

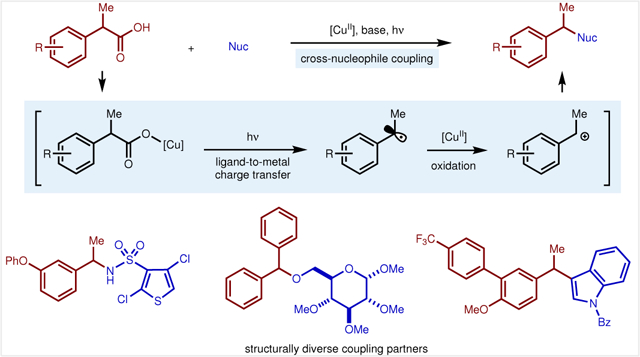
Abstract
Reactions that enable carbon–nitrogen, carbon–oxygen, and carbon–carbon bond formation lie at the heart of synthetic chemistry. However, substrate prefunctionalization is often needed to effect such transformations without forcing reaction conditions. Development of direct coupling methods of abundant feedstock chemicals is therefore highly desirable for the rapid construction of complex molecular scaffolds. We report herein a copper-mediated, net-oxidative decarboxylative coupling of carboxylic acids with diverse nucleophiles under visible light irradiation. Preliminary mechanistic studies suggest that the relevant chromophore in this reaction is a Cu(II) carboxylate species assembled in situ. We propose that visible light excitation to a ligand-to-metal charge transfer (LMCT) state results in a radical decarboxylation process that initiates the oxidative cross-coupling. The reaction is applicable to a wide variety of coupling partners, including complex drug molecules, suggesting that this strategy for cross-nucleophile coupling would facilitate rapid compound library synthesis for the discovery of new pharmaceutical agents.
Graphical Abstract
Decarboxylative coupling reactions are increasingly recognized as powerful tools for complex molecule synthesis1–4. These reactions are attractive because carboxylic acids are feedstock chemicals with significant structural diversity and generally high chemical stability5,6. The most common current strategies for decarboxylative couplings are redox-neutral in nature, pairing a carboxylate pronucleophile with an electrophilic reaction partner (Fig. 1a). This approach is particularly powerful in C–C bond-forming reactions with organohalides, which are widely available electrophiles whose reactivity in cross-coupling reactions is well understood2,7–11. The construction of polar C–N and C–O bonds, in contrast, has generally required prefunctionalization of the acid with an internal oxidant such as a redox-active hydroxyphthalimide (PINO) or iodinane ester (Fig. 1b)3,12,13. These approaches increase step count, complicate purification of the products, and hinder the prosecution of large libraries.
Fig. 1 |. Common strategies for decarboxylative coupling usually involve prefunctionalizations of either or both reacting partners while the current strategy enables direct decarboxylative coupling.
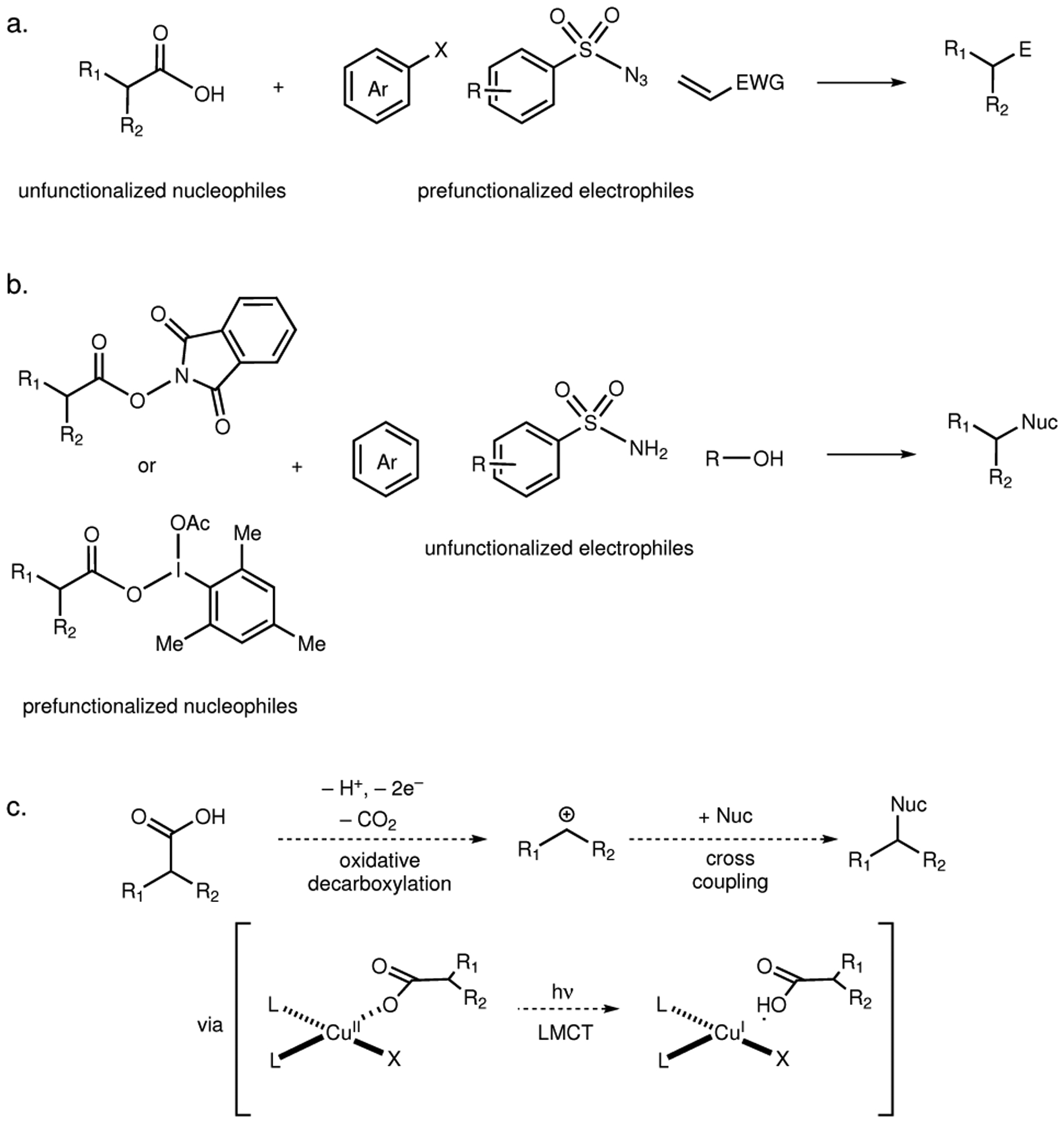
a, Redox-neutral cross-couplings of carboxylic acid feedstocks. b, Use of redox-active PINO and iodinane esters for formal decarboxylative coupling reactions. c, Proposed design plan for oxidative decarboxylative cross-couplings employing visible-light photoactive Cu(II) carboxylates.
Net-oxidative decarboxylative cross-couplings avoid the need for prefunctionalization and offer a direct strategy for installation of simple nitrogen and oxygen nucleophiles. The classical approach to this transformation is decarboxylative Kolbe electrolysis; these reactions, however, require solvent quantities of the heteronucleophile to serve as a sacrificial oxidant and to outcompete Kolbe dimerization14 , 15. Baran recently reported an improved electrochemical decarboxylative ether synthesis that utilized stoichiometric Ag(I) terminal oxidants to prevent nucleophile decomposition16. Electrochemical decarboxylative Ritter reactions have also been reported; however, they are relatively underdeveloped and are consequently limited in scope17. Transition metal mediated oxidative decarboxylative couplings under thermal conditions have also been reported, but typically require high temperatures that lead to poor chemoselectivity and functional group tolerance18 – 22. Lundgren recently reported Cu-catalyzed oxidative decarboxylative C–N bond formation under mild conditions; this method, however, is limited to electron-deficient substrates that support anionic decarboxylation23.
We imagined that a photochemical strategy could provide a general alternative for decarboxylative cross-nucleophile coupling reactions. Although many examples of redox-neutral photoredox couplings of carboxylates1–3, 7–11 or prefunctionalized PINO12,13 and hypervalent iodine esters24 , 25 have been reported, net-oxidative decarboxylative couplings between unfunctionalized acids and heteronucleophiles are underdeveloped23, 26. Recently, Larionov disclosed a remarkable metallophotoredox system for the decarboxylative coupling of diverse carboxylic acids with arylamines;27 this system, however, proceeds via an inner-sphere C–N bond-forming mechanism that was not shown to be applicable to nucleophiles beyond arylamines. We imagined that a different mechanistic concept might be required to enable oxidative photochemical decarboxylative couplings with a broader nucleophile scope.
We have previously argued that Cu(II) salts are ideal terminal oxidants for a variety of net-oxidative photoredox reactions initiated by organic or Ir photoredox catalysts28–32. They are relatively inexpensive, terrestrially abundant, and offer relatively low toxicity concerns for applications in pharmaceutical chemistry33. Because Cu(II) carboxylate complexes are often colored, we wondered whether their intrinsic photochemistry could be harnessed to enable decarboxylative couplings in the absence of an exogenous photosensitizer via a photoinduced ligand-to-metal charge transfer (LMCT) excitation (Fig. 1c). Oxidative decarboxylative substitutions initiated by UV irradiation of the charge-transfer bands of Cu(II) carboxylates were initially studied by DeGraff34 and Faust35,36, but applications of this phenomenon in synthesis are quite limited37. If this reactivity could be accessed with visible light irradiation, the resulting method would enable a versatile and operationally simple oxidative coupling reaction.
Results and Discussion:
As an initial model system, we elected to study the oxidative decarboxylative coupling of 1-phenylpropanoic acid (1a) with 4-methoxybenzenesulfonamide (1b), which was motivated by the demonstrated importance of sulfonamides as important pharmacophores in medicinal chemistry38. We speculated this coupling could be promoted by irradiation with a 427 nm LED lamp in the presence of Cu(OTf)2 and a base to promote in situ formation of the Cu(II) carboxylate chromophore (Table 1). However, only trace reaction was observed in a variety of common organic solvents (Entry 1). We hypothesized that this could be the result of speciation to higher-order Cu(II) carboxylate aggregates that might be either insoluble or photoinactive at these wavelengths39. We thus examined the effect of several Lewis basic ligands that we hypothesized might modulate the aggregation state of Cu(II). Although a range of mono- and bidentate ligands common in Cu-mediated transformations resulted in poor reactivity (Entries 2–3), addition of 2.0 equivalents of MeCN improved the yield of coupling product 2 (Entry 4). The reaction proved sensitive to the concentration of the nitrile; higher loading of MeCN resulted in increased incorporation of the nitrile to afford Ritter adduct 3 (Entry 6), and optimal chemoselectivity was achieved using 5.0 equivalents (Entry 5). The structure of the nitrile ligand also influences reaction performance, and i-PrCN was found to be most effective at 5.5 equivalents (Entries 7–11). Under these conditions (Entry 11), 2 could be isolated in 73% yield. Control experiments indicated that no reaction occurs when the reaction is performed in a flask wrapped with aluminum foil, confirming that this reaction is indeed promoted by light and not by heating from the thermal output of the LED (Entry 12). Finally, conducting the reaction under ambient atmosphere with unpurified solvent afforded a lower but still serviceable yield of 2, highlighting the operational simplicity of this coupling reaction (Entry 13).
Table 1 |.
Optimization studies of decarboxylative sulfonamidation.

| ||||
|---|---|---|---|---|
| entry | solvent | ligand | 2 (%) | 3 (%) |
| 1 | THF, Et2O, EtOAc, toluene, CH2Cl2, DMF | - | < 2 | |
| 2 | CH2Cl2 | PPh3, DABCO, pyridine dtbbpy (0.20 equiv) | < 5 | < 5 |
| 3 | CH2Cl2 | dtbbpy (2.0 equiv.) | 6 | 4 |
| 4 | CH2Cl2 | MeCN (2.0 equiv.) | 18 | 5 |
| 5 | CH2Cl2 | MeCN (5.0 equiv.) | 25 | 43 |
| 6 | CH2Cl2 | EtCN (5.0 equiv.) | 40 | 0 |
| 7 | CH2Cl2 | PhCN (5.0 equiv.) | 26 | 0 |
| 8 | CH2Cl2 | CyCN (5.0 equiv.) | 34 | 0 |
| 9 | CH2Cl2 | i-PrCN (5.0 equiv.) | 58 | 0 |
| 10 | CH2Cl2 | i-PrCN (5.5 equiv.) | 59 | 2 |
| 11 | CH2Cl2 | i-PrCN (5.5 equiv.) | 66(73a) | 0 |
| 12b | CH2Cl2 | i-PrCN (5.5 equiv.) | 0 | 0 |
| 13c | CH2Cl2 | i-PrCN (5.5 equiv.) | 58 | 6 |
0.10 mmol screening conditions: Cu(OTf)2 (2.5 equiv.), Na3PO4 (3.0 equiv.), 1a (1.0 equiv.), 1b (3.0 equiv.), ligand, and solvent (0.10 M) are added to a 1-dram reaction vial equipped with a stir bar in a glovebox. The vial is stirred at rt with irradiation by a 34 W blue LED. In situ yield determined by GC or 1H NMR with 1-methylnaphthalene as an internal standard.
Isolated yield on 0.20 mmol scale.
Reaction conducted without light.
Reaction set up under air using unpurified solvent.
Substrate Scope:
Studies of the scope of this decarboxylative sulfonamidation are summarized in Table 2. A survey of arylacetic acids revealed that yields are highest with electron-donating aromatic substituents (2, 5–8), consistent with the greater facility with which they are oxidized by Cu(II). Steric hindrance is readily tolerated by the Cu(II) oxidant, including o-tolyl substitution and α-branching (14, 18–20). Highly hindered C–N bonds can be forged from tertiary acids (21), including those containing substructures of contemporary pharmaceutical interest (22). A primary arylacetic acid undergoes smooth sulfonamidation (17), as does an α-amino acid (24). Unstabilized aliphatic carboxylic acids preferentially undergo oxidative elimination rather than nucleophilic substitution and represent a current limitation of the method (28)19,40. A geometrically constrained adamantane carboxylic acid that cannot undergo oxidative elimination, however, affords 25 in good yield. The scope of this reaction with respect to the sulfonamide coupling partner is also broad. The electronic properties of the sulfonamide exert a modest influence, with electron-deficient sulfonamides affording slightly lower yields (9–13). Primary sulfonamides substituted with aryl, heteroaryl (15, 34), and alkyl groups (19, 30, 31) react smoothly. Hindered (17, 30) and secondary sulfonamides (23) also react in good yields. A variety of functional groups are easily tolerated on either reaction partner under these conditions, including aryl halides (7, 11–13, 34), esters (16), carbamates (24), sulfamates (26), acetals (27), and polycyclic arenes (26, 27). As a demonstration of the potential utility of this method in the discovery of novel pharmaceutical candidates, we examined decarboxylative functionalizations involving a set of common anti-inflammatory drugs (29–34). The functional groups embedded in these bioactive compounds are well-tolerated, including aliphatic and aryl ketones (30, 33), diaryl ethers (34), aryl fluorides (31), and Lewis basic heterocycles (29).
Table 2 |.
Scope of decarboxylative cross-coupling with sulfonamide nucleophiles.
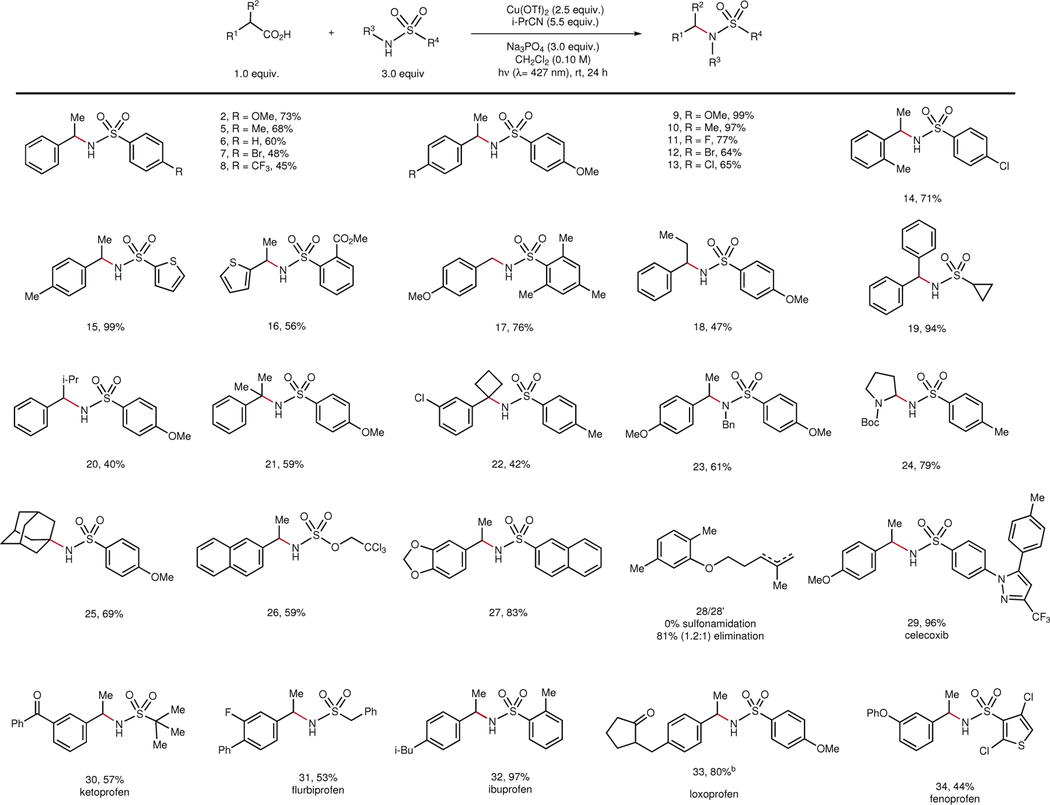
|
0.20 mmol scale isolation conditions: Cu(OTf)2 (2.5 equiv.), Na3PO4 (3.0 equiv.), sulfonamide (3.0 equiv.), carboxylic acid (1.0 equiv.), CH2Cl2 (0.10 M), and i-PrCN (5.5 equiv.) are added to a 1.5-dram reaction vial equipped with a stir bar in a glovebox. The vial is irradiated by two 34 W blue LEDs at a distance of 10 cm.
The 1.2:1 ratio refers to the trisubstited:1,1-disubstituted olefin product distribution as determined by 1H NMR of the crude reaction mixture.
Diastereomers were not detected in NMR analysis.
We propose that this oxidative decarboxylation strategy could provide a general platform for oxidative cross-coupling of carboxylic acid building blocks with diverse nucleophiles. Preliminary investigations of generality across different nucleophile classes are summarized in Table 3. Decreasing the nucleophile loading to 1.5 equivalents and conducting the reaction in acetonitrile enabled the smooth coupling of a range of carbamate (35–38) and amide (39) nucleophiles. High-yielding Ritter amidation can also be accomplished by performing the photodecarboxylation in a nitrile solvent without an added nucleophile (40–46). This provides an attractive alternative to the Curtius rearrangement for arylacetic acids that might usually require azides and problematic high-energy intermediates. Alcohol nucleophiles, however, coupled less readily. Upon optimization, the reaction performance could be significantly improved by using toluene as solvent, MeCN as ligand, and pyridine as a basic additive. These conditions constitute a method for decarboxylative etherification with similarly broad scope as the sulfonamidation reaction (47–71). Functional groups including terminal alkenes (64, 66), heterocycles (63, 69, 70), protected sugars (70), sulfonamides (69), alkyl halides (67), and alkynes (68) were readily tolerated. Finally, this strategy is not limited to carbon–heteroatom bond-forming reactions. The decarboxylative Friedel–Crafts alkylation of heteroarenes (72–74) and electron-rich arenes (75) can be conducted in good yields, indicating that this oxidative coupling strategy is also applicable to the formation of carbon–carbon bonds.
Table 3 |.
Scope of decarboxylative cross-coupling with different classes of nucleophiles.
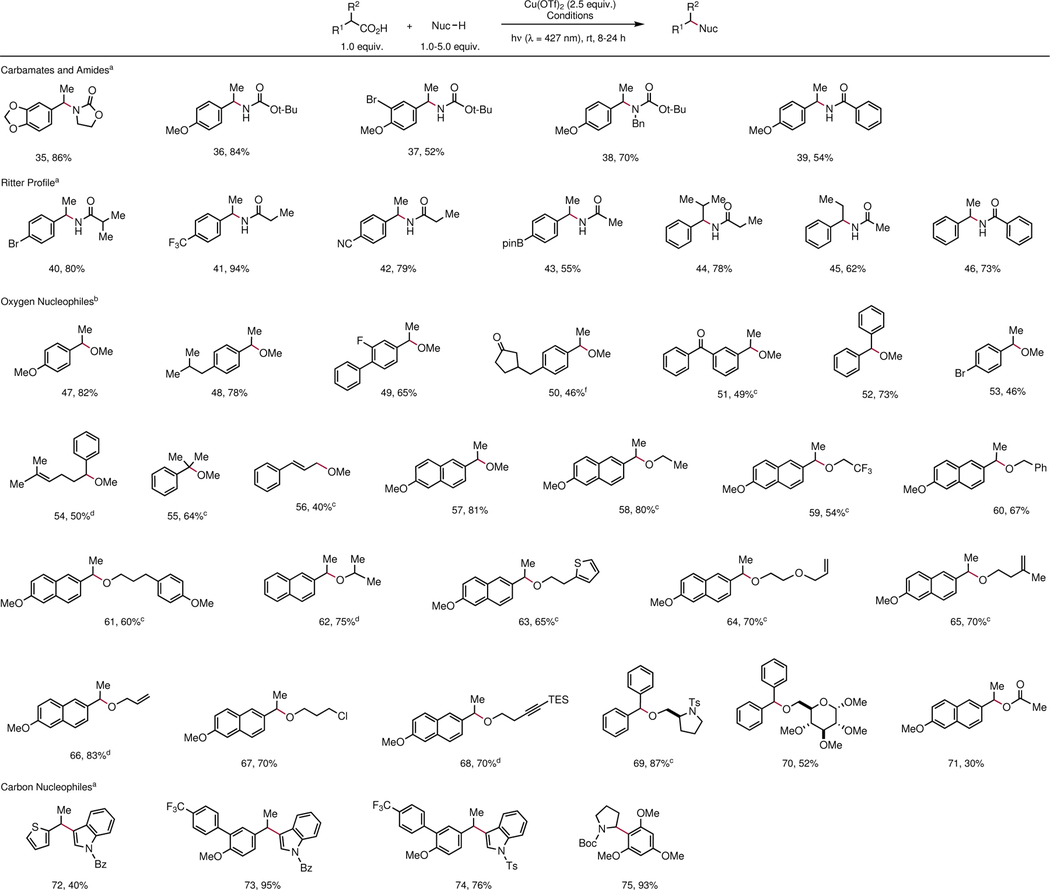
|
Conditions: Cu(OTf)2 (2.5 equiv.), Na3PO4 (3.0 equiv.), nucleophile (1.5 equiv.), carboxylic acid (1.0 equiv.), and MeCN (0.20 M), and are added to a 1.5-dram reaction vial equipped with a stir bar in a glovebox. The vial is irradiated by two 34 W blue LEDs at a distance of 10 cm.
Conditions: Cu(OTf)2 (2.5 equiv.), Na2CO3 (1.0 equiv.), carboxylic acid (1.0 equiv.), and MeCN (0.10 M), and are added to a 1.5-dram reaction vial equipped with a stir bar in a glovebox. The vial is irradiated by two 34 W blue LEDs at a distance of 10 cm.
Diastereomers were not detected in NMR analysis.
Conditions: acid (1.0 equiv.), alcohol (1.0 equiv.), Cu(OTf)2 (2.5 equiv.), and pyridine (3.0 equiv.) are added to Schlenk tubes of 15 cm diameter along with toluene (2.85 mL) and MeCN (150 μL), and the reaction mixture is degassed by freeze-pump-thaw (four 4 min cycles) before irradiation by 34 W blue LED for 8–24 h.
Same conditions in (d), but with 3.0 equiv. alcohol.
Same conditions in (c), but with 5.0 equiv. alcohol. Bpin = pinacolatoboryl.
Mechanistic Studies:
The mechanistic hypothesis guiding our investigations of these reactions is summarized in Fig. 2a. We propose that a photoactive Cu(II) carboxylate chromophore is assembled in situ through the base-mediated reaction of the carboxylic acid and Cu(OTf)2. Photoexcitation to an LMCT state would result in the dissociative formation of a carboxyl radical that can rapidly undergo decarboxylation. The ability of Cu(II) salts to oxidize the resulting carbon-centered radical is well-established, and subsequent nucleophilic substitution would afford the observed products40.
Fig. 2 |. Mechanistic studies.
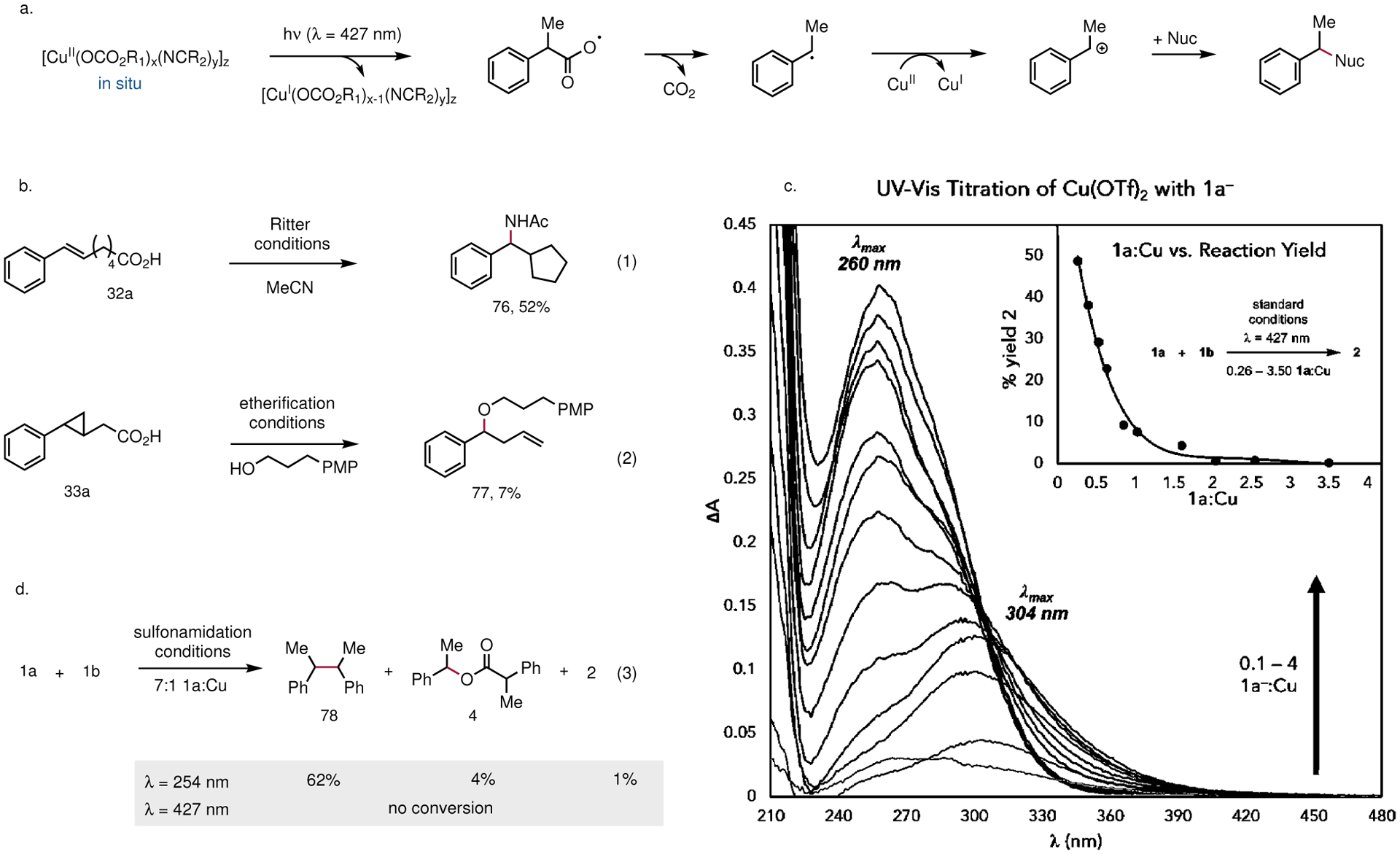
a, The guiding mechanistic hypothesis for this reaction involves MLCT photoexcitation of a preassembled Cu(II) carboxylate complex, spontaneous decarboxylation of the resulting carboxyl radical, and Cu(II)-mediated oxidative coupling of the corresponding radical. b, Decarboxylative cyclization under Ritter amidation conditions is consistent with a putative radical intermediate. c, Decarboxylative ring-opening of a radical clock substrate. d, UV-vis titration study to interrogate the interaction between the carboxylate derived from 1a and Cu(II). An initial species is formed with λmax = 304 nm at low carboxylate loadings, which transitions to a blue-shifted species (λmax = 260 nm) at high carboxylate loadings. The species formed at high carboxylate loadings is photochemically inactive at 427 nm, in line with the empirically optimized conditions. e, UV irradiation at high acid equivalents still leads to radical generation, but chemoselective formation of dimer 78 is observed in lieu of the expected product 2.
To obtain experimental support for the radical nature of the decarboxylative coupling reactions, we investigated the reaction of primary carboxylic acid 2a under conditions for Ritter amidation (Fig. 2b). In this experiment, we observed exclusively cyclopentane product 76 that presumably arises from 5-exo-trig cyclization of the decarboxylated radical intermediate. We also examined the reaction of cyclopropyl-substituted acetic acid 3a under the conditions of oxidative etherification. While the yield of this reaction was low, ring-opened benzylic ether 77 was the exclusive identifiable product (Fig. 2c). Together, these results provide evidence for the involvement of carbon-centered organoradical intermediates.
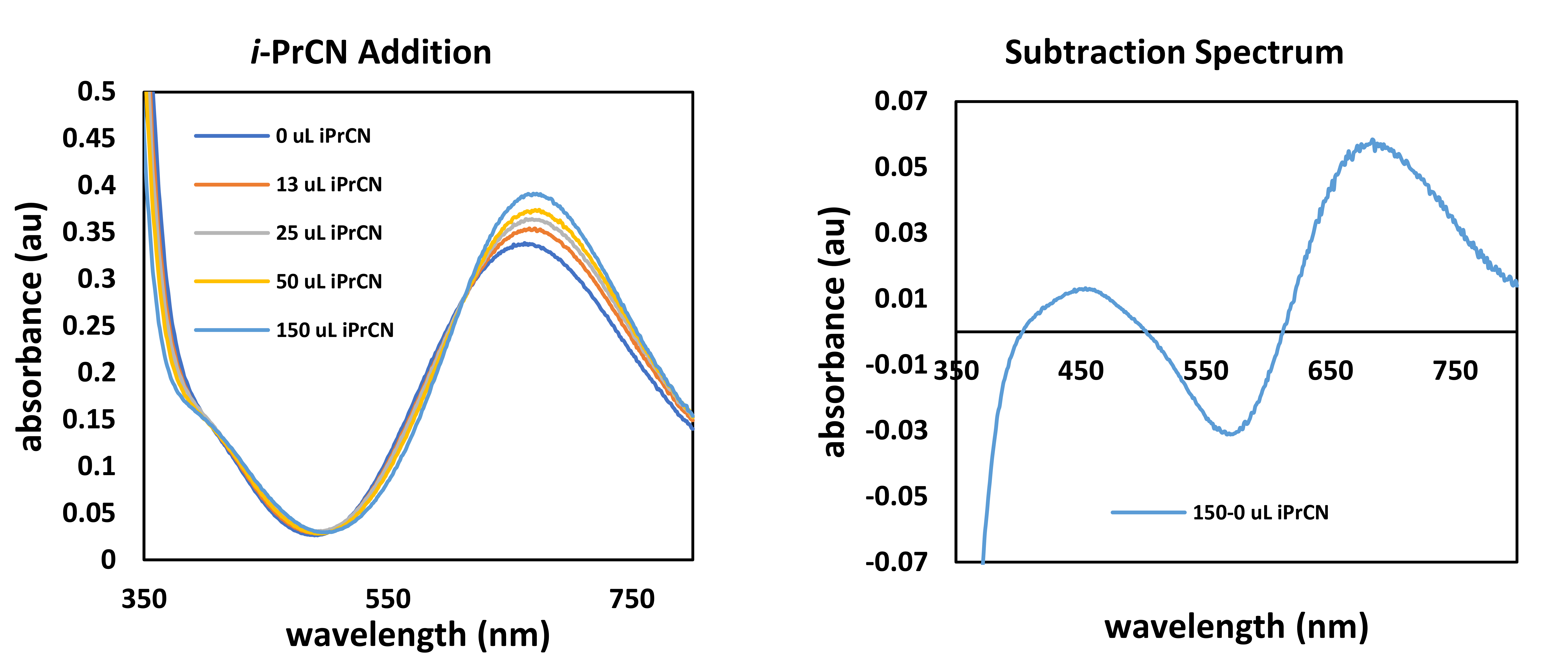
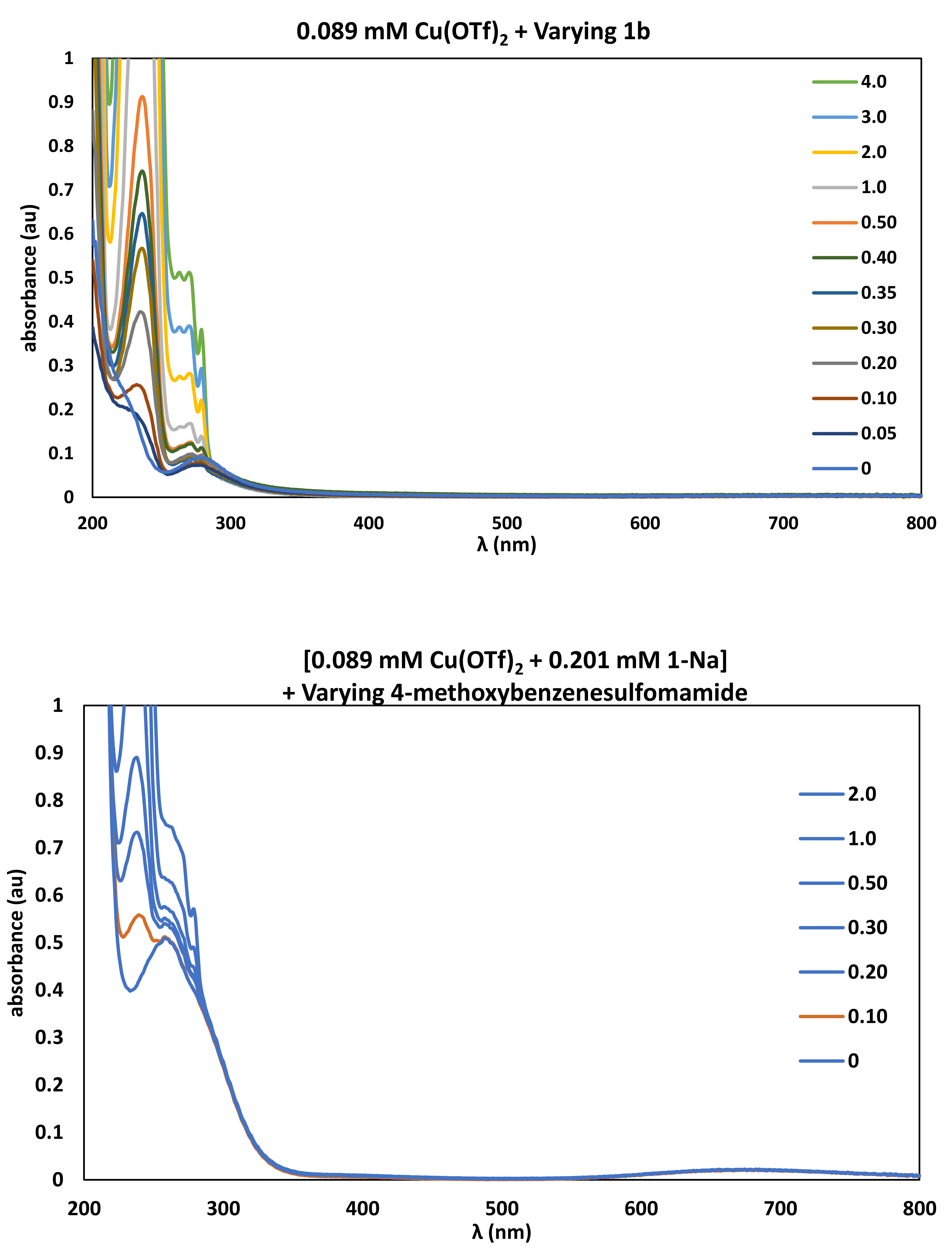
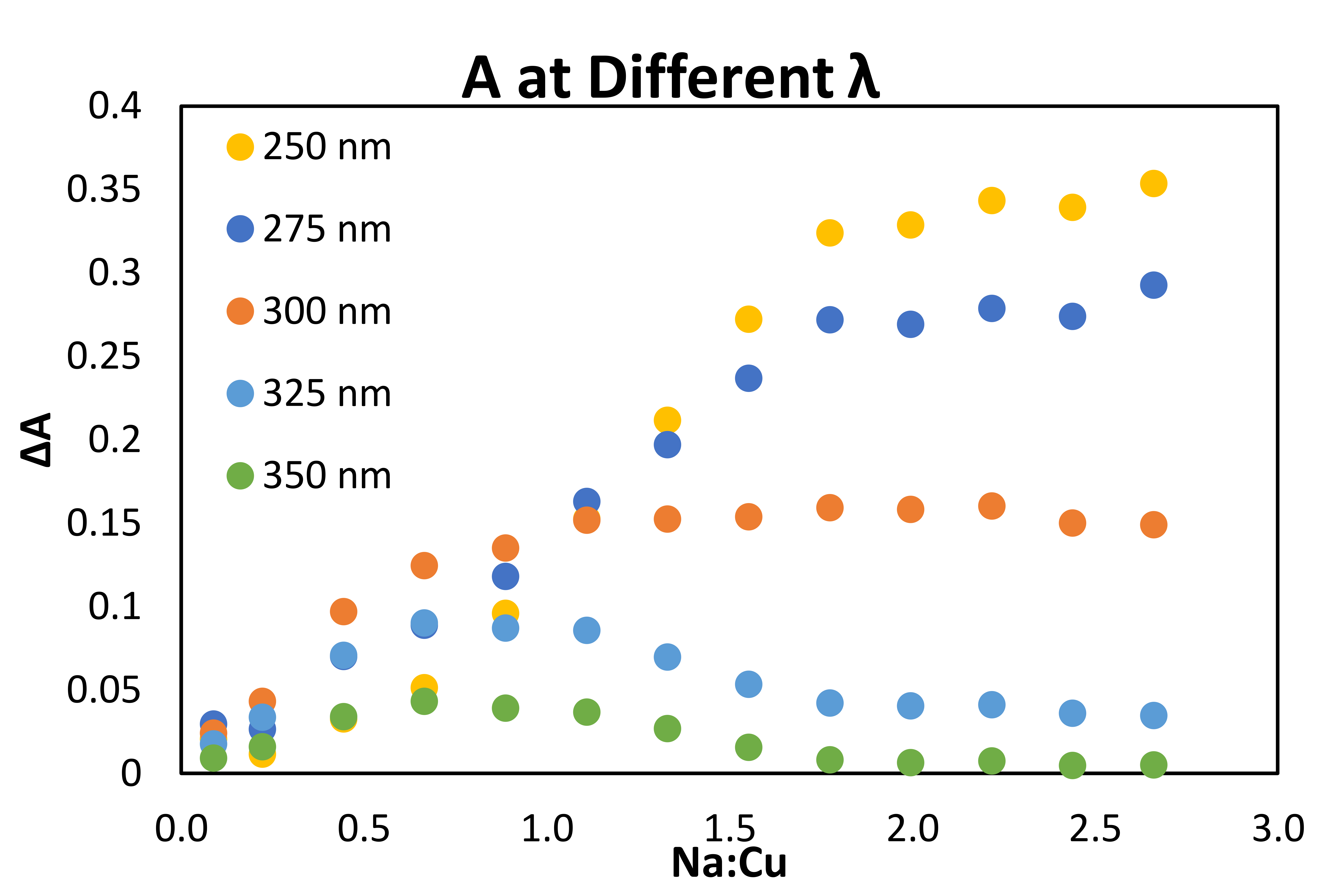
A series of UV-vis titration studies were conducted to gain insight into the nature of the photoactive complex responsible for the decarboxylative coupling (Fig. 2d, Supplementary Figs. 1–4). Addition of the sodium carboxylate derived from 1a (1a–) to a solution of Cu(OTf)2 in MeCN results in substantial changes to the absorbance spectrum. At low concentrations of added 1a–, we observe the growth of an absorption band with λmax = 304 nm. The position of this feature is comparable to the LMCT band of other monomeric Cu(II) carboxylate complexes reported in the literature41. Notably, this feature tails beyond 400 nm and overlaps with the emission spectrum of the 427 nm LED source utilized in the preparative reactions above. Concentrations of 1a– greater than 1 equivalent with respect to Cu(II) result in a depletion of this feature and concomitant growth of a higher energy band with λmax = 260 nm that overlaps less effectively with the emission spectrum of the blue LED. Absorption features with similar energy have been assigned as the LMCT bands of paddlewheel Cu(II) carboxylate dimers41–43. Of note, no meaningful interaction was observed between Cu(OTf)2 and 1b (Supplementary Figs. 5–7).
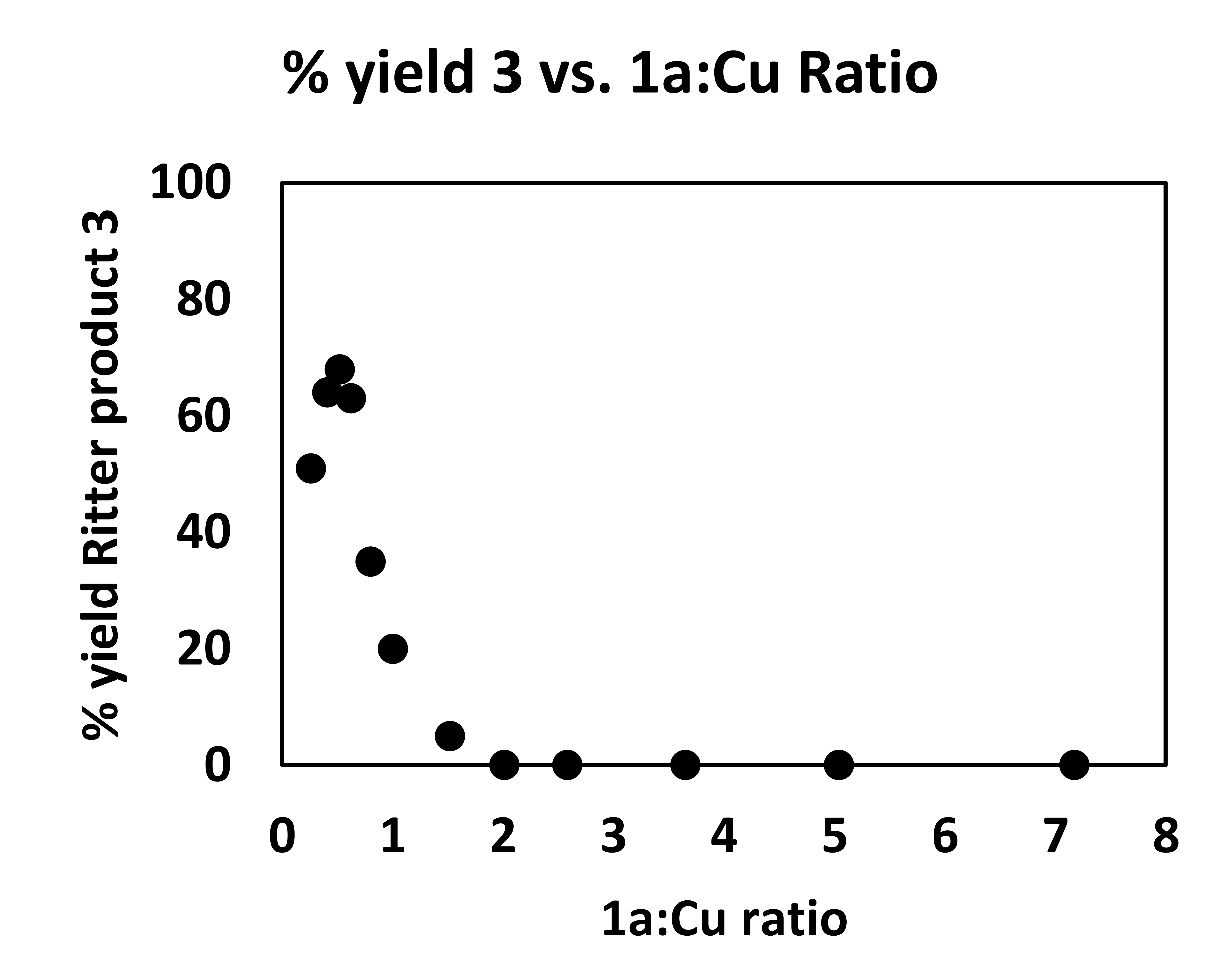
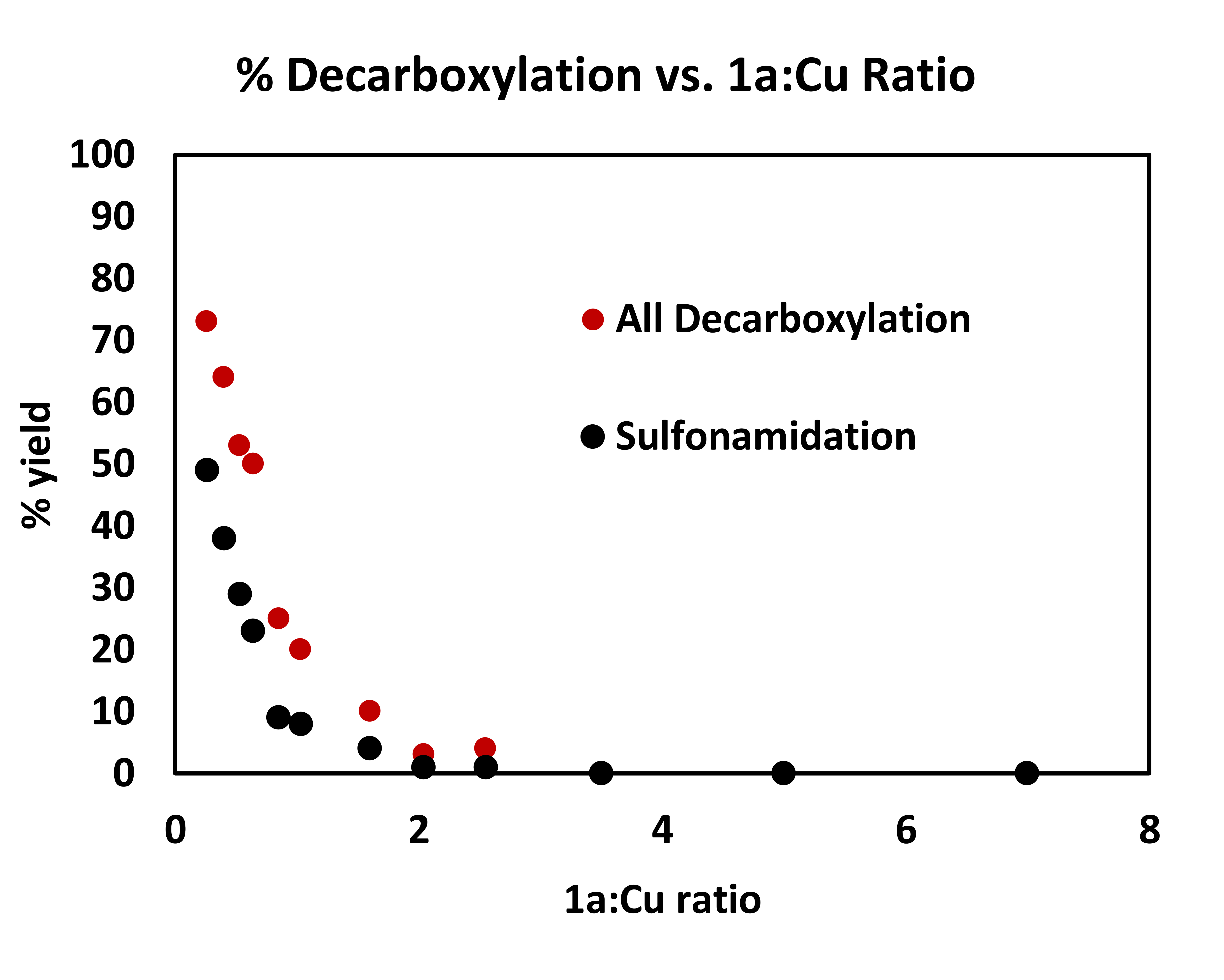
We propose, therefore, that a monomeric species corresponding to the lower-energy absorption feature is responsible for the decarboxylative coupling reaction. This proposal aligns well with the empirically optimized reaction conditions, in which 2.5 equivalents of Cu(OTf)2 relative to 1a provides the highest yield of the coupling product. It is also supported by studies examining the yield of decarboxylative sulfonamidation as a function of the ratio of Cu(II) to 1a (Fig. 2d, inset plot, Supplementary Figs. 8–9). As the concentration of 1a increases, the yield of the reaction drops rapidly, with unreacted 1a accounting for the majority of the mass balance. This result is consistent with the hypothesis that high carboxylate loadings lead to the formation of a blue-shifted dimer that is not significantly photoexcited using the 427 nm LED source. Interestingly, irradiation with a 254 nm light source, closer to λmax of the higher-energy feature, results in only trace formation of 2. Instead, compound 78, consistent with homodimerization of the benzylic radical, is the major product formed (Fig. 2e). Together, these results suggest that while radical generation can occur from multiple Cu(II) carboxylate species, the oxidation and subsequent nucleophilic coupling occurs only from the lower-energy visible light activated complex. Studies to further interrogate the mechanism of this process, including the role of the nitrile ligand (Supplementary Fig. 10 for preliminary insight) are a topic of current research in our laboratory.
Conclusion:
We have developed a strategy for the oxidative cross-nucleophile coupling of carboxylic acids with diverse nitrogen, oxygen, and carbon nucleophiles. This photoreaction exploits the intrinsic photochemical reactivity of a first-row transition metal coordination complex formed in situ and does not require sensitization by an exogenous precious metal photoredox catalyst. Importantly, an investigation of the scope of this reaction indicates that this concept is applicable to diverse carboxylate feedstocks and nucleophilic coupling partners. This process could therefore be a powerful addition to the toolbox of coupling methods for the synthesis and late-stage functionalization of complex pharmaceutical candidates.
Methods:
General procedure for sulfonamidation:
An oven-dried 6-mL vial equipped with a stir bar is brought into a nitrogen-filled glovebox and charged with Cu(OTf)2 (180.8 mg, 2.5 equiv., 0.50 mmol), Na3PO4 (98.2 mg, 3.0 equiv., 0.60 mmol), the sulfonamide nucleophile (1.5–3.0 equiv.), the carboxylic acid (1.0 equiv., 0.20 mmol), methylene chloride (2.0 mL, 0.10 M), and isobutyronitrile (100 μL, 5.5 equiv., 1.1 mmol). The vial is sealed with a screwcap bearing a teflon septum, removed from the glovebox, and placed on a stir plate. The vial is irradiated at 427 nm with two 40 W Kessil Lamp PR160 lamps at a distance of 10 cm with stirring at 800 rpm. A fan is used to maintain the vial at room temperature. After 24 h, the crude reaction mixture is diluted with 1.5 mL EtOAc and adsorbed directly on diatomaceous earth (Celite®). The product is purified by flash chromatography on silica gel, eluting with mixtures of ethyl acetate and hexanes.
General procedure for etherification:
To an oven-dried Schlenk tube of 15 cm diameter, carboxylic acid (0.3 mmol) and alcohol (0.3 mmol – 1.5 mmol, 1 – 5 equiv.) and Cu(OTf)2 (0.75 mmol, 271.25 mg) were added, followed by freshly distilled pyridine (0.9 mmol, 73 μL) and acetonitrile (0.15 mL). The reaction mixture was degassed by freeze-pump-thaw for four 4-min cycles and refilled with nitrogen. It was irradiated for variable periods of time in front of a 40 W blue LED lamp. The reaction mixture was diluted with ethyl acetate or diethyl ether (2 mL) and then washed with deionized water (2 × 5 mL). The aqueous layer was extracted with ethyl acetate or diethyl ether (5 mL). The combined organic layers were dried over Na2SO4 and concentrated and purified by flash column chromatography.
General procedure for other nitrogen nucleophiles and carbon nucleophiles:
An oven-dried 6-mL vial equipped with a stir bar is brought into a nitrogen-filled glovebox and charged with Cu(OTf)2 (180.8 mg, 2.5 equiv., 0.50 mmol), Na3PO4 (98.2 mg, 3.0 equiv., 0.60 mmol) or K3PO4 (127.4 mg, 3.0 equiv., 0.60 mmol), the nucleophile (1.5–3.0 equiv.), the carboxylic acid (1.0 equiv., 0.20 mmol), and acetonitrile (2.0 mL, 0.10 M). The vial is sealed with a screwcap bearing a teflon septum, removed from the glovebox, and placed on a stir plate. The vial is irradiated at 427 nm with two 40 W Kessil Lamp PR160 lamps at a distance of 10 cm with stirring at 800 rpm. A fan is used to maintain the vial at room temperature. After 24 h, the crude reaction mixture is diluted with 1.5 mL EtOAc and adsorbed directly on diatomaceous earth (Celite®). The product is purified by flash chromatography on silica gel, eluting with mixtures of ethyl acetate and hexanes.
General procedure for Ritter amidation:
An oven-dried 6-mL vial equipped with a stir bar is brought into a nitrogen-filled glovebox and charged with Cu(OTf)2 (180.8 mg, 2.5 equiv., 0.50 mmol), Na2CO3 (21.2 mg, 1.0 equiv., 0.20 mmol), the carboxylic acid (1.0 equiv., 0.20 mmol), and the nitrile (2.0 mL, 0.10 M). The vial is sealed with a screwcap bearing a teflon septum, removed from the glovebox, and placed on a stir plate. The vial is irradiated at 427 nm with two 40 W Kessil Lamp PR160 lamps at a distance of 10 cm with stirring at 800 rpm. A fan is used to maintain the vial at room temperature. After 24 h, the crude reaction mixture is diluted with 1.5 mL EtOAc and adsorbed directly on diatomaceous earth (Celite®). The product is purified by flash chromatography on silica gel, eluting with mixtures of ethyl acetate and hexanes.
Data availability
All data supporting the findings of this study are available within the paper and its supplementary files.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank W. B. Swords for helpful discussions.
Funding:
Funding for this work was provided by the NIH (R01GM095666, TPY), an ACS GCI Pharmaceutical Roundtable Research Grant (TPY), and Pfizer (TPY). SNG thanks the NIH for a fellowship grant (F32GM139373), and GAL is the recipient of a 3M Science & Technology Fellowship. Analytical facilities at UW–Madison are funded by the NIH (S10OD012245), NSF (CHE-9304546), and a generous gift from the Paul J. and Margaret M. Bender Fund.
Footnotes
Competing interest declaration
NJB, MWB, JWT and SWB are employees and shareholders of Pfizer, Inc. The remaining authors declare no competing interests.
Supplementary information is available for this paper.
References:
- 1.Xuan J, Zhang Z-G & Xiao W-J. Visible-light-induced decarboxylative functionalization of carboxylic acids and their derivatives. Angew. Chem. Int. Ed 54, 15632–15641 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Rodríguez N & Goossen LJ Decarboxylative coupling reactions: A modern strategy for C–C-bond formation. Chem. Soc. Rev 40, 5030–5048 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Arshadi S, Ebrahimiasl S, Hosseinian A, Monfared A & Vessally E Recent developments in decarboxylative cross-coupling reactions between carboxylic acids and N–H compounds. RSC. Adv 9, 8964–8976 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zeng Z, Feceu A, Sivendran N & Gooßen LJ Decarboxylation - initiated intermolecular carbon–heteroatom bond formation. Adv. Synth. Catal 363, 2678–2722 (2021). [Google Scholar]
- 5.Scott E, Peter F & Sanders J Biomass in the manufacture of industrial products — the use of proteins and amino acids. Appl. Microbiol. Biotechnol 75, 751–762 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gallezot P Conversion of biomass to selected chemical products. Chem. Soc. Rev 41, 1538–1558 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Patra T & Maiti D Decarboxylation as the key step in C–C bond-forming reactions. Chem. Eur. J 23, 7382–7401 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Gooßen LJ, Deng G & Levy LM Synthesis of biaryls via catalytic decarboxylative coupling. Science 313, 662–664 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Moon PJ & Lundgren RJ Metal-catalyzed ionic decarboxylative cross-coupling reactions of C(sp3) acids: reaction development, mechanisms, and application. ACS. Catal 10, 1742–1753 (2010). [Google Scholar]
- 10.Zuo Z & MacMillan DWC Decarboxylative arylation of α-amino acids via photoredox catalysis: A one-step conversion of biomass to drug pharmacophore. J. Am. Chem. Soc 136, 5257–5260 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zuo Z, Cong H, Li W, Choi J, Fu GC & MacMillan DWC Enantioselective decarboxylative arylation of α-amino acids via the merger of photoredox and nickel catalysis. J. Am. Chem. Soc 138, 1832–1835 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shibutani S, Kodo T, Takeda M, Nagao K, Tokunaga N, Sasaki Y & Ohmiya H Organophotoredox-catalyzed decarboxylative C(sp3)−O bond formation. J. Am. Chem. Soc 142, 1211–1216 (2020). [DOI] [PubMed] [Google Scholar]
- 13.Mao R, Frey A, Balon J & Hu X Decarboxylative C(sp3)–N cross-coupling via synergetic photoredox and copper catalysis. Nat. Catal 1, 120–126 (2018). [Google Scholar]
- 14.Kolbe H Beobachtungen über die oxydirende Wirkung des Sauerstoffs, wenn derselbe mit Hülfe einer elektrischen Säule entwickelt wird. J. Prakt. Chem 41, 137–139 (1847). [Google Scholar]
- 15.Hofer H & Moest M Ueber die Bildung von Alkoholen bei der Elektrolyse fettsaurer Salze. Justus Liebigs Ann. Chem 323, 284–323 (1902). [Google Scholar]
- 16.Xiang J et al. Hindered dialkyl ether synthesis with electrogenerated carbocations. Nature 573, 398–402 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eberson L & Nyberg K Studies on the Kolbe electrolytic synthesis. V. An electrochemical analogue of the Ritter reaction. Acta. Chem. Scand 18, 1567–1568 (1964). [Google Scholar]
- 18.Kochi JK A new method for halodecarboxylation of acids using lead(IV) acetate. J. Am. Chem. Soc 87, 2500–2502 (1965). [Google Scholar]
- 19.Bacha J & Kochi JK Alkenes from acids by oxidative decarboxylation. Tetrahedron 24, 2215–2226 (1968). [Google Scholar]
- 20.Wang Z in Comprehensive Organic Name Reactions and Reagents (ed Wang Z) 1646–1649 (John Wiley & Sons, 2010). [Google Scholar]
- 21.Agterberg FPW, Driessen WL, Reedijk J, Oeveringb H & Buijs W Copper-catalyzed oxidative decarboxylation of aliphatic carboxylic acids. Stud. Surf. Sci. Catal 82, 639–646 (1994). [Google Scholar]
- 22.Serguchev YA & Beletskaya IP Oxidative decarboxylation of carboxylic acids. Uspekhi Khimii 49, 2257–2285 (1980) [Google Scholar]
- 23.Kong D, Moon PJ, Bsharat O& Lundgren RL Direct catalytic decarboxylative amination of aryl acetic acids. Angew. Chem. Int. Ed 59, 1313–1319 (2020). [DOI] [PubMed] [Google Scholar]
- 24.Liang Y, Zhang X & MacMillan DWC Decarboxylative sp3 C–N coupling via dual copper and photoredox catalysis. Nature 559, 83–88 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sakakibara Y, Ito E, Fukushima T, Murakami K & Itami K Late-stage functionalization of arylacetic acids by photoredox-catalyzed decarboxylative carbon–heteroatom bond formation. Chem. Eur. J 24, 9254–9258 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Drapeau MP, Bahri J, Lichte D & Gooßen LJ Decarboxylative ipso amination of activated benzoic acids. Angew. Chem. Int. Ed 58, 892–896 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Nguyen VT, Nguyen D, Haug GC, Vuong NTH, Dang HT, Arman HD & Larionov OV Visible light-enabled direct decarboxylative N-alkylation. Angew. Chem. Int. Ed 59, 7921–7927 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reed NL & Yoon TP Oxidase reactions in photoredox catalysis. Chem. Soc. Rev 50, 2954–2967 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reed NL, Herman MI, Miltchev VP & Yoon TP Photocatalytic oxyamination of alkenes: Copper(II) salts as terminal oxidants in photoredox catalysis. Org. Lett 20, 7345–7350 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reed NL, Herman MI, Miltchev VP & Yoon TP Tandem copper and photoredox catalysis in photocatalytic alkene difunctionalization reactions. Beilstein J. Org. Chem 15, 351–356 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee BJ, DeGlopper KS & Yoon TP Site-selective alkoxylation of benzylic C–H bonds via photoredox catalysis. Angew. Chem. Int. Ed 59, 197–202 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reed NL, Lutovsky GA & Yoon TP Copper-Mediated radical–polar crossover enables photocatalytic oxidative functionalization of sterically bulky alkenes. J. Am. Chem. Soc 143, 6065–6070 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guideline for Elemental Impurities Q3D, Current Step 4 version, dated Dec 16, 2014
- 34.Morimoto JY & DeGraff BA Photochemistry of copper complexes. The copper(ll) malonate system. J. Phys. Chem 79, 326–331 (1975). [Google Scholar]
- 35.Sun L, Wu C-H & Faust BC Photochemical redox reactions of inner-sphere copper(II)-dicarboxylate complexes: Effects of the dicarboxylate ligand structure on copper(I) quantum yields. J. Phys. Chem. A 102, 8664–8672 (1998). [Google Scholar]
- 36.Sun L, Wu C-H & Faust BC Photochemical formation of copper(I) from copper(II)-dicarboxylate complexes: effects of outer-sphere versus inner-sphere coordination and of quenching by malonate. J. Phys. Chem. A 104, 4989–4996 (2000). [Google Scholar]
- 37.Xu P, López–Rojas P & Ritter T Radical decarboxylation carbometalation of benzoic acids: A solution to aromatic decarboxylative fluorination. J. Am. Chem. Soc 143, 5349–5354 (2021). [DOI] [PubMed] [Google Scholar]
- 38.Zhao C, Rakesh KP, Ravidar L, Fang W-Y & Qin H-L Pharmaceutical and medicinal significance of sulfur (SVI)-containing motifs for drug discovery: A critical review. Eur. J. Med. Chem 162, 679–734 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsybizova A, Ryland BL, Tsierkezos N, Stahl SS, Roithová J & Schröder D Speciation behavior of copper(II) acetate in simple organic solvents – revealing the effect of trace water. Eur. J. Inorg. Chem 1407–1412 (2014). [Google Scholar]
- 40.Kochi JK, Bemis A & Jenkins CL Mechanism of electron transfer oxidation of alkyl radicals by copper(II) complexes. J. Am. Chem. Soc 90, 4616–4625 (1968). [Google Scholar]
- 41.Dendrinou-Samara C, Jannakoudakis PD, Kessissoglou DP, Manoussakis GE, Mentzafos D & Terzis A Copper(II) complexes with anti-inflammatory drugs as ligands. Solution behaviour and electrochemistry of mono- and bi-nuclear complexes. J. Chem. Soc., Dalton Trans 3259–3264 (1992). [Google Scholar]
- 42.Graddon DP The absorption spectra of complex salts — IV cupric alkanoates. J. Inorg. Nucl. Chem 17, 222–231 (1961). [Google Scholar]
- 43.Kyuzou M, Mori W & Tanaka J Electronic structure and spectra of cupric acetate mono-hydrate revisited. Inorganica Chimica Acta 363, 930–934 (2010). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings of this study are available within the paper and its supplementary files.