

Abstract
Purpose:
Therapeutic strategies against hormonal receptor-positive (HR+)/HER2+ breast cancers with poor response to trastuzumab need to be optimized.
Experimental Design:
Two HR+/HER2+ patient-derived xenograft (PDX) models named as COH-SC1 and COH-SC31 were established to explore targeted therapies for HER2+ breast cancers. RNA sequencing and RPPA (reverse phase protein array) analyses were conducted to decipher molecular features of the two PDXs and define the therapeutic strategy of interest, validated by in vivo drug efficacy examination and in vitro cell proliferation analysis.
Results:
Estrogen acted as a growth-driver of trastuzumab-resistant COH-SC31 tumors, but an accelerator in trastuzumab-sensitive COH-SC1 model. In vivo trastuzumab efficacy examination further confirmed the consistent responses between PDXs and the corresponding tumors. Integrative omics analysis revealed that mammalian target of rapamycin (mTOR) and ERα signaling predominantly regulate tumor growth of the two HR+/HER2+ PDXs. Combination of dual mTOR complex inhibitor MLN0128 and anti-HER2 trastuzumab strongly suppressed tumor growth of COH-SC1 PDX accompanied with increasing ER-positive cell population in vivo. Instead, MLN0128 in combination with anti-estrogen fulvestrant significantly halted the growth of HR+/HER2+ cancer cells in vitro and trastuzumab-resistant COH-SC31 as well as trastuzumab-sensitive COH-SC1 tumors in vivo.
Conclusion:
Compared to the standard trastuzumab treatment, this study demonstrates alternative therapeutic strategies against HR+/HER2+ tumors through establishment of two PDXs coupling with integrative omics analyses and in vivo drug efficacy examination. This work presents a prototype of future “co-clinical” trials to tailor personalized medicine in clinical practice.
Keywords: HR+/HER2+ breast cancers, patient-derived xenografts, Trastuzumab, MLN0128, Fulvestrant
INTRODUCTION
Hormone receptor-positive (HR+)/HER2+ tumors account for approximately 10% of all breast cancers. Activation of estrogen receptor alpha (ERα) promotes the growth of HR+ tumors. HR+/HER2− patients are treated with endocrine therapies, such as aromatase inhibitors (AIs) and ERα antagonists (1–3). HER2 belongs to receptor tyrosine kinase (RTK) family. In the clinical setting, trastuzumab is the current standard treatment for HR−/HER2+ patients. A combination of HER2-targeting agents and chemotherapies is recommended for HER2+ advanced breast cancer (4). HR+/HER2+ breast cancer is heterogeneous and bears the concurrent activity of ER and HER2 (3–6). The HR+ status implies that ER pathway plays a critical role in directing the clinical features of HR+/HER2+ breast cancer. While chemotherapy is typically applied, the increased HR level in HR+/HER2+ tumors is correlated to a better response to endocrine therapies (4–6). Therefore, the use of chemotherapy may over-treat this subgroup of breast cancer patients, causing potentially unnecessary adverse events and economic cost (6). Additionally, tumor response to trastuzumab may be reduced by concurrent activity of the ER pathway (7). Clinical studies show that the addition of anti-HER2 therapies significantly improves the clinical outcomes of HR+/HER2+ patients treated with AIs (8). Clinical and laboratory studies also suggest that cross-talk between the ER and RTK signaling pathways plays a critical role in resistance to endocrine therapies and anti-HER2 agents in HR+/HER2+ breast cancers (3, 5). The molecular mechanisms of acquired and de novo resistance to anti-HER2 agents vary in different biological contexts. Several studies reveal that ERα signaling is augmented in the presence of anti-HER2 agents (9, 10). Notably, cancer cell lines resistant to anti-HER2 agents are often sensitive to the pure anti-estrogen- fulvestrant. In addition, activation of the PI3K/AKT/mTOR signaling has been reported in the establishment of endocrine resistance, leading to estrogen-independent ERα activation. In clinic, the mTORC1 inhibitor everolimus has been prescribed with endocrine agents for advanced and AI-resistant HR+ breast cancers (11–16).
The PI3K/AKT/mTOR axis, as the downstream of the RTK cascade, is considered to be a promising therapeutic target in HR+ and HER2+ breast cancers (14–16). The protein kinase mTOR, which exists in two complexes- mTORC1 and mTORC2 and serves as the catalytic subunit, transduces proliferation cues to the signaling components of the PI3K/AKT/mTOR axis. While activation, PI3K and mTORC2 activate AKT, leading to up-regulation of mTORC1 and the downstream S6K1 and 4EBP1 for promoting cell proliferation and survival (16–20). Due to the PI3K/AKT rebound effect, long-term treatment of mTORC1 inhibition has been known to alleviate AKT activation by mTORC2. Therefore, allosteric mTOR inhibitors (e.g. everolimus) do not fully inhibit mTORC1 activity (19). To overcome this barrier, the ATP-competitive dual mTOR kinase inhibitors, such as MLN0128, are developed to simultaneously target both mTORC1 and mTORC2 (20).
Presently, there are a few HR+/HER2+ breast cancer cell lines available for preclinical research (9), which are limits in understanding the clinical features of patient tumors. Instead, PDXs mimicking the features from the tumors of origin can offer a realistic solution (21). In this study, we established two PDX models from two individual tumors to gain insight on HR+/HER2+ breast cancers. Via the two PDXs with distinct clinical features, we investigated the role of estrogen/ER in controlling tumor growth and dissected molecular characteristics of this breast cancer subtype. Integrative omics analyses coupled with in vivo drug efficacy examination further defined the therapeutic effects of simultaneous suppression on ER signaling and mTOR pathway by fulvestrant and MLN0128, respectively, in HR+/HER2+ breast cancers.
MATERIALS AND METHODS
Patient-derived xenografts and in vivo treatment study
Surgical resections (2×2 mm2) from consented HR+/HER2+ breast cancer patients were orthotopically engrafted into the mammary fat pad of 6~8-week-old female NOD-scid/IL2Rγ−/− (NSG) mice to derive parental tumors. To examine estrogen-dependent growth, tumor slices (2~3 mm) from the parental tumors and in-house-made estrogen pellets containing 1 mg of E2 each were simultaneously implanted into the mammary fat pad of 6-week-old ovariectomized NSG mice (22). For in vivo drug efficacy examination, tumor slices (2~3 mm) from the parental tumors were engrafted into the intact NSG mice. Once tumors were established to be 100~200 mm3 in size, mice were randomized and then subjected to four-week treatment of control (150 μL sterile saline, twice weekly of intraperitoneal injection), trastuzumab (10 mg/kg in sterile saline, twice weekly of intraperitoneal injection), fulvestrant (5 mg in 100 μL sterile saline, once weekly/subcutaneous injection) and/or MLN0128 (0.3 mg/kg in 5% N-methyl-2-pyrrolidone, 15% polyvinyl pyrrolidone in water, six days weekly of gavage) (15). Tumor growth and body weight were monitored twice per week and tumor volume was calculated as length × width2 × π/6 (23). Fulvestrant (Faslodex, AstraZeneca, London, England) and trastuzumab (Genentech, San Francisco, CA, USA) were obtained from the City of Hope National Medical Center Pharmacy. The City of Hope Institutional Review Board approved this study and all patients provided written informed consent prior to tissue collection. All animal experiments were done under a protocol approved by the Institutional Animal Care and Use Committee. Facilities are credited by Association for Assessment and Accreditation of Laboratory Animal Care and operated according to NIH guidelines.
Histological analysis
Immunohistochemistry (IHC) and H&E (hematoxylin and eosin) staining of formalin-fixed tumor tissues were performed by the Pathology Core facility at City of Hope. Antibodies used in IHC included ERα (ab1660; Abcam, Cambridge MA), PR (PA0312; Leica Biosystems Inc., Buffalo Grove IL), HER2 (A0485; DAKO/Agilent Technologies, Santa Clara CA), and Ki-67 (M7240; DAKO). Hormone receptors were interpreted and scored according to joint American Society of Clinical Oncology/College of American Pathologists guidelines (24, 25). Slides were reviewed first at 10X magnification to identify areas of positive staining, followed by confirmation and quantitation at 20~40X magnifications. Ki-67 was scored by identifying areas of most abundant positivity at low magnification, followed by manual counting of 10 high-power (40X) fields. Representative images and scoring were acquired using an Olympus BX46 microscope with DP27 camera and Olympus CellSens software (Olympus, Shinjuku, Japan).
RNA sequencing and data analysis
Total RNA from six and three COH-SC1 and COH-SC31 PDX tumors, respectively, and two biological replicates from an ER+/HER2− PDX established in our laboratory (indicated as REF in Figure 3A) were extracted using RNeasy Extract Kit (Qiagen, Alameda, CA) and then subjected to RNA sequencing conducted by the Integrative Genomics Core at City of Hope. The description in detail of sequencing processing, sequence read mapping, and further systems-level pathway analyses using Ingenuity Pathway Analysis (IPA; Ingenuity® Systems) and Gene Set Enrichment Analysis (GSEA) were summarized in SUPPLEMENTARY MATERIALS AND METHODS. All sequencing data were submitted to GEO database.
Figure 3. Deciphering molecular features of HR+/HER2+ PDXs.
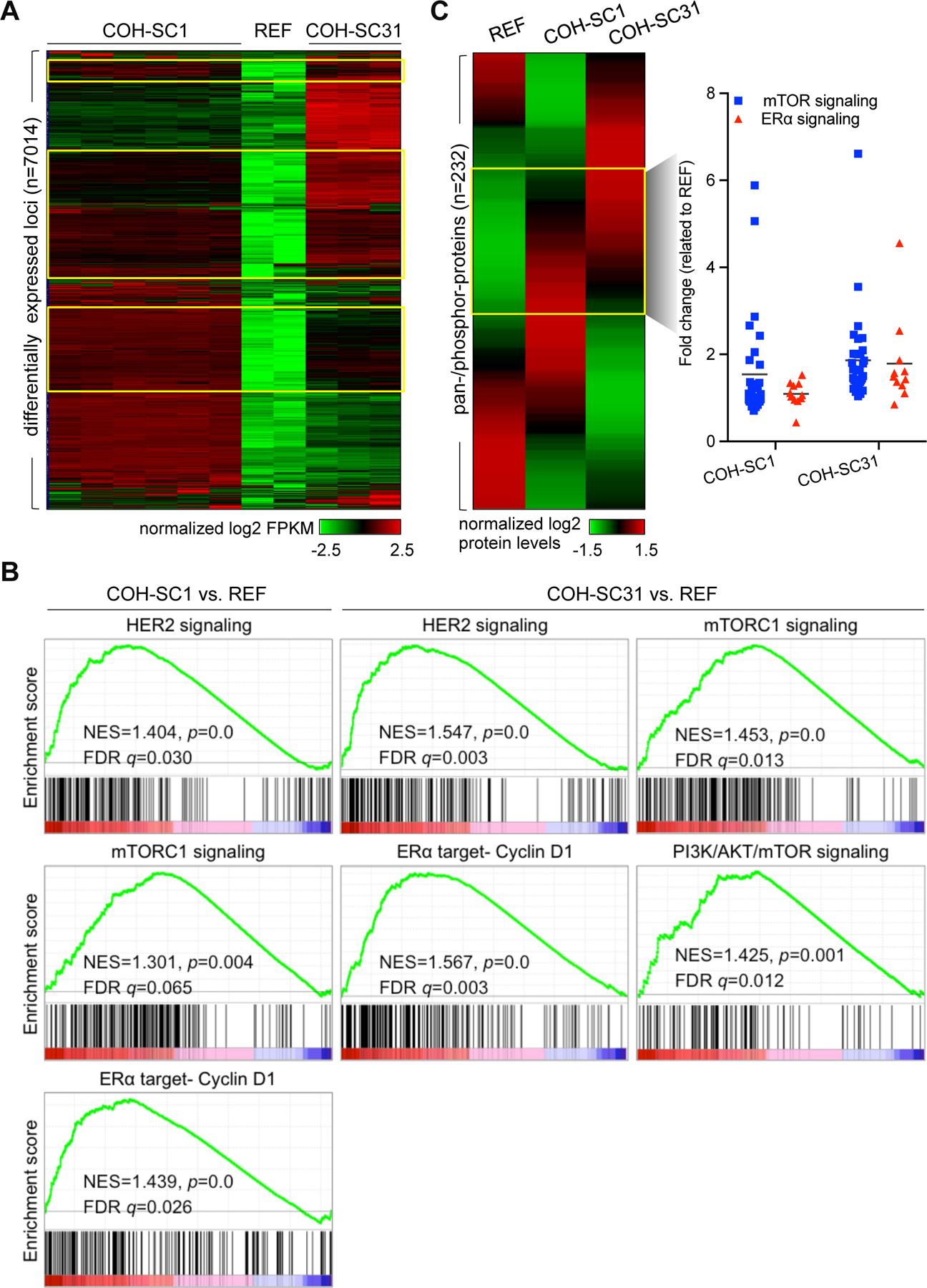
(A) Transcriptome analysis of six COH-SC1 and three COH-SC31 tumors and two samples from the reference ER+/HER2− PDX (named as REF) using RNA-seq. A heat map was plotted to illustrate 7014 differentially expressed loci (p<0.05) in either COH-SC1 or COH-SC31 transcriptome in relation to the REF PDX. Yellow boxes, 549 common up-regulated loci in the two examined PDXs compared to the REF transcriptome. Intensity bar, normalized log2 FPKM (fragments per kilobase per million reads) value. (B) Gene set enrichment analysis (GSEA) of differentially expressed loci between COH-SC1 or COH-SC31 and the REF transcriptomes. HER2, mTOR signaling, and ERα target, Cyclin D1, were significantly and positively enriched in COH-SC1 and COH-SC31 transcriptomes. NES, normalized enrichment score. See also Supplementary Figure 4 for another systems-level pathway analysis using IPA. (C) Proteome analysis of COH-SC1 and COH-SC31 tumors using RPPA. A heat map was plotted to illustrate 232 pan-/phosphor-protein signatures in COH-SC1, COH-SC31, and the reference ER+/HER2− PDXs (left panel). Compared to the reference dataset (REF), pan-/phosphor-proteins participated in PI3K/AKT/mTOR and ERα signaling axis were relatively activated in both two PDXs as shown in yellow box and summarized in a scatter plot (right panel). Intensity bar, normalized log2 value. See also Supplementary Figure 5 for the expression levels of individual pan-/phosphor-proteins involved in HER2/ER and PI3K/AKT/mTOR signaling and the experimental validation of AKT and ER phosphorylation status using Western blot analysis.
Reverse phase protein array (RPPA) analysis
Three snap-frozen tumor samples from COH-SC1, COH-SC31, and an ER+/HER2− PDXs were subjected to RPPA analysis probed a total of 232 antibodies conducted by the MD Anderson Cancer Center Functional Proteomics RPPA Facility as described previously (26). To define the relatively activated signaling in either COH-SC1 or COH-SC31 model, RPPA data of an ER+/HER2− PDX was used as the reference (indicated as REF in Figure 3C).
RESULTS
Establishment and characterization of two HR+/HER2+ breast PDX models
To decipher the biology and potential therapeutic strategy of HR+/HER2+ breast cancers, two PDXs named as COH-SC1 and COH-SC31 were established. As shown in Figure 1A, surgical resections from consented HR+/HER2+ breast cancer patients were engrafted into the mammary fat pad of immunodeficient NSG mice to derive parental tumors (Step 1). Once established, parental tumors were divided into pieces and subsequently implanted to 30~40 mice for the following molecular and pharmacologic analyses. Samples from individual clones were subjected to RNA-seq transcriptome and RRPA proteome analyses to dissect the molecular features of the two PDXs. “Same-patient-on-all-arms” testing cohorts were generated for drug efficacy examination (Step 2). Drug-mediated tumor growth and phenotypic/molecular changes were then investigated (Step 3).
Figure 1. Differential estrogen response in COH-SC1 and COH-SC31 PDXs.
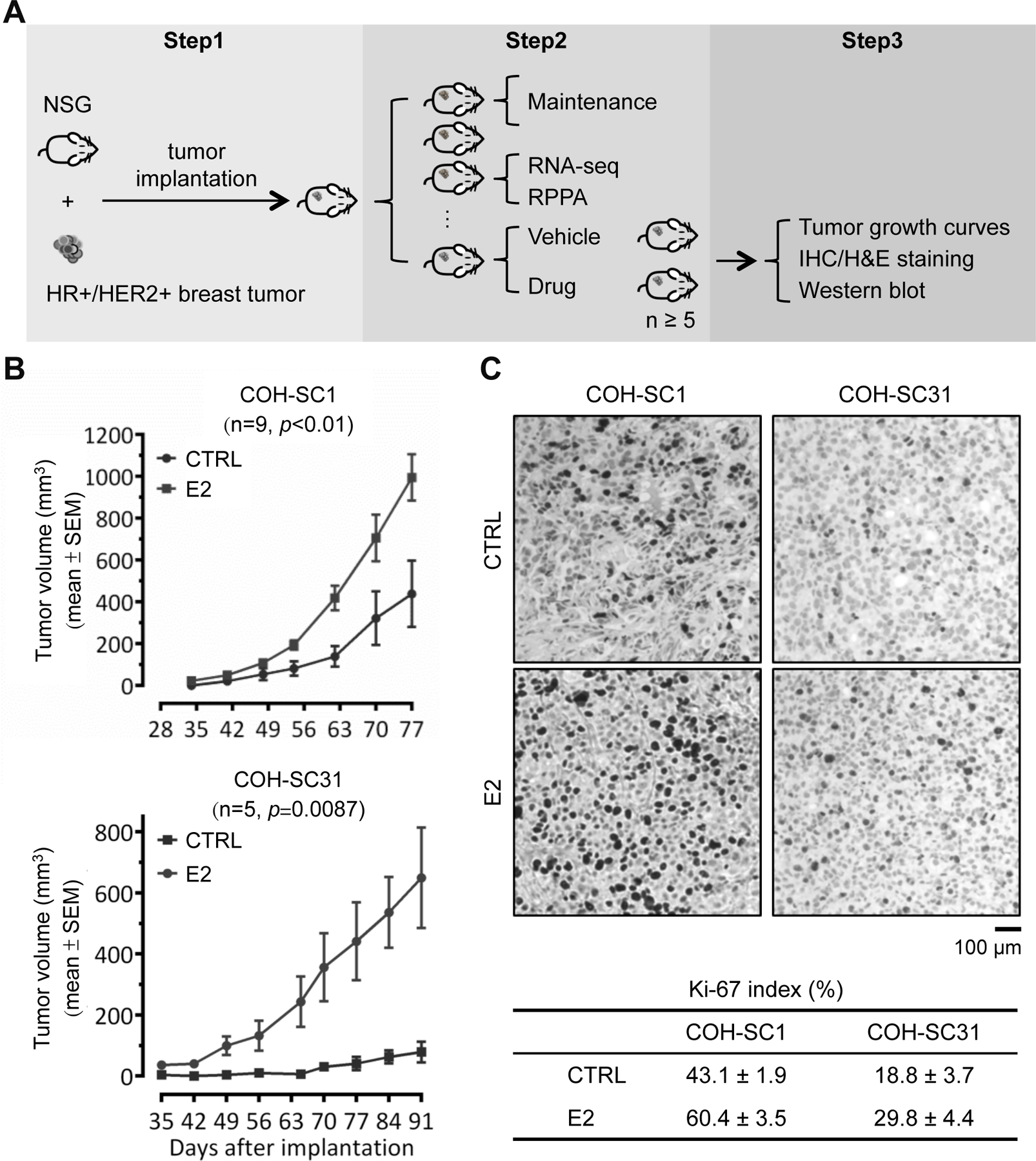
(A) Scheme of breast PDX establishment and utilization. Step 1, establishment of PDXs from breast cancer patients’ tumors; Step 2, stock expansion and molecular feature dissection of the PDXs; Step 3, in vivo drug efficacy examination and the subsequent histological and biochemical validation. (B) Estrogen-involved tumor growth in COH-SC1 and COH-SC31 PDXs. Ovariectomized mice were randomized and implanted COH-SC1 (n=9; upper panel) or COH-SC31 (n=5; lower panel) tumor accompanied with 1 mg estrogen (E2) or vehicle (CTRL) pellets. Tumor growth curves were monitored and plotted after implantation as indicated and p value was determined by two-way ANOVA analysis. (C) Ki-67 expression in COH-SC1 and COH-SC31 PDXs responded to estrogen treatment. Representative images of Ki-67 IHC in COH-SC1 and COH-SC31 PDXs upon E2 treatment were shown in upper panel and the associated scoring was summarized as Mean ± SEM in lower panel. Scale bar, 100 μm.
COH-SC1 was derived from an ER+/PR−/HER2+ breast tumor prior to treatment and the patient later responded well to trastuzmab plus chemotherapies; COH-SC31 was established from a trastuzumab-resistant ER+/PR+/HER2+ metastatic breast tumor (Supplementary Table 1). H&E staining indicated that cell morphologies between PDXs and the corresponding patients’ samples are similar in both models, which COH-SC1 tumors are moderately differentiated but poor-differentiation in COH-SC31 (Supplementary Figure 1A). IHC analyses of ER, PR, and HER2 expression patterns independently supported the recapitulation capability of PDXs by showing that COH-SC1 tumors harbor 1~2% of ER+, no PR expression, and strong HER2+ expression (scored as 3+); 40–50% of ER+, 2–5% of PR+, and scored as 3+ of HER2+ staining in COH-SC31 (Supplementary Figure 1B). To further confirm the differential expression levels of ERα between the two PDXs, Western blot analysis conducted on three biological replicates per model and MCF-7 cancer cells as the positive control pointed out the relatively higher ERα expression in COH-SC31 tumors (Supplementary Figure 1C). In addition, there was no mutation found in ESR1 locus in both COH-SC1 and COH-SC31. Taken together, the above pathological observations on PDXs and the corresponding patients’ tumors were similar, supporting that the established PDXs is capable of recapitulating the clinical features of individual breast tumors.
Differential estrogen response in COH-SC1 and COH-SC31 models
To clarify whether estrogen/ERα signaling drives HR+/HER2+ tumor growth, PDX tumors were implanted into ovariectomized mice accompanied with or without estrogen pellets. Uterus weight was then measured to assess in vivo estrogen responses and showed significant enlargement of uteri in the presence of estrogen pellets (n=9, p<0.001; Supplementary Figure 2A), in supportive of the implanted estrogen pellets as the main source of estrogen supplementation. Tumor volumes and weights were simultaneously monitored to indicate that estrogen remarkably contributes to tumor growth of both two PDXs (p≤0.01; Figure 1B and Supplementary Figure 2B). Additional estrogen-dependency comparison verified that COH-SC31 tumor growth relies on estrogen (n=5, p=0.0087; Figure 1B, lower panel), but the presence of estrogen potentially acts as a growth accelerator in COH-SC1 tumors (n=9, p<0.01; upper panel). For example, at 77th day after tumor implantation (Figure 1B, upper panel), tumor volumes of COH-SC1 in the absence and presence of estrogen were 438.5±159.1 mm3 (see “CTRL”) and 994.8±333.2 mm3 (see “E2”), respectively. Contrastingly, after 91 days in posterior to COH-SC31 tumor implantation (Figure 1B, lower panel), tumor volume was ~100 mm3 under estrogen deprivation (see “CTRL”) and 619.0±164.6mm3 upon E2 supplement (see “E2”). IHC of Ki-67 (a cellular proliferation marker; ref. 27) expression levels was subsequently determined to verify estrogen-driven proliferation by showing approximately 1.4 and 1.6 times higher in Ki-67-positive staining in COH-SC1 (60.4±3.5% vs. 43.1±1.9%; left panels) and COH-SC31 (29.8±4.4% vs. 18.8±3.7%; right panels), respectively (Figure 1C). Notably, Ki-67-positive cells in COH-SC1 (43.1±1.9%) were relatively higher than that of COH-SC31 (18.8±3.7%) under estrogen-deprived conditions (see “CTRL” in Figure 1C), supporting the estrogen-independency in COH-SC1 tumor initiation. These findings demonstrated that estrogen/ERα signaling modulates HR+/HER2+ tumor growth in different manners, which it acted as a crucial initiator in controlling tumor growth of COH-SC31 with higher ER expression, but an accelerator in facilitating growing of COH-SC1 tumors with lower-expressed ER (Figure 1B & 1C and Supplementary Figure 1C).
Distinct trastuzumab response in COH-SC1 and COH-SC31 PDXs
Trastuzumab is the first-line monotherapy of HER2+ breast cancer (4–7). To validate its efficacy against HR+/HER2+ tumors in vivo, the intact NSG mice bearing either COH-SC1 or COH-SC31 tumors were subjected to four-week drug treatment. As shown in Supplementary Figure 3A, either trastuzumab (TRA) or placebo (CTRL) treatment triggered less than 15% reduction in body weights of COH-SC1 and COH-SC31 PDXs, in supportive of the well-tolerated treatment scheme in mice. In COH-SC1 model (n=7), compared to CTRL group, TRA treatment significantly caused ~45% reduction in tumor volume (p=0.006; Figure 2A, upper panel and Supplementary Figure 6A), but not tumor weight (p=0.62; Figure 2B, upper panel and Supplementary Figure 6C). Consisting with the clinico-pharmacological history of the corresponding patient (Supplementary Table 1), COH-SC31 PDX derived from a trastuzumab-relapsed breast tumor was found to be resistant to TRA treatment with no considerable changes in either tumor volume (p=0.0436) or weight (p=0.8889) (n=5; Figure 2, lower panels). These findings exhibited the distinct trastuzumab response in the two established HR+/HER2+ PDXs as well as suggested the insufficiency of trastuzumab monotherapy in treating HR+/HER2+ tumors and the need of defining alternative therapeutic optimization.
Figure 2. In vivo efficacy examination of trastuzumab on HR+/HER2+ PDXs.
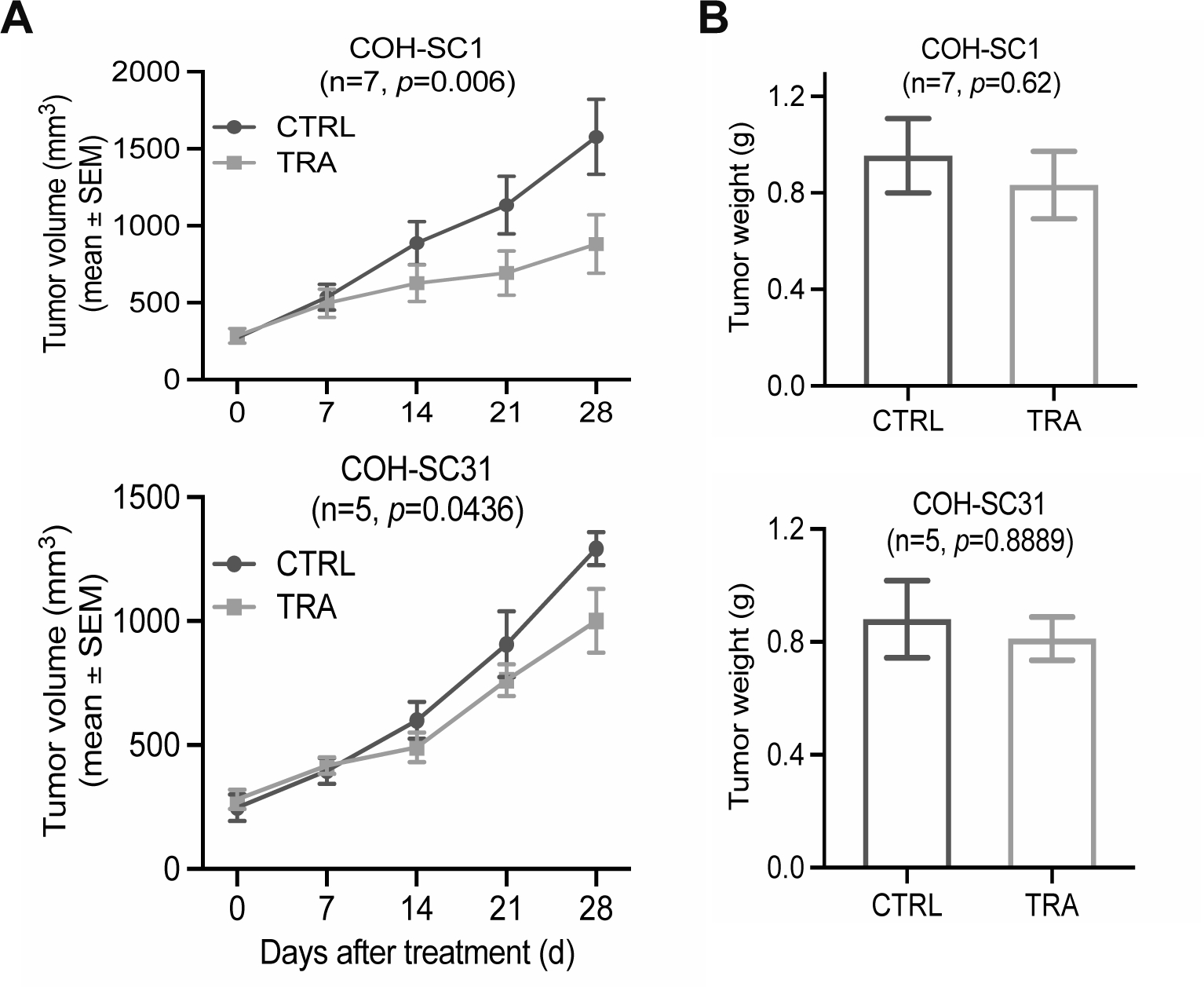
Four-week treatment of trastuzumab (10 mg/kg/twice intraperitoneal injection per week; TRA) or sterile saline (CTRL) was performed on the intact mice bearing COH-SC1 (n=7) or E2-supplemented COH-SC31 (n=5) tumors. Tumor volume (A) and tumor weight (B) were monitored and summarized as Mean±SEM with two-way ANOVA analysis for p value. See also Supplementary Figure 3A for body weight observations.
Characterization of molecular features and clinical relevance of two PDXs
To decode molecular features of HR+/HER2+ breast cancers and explore potential therapeutic targets, whole-genome RNA-seq transcriptome and RPPA proteome analyses were performed on COH-SC1 and COH-SC31 tumors. To identify HR+/HER2+ PDX-specific gene signature, another RNA-seq dataset from an ER+/HER2− PDX tumor was included as the reference (see “REF” in Figure 3A). Compared to two biological replicates of REF results, a heat map plotted to summarize the expression patterns of 7014 differentially expressed loci (p<0.05) in either COH-SC1 (n=6) or COH-SC31 (n=3) datasets individually showed a distinct transcriptome pattern of these two PDX models (Figure 3A). To explore the major signaling controlling HR+/HER2+ tumor growth, a total of 549 common up-regulated loci ([fold-change] ≥1.5, p<0.05) shown in the box areas were subjected to IPA to uncover that RTK (i.e. EGFR, HER2, and HER3) and PI3K/AKT/mTOR pathways are the mostly significant networks in the two studied models (p<0.001; Supplementary Figure 4A). Differential expression profiling between COH-SC1 or COH-31 and REF analyzed by GSEA supportively identified that HER2, mTOR signaling, and ERα target- Cyclin D1, are remarkably activated in the two examined PDXs (Figure 3B). RPPA results of 232 pan-/phosphor-proteins independently revealed that expression levels of the active forms of signaling components in PI3K/AKT/mTOR pathway (e.g. AKT, 4E-BP1, and S6K), as well as those of HER2 (e.g. HER2 and HER3) and ER signaling (e.g. Cyclin D1, c-MYC, and PR) are relatively higher at the post-translational level (p<0.05; Figure 3C and Supplementary Figure 5A & 5B). Western blot analysis of phosphorylation status of AKT at serine 473 and ERα at serine 118 further validated activated AKT signaling in the two examined PDXs, but relatively low levels of activated ERα (Supplementary Figure 5C). RPPA comparison and IPA of differential gene expression between COH-SC1 and COH-SC31 tumors additionally pointed out that expression levels of ERα signaling-involved loci or proteins are relatively higher in COH-SC31 (Supplementary Figures 4B, 5A, and 5B). For example, expression levels of ESR1 (10.64 fold) and well-known ERα-targeted loci, such as TFF1 (657.11 fold) and BMP7 (203.42 fold), were higher in COH-SC31 in relation to that of COH-SC1 (Supplementary Table 2). Summarization of these findings proposed that PI3K/AKT/mTOR signaling is a critical oncogenic pathway in both PDXs but ERα signaling plays a more dominated role in COH-SC31 model.
To address the clinical relevance of COH-SC1 and COH-SC31 PDXs, we conducted in silico expression analysis of a published breast cancer cohort containing 266 samples (Figure 4; ref. 28). Based on PAM50 (prediction analysis of microarray 50) subtype characterization (29), the transcriptome profiling of COH-SC1 tumors was similar to that in luminal-B/basal-like tumors (Figure 4A). Nine genes highlighted in luminal-B/basal-like patient samples were coincidently highly expressed in COH-SC1 model and involved in the PI3K/AKT/mTOR signaling. Kaplan-Meier analysis of those identified gene signature found that activated PI3K/AKT/mTOR axis is correlated to poor prognosis of luminal-B breast cancer patients (p=0.0136; Figure 4C). However, PAM50 analysis of COH-SC31 transcriptome elucidated the association with luminal-A breast cancer subtype (Figure 4B). The highlighted sections of twenty-one highly expressed loci in luminal-A samples were associated with ER and PI3K/AKT/mTOR signaling components, which were also highly expressed in COH-SC31 tumors. Kaplan-Meier survival analysis of those identified genes with increased expression signature was linked to poor prognosis of luminal-A breast cancer patients (p=0.0393; Figure 4D). These clinically relevant analyses suggested that: 1) blockade of the PI3K/AKT/mTOR signaling would potentially suppress COH-SC1 tumors; 2) simultaneous inhibition of ER and PI3K/AKT/mTOR pathways can be a more efficient strategy to attenuate COH-SC31 tumor progression.
Figure 4. Dissection of intrinsic subtypes and clinical relevance in COH-SC1 and COH-SC31.
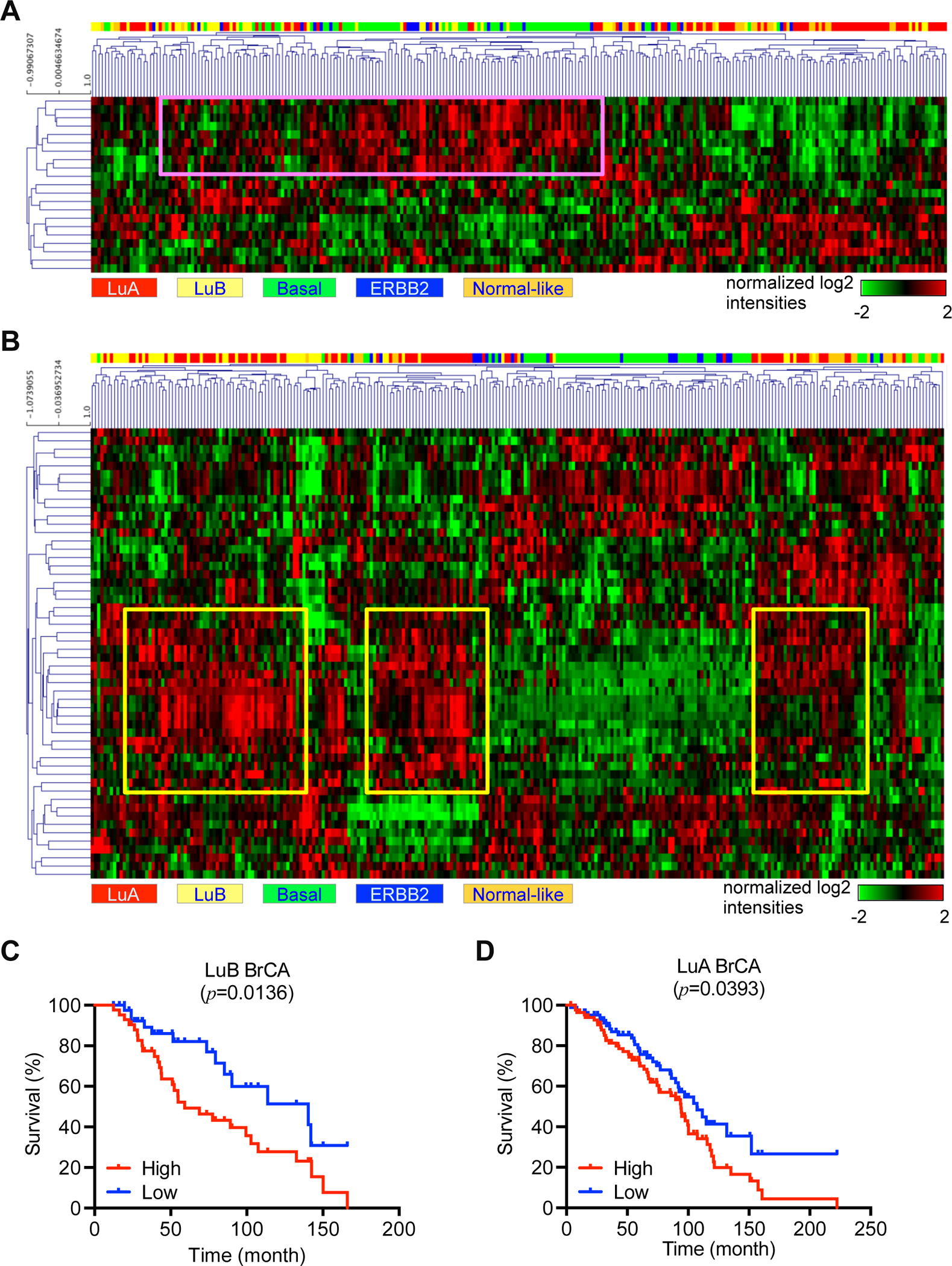
In silico analysis of the activated genes in COH-SC1 or COH-SC31 transcriptomes as aforementioned in Figure 3A in a breast cancer cohort harboring 266 samples within PAM50-classified subtype information was illustrated as heat maps with hierarchical clustering (28). Rows, activated genes; Columns, patient samples. (A) COH-SC1 was associated with luminal-B/basal breast cancers. Pink box, nine mTOR signaling-associated loci. (B) COH-SC31 was linked to luminal-A breast cancers. Yellow boxes, twenty-one mTOR and/or estrogen/ERα pathway-related loci. Kaplan-Meier analysis of luminal-B (C) or luminal-A (D) cancer patients within expression of the boxed genes identified in COH-SC1 (A) or COH-SC31 (B) models, respectively. Log-rank test was used to determine statistical significance.
Dual-therapy treatment strategies efficiently halt HR+/HER2 tumor growth
Integration of transcriptome and proteome results discovered activated PI3K/AKT/mTOR pathway as the oncogenic driver in both HR+/HER2+ PDXs (Figure 3 and Supplementary Figures 4 & 5). To assess its therapeutic potential, in vivo efficacy examination of a dual mTOR inhibitor MLN0128 (MLN) was performed on the two preclinical models using the intact NSG mice bearing either COH-SC1 or COH-SC31 tumors. After four-week oral administration, less than 5% decline in body weight observed in the two PDXs supported the well-tolerated toxicity of treatment dosage (see “MLN” in Supplementary Figures 3B & 6B). MLN treatment profoundly caused 41~66% and 38% decrease in tumor volumes of COH-SC1 (n=7, p≤0.0021) and COH-SC31 (n=5, p=0.0042) PDXs, respectively (Figure 5A and Supplementary Figure 6A). These observations demonstrated that MLN0128 monotherapy results in partial growth inhibition of both HR+/HER2+ PDXs, suggesting that alternative therapeutic strategies are needed for more efficient against HR+/HER2+ tumors.
Figure 5. Efficacy of MLN0128 and/or fulvestrant on treating HR+/HER2+ PDXs in vivo.
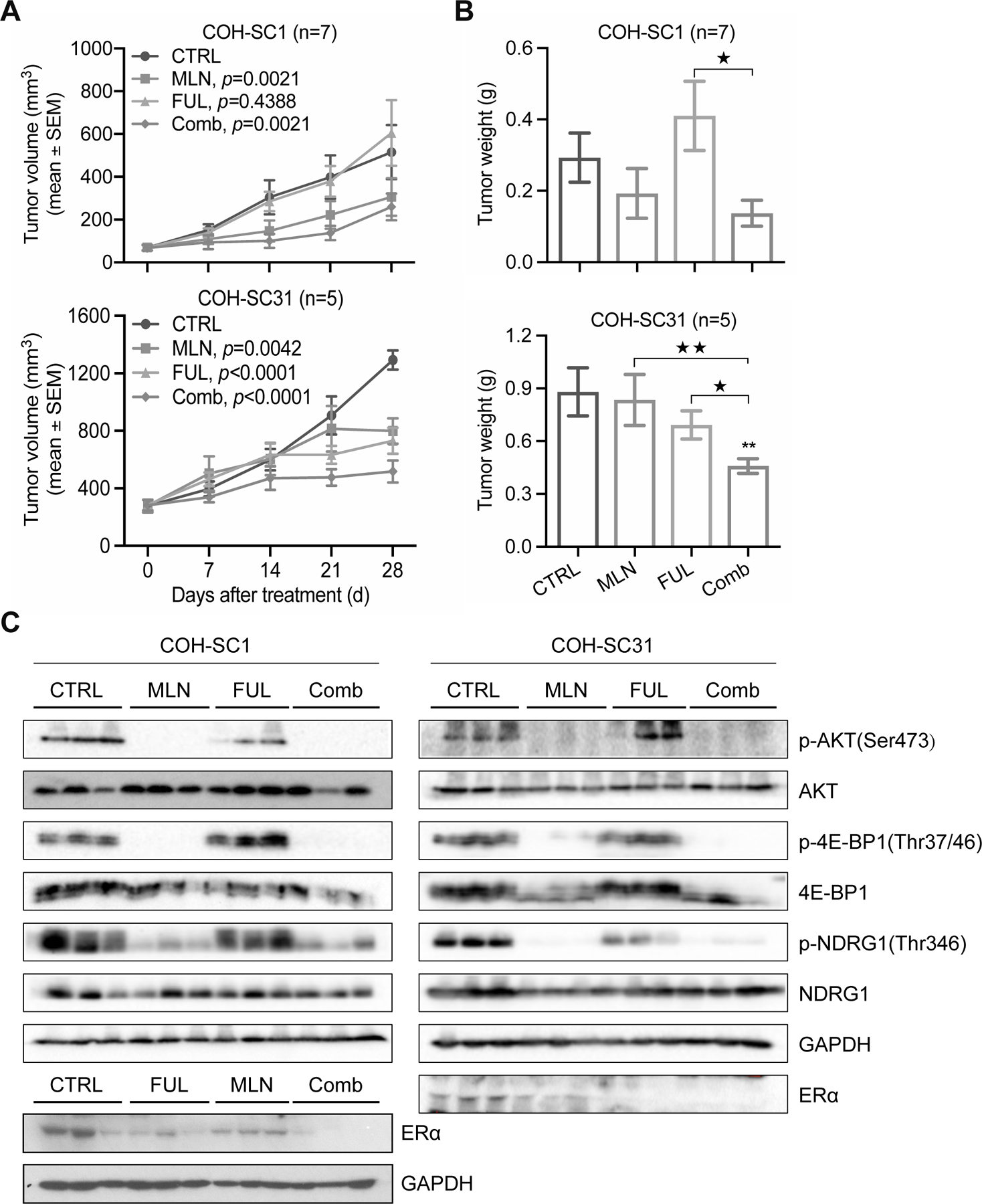
The intact mice bearing COH-SC1 (n=7) or E2-supplemented COH-SC31 (n=5) tumors were randomized for four-week treatment of MLN0128 (0.3 mg/kg/six days of gavage per week; MLN), fulvestrant (5 mg/once subcutaneous injection per week; FUL), and combination (Comb). Tumor volume (A) and tumor weight (B) were measured and summarized as Mean±SEM. P value between CTRL and treated group(s) was addressed by two-way ANOVA analysis. **, p<0.001, compared to CTRL group; ★, p<0.05, ★★, p<0.001, compared to Comb group, determined by Mann Whitney test. See also Supplementary Figure 3B for body weight observations. (C) Validation of treatment efficiency by Western blot analysis. Pan/phosphor-proteins involved in mTOR or ERα signaling as indicated were examined in COH-SC1 (left panel) and COH-SC31 (right panel) samples with three days of MLN and/or FUL treatment. GAPDH, the internal and loading control. Three biological replicates per treatment were assayed.
While TRA single-drug treatment triggered ~45% tumor volume reduction in COH-SC1 PDX (Figure 2A, upper panel and Supplementary Figure 6A), we hypothesized that multi-drug treatment scheme brings better therapeutic efficacy against trastuzumab-sensitive HR+/HER2+ cancers. To verify this hypothesis, we used the same preclinical cohort, which was applied to test trastuzumab efficacy, to evaluate the effectiveness of TRA-plus-MLN combination treatment in vivo. Compared to TRA alone, dual-therapy scheme with the acceptable drug tolerance (~14.9% decrease in body weight; Supplementary Figure 6B) efficiently prohibited the majority of tumor growth (~80.5%, p<0.0001) and reduced tumor weight (p<0.001) (n=7; see “Comb” in Supplementary Figure 6A & 6C). Interestingly, MLN single-drug treatment in the same cohort induced ~66% growth inhibition (p<0.0001) in relation to CTRL group (Supplementary Figure 6A), implying the better potency of MLN against trastuzumab-sensitive COH-SC1 tumors in relation to that in TRA group. However, monotherapy using either TRA or MLN cannot completely attenuate mitogenic signaling (see “Mitotic counts” in Supplementary Figure 6D), suggesting the superior therapeutic potential of combination strategy against HR+/HER2+ tumors.
To corroborate this concept, we examined the drug efficacy in vivo on both COH-SC1 and COH-SC31 PDXs. In trastuzumab-sensitive COH-SC1 model, TRA-plus-MLN combination significantly suppressed ~80.5% tumor growth (see “Comb” in Supplementary Figure 6A). But, it concurrently increased ER-expressing cell population (see “Comb” in Supplementary Figure 6D), implying that simultaneous blockade of ERα and mTOR pathway is another therapeutic scheme against trastuzumab-sensitive HR+/HER2+ cancer, which can potentially lower the recurrence risk resulting from the increasing ER expression. Since activated PI3K/AKT/mTOR and ERα pathways were found to be involved in tumor progression of HR+/HER2+ cancers via integrative omics analysis (Figures 3 and Supplementary Figures 4 and 5), we, thus, tested the efficacy of combined MLN with anti-estrogen fulvestrant (FUL) on the two preclinical cohorts bearing COH-SC1 or COH-SC31 tumors as aforementioned in Figure 5. Within the acceptable drug tolerance (p<0.8933; Supplementary Figure 3B), MLN-plus-FUL combination resulted in significant 50~60% tumor volume reduction in COH-SC1 (60%, p=0.0021) and COH-SC31 (50%, p<0.0001) models and down-regulation of the associated PI3K/AKT/mTOR and ERα signaling (see “Comb” in Figure 5A & 5C), whereas either MLN (41% and 38% decrease in COH-SC1 and COH-SC31, respectively) or FUL (17% increase in COH-SC1 and 43% decrease in COH-SC31) single-drug treatment induced less than 50% suppression. To validate these in vivo observations, MLN or FUL treatment was independently applied on an in vitro culture system including three HR+/HER2+ breast cancer cell lines- MCF-7aro/HER2 derived from MCF-7aro cells with stable overexpression of HER2 (30), BT-474 (HR+/amplified HER2), and BT/Her0.2R derived from BT-474 cells with long-term trastuzumab exposure (31), which was previously found to be sensitive to trastuzumab treatment in the presence of tamoxifen (32), and HR+ MCF-7aro cancer cells. We observed that the four examined cancer cell lines respond to either MLN or FUL in a dose-dependent manner (Figure 6A & 6B). Compared to HR+ MCF-7aro cancer cells, HER2-overexpressing MCF-7aro/HER2 cancer cells were less sensitive to either MLN or FUL treatment (Figure 6C), implying that HER2 overexpression likely activates mTOR and ER signaling as consistent as the integrative omics observations. Though, MCF-7/aroHER2 and BT-474 cancer cells were more sensitive to FUL treatment in relation to that in BT/Her0.2R cells (Figure 6C). Further combination treatment performed on BT-474 and BT/HER0.2R cancer cells revealed that the presence of MLN enhances the potency of FUL in treating HR+/HER2+ cancer cells (Figure 6D & 6E), supporting that dual-therapy treatment using MLN-plus-FUL potentially increase drug potency against trastuzumab-sensitive as well as -resistant HR+/HER2+ tumors.
Figure 6. In vitro response of MLN0128 and fulvestrant in HR+/HER2+ breast cancer cell lines.
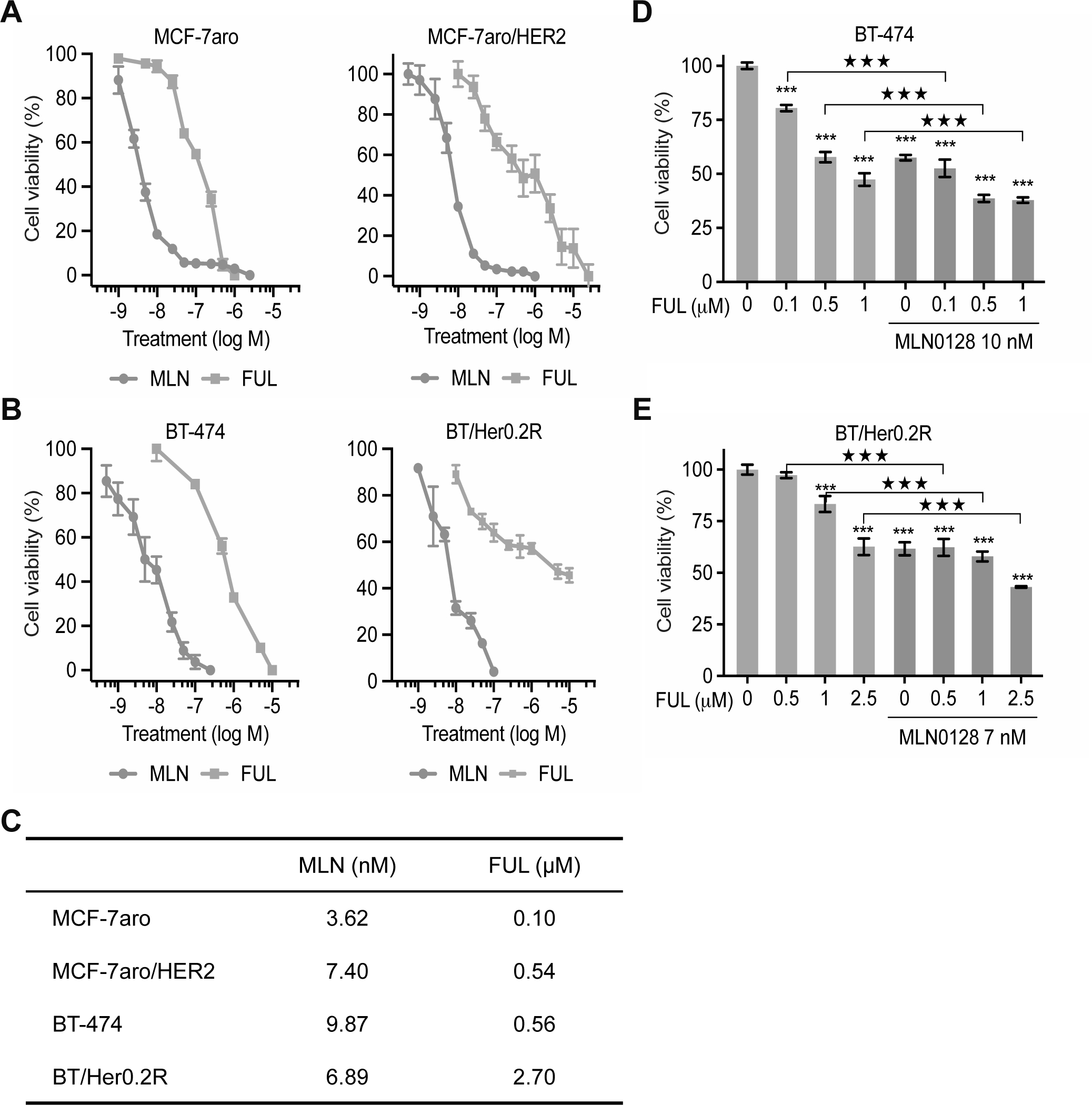
Estrogen-deprived HR+ and HR+/HER2+ cancer cells treated with MLN0128 (MLN) or fulvestrant (FUL) at escalating dosage for six days. Dose-response curves were then plotted as Mean±SD (n=5) in HR+ MCF-7aro, HER2-overexpressing MCF-7/aro (MCF-7aro/HER2) (A), HR+/HER2+ BT-474 cancer cells, and trastuzumab-resistant BT-474 derivate (BT/Her0.2R) (B). The IC50 values of individual cell lines to single-drug treatment were summarized in (C). (D-E) MLN0128 sensitized both trastuzumab-sensitive BT-474 (D) and trastuzumab-resistant BT/HER0.2R (E) cancer cells to fulvestrant treatment. Cells with 72-hr treatment of fulvestrant at different doses as indicated with/out MLN0128 at indicated dose were subjected to cell viability assay and data was shown as Mean±SD (n=6) in a histogram graph. ***, p<0.0001, compared to FUL “0”; ★★★, p<0.0001, compared to FUL single treatment, determined by t test.
DISCUSSION
Despite our knowledge of the apparent therapeutic targets of HR+/HER2+ breast cancer, the optimal therapeutic approach for this subtype remains evolutionary. Currently, endocrine therapy is the preferred option for HR+ cancer (2–5). While utilizing a collection of HR+ breast PDXs to identify therapeutic targets and evaluate drug response has been reported (33), HR+/HER2+ subgroup has not been specifically characterized. In this study, we molecularly established two PDXs from two individual tumors to comprehensively translate the biology of HR+/HER2+ breast cancers and subsequently explore targeted-therapy strategies. COH-SC1 PDX was derived from a tumor removed from a patient with positive response to trastuzumab plus chemotherapy, whereas COH-SC31 model was established from a trastuzumab-resistant tumor. According to dissection of molecular features using omics approaches and PAM50 analyses, we found that COH-SC1 PDX with high expression of MKI167 (encoding Ki-67) and low levels of ERα is associated with luminal-B subtype (Figure 4A); COH-SC31 with high expression of ESR1 (encoding ERα) and BCL2 presented as luminal-A subtype (Figure 4B). Taken together, the two PDXs revealed that different levels of ERα and dissimilar trastuzumab responses in HR+/HER2+ breast cancers might link to diverse treatment scheme.
ER signaling has been reported to drive HR+/HER2+ breast tumorigenesis in cancer cell lines (3–5). Here, we used COH-SC31 PDX harboring high expression of ERα to confirm the crucial role of ER signaling in modulating HR+/HER2+ tumor growth. Long-term estrogen exposure as the driving force of estrogen/ERα signaling directed COH-SC31 tumor growth at a larger extent than that in COH-SC1 (Figure 1B). The transcriptome analysis additionally pointed to the relative elevation of ER signaling and it function readouts, such as SERPINA1 (34), in COH-SC31 model compared to that of COH-SC1 (Supplementary Figures 1C & 4B). Moreover, the expression patterns of SERPINA1, SIAH2 and EZH2 identified from the transcriptome profiling reflected the pharmacological behaviors of COH-SC1 and COH-SC31 to fulvestrant treatment. It has been documented that high levels of SERPINA1 predict good response to endocrine therapy as similar as our previous observation (34); low expression of SIAH2 is associated with resistance to endocrine therapies in vitro and in clinic (35, 36); reduction of EZH2 expression re-sensitizes cancer cells to anti-estrogens (37). In vivo drug efficacy examination supportively confirmed that COH-SC31 is more responsive to fulvestrant in relation to COH-SC1 does (see “FUL” in Figure 5A). As a result, endocrine therapies should be included to treat HR+/HER2+ breast cancers expressing high levels of ERα or activated estrogen-dependent ERα signaling, especially for those developed trastuzumab resistance.
The bidirectional interaction between ER and RTK pathways frequently conferred resistance to single-drug treatment of endocrine therapy or HER2-targeted therapy. PI3K/AKT/mTOR signaling contained multiple components (e.g. AKT) involved in these processes (3–5, 9). Compared to expression profiling of the reference samples in transcriptional or translational levels (Figure 3 and Supplementary Figures 4 & 5), highly activated PI3K/AKT/mTOR axis was found in the two examined HR+/HER2+ PDXs. Further in silico expression and Kaplan-Meier survival analyses supported the therapeutic value in targeting PI3K/AKT/mTOR signaling (Figure 4C & 4D). Two phase-III clinical trials (BOLERO-2 and HORIZON) evaluated the addition of mTORC1 inhibitors to endocrine therapies in treating advanced or metastatic HR+/HER2− breast cancers (13, 38, 39). But, overall survival (OS) was not benefited from the addition of mTORC1 inhibitors, which may be due to the PI3K/AKT rebound effect, resulting in the alleviation of AKT (40). Instead, MLN0128 targeting to both mTORC1 and mTORC2 was developed to treat HR+ as well as HER2+ breast cancers in vitro and in vivo (15, 20, 41). In ER+ breast cancer, dual mTORC inhibitors (e.g. AZD8055 and AZD2014) combined with endocrine agents exhibited superior inhibition effects on in vitro culture system or cell-based xenografts compared to that of single-drug treatment (14, 16). Additionally, MLN combined with and the dual tyrosine kinase inhibitor- lapatinib has synergistic effects on HER2+ cancer cell lines and PDXs, which are resistant to anti-HER2 drugs (15). At the present time, fulvestrant with either MLN or AZD2014 are being investigated in HR+/HER2− advanced metastatic breast cancer in phase II clinical trials. To sum up, combination strategy with utilization of MLN may shed a light to treat HR+/HER2+ cancers.
In this study, we first observed that COH-SC1 PDX is more responsive to the first-line TRA treatment compared to COH-SC31 model (Figure 2A), consisting with the pharmacological features of the corresponding tumors. Even, TRA therapy only contributed to ~45% abolishment of tumor volumes, suggesting alternative treatment schemes are in need to treat trastuzumab-sensitive HR+/HER2+ breast cancers. Via integrative omics analysis, we uncovered that, instead of HER2 pathway, PI3K/AKT/mTOR and ER signaling are relatively up-regulated in the two studied PDXs and attenuation of these two pathways has therapeutic value against both trastuzumab-sensitive and -resistant HR+/HER2+ breast cancers (Figures 3 and Supplementary Figures 4 & 5). In trastuzumab-sensitive and lower ER expressed COH-SC1 PDX (Figure 2A and Supplementary Figure 1C), we conducted a dual-therapy treatment scheme using TRA and MLN to prove that combination therapy is more potent against tumor growth in relation to TRA or MLN monotherapy does (Supplementary Figure 6A). However, TRA-plus-MLN combination treatment as same as TRA triggered an increase of ER-positive cell population (see “Comb” in Supplementary Figure 6D), indicating that diminishing both PI3K/AKT/mTOR and ER pathways provides better therapeutic potential against HR+/HER2+ cancer. To address this assumption, in vivo drug efficacy examination using MLN and/or FUL revealed that MLN alone as like MLN-plus-FUL combination potently reduces COH-SC1 tumor growth (Figure 5A, upper panel), whereas dual-therapy treatment scheme results in more significant tumor growth inhibition of COH-SC31 PDX (p<0.0001; Figure 5A, lower panel), supporting the applicable potential of MLN in treating HR+/HER2+ tumors. Moreover, we simultaneously targeted ER signaling and one of the major signaling outputs in HER2 pathway- PI3K/AKT/mTOR axis, which proved the concept of treating HR+/HER2+ breast cancers via dual inhibition of ER and HER2 signaling cross talk. The different pharmacological behaviors of the two HR+/HER2+ PDX models with different molecular signatures suggested the importance of estrogen response and ER function/levels in guiding treatment decisions. Due to the complicated and redundant signaling transduction in HER2 signaling network, the treatment backbone may need to be further fine-tuned for a more complete suppression of ER and RTK signaling. However, it is important to figure out the possibility to treat HR+/HER2+ cancers with targeted strategies such as drugs targeting ER, HER2, and PI3K/AKT/mTOR, rather than chemotherapies associated with severe side effects. Clearly, mTOR-targeting drugs, such as MLN0128, works synergistically together with anti-HER2 drugs (e.g. trastuzumab) or endocrine therapeutic agents (e.g. fulvestrant) against ER+/HER2+ breast cancers.
In HR+/HER2+ breast cancers, ER signaling pathway is frequently activated while suppressing HER2 signaling (9, 10, 42), which potentially leads to resistance to anti-HER2 drugs and re-sensitization to endocrine therapies; this scenario was similar to that observed in COH-SC1 and COH-SC31 models (Figure 5A and Supplementary Figure 6D). We considered poorly differentiated COH-SC31 as a good trastuzumab-resistant model in which estrogen-mediated ER activation became the major driving force of growth (Figure 1B). Previous studies had shown that the concurrent pharmacological inhibition of the ER and RTK pathways achieved better effect on tumor regression than the anti-HER2 regimens alone in HR+/HER2+ cells that are resistant to anti-HER2 regimens (9, 10, 42). Our in vivo study showed the MLN-plus-FUL combination treatment synergistically inhibits COH-SC31 tumor growth, providing clinical implication that this therapeutic strategy can serve as a second line treatment for HR+/HER2+ breast cancer patients relapsed from trastuzumab therapy.
With the advance of genomic profiling studies on breast cancers, an increasing number of druggable genomic aberrations are discovered, in suggestive of the importance of personalized medicine in effective breast cancer therapy (43). However, cancer cell line models are limited in adequately reflecting tumor heterogeneity and morphology in vivo, which depreciates the predictive value of these models in drug discovery (21). Alternatively, we utilize PDX technology to explore therapeutic strategies for individual HR+/HER2+ breast cancer. As similar observation reported by Cottu et al (44), COH-SC1 and COH-SC31 PDXs retain biomarker status and pathological characteristics of their original patient tumors. The preclinical phenotype of COH-SC31 is consistent with its clinical history, supporting that this PDX model can recapitulate patient clinical features (Supplementary Figure 1A & 1B). This study presents an example of examining drug efficacy in the ‘same-patient-on-all-arms’ trials and provides a prototype for designing future ‘co-clinical’ trials to tailor personalized therapeutic decisions in clinical practice (45). One caveat is that the inclusion of more PDX breast samples would enhance the clinical implication of this study in understanding and managing HR+/HER2+ breast cancers. Though, low success rate to generate HR+/HER2+ breast PDXs and time-consuming endpoint evaluation in testing drug response of individual PDXs hamper the scaling up of this research module. Moreover, since PDXs are expected to be heterogeneous by nature, molecular characterizations need to be performed with multiple biological replicates as same as what we experienced in this study. Also, while it is difficult to expand the PDX lines in a timely manner, the study has to be carefully prioritized.
In summary, we have generated two HR+/HER2+ breast PDX models that exhibit different molecular and pharmacological features, compared to the currently available cell line models. The establishment and characterization of these two PDX models broadens the representation of HR+/HER2+ breast cancer research tools with more diverse genetic profiles and clinical relevance. We have provided new preclinical models to clinicians and scientists in tailoring therapeutic strategies for this 10% of the total breast cancer population, by demonstrating that 1) dual mTORC inhibitor MLN sensitizes HR+/HER2+ breast cancers to TRA or FUL treatment; 2) the level of estrogen-mediated ER activity in affecting pharmacological behaviors in HR+/HER2+ breast cancers; 3) tumors with higher ER activity in tending to be more responsive to FUL. Noteworthy, targeting to trastuzumab-resistant breast cancers with high ERα expression, MLN in combination with MLN approves to be a potential therapeutic solution. Furthermore, PDX technology has the potential to be implemented into breast cancer disease management. Although performing therapeutic screening in PDXs is labor-intense and time-consuming, it can provide invaluable information in guiding therapeutic strategies for late stage breast cancer patients.
Supplementary Material
TRANSLATIONAL RELEVANCE:
Utilization of patient-derived xenograft (PDX) models in preclinical breast cancer research has been recognized as a more realistic solution to accurately recapitulate the features of patient tumors. In this study, we established two PDX models from two individual hormone receptor-positive (HR+)/HER2+ tumors for preclinical drug examination. Integration of transcriptome and proteome analyses indicated that mTOR and ERα signaling are profoundly elevated for prompting tumor growth of both two PDXs. Furthermore, PDX models facilitate in creating ‘same-patient-on-all-arms’ trial to demonstrate that dual mTOR inhibitor MLN0128 in combination with either trastuzumab or anti-estrogen fulvestrant is more potent to treat HR+/HER2+ breast cancers. Notably, dual-therapy using MLN0128 and fulvestrant is beneficial in treating such breast cancers bearing significant ERα activity, particularly the trastuzumab-relapsed patients. Our research models can be scaled up to tailor personal therapeutic strategies, especially for the late stage patients.
ACKNOWLEDGEMENT
The authors thank Dr. Susan Kane for providing the BT/Her0.2R cancer cell line and Dr. Nicola Solomon for assistance with editing the manuscript.
Financial support, including the source and the number of grants, for each author: This work was supported by Panda Charitable Foundation (to S.C.) and the National Cancer Institute (P30 CA033572). Research reported in this publication included work performed in the Integrative Genomics Core, Pathology Research Service Core, and Animal Resource Center supported by the National Cancer Institute (P30CA033572).
Footnotes
Conflicts of interest: The authors declare no potential conflicts of interest.
REFERENCES
- 1.DeSantis CE, Fedewa SA, Goding Sauer A, Kramer JL, Smith RA, Jemal A. Breast cancer statistics, 2015: Convergence of incidence rates between black and white women. CA Cancer J Clin 2016;66:31–42. [DOI] [PubMed] [Google Scholar]
- 2.Burstein HJ, Prestrud AA, Seidenfeld J, Anderson H, Buchholz TA, Davidson NE, et al. American Society of Clinical Oncology clinical practice guideline: update on adjuvant endocrine therapy for women with hormone receptor-positive breast cancer. J Clin Oncol 2010;28:3784–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu VS, Kanaya N, Lo C, Mortimer J, Chen S. From bench to bedside: What do we know about hormone receptor-positive and human epidermal growth factor receptor 2-positive breast cancer? J Steroid Biochem Mol Biol 2015;153:45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Giordan SH, Temin S, Kirshner JJ, Chandarlapaty S, Crews JR, Davidson NE, et al. American Society of Clinical Oncology. Systemic therapy for patients with advanced human epidermal growth factor receptor 2-positive breast cancer: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol 2014; 32:2078–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Montemurro F, Di Cosimo S, Arpino G. Human epidermal growth factor receptor 2 (HER2)-positive and hormone receptor-positive breast cancer: new insights into molecular interactions and clinical implications. Ann Oncol 2013;24:2715–24. [DOI] [PubMed] [Google Scholar]
- 6.Strasser-Weippl K, Horick N, Smith IE, O’Shaughnessy J, Ejlertsen B, Boyle F, et al. Long-term hazard of recurrence in HER2+ breast cancer patients untreated with anti-HER2 therapy. Breast Cancer Res 2015;17:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rimawi MF, Mayer IA, Forero A, Nanda R, Goetz MP, Rodriguez AA, et al. Multicenter phase II study of neoadjuvant lapatinib and trastuzumab with hormonal therapy and without chemotherapy in patients with human epidermal growth factor receptor 2-overexpressing breast cancer: TBCRC 006. J Clin Oncol 2013;31:1726–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huober J, Fasching PA, Barsoum M, Petruzelka L, Wallwiener D, Thomssen C, et al. Higher efficacy of letrozole in combination with trastuzumab compared to letrozole monotherapy as first-line treatment in patients with HER2-positive, hormone-receptor-positive metastatic breast cancer - results of the eLEcTRA trial. Breast 2012;21:27–33. [DOI] [PubMed] [Google Scholar]
- 9.Wang YC, Morrison G, Gillihan R, Guo J, Ward RM, Fu X, et al. Different mechanisms for resistance to trastuzumab versus lapatinib in HER2-positive breast cancers- role of estrogen receptor and HER2 reactivation. Breast Cancer Res 2011;13:R121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xia W, Bacus S, Hegde P, Husain I, Strum J, Liu L, et al. A model of acquired autoresistance to a potent ErbB2 tyrosine kinase inhibitor and a therapeutic strategy to prevent its onset in breast cancer. Proc Natl Acad Sci U S A 2006;103:7795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paplomata E, O’Regan R. The PI3K/AKT/mTOR pathway in breast cancer: targets, trials and biomarkers. Ther Adv Med Oncol 2014;6:154–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun M, Paciga JE, Feldman RI, Yuan Z, Coppola D, Lu YY, et al. Phosphatidylinositol-3-OH Kinase (PI3K)/AKT2, activated in breast cancer, regulates and is induced by estrogen receptor alpha (ERalpha) via interaction between ERalpha and PI3K. Cancer Res 2001;61:5985–91. [PubMed] [Google Scholar]
- 13.Yardley DA, Noguchi S, Pritchard KI, Burris HA 3rd, Baselga J, Gnant M, et al. Everolimus plus exemestane in postmenopausal patients with HR(+) breast cancer: BOLERO-2 final progression-free survival analysis. Adv Ther 2013;30:870–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guichard SM, Curwen J, Bihani T, D’Cruz CM, Yates JW, Grondine M, et al. AZD2014, an Inhibitor of mTORC1 and mTORC2, Is Highly Effective in ER+ Breast Cancer When Administered Using Intermittent or Continuous Schedules. Mol Cancer Ther 2015;14:2508–18. [DOI] [PubMed] [Google Scholar]
- 15.Garcia-Garcia C, Ibrahim YH, Serra V, Calvo MT, Guzman M, Grueso J, et al. Dual mTORC1/2 and HER2 blockade results in antitumor activity in preclinical models of breast cancer resistant to anti-HER2 therapy. Clin Cancer Res 2012;18:2603–12. [DOI] [PubMed] [Google Scholar]
- 16.Jordan NJ, Dutkowski CM, Barrow D, Mottram HJ, Hutcheson IR, Nicholson RI, et al. Impact of dual mTORC1/2 mTOR kinase inhibitor AZD8055 on acquired endocrine resistance in breast cancer in vitro. Breast Cancer Res 2014;16:R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wander SA, Hennessy BT, Slingerland JM. Next-generation mTOR inhibitors in clinical oncology: how pathway complexity informs therapeutic strategy. J Clin Invest 2011;121:1231–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 2006;22:159–68. [DOI] [PubMed] [Google Scholar]
- 19.Dowling RJ, Topisirovic I, Fonseca BD, Sonenberg N. Dissecting the role of mTOR: lessons from mTOR inhibitors. Biochim Biophys Acta 2010;1804:433–9. [DOI] [PubMed] [Google Scholar]
- 20.Ingels A, Zhao H, Thong AE, Saar M, Valta MP, Nolley R, et al. Preclinical trial of a new dual mTOR inhibitor, MLN0128, using renal cell carcinoma tumorgrafts. Int J Cancer 2014;134:2322–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Landis MD, Lehmann BD, Pietenpol JA, Chang JC. Patient-derived breast tumor xenografts facilitating personalized cancer therapy. Breast Cancer Res 2013;15:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DeRose YS, Gligorich KM, Wang G, Georgelas A, Bowman P, Courdy SJ, et al. Patient-derived models of human breast cancer: protocols for in vitro and in vivo applications in tumor biology and translational medicine. Curr Protoc Pharmacol 2013;Chapter 14:Unit14.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tomayko MM, Reynolds CP. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother Pharmacol 1989;24:148–54. [DOI] [PubMed] [Google Scholar]
- 24.Wolff AC, Hammond ME, Hicks DG, Dowsett M, McShane LM, Allison KH, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol 2013;31:3997–4013. [DOI] [PubMed] [Google Scholar]
- 25.Hammond ME, Hayes DF, Dowsett M, Allred DC, Hagerty KL, Badve S, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for Immuno-histochemical testing of estrogen and progesterone receptors in breast cancer (unabridged version). Arch Pathol Lab Med 2010;134:e48–72. [DOI] [PubMed] [Google Scholar]
- 26.Hennessy BT, Lu Y, Gonzalez-Angulo AM, Carey MS, Myhre S, Ju Z, et al. A Technical Assessment of the Utility of Reverse Phase Protein Arrays for the Study of the Functional Proteome in Non-microdissected Human Breast Cancers. Clin Proteomics 2010;6:129–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dowsett M, Nielsen TO, A’Hern R, Bartlett J, Coombes RC, Cuzick J, et al. Assessment of Ki67 in breast cancer: recommendations from the International Ki-67 in Breast Cancer working group. J Natl Cancer Inst 2011;103:1656–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sabatier R, Finetti P, Adelaide J, Guille A, Borg JP, Chaffanet M, et al. Down-regulation of ECRG4, a candidate tumor suppressor gene, in human breast cancer. PLoS One 2011;6:e27656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 2009;27:1160–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wong C, Wang X, Smith D, Reddy K, Chen S. AKT-aro and HER2-aro, models for de novo resistance to aromatase inhibitors; molecular characterization and inhibitor response studies. Breast Cancer Res Treat 2012;134:671–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chan CT, Metz MZ, Kane SE. Differential sensitivities of trastuzumab (Herceptin)-resistant human breast cancer cells to phosphoinositide-3 kinase (PI-3K) and epidermal growth factor receptor (EGFR) kinase inhibitors. Breast Cancer Res Treat 2005;91:187–201. [DOI] [PubMed] [Google Scholar]
- 32.Chen B, Wang Y, Kane SE, Chen S. Improvement of sensitivity to tamoxifen in estrogen receptor-positive and Herceptin-resistant breast cancer cells. J Mol Endocrinol 2008;41:367–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li S, Shen D, Shao J, Crowder R, Liu W, Prat A, et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep 2013;4:1116–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chan HJ, Li H, Liu Z, Yuan YC, Mortimer J, Chen S. SERPINA1 is a direct estrogen receptor target gene and a predictor of survival in breast cancer patients. Oncotarget 2015;6:25815–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Interiano RB, Yang J, Harris AL, Davidoff AM. Seven In Absentia Homolog 2 (SIAH2) downregulation is associated with tamoxifen resistance in MCF-7 breast cancer cells. J Surg Res 2014;190:203–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jansen MP, Ruigrok-Ritstier K, Dorssers LC, van Staveren IL, Look MP, Meijer-van Gelder ME, et al. Downregulation of SIAH2, an ubiquitin E3 ligase, is associated with resistance to endocrine therapy in breast cancer. Breast Cancer Res Treat 2009;116:263–71. [DOI] [PubMed] [Google Scholar]
- 37.Reijm EA, Jansen MP, Ruigrok-Ritstier K, van Staveren IL, Look MP, van Gelder ME, et al. Decreased expression of EZH2 is associated with upregulation of ER and favorable outcome to tamoxifen in advanced breast cancer. Breast Cancer Res Treat 2011;125:387–94. [DOI] [PubMed] [Google Scholar]
- 38.Piccart M, Hortobagyi GN, Campone M, Pritchard KI, Lebrun F, Ito Y, et al. Everolimus plus exemestane for hormone-receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: overall survival results from BOLERO-2. Ann Oncol 2014;25:2357–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vicier C, Dieci MV, Arnedos M, Delaloge S, Viens P, Andre F. Clinicla development of mTOR inhibitors in breast cancer. Breast Can Res 2014;16:203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem 2009;284:8023–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gokmen-Polar Y, Liu Y, Toroni RA, Sanders KL, Mehta R, Badve S, et al. Investigational drug MLN0128, a novel TORC1/2 inhibitor, demonstrates potent oral antitumor activity in human breast cancer xenograft models. Breast Cancer Res Treat 2012;136:673–82. [DOI] [PubMed] [Google Scholar]
- 42.Munzone E, Curigliano G, Rocca A, Bonizzi G, Renne G, Goldhirsch A, et al. Reverting estrogen-receptor-negative phenotype in HER-2-overexpressing advanced breast cancer patients exposed to trastuzumab plus chemotherapy. Breast Cancer Res 2006;8:R4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carels N, Spinasse LB, Tilli TM, Tuszynski JA. Toward precision medicine of breast cancer. Theor Biol Med Model 2016;13:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cottu P, Marangoni E, Assayag F, de Cremoux P, Vincent-Salomon A, Guyader C, et al. Modeling of response to endocrine therapy in a panel of human luminal breast cancer xenografts. Breast Cancer Res Treat 2012;133:595–606. [DOI] [PubMed] [Google Scholar]
- 45.Clohessy JG, Pandolfi PP. Mouse hospital and co-clinical trial project--from bench to bedside. Nat Rev Clin Oncol 2015;12:491–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.