

Abstract
Extensive efforts, through cell line-based models, have been made to characterize the androgen receptor (AR) signaling pathway in triple-negative breast cancer (TNBC). However, these efforts have not yet reached a consensus with regards to the mechanism of AR in TNBC. Considering that patient-derived xenografts (PDXs) are more appropriate than cell line-based models for recapitulating the structural and molecular features of a patient’s tumor, we have identified and molecularly characterized two new AR-positive TNBC PDX models and assessed the impacts of AR agonist [dihydrotestosterone (DHT)] and antagonist (enzalutamide) on tumor growth and gene expression profiles by utilizing immunohistochemistry, western blots, and RNA-Seq analyses. Two PDX models, termed TN1 and TN2, were derived from two grade-3 TNBC tumors, each harboring 1~5% of AR nuclear positive cancer cells. DHT activated AR in both PDX tumors by increasing nuclear localization and AR protein levels. However, the endpoint tumor volume of DHT-treated TN1 was 3-folds smaller than that of non-treated TN1 tumors. Conversely, the endpoint tumor volume of DHT-treated TN2 was 2-folds larger than that of non-treated TN2. Moreover, enzalutamide failed to antagonize DHT-induced tumor growth in TN2. The RNA-Seq analyses revealed that DHT mainly suppressed gene expression in TN1 (961 down-regulated genes versus 149 up-regulated genes), while DHT promoted gene expression in TN2 (673 up-regulated genes versus 192 down-regulated genes). RNA-Seq data predicted distinct TNBC molecular subtypes for TN1 and TN2. TN1 correlated to a basal-like 1 (BL1) subtype, and TN2 correlated to a basal-like 2 (BL2) subtype. These analyses suggest that TN1 and TN2, which both express functional AR, are two molecularly distinct PDX models. The molecular characterization of these PDX models expands our current knowledge on AR-positive TNBC. Our results do not support that AR is a suitable therapeutic target in TNBC. To our best knowledge, the molecular mechanisms of AR in TNBC are equivocal and should be evaluated using clinically relevant models, considering both the heterogeneous expression of AR in TNBC and the general complexities of AR signaling.
Keywords: Androgen Receptor, Triple Negative Breast Cancer, Patient-derived Xenograft, RNA Sequencing
1. Introduction
Triple-negative breast cancer (TNBC) is an aggressive and heterogenous subtype of breast tumors that lacks the expression of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor HER2/neu (HER2). Targeted therapies against these signaling molecules are not effective for TNBC [1]. According to extensive immunohistochemistry (IHC) studies, one subset of TNBCs positively expresses androgen receptor (AR). The size of this AR positive subset (10~50% of TNBCs) depends on the cut-off score for AR positivity (>1% or >10% of cell nuclear positivity) [2]. In this subset, AR has been considered as a potential therapeutic target [3].
The AR signaling pathway is an oncogenic driver in prostate cancer. This pathway has been targeted by different mechanisms using various pharmacological agents [4]. Thus, considerable efforts have been made to characterize the AR signaling pathway in AR-positive TNBC [3]. In the reported preclinical studies, AR-positive TNBC has been modeled using cell lines from in vitro cultures and cell line-derived xenografts [5, 6]. Importantly, these studies were not able to reach an agreement on the biological significance of AR in TNBC. Unsurprisingly, no criteria has been standardized to select patients and predict their responses to therapy [7, 8]. In three recent clinical trials, patients with AR-positive TNBC had a modest response to AR inhibitors: abiraterone acetate [9], bicalutamide [10], and enzalutamide [11]. To clarify AR signaling in TNBC, and ultimately improve outcomes, more clinically representative model systems are urgently needed [12, 13].
Compared to cell line models, patient-derived-xenografts (PDX) may more accurately reproduce breast cancer diversity, tumor behavior, and metastatic potential [14]. Several PDX models of TNBC have been developed and applied to drug development [15]. However, merely a few of them were reported as AR-positive TNBC. In a review, Christenson et al. described two TNBC PDXs (3561 and PK49) with low levels of functional AR, where functionality was confirmed using dihydrotestosterone (DHT) [3]. In a conference abstract, Reese et al. described an AR-positive TNBC PDX (HCI-009, originally generated by Dr. Alana Welm [16]) that experienced a significant increase in tumor volume when DHT was administered. As expected, HCI-009 tumor volume decreased when treated with the antagonist Seviteronel [17]. To the best of our knowledge, these three examples represent the whole breadth of reported AR-positive PDX models of TNBC, however, none of them were fully molecular and biological characterized.
In our study, we identified and molecularly evaluated two new PDX models of TNBC. The remaining aims of this study were 1) to assess the PDXs as tools for preclinical studies and 2) probe the molecular functions of AR in TNBC. We focused on how tumor growth and gene expression profiles were influenced by AR activation. Our goal was to better define the mechanistic actions of AR in these two PDX models, which may ultimately lead to a better understanding of the roles of AR in TNBC.
2. Materials and Methods
2.1. Patients and tumor specimens
Primary breast tumor samples were collected from patients who were diagnosed with stage 3–4 breast cancers and received treatment at City of Hope. Tumor samples negative for ER, PR, and HER2 were defined as TNBC. Six PDX tumors of TNBC were checked for AR expression by western blot analysis. Clinical and pathological information of two AR-positive samples (COH_98 and COH_53) were summarized in Table 1. Tumor-stroma ratio (TSR) was evaluated. Stroma-high is defined as > 50% stromal area, and stroma-low is defined as ≤ 50% stromal area in the histological section [18]. IHC staining for AR was applied to those two samples to confirm the AR positivity and histological distributions. This study was approved by the City of Hope Institutional Review Board and all patients provided informed, written consent prior to tissue collection. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Table 1.
Characteristics of the clinical-pathological features of patients and tumors for establishing PDX models
| Sample ID | COH 98 | COH 53 | |
|
| |||
| Clinical information | Age | 45 | 43 |
| Ethnicity | Hispanic | Hispanic | |
| Tumor Size (cm) | ≥2.3 | ≥3.3 | |
| Tumor Stage (TNM) | T2N2aM1 | T2N0M0 | |
| Chemotherapy | Neoadjuvant response | Neoadjuvant response | |
| Radiotherapy | N/A | N/A | |
| Anatomy-Pathological information | Sample Site | Breast | Breast |
| Histological type | IDC | IDC | |
| Pathological grade | 3 | 3 | |
| ER | - | − (<1%) | |
| PR | - | − (<1%) | |
| HER2 (IHC) | − (<1%) | N/A | |
| HER2 (FISH) | N/A | - | |
|
| |||
| PDXs ID | TN1 | TN2 | |
|
| |||
| PDX sample information | Growth to 500mm3 | 40 days | 40 days |
| DHT induced growth | NO | Yes | |
| AR upregulated by DHT | Yes | Yes | |
| AR translocation by DHT | Yes | Yes | |
IDC: invasive ductal carcinoma;
N/A: not applicable, not available or no answer
2.2. Establishment of PDX models and treatment
Two AR-positive PDX tumors were implanted into intact female NSG mice (6~8 weeks) and PDX models were established as previously described [19]. Two PDXs were named TN1 and TN2, corresponding to COH_98 and COH_53 respectively (Table 1). For initial implantation, both PDX tumors took as long as 90 days before they were of sufficient size for passaging. For experimental implants, both PDX tumors were slow-growing and took nearly 40 days to reach 500 mm3 (10 mm × 10 mm). When tumor size reached approximately 500 mm3, NSG mice were euthanized via CO2 asphyxiation, and tumors samples were collected for characterization. Within five to ten minutes of resection, tumors were flash-frozen in liquid nitrogen and/or fixed with 4% paraformaldehyde. Flash-frozen tumors were stored at −80°C for Reverse Phase Protein Array (RPPA) [19, 20] and whole exome sequencing (WESeq). Fixed samples were sent to the Pathology Core at City of Hope for H&E staining and IHC analysis of AR, ER, PR, HER2, and Ki67.
For the DHT treatment, PDX tumors of TN1 and TN2 were implanted into NSG female mice (6~8 weeks). On the same day of tumor implantation, DHT pellets (12.5 mg/60 days, Innovative Research of America, Sarasota, FL) were subcutaneously grafted into both treatment groups (TN1/2-DHT). Placebo pellets (Innovative Research of America, Sarasota, FL) were similarly implanted into both control groups (TN1/2-Ctrl). The TN1 group comprised of 4 mice (2 TN1-Ctrl and 2 TN1-DHT), and the TN2 group comprised of 5 mice (2 TN2-Ctrl and 3 TN2-DHT). Tumor growth was measured as previously described [19]. When tumor size reached approximately 500 mm3, mice were euthanized and tumors samples were collected for western blotting, IHC, and RNA Sequencing (RNA-Seq) analyses.
For the enzalutamide treatment, PDX tumors of TN2, for which DHT induced the tumor growth, were grafted into NSG female mice (6~8 weeks) and randomized into two groups when the tumor size reached approximately 500 mm3. On the same day of randomization, DHT pellets (12.5 mg/60 days, Innovative Research of America, Sarasota, FL) were subcutaneously implanted into all mice. The control group comprised of 4 mice that received PBS through daily gavage. The treatment group comprised of 5 mice that received enzalutamide (50 mg/kg/day, Selleckchem, Pittsburgh, PA) via daily gavage. The tumor volume was measured every 5 days. After 15 days of treatment, tumor samples were collected for IHC analyses of AR and Ki67.
NSG mice were bred and housed at the City of Hope Animal Resources Center in ventilated cage racks with free access to water and food. Mice were maintained on a 12 h light/dark cycle. All animal experiments were performed under a protocol (91051) approved by the Institutional Animal Care and Use Committee at City of Hope. Facilities are accredited by the association for assessment and accreditation of laboratory animal care and operated according to NIH guidelines.
2.3. RNA-Seq and Data analysis
Using the RNeasy Extract Kit (Qiagen, Germantown, MD), total RNA was extracted from the flash-frozen PDX tumors of the TN1 group (2 TN1-Ctrl and 2 TN1-DHT) and TN2 group (2 TN2-Ctrl and 3 TN2-DHT). Total RNA extract was sequenced by the Integrative Genomics Core at City of Hope. Principal component analysis (PCA) was used to display transcriptome distance, the relatedness between the two PDX tumors, and the relatedness between the control and DHT groups. Ingenuity Pathway Analysis (IPA) and Gene Set Enrichment Analysis (GSEA) were used to analyze systems-level pathways. Venn diagram analysis was conducted to display the number of overlapping AR responsive genes in datasets of TN1-DHT and TN2-DHT versus three RNA-Seq datasets that describe AR responsive genes in breast cancer tissue and/or cells lines. Two datasets were generated from DHT-treated MDA-MB-453 cells [21] and testosterone-treated MCF-7 cells [22]. One dataset was generated from The Cancer Genome Atlas (TCGA) expression data on the basal subtype of breast invasive carcinoma [23].
2.4. TNBC molecular subtyping via TNBCtype™
RNA-Seq data from TN1 and TN2 were uploaded via TNBCtype™ online tool (http://cbc.mc.vanderbilt.edu/tnbc/prediction.php) for molecular subtyping predictions [24]. The predicted subtype of each sample was displayed with the corresponding correlation coefficient (CC) and permutation p value.
2.5. Immunohistochemistry and histopathological analysis
Immunohistochemistry (IHC) and hematoxylin and eosin (H&E) staining of formalin-fixed tumor tissues were performed by the Pathology Core at City of Hope. Antibodies used in IHC included ER (ab16660, Abcam), PR (PA0312, Leica), HER2 (A0485, DAKO), AR (SP107, Sigma-Aldrich), and Ki67 (MIB-1, DAKO). Slides were reviewed first at 4X magnification to identify areas of positive staining, followed by confirmation and quantitation at 20X magnification. AR and Ki67 were scored by identifying areas of most abundant positivity at low magnification. Then, ten high-power (20X) fields were counted by QuPath software (version 0.2). Representative images and scoring were acquired using an Olympus BX46 microscope with a DP27 camera and Olympus CellSens software (Olympus).
2.6. Immunological blotting
PDX tissues were lysed in Nonidet P-40 buffer (50 mM Tris-Cl, pH 7.5, 1% Nonident P40, 150 mM NaCl, 0.5% sodium deoxycholate and complete protease inhibitor cocktail) on ice for 20 min. Then, tissues were sonicated for 30 s. After centrifugation, the supernatants were collected, mixed with 4×SDS sample buffer, and boiled for 5 min. Protein concentration was quantified using the BioRad BCA Protein Assay. Anti-AR antibodies (SC-7305, Santa-Cruz Biotech) were applied to detect AR. Western blotting was performed as previously described [19].
2.7. Statistical analysis
Results are shown as means ± standard deviation (SD). All statistical analyses were performed using GraphPad Prism software (version 8.0). The significance of the differences between mean values was determined by multiple Student’s t-tests. All tests were two-tailed. Benjamini and Hochberg’s method was used to compute the adjusted p value and control the false discovery rate (FDR) for RNA-Seq analysis.
3. Results
3.1. Establishment and characterization of two AR-positive TNBC PDX models
We checked the expression of AR in six TNBC PDXs established at City of Hope. Western blotting analyses of six PDX tumors detected two of them to be AR-positive TNBC specimens: COH_98 and COH_53. The other four did not considerably exhibit levels of AR (Figure 1B). As shown in Table 1, COH_98 was a grade 3 TNBC breast tumor under neoadjuvant therapy that exhibited lymph node involvement and distant metastasis (T2N2aM1). COH_98 was a tumor with a high TSR score (>90%), due to treatment effects, whose percentage of cancer cells was at most 10%. Ki67 index in COH_98 was as high as 80% in cancer cells. Among them, 1~5% of cancer cells were classified as AR immunoreactive cells by IHC, while stromal regions were negative to AR (Supplementary Figure 1A). COH_53 was also a grade 3 TNBC breast tumor under neoadjuvant therapy, but without distant metastasis (T2N0M0). COH_53 was a tumor with low TSR score (<40%) whose percentage of cancer cells was as high as 60%. 60~70% of cancer cells were positive for Ki67, while nearly 5~8% of them were AR immunoreactive cells (Supplementary Figure 1B). The two AR-positive PDXs were then termed TN1 (COH_98) and TN2 (COH_53), respectively.
Figure 1. Establishment of two AR-positive TNBC PDX models with functional AR upon DHT treatment.
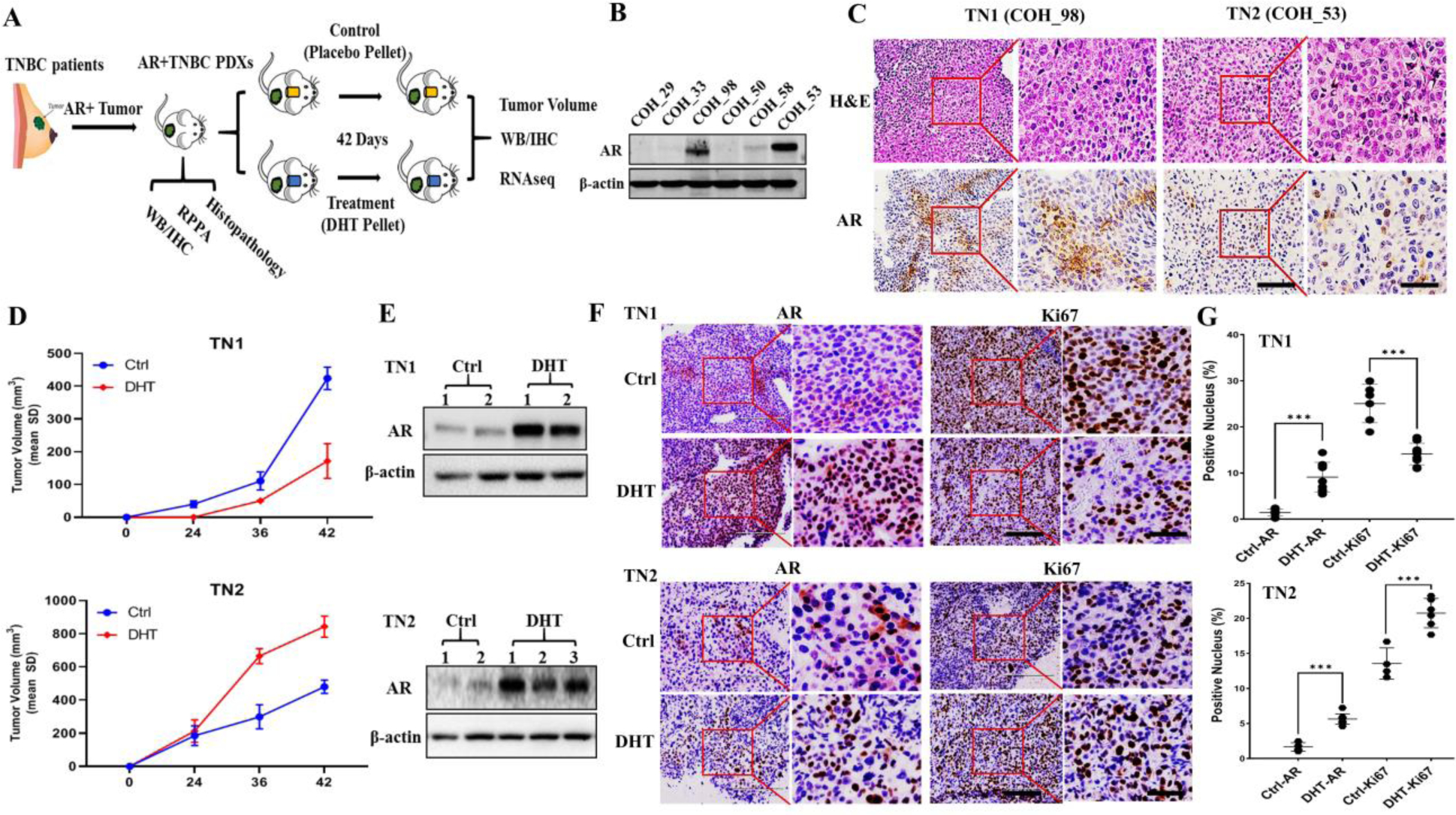
A) Scheme of AR-positive TNBC PDX establishment and utilization; B) Western blot identified AR expression in patients’ TNBC samples; C) Histopathological assessment of nuclei grading by H&E staining and AR positivity in PDXs tumors of TN1 and TN2 under 20X and 40X magnification. Scale bar for 20X and 40X magnification images are 200 μm and 100 μm, respectively. D) Tumor growth curve of PDXs TN1 & TN2 upon DHT treatment. Intact mice were randomized and implanted with a tumor from TN1 (2 Ctrl versus 2 DHT) and TN2 (2 Ctrl versus 3 DHT) by placebo pellets in control (Ctrl) and DHT pellets in treatment group; E) Western blot identified AR expression in PDX tumor of TN1 (2 Ctrl versus 2 DHT) and TN2 (2 Ctrl versus 3 DHT); F) AR and Ki67 expression pattern in PDX tumor of TN1 (2 Ctrl versus 2 DHT) and TN2 (2 Ctrl versus 3 DHT) by IHC at 20X & 40X magnification with a scale bar of 200 μm and 100 μm, respectively. G) Nuclear Score of AR and Ki67 in PDX tumors of TN1 and TN2 (***, p<0.001).
As summarized in Table 1, PDX tumors of TN1 and TN2 were grade 3 TNBC tumors. Cell morphologies of TN1 and TN2 were assessed by H&E staining. Both PDX tumors displayed highly pleomorphic nuclei with one or more eosinophilic macronuclei (Figure 1C, H&E Staining, Supplementary Figure 1A and B). According to the IHC analyses of TN1 and TN2, which were assessed in accordance with American Society of Clinical Oncology / College of American Pathologists (ASCO/CAP) guidelines, both tumors were ER, PR, and HER2 negative tumors (Supplementary Figure 2) with 1~5% of AR nuclear positive cells (Figure 1C, IHC-AR). Ki67 indexes in both PDX tumors (Supplementary Figure 2) were lower than that in the original patients’ specimen. Many PDX models grow slowly due to the immune surveillance mechanisms found in mice [25].
3.2. Differential DHT response in tumor growth of TN1 and TN2 via activation of AR
We next investigated the potential roles of AR in two TNBC PDXs by DHT exposure. On the same day that TN1 or TN2 was implanted, mice were treated with DHT or placebo. We then measured tumor volumes, performed western blots, and assessed tumors by IHC and RNA-Seq analyses (Figure 1A). According to our results, DHT induced growth of TN2 tumors. After 42 days of treatment, the sizes of TN2-DHT tumors were 2-folds larger than that of TN2-Ctrl. Conversely, when TN1 was treated with DHT, growth of the PDX was impeded. Specifically, the endpoint tumor volume of TN1-DHT was 3-folds smaller than that of TN1-Ctrl (Figure 1D).
We subsequently assessed how treatment of the PDXs with DHT affected AR function. When both TN1 and TN2 tumors were treated with DHT, western blots revealed increased expression of AR protein (Figure 1E). IHC staining also detected functional activation of AR; DHT induced translocation of AR into the nucleus and also increased AR expression level (Figure 1F and G). Considering the size of the tumors to be relatively variable (Figure 1D), we checked the Ki67 levels by IHC. The Ki67 index supported our results on tumor growth: while TN1-DHT had a lower index than TN1-Ctrl, the index for TN2-DHT was significantly higher than TN2-Ctrl (Figure 1 F and G).
In order to further assess the AR antagonist effects on PDX model, we carried out an experimental therapy with enzalutamide on the PDX tumor of TN2, for which DHT induced the tumor growth. As shown in Supplementary Figure 3, the tumor growth curve indicated that DHT promoted the growth of TN2 tumor. However, enzalutamide failed to restrain DHT-induced tumor growth in TN2, even though IHC of AR suggested that enzalutamide was able to suppress AR activation in those tumors.
3.3. Distinct transcriptional profiles induced by DHT in TN1 and TN2
As both TN1 and TN2 are new models for AR-positive TNBC, we comprehensively characterized their transcriptional profiles. The resulting data was assessed by PCA, TNBC subtyping, IPA, and GSEA. PCA analysis well separated TN1 from TN2 (Figure 2A). TN1 and TN2 thus appear to capture different transcript profiles. Similarly, TNBC subtyping analyses also predicted distinct molecular subtypes for TN1 and TN2; TN1 correlated to a basal-like 1 (BL1) subtype, and TN2 correlated to a basal-like 2 (BL2) subtype (Figure 2C). These evaluations suggest that TN1 and TN2, which both express functional AR, are molecularly distinct PDX models.
Figure 2. Distinct transcription profiles induced by DHT in PDX tumors of TN1 and TN2.
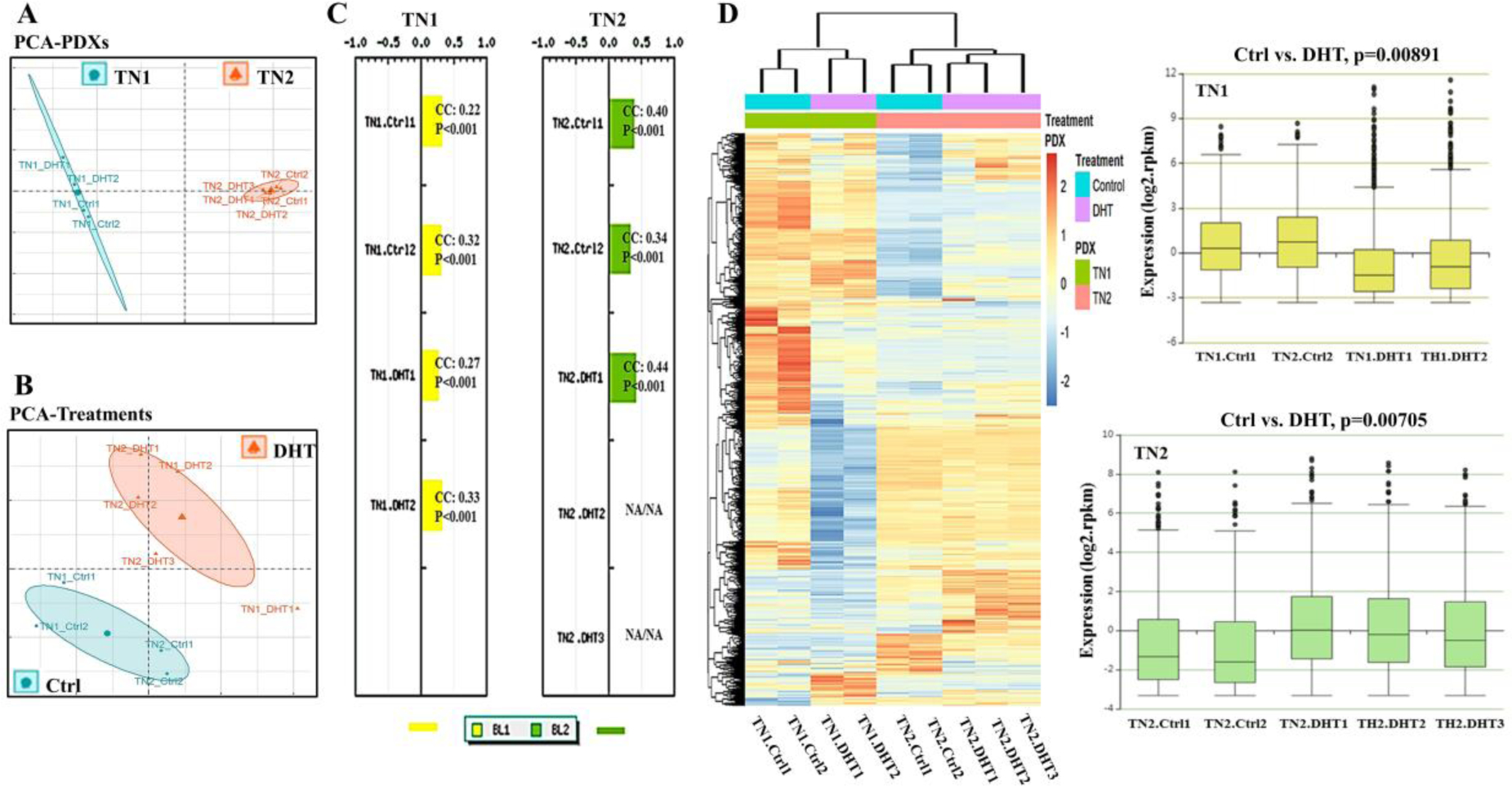
A) Distribution of transcription profiles between TN1 (2 Ctrl versus 2 DHT) and TN2 (2 Ctrl versus 3 DHT) by principal component analysis (PCA); B) Distribution of transcription profiles between Ctrl (2 TN1-Ctrls versus 2 TN2-Ctrls) and DHT (2 TN1-DHTs versus 3 TN2-DHTs) by principal component analysis (PCA); C) TNBC molecular subtyping via TNBCtype™ with TN1 (2 Ctrl versus 2 DHT) and TN2 (2 Ctrl versus 3 DHT). The predicted subtype of each sample was indicated with a corresponding correlation coefficient (CC) and permutation p value (P). A CC value between 0.2–0.4 indicates a slightly positive correlation and a CC value between 0.4–0.7 indicates a fairly positive correlation. D) Hierarchical clustering analysis with a heat map and a boxplot expression diagram of TN1 (2 Ctrl versus 2 DHT, p=0.00891) and TN2 (2 Ctrl versus 3 DHT, p=0.00705).
We also compared TN1 with TN2 in terms of DHT-induced gene transcription. In both PDXs, DHT induced significant transcriptional changes (Figure 2B). However, gene expression profiles of TN1-DHT and TN2-DHT were different. Hierarchical clustering analysis along with Boxplot expression diagrams indicated that genes were generally down-regulated by DHT in TN1, whereas they were up-regulated by DHT in TN2 (Figure 2D). Taken altogether, treatment of the two PDXs with DHT appears to yield significantly distinct transcriptomes.
3.4. Functional characterization of transcription profiles induced by DHT in TN1 and TN2
For our transcription profile characterization, significantly regulated genes were defined as up-regulated genes (Fold-change ≥ 2, p<0.05) versus down-regulated genes (Fold-change ≤ 0.5, p<0.05). A total of 1110 genes were identified in TN1 (961 down-regulated versus 149 up-regulated). The total genes identified was 865 in TN2 (192 down-regulated versus 673 up-regulated). TN1 and TN2 shared 184 common genes. While 61 of these common genes were regulated in the same direction, 123 of them were regulated in the opposite direction. According to these results, the two AR-positive PDXs are very disparate models of TNBC (Figure 3A).
Figure 3. Functional characterization of transcriptional profiles induced by DHT in PDX tumors of TN1 and TN2.
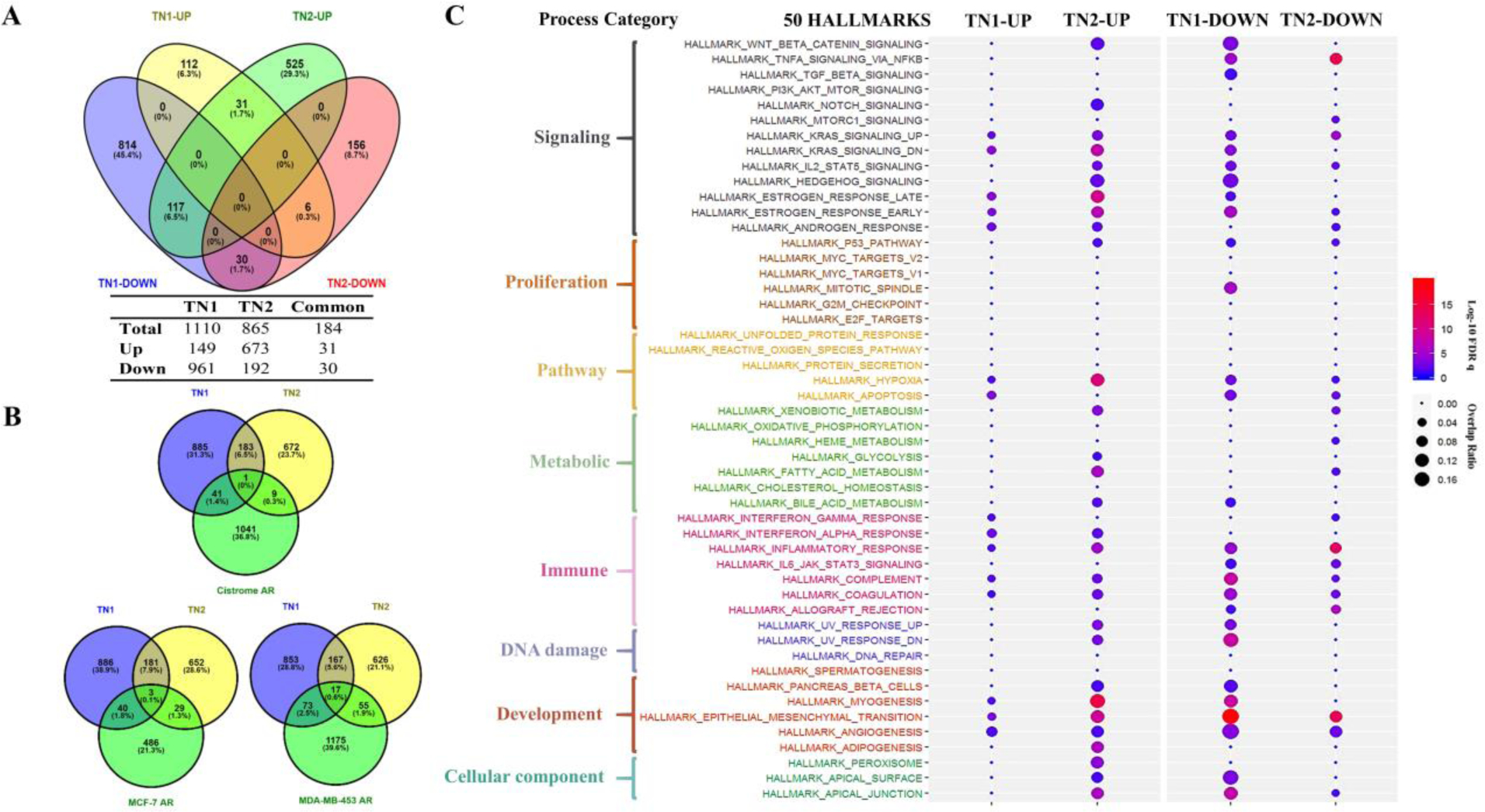
A) Venn’s four-set diagram displayed the number of overlapping and unique genes sets in TN1 and TN2 after DHT treatment. TN1-UP/TN2-UP represented number of up-regulated genes in TN1/TN2-DHTs versus TH1/TH2-Ctrls and vice versa; B) Venn’s three-set diagram displayed the number of overlapping and unique AR responsive genes sets in TN1 and TN2 versus three AR responsive gene sets (Cistrome AR, MCF-7 AR and MDA-MB-453 AR); C) Distribution of GSEA enriched Hallmarks in TN1 (up versus down) and TN2 (up versus down). The size of spot indicated K/K value (overlap ratio) and the color of spot indicated significance by log-10 FDR q value (log-10 FDR q value>1.3, FDR q value<0.05 is the cutoff value for significance).
We next analyzed AR-regulated transcripts in these PDXs after DHT treatment. Venn diagram analysis shown the number of overlapping AR responsive genes among datasets of TN1 and TN2 versus other three datasets. Dataset of Cistrome-AR was derived from breast cancer tissue (Cistrome Cancer-TCGA-Breast invasive carcinoma-basal). Dataset of MCF-7-AR was generated from testosterone-treated MCF-7 cell lines. Dataset of MDA-MB-453-AR was derived from DHT-treated MDA-MB-453 cell lines. For dataset of TN1, Cistrome-AR comparison yielded 42 common transcripts and MCF-7 AR comparison yielded 43 common genes. The same comparisons of TN2 yielded 10 common genes with Cistrome-AR versus 32 common genes with MCF-7 AR. In contrast, more common transcripts were identified with dataset of MDA-MB-453-AR: 90 common genes in TN1 and 72 common genes in TN2. (Figure 3B).
Using GSEA enrichment analysis by 50 Hallmark Gene Sets, we generated a landscape of hallmarks in DHT-treated TN1 and TN2 (Figure 3C). In this figure, the size of the spots is used to convey the relative magnitude of the K/K value (overlap ratio). Spots are also color coded to convey significance by log-10 FDR q value. The upper cutoff boundary for significance is log-10 FDR q >1.3; the lower cutoff is log-10 FDR q <0.05. Treatment of TN1 with DHT down-regulated genes in hallmarks of epithelial-mesenchymal transition, apical junction, myogenesis, angiogenesis, Wnt-beta-catenin, and etc. In TN2, treatment with DHT up-regulated genes in hallmarks of myogenesis, hypoxia, Wnt-beta-catenin, and etc.
IPA analysis was applied to functionally characterize the transcriptomic profiles induced by DHT in TN1 and TN2. Table 2 and Table 3 list the top 5 canonical pathways (Z score >2, p<0.05) and top 5 upstream regulators (Z score >2, p<0.05). The results from IPA analysis agree with the GSEA results (Figure 3C). DHT treatment of TN1 and TN2 induced different canonical pathways and upstream regulators. Specifically, treatment of TN1 with DHT appears to suppress multiple upstream regulators involved in epithelial-mesenchymal transition, whereas treatment of TN2 with DHT appears to activate upstream regulators involved in hypoxia and Wnt-beta-catenin signaling.
Table 2.
Top 5 canonical pathways (Z score >2, p<0.05) of DHT induced transcriptomic profiles in PDX tumors of TN1 and TN2.
| TN1 | TN2 | ||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Ingenuity Canonical Pathways | z-score | p-value | Overlap Ratio | Ingenuity Canonical Pathways | z-score | p-value | Overlap Ratio |
|
| |||||||
| Rho-GDI Signaling | 3.441 | 1.22E-03 | 0.138 (22/159) | Aryl Hydrocarbon Receptor Signaling | −2.212 | 2.69E-07 | 0.157 (22/140) |
| Rho-GTPases Signaling | −3.674 | 1.73E-03 | 0.125 (27/216) | IL-1 Mediated RXR Inhibition | −2.121 | 6.92E-07 | 0.127 (28/221) |
| Actin Cytoskeleton Signaling | −3.30 | 7.93E-03 | 0.119 (18/185) | Planar Cell Polarity Signaling | 2.121 | 8.19E-03 | 0.123 (8/65) |
| Corticotropin Releasing Hormone Signaling | −2.714 | 6.12E-03 | 0.152 (14/92) | Wnt/GSK-3β Signaling | 2.236 | 5.62E-03 | 0.091 (7/396) |
| Glutamate Receptor Signaling | −2 | 3.00E-03 | 0.128 (18/141) | Wnt/Ca+ pathway | 2.449 | 4.43E-03 | 0.103 (6/58) |
Table 3.
Top 5 up-stream regulator (Z score >2, p<0.05) in DHT induced transcriptomic profiles in PDX tumors of TN1 and TN2.
| TN1 | TN2 | |||||||
|---|---|---|---|---|---|---|---|---|
| Top-5 | Upstream Regulator | Activation z-score | p-value | Predicted Activation | Upstream Regulator | Activation z-score | p-value | Predicted Activation |
|
| ||||||||
| Up-regulation | CDH1 | 2.425 | 2.5E-06 | Activated | SMARCA4 | 5.724 | 8.47E-06 | Activated |
| EIF2AK2 | 2.414 | 3.18E-05 | Activated | HIF1A | 4.624 | 6.73E-08 | Activated | |
| PLAG1 | 2.213 | 7.37E-05 | Activated | STAT3 | 4.177 | 4.90E-05 | Activated | |
| IL1B | 2.158 | 3.61E-06 | Activated | TP73 | 3.726 | 2.34E-05 | Activated | |
| EHF | 2.121 | 1.90E-08 | Activated | TGFB1 | 3.691 | 9.06E-07 | Activated | |
|
| ||||||||
| Down-regulation | TGFB1 | −7.092 | 3.12E-11 | Inhibited | TNF | −10.361 | 5.09E-08 | Inhibited |
| SMARCA4 | −5.399 | 5.59E-06 | Inhibited | SYVN1 | −8.641 | 9.12E-11 | Inhibited | |
| CTNNB1 | −5.362 | 1.96E-09 | Inhibited | IL1A | −6.123 | 4.03E-09 | Inhibited | |
| TGFB3 | −3.894 | 2.72E-06 | Inhibited | PRKCD | −6.033 | 9.06E-06 | Inhibited | |
| BMP2 | −3.583 | 4.12E-06 | Inhibited | ERG | −6.016 | 3.14E-06 | Inhibited | |
4. Discussion
Preclinical studies with cell line-based models have revealed highly differential effects of AR in TNBC. Several TNBC cell lines endogenously expressed AR protein at different levels [26]. Among them, MDA-MB-453 was shown to have higher expression levels of endogenous AR and exhibit DHT-dependent cell growth [21, 26]. However, according to other studies on the same cell line, DHT arrests the cell cycle [27] and causes apoptosis [28]. In other AR-positive TNBC cell lines, particularly ones with low levels of endogenous AR (MDA-MB-231), DHT produced a modest effect on both cell proliferation and apoptosis [28]. It was also reported that when AR was overexpressed through transfection in MDA-MB-231 cells, a synthetic androgen (R1881) suppressed cell cycle via p21 activation [29]. Such conflicting data may be partially indicative of the inherent limitations of cell line-based models: adaptation of cells to artificial culture system and treatment, clonal selection, and homogeneity [15]. As such, new models are required to clarify the role AR may play in TNBC [13]. In the present study, we identified and molecularly characterized two new PDX models of AR-positive TNBC.
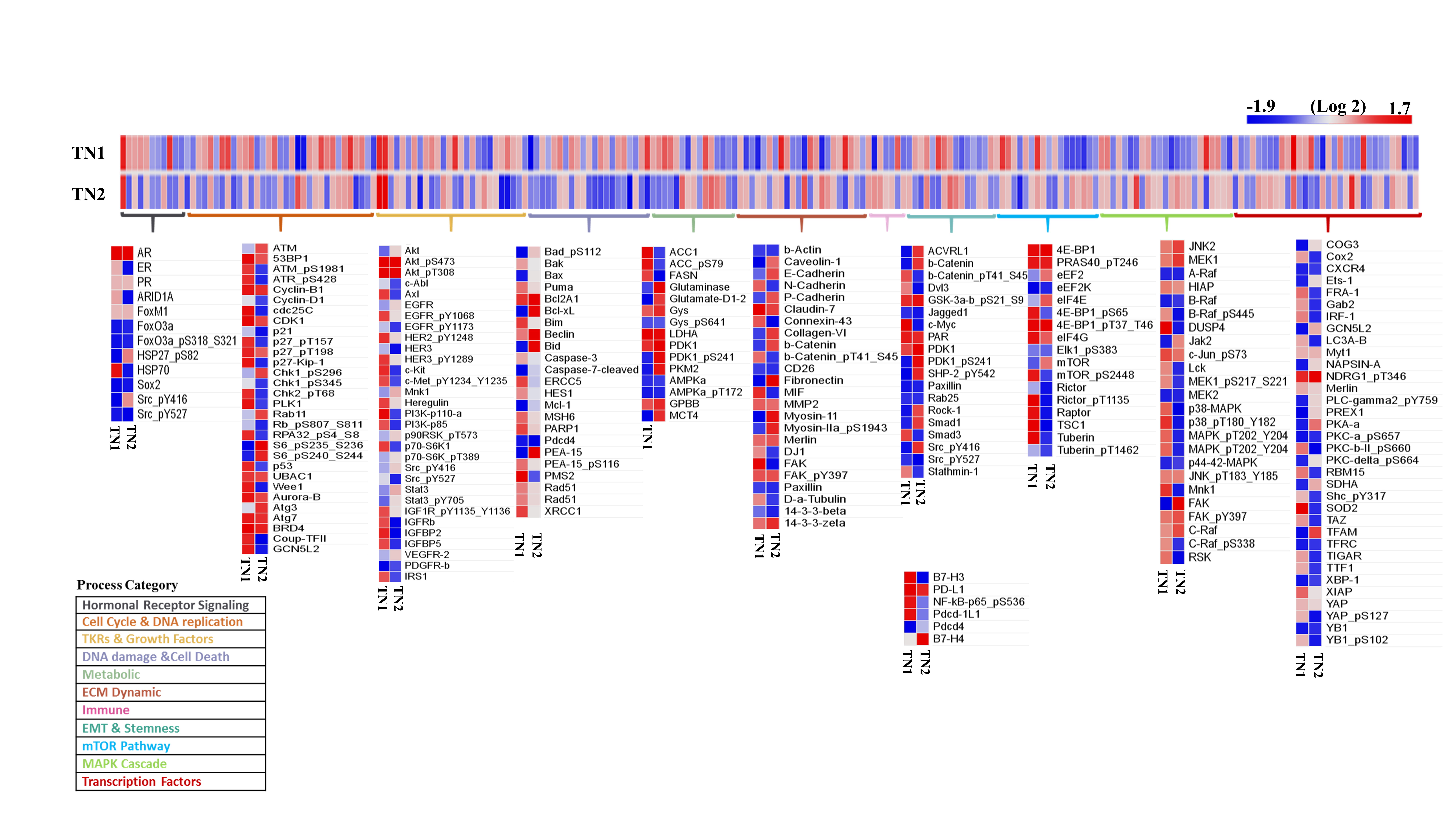
Current practices to characterize AR in breast cancer mainly employ IHC. However, guidelines for these assessments have not been established to help standardize the methodology. For example, when AR-positive TNBC is defined by IHC, the rate of positivity depends on the cut off score, which may either be 1% or 10% of cell nuclear positivity. In a clinical study by Astvatsaturyan et al., 1% of cell nuclei positivity by IHC was determined to be the appropriate threshold for patient selection and therapeutic prediction [30]. Here, we used a combination of western blots and IHC to detect AR positivity in our PDX models. Two new models of AR-positive TNBC, TN1 and TN2, were identified and established. AR expression was not indicated in pathological reports of the original tumors. Thus, we conducted western blots to detect AR positivity in the patients’ tumors. According to the western analysis, AR was detectable in tumors of COH_98 and COH_53 (Figure 1B). We then used IHC to confirm that both specimens displayed low expression of AR protein (1~5% positivity for COH_98 versus 5~8% positivity for COH_53) (Supplementary Figure 1). Such expression pattern was also confirmed by RPPA analysis: both TN1 and TN2 harbor AR protein (Supplementary Figure 4). The whole exome sequencing (WESeq) found that the PDXs of TN1 and TN2 harbor wild-type AR (Supplementary Figure 5).
We used TN1 and TN2 to demonstrate how treatment with an AR agonist (DHT) affected AR activity, tumor growth, and transcription profiles. When either TN1 or TN2 was treated with DHT, western blot and IHC detected significant activation of AR. Compared to the placebo, DHT dramatically increased the total amount of AR protein (Figure 1E) and nuclear AR positivity (Figure 1F/G). Both effects are typical hallmarks of AR transactivation [31, 32]. Our biochemical characterization suggests that treatment of TN1 or TN2 with DHT functionally activates AR. Although AR was functionally activated by DHT in both PDXs, TN1 and TN2 unexpectedly displayed a dissimilar pattern of tumor growth. Treatment of TN2 with DHT increased tumor growth (Figure 1D-TN2) and expression of Ki67 (Figure 1F/G-TN2). In stark contrast, treatment of TN1 with DHT appeared to abrogate growth (Figure 1D-TN1) and reduce expression of Ki67 (Figure 1F/G-TN1). We further assessed the AR antagonist’s (enzalutamide) effect on TN2, a PDX in which DHT induces tumor growth. As shown in Supplementary Figure 3, enzalutamide failed to restrain DHT-induced tumor growth, even though IHC of AR suggested that enzalutamide was able to suppress AR activation in those tumors. To date, only a handful of AR-positive TNBC PDX models have been reported, but not fully characterized. As reported in a conference abstract, the tumor volume of PDX HCI-009 significantly increased under DHT treatment and such induction was effectively suppressed when exposed to Seviteronel, a CYP17 lyase inhibitor and AR antagonist [17]. A short-term (48 h) treatment of ex vivo HCI-009 cultures with enzalutamide also blocked activation of AR [33]. In addition to HCI-009, two other models of AR-positive TNBCs (3561 and PK49) have been described in a review. These PDXs exhibit low AR expression (<1% nuclear positive), but functionally respond to DHT by increased nuclear AR. However, their growth pattern upon treatment, with DHT or enzalutamide, has not been presented [3].
We then performed TNBC molecular subtyping using gene expression metadata with Lehmann’s classification methods [6, 34]. Tumor of TN1 correlated with a basal-like 1 (BL1) subtype of TNBC (Figure 2C). This subtype enriches cell cycle and cell division pathways such as DNA replication, DNA damage response, and RNA polymerase activity. In contrast, tumor of TN2 correlated to a basal-like 2 (BL2) subtype of TNBC. This subtype displays unique gene ontologies involved with growth factor signaling, such as the EGF and MET pathways [6]. Interestingly, neither TN1 nor TN2 were characterized as a subtype of luminal androgen receptor (LAR). It is not known whether a PDX model of LAR subtype is available for comparison. The LAR subtype is characterized by heavily enriched hormonal regulated pathways, which include steroid synthesis and androgen metabolism. The LAR subtype of TNBC generally exhibits high AR expression (9~10 folds higher than other subtypes), while the BL subtypes express AR mRNA and protein at a relatively low level [6, 34]. Regarding to the ongoing controversies as to whether androgen signaling should be blocked or stimulated as a therapy for TNBC patients, to our best knowledge, the molecular mechanisms of AR in TNBC should be taken into consideration along with the TNBC sub-classification, considering both the heterogeneous expression of AR in TNBC and the general complexities of AR signaling [3, 8, 13].
IPA and GSEA were conducted on TN1 and TN2 to functionally characterize the impact of DHT on their transcriptomes. When we compared TN1 to TN2, DHT induced very distinct transcriptional profiles and canonical pathways (Table 2 and 3). In TN1, DHT was predicted to suppress the transcriptions associated with TGFB1, CTNNB1, and BMP2. In TN2, DHT was predicted to activate transcriptional profiles related toTGFB1 and HIF1A (Table 3). Additionally, SMARCA4 regulation was predicted to be down-regulated in TN1 versus up-regulated in TN2. SMARCA4 is a member of the SWItch/Sucrose Non-Fermentable (SWI/SNF) complex, which is needed for AR functionality and serves as a transcriptional coactivator of AR [35]. GSEA analysis also suggested that treatment of TN1 with DHT down-regulated genes involved in apical junction, myogenesis, angiogenesis, Wnt-beta-catenin, and etc. Conversely, treatment of TN2 with DHT up-regulated genes involved in myogenesis, hypoxia, Wnt-beta-catenin, and etc. (Figure 3C). As a nuclear receptor, activated AR can function as a transcription factor and control specific genes that are involved in different, and sometimes opposite, biological processes. In both cell- and tissue-specific manners, AR can either stimulate or suppress cell proliferation and apoptosis, metastasis, EMT/stemness, etc [36]. This type of bidirectional regulation of a specific biological process depends on concurrent signaling pathways being either activated or repressed via androgen-bound AR [37]. Many studies have revealed how AR mediates gene activation, however, more information is needed on AR-mediated gene repression [38]. Altogether, our results suggest that activities of AR in TN1 and TN2 are different and appear to depend on the signaling pathways that are activated or repressed.
In conclusion, we have identified two TNBC PDX models expressing functionally active AR. When AR is activated by DHT, these models exhibit different molecular and phenotypical features. The establishment and characterization of these two PDX models expand the current repertoire of AR-positive TNBC research tools. Our detailed molecular characterization of these PDXs does not support AR as an independent marker for TNBC. It has been recently reported by a multi-institutional study that AR, on its own, is not a prognostic marker for TNBC [39]. Our preclinical studies using biologically relevant models (TN1 and TN2) demonstrate that it is an essential step to produce critical mechanistic information and that they are needed to evaluate a new therapeutic approach before practice in patients. A detection of AR by IHC, without functional assessment, is not sufficient to predict the effectiveness of AR-targeting therapy in AR-expressing TNBC.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Highlights.
Two AR-positive TNBC PDX models were derived and molecularly characterized;
Two PDXs harbored low levels of functional AR, which was transcriptionally activated by AR agonist (DHT);
Two PDXs exhibited highly different patterns of tumor growth and distinct transcriptional profiles upon DHT treatment;
Two PDXs represented different molecular subtypes of TNBC;
Our results do not support that AR is a suitable therapeutic target in TNBC.
7. Acknowledgements
We are grateful to the individuals who donated tissue towards this research. The authors would like to thank Ian Talisman, PhD, for assistance with writing and editing the manuscript. We acknowledged Daniel Schmolze, MD, for assistance with assessment of tumor-stroma ratio in patients’ specimens. This work was supported by Panda Charitable Foundation and Lester M. and Irene C. Finkelstein endowment. This work was also sponsored by the National Cancer Institute (P30CA033572) which supported the service by the Animal Resource Center, the Integrative Genomics Core, and Pathology Research Services Core.
Author Statement of Conflict of Interest
This work was supported by Panda Charitable Foundation and Lester M. and Irene C. Finkelstein endowment. This work was also sponsored by the National Cancer Institute (P30CA033572) which supported the service by the Animal Resource Center, the Integrative Genomics Core, and Pathology Research Services Core at City of Hope.
Abbreviations
- AR
androgen receptor
- ASCO/CAP
American Society of Clinical Oncology / College of American Pathologists
- BL1
basal-like 1
- BL2
basal-like 2
- DHT
dihydrotestosterone
- ER
estrogen receptor
- FDR
false discovery rate
- FISH
fluorescence in situ hybridization
- GSEA
Gene Set Enrichment Analysis
- HER2
human epidermal growth factor
- H&E
hematoxylin and eosin
- IHC
immunohistochemistry
- IPA
Ingenuity Pathway Analysis
- LAR
luminal androgen receptor
- PCA
principal component analysis
- PDXs
patient-derived xenografts
- PR
progesterone receptor
- RNA-Seq
RNA sequencing
- RPPA
reverse phase protein array
- SWI/SNF
SWItch/Sucrose Non-Fermentable
- TCGA
the Cancer Genome Atlas
- TNBC
triple negative breast cancer
- TSR
tumor-stroma ratio
- WESeq
whole exome sequencing
Footnotes
6. Conflict of interest
The authors declare that they have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Kalimutho M, Parsons K, Mittal D, López JA, Srihari S, Khanna KK. Targeted Therapies for Triple-Negative Breast Cancer: Combating a Stubborn Disease. Trends Pharmacol Sci. 2015. 36(12):822–846. [DOI] [PubMed] [Google Scholar]
- [2].Barton VN, D’Amato NC, Gordon MA, Christenson JL, Elias A, Richer JK. Androgen Receptor Biology in Triple Negative Breast Cancer: a Case for Classification as AR+ or Quadruple Negative Disease. Horm Cancer. 2015. 6(5–6):206–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Christenson JL, Trepel JB, Ali HY, Lee S, Eisner JR, Baskin-Bey ES, Elias AD, Richer JK. Harnessing a Different Dependency: How to Identify and Target Androgen Receptor-Positive versus Quadruple-Negative Breast Cancer. Horm Cancer. 2018. 9(2):82–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Proverbs-Singh T, Feldman JL, Morris MJ, Autio KA, Traina TA. Targeting the androgen receptor in prostate and breast cancer: several new agents in development. Endocr Relat Cancer. 2015. 22(3):R87–R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chavez KJ, Garimella SV, Lipkowitz S. Triple negative breast cancer cell lines: one tool in the search for better treatment of triple negative breast cancer. Breast Dis. 2010. 32(1–2):35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011.121(7):2750–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].McNamara KM, Yoda T, Takagi K, Miki Y, Suzuki T, Sasano H. Androgen receptor in triple negative breast cancer. J Steroid Biochem Mol Biol. 2013.133:66–76. [DOI] [PubMed] [Google Scholar]
- [8].McNamara KM, Moore NL, Hickey TE, Sasano H, Tilley WD. Complexities of androgen receptor signaling in breast cancer. Endocr Relat Cancer. 2014.21(4):T161–81. [DOI] [PubMed] [Google Scholar]
- [9].Bonnefoi H, Grellety T, Tredan O, Saghatchian M, Dalenc F, Mailliez A, L’Haridon T, Cottu P, Abadie-Lacourtoisie S, You B, Mousseau M, Dauba J, Del Piano F, Desmoulins I, Coussy F, Madranges N, Grenier J, Bidard FC, Proudhon C, MacGrogan G, Orsini C, Pulido M, Gonçalves A. A phase II trial of abiraterone acetate plus prednisone in patients with triple-negative androgen receptor positive locally advanced or metastatic breast cancer (UCBG 12–1). Ann Oncol. 2016. 27(5):812–8. [DOI] [PubMed] [Google Scholar]
- [10].Gucalp A, Tolaney S, Isakoff SJ, Ingle JN, Liu MC, Carey LA, Blackwell K, Rugo H, Nabell L, Forero A, Stearns V, Doane AS, Danso M, Moynahan ME, Momen LF, Gonzalez JM, Akhtar A, Giri DD, Patil S, Feigin KN, Hudis CA, Traina TA; Translational Breast Cancer Research Consortium. Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic Breast Cancer. Clin Cancer Res. 2013. 19(19):5505–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Traina TA, Miller K, Yardley DA, Eakle J, Schwartzberg LS, O’Shaughnessy J, Gradishar W, Schmid P, Winer E, Kelly C, Nanda R, Gucalp A, Awada A, Garcia-Estevez L, Trudeau ME, Steinberg J, Uppal H, Tudor IC, Peterson A, Cortes J. Enzalutamide for the Treatment of Androgen Receptor-Expressing Triple-Negative Breast Cancer. J Clin Oncol. 2018. 36(9):884–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Barton VN, D’Amato NC, Gordon MA, Lind HT, Spoelstra NS, Babbs BL, Heinz RE, Elias A, Jedlicka P, Jacobsen BM, Richer JK. Multiple molecular subtypes of triple-negative breast cancer critically rely on androgen receptor and respond to enzalutamide in vivo. Mol Cancer Ther. 2015. 14(3):769–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gerratana L, Basile D, Buono G, De Placido S, Giuliano M, Minichillo S, Coinu A, Martorana F, De Santo I, Del Mastro L, De Laurentiis M, Puglisi F, Arpino G. Androgen receptor in triple negative breast cancer: A potential target for the targetless subtype. Cancer Treat Rev. 2018. 68:102–110. [DOI] [PubMed] [Google Scholar]
- [14].Whittle JR, Lewis MT, Lindeman GJ, Visvader JE. Patient-derived xenograft models of breast cancer and their predictive power. Breast Cancer Res. 2015.10;17(1):17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sulaiman A, Wang L. Bridging the divide: preclinical research discrepancies between triple-negative breast cancer cell lines and patient tumors. Oncotarget. 2017.4;8(68):113269–113281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].DeRose YS, Wang G, Lin YC, Bernard PS, Buys SS, Ebbert MT, Factor R, Matsen C, Milash BA, Nelson E, Neumayer L, Randall RL, Stijleman IJ, Welm BE, Welm AL. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat Med. 2011. 17(11):1514–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Reese JM, Babbs BL, Christenson JL, Spoelstra NS, Elias A, Eisner JR, Baskin-Bey ES, Gertz J, Richer JK.. Targeting the androgen receptor with seviteronel, a CYP17 lyase and AR inhibitor, in triple negative breast cancer [abstract]. In: Proceedings of the 2018 San Antonio Breast Cancer Symposium; (2018) Dec 4–8; San Antonio, TX. Philadelphia (PA): AACR. Cancer Res 79(4 Suppl): 2019. Abstract nr P5–05–05. [Google Scholar]
- [18].Mesker WE, Junggeburt JM, Szuhai K, de Heer P, Morreau H, Tanke HJ, Tollenaar RA. The carcinoma-stromal ratio of colon carcinoma is an independent factor for survival compared to lymph node status and tumor stage. Cell Oncol. 2007;29(5):387–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kanaya N, Somlo G, Wu J, Frankel P, Kai M, Liu X, Wu SV, Nguyen D, Chan N, Hsieh MY, Kirschenbaum M, Kruper L, Vito C, Badie B, Yim JH, Yuan Y, Hurria A, Peiguo C, Mortimer J, Chen S. Characterization of patient-derived tumor xenografts (PDXs) as models for estrogen receptor positive (ER+HER2− and ER+HER2+) breast cancers. J Steroid Biochem Mol Biol. 2017. 170:65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Creighton CJ, Huang S. Reverse phase protein arrays in signaling pathways: a data integration perspective. Drug Des Devel Ther. 2015. 9: 3519–3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ni M, Chen Y, Lim E, Wimberly H, Bailey ST, Imai Y, Rimm DL, Liu XS, Brown M. Targeting androgen receptor in estrogen receptor-negative breast cancer. Cancer Cell. 2011. 20(1):119–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Itoh T, Karlsberg K, Kijima I, Yuan YC, Smith D, Ye J, Chen S. Letrozole-, anastrozole-, and tamoxifen-responsive genes in MCF-7aro cells: a microarray approach. Mol Cancer Res. 2005. 3(4):203–18. [DOI] [PubMed] [Google Scholar]
- [23].Mei S, Meyer CA, Zheng R, Qin Q, Wu Q, Jiang P, Li B, Shi X, Wang B, Fan J, Shih C, Brown M, Zang C, Liu XS. Cistrome Cancer: A Web Resource for Integrative Gene Regulation Modeling in Cancer. Cancer Res. 2017. 77(21):e19–e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chen X, Li J, Gray WH, Lehmann BD, Bauer JA, Shyr Y, Pietenpol JA. TNBCtype: A Subtyping Tool for Triple-Negative Breast Cancer. Cancer Inform. 2012. 11:147–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Meehan TF, Conte N, Goldstein T, Inghirami G, Murakami MA, Brabetz S, Gu Z, Wiser JA, Dunn P, Begley DA, Krupke DM, Bertotti A, Bruna A, Brush MH, Byrne AT, Caldas C, Christie AL, Clark DA, Dowst H, Dry JR, Doroshow JH, Duchamp O, Evrard YA, Ferretti S, Frese KK, Goodwin NC, Greenawalt D, Haendel MA, Hermans E, Houghton PJ, Jonkers J, Kemper K, Khor TO, Lewis MT, Lloyd KCK, Mason J, Medico E, Neuhauser SB, Olson JM, Peeper DS, Rueda OM, Seong JK, Trusolino L, Vinolo E, Wechsler-Reya RJ, Weinstock DM, Welm A, Weroha SJ, Amant F, Pfister SM, Kool M, Parkinson H, Butte AJ, Bult CJ. PDX-MI: Minimal Information for Patient-Derived Tumor Xenograft Models. Cancer Res. 2017. 1;77(21):e62–e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Caiazza F, Murray A, Madden SF, Synnott NC, Ryan EJ, O’Donovan N, Crown J, Duffy MJ. Preclinical evaluation of the AR inhibitor enzalutamide in triple-negative breast cancer cells. Endocr Relat Cancer. 2016. 23(4):323–34. [DOI] [PubMed] [Google Scholar]
- [27].Wang Y, Romigh T, He X, Tan MH, Orloff MS, Silverman RH, Heston WD, Eng C. Differential regulation of PTEN expression by androgen receptor in prostate and breast cancers. Oncogene. 2011. 30(42):4327–38. [DOI] [PubMed] [Google Scholar]
- [28].Wang Y, He X, Yu Q, Eng C. Androgen receptor-induced tumor suppressor, KLLN, inhibits breast cancer growth and transcriptionally activates p53/p73-mediated apoptosis in breast carcinomas. Hum Mol Genet. 2013. 22(11):2263–72. [DOI] [PubMed] [Google Scholar]
- [29].Garay JP, Karakas B, Abukhdeir AM, Cosgrove DP, Gustin JP, Higgins MJ, Konishi H, Konishi Y, Lauring J, Mohseni M, Wang GM, Jelovac D, Weeraratna A, Sherman Baust CA, Morin PJ, Toubaji A, Meeker A, De Marzo AM, Lewis G, Subhawong A, Argani P, Park BH. The growth response to androgen receptor signaling in ERα-negative human breast cells is dependent on p21 and mediated by MAPK activation. Breast Cancer Res. 2012. 14(1):R27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Astvatsaturyan K, Yue Y, Walts AE, Bose S. Androgen receptor positive triple negative breast cancer: Clinicopathologic, prognostic, and predictive features. PLoS One. 2018. 13(6):e0197827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lee DK, Chang C. Endocrine mechanisms of disease: Expression and degradation of androgen receptor: mechanism and clinical implication. J Clin Endocrinol Metab. 2003. 88(9):4043–54. [DOI] [PubMed] [Google Scholar]
- [32].Yeap BB, Krueger RG, Leedman PJ. Differential posttranscriptional regulation of androgen receptor gene expression by androgen in prostate and breast cancer cells. Endocrinology. 1999. 140(7):3282–91. [DOI] [PubMed] [Google Scholar]
- [33].Gordon MA, D’Amato NC, Gu H, Babbs B, Wulfkuhle J, Petricoin EF, Gallagher I, Dong T, Torkko K, Liu B, Elias A, Richer JK. Synergy between Androgen Receptor Antagonism and Inhibition of mTOR and HER2 in Breast Cancer. Mol Cancer Ther. 2017. 16(7):1389–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lehmann BD, Jovanović B, Chen X, Estrada MV, Johnson KN, Shyr Y, Moses HL, Sanders ME, Pietenpol JA. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS One. 2016. 11(6):e0157368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Marshall TW, Link KA, Petre-Draviam CE, Knudsen KE. Differential requirement of SWI/SNF for androgen receptor activity. J Biol Chem. 2003. 278(33):30605–13. [DOI] [PubMed] [Google Scholar]
- [36].De Gendt K, Verhoeven G. Tissue- and cell-specific functions of the androgen receptor revealed through conditional knockout models in mice. Mol Cell Endocrinol. 2012. 352(1–2):13–25. [DOI] [PubMed] [Google Scholar]
- [37].Pihlajamaa P, Sahu B, Jänne OA. Determinants of Receptor- and Tissue-Specific Actions in Androgen Signaling. Endocr Rev. 2015. 36(4):357–84. [DOI] [PubMed] [Google Scholar]
- [38].Grosse A, Bartsch S, Baniahmad A. Androgen receptor-mediated gene repression. Mol Cell Endocrinol. 2012. 352(1–2):46–56. [DOI] [PubMed] [Google Scholar]
- [39].Bhattarai S, Klimov S, Mittal K, Krishnamurti U, Li XB, Oprea-Ilies G, Wetherilt CS, Riaz A, Aleskandarany MA, Green AR, Ellis IO, Cantuaria G, Gupta M, Manne U, Agboola J, Baskovich B, Janssen EAM, Callagy G, Walsh EM, Mehta A, Dogra A, Shet T, Gajaria P, Traina T, Nggada HA, Omonisi A, Ahmed SA, Rakha EA, Rida P, Aneja R. Prognostic Role of Androgen Receptor in Triple Negative Breast Cancer: A Multi-Institutional Study. Cancers (Basel). 2019, 11(7). pii: E995. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.