

Abstract
Maternal obesity is associated with pregnancy complications and increases the risk for the infant to develop obesity, diabetes and cardiovascular disease later in life. However, the mechanisms linking the maternal obesogenic environment to adverse short- and long term outcomes remain poorly understood. As compared with pregnant women with normal BMI, women entering pregnancy obese have more pronounced insulin resistance, higher circulating plasma insulin, leptin, IGF-1, lipids and possibly proinflammatory cytokines and lower plasma adiponectin. Importantly, the changes in maternal levels of nutrients, growth factors and hormones in maternal obesity modulate placental function. For example, high insulin, leptin, IGF-1 and low adiponectin in obese pregnant women are expected to activate mTOR signalling in the placenta, promoting protein synthesis, mitochondrial function and nutrient transport. These changes are believed to increase fetal nutrient supply and contribute to fetal overgrowth and/or adiposity in offspring, which increases the risk to develop disease later in life. Interventions that specifically target placental function, such as activation of placental adiponectin receptors, may prevent the transmission of metabolic disease from obese women giving birth to large babies to the next generation. The majority of obese women give birth to normal sized infants and these pregnancies are associated with activation of inflammatory signalling pathways, oxidative stress, decreased oxidative phosphorylation and lipid accumulation in the placenta. Recent bioinformatics approaches expand these alterations to include novel targets, however how placental changes in obese women giving birth to normal sized babies are linked to poor short and long-term infant outcomes is unclear.
Keywords: Maternal-fetal exchange, syncytiotrophoblast, placental transport, fetal growth, human, fetal development
Introduction
High body mass index (BMI) is increasingly prevalent in reproductive aged women around the world. For example, almost 2/3 of American women now enter pregnancy either overweight (BMI 25 – 29.9 kg/m2) or obese (BMI ≥ 30 kg/m2; (1–3)). Maternal overweight in pregnancy is associated with adverse short- and long-term outcomes (4–6), however reports in the literature have predominantly addressed maternal obesity, which will be the focus of this review. Pregnancies complicated by obesity are associated with an array of obstetric complications, including gestational diabetes and preeclampsia that increase infant morbidity and mortality (7,8). Infant adverse outcomes include fetal overgrowth, altered body composition, and neural tube defects (4,9–11). The long-term consequences of maternal obesity in offspring are well documented and include an increased risk to develop cardiovascular disease, metabolic syndrome, diabetes, cancer, and psychiatric disorders (12–18). The strong association between maternal obesity and metabolic syndrome in childhood is of particular concern because it creates a vicious, detrimental cycle of intrauterine transmission of metabolic disease from the mother to her children (17–19). Thus, maternal obesity in pregnancy is a daunting public health problem with a profound impact on the health of the next generation.
Maternal obesity is associated with characteristic changes in circulating levels of nutrients, hormones, growth factors, cytokines and inflammatory mediators, including elevated lipids, leptin and IL-6 and low adiponectin levels. However, the mechanisms linking this ‘obesogenic’ metabolic environment to adverse short- and long-term fetal outcomes remain elusive. It is likely that increased levels of, for example, glucose and lipids in the maternal circulation are transmitted across the placental barrier, leading to fetal hyperglycemia and hyperlipidemia, which may adversely affect the developing fetus. In addition, placental function is regulated by an array of maternal metabolic signals (20,21) many of which are influenced by maternal obesity. Therefore, emerging evidence indicates that maternal obesity causes extensive changes in placental function, which have been suggested to mediate the adverse effects of maternal obesity on fetal development.
This review will focus on recent work determining the impact of obesity in human pregnancy on placental function. Experimental data in relevant animal models will be briefly discussed when providing compelling mechanistic insights. After introducing the short- and long-term consequences of maternal obesity in pregnancy and the characteristic metabolic alterations associated with this condition, we will summarize the findings from recent studies employing ‘omics’ approaches in placentas from obese women and review changes in placental signaling, metabolism and nutrient transport in response to maternal obesity. The review makes the distinction between obese women giving birth to large for gestational age (LGA) infants and maternal obesity associated with delivering an appropriate for gestational age (AGA) infant for several reasons. First, the evidence for adverse metabolic and cardiovascular long-term outcomes is more compelling for large LGA infants born to obese mothers than AGA infants of obese mothers. Second, changes in placental function in obese women delivering LGA infants are likely distinct as compared to obese women delivering AGA babies. Third, Whereas developing a model linking maternal obesity, changes in placental function, fetal overgrowth and long-term adverse consequences is relatively straightforward, it is less clear how placental changes in obese women giving birth to normal sized babies are linked to poor short and long-term infant outcomes. Finally, we will identify priority areas for future studies and speculate on novel strategies for interventions targeting the placenta to prevent poor infant outcomes in pregnancies complicated by obesity.
The Clinical Problem of Maternal Obesity
The prevalence of obesity has increased markedly around the world over the past several decades. In France, one in five women of childbearing age is overweight, and about 15% of women are obese (22). In the United Kingdom and the United States, more than 30% of reproductive age females are obese and another 5% have severe obesity (BMI ≥40) (23,24). The prevalence of obesity also differs in regional and ethnic populations (23,25,26). For example, in the US, over 50% of African-American adults are reported to be obese whereas the prevalence of obesity in Asian-Americans is ~10% (22,25).
As in non-pregnant obese individuals, obesity in pregnancy is associated not only with marked hyperinsulinemia and dyslipidemia but also with impaired endothelial function, higher blood pressure, and low grade-inflammation (27). Maternal obesity increases the risk for pregnancy complications such as gestational hypertension, preeclampsia, stroke, venous thromboembolism, gestational diabetes, and caesarian delivery (6,11,28). Obesity in pregnancy also has a detrimental impact on the health of the offspring. Short-term adverse fetal outcomes in infants of obese mothers include increased risk of fetal overgrowth, still birth (29) and neonatal hypoglycemia (30). Fetal overgrowth is a major contributor to the increased rates of caesarian delivery as well as complications during delivery such as shoulder dystocia (31,32). Severe neonatal hypoglycemia affects 10–15% of newborns and has been associated with neurodevelopmental sequelae (30). In maternal obesity neonatal hypoglycemia is typically transient and stems from maladaptive, persistent hyperinsulinemia initiated by higher glucose concentrations in utero (33). Fetal hyperinsulinemia, measured by proxy in cord blood at delivery, is positively correlated with birth weight and neonatal adiposity but is inversely associated with weight gain up to 2 years of age, particularly in girls (34). Similarly, umbilical cord C-peptide levels are inversely associated with infant weight gain over the first year of life in girls, but not in boys (35). These data demonstrate that maternal obesity disrupts the normal transition in glucose metabolism occurring at birth and suggests that the effect of maternal obesity on offspring risk to develop obesity in childhood is influenced by infant sex.
In addition to the short-term consequences of maternal obesity on maternal and fetal health, infants of obese mothers are more likely to develop a range of health problems later in life, which has been reviewed extensively (5,36). For example, children of obese mothers have an increased risk for developing asthma (37) and neurocognitive disorders (38,39). In addition, maternal obesity, in particular if the infant is large-for-gestational age, is associated with the development of obesity and type-2 diabetes in childhood (14,19), perpetuating a vicious cycle where obese mothers transmit metabolic disease to future generations, and their daughters further propagate the disease to their children. Disease risk persists across the lifespan with strong associations between exposure to maternal obesity in pregnancy and adult chronic inflammatory disorders (40,41), metabolic syndrome (42), diabetes (43), and hypertensive/cardiovascular disease (16).
Metabolism and Hormone Levels in Maternal Obesity
Maternal metabolism adapts to normal pregnancy to allow for the allocation of nutrients for placental and fetal growth. This carefully regulated metabolic adaptation is perturbed when the mother is obese resulting in a less optimal ‘metabolic environment’ that is likely linked to changes in placental function, fetal growth and development. The characteristic metabolic phenotype of women who enter pregnancy obese is related to glucose and lipid homeostasis, metabolic hormones and inflammatory mediators.
Glucose Metabolism
Glucose is the primary substrate for placental and fetal energy metabolism and normal pregnancy induces marked changes in maternal glucose metabolism, including insulin resistance, activation of hepatic glucose production and increased β-cell insulin release with higher plasma C-peptide (44), to promote placental and fetal glucose delivery. Obese women have 50-60% higher postprandial insulin concentrations than normal weight women in both early and late gestation (45). In addition, obese women are more glucose intolerant than pregnant women with normal BMI, as reflected by higher fasting, 1-hour and 2-hour glucose levels following an oral glucose tolerance test (OGTT) (45). Indeed, albeit not meeting the criteria for GDM, the abnormal response to OGTT in obese women is associated with the risk of delivering a LGA infant (46). Increased adiposity persists throughout the lifespan with increased frequency of high BMI in children of obese mothers. Interestingly, maternal pre-pregnant BMI is a stronger predictor for childhood obesity than gestational diabetes (47).
Lipids
While circulating lipids are well known to be elevated in pregnancy in all women, maternal obesity is associated with an altered maternal lipid profile (48). As compared to pregnant women with normal pre-pregnancy BMI, maternal obesity is associated with lower high-density lipoprotein (HDL) levels in first trimester and higher maternal triglyceride (TG) levels in the second and third trimesters. In addition, near term obese women have lower cholesterol and low-density lipoprotein (LDL) levels than women with normal pre-pregnancy BMI (27,49,50). TGs are hydrolyzed to non-esterified (free) fatty acids (NEFA) which are also elevated in maternal plasma throughout gestation in pregnancies complicated with obesity (48). In addition, high maternal TG levels in late pregnancy are associated with an increased risk of delivering a LGA infant (51).
Adipokines
Maternal obesity is associated with characteristic changes in the release of adipokines such as leptin and adiponectin, which have systemic effects on metabolism and energy homeostasis (52,53). Obese women have lower plasma adiponectin levels than normal BMI women throughout pregnancy (27,54–57) and maternal adiponectin is inversely correlated with maternal fat mass, insulin resistance, and glucose production as well as with fetal growth, implicating a role for adiponectin in regulation of maternal metabolism, placental function, and fetal development (54,58,59). Leptin regulates satiety and energy expenditure and, as in non-pregnant individuals, circulating levels of leptin are elevated in obese pregnant women (60). Leptin is positively correlated with both maternal insulin concentrations and BMI in the first and third trimester and with BMI at term (54,57). Furthermore, maternal leptin levels correlate positively to fetal circulating leptin concentrations, and elevated cord leptin levels have been linked to fetal insulin resistance (52,61).
Growth Factors
Insulin-like-growth factors (IGFs) promote protein and carbohydrate metabolism and regulate fetal growth (62). The IGF system consists of IGF-1 and IGF-2, their six binding proteins (IGFBPs), and receptors. Maternal circulating IGFs predominantly originate from the liver and are, to a large extent, bound to IGFBPs (63). In early and late pregnancy, maternal IGF-1 is positively correlated and circulating IGFBP-1 concentrations are inversely correlated to maternal BMI (54,64). At term, cord concentrations of IGFBP-1 and IGFBP-6 are lower (65) in obese compared to lean women. Maternal IGF-1 was positively correlated and IGFBP-1 was inversely correlated to birth weight in infants from obese mothers (66).
Pro- and Anti-inflammatory Cytokines
In normal pregnancy, most inflammatory cytokines in the maternal circulation increase across pregnancy, in part due to cytokine secretion by the placenta (25). Numerous investigators have reported that maternal obesity further increases plasma concentrations of pro-inflammatory cytokines such as IL-6 (27), TNF-α (10), monocyte chemoattractant protein 1 (MCP-1) (68), IL-8, and C-reactive protein (10,27), supporting the concept that the mild pro-inflammatory state associated with normal pregnancy is exacerbated in maternal obesity. However, the literature is not consistent with a number of reports finding no significant elevation in circulating maternal cytokine levels in obese pregnant women as reviewed by Pendeloski and coworkers (69). There are a multitude of potential reasons for these discrepancies. However, the inconsistency in this data suggests that heightened inflammation is not a general phenomenon in pregnancies complicated by obesity but may occur in specific subgroups of obese women. The biological effects of pro-inflammatory cytokines are balanced by anti-inflammatory cytokines, such as interleukin (IL)-1 receptor antagonist, IL-4, IL-6, IL-10, IL-11 and IL-22 (70). Although the effect of obesity on the levels of anti-inflammatory cytokines remains to be fully established, it has been suggested that a lack of the normal increase in IL-10 contributes to a pro-inflammatory environment in obese women (71).
The Human Placenta
The human placenta develops from fetal trophectoderm, which differentiates into trophoblast, and extraembryonic mesoderm, from which fibroblasts, endothelial cells, and macrophages in the villous core develop. As illustrated in Figure 1, the functional unit of the human placenta is the trophoblast villous tree, containing fetal blood vessels and covered by the syncytiotrophoblast, which is directly exposed to maternal blood entering the intervillous space from the spiral arteries (72). There are three subtypes of trophoblast cells in the human placenta; cytotrophoblasts, extravillous trophoblasts and the syncytiotrophoblast (73). Cytotrophoblast cells either undergo fusion to form the multinucleated syncytiotrophoblast or differentiate into extravillous trophoblasts, which invade the spiral arteries in the decidua and myometrium. Trophoblast invasion into the spiral arteries is believed to be critical for the normal gestational increase in utero-placental blood flow by replacing the endothelial cells and degrading smooth muscle in the vessel walls (74,75).
Figure 1. The human placental barrier at term.
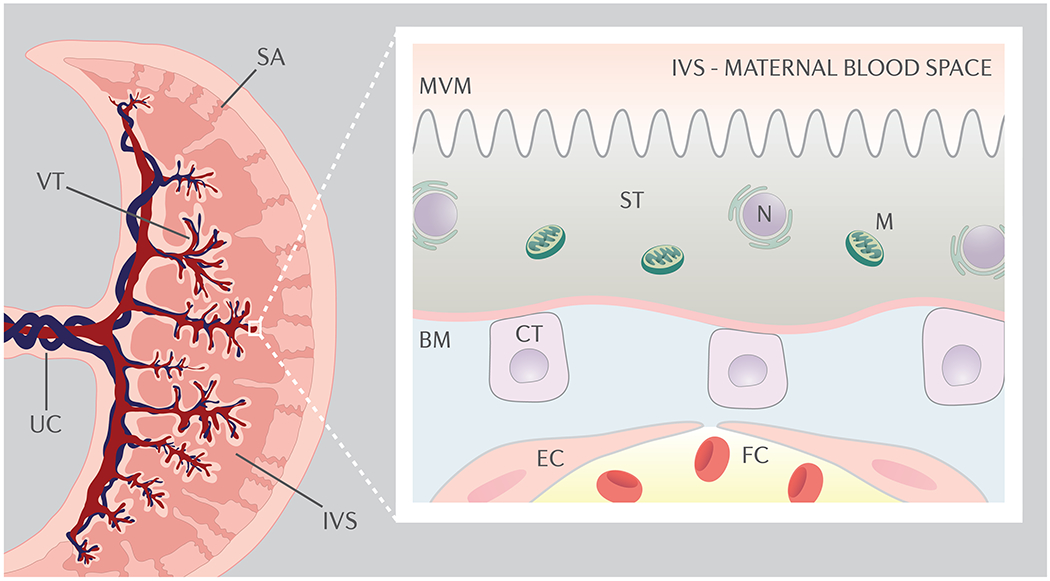
The functional unit of the human placenta is the trophoblast villous tree (VT), containing fetal blood vessels and covered by the syncytiotrophoblast (ST), the multinuclear transporting and hormone producing epithelium of the human placenta, which is generated from mononuclear cytotrophoblast cells (CT). The syncytiotrophoblast is directly exposed to maternal blood entering the intervillous space (IVS) from the spiral arteries (SA). At term, the syncytiotrophoblast and the fetal capillary (FC) endothelial cells (EC) are the only largely continuous cell layers between maternal and fetal blood. The syncytiotrophoblast represents the primary barrier for movement of most solutes from the maternal to the fetal circulations. Specifically, transfer across the two polarized plasma membranes of the syncytiotrophoblast, the apical or microvillous plasma membrane (MVM) directed toward maternal blood in the intervillous space and the basal plasma membrane (BM) facing the fetal capillaries constitute the limiting steps for net flux from maternal to fetal circulations. UC, umbilical cord; N, nucleus, M; mitochondrion.
The syncytiotrophoblast is the transporting and hormone producing epithelium of the human placenta. At term, the syncytiotrophoblast and the fetal capillary endothelium are the only largely continuous cell layers between maternal and fetal blood (Figure 1). Because the fetal-placental capillary endothelial cells allow a relatively unrestricted transfer of small molecules such as glucose and amino acids through intercellular junctions, the syncytiotrophoblast represents the primary barrier for movement of most solutes from the maternal to the fetal circulations. Specifically, transfer across the two polarized plasma membranes of the syncytiotrophoblast, the apical or microvillous plasma membrane (MVM) directed toward maternal blood in the intervillous space and the basal plasma membrane (BM) facing the fetal capillaries constitute the limiting steps for net flux from maternal to fetal circulations (Figure 1). Maternal-fetal exchange of many nutrients and ions occurs by mediated transfer involving transporter proteins expressed in the syncytiotrophoblast plasma membranes.
Maternal Factors Altered by Obesity Regulate Placental Function
Many maternal metabolites, hormones, growth factors and cytokines that are altered in maternal obesity have well established effects on placental function and may mediate the effects of maternal obesity on the placenta.
Insulin
Maternal obesity is associated with hyperinsulinemia and insulin activates placental glucose (76,77) and System A amino acid transporters (77–80). Importantly, despite peripheral insulin resistance, the placenta appears to maintain normal insulin responsiveness in maternal obesity (81). Therefore, the hyperinsulinemia that maintains euglycemia in the face of increasing insulin resistance in obese pregnant women is likely to have significant effects on placental growth and function. For example, if the placental insulin sensitivity is unaffected in maternal obesity as recently reported (81), elevated maternal insulin levels are expected to activate placental insulin and mTOR signaling as well as glucose and amino acid transport.
Adipokines
Maternal obesity is associated with low plasma adiponectin and elevated levels of leptin, changes that are believed to influence placental function and fetal growth (54,59). Leptin promotes placental lipolysis without affecting lipid synthesis (82), potentially resulting in decreased placental TG and cholesterol levels in pregnancies complicated by maternal obesity. In addition, leptin increases system A amino acid transport activity in villous fragments (78,83) and stimulates the release of IL-6, nitric oxide, and human chorionic gonadotrophin in cultured primary human trophoblast cells (78,83). Adiponectin decreases amino acid uptake in term primary human trophoblast cells (84,85). Adiponectin also decreases the gene expression of glucose and System A amino acid transporters in first trimester primary trophoblast (86). Animal experiments demonstrate that adiponectin has similar effects on placental function in vivo. For example, chronic administration of adiponectin in normal weight pregnant mice inhibits placental function, including nutrient transport, and results in intrauterine growth restriction (87). In contrast, low maternal adiponectin, as observed in maternal obesity, is expected to promote placental nutrient transport. A mouse model of maternal obesity in pregnancy, characterized by low maternal adiponectin levels, increased placental insulin signaling and nutrient transport and fetal overgrowth (88) has provided direct evidence to support this hypothesis. Specifically, chronic infusion of adiponectin in obese dams, in rates that increased adiponectin levels to those observed in normal pregnant mice, normalized placental insulin and placental nutrient transport and fetal growth (58). These findings were recently confirmed in elegant studies in which knockdown of the maternal adiponectin gene causing lower circulating levels resulted in increased fetal growth (89,90).
Growth Factors
Maternal obesity has been reported to be associated with increased concentrations of IGF-1 in the maternal circulation (91), which may influence placental development and function. IGF-1 promotes trophoblast proliferation (92) and stimulates glucose transport across a monolayer of BeWo cells, a human choriocarcinoma cell line, by increasing the expression of glucose transporter 1 (GLUT1) in the BM (93). Moreover, IGF-1 increases glucose and amino acid uptake in cultured first trimester primary human trophoblast cells (94). Increased maternal IGF-1 bioavailability (high IGF-1 and low IGFBPs) in obese mothers may therefore promote placental nutrient uptake and transfer to the fetus.
Pro-inflammatory Cytokines
Specific cytokines have been shown to be elevated in the maternal circulation of obese mothers, in particular IL-6 (95,96) and TNF-α (10) and a positive correlation between maternal IL-6 and neonatal fat mass has been demonstrated (96), potentially mediated by effects on placental function. These two cytokines have been studied in cultured trophoblast cells and found to influence key placental functions including lipid and amino acid transport as well as placental metabolism. TNF-α activated phospholipase A2 (PLA2G2A) in cultured primary human trophoblast cells (97). Moreover, placental PLA2G2A expression was found to be increased in maternal obesity and associated with neonatal adiposity and it was proposed that PLA2G2A stimulation by TNF-α and leptin represents a key mechanism to favor excess fetal fat accretion (97). Incubation of primary human trophoblast cells with TNF-α and IL-1β decreased Slit2 expression, which is mechanistically linked to increased secretion of IL-6 and IL-8 and elevated expression of matrix metallopeptidase 9 (MMP-9; (98)). Using primary human trophoblast cells, Jones and co-workers reported that IL-6 stimulates system A amino acid transporter activity mediated by STAT3 signaling and increased expression of SNAT2 (99,100).TNF-α also stimulates System A amino acid transport in primary human trophoblast cells mediated by p38 MAPK signaling (99,100). However, other cytokines likely have the opposite effect because conditioned media from monocyte/P. falciparum-infected erythrocyte co-culture, which contains an array of cytokines, inhibited System A amino acid uptake in primary human trophoblast cells (101). Furthermore, using the same experimental system Liong and co-workers reported that the pro-inflammatory agents lipopolysaccharide (LPS) endotoxin and the viral mimetic polyinosinic:polycytidylic acid (poly(I:C)) decreased the activity of the trophoblast insulin signaling pathway and glucose uptake but increased expression of SNAT1 and 2 and System A uptake of amino acids (102). Finally, some evidence suggests that IGF-I/insulin hybrid receptors are present in cultured primary human trophoblast cells and placenta (103,104) and that TNF-a inhibits the signaling associated with this receptor (104), providing a potential mechanisms by which TNF-a may inhibit insulin actions on the trophoblast. Collectively, these data suggest that, albeit pro-inflammatory cytokines have powerful effects on placental function, the specific effects differ between cytokines and their role in pregnancies complicated by maternal obesity is not yet clear.
Lipids
The effects of fatty acids on trophoblast cells depend on chain length and/or saturation. For example, physiological levels of oleic acid (18:1, OA) stimulate mTOR signaling and System A amino acid uptake in cultured primary human trophoblast cells mediated by toll-like receptor (TLR) 4, whereas the long chain polyunsaturated fatty acid, docosahexaenoic acid (22:6, DHA), had the opposite effect (105). Moreover, OA increased the expression of the lipase coactivator alpha-beta hydrolase 5 CGI-58 and perilipin-2 (PLIN2) in cultured primary human trophoblast cells, suggesting that OA may regulate turnover of placental lipids (106). PLIN2 is essential for trophoblast lipid accumulation in lipid droplets, which protects trophoblast cells from apoptosis during hypoxia (107). The protein expression of placental CGI-58 and PLIN2 was reported to be increased in obese mothers at term (106,107). Additionally, oxidized LDL inhibits trophoblast invasion (108) through oxysterol activation of LXR (109). Palmitic acid (16:0, PA), a saturated ‘lipotoxic’ fatty acid (110), stimulated IL-6, IL-8, PLIN2, and TLR expression and increased release of IL-8 in culture media in primary human trophoblast cells while OA did not have this effect (105,110,111). In general agreement with these findings, PA and TNF-α increased the expression of proinflammatory cytokines (IL-6, TNF-α, and IL-8) through JNK/EGR-1 signaling in human trophoblast cell lines, and placental JNK/EGR1 protein expression is elevated in maternal obesity (112).
Placental Omics in Maternal Obesity
The number of studies reporting placental ‘omics’ signatures in obese women is limited (Table 1), however two common themes emerge from these studies: placental transcripts, proteins and metabolites associated with lipid metabolism and inflammation/immune responses are differentially expressed in placentas exposed to maternal obesity.
Table 1:
Human placental omics studies in maternal obesity.
| Clinical Characteristics of Study Subjects | Tissue Type | Gestational Age/ Delivery Mode | Omics Approach |
|---|---|---|---|
| Proteins and Metabolites | |||
| Normal BMI | Placental tissue | Term; C/S | Proteome (253) |
| Obese (Normoglycemic) and normal BMI | Placental biopsies | Term; C/S | Metabolome (GC-MS) (136) |
| mRNA and miRNA | |||
| Obese, overweight, and normal BMI | Placental tissue | Term | Microarray (254) |
| Obese and normal BMI | Placental tissue | Term | Microarray (133) |
| Obese and normal BMI | Isolated primary trophoblasts | 7-12 weeks; elective termination | Microarray (138) |
| GDM, obese, and normal BMI | Isolated trophoblast | Term; C/S | Laser microdissection prior to microarray (139) |
| Obese and normal BMI | Placental tissue | Term | RNAseq (137) |
| Obese and normal BMI | Placental tissue and placental microbiome | Term | RNAseq and 16S-seq (130) |
| Non-diabetic women delivering large for gestational age infants | Placental tissue | Term | lncRNA and mRNA Array (255) |
| DNA methylation | |||
| Obese and normal BMI | Villus tissue | Term; C/S | Methylation MeDIP/hMeDIP (144) |
| Obese, GDM, pre-eclamptic, and normal BMI | Placental tissue | Term | Methylation (143) |
| Overweight/obese (BMI>25) and normal BMI | Placental tissue | Term | Methylation, Unpublished: GSE120062 |
Normal BMI defined as BMI<25. GDM, Gestational Diabetes; C/S Cesarean Section; GC-MS, Gas Chromatography-Mass Spectrometry; lncRNA, long non-coding RNAs; MeDIP/hMeDIP, Methy/ 5-hydroxymethylcytosine DNA Immunoprecipitation.
Lipid metabolism is an enriched pathway common to multiple placental ‘omics’ studies in maternal obesity. The transcriptome of term placenta from obese women compared to placenta from normal BMI women revealed differential expression of genes uniquely associated with lipid metabolism including decreased APOE, DKK1 (113), ANGPTL4 (114), and NRIP1 (115) and increased lipid droplet-associated protein CIDEA (116), consistent with functional studies indicating increased TG content in cultured trophoblasts from obese women (117). Maternal obesity has also been associated with decreased placental expression of genes involved in retinoic acid (Vitamin A) transport and metabolism (GPC4, ALDH1A1, ALDH1A2, CRABP2, RBP1, RBP4, SDC4, and PTGES) (118), which in placenta are likely to be important for binding of maternal chylomicrons and lipoproteins (119). Reduced placental mTOR gene expression and up-regulation of the genes encoding proteins involved in oxidative stress and mitochondrial function, such as increased sirtuin 1 (SIRT1) and uncoupling protein 2 (UCP2), have been reported in maternal obesity (120). Moreover, placental liver X receptor (LXR) signaling pathway was enriched in placental transcripts from obese mothers, consistent with higher plasma levels of palmitic and oleic acid in the same cohort (121). LXR and the ATP-binding-cassette-transporter-A1 (ABCA1) have been proposed to transport maternal cholesterol at the MVM surface of the syncytiotrophoblast (122) and physiological studies have shown that LXR agonists increase cholesterol transport in trophoblast cells (123). Perturbations in placental lipid metabolism induced by maternal obesity are further supported by proteomic and metabolomic studies. In a recent report, the placental proteomic signature was consistent with increased lipid synthesis and energy production and altered antioxidant capacity in placenta from normoglycemic, obese women compared to normal weight mothers (124). These findings, in concert with data demonstrating higher lipid content and evidence of decreased antioxidant capacity in cultured trophoblasts from obese women, are in general agreement with the proposal that maternal obesity is associated with placental lipotoxicity (112,125).
Enrichment of inflammation and immune response pathways is also a common finding in the placenta in maternal obesity, consistent with the concept that maternal obesity is associated with mild placental inflammation (48,121,126). Placental cytokine-receptor signaling was consistently found to be enriched across multiple placental omics studies (121,125,127). Recent next-generation sequencing studies have demonstrated decreased mRNA expression of LEP, ADIPOR1, IGFBP1, CCK, CRH, IL1R1, IL1R2 and accessory proteins IL1RAP and IL1RAPL2 in maternal obesity as compared to normal weight women at term (121,125).
Untargeted approaches have been used to define a placental methylome (128,129) and recently it was shown that obesity influences DNA methylation patterns in the human placenta (130). Obese women had the highest levels of placental global methylation as compared to normal pregnancy and pregnancies complicated by GDM or preeclampsia (131). Placental transcripts that encode key proteins central to placental growth and metabolism, such as PPARα, IGF-2, and sirtuins, have also been shown to be differentially methylated in maternal obesity (128,130,132).
These studies provide a compelling resource of discovery data, identifying novel placental genes and signaling pathways that are influenced by maternal obesity and therefore may play a role in mediating changes in placental function Recently, approaches to integrate different modalities of ‘omics’ data have been employed. For example, integrative analysis of the transcriptome and metabolome in the BeWo trophoblast cell line in response to high glucose, reflecting one isolated aspect of the obesogenic environment, demonstrated changes fatty acid and phospholipid metabolism (133,134). However, more targeted and mechanistic studies are required to confirm cause-and-effect relationships (135). The heterogeneity of placental tissue precludes assigning ‘omics’ signatures to specific cell types in studies using placental tissue and difficulties to isolate intact syncytium from human placenta has limited the interrogation of the syncytiotrophoblast transcriptome, proteome and metabolome. However, emerging single cell sequencing approaches developed to study the transcriptome of the normal trophoblast (136–140) will allow cell specific placental ‘omics’ approaches to be used in relation to maternal obesity and other pregnancy complications in the near future. Future use of approaches that employ barcoding strategies for single cell sequencing (MATQ-seq) as well as more functional ‘omics’ integrations such as chromatin immunoprecipitation sequencing in concert with selective isolation of chromatin-associated proteins (ChIP-SICAP; (141,142)) represent powerful tools to further explore placental ‘omics’ in maternal obesity.
Placental Inflammation
As discussed above, multiple reports indicate higher circulating levels of lipids, leptin, TNF-α, IL-1β, IL-6, IL-8 and reduced levels of adiponectin in obese women as compared to normal BMI pregnant women. The question to what extent there is placental inflammation in maternal obesity has been studied in some detail. At the RNA level, several studies have found that maternal obesity is associated with higher placental expression of TNFα, IL-1β, IL-8, MCP-1, and cytokine receptors such as CXCR2 at term (100,143–145). Placental TNF-α levels were reported to be elevated in female, but not male, placentas in maternal obesity, suggesting fetal sex differences in the placental inflammatory response to obesity (146). Moreover, maternal obesity is associated with the activation of distinct intracellular placental inflammatory pathways, including signal transducer activated transcription factor 3 (STAT3) and the stress/mitogen activated protein kinases (MAPK) p38 (68,147,148). The number, identity, and activity of immune cells within the placenta are likely to influence the degree of placental inflammation when exposed to maternal obesity. However, available information in this area remains limited. Challier and co-workers demonstrated increased number of CD14+ and CD68+ macrophages in the placenta in maternal obesity as compared to normal BMI women (10). Yet Roberts et al. reported a different placental immune response to maternal obesity, characterized by increased neutrophils and no change in CD14+ and CD68+ macrophages (143). Although placental inflammation in maternal obesity has been suggested to alter placental functions such fatty acid and amino acid transport, metabolism and insulin resistance (100,144,145), the molecular mechanisms linking inflammatory mediators to placental dysfunction in maternal obesity have been described for only a handful of inflammatory mediators (68,99,100,149) and those mediators were studied in isolation when a combination approach may be more physiological.
Placental Metabolism and Mitochondrial Function
As most other tissues, the human placenta relies on glycolysis and oxidative phosphorylation for the production of ATP (150), however the precise contribution to the overall energy needs of the two sources of ATP remains to be clearly established. Elevated circulating concentrations of glucose and lipids in maternal obesity likely affect not only placental uptake but also its metabolism. Glucose has long been of interest for studies aimed at understanding fetal and placental growth. Under low oxygen conditions placental metabolism does not shift to anaerobic glycolysis (151) and hypoxia stimulates lipid accumulation in trophoblasts (152). The relatively high density of mitochondria and the presence of specialized long chain fatty acid uptake transporters (153,154) in human trophoblasts has led to the proposal that placental metabolism is highly oxidative and ‘prefers’ oxidative metabolism to glycolysis (155). Mele and colleagues reported that maternal obesity is associated with decreased placental ATP generation by oxidative phosphorylation without concomitant up-regulation of glycolysis (156). These findings were interpreted to indicate impaired placental mitochondrial function in maternal obesity (156). However, an alternative explanation might be a homeostatic down-regulation of mitochondrial function due to increased glucose availability in vivo, and therefore increased glycolytic glucose flux when the mother is obese and glucose levels are elevated.
β-oxidation is a key energy source for placental metabolism and circulating TGs, the source of placental lipids, are typically increased in maternal obesity (50). Placental β-oxidation, as measured using radiolabeled palmitate was found to be decreased and esterification and storage of lipids were increased in maternal obesity (117). However, when β-oxidation was prevented through chemical inhibition of long chain fatty acid carrier CPT-1, the rate limiting enzyme for fatty acids entry into the mitochondria, non-mitochondrial fatty acid oxidation was greater in placenta from obese compared to normal BMI women (117). Given that placental peroxisomal activity was enhanced in maternal obesity as compared to normal BMI women, the authors concluded that ~10% of trophoblast fatty acid oxidation was non-mitochondrial in maternal obesity, which may exacerbate ROS generation (117). Based on these considerations, it has been proposed that an oversupply of lipids to the placenta exceeds the placental mitochondrial capacity for β-oxidation, resulting in elevated intracellular placental lipid levels, increased lipid storage and promotion of fatty acid transfer to the fetus (106,117,125). Whether or not the infants of obese mothers are hyperlipidemic at birth has not been resolved, with some studies reporting no correlation between maternal BMI and cord or neonatal lipid profile (51,157,158) with other reports showing maternal BMI influenced neonatal TG levels (159).
In addition to glucose and fatty acids, emerging evidence suggest that glutamine, which enters the TCA cycle as α-ketoglutarate, is a major substrate for baseline oxidative phosphorylation in trophoblast cells from normal healthy pregnancy (160). Male, but not female, cultured primary human trophoblast cells isolated from placentas of obese mothers displayed increased preference for fatty acid and glucose at baseline but this preference was accompanied by a decrease in the ability to switch between glucose, fatty acid, and glutamine when oxidation demands increased (160). This apparent plasticity of placental mitochondria, together with differences in experimental buffers and culture conditions (161), in vitro oxidative stress or cell death, differentiation status (155,162), and delivery (labor) method (163,164), as well as the possibility that prolonged cell culture activates anaerobic glycolysis (165), are all contributing factors to the considerable variability in published basal mitochondrial respiration rates in human placental studies (163,166). These considerations complicate interpretation of recent reports on the effect of maternal obesity on placental mitochondria and additional studies in this area are warranted.
Placental mitochondrial density increases across normal gestation (163). Mitochondrial biogenesis is regulated by a multitude of factors, including growth factor and mTOR signaling. For example, inhibition of mTORC1 signaling reduces mitochondrial biogenesis and decreases oxidative phosphorylation in cultured primary human trophoblast (167). Likewise, epidermal growth factor (EGF) is implicated in regulating mitochondrial density in placentas exposed to maternal obesity (155). Maternal obesity has been shown to reduce mitochondrial density in placenta or isolated trophoblasts (117,168). However, Mando and colleagues reported that maternal obesity, but not GDM, was associated with increased mitochondrial DNA as well as normal mitochondrial morphology in syncytiotrophoblast from obese women (169). Recently, mitochondrial density was linked to different trophoblast cell types in human pregnancies (155). Specifically, chemical prevention of primary human trophoblast syncytialization in vitro revealed that cytotrophoblasts have a higher mitochondrial density, rely more on oxidative phosphorylation and have a preference for fatty acids as energy substrate as compared to syncytialized cultured trophoblasts (155). These data suggest that cytotrophoblasts may play a more important role than previously recognized and that increased lipid availability may contribute to cytotrophoblast hyperplasia. The mechanistic links between maternal obesity and changes in placental mitochondrial function remain to be established.
Placental Reactive Oxygen Species and Oxidative Stress in Maternal Obesity
The levels of reactive oxygen species (ROS) typically refer to the abundance of chemically reactive molecules containing oxygen, including peroxides, superoxide and hydroxyl radical. Higher levels of ROS may or may not be associated with oxidative stress (i.e., higher levels of ROS that cause some type of damage) depending on to what extent antioxidant defense mechanisms are activated. In non-pregnant individuals, obesity is associated with higher levels of reactive oxygen species (ROS) and oxidative stress (170). ROS species relevant for obesity include endogenous free radicals (O2− and HO), hydrogen peroxide, and ozone originating from mitochondria but also peroxisomal degradation of branched chain fatty acids, NADPH oxidases, purine degradation, and eicosanoid metabolism. Maternal obesity has been associated with increased maternal ROS including higher levels of maternal malondialdehyde, carbonyl proteins, nitric oxide and superoxide anion with lower glutathione concentrations and superoxide dismutase (SOD) activity (168,171). Moreover, ROS production (156), glutathione concentrations and SOD activity (171) in the placenta is reported to be increased in maternal obesity, which may impair mitochondrial function and explain lower ATP production (156). This is supported by studies in other tissues where prolonged ROS exposure impairs mitochondrial function through mechanisms such as reduced ability to replicate mitochondrial DNA (172) and activation of ROS-dependent cell death mechanisms (173). In situations of nutrient excess, such as maternal obesity, mechanisms to uncouple substrate metabolism to ATP synthesis, including increased expression of uncoupling proteins (UCP2) and antioxidants, normally limit ROS accumulation. In general agreement with this concept, placental activity of superoxide dismutase and catalase is increased in maternal obesity compared to normal BMI mothers (171). Moreover, a recent study linked decreased placental expression of the antioxidant glutathione peroxidase 4 in maternal obesity to markers of oxidative stress in the newborn (174), suggesting that placental oxidative stress may be transmitted to the fetus with possible negative effects on fetal development.
Fatty acids reflect an additional source of placental ROS accumulation through processes in the mitochondria (during anaplerosis) and in the cytoplasm through the action of NADPH oxidase (175). The high circulating maternal lipid levels and higher placental ROS with dysfunctional mitochondria in maternal obesity likely results in the production of oxidized lipid products, including lipid peroxides (176), oxidized lipoproteins (177) and oxysterols, that may adversely impact trophoblast function. Because oxysterols are ligands for LXR, which increases expression of genes involved cholesterol and lipid metabolism, these changes may influence placental lipid transport and/or metabolism. Furthermore, the placenta produces nitric oxide (NO) that can form peroxynitrite, a pro-oxidant that causes excessive protein nitration (nitrative stress). Placental nitrative stress, measured by nitrotyrosine protein modifications, is increased in obese compared to normal weight women (178) and represents a potential link between ROS and redox dysfunction and intracellular signaling pathways (179).
Placental signaling
Receptors for a range of hormones and growth factors, including receptors for adiponectin, insulin (76), leptin (180), and IGF-1 (71), are highly expressed in the maternal facing MVM of the syncytiotrophoblast, consistent with maternal regulation of placental function. In maternal obesity, the effects of changes in maternal levels of hormones and growth factors and increased nutrient levels are believed to modulate intracellular signaling cascades that converge on key nutrient sensing pathways in the placenta. The extent to which the activity of placental growth-promoting signaling pathways are affected by maternal obesity seems to be dependent the degree of excess maternal body fat mass and whether fetal growth is increased, with more pronounced changes found in women with the high BMI giving birth to LGA babies (120,144,181,182).
The expression and/or phosphorylation of the placental insulin/IGF signaling machinery, including down-stream targets IRS-1 and Akt are increased in obese women delivering LGA babies (144). Activation of placental insulin/IGF signaling in maternal obesity is likely to be caused, in part, by the low maternal levels of circulating adiponectin because adiponectin inhibits trophoblast insulin signaling at the level of IRS-1, mediated by activation of PPARα and ceramide synthesis (84,85)(Figure 2). Furthermore, the activity of placental AMPK, a primary energy sensor that is phosphorylated when ATP levels are low, was reported to be markedly decreased in association with maternal obesity and fetal overgrowth (144) indicating ample availability of energy substrates in the placenta of obese mothers in which fetal growth acceleration occurs. mTOR signaling integrates a large number of metabolic factors, including hormone and growth factor signaling, such as insulin, IGF-1 and EGF, ATP/energy levels, amino acids, glucose, and fatty acids levels, in order to coordinate cellular metabolism, growth, and proliferation (183) in response to the availability of substrates. There is now compelling evidence that mTOR serves as a critical node in coordinating placental function in response to maternal factors to match fetal growth to the ability of the mother to provide adequate nutritional support (184). AMPK inhibits mTOR Complex 1 and decreased AMPK signaling in combination with activation of insulin/IGF signaling and a high availability of nutrients may contribute to an activation of placental mTOR signaling in maternal obesity (144). Because mTOR signaling is a positive regulator of an array of key placental functions, including amino acid transport (185,186), folate transport (187) and mitochondrial biogenesis (167), it has been proposed that activation of placental mTOR signaling may contribute to enhanced fetal nutrient delivery and fetal overgrowth (Figure 2), which occurs more commonly in obese women (144).
Figure 2. Trophoblast signaling in obese women delivering large for gestational age babies.
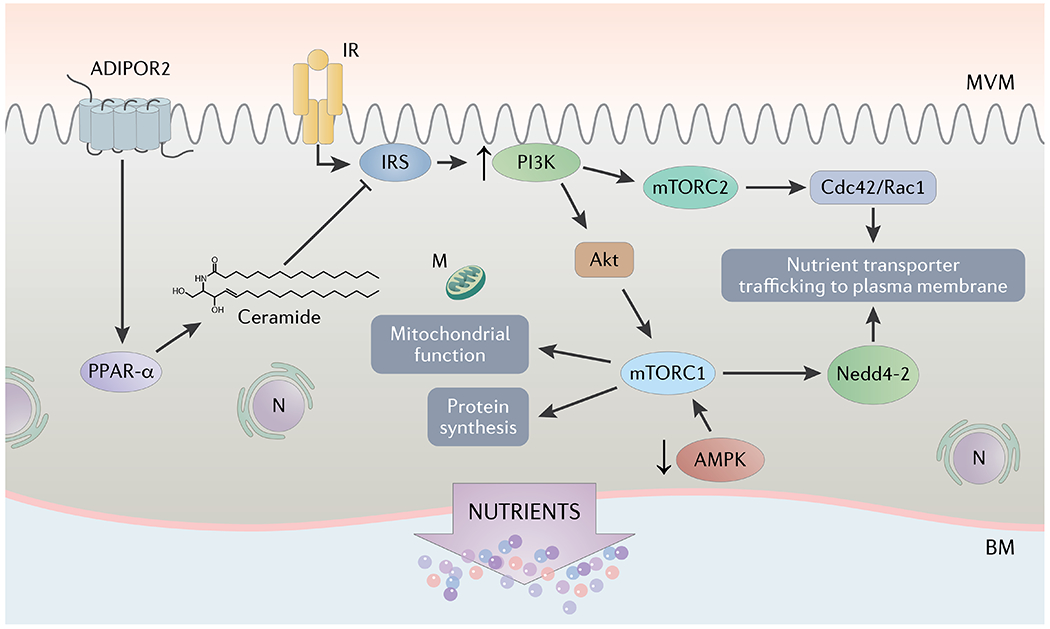
Placental insulin/IGF signaling is activated in obese women delivering large babies, likely due to maternal hyperinsulinemia with maintained placental insulin responsiveness. Moreover, adiponectin inhibits trophoblast insulin signaling at the level of IRS-1, mediated by activation of PPARα and ceramide synthesis. Thus, the low maternal levels of circulating adiponectin in maternal obesity are likely to contribute to the activation of placental insulin signaling. AMPK inhibits mTOR Complex 1 (mTORC1) and decreased AMPK signaling in combination with activation of insulin/IGF signaling and a high availability of nutrients may contribute to an activation of trophoblast mTOR signaling in maternal obesity. Activation of mTORC1 and mTOR complex 2 (mTORC2) independently stimulate trophoblast amino acid transport mediated by distinct mechanisms. Whereas mTORC1 influences amino acid transport by Nedd4-2 mediated ubiquitination, mTORC2 promotes trophoblast amino acid transport by activating Cdc 42/Rac 1. Because mTOR signaling is a positive regulator of an array of key placental functions, including amino acid and folate transport and mitochondrial biogenesis, it has been proposed that activation of placental mTOR signaling may promote increased nutrient delivery to the fetus, contributing to fetal overgrowth. ADIPOR2, adiponectin receptor 2; IR, insulin receptor; PPAR-α, peroxisome proliferator-activated receptor alpha; IRS, insulin receptor substrate; mTORC1, mechanistic target of rapamycin complex 1; mTORC2, mechanistic target of rapamycin complex 2; Cdc42, cell division control protein 42; Rac1, Ras-related C3 botulinum toxin substrate 1; Nedd4-2, neuronal precursor cell-expressed, developmentally downregulated gene 4 isoform 2; AMPK, AMP-activated protein kinase; N, nucleus, M; mitochondrion.
Placental nuclear receptors, such as PPARγ and RXR, are also influenced by maternal obesity. For example, placental PPARγ RNA and protein levels are increased in maternal obesity (117), which may modulate placental development and function given the well-established effects of PPARγ on trophoblast invasion, fatty acid metabolism, and inflammatory responses (188). Both activation of PPARγ and RXR increased fatty acid uptake in primary human trophoblast cells through increased expression fatty acid transport protein 4 (FATP4 (189)).
Nutrient Transport
Placental nutrient transport capacity is one key determinant of fetal growth and there is evidence from human placenta of increased expression/activity of transporters for glucose, amino acids and lipids in pregnancies complicated by maternal obesity, in particular in cases of fetal overgrowth.
Glucose
Glucose is a major metabolic substrate for the placenta and the fetus and the majority of glucose taken up from the mother is transported to the fetus (190). Placental glucose uptake from the maternal circulation is believed to be mediated predominantly by glucose transporter 1 (GLUT-1) expressed in the MVM. Transport to the fetal circulation across the BM, which is traditionally considered the rate-limiting step in maternal-fetal glucose transfer, is also accomplished by GLUT-1. While glucose moves across the placenta by facilitated diffusion and higher postprandial maternal glucose levels in maternal obesity result in increased fetal glucose to support accelerated fetal growth, some evidence suggests that placental glucose transport capacity, as reflected by an increased expression of glucose transporters in the placental barrier, is increased in maternal obesity. For example, BM GLUT-1 expression correlated with birth weight in obese mothers without diabetes (191), suggesting the capacity of the placenta to transfer glucose modulates fetal growth in these pregnancies. Moreover, GLUT1 expression was increased in the BM in primary human trophoblast cells isolated from pregnancies complicated with maternal obesity (191,192). This increased placental glucose transport capacity when exposed to maternal obesity may contribute to increased glucose delivery to the fetus and promote fetal overgrowth even if the mother is euglycemic.
Amino Acids
Placental amino acid transporter systems A and L have been studied extensively. The System A transporter, predominantly expressed in the MVM, mediates the uptake of non-essential neutral amino acids from maternal blood in the intervillous space into the cytosol of the syncytiotrophoblast, energized by the inwardly directly sodium gradient (20,193,194). As a result, the syncytium has high intracellular concentrations of amino acids. System L exchanges essential amino acids such as leucine in the maternal blood for non-essential amino acids, which are accumulated in the cytoplasm by System A. In this way the two transporters work in concert to increase the intracellular concentrations of both essential and non-essential amino acids, which then diffuse across the BM to the fetal circulation. System A activity but not System L was positively correlated with birth weight in a cohort of normal and obese women (144). Unlike system A amino acid transport, placental transport of taurine, a β-amino acid, was lower in obese compared to normal BMI women (195). The inverse relationship between taurine transporter and maternal BMI, suggests obesity impacts placenta taurine consumption and ultimately fetal efflux (195). Placental mTOR signaling is activated in obese women giving birth to LGA babies (144), likely due to a combination of multiple factors, including elevated maternal insulin and leptin as well as low adiponectin levels and elevated levels of nutrients. Activation of mTORC1 and mTORC2 independently activate trophoblast System A and System L amino acid transporter activity by modulation of plasma membrane trafficking of two key transporter isoforms, SNAT2 and LAT1 (185,196). Whereas mTORC1 modulates SNAT2 and LAT1 trafficking by Nedd4-2-regulated ubiquitination (197), mTORC2 regulates amino acid transporters trafficking mediated by Cdc42 and Rac1 and effects on the actin skeleton ((196); Figure 2)). Therefore, as with glucose, the increased capacity to transport amino acids may promote increased fetal growth in utero in some pregnancies complicated by maternal obesity.
Lipids
Maternal circulating TGs and NEFAs are elevated in pregnancy, providing the necessary fatty acids for transport to the fetus. Placental uptake of NEFA occurs primarily via several isoforms of fatty acid transport proteins, FAT/CD36 and specific fatty acid binding proteins localized in the MVM (198). Maternal obesity may have distinct effects on the expression of different fatty acid transporters in the placenta. High BMI women had decreased mRNA expression of FATP1 and FATP4 but increased protein expression of FATP6 and FAT/CD36 in placenta compared to normal BMI women (199). Lager and colleagues, using isolated syncytiotrophoblast plasma membranes, reported higher FATP2 and FATP4 in BM compared to MVM and FATP2 protein abundance in the BM correlated to maternal BMI, suggesting an increase capacity to transfer NEFAs to the fetus (200). In addition, maternal obesity is associated with increased placental lipid accumulation (117) but others have reported lower saturated fatty acid content (199) and impaired ability of the placenta to deliver long chain polyunsaturated fatty acid (LCPUFA) to the fetus (124). There is limited knowledge on what factors regulate expression of placental fatty acid transporters, re-esterification pathways and β-oxidation in the placenta of obese mothers. Szabo and co-workers have proposed that lipid transfer to the fetus is increased in pregnancies complicated by maternal obesity, resulting in greater fetal adipogenesis (201). However, the effect of maternal obesity on placental lipid transport and storage remains to be fully established and represents a high priority area for future research.
Effect of Clinical Interventions on Placental Function in Maternal Obesity
Exercise and lifestyle interventions in prospective trials have been explored in maternal obesity as a way to decrease adverse maternal (such as gestational weight gain, hypertensive disorders, GDM and dysfunctional labor) and infant outcomes (including fetal overgrowth). Although these interventions have been shown to reduce maternal weight gain in pregnancy, no effect on birth weight or incidence of fetal overgrowth have been demonstrated (202,203). Moreover, exercise and lifestyle interventions may have a positive effects on infant body composition and maternal health (9,204). The impact of exercise and lifestyle interventions on placental function in obese women remain largely unknown, although effects on placental development and function have been proposed to underlie ‘paradoxical’ findings of increased fetal weights following an exercise intervention (205).
Omega 3 polyunsaturated fatty acids (n-3 LCPUFAs) have been proposed to be a safe anti-inflammatory mediator to improve outcomes in maternal obesity (206). A recent meta-analysis indicated lower risk for LGA with n-3 LCPUFAs supplementation (207). Moreover, Lager and coworkers demonstrated improved placental function following 800 mg per day DHA supplementation in the second half of pregnancy in obese women. They found that higher levels of placental DHA were associated with decreased amino acid transporter expression and reduced inflammatory markers but also an increase in placental fatty acid transporter expression (208). A follow-up study of a small number of infants from the trial indicated reduced adiposity in the children of supplemented mothers at 2 and 4 years of age, an effect that was not due to differences in duration or exclusivity of breast feeding (209).
Given the relatively limited success of traditional dietary and lifestyle interventions in alleviating adverse pregnancy outcomes in obese women, there is a significant interest in exploring more targeted intervention strategies. Anti-inflammatory agents such as resveratrol have been explored in placental explants treated with LPS or poly(I:C). Resveratrol reduced the mRNA expression of TNF-α, IL-1β, IL-6, IL8 as well as culture media concentrations of IL-6, IL-8, and MCP-1 (210), increased AMPK phosphorylation and decreased uptake of glucose as well as DHA and arachidonic acid (AA) but decreased β-oxidation and did not affect rates of FA esterification (211). However, a recent study suggests caution because administration of resveratrol in pregnant nonhuman primates was associated with negative effects on the fetus (212). In a recent study in isolated primary trophoblasts from normal BMI and obese women, melatonin effectively reduced expression of some antioxidants and increased total respiratory capacity of trophoblasts from obese women but not from normal BMI mothers (213).
Integrated Model
Maternal obesity increases the risk of fetal overgrowth, which is associated with poor maternal outcomes including emergency Caesarean section, obstetrical trauma, postpartum hemorrhage and diabetes as well as risks for the infants, such as shoulder dystocia, brachial plexus injury, skeletal injuries, meconium aspiration, perinatal asphyxia, hypoglycemia, and fetal death (31,214). Infants of obese mothers also tend to have increased adiposity (215,216) and/or insulin resistance at birth (61). A series of recent alarming reports link fetal exposure to the adverse metabolic environment of the obese mother with later development of the metabolic syndrome and cardiovascular disease, in particular if the infants were large at birth (12–18,47,217–228). Collectively, these studies suggest that infants of obese mothers that are large and/or have increased adiposity at birth are particularly susceptible to poor short- and long-term outcomes. Therefore, preventing fetal overgrowth and/or increased fat mass in infants of obese mothers is an important objective in the development of novel intervention strategies.
Available literature strongly suggests that the mechanistic link between maternal obesity, fetal overgrowth/increased infant adiposity and programming of adult disease involves specific changes in the placenta (Figure 3). High insulin, leptin, IGF-1 and nutrient levels and low adiponectin in the maternal circulation are key examples of factors that converge to activate placental mTOR signalling, a positive regulator of an array of key placental functions, including amino acid transport (185,186), folate transport (187) and mitochondrial biogenesis (167). These changes are proposed to promote nutrient delivery to the fetus, increased fetal growth and/or adiposity, which are strongly linked to the development of metabolic and cardiovascular disease in childhood and adult life (Figure 3). We recently reported that normalization of maternal circulating adiponectin in a mouse model of obesity prevented the activation of placental mTOR signaling and nutrient transport, fetal overgrowth and programming of metabolic and cardiovascular disease in the offspring (58,229,230), supporting the concept that low maternal adiponectin, activation of placental mTOR signaling and nutrient transport are not only important in accelerating fetal growth but may also be critical for the programming of adult disease in offspring of obese mothers. Given that the majority of obese women give birth to normal sized infants (11,219,231,232), studies of these pregnancies are essential. Changes in placental function in these pregnancies include activation of inflammatory signalling pathways (10,68,112,143), signs of oxidative stress (171,174,178), decreased oxidative phosphorylation (145,156,168,169), and possibly lipid accumulation (49,106,117,125,199,217), however whether these alterations are linked to poor short and long-term infant outcomes remains to be fully established.
Figure 3. Proposed model of mechanistic links between maternal obesity, fetal overgrowth/increased infant adiposity and fetal programming of adult disease.
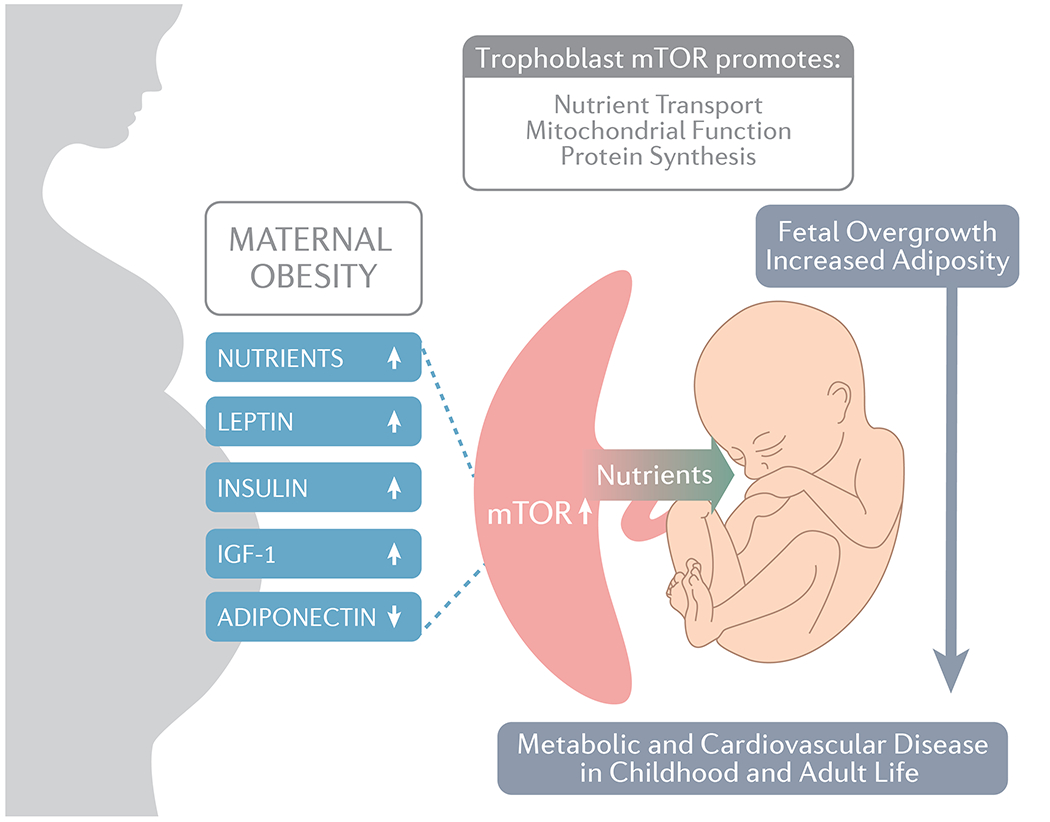
We propose that the mechanistic link between maternal obesity, fetal overgrowth/increased infant adiposity and programming of adult disease involves specific changes in the placenta from increased mTOR signaling. High insulin, leptin, IGF-1 and nutrient levels and low adiponectin in the maternal circulation are key examples of factors that converge to activate placental mTOR signalling, a positive regulator of an array of key placental functions, including amino acid transport and mitochondrial biogenesis. Some of the proposed involved signaling mechanisms are depicted in more detail in Figure 2. These changes are proposed to promote the delivery of nutrients (which may include glucose, amino acids and lipids) to the fetus, increased fetal growth and/or adiposity, which are strongly linked to the development of metabolic and cardiovascular disease in childhood and adult life. IGF-1, insulin-like growth factor 1; mTOR, mechanistic target of rapamycin.
Conclusion and Future perspectives
Given the rapidly increased prevalence of maternal obesity in pregnancy worldwide, the poor long- and short-term outcomes in infants of obese mothers represent a major public health problem in the 21st century. A recent meta-analysis shows that dietary and lifestyle interventions in overweight and obese women have only limited beneficial effects on birth weight and other key obstetrical outcomes (233). Furthermore, lifestyle changes and anti-obesity drugs in children and adults remain largely unsuccessful, highlighting the urgent need to better understand the molecular underpinnings linking maternal obesity to poor short- and long-term outcomes in order to allow for the development of specific interventions during pregnancy to prevent metabolic programming. Although an individuals’ life-long health trajectory may be determined by the first 1000 days of life (i.e., from conception up to two years of postnatal life) (234), fetal life appears to be of particular importance for the programming effect of maternal obesity. Thus, in utero intervention is an attractive and likely efficient approach to prevent childhood obesity and metabolic syndrome in the next generation.
A large body of epidemiological data suggests that altered placental structure and function increases the risk of developing diseases such as obesity, diabetes, cardiovascular disease, and cancer in adult life (235–242). Emerging evidence in mice demonstrates that the placenta directly influences fetal brain development and that changes in placental function mediate the link between maternal obstetrical complications and adverse neurodevelopmental outcomes (243–247). Thus, the placenta determines life-long metabolic and mental health and understanding the functions of the placenta may hold the key to unravelling the molecular pathways underpinning developmental programming in response to maternal obesity. Well-designed mechanistic experiments in relevant animal models guided by observational data obtained in pregnant women will be instrumental in this area. Of particular interest is to determine the molecular pathways causing placental oxidative stress, altered mitochondrial function and lipid handling and low-grade inflammation in maternal obesity and how these placental changes specifically leads to poor short- and long-term outcomes. This will provide a critical foundation for developing interventions that specifically target placental function in women who enter pregnancy obese. We speculate that activation of placental adiponectin receptors by maternal adiponectin supplementation, interventions enhancing endogenous adiponectin secretion or administration of adiponectin receptor agonists represent a promising future clinical intervention in pregnancies complicated by obesity in which maternal adiponectin levels are low.
Acknowledgements
We thank KIMEN Design4Research (kimendesign4research.com) for the graphic design of figures.
Funding
Supported by grants from NIH (HD089980, HD093950 HD065007, HD068370, HD078376, and T32HD007186).
Footnotes
Competing Interests
The authors declare that there are no competing interests associated with the manuscript.
Abbreviations: Gene abbreviations adhere to the HUGO Gene Nomenclature Committee recommendations
References
- 1.Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011-2012. JAMA. 2014. Feb 26;311(8):806–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of obesity in the United States, 2009–2010. NCHS Data Brief. 2012. Jan;(82):1–8. [PubMed] [Google Scholar]
- 3.Fisher SC, Kim SY, Sharma AJ, Rochat R, Morrow B. Is obesity still increasing among pregnant women? Prepregnancy obesity trends in 20 states, 2003–2009. Prev Med. 2013. Jun;56(6):372–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Catalano P, Ehrenberg H. Review article: The short- and long-term implications of maternal obesity on the mother and her offspring. BJOG Int J Obstet Gynaecol. 2006. Jul 7;113(10):1126–33. [DOI] [PubMed] [Google Scholar]
- 5.Hemond J, Robbins RB, Young PC. The Effects of Maternal Obesity on Neonates, Infants, Children, Adolescents, and Adults. Clin Obstet Gynecol. 2016. Mar;59(1):216–27. [DOI] [PubMed] [Google Scholar]
- 6.Fuchs F, Senat M-V, Rey E, Balayla J, Chaillet N, Bouyer J, et al. Impact of maternal obesity on the incidence of pregnancy complications in France and Canada. Sci Rep. 2017. July;7(1):10859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yogev Y, Langer O, Xenakis EMJ, Rosenn B. The association between glucose challenge test, obesity and pregnancy outcome in 6390 non-diabetic women. J Matern-Fetal Neonatal Med Off J Eur Assoc Perinat Med Fed Asia Ocean Perinat Soc Int Soc Perinat Obstet. 2005. Jan;17(1):29–34. [DOI] [PubMed] [Google Scholar]
- 8.Mission JF, Marshall NE, Caughey AB. Obesity in pregnancy: a big problem and getting bigger. Obstet Gynecol Surv. 2013. May;68(5):389–99. [DOI] [PubMed] [Google Scholar]
- 9.Briley AL, Barr S, Badger S, Bell R, Croker H, Godfrey KM, et al. A complex intervention to improve pregnancy outcome in obese women; the UPBEAT randomised controlled trial. BMC Pregnancy Childbirth. 2014. Feb 18;14:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Challier JC, Basu S, Bintein T, Minium J, Hotmire K, Catalano PM, et al. Obesity in pregnancy stimulates macrophage accumulation and inflammation in the placenta. Placenta. 2008. Mar;29(3):274–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ehrenberg HM, Durnwald CP, Catalano P, Mercer BM. The influence of obesity and diabetes on the risk of cesarean delivery. Am J Obstet Gynecol. 2004. Sep;191(3):969–74. [DOI] [PubMed] [Google Scholar]
- 12.Hochner H, Friedlander Y, Calderon-Margalit R, Meiner V, Sagy Y, Avgil-Tsadok M, et al. Associations of maternal prepregnancy body mass index and gestational weight gain with adult offspring cardiometabolic risk factors: the Jerusalem Perinatal Family Follow-up Study. Circulation. 2012. Mar 20;125(11):1381–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parsons TJ, Power C, Manor O. Fetal and early life growth and body mass index from birth to early adulthood in 1958 British cohort: longitudinal study. BMJ. 2001. Dec 8;323(7325):1331–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boney CM, Verma A, Tucker R, Vohr BR. Metabolic syndrome in childhood: association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics. 2005. Mar;115(3):e290–296. [DOI] [PubMed] [Google Scholar]
- 15.Whitaker RC. Predicting preschooler obesity at birth: the role of maternal obesity in early pregnancy. Pediatrics. 2004. Jul;114(1):e29–36. [DOI] [PubMed] [Google Scholar]
- 16.Reynolds RM, Allan KM, Raja EA, Bhattacharya S, McNeill G, Hannaford PC, et al. Maternal obesity during pregnancy and premature mortality from cardiovascular event in adult offspring: follow-up of 1 323 275 person years. BMJ. 2013. Aug 13;347:f4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cnattingius S, Villamor E, Lagerros YT, Wikström A-K, Granath F. High birth weight and obesity--a vicious circle across generations. Int J Obes 2005. 2012. Oct;36(10):1320–4. [DOI] [PubMed] [Google Scholar]
- 18.Lawlor DA, Smith GD, O’Callaghan M, Alati R, Mamun AA, Williams GM, et al. Epidemiologic evidence for the fetal overnutrition hypothesis: findings from the mater-university study of pregnancy and its outcomes. Am J Epidemiol. 2007. Feb 15;165(4):418–24. [DOI] [PubMed] [Google Scholar]
- 19.Catalano PM. Obesity and pregnancy--the propagation of a viscous cycle? J Clin Endocrinol Metab. 2003. Aug;88(8):3505–6. [DOI] [PubMed] [Google Scholar]
- 20.Vaughan OR, Rosario FJ, Powell TL, Jansson T. Regulation of Placental Amino Acid Transport and Fetal Growth. Prog Mol Biol Transl Sci. 2017;145:217–51. [DOI] [PubMed] [Google Scholar]
- 21.Jansson T, Powell TL. Role of placental nutrient sensing in developmental programming. Clin Obstet Gynecol. 2013. Sep;56(3):591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eschwege E, Basdevant A, Crine A, Moisan C, Charles M-A. Type 2 diabetes mellitus in France in 2012: results from the ObEpi survey. Diabetes Metab. 2015. Feb;41(1):55–61. [DOI] [PubMed] [Google Scholar]
- 23.Hales CM, Fryar CD, Carroll MD, Freedman DS, Ogden CL. Trends in Obesity and Severe Obesity Prevalence in US Youth and Adults by Sex and Age, 2007-2008 to 2015-2016. JAMA. 2018. Apr 24;319(16):1723–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Conolly Anne, Davies Byron, NatCen Social Research. Health Survey for England 2017 Adult and child overweight and obesity [Internet]. National Health Service; 2017. Dec. Available from: https://files.digital.nhs.uk/3F/6971DC/HSE17-Adult-Child-BMI-rep.pdf [Google Scholar]
- 25.Ford ES, Maynard LM, Li C. Trends in mean waist circumference and abdominal obesity among US adults, 1999–2012. JAMA. 2014. Sep 17;312(11):1151–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Daigre J-L, Atallah A, Boissin J-L, Jean-Baptiste G, Kangambega P, Chevalier H, et al. The prevalence of overweight and obesity, and distribution of waist circumference, in adults and children in the French Overseas Territories: the PODIUM survey. Diabetes Metab. 2012. Nov;38(5):404–11. [DOI] [PubMed] [Google Scholar]
- 27.Ramsay JE, Ferrell WR, Crawford L, Wallace AM, Greer IA, Sattar N. Maternal obesity is associated with dysregulation of metabolic, vascular, and inflammatory pathways. J Clin Endocrinol Metab. 2002. Sep;87(9):4231–7. [DOI] [PubMed] [Google Scholar]
- 28.Ornaghi S, Mueller M, Barnea ER, Paidas MJ. Thrombosis during pregnancy: Risks, prevention, and treatment for mother and fetus—harvesting the power of omic technology, biomarkers and in vitro or in vivo models to facilitate the treatment of thrombosis. Birth Defects Res Part C Embryo Today Rev. 2015;105(3):209–25. [DOI] [PubMed] [Google Scholar]
- 29.Flenady V, Koopmans L, Middleton P, Frøen JF, Smith GC, Gibbons K, et al. Major risk factors for stillbirth in high-income countries: a systematic review and meta-analysis. Lancet Lond Engl. 2011. Apr 16;377(9774):1331–40. [DOI] [PubMed] [Google Scholar]
- 30.Turner D, Monthé-Drèze C, Cherkerzian S, Gregory K, Sen S. Maternal obesity and cesarean section delivery: additional risk factors for neonatal hypoglycemia? J Perinatol Off J Calif Perinat Assoc. 2019. Aug;39(8):1057–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jolly MC, Sebire NJ, Harris JP, Regan L, Robinson S. Risk factors for macrosomia and its clinical consequences: a study of 350,311 pregnancies. Eur J Obstet Gynecol Reprod Biol. 2003. Nov;111(1):9–14. [DOI] [PubMed] [Google Scholar]
- 32.Nesbitt TS, Gilbert WM, Herrchen B. Shoulder dystocia and associated risk factors with macrosomic infants born in California. Am J Obstet Gynecol. 1998. Aug;179(2):476–80. [DOI] [PubMed] [Google Scholar]
- 33.Stanley CA, Rozance PJ, Thornton PS, De Leon DD, Harris D, Haymond MW, et al. Re-evaluating “transitional neonatal hypoglycemia”: mechanism and implications for management. J Pediatr. 2015. Jun;166(6):1520–1525.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brunner S, Schmid D, Hüttinger K, Much D, Heimberg E, Sedlmeier E-M, et al. Maternal insulin resistance, triglycerides and cord blood insulin in relation to post-natal weight trajectories and body composition in the offspring up to 2 years. Diabet Med J Br Diabet Assoc. 2013. Dec;30(12):1500–7. [DOI] [PubMed] [Google Scholar]
- 35.Regnault N, Botton J, Heude B, Forhan A, Hankard R, Foliguet B, et al. Higher Cord C-Peptide Concentrations Are Associated With Slower Growth Rate in the 1st Year of Life in Girls but Not in Boys. Diabetes. 2011. Aug;60(8):2152–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stang J, Huffman LG. Position of the Academy of Nutrition and Dietetics: Obesity, Reproduction, and Pregnancy Outcomes. J Acad Nutr Diet. 2016. Apr 1;116(4):677–91. [DOI] [PubMed] [Google Scholar]
- 37.Forno E, Young OM, Kumar R, Simhan H, Celedón JC. Maternal obesity in pregnancy, gestational weight gain, and risk of childhood asthma. Pediatrics. 2014. Aug;134(2):e535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gaillard R, Santos S, Duijts L, Felix JF. Childhood Health Consequences of Maternal Obesity during Pregnancy: A Narrative Review. Ann Nutr Metab. 2016;69(3–4):171–80. [DOI] [PubMed] [Google Scholar]
- 39.Basatemur E, Gardiner J, Williams C, Melhuish E, Barnes J, Sutcliffe A. Maternal prepregnancy BMI and child cognition: a longitudinal cohort study. Pediatrics. 2013. Jan;131(1):56–63. [DOI] [PubMed] [Google Scholar]
- 40.Nguyen MU, Wallace MJ, Pepe S, Menheniott TR, Moss TJ, Burgner D. Perinatal inflammation: a common factor in the early origins of cardiovascular disease? Clin Sci. 2015. Oct 1;129(8):769–84. [DOI] [PubMed] [Google Scholar]
- 41.Rizzo GS, Sen S. Maternal obesity and immune dysregulation in mother and infant: A review of the evidence. Paediatr Respir Rev. 2015. Sep;16(4):251–7. [DOI] [PubMed] [Google Scholar]
- 42.Armitage JA, Poston L, Taylor PD. Developmental origins of obesity and the metabolic syndrome: the role of maternal obesity. Front Horm Res. 2008;36:73–84. [DOI] [PubMed] [Google Scholar]
- 43.Godfrey KM, Reynolds RM, Prescott SL, Nyirenda M, Jaddoe VWV, Eriksson JG, et al. Influence of maternal obesity on the long-term health of offspring. Lancet Diabetes Endocrinol. 2017. Jan;5(1):53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barbour LA. Metabolic Culprits in Obese Pregnancies and Gestational Diabetes Mellitus: Big Babies, Big Twists, Big Picture: The 2018 Norbert Freinkel Award Lecture. Diabetes Care. 2019. May;42(5):718–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barbour LA, Farabi SS, Friedman JE, Hirsch NM, Reece MS, Van Pelt RE, et al. Postprandial Triglycerides Predict Newborn Fat More Strongly than Glucose in Women with Obesity in Early Pregnancy. Obes Silver Spring Md. 2018;26(8):1347–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.HAPO Study Cooperative Research Group, Metzger BE, Lowe LP, Dyer AR, Trimble ER, Chaovarindr U, et al. Hyperglycemia and adverse pregnancy outcomes. N Engl J Med. 2008. May 8;358(19):1991–2002. [DOI] [PubMed] [Google Scholar]
- 47.Catalano PM, Farrell K, Thomas A, Huston-Presley L, Mencin P, de Mouzon SH, et al. Perinatal risk factors for childhood obesity and metabolic dysregulation123. Am J Clin Nutr. 2009. Nov;90(5):1303–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hellmuth C, Lindsay KL, Uhl O, Buss C, Wadhwa PD, Koletzko B, et al. Association of maternal prepregnancy BMI with metabolomic profile across gestation. Int J Obes 2005. 2017;41(1):159–69. [DOI] [PubMed] [Google Scholar]
- 49.Dubé E, Gravel A, Martin C, Desparois G, Moussa I, Ethier-Chiasson M, et al. Modulation of fatty acid transport and metabolism by maternal obesity in the human full-term placenta. Biol Reprod. 2012. Jul;87(1):14, 1–11. [DOI] [PubMed] [Google Scholar]
- 50.Vahratian A, Misra VK, Trudeau S, Misra DP. Prepregnancy body mass index and gestational age-dependent changes in lipid levels during pregnancy. Obstet Gynecol. 2010. Jul;116(1):107–13. [DOI] [PubMed] [Google Scholar]
- 51.Geraghty AA, Alberdi G, O’Sullivan EJ, O’Brien EC, Crosbie B, Twomey PJ, et al. Maternal and fetal blood lipid concentrations during pregnancy differ by maternal body mass index: findings from the ROLO study. BMC Pregnancy Childbirth. 2017. Oct 16;17(1):360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tessier DR, Ferraro ZM, Gruslin A. Role of leptin in pregnancy: consequences of maternal obesity. Placenta. 2013. Mar;34(3):205–11. [DOI] [PubMed] [Google Scholar]
- 53.Haghiac M, Basu S, Presley L, Serre D, Catalano PM, Hauguel-de Mouzon S. Patterns of adiponectin expression in term pregnancy: impact of obesity. J Clin Endocrinol Metab. 2014. Sep;99(9):3427–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jansson N, Nilsfelt A, Gellerstedt M, Wennergren M, Rossander-Hultheén L, Powell TL, et al. Maternal hormones linking maternal body mass index and dietary intake to birth weight. Am J Clin Nutr. 2008. Jun 1;87(6):1743–9. [DOI] [PubMed] [Google Scholar]
- 55.Hendler I, Blackwell SC, Mehta SH, Whitty JE, Russell E, Sorokin Y, et al. The levels of leptin, adiponectin, and resistin in normal weight, overweight, and obese pregnant women with and without preeclampsia. Am J Obstet Gynecol. 2005. Sep;193(3 Pt 2):979–83. [DOI] [PubMed] [Google Scholar]
- 56.Nien JK, Mazaki-Tovi S, Romero R, Erez O, Kusanovic JP, Gotsch F, et al. Plasma adiponectin concentrations in non-pregnant, normal and overweight pregnant women. J Perinat Med. 2007;35(6):522–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vernini JM, Moreli JB, Costa RAA, Negrato CA, Rudge MVC, Calderon IMP. Maternal adipokines and insulin as biomarkers of pregnancies complicated by overweight and obesity. Diabetol Metab Syndr. 2016;8(1):68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aye ILMH, Rosario FJ, Powell TL, Jansson T Adiponectin supplementation in pregnant mice prevents the adverse effects of maternal obesity on placental function and fetal growth. Proc Natl Acad Sci U S A. 2015. Oct 13;112(41):12858–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ahlsson F, Diderholm B, Ewald U, Jonsson B, Forslund A, Stridsberg M, et al. Adipokines and their relation to maternal energy substrate production, insulin resistance and fetal size. Eur J Obstet Gynecol Reprod Biol. 2013. May;168(1):26–9. [DOI] [PubMed] [Google Scholar]
- 60.Triantafyllou GA, Paschou SA, Mantzoros CS. Leptin and Hormones: Energy Homeostasis. Endocrinol Metab Clin. 2016. Sep 1;45(3):633–45. [DOI] [PubMed] [Google Scholar]
- 61.Catalano PM, Presley L, Minium J, Hauguel-de Mouzon S. Fetuses of obese mothers develop insulin resistance in utero. Diabetes Care. 2009. Jun;32(6):1076–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sferruzzi-Perri AN, Sandovici I, Constancia M, Fowden AL. Placental phenotype and the insulin-like growth factors: resource allocation to fetal growth. J Physiol. 2017. Aug 1;595(15):5057–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chard T Insulin-like growth factors and their binding proteins in normal and abnormal human fetal growth. Growth Regul. 1994. Sep;4(3):91–100. [PubMed] [Google Scholar]
- 64.Åsvold BO, Eskild A, Jenum PA, Vatten LJ. Maternal concentrations of insulin-like growth factor I and insulin-like growth factor binding protein 1 during pregnancy and birth weight of offspring. Am J Epidemiol. 2011. Jul 15;174(2):129–35. [DOI] [PubMed] [Google Scholar]
- 65.Lappas M Insulin-like growth factor-binding protein 1 and 7 concentrations are lower in obese pregnant women, women with gestational diabetes and their fetuses. J Perinatol Off J Calif Perinat Assoc. 2015. Jan;35(1):32–8. [DOI] [PubMed] [Google Scholar]
- 66.Patel N, Hellmuth C, Uhl O, Godfrey K, Briley A, Welsh P, et al. Cord Metabolic Profiles in Obese Pregnant Women: Insights Into Offspring Growth and Body Composition. J Clin Endocrinol Metab. 2018. January;103(1):346–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kirwan JP, Hauguel-De Mouzon S, Lepercq J, Challier J-C, Huston-Presley L, Friedman JE, et al. TNF-alpha is a predictor of insulin resistance in human pregnancy. Diabetes. 2002. Jul;51(7):2207–13. [DOI] [PubMed] [Google Scholar]
- 68.Aye ILMH, Lager S, Ramirez VI, Gaccioli F, Dudley DJ, Jansson T, et al. Increasing maternal body mass index is associated with systemic inflammation in the mother and the activation of distinct placental inflammatory pathways. Biol Reprod. 2014. Jun;90(6):129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pendeloski KPT, Ono E, Torloni MR, Mattar R, Daher S. Maternal obesity and inflammatory mediators: A controversial association. Am J Reprod Immunol N Y N 1989. 2017;77(5). [DOI] [PubMed] [Google Scholar]
- 70.Azizian M, Mahdipour E, Mirhafez SR, Shoeibi S, Nematy M, Esmaily H, et al. Cytokine profiles in overweight and obese subjects and normal weight individuals matched for age and gender. Ann Clin Biochem. 2016. Nov;53(6):663–8. [DOI] [PubMed] [Google Scholar]
- 71.Fang J, Furesz TC, Lurent RS, Smith CH, Fant ME. Spatial polarization of insulin-like growth factor receptors on the human syncytiotrophoblast. Pediatr Res. 1997. Feb;41(2):258–65. [DOI] [PubMed] [Google Scholar]
- 72.Burton GJ. The fine structure of the human placental villus as revealed by scanning electron microscopy. Scanning Microsc. 1987. Dec;1(4):1811–28. [PubMed] [Google Scholar]
- 73.Burton GJ, Fowden AL. The placenta: a multifaceted, transient organ. Philos Trans R Soc Lond B Biol Sci. 2015. Mar 5;370(1663):20140066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Burton GJ, Woods AW, Jauniaux E, Kingdom JCP. Rheological and Physiological Consequences of Conversion of the Maternal Spiral Arteries for Uteroplacental Blood Flow during Human Pregnancy. Placenta. 2009. Jun 1;30(6):473–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pollheimer J, Vondra S, Baltayeva J, Beristain AG, Knöfler M. Regulation of Placental Extravillous Trophoblasts by the Maternal Uterine Environment. Front Immunol. 2018;9:2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.James-Allan LB, Arbet J, Teal SB, Powell TL, Jansson T. Insulin stimulates GLUT4 trafficking to the syncytiotrophoblast basal plasma membrane in the human placenta. J Clin Endocrinol Metab. 2019. May 21; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ericsson A, Hamark B, Jansson N, Johansson BR, Powell TL, Jansson T. Hormonal regulation of glucose and system A amino acid transport in first trimester placental villous fragments. Am J Physiol Regul Integr Comp Physiol. 2005. Mar;288(3):R656–662. [DOI] [PubMed] [Google Scholar]
- 78.Jansson N, Greenwood SL, Johansson BR, Powell TL, Jansson T. Leptin stimulates the activity of the system A amino acid transporter in human placental villous fragments. J Clin Endocrinol Metab. 2003. Mar;88(3):1205–11. [DOI] [PubMed] [Google Scholar]
- 79.Roos S, Kanai Y, Prasad PD, Powell TL, Jansson T. Regulation of placental amino acid transporter activity by mammalian target of rapamycin. Am J Physiol Cell Physiol. 2009. Jan;296(1):C142–150. [DOI] [PubMed] [Google Scholar]
- 80.Karl PI, Alpy KL, Fisher SE. Amino acid transport by the cultured human placental trophoblast: effect of insulin on AIB transport. Am J Physiol. 1992. Apr;262(4 Pt 1):C834–839. [DOI] [PubMed] [Google Scholar]
- 81.Castillo-Castrejon M, Jansson T, Powell TL. No evidence of attenuation of placental insulin-stimulated Akt phosphorylation and amino acid transport in maternal obesity and gestational diabetes mellitus. Am J Physiol Endocrinol Metab. 2019. Dec 1;317(6):E1037–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.White V, González E, Capobianco E, Pustovrh C, Martínez N, Higa R, et al. Leptin modulates nitric oxide production and lipid metabolism in human placenta. Reprod Fertil Dev. 2006;18(4):425–32. [DOI] [PubMed] [Google Scholar]
- 83.Cameo P, Bischof P, Calvo JC. Effect of leptin on progesterone, human chorionic gonadotropin, and interleukin-6 secretion by human term trophoblast cells in culture. Biol Reprod. 2003. Feb;68(2):472–7. [DOI] [PubMed] [Google Scholar]
- 84.Jones HN, Jansson T, Powell TL. Full-length adiponectin attenuates insulin signaling and inhibits insulin-stimulated amino Acid transport in human primary trophoblast cells. Diabetes. 2010. May;59(5):1161–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Aye ILMH, Gao X, Weintraub ST, Jansson T, Powell TL. Adiponectin inhibits insulin function in primary trophoblasts by PPARα-mediated ceramide synthesis. Mol Endocrinol Baltim Md. 2014. Apr;28(4):512–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Duval F, Santos ED, Poidatz D, Sérazin V, Gronier H, Vialard F, et al. Adiponectin Inhibits Nutrient Transporters and Promotes Apoptosis in Human Villous Cytotrophoblasts: Involvement in the Control of Fetal Growth. Biol Reprod. 2016;94(5):111. [DOI] [PubMed] [Google Scholar]
- 87.Rosario FJ, Schumacher MA, Jiang J, Kanai Y, Powell TL, Jansson T. Chronic maternal infusion of full-length adiponectin in pregnant mice down-regulates placental amino acid transporter activity and expression and decreases fetal growth. J Physiol. 2012. Mar 15;590(6):1495–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rosario FJ, Kanai Y, Powell TL, Jansson T. Increased placental nutrient transport in a novel mouse model of maternal obesity with fetal overgrowth. Obesity. 2015;23(8):1663–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Qiao L, Wattez J-S, Lee S, Guo Z, Schaack J, Hay WW, et al. Knockout maternal adiponectin increases fetal growth in mice: potential role for trophoblast IGFBP-1. Diabetologia. 2016;59(11):2417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Qiao L, Wattez J-S, Lee S, Nguyen A, Schaack J, Hay WW, et al. Adiponectin Deficiency Impairs Maternal Metabolic Adaptation to Pregnancy in Mice. Diabetes. 2017;66(5):1126–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Olausson H, Löf M, Brismar K, Forsum E, Sohlström A. Maternal serum concentrations of insulin-like growth factor (IGF)-I and IGF binding protein-1 before and during pregnancy in relation to maternal body weight and composition and infant birth weight. Br J Nutr. 2010. Sep;104(6):842–8. [DOI] [PubMed] [Google Scholar]
- 92.Bhaumick B, George D, Bala RM. Potentiation of epidermal growth factor-induced differentiation of cultured human placental cells by insulin-like growth factor-I. J Clin Endocrinol Metab. 1992. May;74(5):1005–11. [DOI] [PubMed] [Google Scholar]
- 93.Baumann MU, Schneider H, Malek A, Palta V, Surbek DV, Sager R, et al. Regulation of human trophoblast GLUT1 glucose transporter by insulin-like growth factor I (IGF-I). PloS One. 2014;9(8):e106037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kniss DA, Shubert PJ, Zimmerman PD, Landon MB, Gabbe SG. Insulinlike growth factors. Their regulation of glucose and amino acid transport in placental trophoblasts isolated from first-trimester chorionic villi. J Reprod Med. 1994. Apr;39(4):249–56. [PubMed] [Google Scholar]
- 95.Farah N, Hogan AE, O’Connor N, Kennelly MM, O’Shea D, Turner MJ. Correlation between maternal inflammatory markers and fetomaternal adiposity. Cytokine. 2012. Oct;60(1):96–9. [DOI] [PubMed] [Google Scholar]
- 96.Radaelli T, Uvena-Celebrezze J, Minium J, Huston-Presley L, Catalano P, Hauguel-de Mouzon S. Maternal interleukin-6: marker of fetal growth and adiposity. J Soc Gynecol Investig. 2006. Jan;13(1):53–7. [DOI] [PubMed] [Google Scholar]
- 97.Varastehpour A, Radaelli T, Minium J, Ortega H, Herrera E, Catalano P, et al. Activation of phospholipase A2 is associated with generation of placental lipid signals and fetal obesity. J Clin Endocrinol Metab. 2006. Jan;91(1):248–55. [DOI] [PubMed] [Google Scholar]
- 98.Lim R, Lappas M. Slit2 exerts anti-inflammatory actions in human placenta and is decreased with maternal obesity. Am J Reprod Immunol N Y N 1989. 2015. Jan;73(1):66–78. [DOI] [PubMed] [Google Scholar]
- 99.Jones HN, Jansson T, Powell TL. IL-6 stimulates system A amino acid transporter activity in trophoblast cells through STAT3 and increased expression of SNAT2. Am J Physiol Cell Physiol. 2009. Nov;297(5):C1228–1235. [DOI] [PubMed] [Google Scholar]
- 100.Aye ILMH, Jansson T, Powell TL. TNF-α stimulates System A amino acid transport in primary human trophoblast cells mediated by p38 MAPK signaling. Physiol Rep. 2015;3(10):e12594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dimasuay KG, Aitken EH, Rosario F, Njie M, Glazier J, Rogerson SJ, et al. Inhibition of placental mTOR signaling provides a link between placental malaria and reduced birthweight. BMC Med. 2017. Jan 3;15(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liong S, Lappas M. Lipopolysaccharide and double stranded viral RNA mediate insulin resistance and increase system a amino acid transport in human trophoblast cells in vitro. Placenta. 2017;51:18–27. [DOI] [PubMed] [Google Scholar]
- 103.Federici M, Porzio O, Zucaro L, Fusco A, Borboni P, Lauro D, et al. Distribution of insulin/insulin-like growth factor-I hybrid receptors in human tissues. Mol Cell Endocrinol. 1997. May 16;129(2):121–6. [DOI] [PubMed] [Google Scholar]
- 104.Hashimoto R, Sakai K, Matsumoto H, Iwashita M. Tumor necrosis factor-alpha (TNF-alpha) inhibits insulin-like growth factor-I (IGF-I) activities in human trophoblast cell cultures through IGF-I/insulin hybrid receptors. Endocr J. 2010;57(3):193–200. [DOI] [PubMed] [Google Scholar]
- 105.Lager S, Jansson T, Powell TL. Differential regulation of placental amino acid transport by saturated and unsaturated fatty acids. Am J Physiol Cell Physiol. 2014. Oct 15;307(8):C738–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hirschmugl B, Desoye G, Catalano P, Klymiuk I, Scharnagl H, Payr S, et al. Maternal obesity modulates intracellular lipid turnover in the human term placenta. Int J Obes 2005. 2017;41(2):317–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bildirici I, Schaiff WT, Chen B, Morizane M, Oh S-Y, O’Brien M, et al. PLIN2 Is Essential for Trophoblastic Lipid Droplet Accumulation and Cell Survival During Hypoxia. Endocrinology. 2018. January;159(12):3937–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Pavan L, Tsatsaris V, Hermouet A, Therond P, Evain-Brion D, Fournier T. Oxidized low-density lipoproteins inhibit trophoblastic cell invasion. J Clin Endocrinol Metab. 2004. Apr;89(4):1969–72. [DOI] [PubMed] [Google Scholar]
- 109.Pavan L, Hermouet A, Tsatsaris V, Thérond P, Sawamura T, Evain-Brion D, et al. Lipids from oxidized low-density lipoprotein modulate human trophoblast invasion: involvement of nuclear liver X receptors. Endocrinology. 2004. Oct;145(10):4583–91. [DOI] [PubMed] [Google Scholar]
- 110.Colvin BN, Longtine MS, Chen B, Costa ML, Nelson DM. Oleate attenuates palmitate-induced endoplasmic reticulum stress and apoptosis in placental trophoblasts. Reprod Camb Engl. 2017;153(4):369–80. [DOI] [PubMed] [Google Scholar]
- 111.Yang X, Haghiac M, Glazebrook P, Minium J, Catalano PM, Hauguel-de Mouzon S. Saturated fatty acids enhance TLR4 immune pathways in human trophoblasts. Hum Reprod Oxf Engl. 2015. Sep;30(9):2152–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Saben J, Zhong Y, Gomez-Acevedo H, Thakali KM, Borengasser SJ, Andres A, et al. Early growth response protein-1 mediates lipotoxicity-associated placental inflammation: role in maternal obesity. Am J Physiol Endocrinol Metab. 2013. Jul 1;305(1):E1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Strakovsky RS, Pan Y-X. A decrease in DKK1, a WNT inhibitor, contributes to placental lipid accumulation in an obesity-prone rat model. Biol Reprod. 2012. Mar;86(3):81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liu L, Zhuang X, Jiang M, Guan F, Fu Q, Lin J. ANGPTL4 mediates the protective role of PPARγ activators in the pathogenesis of preeclampsia. Cell Death Dis. 2017. Sep;8(9):e3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Vaiman D, Calicchio R, Miralles F. Landscape of transcriptional deregulations in the preeclamptic placenta. PloS One. 2013;8(6):e65498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Inohara N, Koseki T, Chen S, Wu X, Núñez G. CIDE, a novel family of cell death activators with homology to the 45 kDa subunit of the DNA fragmentation factor. EMBO J. 1998. May 1;17(9):2526–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Calabuig-Navarro V, Haghiac M, Minium J, Glazebrook P, Ranasinghe GC, Hoppel C, et al. Effect of Maternal Obesity on Placental Lipid Metabolism. Endocrinology. 2017. January;158(8):2543–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sureshchandra S, Marshall NE, Wilson RM, Barr T, Rais M, Purnell JQ, et al. Inflammatory Determinants of Pregravid Obesity in Placenta and Peripheral Blood. Front Physiol. 2018;9:1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sapin V, Chaïb S, Blanchon L, Alexandre-Gouabau M-C, Lémery D, Charbonne F, et al. Esterification of Vitamin A by the Human Placenta Involves Villous Mesenchymal Fibroblasts. Pediatr Res. 2000. Oct;48(4):565–72. [DOI] [PubMed] [Google Scholar]
- 120.Martino J, Sebert S, Segura MT, García-Valdés L, Florido J, Padilla MC, et al. Maternal Body Weight and Gestational Diabetes Differentially Influence Placental and Pregnancy Outcomes. J Clin Endocrinol Metab. 2016. Jan;101(1):59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Altmäe S, Segura MT, Esteban FJ, Bartel S, Brandi P, Irmler M, et al. Maternal Pre-Pregnancy Obesity Is Associated with Altered Placental Transcriptome. PloS One. 2017;12(1):e0169223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Plösch T, Gellhaus A, van Straten EME, Wolf N, Huijkman NCA, Schmidt M, et al. The liver X receptor (LXR) and its target gene ABCA1 are regulated upon low oxygen in human trophoblast cells: a reason for alterations in preeclampsia? Placenta. 2010. Oct;31(10):910–8. [DOI] [PubMed] [Google Scholar]
- 123.Kallol S, Huang X, Müller S, Ontsouka CE, Albrecht C. Novel Insights into Concepts and Directionality of Maternal−Fetal Cholesterol Transfer across the Human Placenta. Int J Mol Sci. 2018. Aug 9;19(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Fattuoni C, Mandò C, Palmas F, Anelli GM, Novielli C, Parejo Laudicina E, et al. Preliminary metabolomics analysis of placenta in maternal obesity. Placenta. 2018;61:89–95. [DOI] [PubMed] [Google Scholar]
- 125.Saben J, Lindsey F, Zhong Y, Thakali K, Badger TM, Andres A, et al. Maternal obesity is associated with a lipotoxic placental environment. Placenta. 2014. Mar;35(3):171–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lassance L, Haghiac M, Leahy P, Basu S, Minium J, Zhou J, et al. Identification of early transcriptome signatures in placenta exposed to insulin and obesity. Am J Obstet Gynecol. 2015. May;212(5):647.e1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bari MF, Ngo S, Bastie CC, Sheppard AM, Vatish M. Gestational diabetic transcriptomic profiling of microdissected human trophoblast. J Endocrinol. 2016;229(1):47–59. [DOI] [PubMed] [Google Scholar]
- 128.Gamage TKJB, Schierding W, Hurley D, Tsai P, Ludgate JL, Bhoothpur C, et al. The role of DNA methylation in human trophoblast differentiation. Epigenetics. 2018;13(12):1154–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Schroeder DI, Blair JD, Lott P, Yu HOK, Hong D, Crary F, et al. The human placenta methylome. Proc Natl Acad Sci U S A. 2013. Apr 9;110(15):6037–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ou X-H, Zhu C-C, Sun S-C. Effects of obesity and diabetes on the epigenetic modification of mammalian gametes. J Cell Physiol. 2019. Jun;234(6):7847–55. [DOI] [PubMed] [Google Scholar]
- 131.Nomura Y, Lambertini L, Rialdi A, Lee M, Mystal EY, Grabie M, et al. Global methylation in the placenta and umbilical cord blood from pregnancies with maternal gestational diabetes, preeclampsia, and obesity. Reprod Sci Thousand Oaks Calif. 2014. Jan;21(1):131–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mitsuya K, Parker AN, Liu L, Ruan J, Vissers MCM, Myatt L. Alterations in the placental methylome with maternal obesity and evidence for metabolic regulation. PloS One. 2017;12(10):e0186115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hulme CH, Stevens A, Dunn W, Heazell AEP, Hollywood K, Begley P, et al. Identification of the functional pathways altered by placental cell exposure to high glucose: lessons from the transcript and metabolite interactome. Sci Rep. 2018. 27;8(1):5270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hulme CH, Nicolaou A, Murphy SA, Heazell AEP, Myers JE, Westwood M. The effect of high glucose on lipid metabolism in the human placenta. Sci Rep. 2019. Oct 1;9(1):14114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Albrecht C, Baker JC, Blundell C, Chavez SL, Carbone L, Chamley L, et al. IFPA meeting 2016 workshop report I: Genomic communication, bioinformatics, trophoblast biology and transport systems. Placenta. 2017. Dec 1;60:S5–9. [DOI] [PubMed] [Google Scholar]
- 136.Vento-Tormo R, Efremova M, Botting RA, Turco MY, Vento-Tormo M, Meyer KB, et al. Single-cell reconstruction of the early maternal-fetal interface in humans. Nature. 2018;563(7731):347–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Pavličev M, Wagner GP, Chavan AR, Owens K, Maziarz J, Dunn-Fletcher C, et al. Single-cell transcriptomics of the human placenta: inferring the cell communication network of the maternal-fetal interface. Genome Res. 2017;27(3):349–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Suryawanshi H, Morozov P, Straus A, Sahasrabudhe N, Max KEA, Garzia A, et al. A single-cell survey of the human first-trimester placenta and decidua. Sci Adv. 2018;4(10):eaau4788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lv B, An Q, Zeng Q, Zhang X, Lu P, Wang Y, et al. Single-cell RNA sequencing reveals regulatory mechanism for trophoblast cell-fate divergence in human peri-implantation conceptuses. PLoS Biol. 2019. Oct;17(10):e3000187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Robinson JF, Kapidzic M, Gormley M, Ona K, Dent T, Seifikar H, et al. Transcriptional Dynamics of Cultured Human Villous Cytotrophoblasts. Endocrinology. 2017. January;158(6):1581–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Rafiee M-R, Girardot C, Sigismondo G, Krijgsveld J. Expanding the Circuitry of Pluripotency by Selective Isolation of Chromatin-Associated Proteins. Mol Cell. 2016. March;64(3):624–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sheng K, Cao W, Niu Y, Deng Q, Zong C. Effective detection of variation in single-cell transcriptomes using MATQ-seq. Nat Methods. 2017;14(3):267–70. [DOI] [PubMed] [Google Scholar]
- 143.Roberts KA, Riley SC, Reynolds RM, Barr S, Evans M, Statham A, et al. Placental structure and inflammation in pregnancies associated with obesity. Placenta. 2011. Mar;32(3):247–54. [DOI] [PubMed] [Google Scholar]
- 144.Jansson N, Rosario FJ, Gaccioli F, Lager S, Jones HN, Roos S, et al. Activation of Placental mTOR Signaling and Amino Acid Transporters in Obese Women Giving Birth to Large Babies. J Clin Endocrinol Metab. 2013. Jan;98(1):105–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Myatt L, Maloyan A. Obesity and Placental Function. Semin Reprod Med. 2016. Jan;34(1):42–9. [DOI] [PubMed] [Google Scholar]
- 146.Muralimanoharan S, Guo C, Myatt L, Maloyan A. Sexual dimorphism in miR-210 expression and mitochondrial dysfunction in the placenta with maternal obesity. Int J Obes 2005. 2015. Aug;39(8):1274–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Levy DE, Lee C. What does Stat3 do? J Clin Invest. 2002. May;109(9):1143–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Whitmarsh AJ. A central role for p38 MAPK in the early transcriptional response to stress. BMC Biol. 2010. Apr 27;8:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Aye ILMH, Jansson T, Powell TL. Interleukin-1β inhibits insulin signaling and prevents insulin-stimulated system A amino acid transport in primary human trophoblasts. Mol Cell Endocrinol. 2013. Dec 5;381(1–2):46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bax BE, Bloxam DL. Energy metabolism and glycolysis in human placental trophoblast cells during differentiation. Biochim Biophys Acta. 1997. Apr 11;1319(2–3):283–92. [DOI] [PubMed] [Google Scholar]
- 151.Bloxam DL, Bobinski PM. Energy metabolism and glycolysis in the human placenta during ischaemia and in normal labour. Placenta. 1984. Oct;5(5):381–94. [DOI] [PubMed] [Google Scholar]
- 152.Biron-Shental T, Schaiff WT, Ratajczak CK, Bildirici I, Nelson DM, Sadovsky Y. Hypoxia regulates the expression of fatty acid-binding proteins in primary term human trophoblasts. Am J Obstet Gynecol. 2007. Nov;197(5):516.e1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Shekhawat P, Bennett MJ, Sadovsky Y, Nelson DM, Rakheja D, Strauss AW. Human placenta metabolizes fatty acids: implications for fetal fatty acid oxidation disorders and maternal liver diseases. Am J Physiol Endocrinol Metab. 2003. Jun;284(6):E1098–1105. [DOI] [PubMed] [Google Scholar]
- 154.Rakheja D, Bennett MJ, Foster BM, Domiati-Saad R, Rogers BB. Evidence for fatty acid oxidation in human placenta, and the relationship of fatty acid oxidation enzyme activities with gestational age. Placenta. 2002. May;23(5):447–50. [DOI] [PubMed] [Google Scholar]
- 155.Kolahi KS, Valent AM, Thornburg KL. Cytotrophoblast, Not Syncytiotrophoblast, Dominates Glycolysis and Oxidative Phosphorylation in Human Term Placenta. Sci Rep. 2017. 23;7:42941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mele J, Muralimanoharan S, Maloyan A, Myatt L. Impaired mitochondrial function in human placenta with increased maternal adiposity. Am J Physiol Endocrinol Metab. 2014. Sep 1;307(5):E419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Nayak CD, Agarwal V, Nayak DM. Correlation of cord blood lipid heterogeneity in neonates with their anthropometry at birth. Indian J Clin Biochem IJCB. 2013. Apr;28(2):152–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ramy N, Zakaria M, El Kafoury M, Kamal M. Cord blood lipid profile in relation to anthropometric measures of newborns. Kasr Al Ainy Med J. 23(1):54. [Google Scholar]
- 159.Gunes T, Koklu E, Ozturk MA. Maternal and cord serum lipid profiles of preterm infants with respiratory distress syndrome. J Perinatol Off J Calif Perinat Assoc. 2007. Jul;27(7):415–21. [DOI] [PubMed] [Google Scholar]
- 160.Wang Y, Bucher M, Myatt L. Use of Glucose, Glutamine and Fatty Acids for Trophoblast Respiration in Lean, Obese and Gestational Diabetic Women. J Clin Endocrinol Metab. 2019. May 22; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sferruzzi-Perri AN, Higgins JS, Vaughan OR, Murray AJ, Fowden AL. Placental mitochondria adapt developmentally and in response to hypoxia to support fetal growth. Proc Natl Acad Sci. 2019. Jan 29;116(5):1621–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wakeland AK, Soncin F, Moretto-Zita M, Chang C-W, Horii M, Pizzo D, et al. Hypoxia Directs Human Extravillous Trophoblast Differentiation in a Hypoxia-Inducible Factor-Dependent Manner. Am J Pathol. 2017. Apr;187(4):767–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Holland OJ, Hickey AJR, Alvsaker A, Moran S, Hedges C, Chamley LW, et al. Changes in mitochondrial respiration in the human placenta over gestation. Placenta. 2017. Sep;57:102–12. [DOI] [PubMed] [Google Scholar]
- 164.Holland O, Dekker Nitert M, Gallo LA, Vejzovic M, Fisher JJ, Perkins AV. Review: Placental mitochondrial function and structure in gestational disorders. Placenta. 2017;54:2–9. [DOI] [PubMed] [Google Scholar]
- 165.Watson AL, Skepper JN, Jauniaux E, Burton GJ. Susceptibility of human placental syncytiotrophoblastic mitochondria to oxygen-mediated damage in relation to gestational age. J Clin Endocrinol Metab. 1998. May;83(5):1697–705. [DOI] [PubMed] [Google Scholar]
- 166.Maloyan A, Mele J, Muralimanohara B, Myatt L. Measurement of mitochondrial respiration in trophoblast culture. Placenta. 2012. May;33(5):456–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Rosario FJ, Gupta MB, Myatt L, Powell TL, Glenn JP, Cox L, et al. Mechanistic Target of Rapamycin Complex 1 Promotes the Expression of Genes Encoding Electron Transport Chain Proteins and Stimulates Oxidative Phosphorylation in Primary Human Trophoblast Cells by Regulating Mitochondrial Biogenesis. Sci Rep. 2019. Jan 22;9(1):246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Hastie R, Lappas M. The effect of pre-existing maternal obesity and diabetes on placental mitochondrial content and electron transport chain activity. Placenta. 2014. Sep;35(9):673–83. [DOI] [PubMed] [Google Scholar]
- 169.Mandò C, Anelli GM, Novielli C, Panina-Bordignon P, Massari M, Mazzocco MI, et al. Impact of Obesity and Hyperglycemia on Placental Mitochondria. Oxid Med Cell Longev. 2018;2018:2378189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Molenaar EA, Massaro JM, Jacques PF, Pou KM, Ellison RC, Hoffmann U, et al. Association of lifestyle factors with abdominal subcutaneous and visceral adiposity: the Framingham Heart Study. Diabetes Care. 2009. Mar;32(3):505–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Malti N, Merzouk H, Merzouk SA, Loukidi B, Karaouzene N, Malti A, et al. Oxidative stress and maternal obesity: feto-placental unit interaction. Placenta. 2014. Jun;35(6):411–6. [DOI] [PubMed] [Google Scholar]
- 172.Graziewicz MA, Day BJ, Copeland WC. The mitochondrial DNA polymerase as a target of oxidative damage. Nucleic Acids Res. 2002. Jul 1;30(13):2817–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell. 2012. Oct 26;48(2):158–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Ballesteros-Guzmán AK, Carrasco-Legleu CE, Levario-Carrillo M, Chávez-Corral DV, Sánchez-Ramírez B, Mariñelarena-Carrillo EO, et al. Prepregnancy Obesity, Maternal Dietary Intake, and Oxidative Stress Biomarkers in the Fetomaternal Unit. BioMed Res Int. 2019;2019:5070453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Schönfeld P, Wojtczak L. Fatty acids as modulators of the cellular production of reactive oxygen species. Free Radic Biol Med. 2008. Aug 1;45(3):231–41. [DOI] [PubMed] [Google Scholar]
- 176.Walsh SW, Wang Y. Secretion of lipid peroxides by the human placenta. Am J Obstet Gynecol. 1993. Dec;169(6):1462–6. [DOI] [PubMed] [Google Scholar]
- 177.Bonet B, Chait A, Gown AM, Knopp RH. Metabolism of modified LDL by cultured human placental cells. Atherosclerosis. 1995. Jan 20;112(2):125–36. [DOI] [PubMed] [Google Scholar]
- 178.Roberts VHJ, Smith J, McLea SA, Heizer AB, Richardson JL, Myatt L. Effect of Increasing Maternal Body Mass Index on Oxidative and Nitrative Stress in the Human Placenta. Placenta. 2009. Feb;30(2):169–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Seo J, Lee K-J. Post-translational modifications and their biological functions: proteomic analysis and systematic approaches. J Biochem Mol Biol. 2004. Jan 31;37(1):35–44. [DOI] [PubMed] [Google Scholar]
- 180.Ebenbichler CF, Kaser S, Laimer M, Wolf HJ, Patsch JR, Illsley NP. Polar expression and phosphorylation of human leptin receptor isoforms in paired, syncytial, microvillous and basal membranes from human term placenta. Placenta. 2002. Jul;23(6):516–21. [DOI] [PubMed] [Google Scholar]
- 181.Brett KE, Ferraro ZM, Holcik M, Adamo KB. Placenta nutrient transport-related gene expression: the impact of maternal obesity and excessive gestational weight gain. J Matern-Fetal Neonatal Med Off J Eur Assoc Perinat Med Fed Asia Ocean Perinat Soc Int Soc Perinat Obstet. 2016;29(9):1399–405. [DOI] [PubMed] [Google Scholar]
- 182.Mina TH, Räikkönen K, Riley SC, Norman JE, Reynolds RM. Maternal distress associates with placental genes regulating fetal glucocorticoid exposure and IGF2: Role of obesity and sex. Psychoneuroendocrinology. 2015. Sep;59:112–22. [DOI] [PubMed] [Google Scholar]
- 183.Kim J, Guan K-L. mTOR as a central hub of nutrient signalling and cell growth. Nat Cell Biol. 2019;21(1):63–71. [DOI] [PubMed] [Google Scholar]
- 184.Gupta MB, Jansson T. Novel roles of mechanistic target of rapamycin signaling in regulating fetal growth†. Biol Reprod. 2019. Apr 1;100(4):872–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Rosario FJ, Kanai Y, Powell TL, Jansson T. Mammalian target of rapamycin signalling modulates amino acid uptake by regulating transporter cell surface abundance in primary human trophoblast cells. J Physiol. 2013. Feb 1;591(3):609–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Rosario FJ, Nathanielsz PW, Powell TL, Jansson T. Maternal folate deficiency causes inhibition of mTOR signaling, down-regulation of placental amino acid transporters and fetal growth restriction in mice. Sci Rep. 2017. 21;7(1):3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Rosario FJ, Powell TL, Jansson T. mTOR folate sensing links folate availability to trophoblast cell function. J Physiol. 2017. January;595(13):4189–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Kadam L, Kohan-Ghadr HR, Drewlo S. The balancing act- PPAR-γ’s roles at the maternal-fetal interface. Syst Biol Reprod Med. 2015. Apr;61(2):65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Schaiff WT, Bildirici I, Cheong M, Chern PL, Nelson DM, Sadovsky Y. Peroxisome proliferator-activated receptor-gamma and retinoid X receptor signaling regulate fatty acid uptake by primary human placental trophoblasts. J Clin Endocrinol Metab. 2005. Jul;90(7):4267–75. [DOI] [PubMed] [Google Scholar]
- 190.Michelsen TM, Holme AM, Holm MB, Roland MC, Haugen G, Powell TL, et al. Uteroplacental Glucose Uptake and Fetal Glucose Consumption: A Quantitative Study in Human Pregnancies. J Clin Endocrinol Metab. 2019. January;104(3):873–82. [DOI] [PubMed] [Google Scholar]
- 191.Acosta O, Ramirez VI, Lager S, Gaccioli F, Dudley DJ, Powell TL, et al. Increased glucose and placental GLUT-1 in large infants of obese nondiabetic mothers. Am J Obstet Gynecol. 2015. Feb;212(2):227.e1–7. [DOI] [PubMed] [Google Scholar]
- 192.Gaither K, Quraishi AN, Illsley NP. Diabetes alters the expression and activity of the human placental GLUT1 glucose transporter. J Clin Endocrinol Metab. 1999. Feb;84(2):695–701. [DOI] [PubMed] [Google Scholar]
- 193.Godfrey KM, Matthews N, Glazier J, Jackson A, Wilman C, Sibley CP. Neutral amino acid uptake by the microvillous plasma membrane of the human placenta is inversely related to fetal size at birth in normal pregnancy. J Clin Endocrinol Metab. 1998. Sep;83(9):3320–6. [DOI] [PubMed] [Google Scholar]
- 194.Glazier JD, Cetin I, Perugino G, Ronzoni S, Grey AM, Mahendran D, et al. Association between the activity of the system A amino acid transporter in the microvillous plasma membrane of the human placenta and severity of fetal compromise in intrauterine growth restriction. Pediatr Res. 1997. Oct;42(4):514–9. [DOI] [PubMed] [Google Scholar]
- 195.Ditchfield AM, Desforges M, Mills TA, Glazier JD, Wareing M, Mynett K, et al. Maternal obesity is associated with a reduction in placental taurine transporter activity. Int J Obes 2005. 2015. Apr;39(4):557–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Jansson T, Castillo-Castrejon M, Gupta MB, Powell TL, Rosario FJ. Down-regulation of placental Cdc42 and Rac1 links mTORC2 inhibition to decreased trophoblast amino acid transport in human intrauterine growth restriction. Clin Sci Lond Engl 1979. 2019. Dec 11; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Rosario FJ, Dimasuay KG, Kanai Y, Powell TL, Jansson T. Regulation of amino acid transporter trafficking by mTORC1 in primary human trophoblast cells is mediated by the ubiquitin ligase Nedd4-2. Clin Sci Lond Engl 1979. 2016. Apr 1;130(7):499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Campbell FM, Bush PG, Veerkamp JH, Dutta-Roy AK. Detection and cellular localization of plasma membrane-associated and cytoplasmic fatty acid-binding proteins in human placenta. Placenta. 1998. Aug;19(5–6):409–15. [DOI] [PubMed] [Google Scholar]
- 199.Segura MT, Demmelmair H, Krauss-Etschmann S, Nathan P, Dehmel S, Padilla MC, et al. Maternal BMI and gestational diabetes alter placental lipid transporters and fatty acid composition. Placenta. 2017. Sep;57:144–51. [DOI] [PubMed] [Google Scholar]
- 200.Lager S, Ramirez VI, Gaccioli F, Jang B, Jansson T, Powell TL. Protein expression of fatty acid transporter 2 is polarized to the trophoblast basal plasma membrane and increased in placentas from overweight/obese women. Placenta. 2016. Apr;40:60–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Szabo AJ. Transferred maternal fatty acids stimulate fetal adipogenesis and lead to neonatal and adult obesity. Med Hypotheses. 2019. Jan;122:82–8. [DOI] [PubMed] [Google Scholar]
- 202.Flynn AC, Dalrymple K, Barr S, Poston L, Goff LM, Rogozińska E, et al. Dietary interventions in overweight and obese pregnant women: a systematic review of the content, delivery, and outcomes of randomized controlled trials. Nutr Rev. 2016. May;74(5):312–28. [DOI] [PubMed] [Google Scholar]
- 203.Renault KM, Nørgaard K, Nilas L, Carlsen EM, Cortes D, Pryds O, et al. The Treatment of Obese Pregnant Women (TOP) study: a randomized controlled trial of the effect of physical activity intervention assessed by pedometer with or without dietary intervention in obese pregnant women. Am J Obstet Gynecol. 2014. Feb;210(2):134.e1–9. [DOI] [PubMed] [Google Scholar]
- 204.Patel N, Godfrey KM, Pasupathy D, Levin J, Flynn AC, Hayes L, et al. Infant adiposity following a randomised controlled trial of a behavioural intervention in obese pregnancy. Int J Obes 2005. 2017;41(7):1018–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Vinter CA, Jensen DM, Ovesen P, Beck-Nielsen H, Jørgensen JS. The LiP (Lifestyle in Pregnancy) Study. Diabetes Care. 2011. Dec;34(12):2502–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Muhlhausler BS, Gibson RA, Makrides M. Effect of long-chain polyunsaturated fatty acid supplementation during pregnancy or lactation on infant and child body composition: a systematic review. Am J Clin Nutr. 2010. Oct;92(4):857–63. [DOI] [PubMed] [Google Scholar]
- 207.Middleton P, Gomersall JC, Gould JF, Shepherd E, Olsen SF, Makrides M. Omega-3 fatty acid addition during pregnancy. Cochrane Database Syst Rev. 2018. 15;11:CD003402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Lager S, Ramirez VI, Acosta O, Meireles C, Miller E, Gaccioli F, et al. Docosahexaenoic Acid Supplementation in Pregnancy Modulates Placental Cellular Signaling and Nutrient Transport Capacity in Obese Women. J Clin Endocrinol Metab. 2017. January;102(12):4557–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Foster BA, Escaname E, Powell TL, Larsen B, Siddiqui SK, Menchaca J, et al. Randomized Controlled Trial of DHA Supplementation during Pregnancy: Child Adiposity Outcomes. Nutrients. 2017. Jun 2;9(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Tran HT, Liong S, Lim R, Barker G, Lappas M. Resveratrol ameliorates the chemical and microbial induction of inflammation and insulin resistance in human placenta, adipose tissue and skeletal muscle. PloS One. 2017;12(3):e0173373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Landau D, Haghiac M, Minium J, Skomorovska-Prokvolit Y, Calabuig-Navarro V, O’Tierney-Ginn P. Activation of AMPK in Human Placental Explants Impairs Mitochondrial Function and Cellular Metabolism. Reprod Sci Thousand Oaks Calif. 2019;26(4):487–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Roberts VHJ, Pound LD, Thorn SR, Gillingham MB, Thornburg KL, Friedman JE, et al. Beneficial and cautionary outcomes of resveratrol supplementation in pregnant nonhuman primates. FASEB J Off Publ Fed Am Soc Exp Biol. 2014. Jun;28(6):2466–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Ireland KE, Maloyan A, Myatt L. Melatonin Improves Mitochondrial Respiration in Syncytiotrophoblasts From Placentas of Obese Women. Reprod Sci Thousand Oaks Calif. 2018;25(1):120–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Boulet SL, Salihu HM, Alexander GR. Mode of delivery and birth outcomes of macrosomic infants. J Obstet Gynaecol J Inst Obstet Gynaecol. 2004. Sep;24(6):622–9. [DOI] [PubMed] [Google Scholar]
- 215.HAPO Study Cooperative Research Group. Hyperglycaemia and Adverse Pregnancy Outcome (HAPO) Study: associations with maternal body mass index. BJOG Int J Obstet Gynaecol. 2010. Apr;117(5):575–84. [DOI] [PubMed] [Google Scholar]
- 216.Sewell MF, Huston-Presley L, Super DM, Catalano P. Increased neonatal fat mass, not lean body mass, is associated with maternal obesity. Am J Obstet Gynecol. 2006. Oct;195(4):1100–3. [DOI] [PubMed] [Google Scholar]
- 217.Evagelidou EN, Kiortsis DN, Bairaktari ET, Giapros VI, Cholevas VK, Tzallas CS, et al. Lipid profile, glucose homeostasis, blood pressure, and obesity-anthropometric markers in macrosomic offspring of nondiabetic mothers. Diabetes Care. 2006. Jun;29(6):1197–201. [DOI] [PubMed] [Google Scholar]
- 218.Hirschler V, Roque MI, Calcagno ML, Gonzalez C, Aranda C. Maternal waist circumference and the prediction of children’s metabolic syndrome. Arch Pediatr Adolesc Med. 2007. Dec;161(12):1205–10. [DOI] [PubMed] [Google Scholar]
- 219.Catalano PM, Ehrenberg HM. The short- and long-term implications of maternal obesity on the mother and her offspring. BJOG Int J Obstet Gynaecol. 2006. Oct;113(10):1126–33. [DOI] [PubMed] [Google Scholar]
- 220.Hediger ML, Overpeck MD, McGlynn A, Kuczmarski RJ, Maurer KR, Davis WW. Growth and fatness at three to six years of age of children born small- or large-for-gestational age. Pediatrics. 1999. Sep;104(3):e33. [DOI] [PubMed] [Google Scholar]
- 221.Wang X, Liang L, Junfen FU, Lizhong DU. Metabolic syndrome in obese children born large for gestational age. Indian J Pediatr. 2007. Jun;74(6):561–5. [DOI] [PubMed] [Google Scholar]
- 222.Curhan GC, Chertow GM, Willett WC, Spiegelman D, Colditz GA, Manson JE, et al. Birth weight and adult hypertension and obesity in women. Circulation. 1996. Sep 15;94(6):1310–5. [DOI] [PubMed] [Google Scholar]
- 223.Curhan GC, Willett WC, Rimm EB, Spiegelman D, Ascherio AL, Stampfer MJ. Birth weight and adult hypertension, diabetes mellitus, and obesity in US men. Circulation. 1996. Dec 15;94(12):3246–50. [DOI] [PubMed] [Google Scholar]
- 224.Poston L Developmental programming and diabetes - The human experience and insight from animal models. Best Pract Res Clin Endocrinol Metab. 2010. Aug;24(4):541–52. [DOI] [PubMed] [Google Scholar]
- 225.Alfaradhi MZ, Ozanne SE. Developmental programming in response to maternal overnutrition. Front Genet. 2011;2:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Eriksson JG, Forsen TJ, Osmond C, Barker DJP. Pathways of infant and childhood growth that lead to type 2 diabetes. Diabetes Care. 2003. Nov;26(11):3006–10. [DOI] [PubMed] [Google Scholar]
- 227.Eriksson J, Forsén T, Osmond C, Barker D. Obesity from cradle to grave. Int J Obes Relat Metab Disord J Int Assoc Study Obes. 2003. Jun;27(6):722–7. [DOI] [PubMed] [Google Scholar]
- 228.Forsén T, Eriksson JG, Tuomilehto J, Teramo K, Osmond C, Barker DJ. Mother’s weight in pregnancy and coronary heart disease in a cohort of Finnish men: follow up study. BMJ. 1997. Oct 4;315(7112):837–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Paulsen ME, Rosario FJ, Wesolowski SR, Powell TL, Jansson T. Normalizing adiponectin levels in obese pregnant mice prevents adverse metabolic outcomes in offspring. FASEB J Off Publ Fed Am Soc Exp Biol. 2019;33(2):2899–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Vaughan OR, Rosario FJ, Powell TL, Jansson T. Normalisation of circulating adiponectin levels in obese pregnant mice prevents cardiac dysfunction in adult offspring . Int J Obes 2005. 2019. May 10; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Spada E, Chiossi G, Coscia A, Monari F, Facchinetti F. Effect of maternal age, height, BMI and ethnicity on birth weight: an Italian multicenter study. J Perinat Med. 2018. Nov 27;46(9):1016–21. [DOI] [PubMed] [Google Scholar]
- 232.Zhang C, Hediger ML, Albert PS, Grewal J, Sciscione A, Grobman WA, et al. Association of Maternal Obesity With Longitudinal Ultrasonographic Measures of Fetal Growth: Findings From the NICHD Fetal Growth Studies-Singletons. JAMA Pediatr. 2018. January;172(1):24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Thangaratinam S, Rogozinska E, Jolly K, Glinkowski S, Roseboom T, Tomlinson JW, et al. Effects of interventions in pregnancy on maternal weight and obstetric outcomes: meta-analysis of randomised evidence. BMJ. 2012. May 16;344:e2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Schwarzenberg SJ, Georgieff MK, COMMITTEE ON NUTRITION. Advocacy for Improving Nutrition in the First 1000 Days to Support Childhood Development and Adult Health. Pediatrics. 2018;141(2). [DOI] [PubMed] [Google Scholar]
- 235.Thornburg KL, O’Tierney PF, Louey S. Review: The placenta is a programming agent for cardiovascular disease. Placenta. 2010. Mar;31 Suppl:S54–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Ursini G, Punzi G, Chen Q, Marenco S, Robinson JF, Porcelli A, et al. Convergence of placenta biology and genetic risk for schizophrenia. Nat Med. 2018;24(6):792–801. [DOI] [PubMed] [Google Scholar]
- 237.Barker D, Osmond C, Grant S, Thornburg KL, Cooper C, Ring S, et al. Maternal cotyledons at birth predict blood pressure in childhood. Placenta. 2013. Aug;34(8):672–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Barker DJP, Osmond C, Thornburg KL, Kajantie E, Eriksson JG. The shape of the placental surface at birth and colorectal cancer in later life. Am J Hum Biol Off J Hum Biol Counc. 2013. Aug;25(4):566–8. [DOI] [PubMed] [Google Scholar]
- 239.Barker DJP, Thornburg KL. Placental programming of chronic diseases, cancer and lifespan: a review. Placenta. 2013. Oct;34(10):841–5. [DOI] [PubMed] [Google Scholar]
- 240.Barker DJP, Osmond C, Thornburg KL, Kajantie E, Eriksson JG. The lifespan of men and the shape of their placental surface at birth. Placenta. 2011. Oct;32(10):783–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Barker DJP, Larsen G, Osmond C, Thornburg KL, Kajantie E, Eriksson JG. The placental origins of sudden cardiac death. Int J Epidemiol. 2012. Oct;41(5):1394–9. [DOI] [PubMed] [Google Scholar]
- 242.Eriksson JG, Kajantie E, Thornburg KL, Osmond C, Barker DJP. Mother’s body size and placental size predict coronary heart disease in men. Eur Heart J. 2011. Sep;32(18):2297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Mikaelsson MA, Constância M, Dent CL, Wilkinson LS, Humby T. Placental programming of anxiety in adulthood revealed by Igf2-null models. Nat Commun. 2013;4:2311. [DOI] [PubMed] [Google Scholar]
- 244.Bonnin A, Goeden N, Chen K, Wilson ML, King J, Shih JC, et al. A transient placental source of serotonin for the fetal forebrain. Nature. 2011. Apr 21;472(7343):347–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Howerton CL, Bale TL. Targeted placental deletion of OGT recapitulates the prenatal stress phenotype including hypothalamic mitochondrial dysfunction. Proc Natl Acad Sci U S A. 2014. Jul 1;111(26):9639–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Howerton CL, Morgan CP, Fischer DB, Bale TL. O-GlcNAc transferase (OGT) as a placental biomarker of maternal stress and reprogramming of CNS gene transcription in development. Proc Natl Acad Sci U S A. 2013. Mar 26;110(13):5169–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Goeden N, Velasquez J, Arnold KA, Chan Y, Lund BT, Anderson GM, et al. Maternal Inflammation Disrupts Fetal Neurodevelopment via Increased Placental Output of Serotonin to the Fetal Brain. J Neurosci Off J Soc Neurosci. 2016. January;36(22):6041–9. [DOI] [PMC free article] [PubMed] [Google Scholar]