

Abstract
In this issue of Structure, Shang and Kojetin (2021) present insights into the binding mechanism of artificial agonists to the PPARγ nuclear receptor. These data support a two-step model, with induced fit and conformational selection aspects. This mechanism may be used among similar compounds and receptors, further advancing drug development.
Peroxisome proliferator-activated receptor γ (PPARγ) is a well-known member of the large superfamily of nuclear receptors (NR), which serve as transcription factors that respond to endocrine signaling. As a type II NR, PPARγ regulates the transcription of metabolic genes in response to changes in the occupancy of small molecule compounds within its ligand binding domain (LBD) (Kim et al., 2015). In the absence of a ligand, PPARγ silences genes through the formation of a corepressor complex. In contrast, binding by any of the wide range of natural and synthetic agonists within LBD facilitates coactivator or secondary NR recruitment, leading to the control of cell type- and signal-specific groups of genes.
Understanding how this ligand-dependent control works is essential to knowing how PPARγ and its respective coregulators regulate the transcription of genes in response to changes in lipid metabolism. Particularly within adipocytes, hepatocytes, and macrophages, PPARγ is a master regulator of cell proliferation, differentiation, immunity, and metabolic function (Kim et al. 2015). In humans, PPARγ loss of function mutations cause lipodystrophy and insulin resistance (Ludtke et al. 2007); gain of function mutations, while quite rare, lead to extreme obesity (Agostini et al. 2018). Most of these mutations affect the LBD, underscoring the importance of small molecule control in this pathway.
Based on this foundation, synthetic agonists of PPARγ have long been sought to treat insulin-resistant diabetes, joined more recently to intervene in overall metabolic syndrome and cancer development. These agonists include the thiazolidinedione (TZD) class of drugs, first used as an insulin sensitizer by promoting adipocyte differentiation through an unknown mechanism. When the receptor of TZDs was discovered to be PPARγ, excitement soared about the potential for this class of drugs to be used as a potential treatment for insulin-resistant diabetes (Lehmann et al. 1995). However, concerns about side effects from TZD treatment, ranging from edema to cardiac disorders, markedly reduced the use of these drugs in the late 2000s. While these concerns have since substantially ameliorated, this has not stopped the quest for alternative scaffolds to be used as PPARγ agonists; a number have been found and used in a variety of clinical settings, with varying benefits and drawbacks (Huang et al. 2021).
Integral to such searches, biophysical and structural studies of PPARγ LBD with small molecule ligands interactions have provided key insights into the protein/ligand interactions necessary for binding and subsequent gene activation. Efficacy requires drug binding to an orthosteric pocket deep within the LBD, followed by conformational change involving an alpha-helix (“helix 12”) adjacent to this pocket to subsequently recruit transcriptional coactivators. Prior studies have supported both induced fit and conformational selection models of this process, with X-ray crystallography data typically supporting the former and solution NMR the latter (Bruning et al. 2010, Shang et al. 2020) In this issue of Structure, Shang and Kojetin (2021) directly tackle this issue with a combination of approaches, supporting the existence of an induced fit model involving a two-step process of ligand binding into a weak encounter complex followed by subsequent conformational change (Figure 1).
Figure 1 – Schematic of agonist binding within PPARγ LBD, based on this issue’s article (Shang and Kojetin 2021).
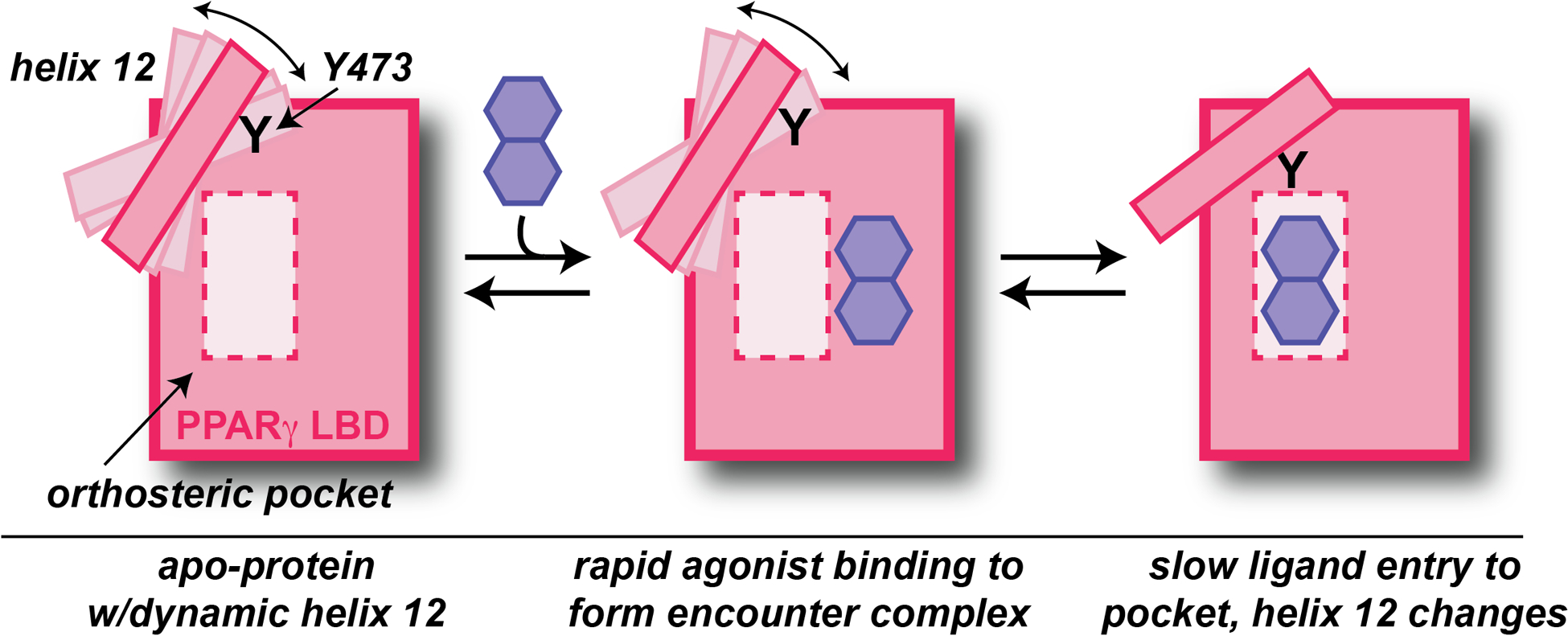
Three states of the PPARγ LBD are depicted: an apo form, an initial rapidly-formed encounter complex with small molecule agonist, and a subsequently-generated complex with the ligand bound within the orthosteric pocket. Conformational and dynamic changes in helix 12 caused in the last step are required for coactivator recruitment, relying critically on contacts between Tyr 473 and agonist.
Central to this manuscript’s advances, the authors present solution NMR titrations of several agonists into 15N-labeled samples of the PPARγ LBD. In general, typical titrations reveal ligand-induced chemical shift perturbations (CSPs) with one of two patterns: either “fast exchange” behavior with a chemical shift of the population-weighted average of the apo- and ligand-bound species, or “slow exchange” behavior with separately observed peaks corresponding to the populations of apo- and ligand-bound species. However, data from three PPARγ LBD/agonist pairs show an intriguing mixed behavior – fast exchange at low ligand:protein stoichiometry, followed by slow exchange at higher concentrations – at backbone amides distributed throughout the domain. When combined with surface plasmon resonance (SPR)-based measurements of binding kinetics, the authors present a strong case that PPARγ LBD agonists generally bind via an initial fast step to generate an encounter complex followed by subsequent conformational change in the protein.
To define the possible structures of complexes corresponding to these states, Shang and Kojetin (2021) creatively took advantage of a fortuitous crystallization feature – apo-PPARγ LBD crystallizes in two conformations in the asymmetric unit, differing by the location of the critical helix 12. Thus, to probe the characteristics of these conformations, the authors first formed crystals of the apo-protein, and subsequently soaked them with several PPARγ agonists for X-ray structure determination. In doing so, the authors obtained pairs of ligand-bound structures from these analyses to get views of both an initial encounter complex, with the ligand sitting just outside a solvent-accessible pore leading into the orthosteric pocket (and helix 12 in an inactive conformation), and an active state structure with the ligand bound deeply into the pocket and helix 12 in a conformation suitable for recruiting coactivators. Notably, these structures agree with prior molecular dynamics simulations suggesting the aforementioned pore as the route of ligand entry into the pocket (Genest et al. 2008).
Shang and Kojetin (2021) close by investigating agonist binding to a PPARγ variant mutated in a key helix 12 residue thought to be involved in coupling ligand binding within the orthosteric pocket to changes in the helix 12 conformation (Y473E). ITC and NMR titrations of agonists into this variant confirm that the compounds can still bind with high affinity via the two-state process described for wild-type protein but fail to produce the conformational changes required for coactivator recruitment, effectively converting the agonists into antagonist-like behavior. Notably, broadening within 15N/1H TROSY peaks for residues in and near helix 12 for the Y473E variant are consistent with substantial dynamics in helix 12, supporting the critical role of Y473 in control of PPARγ LBD activity as well as suggesting that this process involves both structural and dynamic changes in helix 12, consistent with prior reports (Kojetin and Burris 2013).
Many aspects of this work translate well beyond the specific PPARγ/agonist pairs tested here, and there is a high potential for a similar interplay to be occurring in other NRs with both natural and artificial ligands. In particular, the two-step binding process model suggests potential routes for additional protein/ligand contacts to substantially influence binding affinities and rates beyond what might be imagined from solely the final complexes of ligands bound within the orthosteric pocket. This work also provides yet another reminder of the importance of the use of multiple methods – monitoring both compound binding and functional efficacy and doing so with a mix of biophysical approaches – to characterize complex phenomena with the potential to substantially impact drug efficacy.
Acknowledgements
Research in the authors’ laboratory is supported by grants from the NIH (R01 GM106239) and NSF (MCB 1818148).
References
- Agostini M, Schoenmakers E, Beig J, Fairall L, Szatmari I, Rajanayagam O, Muskett FW, Adams C, Marais AD, O’Rahilly S, Semple RK, Nagy L, Majithia AR, Schwabe JWR, Blom DJ, Murphy R, Chatterjee K and Savage DB (2018). “A Pharmacogenetic Approach to the Treatment of Patients With PPARG Mutations.” Diabetes 67(6): 1086–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruning JB, Parent AA, Gil G, Zhao M, Nowak J, Pace MC, Smith CL, Afonine PV, Adams PD, Katzenellenbogen JA and Nettles KW (2010). “Coupling of receptor conformation and ligand orientation determine graded activity.” Nat Chem Biol 6(11): 837–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genest D, Garnier N, Arrault A, Marot C, Morin-Allory L and Genest M (2008). “Ligand-escape pathways from the ligand-binding domain of PPARgamma receptor as probed by molecular dynamics simulations.” Eur Biophys J 37(4): 369–379. [DOI] [PubMed] [Google Scholar]
- Huang R, Zhang C, Wang X and Hu H (2021). “PPARgamma in Ischemia-Reperfusion Injury: Overview of the Biology and Therapy.” Front Pharmacol 12: 600618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Song J and Park KW (2015). “The multifaceted factor peroxisome proliferator-activated receptor gamma (PPARgamma) in metabolism, immunity, and cancer.” Arch Pharm Res 38(3): 302–312. [DOI] [PubMed] [Google Scholar]
- Kojetin DJ and Burris TP (2013). “Small molecule modulation of nuclear receptor conformational dynamics: implications for function and drug discovery.” Mol Pharmacol 83(1): 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM and Kliewer SA (1995). “An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma).” J Biol Chem 270(22): 12953–12956. [DOI] [PubMed] [Google Scholar]
- Ludtke A, Buettner J, Schmidt HH and Worman HJ (2007). “New PPARG mutation leads to lipodystrophy and loss of protein function that is partially restored by a synthetic ligand.” J Med Genet 44(9): e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J and Kojetin DJ (2021). “Structural mechanism underlying ligand binding and activation of PPARgamma.” Structure. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J, Mosure SA, Zheng J, Brust R, Bass J, Nichols A, Solt LA, Griffin PR and Kojetin DJ (2020). “A molecular switch regulating transcriptional repression and activation of PPARgamma.” Nat Commun 11(1): 956. [DOI] [PMC free article] [PubMed] [Google Scholar]