

Abstract
Glucagon‐like peptide‐1 (GLP‐1) receptors belong to the pharmaceutically important Class B family of GPCRs and are involved in many biologically significant signalling pathways. Its incretin peptide ligand GLP‐1 analogues are effective treatments for Type 2 diabetes. Although developing non‐peptide low MW drugs targeting GLP‐1 receptors remains elusive, considerable progress has been made in discovering non‐peptide agonists and positive allosteric modulators (PAMs) of GLP‐1 receptors with demonstrated efficacy. Many of these compounds induce biased signalling in GLP‐1 receptor‐mediated functional pathways. High‐quality structures of GLP‐1 receptors in both inactive and active states have been reported, revealing detailed molecular interactions between GLP‐1 receptors and non‐peptide agonists or PAMs. These progresses raise the exciting possibility of developing non‐peptide drugs of GLP‐1 receptors as alternative treatments for Type 2 diabetes. The insight into the interactions between the receptor and the non‐peptide ligand is also useful for developing non‐peptide ligands targeting other Class B GPCRs.
LINKED ARTICLES
This article is part of a themed issue on GLP1 receptor ligands (BJP 75th Anniversary). To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v179.4/issuetoc
Keywords: glucagon‐like peptide‐1 receptor, high‐throughput screening, nonpeptide agonist, positive allosteric modulator, type 2 diabetes, β‐cell
Abbreviations
- ECD
extracellular N‐terminal domain
- ECL
extracellular loop
- GLP‐1
glucagon‐like peptide‐1
- MCAO
middle cerebral artery occlusion
- NAM
negative allosteric modulator
- OEA
oleoylethanolamide
- PAM
positive allosteric modulator
- SEA
stearoylethanolamide
- TMD
transmembrane domain
1. INTRODUCTION
The glucagon‐like peptide‐1 (GLP) receptor is a member of the Class B GPCR superfamily and is found in both beta‐cells in the pancreas and neurons in the CNS. GLP‐1 receptors are involved in a broad range of physiological processes, including promoting insulin biosynthesis and secretion from pancreatic beta‐cells in a glucose‐dependent manner, inhibiting glucagon release and gastric emptying, and cardioprotection and neuroprotection. GLP‐1 receptors are activated by the binding of a GLP‐1 peptide hormone that can trigger Gαs or other G‐protein‐mediated pathways or β‐arrestin‐mediated pathways (De Graaf et al., 2016). The activation of the G‐protein‐dependent pathways subsequently results in cAMP accumulation, Ca2+ mobilization and the phosphorylation of ERK1/2 (pERK1/2), and in the case of the β‐arrestin‐mediated pathways, also pERK1/2. Given its central role in insulin secretion, GLP‐1 receptors are an effective target for the treatment of Type 2 diabetes mellitus.
Like other Class B GPCRs, GLP‐1 receptors contain an extracellular N‐terminal domain (ECD) of more than 100 amino acid residues (Watkins et al., 2012), and a transmembrane domain (TMD) composed of seven helices connected by extracellular and intracellular loops. GLP‐1 peptides bind at both the ECD and the extracellular half of the TMD (Y. Zhang et al., 2017). Upon GLP‐1 binding, the ECD adopts an open conformation containing an extended peptide‐binding groove where multiple interactions with the C‐terminus of the GLP‐1 form. The binding of the GLP‐1 to the ECD brings the N‐terminus of the GLP‐1 peptide near the TMD, and their interactions cause the conformational change in the helical bundle, which enables interactions of the intracellular half of the TMD with the G‐protein.
1.1. GLP‐1 receptor agonists and peptide drugs
GLP‐1 receptors are activated by five endogenous incretin peptide hormones that include GLP‐1(1–37), GLP‐1(7–37), GLP‐1(1–36)NH2, GLP‐1(7–36)NH2 and GLP‐1(9–36)NH2 and a low‐affinity, structurally analogous peptide, oxyntomodulin. Among them, GLP‐1(7–37) and GLP‐1(7–36)NH2 are two primary forms. After its secretion from L cells, GLP‐1(7–36)NH2 is rapidly metabolized to GLP‐1(9–36)NH2. The latter is considered a weak, partial agonist of GLP‐1 receptors, but its concentration in circulation blood could be fivefold to 10‐fold higher than GLP‐1(7–36)NH2 (Orskov et al., 1994). GLP‐1 receptors are also activated by glucagon, and this cross reactivity is thought to be physiologically important (Capozzi et al., 2019). GLP‐1 binds to GLP‐1 receptors with high specificity over other members of the glucagon subfamily to which GLP‐1 receptors belong. However, GLP‐1 is partly degraded by the enzyme, dipeptidyl peptidase‐4, making GLP‐1 a rather restricted treatment in Type 2 diabetes. Therapeutically, various peptide mimetics with enhanced pharmacokinetic profiles, including exenatide (exendin‐4) (Eng et al., 1992), semaglutide (Lau et al., 2015), dulaglutide (Glaesner et al., 2010) and liraglutide (Knudsen et al., 2000), are approved drugs.
Despite the clinical success of GLP‐1‐mimetic peptides in managing the effects of Type 2 diabetes, there are several limitations with these peptide drugs. The relatively large peptide molecules are expensive to produce and require subcutaneous injection. They can cause adverse side effects such as vomiting and nausea, of which the mechanism is still unknown in some patients. An important development in GLP‐1 therapy is the recent approval of a new formulation of the approved semaglutide with the absorption enhancer, sodium N‐(8‐[2‐hydroxybenzoyl]amino)caprylate, for oral delivery (Davies et al., 2017). However, there are several limitations to this advance in formulation. The tablet must be administered after overnight fasting and at least 30 min before the first food, beverage or other oral medicines with no more than 4 ounces of plain water (U.S. Food and Drug Administration, 2019). In addition, oral semaglutide was found to induce nausea and gastrointestinal side effects at slightly greater severity than those with injectable GLP‐1 mimetics (Pratley et al., 2019).
Hence, developing low MW orally effective drugs targeting GLP‐1 receptors has been pursued. Despite intensive efforts, there are no low MW non‐peptide drugs targeting the GLP‐1 receptors in the market. A number of important factors contribute to such difficulties. For instance, the orthosteric GLP‐1 binding sites across members of the glucagon subfamily are highly conserved, making it difficult to achieve high selectivity for GLP‐1 receptors. Furthermore, the extended nature of the GLP‐1 binding groove and the occurrence of multiple interactions along the groove present a challenge to mimic the peptide–receptor interactions by non‐peptide molecules. In addition, the structure of the GLP‐1 receptor including its TMD had not been available until 2017.
1.2. Positive allosteric modulation of GLP‐1 receptors
GPCRs including the GLP‐1 receptors are naturally allosteric molecules, whose activity can be indirectly regulated by allosteric modulators. Allosteric modulators do not bind to the orthosteric binding site but instead act at another binding site (allosteric site) to regulate the activation of the receptor by its natural ligand (Liu & Nussinov, 2016). It has been observed that the effect of an allosteric modulator could be specific to the orthosteric ligand present, a phenomenon termed ‘probe dependence’ (Kenakin, 2008). Depending on their effects on the receptor activity, allosteric modulators can be classified as positive, negative or neutral. Positive allosteric modulators (PAMs) increase the binding affinity and/or efficacy of the natural agonist towards the receptor. Developing low MW compounds that target allosteric sites on GPCRs has been clinically successful (Dorr et al., 2005; Harrington & Fotsch, 2007). This raises exciting possibilities in the development of GLP‐1 receptor PAMs, as an alternative approach to targeting the orthosteric site.
PAMs targeting the GLP‐1 receptors are considered to have several potential benefits. In general, allosteric sites are less conserved than the orthosteric sites, and targeting them can help achieve better subtype selectivity and reduce side effects (Lazareno et al., 2004). PAMs are shown to selectively activate certain signalling pathways over others and can thus provide desired functional selectivity and novel modes of efficacy (Pupo et al., 2016). Third, without increasing bioavailability, allosteric ligands can augment the efficacy of orthosteric ligands. Hence, developing non‐peptide PAMs of GLP‐1 receptors, which can augment the efficacy of endogenous and exogenous GLP‐1 and its analogues without increased bioavailability, is attractive and significant.
1.3. Biased agonism of GLP‐1 receptors
A GPCR molecule can adopt multiple active conformations, and different ligands can bind to and stabilize different active conformations, thus, selectively activate distinct signal transduction pathways. This concept of biased agonism or ligand‐directed biased signalling or functional selectivity is especially relevant to GLP‐1 receptors, because these receptors have multiple endogenous peptide agonists that may promote the receptor to take diverse conformational states and is involved in several signalling pathways (see reviews in this issue). Allosteric modulation offers an additional means of regulating signalling transduction through receptor activation, thus affecting biased signalling of GLP‐1 receptors.
Most low MW agonists and PAMs of GLP‐1 receptors have been described since 2007 (Chen et al., 2007; Knudsen et al., 2007), and earlier reports have been reviewed comprehensively (Willard, Bueno et al., 2012). Since then, there have been remarkable advances made in the discovery, optimization and clinical development of non‐peptide agonists and PAMs for GLP‐1 receptors. Two novel non‐peptide agonists are currently in preclinical or clinical trials, and several newly reported PAMs show favourable pharmacological profiles. These compounds provide novel models of efficacy and may lead to novel therapeutic agents for the treatment of Type 2 diabetes. In addition, significant breakthroughs have been achieved in the recent years in the elucidation of GLP‐1 receptor structures in both inactive and active states, which provides structural insight into the binding of non‐peptide agonists/PAMs and their interactions with GLP‐1 receptors. All these new developments warrant the need for an updated review. This review covers, primarily, the non‐peptide agonists and PAMs for the GLP‐1 receptor, reported in the literature since 2012. Previously reviewed compounds with new developments are also included. Compounds only reported in patent disclosure and PubChem databases (Kim et al., 2019) are generally not discussed unless they are related to others that have been extensively studied. All reported GLP‐1 receptor structures complexed with either a non‐peptide agonist or a PAM molecule are discussed, as well.
2. NON‐PEPTIDE AGONISTS OF GLP‐1 RECEPTORS
Non‐peptide drugs for the treatment of Type 2 diabetes could offer the convenience of oral administration and reduce side effects currently associated with peptide drugs targeting GLP‐1 receptors. Hence, the development of non‐peptide agonists of these receptors has been pursued for many years, and significant progress has been made in recent years with three compounds entering preclinical or clinical trials. Structural studies indicate that all the non‐peptide agonists of known structure bind primarily in the helix bundle of the receptor overlapping with the orthosteric binding pocket of GLP‐1 peptide. However, these agonists occupy different positions in the orthosteric binding pocket and thereby form different types of interactions with the GLP‐1 receptor.
2.1. S4P and Boc5
Some of the first reported non‐peptide agonists of the GLP‐1 receptors are substituted cyclobutanes exemplified by compound 1 (Boc5) (Figure 1, [1]) (Chen et al., 2007). By high‐throughput (HT) screening a diverse library of 48,160 compounds against HEK293 cells transfected with a rat GLP‐1 receptor vector and a CRE‐luciferase reporter plasmid, two synthetic olefin compounds were identified in promoting luciferase response and increasing cAMP accumulation. It was then realized that these two active compounds dimerize in DMSO solution and form substituted cyclobutanes, S4P and Boc5. In the CRE‐luciferase assay, S4P is a partial agonist, whereas the related Boc5 displayed full agonist activity. Both compounds are selective towards GLP‐1 receptors and neither compound activates cells without GLP‐1 receptors or cells expressing glucagon receptors (GCGR) or GLP‐2 receptors. With exendin(9–39), a GLP‐1 receptor antagonist bound at the orthosteric site, neither S4P or Boc5 activates GLP‐1 receptors. In receptor binding assays, both compounds displace 125I‐GLP‐1. These data suggest that these two compounds are likely to function as GLP‐1 mimetics in vitro.
FIGURE 1.
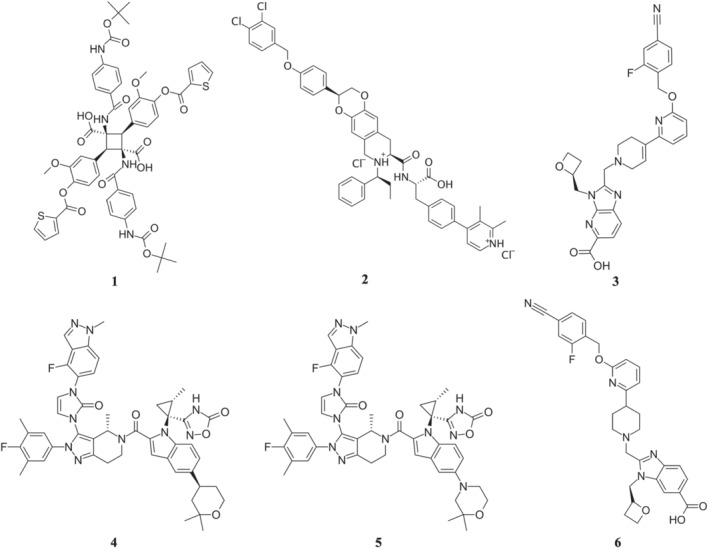
Chemical structures of non‐peptide agonists of GLP‐1 receptors. Compounds shown are (1) Boc5, (2) TT‐OAD2, (3) RGT1383, (4) LY3502970 (OWL833), (5) CHU‐128 and (6) PF06882961
In isolated rat pancreatic islets, Boc5 exhibits glucose‐dependent, insulinotropic effects in a dose‐dependent manner. In vivo studies using female mice (C57BL/6, wild type [WT]) show that Boc5 inhibits food intake in a dose‐dependent manner and the effects of Boc5 can be completely blocked by pretreatment of mice with exendin (9–39). Chronically, the effect of Boc5 administration was tested in db/db mice, a murine model of Type 2 diabetes. The glycated haemoglobin levels in the mice were reduced down to a level comparative to that of non‐diabetic mice. In addition, reduced food intake, lowered body weight and enhanced insulin secretion were also observed.
In a follow‐up study, the in vivo pharmacology of Boc5 was determined in both lean (C57BL/6J) and diabetic (db/db) mice, and a range of pharmacological parameters were investigated including glycaemic control and weight loss (Su et al., 2008). Many of the aspects reported previously were confirmed including dose‐dependent effects on reduced food intake and insulin secretion stimulation. Little effect was observed on normal mice treated in the same manner. In another study, subchronic treatment of diet‐induced obese mice with Boc5 was carried out, and a broad range of antidiabetic effects of the treatment were observed, suggesting Boc5 may produce metabolic benefits via multiple synergistic mechanisms (Su et al., 2008). Through SAR studies, a more potent Boc5 analogue was synthesized which is fourfold to fivefold more potent than Boc5 both in vitro and in vivo. Despite the improvement in potency, the new compound and Boc5 share poor drug‐like properties and have not been further developed as oral drugs (Liu et al., 2012).
2.2. Azoanthracenes
In several patents, Transtech Pharmaceuticals (TTP), later renamed as vTv Therapeutics, disclosed a number of azoanthracene and oxadiazoanthracene derivatives as GLP‐1 receptor agonists, exemplified by compound 2 (TT‐OAD2) (Figure 1, [2]) (Mjalli et al., 2009; Polisetti et al., 2011; Rao, 2009). TT‐OAD2 is a weak agonist of GLP‐1 receptors with slow kinetics. In HEK293 cells that overexpress GLP‐1 receptors, TT‐OAD2 shows biased signalling for cAMP accumulation and displays only weak responses in terms of Ca2+ mobilization and no detectable β‐arrestin‐1 recruitment (Zhao et al., 2020). TT‐OAD2 inhibits GLP‐1 and oxyntomodulin‐mediated cAMP, Ca2+, pERK1/2 and β‐arrestin responses in a concentration‐dependent manner, suggesting TT‐OAD2 is not a PAM of GLP‐1 receptors. In humanized GLP‐1 receptor mice, TT‐OAD2 is insulinotropic, and this effect is dependent on the GLP‐1 receptor.
In a rather recent report, the structure of GLP‐1 receptors in complex with TT‐OAD2 and G‐protein subunit was solved using cryo‐EM techniques (Zhao et al., 2020). TT‐OAD2 binds in the GLP‐1 receptor helical bundle near the extracellular domain and interacts with residues within TM1–TM3 and extracellular loop (ECL) 1–2 (Figure 2). Most of the interactions are considered hydrophobic and others manifesting π–π stacking. In the binding site, TT‐OAD2 adopts a U‐shaped orientation with the 3,4‐dichloro‐benzyl ring of TT‐OAD2 protruding through transmembrane helices 2 and 3 and embedding in the detergent micelle. This TT‐OAD2 binding site overlaps subtly with that of GLP‐1 peptide, and 10 out of 29 residues were found to interact with both TT‐OAD2 and GLP‐1.
FIGURE 2.
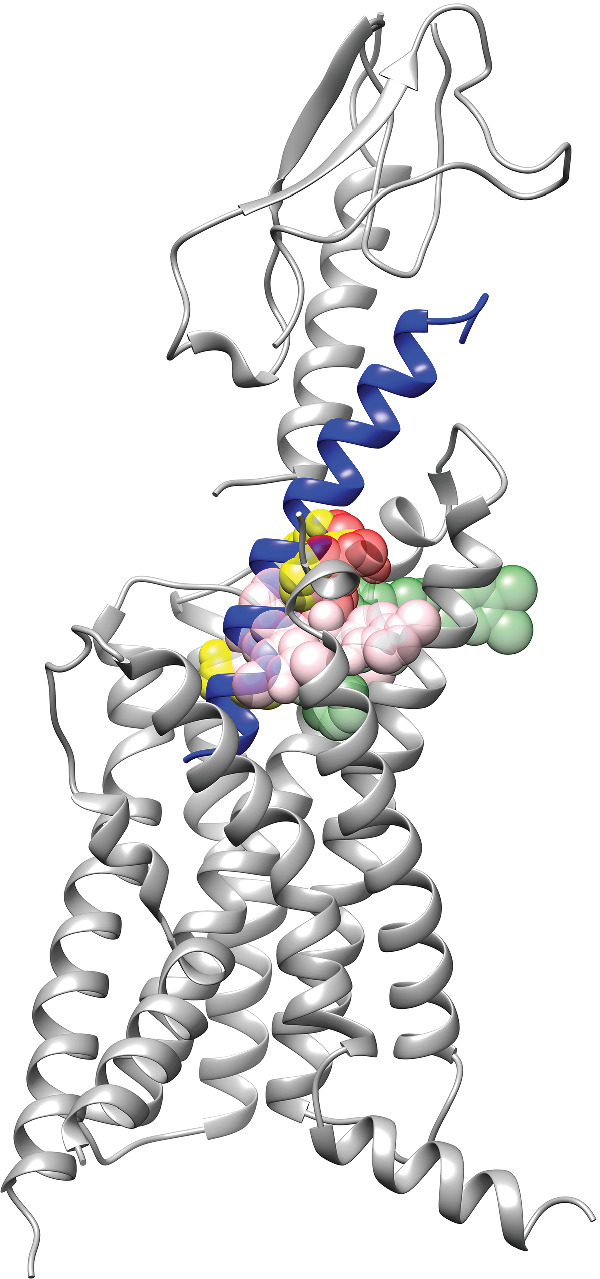
Binding sites of non‐peptide agonists of GLP‐1 receptors. The structure of the GLP‐1 receptor is shown (ECD: PDB ID: 6VCB; TMD, PDB ID: 6VCB), and the GLP‐1 peptide (PDB ID: 6VCB) is coloured in blue. Agonists are shown as coloured spheres. Dark green, TT‐OAD2 (PDB ID: 6ORV); yellow, RGT1383 (PDB ID: 7C2E); pink, LY3502970 (PDB ID: 6XOX); and red, PF06882961 (PDB ID: 6X1A). The opacity for the dark green and red colour was set at 47.5% and for the yellow and pink colour, 60%
The leading molecule in this class, TTP273, whose structure is not disclosed, is an analogue from the same class as TT‐OAD2. TTP273 exhibited biased signalling for cAMP signalling and did not activate ERK pathway significantly at clinically relevant concentrations (vTv Therapeutics, 2016). In mice, TTP273 enhances glucose‐dependent insulin secretion, reduces glucose levels following an oral glucose tolerance test and decreases food intake. TTP273 has completed a 3‐month Phase II clinical trial in patients with Type 2 diabetes (ClinicalTrials.gov Identifier: NCT02653599). According to the company website (https://vtvtherapeutics.com/pipeline/ttp273/), TTP273 is well tolerated by the patients and demonstrates statistically significant reduction in glycated haemoglobin level. The side effects of nausea and vomiting are negligible, suggesting a potential clinical advantage over GLP‐1‐mimetic peptides. However, as previously mentioned (Willard et al., 2012), TT‐OAD2 and other molecules disclosed in these patents are all of high MW and high lipophilicity, which may explain the recent complexity in identifying optimal dosing for TTP273 (Zhao et al., 2020).
2.3. RGT1383
A recent publication disclosed the discovery of a non‐peptide GLP‐1 receptor agonist, RGT1383, compound 3 (Figure 1, [3]). In vitro studies indicate RGT1383 is a full agonist for cAMP signalling with an EC50 value in the range of 0.2 nM and a partial agonist for β‐arrestin recruitment with maximal arrestin recruitment at ~30% (Ma et al., 2020).
In the cryo‐EM determined structure of GLP‐1 receptors with RGT1383 and G‐protein, the agonist RGT1383 binds in a tightly packed orthosteric binding pocket in the TMD (Figure 2) (Ma et al., 2020). RGT1383 interacts with residues from TM1–TM3, TM7 and ECL1–ECL2, as well as the ECD. These interactions between RGT1383 and the receptor were confirmed through mutagenesis studies using cAMP luciferase reporter assays. Compared with GLP‐1 receptors bound to TT‐OAD2, RGT1383 forms more extensive interactions with residues in TM7, which induce inward displacements of the ECL3 and the extracellular ends of TM6–TM7. The binding site of RGT1383 almost completely overlaps with the sites occupied by residues 10–20 of GLP‐1.
2.4. LY3502970 (OWL‐833) and CHU‐128
In search of low MW activators of GLP‐1 receptors, Kawai et al. at Chuai Pharmaceutical and Eli Lilly and Company used a screening method that detects compound‐induced expression of a urokinase‐type plasminogen activator in LLC‐PK1 cells expressing human GLP‐1 receptors (Tamura et al., 2016), followed by multiple rounds of SAR work to optimize affinity and drug‐like properties. These led to the discovery of compound 4 (LY3502970, also known as OWL‐833) (Figure 1, [4]) (Yoshino et al., 2018). In vitro studies using HEK293 cells expressing various densities of human GLP‐1 receptors reveals that LY3502970 is a partial agonist of these receptors, biased towards cAMP accumulation with no detectable β‐arrestin recruitment. It is much more potent than TT‐OAD2 in stimulating GLP‐1 receptor‐mediated cAMP accumulation and is highly selective against other Class B GPCRs. In vivo studies show that oral administration of LY3502970 results in glucose lowering in humanized GLP‐1 receptor transgenic mice (use of these mice is essential due to the species specificity of the molecule; X. Zhang et al., 2020) and insulinotropic and hypophagic effects in non‐human primates. Given its favourable pharmacokinetic profile and efficacy, LY3502970 is currently being evaluated in early stage clinical trial for its potential as an antidiabetic agent (ClinicalTrials.gov Identifier: NCT04426474). From the same patent series by Chugai, compound 5 (CHU‐128) (Figure 1, [5]) has also been studied in vitro (X. Zhang et al., 2020). Given that the chemical structure of CHU‐128 is very close to that of LY3502970, it is not surprising to find that CHU‐128 has a pharmacological profile similar to that of LY3502970.
In the recently reported cryo‐EM structure of GLP‐1 receptors with LY3502970, GsiN18, antibody Nb35 and single‐chain variable fragment scFv16, the compound LY3502970 binds in the helical bundle that partlly overlaps with the area where TT‐OAD2 binds and interacts with residues within the ECD, TM1–TM3, ECL2 and TM7 (Figure 2) (Kawai et al., 2020). The binding of LY3502970 results in a distinct conformation of the ECD and the extracellular portion of the 7TM segments. The overall binding modes between LY3502970 and TT_OAD2 are different. TT_OAD2 adopts a U‐shaped orientation with both ends of its backbone sitting between TM2, TM3 and ECL1 (Zhao et al., 2020). For LY3502970, its 2,2‐dimethyl‐tetrahydropyran moiety occupies a similar position as the 2,3‐dime‐thylpyridine ring of TT‐OAD2, whereas its 4‐fluoro‐1‐methyl‐indazole moiety at the other end extends between TM1 and TM2. Further, its 3,5‐dimethyl‐4‐fluoro‐phenyl ring interacts with TM1 and TM7. Similarly, compound CHU‐128 adopts the same binding mode as LY3502970.
2.5. PF06882961
Using BETP‐sensitized cAMP screening assay for HT screening of 2.8 million compounds from the Pfizer compound collection, followed by lead optimization, Aspnes et al. at Pfizer Inc. reported a series of pyrimidine derivatives represented by compound 6 (PF06882961) (Figure 1, [6]) (Aspnes et al., 2018; Griffith et al., 2020). In vitro studies indicate that the pharmacological profile of PF06882961 is more closer to that of GLP‐1 than some closely related peptides such as exendin‐4, liraglutide and endogenous oxyntomodulin. It is a full agonist for cAMP production but only a partial agonist in Ca2+ mobilization, pERK1/2 and β‐arrestin recruitment. Kinetically, PF06882961 also has a similar kinetic profile to GLP‐1 in inducing Gs‐protein conformational change, Ca2+ signalling and pERK1/2. Overall, PF06882961 represents a particularly attractive structure through both in vitro and in vivo studies (Griffith et al., 2020) and is currently being evaluated in multiple clinical trials (ClinicalTrials.gov Identifier: NCT04552470 and others).
In the recently determined cryo‐EM structure of the GLP‐1 receptor with PF06882961 and Gs protein (X. Zhang et al., 2020), PF06882961 binds in a buried pocket within the helical bundle, adopting an elongated pose principally located within the bound GLP‐1 molecular envelope (Figures 2). The binding of PF06882961 overlaps with the location of GLP‐1 residues G10‐E21 in the GLP‐1‐bound structure. Further, the PF06882961 binding cavity extends deep into the receptor core and overlaps with the binding site of the GLP‐1 N‐terminal H7–E9. This allows PF06882961 to interact with water molecules below the peptide and stabilize the central polar network of the receptor. The binding sites for PF06882961 and LY3502970 or CHU‐128 overlap substantially, although each adopts a very distinct pose. This substantial overlap helps explain the species specificity of both PF06882961 and LY3502970 compounds.
3. PAMS OF GLP‐1 RECEPTORS
PAMs of the GLP‐1 receptor as drugs for the treatment of Type 2 diabetes could provide several potential benefits including augmenting the action of endogenous GLP‐1 receptor peptide agonists. Hence, discovering PAMs of these receptors has attracted intensive research efforts and a number of PAMs with favourable pharmacological profiles have been reported. Unlike non‐peptide agonists of GLP‐1 receptors, the known allosteric binding sites for PAMs appear to occupy many different sites in the structure of the GLP‐1 receptor. As a result, they adopt clearly different binding modes.
3.1. HT screening
The full‐length structures of the GLP‐1 receptor became available in 2017. For a long time, HT screening has been used extensively in various efforts to discover PAMs of GLP‐1 receptors. Among those reported in the early years, the most extensively studied are the Novo Nordisk compound 2 and the Eli Lilly compound BETP (also known as compound B) and its derivatives.
3.1.1. Compound 2 and other quinoxalines
The first reported and also the most extensively studied PAMs of the GLP‐1 receptor are a series of quinoxalines identified by Knudsen et al. (2007) at Novo Nordisk. In a screen for low MW agonists of GLP‐1 receptors, the group first screened 500,000 compounds using a competition‐binding assay and then screened 250,000 compounds using a functional assay, which was followed by structural modifications. This led to the discovery of a series of substituted quinoxalines, as low MW agonists of human GLP‐1 receptors, exemplified by compound 7 (compound 2) (Figure 3, [7]).
FIGURE 3.
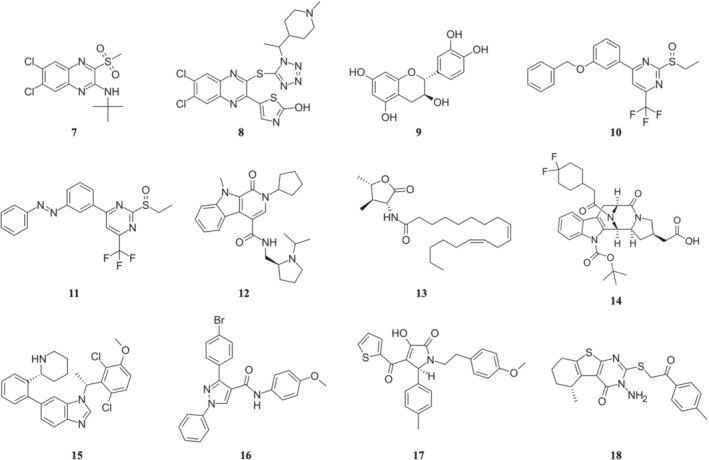
Chemical structures of PAMs of GLP‐1 receptors. Compounds shown are (7) compound 2, (8) DA‐15864, (9) catechin, (10) BETP, (11) PhotoETP, (12) VU0453379, (13) N55, (14) compound 19, (15) LSN3160440, (16) compound 20, (17) M_4 and (18) C‐1
Using a cAMP functional assay, compound 2 was shown to be a full agonist of human GLP‐1 receptors and is specific for the human receptor compared with other glucagon B1 GPCRs. Saturation‐binding and functional experiments showed compound 2 to increase the binding affinity of GLP‐1 to GLP‐1 receptors but not its potency. Further, the agonistic effect of compound 2 was not blocked by exendin(9–39). Additional studies determined that compound 2 stimulates glucose‐dependent insulin release from WT mouse islets but not from GLP‐1 receptor knockout mouse islets, and compound 2 and GLP‐1 have additive effects on insulin release. Collectively, these data demonstrate compound 2 interacts with human GLP‐1 receptors at a site different from the orthosteric binding site for GLP‐1 and functions as an agonistic, PAM (ago‐PAM).
The agonistic effect of compound 2 was independently confirmed by a subsequent report from University of Ulster, which examined the in vitro and in vivo metabolic actions of compound 2 (Irwin et al., 2010). Although less potent than GLP‐1, exenatide and liraglutide, compound 2 can significantly stimulate insulin secretion from BRIN‐BD11 cells in a concentration‐dependent fashion. In mice, compound 2 decreases the glucose concentration compared with controls. In another study using a cell line with recombinant expression for the human GLP‐1 receptor and the rat insulinoma cell line INS‐1E expressing GLP‐1 receptors, compound 2 significantly enhanced efficacy and potency of GLP‐1(9–36)NH2 for cAMP accumulation, Ca2+ signalling and pERK1/2, confirming its positive allosteric effects (Li et al., 2012).
With the discovery of compound 2 as an ago‐PAM of GLP‐1 receptors, systematic SAR studies using the quinoxaline scaffold were carried out at its C‐2 position, C‐3 position and the quinoxaline ring (Teng et al., 2007). The most potent and efficacious compounds are found to be 6,7‐dichlo‐roquinoxalines bearing an alkyl sulfonyl group at the C‐2 position and a secondary alkyl amino group at the C‐3 position. However, the active quinoxalines appear chemically unstable when treated with strong nucleophiles, for example, DTT, or strong bases, for example, KOH, and they also have high microsomal turnover rate. Hence, improvement on chemical stability and pharmacokinetic properties of these compounds is necessary before further in vivo characterization is conducted.
Other efforts to explore the quinoxaline scaffold have followed. A series of 2‐thio‐substituted quinoxalines were reported as weak GLP‐1 receptor agonists (Kopin & Beinborn, 2004). Another study reported the synthesis of two series of 3,6,7‐trisubstituted‐2‐(1H‐imidazol‐2‐ylsulfanyl)‐quinoxalines and 2‐(quinoxalin‐2‐yl)‐isothioureas (Bahekar et al., 2007). The glucose‐dependent insulinotropic activity of these compounds was identical to that of compound 2 using a RIN5F cell‐base assay. Moon et al. (2011) disclosed another 2‐thio‐quinoxaline analogue, compound 8 (DA‐15864) (Figure 3, [8]). In CHO cells, DA‐15864 selectively stimulates human with EC50 values of 163 nM and increases intracellular cAMP levels. In rat insulinoma cells, the compound significantly increases glucose‐stimulated insulin secretion and acts synergistically with GLP‐1. In vivo intravenous glucose tolerance tests using normal mice showed that DA‐15864 increased the peak plasma level of insulin significantly. Thus, DA‐15864 appears to be a promising, orally available, GLP‐1 receptor agonist with the potential for treatment of diabetes.
Apart from their location in pancreatic beta‐cells, GLP‐1 receptors are widely expressed in the brain and are also considered to be an effective therapeutic target for diseases of the CNS. Thus, GLP‐1 mimetics including exendin‐4 and liraglutide have neuroprotective effect on ischaemic stroke (Briyal et al., 2012; Darsalia et al., 2014; Li et al., 2009; Sato et al., 2013). Given the agonistic and modulating effect of compound 2 on GLP‐1 receptors, its potential neuroprotective effects on cerebral ischemia were evaluated (H. Zhang et al., 2016). In vitro, compound 2 increased cortical neuron survival in oxygen–glucose deprivation and reperfusion. In middle cerebral artery occlusion (MCAO) mice, compound 2 with exendin‐4 significantly decreased the neurological deficit following MCAO. Using Western blot, GLP‐1 receptors present in cortical neurons could be activated by compound 2. Measuring the stimulated cAMP level in neuronal cells, compound 2 and exendin‐4 induced activation of GLP‐1 receptors, in an additive manner. Therefore, the neuroprotective effects observed are mediated by the activation of the GLP‐1 receptors through the cAMP‐PKA‐CREB signalling pathway.
3.1.2. Flavonoids
In search for GPCR allosteric modulators, Domain Therapeutics reported a series of quercetin‐like flavonoids as PAMs of GLP‐1 receptors (Schann et al., 2009). These compounds and other flavonoids such as flavones, isoflavones and catechin (compound 9) (Figure 3, [9]) were subsequently characterized by Koole et al., (2010) and Wootten et al. (2011). A series of hydroxyl flavonols and catechin were found to be probe dependent and induce biased signalling. Hydroxyl flavonols do not have an effect on cAMP signalling but selectively modulate Ca2+ signalling in a peptide‐agonist dependent way. The Ca2+ modulation of quercetin was only observed with truncated GLP‐1 peptide or exendin‐4 and not on oxyntomodulin or full‐length peptides. On the other hand, catechin shows no allosteric effects on peptide‐mediated Ca2+ signalling. However, catechin negatively modulates cAMP formation in the presence of truncated GLP‐1 peptide, but not with full‐length GLP‐1, oxyntomodulin or exendin‐4.
Furthermore, the effects of the hydroxyl flavonols and catechin were driven primarily by their effects on orthosteric ligand efficacy. Although the undesirable characteristics of these polyphenolic compounds preclude them from further optimization for pharmacological purposes, the discovery of this series of compounds is further evidence for the allosterically modulation of GLP‐1 receptor function.
3.1.3. Pyrimidines
Using cell‐based and insulin secretion assays with rodent and human islets to screen a small library of compounds, generated through a three‐dimensional pharmacophore model, followed by chemical modification, Sloop et al. (2010) at Eli Lilly and Company reported a series of pyrimidine‐based compounds, represented by compound 10 (Compound B, or BETP) (Figure 3, [10]), that activate GLP‐1 receptors and stimulate glucose‐dependent insulin secretion. Similar to the quinoxalines, these compounds exhibit both agonistic and positive allosteric modulating effects on GLP‐1 receptors. BETP alone induced GLP‐1 receptor‐mediated cAMP signalling in HEK293 cells, leading to increased insulin secretion from rodent and human islets in a dose‐dependent manner. Combined with endogenous GLP‐1 peptide, BETP did not compete with 125I‐GLP‐1 but instead acted in an additive manner to induce both cAMP production and insulin secretion, suggesting BETP binds in an allosteric site and can function as a PAM. Consistent with these results, the activity of BETP was not blocked by the GLP‐1 competitive antagonist exendin‐4(9–39), and BETP can activate even the ECD truncated GLP‐1 receptor. In vivo studies show that BETP can stimulate insulin secretion in Sprague–Dawley rats.
In a subsequent study, the potential modulating effects of BETP on oxyntomodulin were examined (Willard, Wootten, et al., 2012). In vitro studies were carried out using a heterologous system consisting of HEK293 cells expressing human GLP‐1 receptors, BETP potentiated oxyntomodulin‐induced GLP‐1 receptor signalling and increased the binding affinity of oxyntomodulin for GLP‐1 receptors. BETP also enhances the activation of the Gs protein. In vivo studies by performing an IVGTT in Wistar rats indicated that BETP enhanced oxyntomodulin‐stimulated insulin secretion. In line with these observations, BETP induced biased signalling at oxyntomodulin‐mediated GLP‐1 receptors by selectively enhancing cAMP accumulation over other pathways including Ca2+ mobilization, pERK1/2 or β‐arrestin recruitment. In several following studies, the probe dependence of BETP in the presence of truncated GLP‐1(7–36)NH2 and GLP‐1(9–36)NH2 was further confirmed (Bueno et al., 2016; Li et al., 2012).
In another study, the mode of action of BETP was compared with compound 2, and clear differences were observed (Cheong et al., 2012). Both compounds can function as the GLP‐1 receptor agonist and independently activate human GLP‐1 receptors. However, they exert different probe dependence in the presence of full‐length GLP‐1, GLP‐1(9–36) or exenatide. Compound 2 shows an additive effect but did not increase the maximum efficacy when combined with full‐length GLP‐1, whereas BETP increases the maximum efficacy of GLP‐1 in a concentration‐dependent manner. For Ca2+ influx, both compounds slowly increase Ca2+ influx. However, the response induced by BETP lasts longer than that of compound 2 and GLP‐1. A similar study has also confirmed that BETP and compound 2 display different probe dependence and induce distinctive biased signalling (Wootten et al., 2013). Given the evident structural differences between the pyrimidine series and the quinoxaline series which may result in different active conformations, their different action pattern is expected.
To further understand the mechanism of BETP on GLP‐1 receptor activation and signalling, Eng et al. (2013) have determined that BETP covalently modifies Cys 347 located in the third intracellular loop of GLP‐1 receptors and this Cys residue is critical for its allosteric effect (Figure 4). Another study showed that diverse PAMs of GLP‐1 receptors including BETP and compound 2, with their electrophilic nature, all activate the receptor via the same mechanism as BETP (Bueno et al., 2016). Specifically, compound 2 is proposed to be located orthogonally above helix 6 and form multiple interactions with residues at the interface of helices 5 and 6. This binding model was experimentally validated by mutagenesis studies (Song et al., 2017). As both BETP and compound 2 as well as DA‐15864 and several others PAMs, all covalently modify the C347 residue of GLP‐1 receptors (Bueno et al., 2016; Eng et al., 2013) and display poor pharmacokinetic properties, these factors have prevented their further clinical development (Willard, Bueno et al., 2012).
FIGURE 4.
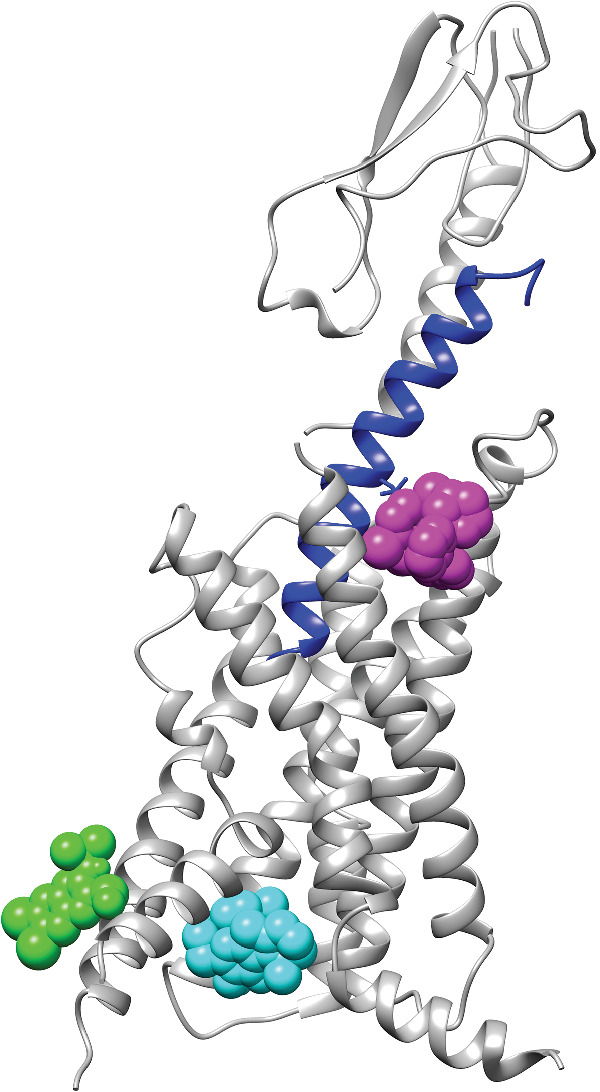
Binding sites for PAMs of GLP‐1 receptors. The GLP‐1 receptor structure is shown as cartoons (PDB ID: 6VCB), and the GLP‐1 peptide is coloured in blue. Positive allosteric modulators (PAMs) are shown as coloured spheres. Green, compound 2 (docking model); cyan, C‐1 (docking model); and magenta, LSN3160440 (PDB ID: 6VCB)
3.1.4. PhotoETP
Both compound 2 and BETP demonstrate several attractive features that may be beneficial to the mechanistic studies of allosteric regulation of GLP‐1 receptors. For instance, both compounds showed probe dependence and both can induce biased signalling of the GLP‐1 receptor pathway in the presence of various GLP‐1 receptor peptides. To fine‐tune the activity of BETP, Broichhagen et al. (2016) designed an optically activated analogue of BETP (PhotoETP) in which the O‐benzyl group of BETP was replaced by an azobenzene group (compound 11) (Figure 3, [11]). This PhotoETP can be activated or inactivated by switching between the trans‐isomer and cis‐isomer by light. Although both the trans‐isomer and the cis‐isomer can augment insulin secretion in response to GLP‐1(7–36)NH2, only the trans‐isomer demonstrates modulating activity profile similar to BETP. In CHO cells expressing GLP‐1 receptors, only the trans‐isomer activates cAMP production in the presence of GLP‐1(9–36)NH2; in beta‐cell islets, only the trans‐isomer increases Ca2+ levels in GLP‐1(7–36)NH2‐induced signalling.
In a more recent report, the same group experimented with varying moieties of the base structures of BETP with restricted degrees of freedom in order to understand the effects of ligand conformation in allosteric binding of GLP‐1 receptors (Jones et al., 2017). Compounds incorporating a trans‐stilbene outperform those with cis‐stilbene, as well as BETP, in enhancing cAMP accumulation and Ca2+ influx.
3.1.5. Compound VU0453379
Morris, Days, et al. (2014) developed an innovative HT screening system based on a primary Ca2+ screen, a secondary cAMP screen and follow‐up testing to identify non‐peptide agonists and PAMs of human GLP‐1 receptors. Out of 175,000 compounds initially screened, 98 compounds showed a variety of activities at GLP‐1 receptors. Among them, VU00056556 and VU0109197, which share a common hexahydroquinolone carboxylate core, were PAMS for GLP‐1 receptors. Both compounds induced GLP‐1‐dependent cAMP accumulation and Ca2+ mobilization within the human 9‐3‐H cells expressing GLP‐1 receptors at a higher level than GLP‐1 alone. In addition, several other compounds can modulate GLP‐1 receptor activities.
Following the HT screening, the group selected one lead compound, separated its racemic isomers and further optimized the structure of its (S)‐enantiomer. This was followed by SAR studies using an analogue library. All these efforts led to the discovery of a novel PAM, compound 12 (VU0453379) (Figure 3, [12]) (Morris, Nance, et al., 2014). VU0453379 exhibits no probe dependence by potentiating endogenous GLP‐1 as well as synthetic peptide agonists, exendin 4 and liragulitide. VU0453379 shows biased signalling with weak effects on β‐arrestin recruitment and is highly selective towards GLP‐1 receptors. In primary mouse pancreatic islets, VU0453379 augmented glucose‐dependent insulin secretion by low‐dose exendin‐4. Using a haloperidol‐induced catalepsy model, VU0453379 significantly reversed catalepsy by potentiating endogenous GLP‐1 receptors. With favourable physiochemical properties, VU0453379 also demonstrates favourable metabolic and pharmokinetic profiles. A rather important observation is that VU0453379 crossed the blood‐brain barrier and was the first CNS‐penetrant GLP‐1 receptor PAM ever reported.
3.1.6. HIT‐465 and HIT‐736
The endogenous partial agonist of GLP‐1 receptors, GLP(9–36)NH2, was distinguished due to its increased bioavailability and longer half‐life than its non‐degraded counterpart GLP‐1(7–36)NH2. It is thus of interest to develop PAMs that can modulate both GLP‐1(7–36)NH2 and GLP‐1(9–36)NH2. Employing a comprehensive screening system consisting of Ca2+ mobilization assay, cAMP accumulation assay, β‐arrestin assay and receptor internalization assay, Nakane et al. (2015) have screened approximately 450,000 low MW compounds. Two compounds, HIT465 and HIT‐736, whose structures are not disclosed, were identified to potentiate the human GLP‐1 receptor response to GLP‐1(9–36)NH2, at the same level as compound 2. Further studies indicated that HIT‐465 and HIT‐736 are biased modulators of human GLP‐1 receptors. To study the mechanism of these two compounds, GLP‐1 receptor mutants that reduce allosteric modulating activity of compound 2 were expressed in CHO‐K1 cells, respectively. The modulatory activities of HIT‐465 and HIT‐736 using these mutants were not affected in the presence of either GLP‐1(7–36)NH2 or GLP‐1(9–36)NH2. Together, these data suggested that the allosteric binding site and mode of mechanism of HIT‐465 and HIT‐736 are different from those of compound 2.
3.1.7. Compound N55
Fenugreek (Trigonella foenum‐graecum) seeds have been observed to reduce glucose and glycated haemoglobin levels, thereby alleviate some effects of Type 2 diabetes. By screening extracts from fenugreek seeds using an intracellular cAMP biosensor and GLP‐1 receptor endocytosis assays, King et al. (2015) found that ethanol extracts enhanced GLP‐1 signalling. Further purification identified an active compound 13 (N55) (Figure 3, [13]), which has a N‐linoleoyl‐2‐amino‐γ‐butyrolactone structure. In vitro studies show that N55 promotes GLP‐1‐dependent cAMP accumulation and GLP‐1 receptor endocytosis in a dose‐dependent manner. N55 also exhibits strong probe dependence in that it specifically enhances activity of GLP‐1(7–36)NH2 but not GIP or exendin‐4.
What is most interesting about compound N55 is its unique mechanism. GLP‐1 is known to bind to signal‐enhanced oleoylethanolamide (OEA) or 2‐oleoylglycerol but not to stearoylethanolamide (SEA) (Cheng et al., 2015). To understand the mechanism of N55, competition‐binding experiments were performed to examine the effect of increasing N55 concentrations on the binding of [3H] OEA to GLP‐1. As the concentration of N55 increased, binding of OEA gradually decreased. This was further confirmed by trypsin cleavage experiments. When the trypsin cleavage reactions were carried out in the presence of 77‐μM N55, the residual GLP‐1 activity was dramatically reduced compared with that without N55. These experiments suggest that rather than binding to an allosteric site on GLP‐1 receptors, like other known PAMs, N55 binds to GLP‐1(7–36)NH2 directly. The binding of N55 to GLP‐1 may induce conformational change in GLP‐1(7–36)NH2, thus slowing its cleavage and digestion by trypsin enzymes. Hence, N55 represents a new class of PAMs of GLP‐1 receptors and targeting GLP‐1 peptides could be a viable approach to modulation of GLP‐1 receptor activity.
3.1.8. Compound 19
In another effort to take advantage of the increased bioavailability and longer half‐life of GLP‐1(9–36)NH2, a group at Sanofi‐Aventis Deutschland GmbH performed HT screening of its own compound collection using an HTRF cAMP assay in a HEK293 cell line overexpressing the human GLP‐1 receptor, which was followed by chemical optimization of the lead compounds (Méndez et al., 2020). These efforts led to the discovery of compound 14 (compound 19) (Figure 3, [14]) based on a 3,4,5,6‐tetrahydro‐1H‐1,5‐epiminoazocino[4,5‐b]indole scaffold. In vitro assay demonstrated that compound 19 is a potent GLP‐1 receptor PAM by clearly potentiating GLP(9–36)NH2 with EC50 value of 5 nM. In the endogenous pancreatic beta‐cell line 1.1B4 cells, compound 19 also shows robust PAM efficacy. In dispersed rat pancreatic islets, compound 19 significantly improved the GLP‐1(9–36)NH2‐mediated glucose‐stimulated insulin secretion. In vivo pharmacokinetic and pharmacological characterization indicated that compound 19 allosterically activated GLP‐1 receptors in the presence of GLP(9–36)NH2. To date, compound 19 is the most potent non‐covalent PAM of GLP‐1 receptors reported, although the exogenous addition of GLP(9–36)NH2 is still needed in order to elicit a significant response on glucose homeostasis in vivo.
3.1.9. LSN3160440
In another effort to take advantage of endogenous GLP‐1(9–36)NH2, the group at Eli Lilly and Company performed the screening of a diverse 220,000 compound library using the HEK293 cells expressing human GLP‐1 receptors, in the presence of a 20%‐maximum effective concentration (EC20) of GLP‐1(9–36). The screening led to the discovery of a known MET kinase inhibitor as a weak potentiator of GLP‐1 receptors (Bueno et al., 2020). Multiple rounds of SAR optimization were conducted on this lead compound which resulted in the discovery of compound 15 (LSN3160440) (Figure 3, [15]). In vitro, LSN3160440 enhanced the potency and efficacy of GLP‐1(9–36) in GLP‐1 receptor‐induced cAMP signalling. Competitive binding studies indicated LSN3160440 cooperatively modulated the binding affinity and efficacy of GLP‐1(9–36) for GLP‐1 receptor activation. LSN3160440 is selective against MET and 33 other diverse kinases, as well as 261 GPCRs. LSN3160440 also exhibits strong probe dependence, being a robust potentiator of GLP‐1(9–36) but not oxyntomodulin or full‐length GLP‐1.
In pancreatic islets from mice, LSN3160440 combined with GLP‐1(9–36) significantly increases glucose‐dependent insulin secretion to the levels comparable to that produced by full‐length GLP‐1. In vivo characterization using Wistar rats indicated that the combination of LSN3160440 and GLP‐1(9–36) elicits an insulinotropic effect similar to that produced by full‐length GLP‐1.
The cryo‐EM structure of GLP‐1 receptors bound to LSN3160440, GLP‐1 and Gs protein was solved which indicates LSN3160440 binds at the extracellular side of the helical bundle in a pocket formed by residues in the interface between TM1 and TM2 (Figure 4) (Bueno et al., 2020). In the complex structure, LSN3160440 interacts with both GLP‐1 peptide and GLP‐1 receptor simultaneously. The 2,6‐dichloro‐3‐methoxyl phenyl moiety of LSN3160440 forms van der Waals (vdW) contacts with the GLP‐1 peptide and the benzimidazole of LSN3160440 forms vdW interactions and π–π stacking with the GLP‐1 receptor. LSN3160440 is likely to be the only reported PAM for a receptor that mediates its positive allostery through simultaneous interactions with both the orthosteric ligand and the receptor. Reciprocal site‐specific mutagenesis studies demonstrate that one varied amino acid between GLP‐1(9–36) and oxyntomodulin, V16 of GLP‐1 and Y10 of oxyntomodulin, confers probe dependency on LSN3160440. The discovery of LSN3160440 and its unique binding mode indicates that developing stabilizers at protein–protein interface to affect cell surface signalling can be achieved.
3.2. Computer‐aided molecule design
Along with HT screening, structure‐based virtual screening has also been used to identify novel PAMs of GLP‐1 receptors. Unlike the former, structure‐based computational approach is much cheaper and provides a rational way to further optimize a lead compound.
3.2.1. Compound 20
The discovery of GLP‐1 receptor PAMs, such as compound 2 and BETP, inspired screening efforts, applying computer‐aided molecular design techniques. The first report employing structure‐based virtual screening adopted the X‐ray crystal structure of Class A rhodopsin receptors to construct 3D models of two Class B GPCRs, the corticotropin‐releasing hormone CRF1 receptor and the glucagon receptor (de Graaf et al., 2011). The model of the glucagon receptor was subsequently used to screen a database of 1.9 million commercially available drug‐like compounds in an effort to identify non‐competitive antagonists. Twenty‐three compounds were selected and evaluated in vitro in a whole cell‐based functional assay for binding to glucagon receptors and human GLP‐1 receptors and for modulation of receptor‐induced cAMP accumulation. Two compounds were confirmed to inhibit glucagon receptor activity in a dose‐dependent manner, and one was characterized as a PAM of GLP‐1 receptors, compound 16 (compound 20) (Figure 3, [16]). Although the potency of compound 20 is modest, this work demonstrates the usefulness of the computational approach in discovering PAMs for Class B GPCRs.
3.2.2. Compound M_4 and C‐1
Following the publication of the structure of two Class B GPCRs, the CRF1 receptor (PDB ID: 4K5Y) and the glucagon receptor (PDB ID: 4L6R), our group constructed the homology models of human GLP‐1 receptors, based on those two crystal structures and carried out structure‐based virtual screening of 5689 compounds from the ZINC database (Redij, Chaudhari, et al., 2019). These compounds have similar physicochemical properties to those of potential low MW agonists of GLP‐1 receptors and were identified through ligand‐based similarity search. Eight top‐ranked compounds from virtual screening were selected and evaluated using the GLP‐1 receptor‐dependent luciferase reporter system. Two compounds were confirmed to activate human GLP‐1 receptors in a dose‐dependent manner and one synergized with GLP‐1 to stimulate GLP‐1 receptor activity, compound 17 (M_4) (Figure 3, [17]). Using in vitro insulin secretion assay in INS‐1832/13 cells, compound M_4 induced glucose‐dependent insulin secretion.
When the cryo‐EM structure of GLP‐1 receptors at the active state became available (Y. Zhang et al., 2017), we carried out another round of structure‐based screening studies using this structure and identified another compound as a PAM for GLP‐1 receptors, compound 18 (C‐1) (Figure 3, [18]) (Redij, Ma, et al., 2019). Using the same GLP‐1 receptor‐dependent luciferase reporter system, compound C‐1 activates human GLP‐1 receptors in a dose‐dependent manner. When combined with GLP‐1, C‐1 improves GLP‐1's affinity and efficacy to human GLP‐1 receptors. Using in vitro insulin secretion assay in INS‐1832/13 cells, compound C‐1 (9.7 μM), induced insulin secretion at the similar level as that of GLP‐1 (181 nM). Combined with GLP‐1, C‐1 again showed the synergistic effect in stimulating insulin secretion. Despite its modest activity, this compound demonstrates favourable drug‐like properties. For instance, with the molecular weight of 399, this compound represents one of the smallest known PAMs for the GLP‐1 receptor.
In our structure‐based screening work based on the cryo‐EM structure of GLP‐1 receptors (Y. Zhang et al., 2017), we have utilized the intracellular region of these receptors near the G‐protein‐binding site to discover C‐1, a GLP‐1 receptor PAM (Figure 4) (Redij, Ma, et al., 2019). To confirm this binding site for C‐1, we have subsequently carried out site‐specific mutagenesis studies using three mutants N406A, S352A and V332W individually. Among them, N406 and S352 are predicted to bind to compound C‐1 directly, and V332 is not present in the predicted binding site and used as the control. Treatment of the cells with C‐1 in combination with GLP‐1 significantly increased the activity of WT GLP‐1 receptors. Under the same conditions, C‐1 failed to activate the N406A and S352A mutant GLP‐1 receptors , but not the V332A mutant. These data support the notion that the intracellular region of GLP‐1 receptors near the G‐protein‐binding site is the binding site for C‐1.
4. CONCLUSIONS AND FUTURE PERSPECTIVES
Since the approval of exendin‐4, the first GLP‐1 receptor peptide drug in 2005, remarkable progress has been made in the discovery, optimization and clinical development of non‐peptide agonists and PAMs for GLP‐1 receptors, as shown by the three non‐peptide agonists currently in the preclinical or clinical trials. Further, recent breakthroughs in structure determination of GLP‐1 receptor complexes have greatly facilitated the identification of agonist and PAM binding sites and binding modes as well as their mechanisms of action.
All the non‐peptide agonists of GLP‐1 receptors of known structure bind primarily in the helix bundle of the receptor with the binding pocket overlapping to that of GLP‐1 peptide and the binding mode either similar or distinct from GLP‐1 (Figure 2). The multiple active conformations of GLP‐1 receptors, induced by different agonist‐binding modes, can transduce a common conformational change at the intracellular side of TMD, which subsequently induces G‐protein binding. On the other hand, these differences in the binding mode contribute to the different efficacy and biased agonism of these agonistic molecules.
The discovery of non‐peptide agonists of GLP‐1 receptors with efficacy similar to that of the endogenous peptide hormone and the structural determination of their binding modes within GLP‐1 receptors clearly demonstrate that non‐peptide agonists are not required to closely mimic the extensive interactions formed between peptides and ECD and TMD of GLP‐1 receptors in order to induce receptor activation. Identifying non‐peptide ligands is the principal bottleneck in drug discovery for Class B GPCRs. The uncovering of the binding pocket for non‐peptide agonists in the TMD of GLP‐1 receptors which overlaps with the binding site of the N‐terminus of peptides provides a novel avenue to the discovery of non‐peptide agonists for other therapeutically important Class B GPCRs.
Although there are no low MW PAMs entering clinical trials, investigations of the effects of low MW modulators present compelling evidence that continuing to explore PAM compounds and their relevant properties related to GLP‐1 receptor signalling should prove beneficial. Unlike non‐peptide agonists of GLP‐1 receptors and like allosteric modulators of other GPCRs (Chan et al., 2019), the allosteric binding sites of PAMs seem to be located at several different sites, all over the GLP‐1 receptor structures, including the GLP‐1 peptide itself, in the extracellular region of the TMD and in the intracellular region of the TMD (Figure 4). Given the fact that their binding sites and binding modes vary markedly, it is understandable that these PAMs affect the affinity and efficacy of orthosteric ligand in a probe‐dependent way and induce biased signalling in GLP‐1 receptor‐mediated pathways.
Binding of a non‐peptide agonist or a PAM molecule to GLP‐1 receptors is essential to the activation and signalling of these receptors. Elucidation of the binding sites for non‐peptide agonists and PAMs and characterization of their interactions with GLP‐1 receptors are important for the understanding of modulating mechanism of non‐peptide agonists and PAMs as well as for the structure‐based design of molecules. Site‐specific mutagenesis data from our lab support the notion that the intracellular region of GLP‐1 receptors near the G‐protein‐binding site is the binding site for C‐1. Interestingly, this allosteric site is also where two NAMs of GLP‐1 receptors (Song et al., 2017) and a few NAMs of other GPCRs (Chan et al., 2019) bind. Given the ubiquitous existence of this binding pocket in all solved GPCR structures, it could be a common allosteric site for all GPCRs. It will be of great interest to determine whether this is the case.
4.1. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in the IUPHAR/BPS Guide to PHARMACOLOGY (http://www.guidetopharmacology.org), and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander, Christopoulos et al., 2019; Alexander, Fabbro et al., 2019).
AUTHOR CONTRIBUTIONS
The manuscript was written through contributions of both authors. Both authors have given approval to the final version of the manuscript. F.M. prepared the figures; L.Z. directed the project and designed the figures.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
ACKNOWLEDGEMENTS
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R15GM140406. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Malik, F. , & Li, Z. (2022). Non‐peptide agonists and positive allosteric modulators of glucagon‐like peptide‐1 receptors: Alternative approaches for treatment of Type 2 diabetes. British Journal of Pharmacology, 179(4), 511–525. 10.1111/bph.15446
Funding information National Institute of General Medical Sciences of the National Institutes of Health, Grant/Award Number: R15GM140406
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article because no new data were created or analysed in this study.
REFERENCES
- Alexander, S. P. H. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Armstrong, J. F. , Faccenda, E. , Harding, S. D. , Pawson, A. J. , Sharman, J. L. , Southan, C. , Davies, J. A. , & CGTP Collaborators . (2019). The concise guide to pharmacology 2019/20: G protein‐coupled receptors. British Journal of Pharmacology, 176, S21–S141. 10.1111/bph.14748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Armstrong, J. F. , Faccenda, E. , Harding, S. D. , Pawson, A. J. , Sharman, J. L. , Southan, C. , Davies, J. A. , & CGTP Collaborators . (2019). The Concise Guide to PHARMACOLOGY 2019/20: Enzymes. British Journal of Pharmacology, 176, S297–S396. 10.1111/bph.14752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspnes, G. E. , Bagley, S. W. , Curto, J. M. , Dowling, M. S. , Edmonds, D. J. , Flanagan, M. E. , Futatsugi, K. , Griffith, D. A. , Huard, K. , Ingle, G. , Jiao, W. , Limberakis, C. , Mathiowetz, A. M. , Piotrowski, D. W. , & Ruggeri, R. B. (2018). Glp‐1 receptor agonists and uses thereof. WO/2018/109607
- Bahekar, R. H. , Jain, M. R. , Gupta, A. A. , Goel, A. , Jadav, P. A. , Patel, D. N. , Prajapati, V. M. , & Patel, P. R. (2007). Synthesis and antidiabetic activity of 3,6,7‐trisubstituted‐2‐(1H‐imidazol‐2‐ylsulfanyl)quinoxalines and quinoxalin‐2‐yl isothioureas. Archiv der Pharmazie, 340(7), 359–366. 10.1002/ardp.200700024 [DOI] [PubMed] [Google Scholar]
- Briyal, S. , Gulati, K. , & Gulati, A. (2012). Repeated administration of exendin‐4 reduces focal cerebral ischemia‐induced infarction in rats. Brain Research, 1427, 23–34. 10.1016/j.brainres.2011.10.026 [DOI] [PubMed] [Google Scholar]
- Broichhagen, J. , Johnston, N. R. , von Ohlen, Y. , Meyer‐Berg, H. , Jones, B. J. , Bloom, S. R. , Rutter, G. A. , Trauner, D. , & Hodson, D. J. (2016). Allosteric optical control of a class B G‐protein‐coupled receptor. Angewandte Chemie International Edition, 55(19), 5865–5868. 10.1002/anie.201600957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueno, A. B. , Showalter, A. D. , Wainscott, D. B. , Stutsman, C. , Marin, A. , Ficorilli, J. , Cabrera, O. , Willard, F. S. , & Sloop, K. W. (2016). Positive allosteric modulation of the glucagon‐like peptide‐1 receptor by diverse electrophiles. The Journal of Biological Chemistry, 291(20), 10700–10715. 10.1074/jbc.M115.696039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueno, A. B. , Sun, B. , Willard, F. S. , Feng, D. , Ho, J. D. , Wainscott, D. B. , Showalter, A. D. , Vieth, M. , Chen, Q. , Stutsman, C. , Chau, B. , Ficorilli, J. , Agejas, F. J. , Cumming, G. R. , Jiménez, A. , Rojo, I. , Kobilka, T. S. , Kobilka, B. K. , & Sloop, K. W. (2020). Structural insights into probe‐dependent positive allosterism of the GLP‐1 receptor. Nature Chemical Biology, 16(10), 1105–1110. 10.1038/s41589-020-0589-7 [DOI] [PubMed] [Google Scholar]
- Capozzi, M. E. , Wait, J. B. , Koech, J. , Gordon, A. N. , Coch, R. W. , Svendsen, B. , Finan, B. , D'Alessio, D. A. , & Campbell, J. E. (2019). Glucagon lowers glycemia when β‐cell s are active. JCI Insight, 5(16), e129954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, H. C. S. , Li, Y. , Dahoun, T. , Vogel, H. , & Yuan, S. (2019). New binding sites, new opportunities for GPCR drug discovery. Trends in Biochemical Sciences, 44(4), 312–330. 10.1016/j.tibs.2018.11.011 [DOI] [PubMed] [Google Scholar]
- Chen, D. , Liao, J. , Li, N. , Zhou, C. , Liu, Q. , Wang, G. , Zhang, R. , Zhang, S. , Lin, L. , Chen, K. , Xie, X. , Nan, F. , Young, A. A. , & Wang, M. W. (2007). A nonpeptidic agonist of glucagon‐like peptide 1 receptors with efficacy in diabetic db/db mice. Proceedings of the National Academy of Sciences of the United States of America, 104(3), 943–948. 10.1073/pnas.0610173104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, Y.‐H. , Ho, M.‐S. , Huang, W.‐T. , Chou, Y.‐T. , & King, K. (2015). Modulation of glucagon‐like peptide‐1 (GLP‐1) potency by endocannabinoid‐like lipids represents a novel mode of regulating GLP‐1 receptor signaling. The Journal of Biological Chemistry, 290(23), 14302–14313. 10.1074/jbc.M115.655662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong, Y.‐H. , Kim, M.‐K. , Son, M.‐H. , & Kaang, B.‐K. (2012). Two small molecule agonists of glucagon‐like peptide‐1 receptor modulate the receptor activation response differently. Biochemical and Biophysical Research Communications, 417(1), 558–563. 10.1016/j.bbrc.2011.12.004 [DOI] [PubMed] [Google Scholar]
- Darsalia, V. , Hua, S. , Larsson, M. , Mallard, C. , Nathanson, D. , Nyström, T. , Sjöholm, Å. , Johansson, M. E. , & Patrone, C. (2014). Exendin‐4 reduces ischemic brain injury in normal and aged type 2 diabetic mice and promotes microglial M2 polarization. PLoS ONE, 9(8), e103114. 10.1371/journal.pone.0103114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies, M. , Pieber, T. R. , Hartoft‐Nielsen, M.‐L. , Hansen, O. K. H. , Jabbour, S. , & Rosenstock, J. (2017). Effect of oral semaglutide compared with placebo and subcutaneous semaglutide on glycemic control in patients with type 2 diabetes: A randomized clinical trial. JAMA, 318(15), 1460–1470. 10.1001/jama.2017.14752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Graaf, C. , Rein, C. , Piwnica, D. , Giordanetto, F. , & Rognan, D. (2011). Structure‐based discovery of allosteric modulators of two related class B G‐protein‐coupled receptors. ChemMedChem, 6(12), 2159–2169. 10.1002/cmdc.201100317 [DOI] [PubMed] [Google Scholar]
- De Graaf, C. , Donnelly, D. , Wootten, D. , Lau, J. , Sexton, P. M. , Miller, L. J. , Ahn, J.‐M. , Liao, J. , Fletcher, M. M. , Yang, D. , Brown, A. J. H. , Zhou, C. , Deng, J. , & Wang, M.‐W. (2016). Glucagon‐like peptide‐1 and its class B G protein–coupled receptors: A long march to therapeutic successes. Pharmacological Reviews, 68(4), 954–1013. 10.1124/pr.115.011395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorr, P. , Westby, M. , Dobbs, S. , Griffin, P. , Irvine, B. , Macartney, M. , Mori, J. , Rickett, G. , Smith‐Burchnell, C. , Napier, C. , Webster, R. , Armour, D. , Price, D. , Stammen, B. , Wood, A. , & Perros, M. (2005). Maraviroc (UK‐427,857), a potent, orally bioavailable, and selective small‐molecule inhibitor of chemokine receptor CCR5 with broad‐spectrum anti‐human immunodeficiency virus type 1 activity. Antimicrobial Agents and Chemotherapy, 49(11), 4721–4732. 10.1128/AAC.49.11.4721-4732.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng, H. , Sharma, R. , McDonald, T. S. , Edmonds, D. J. , Fortin, J.‐P. , Li, X. , Stevens, B. D. , Griffith, D. A. , Limberakis, C. , Nolte, W. M. , Price, D. A. , Jackson, M. , & Kalgutkar, A. S. (2013). Demonstration of the innate electrophilicity of 4‐(3‐[benzyloxy]phenyl)‐2‐(ethylsulfinyl)‐6‐(trifluoromethyl)pyrimidine (BETP), a small‐molecule positive allosteric modulator of the glucagon‐like peptide‐1 receptor. Drug Metabolism and Disposition, 41(8), 1470–1479. 10.1124/dmd.113.052183 [DOI] [PubMed] [Google Scholar]
- Eng, J. , Kleinman, W. A. , Singh, L. , Singh, G. , & Raufman, J. P. (1992). Isolation and characterization of exendin‐4, an exendin‐3 analogue, from Heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas. The Journal of Biological Chemistry, 267(11), 7402–7405. 10.1016/S0021-9258(18)42531-8 [DOI] [PubMed] [Google Scholar]
- Glaesner, W. , Vick, A. M. , Millican, R. , Ellis, B. , Tschang, S.‐H. , Tian, Y. , Bokvist, K. , Brenner, M. , Koester, A. , Porksen, N. , Etgen, G. , & Bumol, T. (2010). Engineering and characterization of the long‐acting glucagon‐like peptide‐1 analogue LY2189265, an Fc fusion protein. Diabetes/Metabolism Research and Reviews, 26(4), 287–296. 10.1002/dmrr.1080 [DOI] [PubMed] [Google Scholar]
- Griffith, D. A. , Edmonds, D. J. , Fortin, J.‐P. , Kalgutkar, A. S. , Kuzmiski, J. B. , Loria, P. M. , Saxena, A. R. , Bagley, S. W. , Buckeridge, C. , Curto, J. M. , Derksen, D. R. , Dias, J. M. , Griffor, M. C. , Han, S. , Jackson, V. M. , Landis, M. S. , Lettiere, D. J. , Limberakis, C. , Liu, Y. , … Tess, D. A. (2020). A small‐molecule oral agonist of the human glucagon‐like peptide‐1 receptor. BioRxiv, 2020.09.29.319483. 10.1101/2020.09.29.319483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington, P. E. , & Fotsch, C. (2007). Calcium sensing receptor activators: Calcimimetics. Current Medicinal Chemistry, 14(28), 3027–3034. 10.2174/092986707782794096 [DOI] [PubMed] [Google Scholar]
- Irwin, N. , Flatt, P. R. , Patterson, S. , & Green, B. D. (2010). Insulin‐releasing and metabolic effects of small molecule GLP‐1 receptor agonist 6,7‐dichloro‐2‐methylsulfonyl‐3‐N‐tert‐butylaminoquinoxaline. European Journal of Pharmacology, 628(1–3), 268–273. 10.1016/j.ejphar.2009.11.022 [DOI] [PubMed] [Google Scholar]
- Jones, B. J. , Scopelliti, R. , Tomas, A. , Bloom, S. R. , Hodson, D. J. , & Broichhagen, J. (2017). Potent prearranged positive allosteric modulators of the glucagon‐like peptide‐1 receptor. ChemistryOpen, 6(4), 501–505. 10.1002/open.201700062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai, T. , Sun, B. , Yoshino, H. , Feng, D. , Suzuki, Y. , Fukazawa, M. , Nagao, S. , Wainscott, D. B. , Showalter, A. D. , Droz, B. A. , Kobilka, T. S. , Coghlan, M. P. , Willard, F. S. , Kawabe, Y. , Kobilka, B. K. , & Sloop, K. W. (2020). Structural basis for GLP‐1 receptor activation by LY3502970, an orally active nonpeptide agonist. Proceedings of the National Academy of Sciences, 117(47), 29959–29967. 10.1073/pnas.2014879117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin, T. (2008). Functional selectivity in GPCR modulator screening. Combinatorial Chemistry & High Throughput Screening, 11(5), 337–343. 10.2174/138620708784534824 [DOI] [PubMed] [Google Scholar]
- Kim, S. , Chen, J. , Cheng, T. , Gindulyte, A. , He, J. , He, S. , Li, Q. , Shoemaker, B. A. , Thiessen, P. A. , Yu, B. , Zaslavsky, L. , Zhang, J. , & Bolton, E. E. (2019). PubChem 2019 update: Improved access to chemical data. Nucleic Acids Research, 47(D1), D1102–D1109. 10.1093/nar/gky1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King, K. , Lin, N.‐P. , Cheng, Y.‐H. , Chen, G.‐H. , & Chein, R.‐J. (2015). Isolation of positive modulator of glucagon‐like peptide‐1 signaling from Trigonella foenum‐graecum (fenugreek) seed. Journal of Biological Chemistry, 290(43), 26235–26248. 10.1074/jbc.M115.672097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen, L. B. , Kiel, D. , Teng, M. , Behrens, C. , Bhumralkar, D. , Kodra, J. T. , Holst, J. J. , Jeppesen, C. B. , Johnson, M. D. , de Jong, J. C. , Jorgensen, A. S. , Kercher, T. , Kostrowicki, J. , Madsen, P. , Olesen, P. H. , Petersen, J. S. , Poulsen, F. , Sidelmann, U. G. , Sturis, J. , … Lau, J. (2007). Small‐molecule agonists for the glucagon‐like peptide 1 receptor. Proceedings of the National Academy of Sciences of the United States of America, 104(3), 937–942. 10.1073/pnas.0605701104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen, L. B. , Nielsen, P. F. , Huusfeldt, P. O. , Johansen, N. L. , Madsen, K. , Pedersen, F. Z. , Thøgersen, H. , Wilken, M. , & Agersø, H. (2000). Potent derivatives of glucagon‐like peptide‐1 with pharmacokinetic properties suitable for once daily administration. Journal of Medicinal Chemistry, 43(9), 1664–1669. 10.1021/jm9909645 [DOI] [PubMed] [Google Scholar]
- Koole, C. , Wootten, D. , Simms, J. , Valant, C. , Sridhar, R. , Woodman, O. L. , Miller, L. J. , Summers, R. J. , Christopoulos, A. , & Sexton, P. M. (2010). Allosteric ligands of the glucagon‐like peptide 1 receptor (GLP‐1R) differentially modulate endogenous and exogenous peptide responses in a pathway‐selective manner: Implications for drug screening. Molecular Pharmacology, 78(3), 456–465. 10.1124/mol.110.065664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopin, A. , & Beinborn, M. (2004). Methods and compositions for the treatment of metabolic disorders. WO/2004/103310.
- Lau, J. , Bloch, P. , Schäffer, L. , Pettersson, I. , Spetzler, J. , Kofoed, J. , Madsen, K. , Knudsen, L. B. , McGuire, J. , Steensgaard, D. B. , Strauss, H. M. , Gram, D. X. , Knudsen, S. M. , Nielsen, F. S. , Thygesen, P. , Reedtz‐Runge, S. , & Kruse, T. (2015). Discovery of the once‐weekly glucagon‐like peptide‐1 (GLP‐1) analogue semaglutide. Journal of Medicinal Chemistry, 58(18), 7370–7380. 10.1021/acs.jmedchem.5b00726 [DOI] [PubMed] [Google Scholar]
- Lazareno, S. , Dolezal, V. , Popham, A. , & Birdsall, N. J. (2004). Thiochrome enhances acetylcholine affinity at muscarinic M4 receptors: Receptor subtype selectivity via cooperativity rather than affinity. Molecular Pharmacology, 65(1), 257–266. 10.1124/mol.65.1.257 [DOI] [PubMed] [Google Scholar]
- Li, N. , Lu, J. , & Willars, G. B. (2012). Allosteric modulation of the activity of the glucagon‐like peptide‐1 (GLP‐1) metabolite GLP‐1 9–36 amide at the GLP‐1 receptor. PLoS ONE, 7(10), e47936. 10.1371/journal.pone.0047936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Perry, T. , Kindy, M. S. , Harvey, B. K. , Tweedie, D. , Holloway, H. W. , Powers, K. , Shen, H. , Egan, J. M. , Sambamurti, K. , Brossi, A. , Lahiri, D. K. , Mattson, M. P. , Hoffer, B. J. , Wang, Y. , & Greig, N. H. (2009). GLP‐1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proceedings of the National Academy of Sciences of the United States of America, 106(4), 1285–1290. 10.1073/pnas.0806720106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J. , & Nussinov, R. (2016). Allostery: An overview of its history, concepts, methods, and applications. PLoS Computational Biology, 12(6), e1004966. 10.1371/journal.pcbi.1004966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Q. , Li, N. , Yuan, Y. , Lu, H. , Wu, X. , Zhou, C. , He, M. , Su, H. , Zhang, M. , Wang, J. , Wang, B. , Wang, Y. , Ma, D. , Ye, Y. , Weiss, H.‐C. , Gesing, E. R. F. , Liao, J. , & Wang, M.‐W. (2012). Cyclobutane derivatives as novel nonpeptidic small molecule agonists of glucagon‐like peptide‐1 receptor. Journal of Medicinal Chemistry, 55(1), 250–267. 10.1021/jm201150j [DOI] [PubMed] [Google Scholar]
- Ma, H. , Huang, W. , Wang, X. , Zhao, L. , Jiang, Y. , Liu, F. , Guo, W. , Sun, X. , Zhong, W. , Yuan, D. , & Xu, H. E. (2020). Structural insights into the activation of GLP‐1R by a small molecule agonist. Cell Research, 30(12), 1140–1142. 10.1038/s41422-020-0384-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méndez, M. , Matter, H. , Defossa, E. , Kurz, M. , Lebreton, S. , Li, Z. , Lohmann, M. , Löhn, M. , Mors, H. , Podeschwa, M. , Rackelmann, N. , Riedel, J. , Safar, P. , Thorpe, D. S. , Schäfer, M. , Weitz, D. , & Breitschopf, K. (2020). Design, synthesis, and pharmacological evaluation of potent positive allosteric modulators of the glucagon‐like peptide‐1 receptor (GLP‐1R). Journal of Medicinal Chemistry, 63(5), 2292–2307. 10.1021/acs.jmedchem.9b01071 [DOI] [PubMed] [Google Scholar]
- Mjalli, A. , Polisetti, D. , Yokum, T. , Santhosh, K. , Guzel, M. , Behme, C. , & Davis, S. (2009). Oxadiazoanthracene compounds for the treatment of diabetes. WO/2009/111700.
- Moon, H.‐S. , Kim, M.‐K. , & Son, M.‐H. (2011). The development of non‐peptide glucagon‐like peptide‐1 receptor agonist for the treatment of type 2 diabetes. Archives of Pharmacal Research, 34(7), 1041–1043. 10.1007/s12272-011-0721-z [DOI] [PubMed] [Google Scholar]
- Morris, L. C. , Days, E. L. , Turney, M. , Mi, D. , Lindsley, C. W. , Weaver, C. D. , & Niswender, K. D. (2014). A duplexed high‐throughput screen to identify allosteric modulators of the glucagon‐like peptide 1 and glucagon receptors. Journal of Biomolecular Screening, 19(6), 847–858. 10.1177/1087057114520971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, L. C. , Nance, K. D. , Gentry, P. R. , Days, E. L. , Weaver, C. D. , Niswender, C. M. , Thompson, A. D. , Jones, C. K. , Locuson, C. W. , Morrison, R. D. , Daniels, J. S. , Niswender, K. D. , & Lindsley, C. W. (2014). Discovery of (S)‐2‐cyclopentyl‐N‐((1‐isopropylpyrrolidin2‐yl)‐9‐methyl‐1‐oxo‐2,9‐dihydro‐1H‐pyrrido[3,4‐b]indole‐4‐carboxamide (VU0453379): A novel, CNS penetrant glucagon‐like peptide 1 receptor (GLP‐1R) positive allosteric modulator (PAM). Journal of Medicinal Chemistry, 57(23), 10192–10197. 10.1021/jm501375c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakane, A. , Gotoh, Y. , Ichihara, J. , & Nagata, H. (2015). New screening strategy and analysis for identification of allosteric modulators for glucagon‐like peptide‐1 receptor using GLP‐1 (9–36) amide. Analytical Biochemistry, 491, 23–30. [DOI] [PubMed] [Google Scholar]
- Orskov, C. , Rabenhøj, L. , Wettergren, A. , Kofod, H. , & Holst, J. J. (1994). Tissue and plasma concentrations of amidated and glycine‐extended glucagon‐like peptide I in humans. Diabetes, 43(4), 535–539. 10.2337/diab.43.4.535 [DOI] [PubMed] [Google Scholar]
- Polisetti, D. , Benjamin, E. , Quada, J. , & Thorsteinsson, T. (2011). Solid compositions comprising an oxadiazoanthracene compound and methods of making and using the same. WO/2011/031620.
- Pratley, R. , Amod, A. , Hoff, S. T. , Kadowaki, T. , Lingvay, I. , Nauck, M. , Pedersen, K. B. , Saugstrup, T. , Meier, J. J. , & PIONEER 4 investigators . (2019). Oral semaglutide versus subcutaneous liraglutide and placebo in type 2 diabetes (PIONEER 4): A randomised, double‐blind, phase 3a trial. Lancet, 394(10192), 39–50. 10.1016/S0140-6736(19)31271-1 [DOI] [PubMed] [Google Scholar]
- Pupo, A. S. , Duarte, D. A. , Lima, V. , Teixeira, L. B. , Parreiras‐e‐Silva, L. T. , & Costa‐Neto, C. M. (2016). Recent updates on GPCR biased agonism. Pharmacological Research, 112, 49–57. 10.1016/j.phrs.2016.01.031 [DOI] [PubMed] [Google Scholar]
- Rao, M. (2009). Ligands for the Glp‐1 receptor and methods for discovery thereof. WO/2009/126709.
- Redij, T. , Chaudhari, R. , Li, Z. , Hua, X. , & Li, Z. (2019). Structural modeling and in silico screening of potential small‐molecule allosteric agonists of a glucagon‐like peptide 1 receptor. ACS Omega, 4(1), 961–970. 10.1021/acsomega.8b03052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redij, T. , Ma, J. , Li, Z. , Hua, X. , & Li, Z. (2019). Discovery of a potential positive allosteric modulator of glucagon‐like peptide 1 receptor through virtual screening and experimental study. Journal of Computer‐Aided Molecular Design, 33(11), 973–981. [DOI] [PubMed] [Google Scholar]
- Sato, K. , Kameda, M. , Yasuhara, T. , Agari, T. , Baba, T. , Wang, F. , Shinko, A. , Wakamori, T. , Toyoshima, A. , Takeuchi, H. , Sasaki, T. , Sasada, S. , Kondo, A. , Borlongan, C. V. , Matsumae, M. , & Date, I. (2013). Neuroprotective effects of liraglutide for stroke model of rats. International Journal of Molecular Sciences, 14(11), 21513–21524. 10.3390/ijms141121513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schann, S. , Mayer, S. , Frauli, M. , Franchet, C. , & Neuville, P. (2009) Sounds of silence: Innovative approach for identification of novel GPCR‐modulator chemical entities. Proceedings of the 238th American Chemical Society National Meeting, Washington, DC, USA.
- Sloop, K. W. , Willard, F. S. , Brenner, M. B. , Ficorilli, J. , Valasek, K. , Showalter, A. D. , Farb, T. B. , Cao, J. X. , Cox, A. L. , Michael, M. D. , Gutierrez Sanfeliciano, S. M. , Tebbe, M. J. , & Coghlan, M. J. (2010). Novel small molecule glucagon‐like peptide‐1 receptor agonist stimulates insulin secretion in rodents and from human islets. Diabetes, 59(12), 3099–3107. 10.2337/db10-0689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, G. , Yang, D. , Wang, Y. , de Graaf, C. , Zhou, Q. , Jiang, S. , Liu, K. , Cai, X. , Dai, A. , Lin, G. , Liu, D. , Wu, F. , Wu, Y. , Zhao, S. , Ye, L. , Han, G. W. , Lau, J. , Wu, B. , Hanson, M. A. , … Stevens, R. C. (2017). Human GLP‐1 receptor transmembrane domain structure in complex with allosteric modulators. Nature, 546(7657), 312–315. 10.1038/nature22378 [DOI] [PubMed] [Google Scholar]
- Su, H. , He, M. , Li, H. , Liu, Q. , Wang, J. , Wang, Y. , Gao, W. , Zhou, L. , Liao, J. , Young, A. A. , & Wang, M.‐W. (2008). Boc5, a non‐peptidic glucagon‐like peptide‐1 receptor agonist, invokes sustained glycemic control and weight loss in diabetic mice. PLoS ONE, 3(8), e2892. 10.1371/journal.pone.0002892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura, T. , Noda, H. , Joyashiki, E. , Hoshino, M. , Watanabe, T. , Kinosaki, M. , Nishimura, Y. , Esaki, T. , Ogawa, K. , Miyake, T. , Arai, S. , Shimizu, M. , Kitamura, H. , Sato, H. , & Kawabe, Y. (2016). Identification of an orally active small‐molecule PTHR1 agonist for the treatment of hypoparathyroidism. Nature Communications, 7, 13384. 10.1038/ncomms13384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng, M. , Johnson, M. D. , Thomas, C. , Kiel, D. , Lakis, J. N. , Kercher, T. , Aytes, S. , Kostrowicki, J. , Bhumralkar, D. , Truesdale, L. , May, J. , Sidelman, U. , Kodra, J. T. , Jørgensen, A. S. , Olesen, P. H. , de Jong, J. C. , Madsen, P. , Behrens, C. , Pettersson, I. , … Lau, J. (2007). Small molecule ago‐allosteric modulators of the human glucagon‐like peptide‐1 (hGLP‐1) receptor. Bioorganic & Medicinal Chemistry Letters, 17(19), 5472–5478. 10.1016/j.bmcl.2007.06.086 [DOI] [PubMed] [Google Scholar]
- U.S. Food and Drug Administration . (2019). Drug approval package: RYBELSUS labeling information. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/213051Orig1s000lbl.pdf.
- vTv Therapeutics . (2016). Oral small molecule GLP‐1 receptor (GLP‐1R) agonists for type 2 diabetes (T2DM) with negligible nausea and vomiting. Keystone Symposia on Molecular and Cellular Biology held in La Jolla, California, USA.
- Watkins, H. A. , Au, M. , & Hay, D. L. (2012). The structure of secretin family GPCR peptide ligands: Implications for receptor pharmacology and drug development. Drug Discovery Today, 17(17–18), 1006–1014. 10.1016/j.drudis.2012.05.005 [DOI] [PubMed] [Google Scholar]
- Willard, F. S. , Bueno, A. B. , & Sloop, K. W. (2012). Small molecule drug discovery at the glucagon‐like peptide‐1 receptor. Experimental Diabetes Research, 2012, 709893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willard, F. S. , Wootten, D. , Showalter, A. D. , Savage, E. E. , Ficorilli, J. , Farb, T. B. , Bokvist, K. , Alsina‐Fernandez, J. , Furness, S. G. B. , Christopoulos, A. , Sexton, P. M. , & Sloop, K. W. (2012). Small molecule allosteric modulation of the glucagon‐like peptide‐1 receptor enhances the insulinotropic effect of oxyntomodulin. Molecular Pharmacology, 82(6), 1066–1073. 10.1124/mol.112.080432 [DOI] [PubMed] [Google Scholar]
- Wootten, D. , Savage, E. E. , Willard, F. S. , Bueno, A. B. , Sloop, K. W. , Christopoulos, A. , & Sexton, P. M. (2013). Differential activation and modulation of the glucagon‐like peptide‐1 receptor by small molecule ligands. Molecular Pharmacology, 83(4), 822–834. [DOI] [PubMed] [Google Scholar]
- Wootten, D. , Simms, J. , Koole, C. , Woodman, O. L. , Summers, R. J. , Christopoulos, A. , & Sexton, P. M. (2011). Modulation of the glucagon‐like peptide‐1 receptor signaling by naturally occurring and synthetic flavonoids. The Journal of Pharmacology and Experimental Therapeutics, 336(2), 540–550. 10.1124/jpet.110.176362 [DOI] [PubMed] [Google Scholar]
- Yoshino, H. , Tsuchiya, S. , Matsuo, A. , Sato, T. , Nishimoto, M. , Oguri, K. , Ogawa, H. , Nishimura, Y. , Furuta, Y. , Kashiwagi, H. , Hori, N. , Kamon, T. , Shiraishi, T. , Yoshida, S. , Kawai, T. , Tanida, S. , & Aoki, M. (2018). Pyrazolopyridine derivative having Glp‐1 receptor agonist effect. WO/2018/056453.
- Zhang, H. , Liu, Y. , Guan, S. , Qu, D. , Wang, L. , Wang, X. , Li, X. , Zhou, S. , Zhou, Y. , Wang, N. , Meng, J. , & Ma, X. (2016). An orally active allosteric GLP‐1 receptor agonist is neuroprotective in cellular and rodent models of stroke. PLoS ONE, 11(2), e0148827. 10.1371/journal.pone.0148827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X. , Belousoff, M. J. , Zhao, P. , Kooistra, A. J. , Truong, T. T. , Ang, S. Y. , Underwood, C. R. , Egebjerg, T. , Šenel, P. , Stewart, G. D. , Liang, Y.‐L. , Glukhova, A. , Venugopal, H. , Christopoulos, A. , Furness, S. G. B. , Miller, L. J. , Reedtz‐Runge, S. , Langmead, C. J. , Gloriam, D. E. , … Wootten, D. (2020). Differential GLP‐1R binding and activation by peptide and non‐peptide agonists. Molecular Cell, 80(3), 485–500. 10.1016/j.molcel.2020.09.020 [DOI] [PubMed] [Google Scholar]
- Zhang, Y. , Sun, B. , Feng, D. , Hu, H. , Chu, M. , Qu, Q. , Tarrasch, J. T. , Li, S. , Sun Kobilka, T. , Kobilka, B. K. , & Skiniotis, G. (2017). Cryo‐EM structure of the activated GLP‐1 receptor in complex with a G protein. Nature, 546(7657), 248–253. 10.1038/nature22394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, P. , Liang, Y. L. , Belousoff, M. J. , Deganutti, G. , Fletcher, M. M. , Willard, F. S. , Bell, M. G. , Christe, M. E. , Sloop, K. W. , Inoue, A. , Truong, T. T. , Clydesdale, L. , Furness, S. G. B. , Christopoulos, A. , Wang, M. W. , Miller, L. J. , Reynolds, C. A. , Danev, R. , Sexton, P. M. , & Wootten, D. (2020). Activation of the GLP‐1 receptor by a non‐peptidic agonist. Nature, 577(7790), 432–436. 10.1038/s41586-019-1902-z [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article because no new data were created or analysed in this study.