Stivers JT. A novel zinc snap motif conveys structural stability to 3‐methyladenine DNA glycosylase I. J Biol Chem. 2003;278:19442–19446. [DOI] [PubMed] [Google Scholar]
- 37. Wan L, Nagata T, Morishita R, Takaori‐Kondo A, Katahira M. Observation by real‐time NMR and interpretation of length‐ and location‐dependent deamination activity of APOBEC3B. ACS Chem Biol. 2017;12:2704–2708. [DOI] [PubMed] [Google Scholar]
- 38. Shlyakhtenko LS, Dutta S, Banga J, Li M, Harris RS, Lyubchenko YL. APOBEC3G interacts with ssDNA by two modes: AFM studies. Sci Rep. 2015;5:15648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kamba K, Nagata T, Katahira M. Catalytic analysis of APOBEC3G involving real‐time NMR spectroscopy reveals nucleic acid determinants for deamination. PloS One. 2015;10:e0124142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bax A, Grzesiek S. Methodological sdvances in protein NMR. Acc Chem Res. 1993;26:131–138. [Google Scholar]
- 41. Clore GM, Gronenborn AM. Determining the structures of large proteins and protein complexes by NMR. Trends Biotechnol. 1998;16:22–34. [DOI] [PubMed] [Google Scholar]
- 42. Yamazaki T, Forman JD, Kay LE. 2‐Dimensional NMR experiments for correlating C‐13‐Beta and H‐1‐Delta/Epsilon chemical‐shifts of aromatic residues in C‐13‐labeled proteins via scalar couplings. J Am Chem Soc. 1993;115:11054–11055. [Google Scholar]
- 43. Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. [DOI] [PubMed] [Google Scholar]
- 44. Goddard TD, Kneller DG. SPARKY 3. San Francisco, CA: University of California, 2001. [Google Scholar]
- 45. Kuszewski J, Clore GM. Sources of and solutions to problems in the refinement of protein NMR structures against torsion angle potentials of mean force. J Magn Reson. 2000;146:249–254. [DOI] [PubMed] [Google Scholar]
- 46. Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. [DOI] [PubMed] [Google Scholar]
- 47. Chen KM, Harjes E, Gross PJ, et al. Structure of the DNA deaminase domain of the HIV‐1 restriction factor APOBEC3G. Nature. 2008;452:116–119. [DOI] [PubMed] [Google Scholar]
- 48. Furukawa A, Nagata T, Matsugami A, et al. Structure, interaction and real‐time monitoring of the enzymatic reaction of wild‐type APOBEC3G. EMBO J. 2009;28:440–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Holden LG, Prochnow C, Chang YP, et al. Crystal structure of the anti‐viral APOBEC3G catalytic domain and functional implications. Nature. 2008;456:121–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lu X, Zhang T, Xu Z, et al. Crystal structure of DNA cytidine deaminase ABOBEC3G catalytic deamination domain suggests a binding mode of full‐length enzyme to single‐stranded DNA. J Biol Chem. 2015;290:4010–4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK‐NMR: Programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting information