

Abstract
To expand protein's covalent bonding ability, latent bioreactive unnatural amino acids have been designed and genetically encoded into proteins, which react with specific natural amino acid residues through proximity‐enabled bioreactivity. The resultant new covalent bonds can be selectively created within and between proteins in vitro, in cells, and in vivo. Offering diverse properties previously unattainable, these covalent linkages have been harnessed to enhance protein properties, to modulate protein function, to probe ligand–receptor binding, to identify elusive protein interactions, and to develop covalent protein drugs. Selective introduction of covalent bonds into proteins is affording novel avenues for biological studies, synthetic biology, and biotherapeutics.
Keywords: covalent drug, genetic code expansion, latent bioreactive unnatural amino acid, protein therapeutics, protein–protein interaction, proximity‐enabled bioreactivity
1. INTRODUCTION
Amino acid side chains of proteins mainly interact with each other and with other biomolecules in noncovalent mode, such as hydrogen bonds, ionic bonds, Van der Waals interaction, and hydrophobic interaction. The disulfide bond is the only covalent bond that forms spontaneously between two cysteine residues. 1 Other covalent bonds, such as the isopeptide bond between Lys and Asn or Asp and the N—O—S bridge between Lys and Cys, have been discovered in some proteins, 2 , 3 , 4 but their formation requires special protein microenvironment, and cannot be generally introduced into different sites of various proteins. Compared with noncovalent bonding, covalent bonding offers stronger interaction and higher selectivity. Indeed, the disulfide bond plays a unique role in the folding, stability, structure, and function of a variety of proteins including antibodies, cytokines, and receptors. However, the disulfide bond is a relatively weak covalent bond. Its reversibility and redox sensitivity, being characteristic, also set limits on protein function, engineering, and applications.
To break this natural barrier, we began to explore whether new covalent bonding ability could be generated for proteins over 10 years ago. 5 Apparently, there has not been evolutionary pressure for proteins to have additional covalent side chain linkages than the disulfide, but new covalent bonding would expand avenues for generating novel protein structure, property, and function. It is possible to introduce two bioorthogonal functional groups into proteins and use them to create covalent bonds. We did not pursue that route because in many applications the target protein is not amenable for chemical or genetic modification, such as a disease‐related target protein in human in vivo. Our approach focuses on introducing a single functional group into protein and using it to selectively target natural amino acid residues. In this article, we briefly review how new covalent bonding ability has been made possible for proteins through this general strategy, and showcase how the new covalent linkages provide innovative ways for protein research, biological studies, and therapeutic applications.
2. PROXIMITY‐ENABLED BIOREACTIVITY AND LATENT BIOREACTIVE UNNATURAL AMINO ACID
Our approach is to genetically introduce into proteins an unnatural amino acid (Uaa), whose side chain bears a functional group to react with the side chain of the target natural residue, so as to build a covalent linkage spontaneously. 5 , 6 Suitable chemical reaction needs to be adequately active to target the amino acid side chains, yet must proceed in mild conditions to be compatible for use in live cells. As natural amino acids and residues are ubiquitous in proteins and in cells, it had been a huge challenge to achieve reaction specificity for a particular residue of a desired protein in the sea of the proteome. In addition, off‐target reactions of a bioreactive Uaa with other proteins or biomolecules could result in cytotoxicity, and side reactions with the translational machinery could stall translation, preventing Uaa incorporation via ribosome.
We hypothesized that the contradiction between bioreactivity, selectivity, and genetic encoding could be overcome via proximity‐enabled bioreactivity (Figure 1). 5 In essence, the reactivity of the Uaa must be fine‐tuned and selectively triggered. On the one hand, the Uaa reactivity should be tuned down so that it does not react with free amino acids, protein residues, and other biomolecules inside cells under physiological conditions, thus permitting genetic incorporation; on the other hand, after the Uaa is incorporated into protein and placed in proximity to its target natural residue, the proximity will reduce entropy of activation and increase the effective concentration of reactants, leading to large increase in reaction rate to facilitate the reaction and form the covalent bond specifically. Therefore, we call this type of Uaa latent bioreactive Uaa to indicate its reactivity toward a biomolecule (in contrast to bioorthogonal) and its hidden property until triggered by proximity.
FIGURE 1.
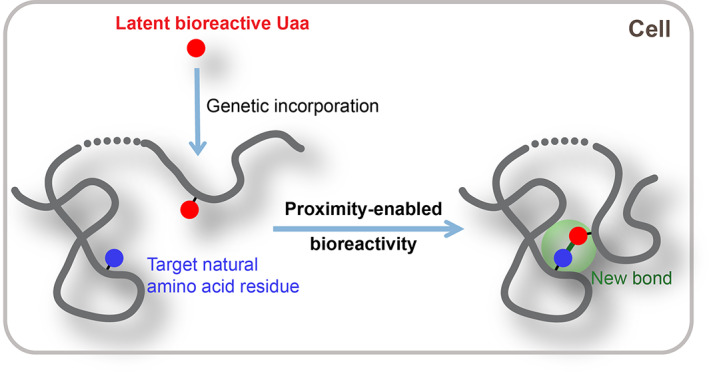
Building new covalent bonds for proteins via proximity‐enabled bioreactivity. Proximity of the latent bioreactive unnatural amino acid (Uaa) to its target natural amino acid residue can be brought by intramolecular protein folding or conformational change, or by intermolecular protein interactions, accelerating the reaction between them to form a covalent bond specifically
The first success was achieved by developing Uaa p‐2′‐fluoroacetyl‐phenylalanine (Ffact) to target Cys selectively (Figure 2a). 5 The sulfhydryl group of Cys has the highest nucleophilicity among natural amino acid side chains, so a weak electrophilic group was expected to selectively react with Cys only when in close proximity. After testing various weak electrophiles, Ffact was identified to react with Cys only when Cys concentration was elevated (10 mM) much higher than intracellular amino acid concentrations (<1 mM). After Ffact was incorporated into proteins using an orthogonal tRNA/synthetase pair via genetic code expansion, 7 , 8 mass spectrometric (MS) analysis of the protein confirmed that there was no adducts of Cys, glutathione, imidazole, or other modifications of Ffact, indicating that Ffact was stable and unmodified during protein biosynthesis and purification. Next, Ffact and Cys were introduced into an affibody and the Z protein, respectively, at their binding interface. Incubation of the purified affibody and Z proteins at mild conditions results in covalent complex formation (63% yield). In addition, when Ffact was replaced with the isosteric Fact, which only lacks the fluorine, or Cys was moved to a different position, no covalent complex could be detected, indicating the reaction is proximity‐dependent and specific. Ffact has also been shown to form a covalent bond with Cys within different fluorescent proteins (FPs) in Escherichia coli cells, and can be incorporated in GPCR in mammalian cells to covalently capture the peptide ligand of the receptor. 9 Therefore, release the latent reactivity of the Uaa via proximity inventively enables the selective formation of a covalent bond between the latent bioreactive Uaa and its target natural residue, offering covalent bonding ability for proteins.
FIGURE 2.
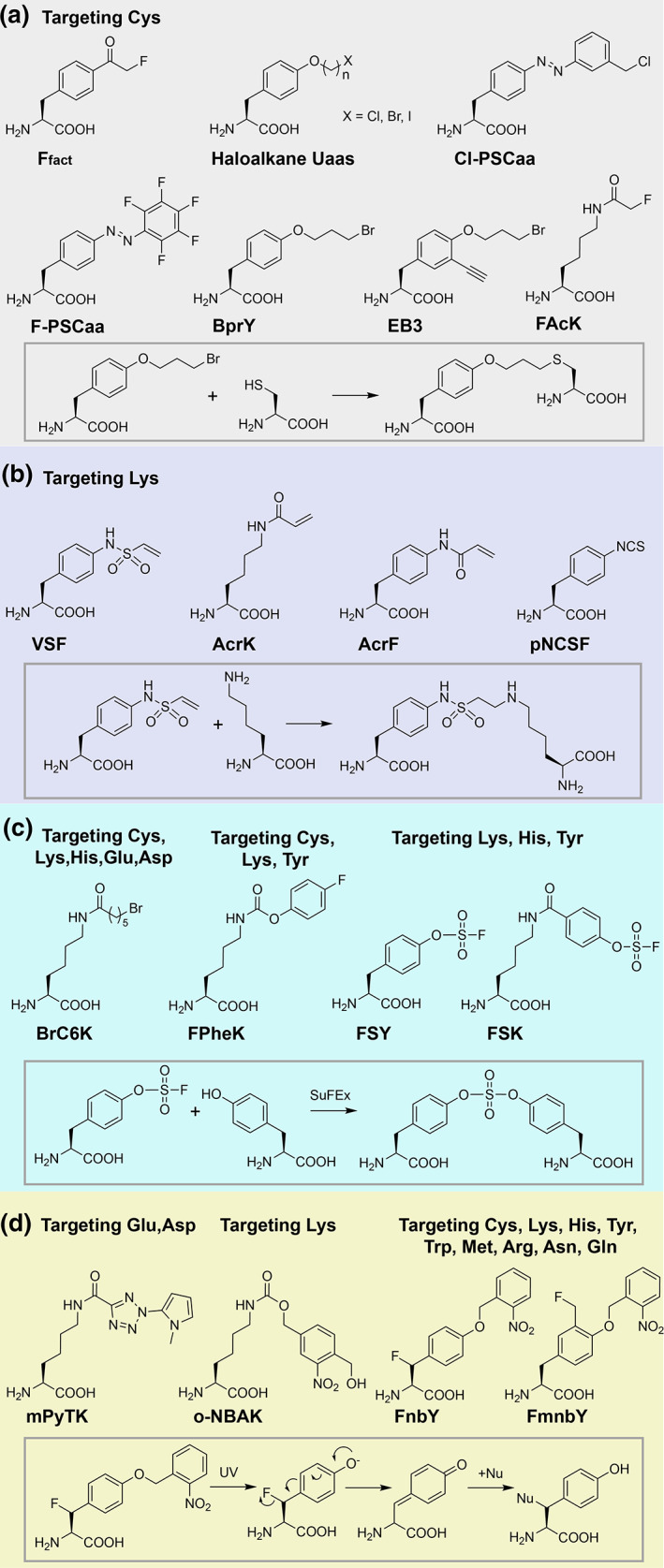
Latent bioreactive unnatural amino acids (Uaas) that have been genetically encoded into proteins to covalently target different natural amino acid residues. A representative reaction is shown in the box for each category. (a) Latent bioreactive Uaas targeting Cys. (b) Latent bioreactive Uaas targeting Lys. (c) Latent bioreactive Uaas capable of targeting multiple residues. (d) Photoactivatable bioreactive Uaas reacting with specific residues in proximity
3. COVALENTLY TARGETING VARIOUS AMINO ACID RESIDUES
3.1. Targeting different amino acid residues
Following the success of Ffact, a range of latent bioreactive Uaas containing other functional groups have been subsequently demonstrated suitable for genetic encoding and targeting Cys via proximity‐enabled bioreactivity. A series of haloalkane Uaas were synthesized and incorporated, which contain different halogen atoms on aliphatic chains of varying length (Figure 2a). 10 Through an evolved Methanosarcina mazei tRNAPyl/XYRS pair, they can be incorporated in proteins in E. coli and mammalian cells and react with a proximal Cys intramolecularly and intermolecularly. Different side chain lengths provide certain flexibility for Cys position. Meanwhile, a benzyl chloride containing Uaa, Cl‐PhotoSwitchable Click amino acids (PSCaa), was also developed and incorporated in E. coli and mammalian cells. 11 It reacts efficiently with Cys in proximity to form a photo‐switchable bridge that isomerizes in response to UV light. Besides these nucleophilic substitution reactions, the F‐PSCaa has later been incorporated and react with nearby Cys via nucleophilic aromatic substitution (SNAr), building an azo bridge on proteins in situ. 12 This bridge can be reversibly switched by visible light.
Cys is not always available at desired positions or in target proteins. To expand the diversity of proteins applicable by proximity‐enabled bioreactivity, we installed alkyl bromide on a long linear alkyl side chain to increase its orientation flexibility. 13 The resultant Uaa BrC6K (Figure 2c), after incorporation into proteins, was able to react with proximal His and Lys in addition to Cys, enabling cross‐linking of an affibody to HER2 receptor on cancer cells. Furman et al. designed and incorporated Uaa VSF (Figure 2b), 14 which contains vinyl sulfonamide and reacts with nearby Lys in high efficiency (86%) in vitro, cross‐linking Herceptin Fab to the HER2 receptor on cells. They also showed that acrylamide containing Uaas, AcrK and AcrF, could react with Lys but with modest yields under basic conditions. In another work, Xuan et al. developed Uaa pNCSF, which uses aryl isothiocyanate to react with proximal Lys within and between proteins. 15 These results demonstrate that the concept of proximity‐enabled bioreactivity can be generally used to target different natural amino acid residues.
3.2. Multitargeting amino acid residues
Lys‐targeting bioreactive Uaas can often react with Cys as well, such as the aforementioned BrC6K and VSF. The multitargeting ability of a Uaa would increase the suitable sites to build covalent bonds in proteins without sacrificing selectivity, since selectivity is based on proximity. In addition to targeting Cys, His, and Lys, Cigler et al. found that BrC6K also reacted with Glu and Asp. 16 Another multitargeting Uaa is FPheK developed by Xuan et al. and containing aryl carbamate (Figure 2c). 17 Incorporation of FPheK allows covalent cross‐linking with nearby Cys, Lys, and Tyr residues. 18
We focused on aryl fluorosulfate due to its exceptional biocompatibility and click nature of sulfur‐fluoride exchange (SuFEx) reactivity when installed on small molecules. 19 , 20 We first designed and genetically encoded fluorosulfate‐l‐tyrosine (FSY) into proteins in E. coli and mammalian cells (Figure 2c). 21 FSY was nontoxic to cells, and the incorporated FSY was able to react with proximal Lys, His, and Tyr in proteins via SuFEx. The reaction occurs in high efficiency both in vitro and in live cells, affording stable linkages resistant to hydrolysis. Interestingly, FSY also reacts with nearby Cys, Ser, and Thr, but the resultant linkages are unstable. Ser is converted to dehydroalanine (Dha) and Thr to dehydrobutyrine (Dhb), which can be used for protein labeling or conjugation. 22 We next designed and genetically encoded fluorosulfonyloxybenzoyl‐l‐lysine (FSK) in E. coli and mammalian cells. 23 FSK has similar reactive specificity as FSY, but it contains a longer and more flexible side chain than FSY, enabling reaction with sites unreachable by FSY. The target residues of FSY and FSK, that is, Lys, His, and Tyr, are often abundant at or near the binding interface of proteins, dramatically expanding the applicability of proximity‐enabled bioreactivity. To date, they are our latent bioreactive Uaa of choice for various applications.
3.3. Photo‐controlled targeting of amino acid residues
The reactivity of a Uaa can also be caged and then released by light. Conventional photo‐cross‐linking Uaas employ azide, benzophenone, or diazirine that are activated by UV light. However, these photo‐cross‐linking Uaas have no reaction specificity for amino acids and thus are unsuitable for precise introduction of covalent bonds into proteins. Photoactivatable bioreactive Uaas have emerged recently, whose reactivity is trigger by light (rather than by proximity) and is chemically specific for certain amino acid residues. Importantly, the reactivity is also limited to residues in proximity. Therefore, we discuss them here since they can be used to generate specific covalent linkages for proteins as well, with the bonus of spatiotemporal resolution afforded by light responsiveness.
Tian et al. reported the incorporation of mPyTK containing a methylpyrroletetrazole (Figure 2d), which cross‐linked GST into dimer and the EGFR‐Grb2 complex upon UV activation. 24 The photogenerated carboxy‐nitrile imine was proposed to react with the carboxylate of a nearby Glu residue. In another work, Hu et al. incorporated o‐NBAK, an o‐nitrobenzyl alcohol derived lysine. 25 UV‐activation of o‐NBAK generates aryl‐nitroso intermediate, which selectively reacts with nearby Lys residues.
We aimed to multitarget a range of amino acid residues through the photoactivation mechanism while keeping the chemical specificity. Two Uaas, FnbY and FmnbY (Figure 2d), were designed and genetically incorporated, which contain photocaged para‐quinone methide (QM) and ortho‐QM, respectively. 26 UV light activation generates QM at the Uaa site, which is highly reactive with nucleophiles through Michael addition. Through GST dimeric cross‐linking, nine amino acid residues with nucleophilic side chains (Cys, Lys, His, Tyr, Trp, Met, Arg, Asn, and Gln) in close proximity were found to covalently linked with the photoactivated FnbY 26 and FmnbY (unpublished results). What is more, when there is no nucleophilic residue nearby, the photoactivated FnbY and FmnbY in proteins also allow rapid (<10 s) conjugation of amine or thiol reagents. 27
In contrast to the conventional photo‐cross‐linking Uaas, these photoactivatable bioreactive Uaas have amino acid specificity, which greatly simplifies the identification of cross‐linked peptides and residues (and thus the captured unknown interacting proteins) through tandem MS. They also make it possible to precisely engineer covalent bonds into proteins through optical control.
4. COVALENT BONDING WITHIN A PROTEIN
As the latent bioreactive Uaa is genetically incorporated into proteins through translation, this approach allows new covalent linkages to be selectively introduced into proteins in a similar manner as the disulfide bond. The new covalent bonds can afford properties not possessed by the disulfide bond (e.g., irreversibility and extended length), and other desired characteristics can be further embedded in the Uaa in addition to the latent reactive group. Applications of new covalent linkages within a protein are proliferating, so we focus on summarizing the initial applications associated with the development of the Uaas.
4.1. Enhancing protein properties
The reaction of Ffact with Cys produces a strong and irreversible covalent bond, which has been utilized to enhance the photostability of FPs (Figure 3a). 5 The fluorophore of FPs has one end covalently attached to the central α‐helix while the other end dangling. By replacing Tyr67 of the fluorophore with Ffact and a nearby Ser146 with Cys, a covalent bond was spontaneously formed between Ffact and Cys to covalently link the dangling end of the fluorophore to the β‐barrel of FP. This bond makes the fluorophore more rigid and increases the lifespan and photon output of two tested FPs (mPlum and mKate2) by approximately twofold, which are beneficial for single molecule imaging.
FIGURE 3.
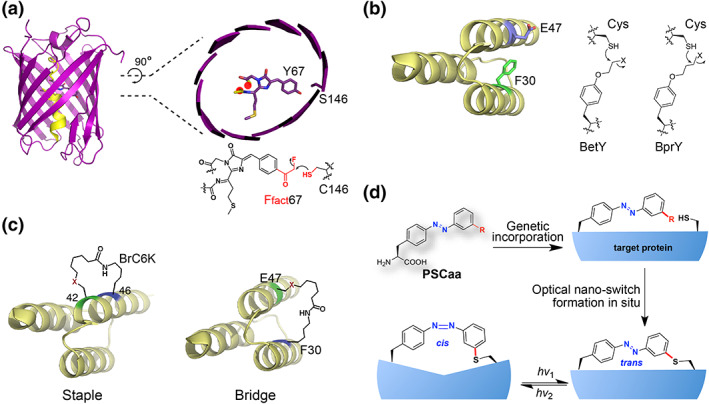
Applications of new covalent bonds built within a protein. (a) A single covalent bond formed between Ffact and Cys rigidly links the fluorophore to the β‐barrel and increases photostability of fluorescent proteins. (b) Covalent linkage formed with haloalkane unnatural amino acid (Uaa) and Cys with appropriate length increases the thermostability of the affibody. (c) Recombinantly build staples or bridges onto proteins through BrC6K reacting with Cys. (d) PhotoSwitchable Click amino acids (PSCaa) reacting with Cys to build optical nano‐switch onto proteins in situ, which allows optical modulation of protein structure and function reversibly with high resolution up to amino acid residue level
One of the important roles the disulfide bond play is to increase protein stability, yet the disulfide bond length cannot be adjusted to fit all positions. Haloalkane Uaas with different side chain lengths have been incorporated into an affibody protein to react with a Cys introduced nearby, covalently cross‐linking two sites at distances further than the disulfide bond allows (Figure 3b). 10 When a crosslink with appropriate distance was generated, the thermostability of the affibody was improved, with melting temperature increased from 46.7 to 60.4°C. Simultaneously engineering of multiple bonds coupled with screening should be able to further enhance the thermostability of proteins.
4.2. Recombinantly stapling and bridging proteins
α‐Helical peptides can be chemically stapled, which has been shown to increase target affinity, proteolytic resistance, serum half‐life, and cell penetration. 28 The latent bioreactive Uaas now allow us to recombinantly staple peptides as well as proteins in cells (Figure 3c). 13 BrC6K was incorporated into an affibody with Cys mutated at the i + 4 site. Expression of the mutant affibody directly in live cells produces affibody with its α‐helix stapled by BrC6K reacting with Cys in 100% efficiency without further treatment. In addition to stapling an α‐helix, one can also install bridges spanning different secondary structures of proteins using this strategy. Recombinant expression of staples and bridges on proteins avoids the use of chemical catalysts, and should allow for the generation of library of different staples for selection and facilitate production in large scale.
4.3. Building optical nano‐switch for molecular optobiology
In addition to forming a covalent linkage, latent bioreactive Uaa can also be designed to harbor extra desired properties, so that such properties can be introduced into proteins site specifically and stably in the covalent bridge. PSCaa are thus developed to contain an azobenzene photo‐switch in addition to a functional group reacting with Cys, offering light response to general proteins (Figure 3d). 11 , 12
The Cl‐PSCaa isomerizes by light of 365/405 nm, and the F‐PSCaa isomerizes by visible light of 405/540 nm. When the F‐PSCaa was incorporated into calmodulin (CaM) at site 76 and a Cys was placed at site 83, an optical nano‐switch was precisely built on CaM by covalently connecting the two sites. 12 Illumination with green light (540 nm) drives the nano‐switch photoisomerize from trans to cis configuration, and subsequent blue light (405 nm) reverts cis to trans yielding the photostationary state of the trans state. Successive illumination with either green or blue light allows for reversible transformation between the two states. The photoisomerization of the nano‐switch was found to drive significant conformational change of CaM reversibly, as detected by circular dichroism. Consequently, the conformational change of CaM, in turn, modulates CaM's binding activity to the CaM‐binding domain of the neuronal nitric oxide synthase.
Compared with light‐sensitive protein domains, 29 optical nano‐switches built with latent bioreactive PSCaas are much smaller in size and have higher site flexibility for installation, which should minimize interference and offer high resolution to the amino acid residue level. The nano‐switch modulates the structure, which can be readily used on proteins with unknown function. With the rapid advancement in accurate protein structure predication, 30 , 31 the nano‐switch can be a versatile optical controller to regulate various positions and secondary features of proteins for research and engineering purposes with spatiotemporal resolution. 32
5. COVALENT BONDING BETWEEN TWO PROTEINS
Covalent bonding between two interacting proteins provides a robust connection of the two proteins for basic research and diverse applications. A notable advantage of the proximity‐enabled bioreactivity of latent bioreactive Uaa is that the target protein does not need to be modified, that is, native proteins in their physiological settings can be addressed. We highlight a few examples harnessing this feature to investigate protein interactions and to develop new protein therapeutics.
5.1. Probing ligand–receptor interaction in mammalian cells
Studying the ligand–receptor interaction directly in live cells can help mitigate concerns such as those associated with artificial membrane composition, in vitro reconstitution, and system incompleteness. The biocompatibility and genetic encodability of latent bioreactive Uaas allow their use for research in live systems. Ffact has been used to probe how a class B GPCR, corticotropin releasing factor receptor type 1 (CRF1R), interacts with its peptide ligand in mammalian cells (Figure 4). 9 To determine what sites of CRF1R interact with its native peptide ligand Urocortin‐1 (Ucn1), the photo‐cross‐linking Uaa p‐azido‐phenylalanine (Azi) was first incorporated into various positions of CRF1R expressed in HEK293T cells to crosslink Ucn1 upon UV light activation. 9 , 33 Detection of the covalent complex of CRF1R‐Ucn1 in Western analysis of the cell lysate indicates that Ucn1 interacts with CRF1R at the Azi incorporation site. After the interaction sites have been mapped onto the receptor, how the peptide ligand is positioned remains unsolvable, because photo‐cross‐linking reaction of Azi is not specific to amino acid residues. The latent bioreactive Uaa Ffact was then incorporated into CRF1R and Cys positioned at different sites of Ucn1. As Ffact‐Cys reaction is both residue and distance dependent, the detection of CRF1R‐Ucn1 covalent complex resulting from the Ffact‐Cys reaction thus provides reciprocal spatial constrains of the receptor–ligand interaction. These constrains were analyzed in combination with the structures of CRF1R transmembrane and extracellular domains, yielding a complete conformational model for the peptide‐receptor complex. The model unveils the binding path of the peptide agonist in the activation domain of the class B receptor, and allows molecular insights into the receptor activation mechanism. Overall, this approach provides panoramic information derived from the full‐length receptor in the native context of the live cell, which complements data derived from crystallographic characterization of isolated receptors in an artificial environment.
FIGURE 4.
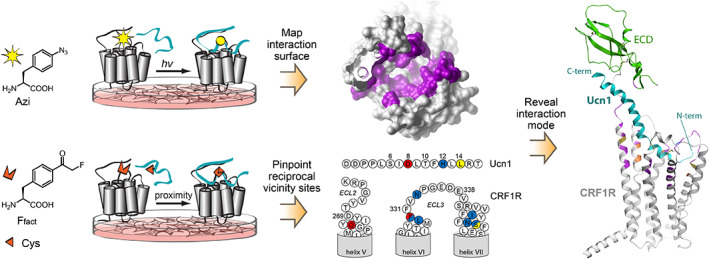
Probing ligand–receptor interaction on mammalian cells. Photo‐cross‐linking unnatural amino acid (Uaa) Azi was incorporated into corticotropin releasing factor receptor type 1 (CRF1R) to reveal the binding pocket (in purple) for its peptide agonist Ucn1; Latent bioreactive Uaa Ffact was incorporated into CRF1R together with Cys in Ucn1 to determine reciprocal spatial constrains (circle with same colors) of the receptor–ligand complex. These data, obtained from intact receptor expressed in live cells, were then integrated to build a conformation model for full‐length CRF1R in complex with Ucn1
5.2. GECX to capture and identify protein–protein interaction in cells
Moving from mammalian cell surface to inside cells, latent bioreactive Uaa allows Genetically Encoded Chemical Cross‐linking (GECX) of interacting proteins in live cells for identification (Figure 5a). 34 Initially, the strategy was demonstrated with Uaas BprY and EB3 (Figure 2a), which contain alkyl bromide to react with Cys in proximity. The Uaa is incorporated into the target protein at a selected site, which will be positioned close to a Cys of the interacting protein upon protein interaction. The reaction of the Uaa with the Cys then captures the interacting protein to the target in cells. After affinity purification of the target protein, the co‐purified cross‐linked protein is digested by protease and analyzed with MS to reveal the protein identity and cross‐linking site. This GECX strategy is able to capture proteins binding in low affinity (affibody with Z protein, K d in μM) and enzyme‐substrate interactions (e.g., ubiquitin‐conjugating enzyme UBE2D3 with substrate PCNA, 34 protein tyrosine phosphatase SHP2 with substrate HER2 35 ). In addition, by placing BprY or EB3 close to the active site of the oxidoreductase thioredoxin (Trx), 91 E. coli proteins have been captured and identified with MS as potential Trx substrate proteins. 34
FIGURE 5.
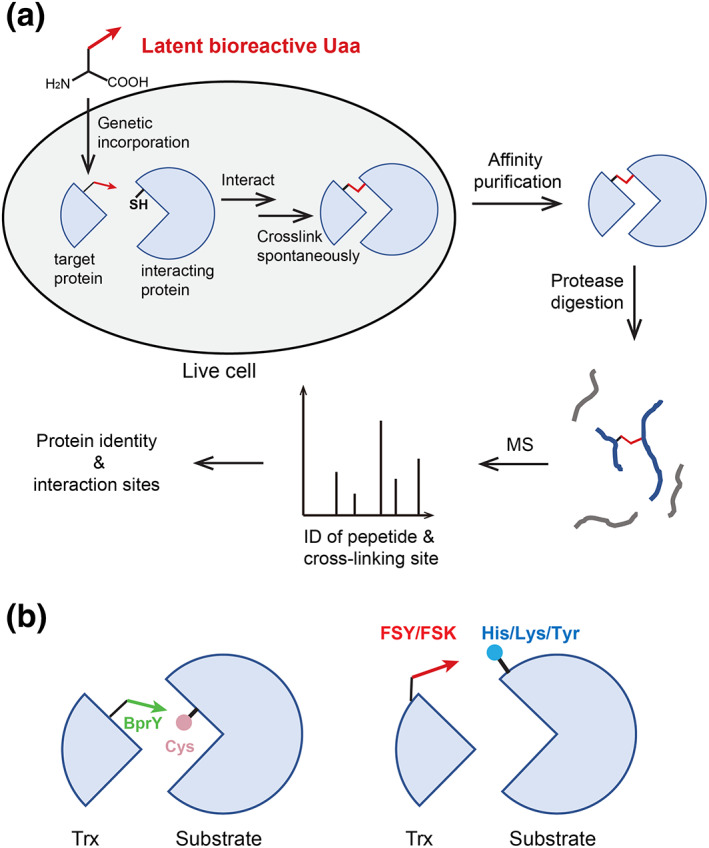
(a) Genetically Encoded Chemical Cross‐linking (GECX) to capture protein–protein interactions in live cells for subsequent identification by MS. Initial development of GECX used unnatural amino acids (Uaas) BprY and EB3 to target Cys residue. (b) Use of fluorosulfate‐l‐tyrosine (FSY) and fluorosulfonyloxybenzoyl‐l‐lysine (FSK) in GECX enables to target His, Lys, or Tyr, and to place the Uaa at the binding periphery, thus expanding the diversity of proteins targetable
In a follow‐up study, 23 FSY and FSK, which are able to target His/Lys/Tyr, have also been successfully used in GECX in live cells, enabling the covalent capture of proteins lacking Cys (Figure 5b). Furthermore, due to the abundance of His/Lys/Tyr at protein surface, FSY and FSK can be incorporated at the periphery of the binding interface, rather than in the active site or inside the binding interface, to minimize potential interference with the protein–protein interaction under study.
GECX spontaneously crosslinks interacting proteins, obviating an exogenous trigger (e.g., light) to activate. Protein interactions can be captured whenever they occur, which is especially suitable for in vivo applications where exogenous trigger is difficult to deliver or timing is hard to decide. The long reaction window of latent bioreactive Uaa also improves cross‐linking efficiency and thus detection sensitivity. The reaction specificity of the Uaa further generates predictable and defined covalent linkage facilitating MS analysis. The MS identification of the cross‐linked peptide also serves as evidence of direct interaction, reducing false positive of indirect binders. Combined with more efficient methods for analysis of protein crosslinks, 36 GECX should enable efficient identification of direct interactomes of various proteins in live cells.
5.3. Evaluating chemical reactivity in protein context
The reactivity between a chemical functional group of a small molecule and that of a large biomolecule cannot be reliably predicted from the reactivity of the corresponding functional groups separately installed on two small molecules, because the latter has no proximity effect that exists in the former. A strategy has been developed to determine the chemical reactivity of two functional groups in the context of close proximity afforded by proteins. 37 The functional groups to be tested were separately installed at the interface of two interacting proteins in amino acid side chains, reaction of which will result in covalent cross‐linking of interacting proteins for detection. We demonstrated that fluoroacetamide, installed on Uaa FAcK (Figure 2a) and previously thought inert in cells, actually reacted with the thiol group of cysteine when in proximity. Introducing the functional group into testing proteins as a Uaa cannot always be successful, so a facile method to attach functionalities onto proteins is later developed. 27 Uaa FnbY and FmnbY are genetically incorporated into proteins at the desired site, which generate QM upon UV activation (Figure 2d). In the absence of nucleophilic residues in proximity, QM then rapidly reacts with the exogenously added amine or thiol derivatives to attach various functionalities to proteins. This protein‐confined proximity strategy will be valuable for accurately evaluating chemical reactivity of small molecules toward biomolecules, to avoid undesired side reactions of drugs and to discover new covalent warheads.
5.4. Proximity‐enabled reactive therapeutics for developing covalent protein drugs
Small molecule covalent drugs provide desirable therapeutic properties over noncovalent ones for treating challenging diseases. 38 The potential of covalent protein drugs, however, remains largely untapped due to protein's inability to bind targets covalently. Encouraged by initial success in developing a covalent peptide inhibitor, 39 a proximity‐enabled reactive therapeutics (PERx) approach has been developed recently to generate covalent protein drugs (Figure 6a). 40 A latent bioreactive Uaa is introduced into a protein drug at or near the binding interface, which reacts with a natural residue of the target protein only when the drug binds to the target, resulting in covalent binding.
FIGURE 6.
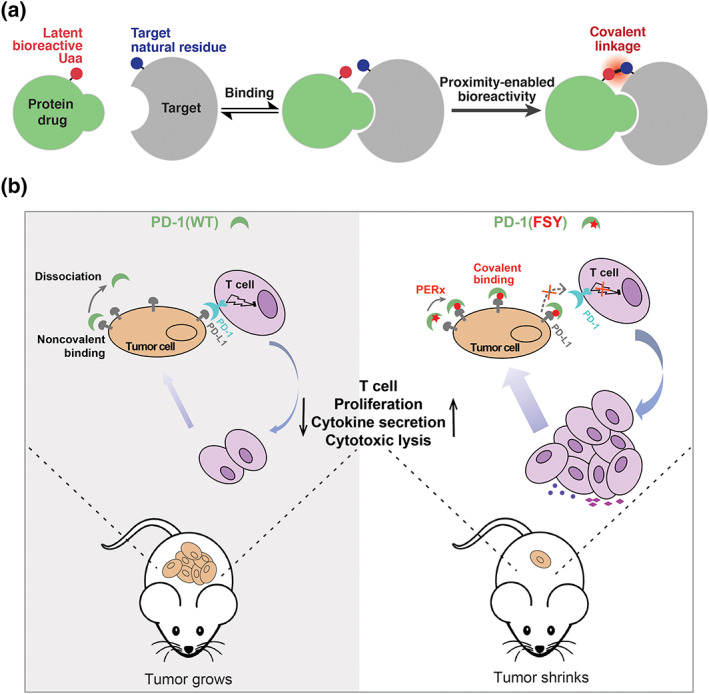
(a) The principle of proximity‐enabled reactive therapeutics (PERx) for developing covalent protein drugs. (b) PERx‐capable programmed cell death‐1 (PD‐1) (fluorosulfate‐l‐tyrosine [FSY]) potently inhibits tumor growth in mice. Using its FSY to react with His69 of PD‐L1, PD‐1(FSY) irreversibly binds with PD‐L1 on tumor cells, efficiently blocking the interaction of PD‐L1 (on tumor cells) with PD‐1 (on T cells). The blockage revives T cells' proliferation and activity to kill tumor cells
The human immune‐checkpoint programmed cell death‐1 (PD‐1) and its ligand PD‐L1 were initially chosen to test this idea (Figure 6b). On the basis of human PD‐1/PD‐L1 complex structure, FSY was incorporated into the ectodomain of human PD‐1 at the binding interface, aiming His69 of PD‐L1. Through FSY reacting with His69 of PD‐L1, the resultant PD‐1(FSY) covalently binds to human PD‐L1 in vitro, on cancer cell surface, and in tumor xenografted in mice with high efficiency and target specificity. Interaction of PD‐1 on T cells with PD‐L1 on cancer cells inhibits T cell proliferation and activity. Irreversible binding of PD‐1(FSY) to PD‐L1 has been found to effectively block the PD‐1/PD‐L1 interaction. As expected, in comparison with the noncovalent PD‐1(WT), PD‐1(FSY) significantly enhances the activities of human T cells and CAR‐T cells in vitro, reaching the same efficiency as atezolizumab, an FDA‐approved anti‐PD‐L1 antibody. When PD‐1(FSY) is administrated into xenograft mouse models immune‐humanized with either human peripheral blood mononuclear cells, human CAR‐T cells, or with the systematic bone marrow–liver–thymus humanization (Hu‐mice), PD‐1(FSY) exhibits strikingly potent antitumor effect over the noncovalent PD‐1(WT), attaining therapeutic efficacy equivalent or superior to atezolizumab.
Besides the native interacting protein pair PD‐1/PD‐L1, engineered protein binders can also be converted into the covalent mode for potential PERx. Incorporation of FSY into HER2‐specific affibodies 40 and of FSY or FSK into EGFR‐specific nanobodies 23 allow the resultant affibody or nanobody mutants to irreversibly bind with the corresponding receptor in vitro and on cancer cell surface, providing a general route to target native receptors covalently.
PERx is straightforward—only a single mutation, the latent bioreactive Uaa, is introduced into the protein drug. This simplifies the procedures for covalent protein drug conversion and should minimize potential immunogenicity. PERx‐capable protein drugs bind their target irreversibly, and thus decouple drug pharmacodynamic effect from pharmacokinetics. Small proteins are usually cleared in vivo quickly, but they are preferred for conditions such as extravasation and tissue penetration. Methods to extend protein half‐lives often increase their sizes and reduce efficacy. PERx now enables the direct use of small proteins in vivo without half‐life extension through other modifications. Off‐target reaction is a critical concern for covalent drugs, but PERx affords unusual target specificity, because its covalent reactivity necessitates both drug‐target binding and unnatural–natural amino acid pairing. In short, PERx is a general platform technology for converting various interacting proteins into covalent binders. We expect it will lead to the rapid development of covalent protein drugs as well as new avenues for biological research.
6. FUTURE OUTLOOK
The concept of proximity‐enabled bioreactivity solves the conflicting requirements of reactivity, selectivity, and biocompatibility of bioreactive Uaas, enabling the genetic encoding of latent bioreactive Uaas in live cells and the introduction of new covalent bonding ability to proteins. Covalent linkages targeting different amino acid residues have been achieved within and between proteins, in vitro, in cells, and in vivo. Expanding beyond the disulfide bond with various properties, these new covalent linkages are enabling novel applications in protein research, engineering, and applications.
At the outset, protein structural information has been important for deciding where to place the latent bioreactive Uaa to generate the covalent bond. However, rapid advancement in predicting protein structures and interactions will make an immense number of proteins amenable to new covalent bonding. Other computational methods such as molecular dynamics simulation may further improve Uaa site selection for enhanced reaction rate and efficiency. Still in its infancy, the impact of covalent proteins on biology and therapeutics awaits more comprehensive and in‐depth investigations, such as idiosyncratic side effect and immunogenicity, which will feed back to the further optimization of the latent bioreactive Uaa. Finally, we expect that the concept of proximity‐enabled bioreactivity can be similarly applied to covalently target other biomolecules inside cells.
AUTHOR CONTRIBUTIONS
Li Cao: Writing – original draft (equal); writing – review and editing (equal). Lei Wang: Funding acquisition (lead); supervision (lead); writing – original draft (equal); writing – review and editing (equal).
ACKNOWLEDGMENTS
The authors acknowledge the support of the National Institutes of Health (R01GM118384). L. W. is grateful to his wonderful group members past and present for making this all possible.
Cao L, Wang L. New covalent bonding ability for proteins. Protein Science. 2022;31:312–322. 10.1002/pro.4228
Lei Wang is the winner of the 2021 Emil Thomas Kaiser Award.
Funding information National Institute of General Medical Sciences, Grant/Award Number: R01GM118384
REFERENCES
- 1. Sevier CS, Kaiser CA. Formation and transfer of disulphide bonds in living cells. Nat Rev Mol Cell Biol. 2002;3:836–847. [DOI] [PubMed] [Google Scholar]
- 2. Kang HJ, Baker EN. Intramolecular isopeptide bonds: Protein crosslinks built for stress? Trends Biochem Sci. 2011;36:229–237. [DOI] [PubMed] [Google Scholar]
- 3. Wensien M, Pappenheim von FR, Funk L‐M, et al. A lysine‐cysteine redox switch with an NOS bridge regulates enzyme function. Nature. 2021;593:460–464. [DOI] [PubMed] [Google Scholar]
- 4. Matthews BW. Recognizing lysine‐cysteine crosslinks in proteins. Protein Sci. 2021;30:1491–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Xiang Z, Ren H, Hu YS, et al. Adding an unnatural covalent bond to proteins through proximity‐enhanced bioreactivity. Nat Methods. 2013;10:885–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang L. Genetically encoding new bioreactivity. N Biotechnol. 2017;38:16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang L, Brock A, Herberich B, Schultz PG. Expanding the genetic code of Escherichia coli . Science. 2001;292:498–500. [DOI] [PubMed] [Google Scholar]
- 8. Wang L. Engineering the genetic code in cells and animals: Biological considerations and impacts. Acc Chem Res. 2017;50:2767–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Coin I, Katritch V, Sun T, et al. Genetically encoded chemical probes in cells reveal the binding path of Urocortin‐I to CRF class B GPCR. Cell. 2013;155:1258–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xiang Z, Lacey VK, Ren H, et al. Proximity‐enabled protein crosslinking through genetically encoding haloalkane unnatural amino acids. Angew Chem Int Ed. 2014;53:2190–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hoppmann C, Lacey VK, Louie GV, Wei J, Noel JP, Wang L. Genetically encoding photoswitchable click amino acids in Escherichia coli and mammalian cells. Angew Chem Int Ed. 2014;53:3932–3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hoppmann C, Maslennikov I, Choe S, Wang L. In situ formation of an azo bridge on proteins controllable by visible light. J Am Chem Soc. 2015;137:11218–11221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen XH, Xiang Z, Hu YS, Lacey VK, Cang H, Wang L. Genetically encoding an electrophilic amino acid for protein stapling and covalent binding to native receptors. ACS Chem Biol. 2014;9:1956–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Furman JL, Kang M, Choi S, et al. A genetically encoded aza‐Michael acceptor for covalent cross‐linking of protein‐receptor complexes. J Am Chem Soc. 2014;136:8411–8417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xuan W, Li J, Luo X, Schultz PG. Genetic incorporation of a reactive isothiocyanate group into proteins. Angew Chem Int Ed. 2016;128:10219–10222. [DOI] [PubMed] [Google Scholar]
- 16. Cigler M, Muller TG, Horn‐Ghetko D, et al. Proximity‐triggered covalent stabilization of low‐affinity protein complexes in vitro and in vivo. Angew Chem Int Ed. 2017;56:15737–15741. [DOI] [PubMed] [Google Scholar]
- 17. Xuan W, Shao S, Schultz PG. Protein crosslinking by genetically encoded noncanonical amino acids with reactive aryl carbamate side chains. Angew Chem Int Ed. 2017;56:5096–5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yu C, Tang J, Loredo A, et al. Proximity‐induced site‐specific antibody conjugation. Bioconjug Chem. 2018;29:3522–3526. [DOI] [PubMed] [Google Scholar]
- 19. Dong J, Krasnova L, Finn MG, Sharpless KB. Sulfur(VI) fluoride exchange (SuFEx): Another good reaction for click chemistry. Angew Chem Int Ed. 2014;53:9430–9448. [DOI] [PubMed] [Google Scholar]
- 20. Chen W, Dong J, Plate L, et al. Arylfluorosulfates inactivate intracellular lipid binding protein(s) through chemoselective SuFEx reaction with a binding site Tyr residue. J Am Chem Soc. 2016;138:7353–7364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang N, Yang B, Fu C, et al. Genetically encoding fluorosulfate‐L‐tyrosine to react with lysine, histidine, and tyrosine via SuFEx in proteins in vivo. J Am Chem Soc. 2018;140:4995–4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yang B, Wang N, Schnier PD, et al. Genetically introducing biochemically reactive amino acids dehydroalanine and dehydrobutyrine in proteins. J Am Chem Soc. 2019;141:7698–7703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu J, Cao L, Klauser PC, et al. A genetically encoded fluorosulfonyloxybenzoyl‐L‐lysine for expansive covalent bonding of proteins via SuFEx chemistry. J Am Chem Soc. 2021;143:10341–10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tian Y, Jacinto MP, Zeng Y, et al. Genetically encoded 2‐aryl‐5‐carboxytetrazoles for site‐selective protein photo‐cross‐linking. J Am Chem Soc. 2017;139:6078–6081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hu W, Yuan Y, Wang C‐H, et al. Genetically encoded residue‐selective photo‐crosslinker to capture protein‐protein interactions in living cells. Chem. 2019;5:2955–2968. [Google Scholar]
- 26. Liu J, Li S, Aslam NA, et al. Genetically encoding photocaged quinone methide to multitarget protein residues covalently in vivo. J Am Chem Soc. 2019;141:9458–9462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu J, Cheng R, Van Eps N, et al. Genetically encoded quinone methides enabling rapid, site‐specific, and photocontrolled protein modification with amine reagents. J Am Chem Soc. 2020;142:17057–17068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bock JE, Gavenonis J, Kritzer JA. Getting in shape: Controlling peptide bioactivity and bioavailability using conformational constraints. ACS Chem Biol. 2013;8:488–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fan LZ, Lin MZ. Optical control of biological processes by light‐switchable proteins. Wiley Interdiscip Rev Dev Biol. 2015;4:545–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Baek M, DiMaio F, Anishchenko I, et al. Accurate prediction of protein structures and interactions using a three‐track neural network. Science. 2021;373:871–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Klippenstein V, Hoppmann C, Ye S, Wang L, Paoletti P. Optocontrol of glutamate receptor activity by single side‐chain photoisomerization. Elife. 2017;6:2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Coin I, Perrin MH, Vale WW, Wang L. Photo‐cross‐linkers incorporated into G‐protein‐coupled receptors in mammalian cells: A ligand comparison. Angew Chem Int Ed. 2011;50:8077–8081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang B, Tang S, Ma C, et al. Spontaneous and specific chemical cross‐linking in live cells to capture and identify protein interactions. Nat Commun. 2017;8:2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tang H, Dai Z, Qin X, et al. Proteomic identification of protein tyrosine phosphatase and substrate interactions in living mammalian cells by genetic encoding of irreversible enzyme inhibitors. J Am Chem Soc. 2018;140:13253–13259. [DOI] [PubMed] [Google Scholar]
- 36. Liu C, Wu T, Shu X, et al. Identification of protein direct interactome with genetic code expansion and search engine OpenUaa. Adv Biol. 2021;5:e2000308. [DOI] [PubMed] [Google Scholar]
- 37. Kobayashi T, Hoppmann C, Yang B, Wang L. Using protein‐confined proximity to determine chemical reactivity. J Am Chem Soc. 2016;138:14832–14835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Rev Drug Discov. 2011;10:307–317. [DOI] [PubMed] [Google Scholar]
- 39. Hoppmann C, Wang L. Proximity‐enabled bioreactivity to generate covalent peptide inhibitors of p53‐Mdm4. Chem Commun. 2016;52:5140–5143. [DOI] [PubMed] [Google Scholar]
- 40. Li Q, Chen Q, Klauser PC, et al. Developing covalent protein drugs via proximity‐enabled reactive therapeutics. Cell. 2020;182:85–97. [DOI] [PubMed] [Google Scholar]