

Abstract
Numerous studies in hepatocellular carcinoma (HCC) have proposed tissue-based gene signatures for individualized prognostic assessments. Here, we develop a novel circulating tumor cell (CTC)–based transcriptomic profiling assay to translate tissue-based messenger RNA (mRNA) signatures into a liquid biopsy setting for noninvasive HCC prognostication. The HCC-CTC mRNA scoring system combines the NanoVelcro CTC Assay for enriching HCC CTCs and the NanoString nCounter platform for quantifying the HCC-CTC Risk Score (RS) panel in enriched HCC CTCs. The prognostic role of the HCC-CTC RS was assessed in The Cancer Genome Atlas (TCGA) HCC cohort (n = 362) and validated in an independent clinical CTC cohort (n = 40). The HCC-CTC RS panel was developed through our integrated data analysis framework of 8 HCC tissue-based gene signatures and identified the top 10 prognostic genes (discoidin domain receptor tyrosine kinase 1 [DDR1], enoyl-CoA hydratase and 3-hydroxyacyl CoA dehydrogenase [EHHADH], androgen receptor [AR], lumican [LUM], hydroxysteroid 17-beta dehydrogenase 6[HSD17B6], prostate transmembrane protein, androgen induced 1 [PMEPA1], tsukushi, small leucine rich proteoglycan [TSKU], N-terminal EF-hand calcium binding protein 2 [NECAB2], ladinin 1 [LAD1], solute carrier family 27 member 5 [SLC27A5]) highly expressed in HCC with low expressions in white blood cells. The panel accurately discriminated overall survival in TCGA HCC cohort (hazard ratio [HR], 2.0; 95% confidence interval [CI], 1.4-2.9). The combined use of the scoring system and HCC-CTC RS panel successfully distinguished artificial blood samples spiked with an aggressive HCC cell type, SNU-387, from those spiked with PLC/PRF/5 cells (P = 0.02). In the CTC validation cohort (n = 40), HCC-CTC RS remained an independent predictor of survival (HR, 5.7; 95% CI, 1.5-21.3; P = 0.009) after controlling for Model for End-Stage Liver Disease score, Barcelona Clinic Liver Cancer stage, and CTC enumeration count. Our study demonstrates a novel interdisciplinary approach to translate tissue-based gene signatures into a liquid biopsy setting. This noninvasive approach will allow real-time disease profiling and dynamic prognostication of HCC.
Hepatocellular carcinoma (HCC) accounts for 85% of all primary liver malignancies(1) and is the fourth leading cause of cancer-related mortality worldwide.(2) With a 5-year survival of less than 20%, HCC is among the most lethal malignancies.(3) For patients presenting with potentially curable disease, current treatment stratification algorithms such as the Barcelona Clinic Liver Cancer (BCLC) staging system(4,5) or transplant selection criteria such as the Milan criteria(6) have limited accuracy in prognosticating posttreatment recurrence and survival. Therefore, adjunct biomarkers that may further stratify prognosis to optimize treatment strategies represent an unmet need.
With the advent of new gene profiling technologies and bioinformatics pipelines during the past decades, numerous studies have elucidated the underlying genetic mutational landscape(7,8) of HCC and proposed several tissue-based prognostic signatures. Integrative bioinformatics analysis of large genomic and transcriptomic data sets(9-11) can be leveraged to provide insights into the underlying cancer biology to allow individualized patient-level prognostic assessments. Despite the robustness of these molecular classification approaches, the procurement of tumor tissues is only available in a small proportion of patients and inherently poses risk because of the requirement of invasive procedures.(12,13)
Circulating tumor cells (CTCs) are regarded as a noninvasive alternative to tissue biopsy, offering the opportunity to gain insight into the underlying cancer using a less-invasive “liquid biopsy” approach.(14,15) Our team has proposed that phenotypic subclassification of HCC CTCs can augment their utility for prognosis.(16,17) Using the NanoVelcro CTC Assay, we identified HCC CTCs with the expression of either vimentin(16) or programmed death ligand 1 (PD-L1),(17) both of which are strongly correlated with an inferior overall survival (OS) in patients across all stages of disease. Furthermore, the presence of vimentin-positive HCC CTCs independently portends a faster time to recurrence(16) in clinically indistinguishable early stage patients undergoing curative intent treatment. Nevertheless, microscopic counting of CTCs among an excess of background white blood cells (WBCs) is time-consuming and inevitably subjective, therefore limiting its application in clinical practice. To overcome these limitations, Kalinich et al. detected HCC CTCs by quantifying 10 HCC-specific genes in blood samples with droplet digital PCR(18); however, their applications focused on early detection and noninvasive monitoring of disease rather than prognostication.
Here we describe an HCC-CTC messenger RNA (mRNA) scoring system (Fig. 1) composed of an upstream NanoVelcro CTC Assay for enrichment of HCC CTCs from blood and a downstream NanoString nCounter platform(19,20) for quantification of the HCC-CTC-derived mRNA. In parallel, an integrated data analysis framework is implemented to establish a prognostic mRNA panel named the HCC-CTC Risk Score (RS) panel by selecting HCC-specific genes from publicly available HCC tissue signatures.(9-11,21-24) Leveraging this scoring system, we validate the prognostic importance of the HCC-CTC RS panel from an independent cohort of patients with HCC. Our results demonstrate the feasibility of translating tissue-based transcriptomic profiling into a liquid biopsy setting, paving the way for noninvasive HCC prognostication.
FIG. 1.

HCC-CTC mRNA scoring system is composed of the following 2 crucial steps: (1) NanoVelcro CTC Assay for enrichment of HCC CTCs in the blood samples collected from patients with HCC and (2) NanoString nCounter platform for quantification of the HCC-CTC RS panel in the enriched HCC CTCs. Following CTC enrichment, the CTCs were lysed in the devices, followed by mRNA extraction and reverse transcription to obtain HCC-CTC-derived cDNA. In parallel, an integrated data analysis framework was developed to select the HCC-CTC RS panel—a final set of 10 genes that carry significant prognostic value and were independently associated with the survival of patients with HCC.
Materials and Methods
INTEGRATED DATA ANALYSIS FRAMEWORK FOR THE DEVELOPMENT OF THE HCC-CTC RS PANEL
Development of the HCC-CTC RS Panel
To develop a panel of genes predicting poor clinical outcome, we curated 8 HCC gene signatures from 7 publicly available HCC tissue-based transcriptome studies (Supporting Table 1), including 2 signatures defined from the analysis of The Cancer Genome Atlas (TCGA) data(11) and the following 6 previously published signatures: Hoshida’s HCC subtyping study,(9) cholangiocarcinoma-like HCC,(10) Hippo pathway inactivation,(21) RS classifier based on the Cox proportional hazards model,(22) National Cancer Institute proliferation (NCIP) signature,(23) and hepatoblastoma-like tumors.(24) We defined the iCluster1 and isocitrate dehydrogenase (IDH) signatures, which are known to be associated with poor prognosis.(11) To define the iCluster1 signature, we selected the genes with significant differential regulation in iCluster1 compared with iCluster2 or iCluster3 using TCGA HCC transcriptome data(11) by an integrated statistical hypothesis testing method.(25) Briefly, T value, rank sum difference, and log2 median ratio were computed for each gene. An empirical distribution of the null hypothesis (ie, the gene is not differentially expressed) was estimated by calculating T values, rank sum differences, and log2 median ratios for the genes after performing the random permutation of the samples. For each gene, the P values for each of the observed statistics were computed using their corresponding empirical distributions by 2-tailed tests. Then, these 3 P values are combined into an adjusted P value using the Stouffer method.(26) The differentially expressed genes (DEGs) were selected with adjusted P values <0.05 and absolute log2 fold change ≥0.58 (1.5-fold). We defined the top 100 genes that demonstrated a large fold change in iCluster1 compared with iCluster2 and iCluster3. Similarly, the IDH-like signature was defined as the top 100 DEGs by the comparison of IDH1/2 mutant and wild type using TCGA HCC transcriptome data.
The genes in the HCC-CTC RS panel were selected with the following steps: (1) selecting genes identified in at least 2 independent HCC prognostic signatures, (2) identifying subsets of genes that have absolute greater than 2-fold changes and P values <0.05 in iCluster1 versus iCluster2 or iCluster3 and IDH mutant versus wild type, (3) selecting genes that are highly expressed in HCC cell lines (median log2 expression >5) from the Cancer Cell Line Encyclopedia (CCLE),(27) and (4) confirming the selected genes have low expressions in the immune cell populations from the Differentiation Map data set (DMAP)(28) to minimize background signals from nonspecifically captured WBCs. Genes with log2 expressions greater than 7 (cutoff value for the absent/present calling) in more than 20 samples (about 10% of total number of samples) were excluded as high expressed genes in immune cells.
Evaluation of the HCC-CTC RS Panel in the TCGA HCC Cohort
We used the TCGA HCC cohort, which included 362 patients with HCC, to evaluate the prognostic ability of the HCC-CTC RS panel. A total of 356 patients with OS data and 357 patients with time to recurrence (TTR) data were included for OS and TTR analysis, respectively. The gene expression and clinical information of the TCGA HCC cohort were acquired from the University of California Santa Cruz Xena Browser.(29) The multivariable Cox proportional hazards model regression analysis was performed to investigate the prognostic association between HCC-CTC RS panel genes and the OS and TTR from the TCGA HCC cohort. The HCC-CTC RS of each patient was calculated with the following equation:
where n is the number of genes in the HCC-CTC RS panel, βi is the coefficient of the ith gene that is modeled by Cox proportional hazards model regression analysis, and xi is the median centered gene expression of the ith gene. The TCGA HCC cohort was stratified into high-risk or low-risk groups at the median of the HCC-CTC RSs from the patients. The associations of the HCC-CTC RS with OS and TTR were analyzed by applying univariate Cox proportional hazards model regression in the TCGA HCC cohort.
WORKFLOW OF THE HCC-CTC mRNA SCORING SYSTEM
The workflow of the HCC-CTC mRNA scoring system in conjunction with the HCC-CTC RS panel is illustrated in Fig. 1. First, after initial blood sample processing (see blood processing details in Supporting Methods), the peripheral blood mononuclear cell (PBMC) samples were processed through the NanoVelcro CTC Assay as previously published(16,17) for enrichment of HCC CTCs. The enriched HCC CTCs were lysed in the device, followed by mRNA extraction and reverse transcription (Supporting Fig. 1; see the experimental details of the NanoVelcro CTC Assay in Supporting Methods). Second, the HCC-CTC-derived complementary DNA (cDNA) was introduced to the NanoString nCounter platform (NanoString Technologies, Inc., WA, USA)(19) to quantify the expression of the HCC-CTC RS panel genes. Finally, the HCC-CTC RS for each sample was calculated using the equation described previously and applied to classify patients into high-risk or low-risk groups.
HCC-CTC ENUMERATION
Blood samples were processed following the protocol of the NanoVelcro CTC Assay for enrichment of HCC CTCs.(16,17) Enriched HCC CTCs were imaged using multicolor immunocytochemistry following the criteria developed previously.(16,17) In brief, WBCs were defined as round/ovoid cells, 4′,6-diamidino-2-phenylindole (DAPI)+/CD45+/cytokeratin−, with sizes ≤6 μm, and HCC CTCs were defined as round/ovoid cells, DAPI+/CD45−/cytokeratin+, with sizes >6 μm on the multichannel immunocytochemistry image.
VALIDATION OF THE HCC-CTC RS PANEL USING HCC CELL LINES
The dynamic range and linearity of signals from the HCC-CTC RS panel were tested with mRNA extracted from the human HCC SNU-387 cell line over a range of cell numbers (n = 5, 10, 50, and 100 HCC cells), mimicking CTC numbers in 2-mL clinical blood samples(16,17,30) (see the experimental details in Supporting Methods).
To demonstrate the specificity of the 10 genes in the HCC-CTC RS panel to HCC, gene expression levels in SNU-387, PLC/PRF/5 HCC cell lines (n = 5, 10, 50, 100 HCC cells), and WBCs from healthy donors (n = 50, 100, 500, 1000 WBCs) were quantified and compared (see the experimental details in Supporting Methods).
VALIDATION OF THE HCC-CTC mRNA SCORING SYSTEM AND THE HCC-CTC RS PANEL USING ARTIFICIAL BLOOD SAMPLES
The feasibility of the combined use of the HCC-CTC mRNA scoring system and the HCC-CTC RS panel to discriminate high-risk from low-risk cancers was tested using triplicate artificial blood samples prepared by spiking 30 SNU-387 or PLC/PRF/5 cells into 2 × 106 PBMCs from the blood of healthy donors (see the experimental details in Supporting Methods). These artificial blood samples were introduced to the HCC-CTC mRNA scoring system (Fig. 1) for quantification of the HCC-CTC RSs.
VALIDATION OF THE HCC-CTC mRNA SCORING SYSTEM AND THE HCC-CTC RS PANEL IN AN INDEPENDENT HCC-CTC COHORT
Patient Recruitment and Blood Collection
Following written informed consent (University of California Los Angeles Institutional Review Board no. 14-001932), study participants were enrolled (February 2016-February 2019) with the collection of peripheral venous blood from healthy volunteers and patients with HCC across all stages with a confirmed pathologic or radiographic diagnosis (Liver Imaging Reporting and Data System category 5). Patients were excluded if they had concomitant neoplasms or histories of extrahepatic malignancy within the past 5 years. Demographic and clinical variables were collected and maintained in a prospective manner. At the time of blood draw, the tumor burden of patients with HCC was characterized by BCLC staging system. OS was calculated as the time from enrollment blood draw to death or last follow-up. Detailed information and patient characteristics are shown in Table 1, and continuous variables were summarized as medians and interquartile ranges (IQRs).
TABLE 1.
Clinical Characteristics of the HCC-CTC Cohort (n = 40)
| Characteristics | Median (IQR) or n (%) |
|---|---|
| Age, years | 68 (60.3-73.8) |
| Male | 28 (70.0) |
| Cirrhosis | 29 (72.5) |
| Child class (% of cirrhosis) | |
| A | 25 (86.2) |
| B | 2 (6.9) |
| C | 2 (6.9) |
| HCC etiology | |
| HBV | 9 (22.5) |
| HCV | 16 (40.0) |
| ALD | 3 (7.5) |
| NAFLD/NASH | 6 (15.0) |
| Others | 6 (15.0) |
| Number of lesions | |
| Single | 26 (65.0) |
| Multiple | 14 (35.0) |
| MELD score | 7 (6-8) |
| BCLC stage | |
| A | 24 (60.0) |
| B | 4 (10.0) |
| C | 11 (27.5) |
| D | 1 (2.5) |
| AJCC stage | |
| IA-IB | 22 (55.0) |
| II | 5 (12.5) |
| IIIA-IIIB | 5 (12.5) |
| IVA-IVB | 8 (20.0) |
| AFP at blood collection, ng/mL | 9.7 (4.5-684.0) |
| Any treatment prior to blood collection | 10 (25.0) |
| Type of first treatment after blood collection | |
| Liver resection | 22 (55.0) |
| Liver transplant | 1 (2.5) |
| Locoregional therapy | 8 (20.0) |
| Sorafenib | 2 (5.0) |
| PD-1 checkpoint inhibitor | 5 (12.5) |
| No treatment | 2 (5.0) |
| HCC CTC count per 4 mL venous blood | 6.5 (2.3-11.0) |
Gene Expression Analysis and HCC-CTC RS Calculation
NanoStringNorm R package (version 1.2.0, https://github.com/cran/NanoStringNorm) was used for expression data normalization. The “housekeeping geometric mean” option, which estimates and adjusts cell input, was used for the normalization of CTC yields. To reduce systematic variance, the quantile normalization method was applied to the log2-transformed expression data. The HCC-CTC RS of each patient was calculated using the equation described previously. Patients in the HCC-CTC cohort were classified into the high-risk or low-risk group using the cutoff value of the HCC-CTC RS derived from the TCGA HCC cohort (the median of the HCC-CTC RSs in the TCGA HCC cohort).
STATISTICAL ANALYSIS
Percentiles of the expressions of the HCC-CTC RS panel (10 genes) in primary HCC tissues (from TCGA), HCC cell lines (from the CCLE(27)), and immune cells (from DMAP(28)) were compared using the Wilcoxon rank sum test. The Kaplan-Meier curve plot was used to estimate OS and TTR in the TCGA HCC cohort, and estimates of OS and TTR were compared using Cox proportional hazards model regression.
Linear regression analysis was performed to assess the linearity of the mRNA expression readout in the HCC cell lines (SNU-387 and PLC/PRF/5) and healthy donor PBMCs. The slopes and coefficient of determination (R2) were calculated. A 2-sample t test was used to assess the difference in HCC-CTC RSs between different HCC cell line spiking samples (SNU-387 versus PLC/PRF/5).
In the HCC-CTC cohort, a penalized spline smoothing method was performed to identify an optimal cutoff value of the HCC-CTC RS to classify the high-risk and low-risk groups for prognostication. The Gaussian mixture model was applied to present the high-risk and low-risk groups in the TCGA HCC cohorts and the HCC-CTC cohort. Principal component analysis (PCA) was used to show the separation of high-risk and low-risk groups in the HCC-CTC cohort. The Kaplan-Meier curve method was used to estimate OS and TTR. Univariate and multivariable analyses were performed using Cox proportional hazards model regression to identify independent predictors of OS. Hazard ratios (HRs) and 95% confidence intervals (CIs) were reported, and statistical significance was assessed based on nonoverlapping 95% CIs. A sensitivity analysis in the subset of patients with BCLC stage A receiving liver resection was performed.
All statistical analyses were performed using R statistical software (version 3.5.3; R Foundation for Statistical Computing, Vienna, Austria) and MATLAB (MathWorks, Natick, MA) with 2-sided tests and a significance level of 0.05.
Results
INTEGRATED DATA ANALYSIS FRAMEWORK FOR THE DEVELOPMENT OF THE HCC-CTC RS PANEL
To develop the HCC-CTC RS panel, we first integrated 8 tissue-based prognostic gene signatures (Figs. 1 and 2A)(9-11,21-24) and included 1535 candidate genes (Supporting Table 2). A stepwise method was adopted to serially pare down genes highly expressed in immune cells and specifically select genes highly expressed in HCC. In this way, we could avoid the signals from background leukocytes nonspecifically captured on the NanoVelcro Chips.(30) We designed a data analysis framework through the integration of HCC signatures and transcriptome data from patients with HCC, cancer cell lines (CCLE, n = 1036), and immune cells (DMAP, n = 211) with a rigorous selection process (Fig. 2A; see details in the Materials and Methods). This bioinformatics approach yielded a final 10 genes, that is, discoidin domain receptor tyrosine kinase 1 (DDR1), enoyl-CoA hydratase and 3-hydroxyacyl CoA dehydrogenase (EHHADH), androgen receptor (AR), lumican (LUM), hydroxysteroid 17-beta dehydrogenase 6 (HSD17B6), prostate transmembrane protein, androgen induced 1 (PMEPA1), tsukushi, small leucine rich proteoglycan (TSKU), N-terminal EF-hand calcium binding protein 2 (NECAB2), ladinin 1 (LAD1), and solute carrier family 27 member 5 (SLC27A5) to be included in our HCC-CTC RS panel (Fig. 2B).
FIG. 2.

Integrated data analysis for selection of the HCC-CTC RS panel and evaluation in the TCGA HCC cohort. (A) Schematic flow of the selection of 10 genes for the HCC-CTC RS panel, that is, DDR1, PMEPA1, LAD1, LUM, NECAB2, AR, EHHADH, TSKU, SLC27A5, HSD17B6. (B) Network analysis showing the association between the HCC-CTC RS panel genes and 8 public HCC tissue-based signatures. The square nodes represent the transcriptome-based signatures, with size of the squares proportional to number of genes from that signature included in the final HCC-CTC RS panel (number of genes from each signature in parentheses). The circle nodes represent each of the 10 genes in the HCC-CTC RS panel, with straight lines demonstrating the association between the 10 genes and the signatures. The colors of the inner circle and outer circle represent the log2 fold change of the iCluster1 versus iCluster2 and iCluster3 and IDH mutation versus IDH wild type, respectively. (C) Box plot showing the percentile of expression level of the HCC-CTC RS panel genes in TCGA tissues from patients with primary HCC, HCC cell lines from the CCLE, and immune cells from DMAP. As is shown, the HCC-CTC RS panel is highly expressed in HCC tissues and cell lines but has very low expression in immune cells, making them ideal for blood-based assessment. (D) Kaplan-Meier curve plot of OS in a TCGA HCC cohort (n = 356) stratified by HCC-CTC RS (P < 0.001). (E) Kaplan-Meier curve plot of TTR in a TCGA HCC cohort (n = 357) stratified by HCC-CTC RS (P = 0.003) with a follow-up time of 2 years. (D,E) High-risk versus low-risk groups were stratified at the median of the HCC-CTC RS for both OS and TTR analyses.
When evaluating the expression levels of these final 10 genes compared with all genes characterized in primary HCC tissues (from TCGA), HCC cell lines (from the CCLE), and immune cells (from DMAP), the 10-gene panel was top ranked in primary tissues from patients with HCC, highly ranked in HCC cell lines, and lowly ranked in immune cells (P < 0.001 between primary tissues and HCC cell lines; P < 0.001 between primary tissues and immune cells; Fig. 2C).
To assign each of these genes a prognostic value, multivariable Cox proportional hazards model regression analysis was performed on the TCGA HCC cohort (n = 362), with the β coefficients from that model incorporated into the HCC-CTC RS (Supporting Table 3; see details in the Materials and Methods). The TCGA HCC cohort was stratified into 2 groups at the median of the HCC-CTC RS of the patients (HCC-CTC RS = 0.0082). The clinical characteristics of the TCGA HCC cohort are summarized in Supporting Table 4, and the HCC-CTC RS of each patient is shown in Supporting Table 5). The HCC-CTC RS accurately discriminated both OS (HR, 2.0; 95% CI, 1.4-2.9; P < 0.001; Fig. 2D) and TTR (HR, 1.7; 95% CI, 1.2-2.4; P = 0.003; Fig. 2E) between the high-risk and low-risk groups, with the discrimination of early postresection HCC recurrence critically important in supporting that the HCC-CTC RS predicts cancer-specific outcomes and not only non-HCC-related death.
VALIDATION OF THE HCC-CTC RS PANEL USING HCC CELL LINES
The dynamic range and linearity of signals from the HCC-CTC RS panel were validated using the SNU-387 HCC cell line. First, we observed excellent linear correlations between the number of SNU-387 cells and the mRNA expression levels of the housekeeping genes (R2 = 0.98; Supporting Fig. 2A) and the 10 genes in the HCC-CTC RS panel (R2 = 0.99; Supporting Fig. 2B). Second, we were able to detect the signals of the HCC-CTC RS panel from as low as 5 SNU-387 cells (Supporting Fig. 2B).
In addition, we demonstrated that the 10 genes are specific to HCC cells by showing that signals from 5 SNU-387 cells are significantly higher than those in 1000 WBCs (P < 0.001; Supporting Fig. 2C). The result indicated that our data analysis framework (Fig. 2A) successfully selects an HCC-specific gene panel and filters out unwanted signals from WBCs.
VALIDATION OF THE HCC-CTC mRNA SCORING SYSTEM AND THE HCC-CTC RS PANEL USING ARTIFICIAL BLOOD SAMPLES
To test the feasibility of the combined use of the HCC-CTC mRNA scoring system and HCC-CTC RS panel to discriminate between high-risk and low-risk HCC, we calculated the HCC-CTC RSs of 28 HCC cell lines (with different degrees of aggressive tumor biology; American Type Culture Collection, Manassas, VA) from the CCLE (Fig. 3A).(27) The resulting HCC-CTC RSs revealed that the SNU-387 HCC cell line exhibited the highest RS, whereas the PLC/PRF/5 HCC cell line had the lowest RS. These HCC-CTC RSs faithfully reflected the underlying tumor biology of individual HCC cell lines.(27,31) We selected SNU-387 and PLC/PRF/5 cell lines for further experiments to validate the HCC-CTC mRNA scoring system in conjunction with the HCC-CTC RS panel. The artificial blood samples, prepared by spiking 30 SNU-387 or PLC/PRF/5 cells into 2 × 106 PBMCs from the blood of healthy donors, were processed following the workflow in Fig. 1. The quantified expression levels of the 10 genes were normalized and the HCC-CTC RSs were calculated (Fig. 3B). Consistent with previous analyses in CCLE data, SNU-387 showed higher HCC-CTC RS compared with PLC/PRF/5 (P = 0.02; Fig. 3C), confirming that the combined use of the HCC-CTC mRNA scoring system and the HCC-CTC RS panel is capable of distinguishing high-risk samples from low-risk samples.
FIG. 3.

Evaluation of the HCC-CTC RS panel using the HCC-CTC mRNA scoring system and HCC cell lines. (A) The HCC-CTC RSs of 28 HCC cell lines using the transcriptome data from the CCLE. (B) The heatmap of the risk activity of the HCC-CTC RS panel and bar plot of the HCC-CTC RS in the artificial blood samples. A total of 3 sets of 30 SNU-387 cells were spiked in 2 × 106 healthy donor PBMCs (red) and 3 sets of PLC/PRF/5 cells were spiked in 2 × 106 healthy donor PBMCs (blue). The risk activity was calculated by multiplying the expression of the HCC-CTC RS panel genes by the coefficient values. (C) The HCC-CTC RS comparison of artificial blood samples from SNU-387 and PLC/PRF/5 (P = 0.02). Of note, assessment of the HCC-CTC RS following (C) artificial spiking into healthy donor PBMCs recapitulates the HCC-CTC RS from (A) transcriptome data from the CCLE.
VALIDATION OF THE HCC mRNA SCORING SYSTEM AND THE HCC-CTC RS PANEL IN AN INDEPENDENT HCC-CTC COHORT
Having demonstrated that our 10-gene HCC-CTC RS panel discriminated the survival of patients in the TCGA HCC cohort when characterized by their tissue-based mRNA expressions, we sought to validate the feasibility of the combined use of the HCC mRNA scoring system and the HCC-CTC RS panel from CTCs in an independent HCC-CTC patient cohort. A total of 40 patients with HCC and 6 healthy donors were enrolled in this validation cohort. At the time of blood draw, the median age of the patients with HCC was 68 years (IQR, 60.3-73.8 years), and 28 patients with HCC (70.0%) were men. The median laboratory Model for End-Stage Liver Disease (MELD) score of patients with HCC was 7 (IQR, 6-8). When categorized by BCLC stage, 24 (60.0%), 4 (10.0%), 11 (27.5%), and 1 (2.5%) patients were stages A, B, C, and D, respectively. HCC CTCs were found in 37/40 (92.5%) patients with HCC (median, 6.5 cells per 4 mL venous blood; IQR, 2.3-11.0 cells). The clinical features of the HCC-CTC cohort are summarized in Table 1.
The blood samples from 40 patients with HCC and 6 healthy donors were processed following the workflow shown in Fig. 1, and the CTC count and heatmap of the HCC-CTC RS panel is depicted in Fig. 4A. The distribution of the HCC-CTC RSs measured from the HCC-CTC cohort (Supporting Fig. 3A; the HCC-CTC RS of each patient was summarized in Supporting Table 6) showed a very similar range and modality when compared with the HCC-CTC RSs measured from the TCGA HCC cohort (Supporting Fig. 3B). Furthermore, as illustrated in Supporting Fig. 4, the cutoff value of the HCC-CTC RS derived from the TCGA HCC cohort (HCC-CTC RS = 0.0082) is similar to that identified by the penalized spline smoothing method in the HCC-CTC cohort (HCC-CTC RS = −0.0329), indicating the role of HCC CTCs as surrogates for HCC tissues.
FIG. 4.

HCC-CTC mRNA scoring system for quantification of the HCC-CTC RS panel using clinical blood samples in an independent HCC-CTC cohort. (A) The HCC-CTC count and the heatmap of the HCC-CTC RS panel expression patterns from 40 patients with HCC and 6 healthy donors. The color bar represents gradients of log2 fold change of the genes between patients with HCC and healthy donors. The patients with HCC are sorted by increasing HCC-CTC RS from left to right, with low-risk and high-risk groups defined by the exact same threshold from the TCGA HCC cohort. (B) PCA result using the risk activity of the HCC-CTC RS panel showing the distribution of 40 patients. The size of node represents the absolute HCC-CTC RS, and the red and blue nodes represent the high-risk and low-risk groups, respectively. (C) Kaplan–Meier curve analysis of OS in the HCC-CTC cohort stratified by HCC-CTC RS (P = 0.02). (D) Box plot showing the distribution of the HCC-CTC RSs in BCLC stage A high-risk group, BCLC stage A low-risk group, BCLC stages B to D high-risk group, and BCLC stages B to D low-risk group.
The PCA of the gene expression of the HCC-CTC RS panel demonstrated a clear separation of high-risk and low-risk groups (Fig. 4B), with further validation demonstrated in the Kaplan-Meier survival curve analysis revealing significantly inferior OS in the high-risk HCC-CTC group compared with the low-risk HCC-CTC group (HR, 4.2; 95% CI, 1.3-13.9; P = 0.02; Fig. 4C). In addition, there was a similar distribution of the HCC-CTC RSs when comparing patients with BCLC stage A and patients with BCLC stages B to D (Fig. 4D), solidifying the role of the HCC-CTC RS for further discrimination of prognosis beyond what is known by BCLC stratification. The CTC counts were not significantly different between the high-risk and low-risk groups (Supporting Fig. 5).
To validate the utility of the HCC-CTC RS for prognostication, Cox proportional hazards model regression analysis was performed (Fig. 5). On univariate analysis, both the HCC-CTC RS (HR, 4.2; 95% CI, 1.3-13.9; P = 0.02; reference: low-risk group) and BCLC stage (HR, 6.7; 95% CI, 1.8-24.6; P = 0.004; reference: BCLC stage A) were associated with OS (Fig. 5A), but the CTC count and MELD score were not. Both the HCC-CTC RS (HR, 5.7; 95% CI, 1.5-21.3; P = 0.009) and BCLC stage (HR, 8.7; 95% CI, 2.2-34.4; P = 0.002) remained independent predictors of OS on multivariable analysis after adjusting for MELD score and CTC count (Fig. 5B).
FIG. 5.
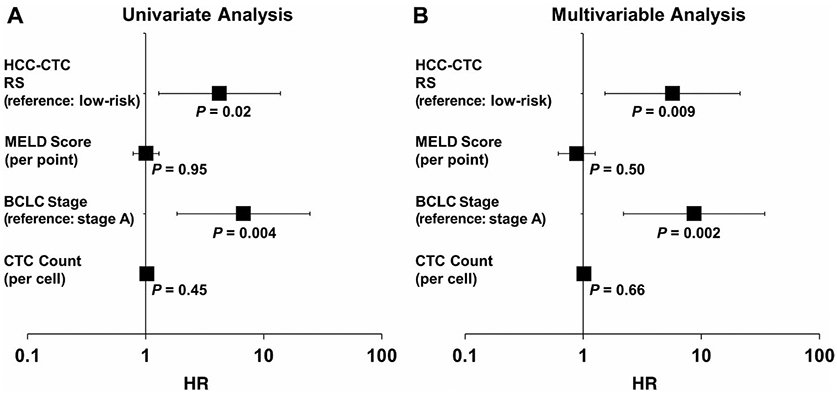
Univariate and multivariable Cox proportional hazards model regression analysis of the HCC-CTC RS in the HCC-CTC cohort. (A) Forest plot of HRs estimated by univariate analysis of the HCC-CTC RS, MELD score, BCLC stage, and CTC count for OS in the HCC-CTC cohort (HR of the HCC-CTC RS = 4.2 [reference: low-risk group], HR of MELD score = 1.0 [per point], HR of BCLC stage = 6.7 [reference: BCLC stage A], and HR of CTC count = 1.0 [per cell]). (B) Forest plot of HRs estimated by multivariable analysis of the HCC-CTC RS, MELD score, BCLC stage, and CTC count for OS in the HCC-CTC cohort (HR of the HCC-CTC RS = 5.7 [reference: low-risk group], HR of MELD score = 0.9 [per point], and HR of BCLC stage = 8.7 [reference: BCLC stage A], and HR of CTC count = 1.0 [per cell]).
Furthermore, we performed subgroup analyses among the patients with BCLC stage A HCC receiving liver resection (n = 20) to evaluate the prognostic value for recurrence beyond BCLC staging. In this subgroup, 11 patients were classified as low risk and 9 patients were classified as high risk. It is noteworthy that 3 patients in the high-risk group (33.33%) experienced HCC recurrence, whereas none of the low-risk group had recurrence during the follow-up period (HR, 12.4; 95% CI, 0.6-279.2; P = 0.11; Supporting Fig. 6).
Discussion
During the past decade, there have been numerous studies defining the underlying genetic mutational landscape in HCC and identifying prognostic transcriptomic signatures. Despite these critical advances, the practical barriers for translating these HCC tissue-based signatures into actionable, clinically relevant information have not been overcome. In this study, we demonstrate a first-in-class, noninvasive method to translate HCC tissue-based gene signatures into a liquid biopsy setting by developing the HCC-CTC mRNA scoring system and the HCC-CTC RS panel. The HCC-CTC mRNA scoring system is composed of a microfluidic NanoVelcro CTC Assay for enrichment of HCC CTCs from blood samples of patients with HCC coupled with the NanoString nCounter platform for quantification of selected mRNA transcripts (ie, the HCC-CTC RS panel) in the enriched HCC CTCs. The HCC-CTC RS panel included 10 highly selected genes derived from tissue-based transcriptomes via an integrated data analysis framework. We showed that this HCC-CTC RS is a significant prognosticator of outcomes in the TCGA HCC cohort as well as an independent validation HCC-CTC cohort through isolation and transcriptome characterization of HCC CTCs.
One of the major limitations in characterizing patients with HCC into the various prognostic molecular subclasses(9-11) is the reliance on the acquisition of tumor tissue for subsequent genomic analysis. In clinical practice, this prognostic information is rarely available before definitive surgical intervention and thus has not been able to guide therapeutic approaches. In addition, repeated molecular characterization for monitoring the disease status or evaluating treatment response is hard to achieve in clinical practice, as this relies on repeated invasive procedures. To address these unmet needs, we developed the HCC-CTC mRNA scoring system and the HCC-CTC RS panel to translate tissue-based transcriptome profiling into a liquid biopsy setting.
Although enumerating CTCs is of prognostic significance,(16,17) recent research efforts are moving toward the molecular characterization(18,32) of CTCs to provide new insights into cancer biology. To fulfill HCC prognostication by a noninvasive CTC assay, it is essential that purified CTCs subjected to molecular analysis are derived from and reflect the underlying tumor. Recently, our team showed that somatic copy number alternation (sCNA) from purified HCC CTCs immediately prior to surgical resection recapitulates with high fidelity the sCNAs observed in the resected primary tumor but not the WBCs and peritumoral normal liver.(32) In our current work, we have shown that the distribution of the HCC-CTC RSs as measured from CTCs in the HCC-CTC cohort showed a similar range and modality when compared with those measured from primary tumor tissues in the TCGA HCC cohort (Supporting Fig. 3). Collectively, these observations lend strong proof that HCC CTCs purified from peripheral blood may serve as a molecular surrogate for the primary tumor.
A critical step to translate tissue-based HCC transcriptomic signatures into a liquid biopsy setting requires a process to efficiently isolate and purify HCC CTCs for downstream mRNA isolation and characterization. Our HCC-CTC mRNA scoring system overcomes the rate-limiting hurdles associated with immunostaining and microscopic imaging and counting of CTCs compared with the conventional CTC enumeration studies,(18) mitigating the significant interobserver variability and personal bias that exist in identifying CTCs. Most important, in contrast to the current CTC mRNA assays,(18,33) which are limited in their ability to quantify small amounts of disease-specific mRNA, the HCC-CTC mRNA scoring system is capable of quantifying the HCC-CTC RS panel with high sensitivity in enriched HCC-CTC samples. By using the NanoVelcro technology and the multimarker antibody cocktail, we have shown that HCC CTCs can be reproducibly enriched.(16,17) In addition, the digital barcode system of the NanoString nCounter platform enables quantification of small amounts of HCC-specific mRNA,(20) which further confers the second layer of sensitivity to the whole system.
Despite the highly sensitive enrichment of CTCs by the scoring system, nonspecifically captured WBCs inevitably pose a challenge to the interpretation of downstream molecular characterization of CTCs. In this regard, we applied an integrated data analysis framework, a novel approach developed by our team,(30) to select prognostic gene signatures specific to HCC cells. We started with 8 independent published HCC signatures representing HCC aggressiveness and disease-relevant pathways. By selecting common genes from 2 or more HCC signatures, we can ensure that the genes could reflect the characteristics of aggressive HCC without being biased in 1 data set. To avoid the unwanted signal caused by nonspecifically captured WBCs, we applied a filter using DMAP to eliminate highly expressed genes in WBCs. In addition, the filter using CCLE transcriptome data allow us to further select genes more specific to HCC. Through this integrated data analysis framework, the final HCC-CTC RS panel was shown to have high expression in HCC cells but very low expression in WBCs (Fig. 2C, Supporting Fig. 2C). Most important, this selected panel retains robust prognostic power to discriminate patients with HCC with poor prognosis (Fig. 2D,E). We then validated that the combined use of the HCC-CTC mRNA scoring system and the HCC-CTC RS panel is capable of discriminating artificial HCC blood samples prepared by spiking HCC cell lines with different aggressiveness (Fig. 3). These results laid the foundation to fulfill the goal to prognosticate HCC in a noninvasive manner.
We sought to validate that the tissue-derived HCC-CTC RS panel was also prognostic in an independent HCC-CTC cohort through CTC transcriptomic characterization. Using our HCC-CTC mRNA scoring system coupled with the HCC-CTC RS panel, expression analysis showed excellent discrimination of patients with HCC from healthy volunteers (Fig. 4A). By applying the same HCC-CTC RS cutoff from the TCGA HCC cohort, there was a clear separation of the HCC-CTC cohort into low-risk and high-risk groups (Fig. 4B), with high-risk patients demonstrating significantly inferior OS compared with low-risk patients (HR, 4.2; P = 0.02; Fig. 4C). In addition, the CTC-based RS characterization was not simply recapitulating the clinical stage that impacts survival, with patients with BCLC stage A and patients with more advanced BCLC stages B to D having a similar proportion of low-risk and high-risk patients (Fig. 4D), supporting the fact that similarly staged patients based on clinical staging systems can be further prognosticated based on their 10 gene signatures. Perhaps most important, the HCC-CTC RS remained an independent predictor of OS in the HCC-CTC cohort even after controlling for BCLC stage, MELD score, and CTC count (Fig. 5B). Furthermore, in the subset of 20 patients with BCLC stage A undergoing resection, all 3 recurrences occurred in the HCC-CTC RS high-risk group (Supporting Fig. 6). Taken collectively, these results demonstrate a proof of principle that prognostic signatures from tissue-based HCC transcriptomes can be translated into a liquid biopsy–based transcriptome profile that adds important prognostic information beyond CTC enumeration.
The HCC-CTC RS is calculated from 10 gene signatures: DDR1, EHHADH, AR, LUM, HSD17B6, PMEPA1, TSKU, NECAB2, LAD1, and SLC27A5. Previous studies have demonstrated some biological characteristics of these genes for prognostication. For example, PMEPA1 is an important target gene of transforming growth factor β (TGF-β) signaling and highly expressed in many types of cancers including colon, breast, lung, and prostate cancers.(34-37) Recently, PMEPA1 is reported to play a key role in the transformation of TGF-β from tumor suppressor to oncogene in HCC.(38) The SLC27A5 gene, a member of the solute carrier 27A gene family, encodes a transport protein for fatty acid transport and bile acid metabolism and is exclusively expressed in the liver.(39) In patients with HCC, reduced SCL27A5 expression contributes to tumor progression and poor prognosis.(40) These discoveries support the fact that our HCC-CTC RS can reflect the biological characteristics of HCC that may be related to tumor aggressiveness. The associations between the expression of these genes and the prognosis of patients merit further validation in future studies.
Despite these encouraging results, we acknowledge several limitations of our study. First, the validation HCC-CTC patient cohort was enrolled from a single institution with a relatively small number of patients with different stages and varying tumor and treatment characteristics. Despite the sample size and heterogeneity, the HCC-CTC RS remained an independent predictor of OS in multivariable analysis, supporting the contention that the assay is assessing a real-time prognostic phenotype of the patient irrespective of past history and treatments. In a small subgroup of patients with BCLC stage A receiving liver resection (n = 20), all 3 recurrence events occurred in the high-risk HCC-CTC group; however, the HR of 12.38 did not reach statistical significance (P = 0.11) undoubtedly because of the small number of events and limited cohort size. Second, this was purely an essential proof-of-principle study to show the for the first time the feasibility of translating tissue-based prognostic signatures into the liquid biopsy setting. Many of the 10 genes included in the HCC-CTC RS panel have a known role in cancer biology, but it is essential to highlight that we believe that further refinement of the panel of genes is necessary to improve the prognostic ability and utility of such a blood-based assay. In addition, further efforts are warranted to show the concordance of mRNA expression between the enriched CTCs and paired tissues. Lastly, we acknowledge that larger multicenter validation studies will be essential to pave the way for blood-based prognostic assays to impact clinical decision making for both early stage patients who are candidates for curative-intent treatments and advanced stage patients where predictors of response to the multitude of new systemic agents remain unavailable.
To our knowledge, this is the first study to successfully translate HCC tissue-based transcriptomic signatures into blood-based CTC mRNA characterization for HCC prognostic stratification. Using this methodology, we are able to noninvasively perform real-time disease profiling, which may help physicians dynamically monitor disease progression, evaluate treatment outcome, and select therapies during the course of disease. Moving forward, our approach could also be used to translate existing or developing tissue-based signatures of other cancers into a liquid biopsy setting. This approach holds great promise to significantly augment current cancer staging systems and allow for tailoring patient-specific treatments, 2 critical goals in precision oncology.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (U01CA198900, U01EB026421, R01CA218356, R21CA235340, R01CA246304, R01CA253651, R21CA240887, and R01CA255727), American College of Gastroenterology Junior Faculty Development Award, Department of Defense Peer Reviewed Cancer Research Program Career Development Award (CA191051), Cedars-Sinai Clinical Scholar Award, and Huiying Foundation.
Richard S. Finn consults and received grants from Bayer, Bristol-Myers Squibb, Eisai, Eli Lilly, Merck, Pfizer, and Roche. He consults for AstraZeneca and Cstone. He received grants from Adaptimmune. Hsian-Rong Tseng owns stock in Cytolumina Techologies Corp. and Pulsar Therapeutics. Ju Dong Yang consults for Exact Sciences, Eisai, and Gilead. Saeed Sadeghi consults for Eisai and Exelixis.
Abbreviations:
- AFP
alpha-fetoprotein
- AJCC
American Joint Committee on Cancer
- ALD
alcohol-related liver disease
- AR
androgen receptor
- BCLC
Barcelona Clinic Liver Cancer
- CCLE
Cancer Cell Line Encyclopedia
- cDNA
complementary DNA
- CI
confidence interval
- CTC
circulating tumor cell
- DAPI
4′,6-diamidino-2-phenylindole
- DDR1
discoidin domain receptor tyrosine kinase 1
- DEG
differentially expressed gene
- DMAP
differentiation Map data set
- EHHADH
enoyl-CoA hydratase and 3-hydroxyacyl CoA dehydrogenase
- HBV
hepatitis B virus
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- HR
hazard ratio
- HSD17B86
hydroxysteroid 17-beta dehydrogenase 6
- IDH
isocitrate dehydrogenase
- IQR
interquartile range
- LAD1
ladinin 1
- LUM
lumican
- MELD
Model for End-Stage Liver Disease
- mRNA
messenger RNA
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- NCIP
National Cancer Institute proliferation
- NECAB2
N-terminal EF-hand calcium binding protein 2
- OS
overall survival
- PBMC
peripheral blood mononuclear cell
- PC
principal component
- PCA
principal component analysis
- PD-L1
programmed death ligand 1
- PMEPA1
prostate transmembrane protein, androgen induced 1
- RS
risk score
- sCNA
somatic copy number alteration
- SLC27A5
solute carrier family 27 member 5
- TCGA
The Cancer Genome Atlas
- TGF-β
transforming growth factor β
- TSKU
tsukushi, small leucine rich proteoglycan
- TTR
time to recurrence
- WBC
white blood cell
REFERENCES
- 1).Nault JC, Villanueva A. Biomarkers for hepatobiliary cancers. Hepatology 2021;73(suppl 1):115–127. [DOI] [PubMed] [Google Scholar]
- 2).Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol 2019;16:589–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3).Global Burden of Disease Liver Cancer Collaboration, Akinyemiju T, Abera S, Ahmed M, Alam N, Alemayohu MA, et al. The burden of primary liver cancer and underlying etiologies from 1990 to 2015 at the global, regional, and national level: results from the Global Burden of Disease Study 2015. JAMA Oncol 1990;2017:1683–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4).Bruix J, Reig M, Sherman M. Evidence-based diagnosis, staging, and treatment of patients with hepatocellular carcinoma. Gastroenterology 2016;150:835–853. [DOI] [PubMed] [Google Scholar]
- 5).Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet 2003;362:1907–1917. [DOI] [PubMed] [Google Scholar]
- 6).Mazzaferro V, Regalia E, Doci R, Andreola S, Pulvirenti A, Bozzetti F, et al. Liver transplantation for the treatment of small hepatocellular carcinomas in patients with cirrhosis. N Engl J Med 1996;334:693–699. [DOI] [PubMed] [Google Scholar]
- 7).Zucman-Rossi J, Villanueva A, Nault J-C, Llovet JM. Genetic landscape and biomarkers of hepatocellular carcinoma. Gastroenterology 2015;149:1226–1239.e4. [DOI] [PubMed] [Google Scholar]
- 8).Schulze K, Imbeaud S, Letouzé E, Alexandrov LB, Calderaro J, Rebouissou S, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet 2015;47:505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9).Hoshida Y, Nijman SMB, Kobayashi M, Chan JA, Brunet J-P, Chiang DY, et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res 2009;69:7385–7392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10).Woo HG, Lee J-H, Yoon J-H, Kim CY, Lee H-S, Jang JJ, et al. Identification of a cholangiocarcinoma-like gene expression trait in hepatocellular carcinoma. Cancer Res 2010;70:3034–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11).Cancer Genome Atlas Research Network. Electronic address wbe, Cancer Genome Cancer Genome Atlas Research Network. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 2017;169:1327–1341.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12).Silva MA, Hegab B, Hyde C, Guo B, Buckels JAC, Mirza DF. Needle track seeding following biopsy of liver lesions in the diagnosis of hepatocellular cancer: a systematic review and meta-analysis. Gut 2008;57:1592–1596. [DOI] [PubMed] [Google Scholar]
- 13).Rockey DC, Caldwell SH, Goodman ZD, Nelson RC, Smith AD. Liver biopsy. Hepatology 2009;49:1017–1044. [DOI] [PubMed] [Google Scholar]
- 14).Ahn JC, Teng PC, Chen PJ, Posadas E, Tseng H-R, Lu SC, Yang JD. Detection of circulating tumor cells and their implications as a novel biomarker for diagnosis, prognostication, and therapeutic monitoring in hepatocellular carcinoma. Hepatology 2021;73:422–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15).Keller L, Pantel K. Unravelling tumour heterogeneity by single-cell profiling of circulating tumour cells. Nat Rev Cancer 2019;19:553–567. [DOI] [PubMed] [Google Scholar]
- 16).Court CM, Hou S, Winograd P, Segel NH, Li QW, Zhu Y, et al. A novel multimarker assay for the phenotypic profiling of circulating tumor cells in hepatocellular carcinoma. Liver Transpl 2018;24:946–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17).Winograd P, Hou S, Court CM, Lee Y-T, Chen P-J, Zhu Y, et al. Hepatocellular carcinoma-circulating tumor cells expressing PD-L1 are prognostic and potentially associated with response to checkpoint inhibitors. Hepatol Commun 2020;4:1527–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18).Kalinich M, Bhan I, Kwan TT, Miyamoto DT, Javaid S, LiCausi JA, et al. An RNA-based signature enables high specificity detection of circulating tumor cells in hepatocellular carcinoma. Proc Natl Acad Sci U S A 2017;114:1123–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19).Veldman-Jones MH, Brant R, Rooney C, Geh C, Emery H, Harbron CG, et al. Evaluating robustness and sensitivity of the NanoString Technologies nCounter platform to enable multiplexed gene expression analysis of clinical samples. Cancer Res 2015;75:2587–2593. [DOI] [PubMed] [Google Scholar]
- 20).Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol 2008;26:317–325. [DOI] [PubMed] [Google Scholar]
- 21).Sohn BH, Shim J-J, Kim S-B, Jang KY, Kim SM, Kim JH, et al. Inactivation of hippo pathway is significantly associated with poor prognosis in hepatocellular carcinoma. Clin Cancer Res 2016;22:1256–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22).Kim SM, Leem S-H, Chu I-S, Park Y-Y, Kim SC, Kim S-B, et al. Sixty-five gene-based risk score classifier predicts overall survival in hepatocellular carcinoma. Hepatology 2012;55:1443–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23).Lee J-S, Chu I-S, Heo J, Calvisi DF, Sun Z, Roskams T, et al. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology 2004;40:667–676. [DOI] [PubMed] [Google Scholar]
- 24).Cairo S, Armengol C, De Reynies A, Wei Y, Thomas E, Renard C-A, et al. Hepatic stem-like phenotype and interplay of Wnt/beta-catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell 2008;14:471–484. [DOI] [PubMed] [Google Scholar]
- 25).Shin J, Kim M, Jung H-J, Cha HL, Suh-Kim H, Ahn S, et al. Characterization of developmental defects in the forebrain resulting from hyperactivated mTOR signaling by integrative analysis of transcriptomic and proteomic data. Sci Rep 2017;7:2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26).Hwang D, Rust AG, Ramsey S, Smith JJ, Leslie DM, Weston AD, et al. A data integration methodology for systems biology. Proc Natl Acad Sci U S A 2005;102:17296–17301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27).Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012;483:603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28).Novershtern N, Subramanian A, Lawton LN, Mak RH, Haining WN, McConkey ME, et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell 2011;144:296–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29).Goldman MJ, Craft B, Hastie M, Repečka K, McDade F, Kamath A, et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol 2020;38:675–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30).Jan YJ, Yoon J, Chen J-F, Teng P-C, Yao NU, Cheng S, et al. A Circulating tumor cell-RNA assay for assessment of androgen receptor signaling inhibitor sensitivity in metastatic castration-resistant prostate cancer. Theranostics 2019;9:2812–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31).Caruso S, Calatayud A-L, Pilet J, La Bella T, Rekik S, Imbeaud S, et al. Analysis of liver cancer cell lines identifies agents with likely efficacy against hepatocellular carcinoma and markers of response. Gastroenterology 2019;157:760–776. [DOI] [PubMed] [Google Scholar]
- 32).Court CM, Hou S, Liu L, Winograd P, DiPardo BJ, Liu SX, et al. Somatic copy number profiling from hepatocellular carcinoma circulating tumor cells. NPJ Precis Oncol 2020;4:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33).Mohamed Suhaimi NA, Foong YM, Lee DY, Phyo WM, Cima I, Lee EXW, et al. Non-invasive sensitive detection of KRAS and BRAF mutation in circulating tumor cells of colorectal cancer patients. Mol Oncol 2015;9:850–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34).Zhang L, Wang X, Lai C, Zhang H, Lai M. PMEPA1 induces EMT via a non-canonical TGF-beta signalling in colorectal cancer. J Cell Mol Med 2019;23:3603–3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35).Vo Nguyen TT, Watanabe Y, Shiba A, Noguchi M, Itoh S, Kato M. TMEPAI/PMEPA1 enhances tumorigenic activities in lung cancer cells. Cancer Sci 2014;105:334–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36).Giannini G, Ambrosini MI, Di Marcotullio L, Cerignoli F, Zani M, MacKay AR, et al. EGF- and cell-cycle-regulated STAG1/PMEPA1/ERG1.2 belongs to a conserved gene family and is overexpressed and amplified in breast and ovarian cancer. Mol Carcinog 2003;38:188–200. [DOI] [PubMed] [Google Scholar]
- 37).Rae FK, Hooper JD, Nicol DL, Clements JA. Characterization of a novel gene, STAG1/PMEPA1, upregulated in renal cell carcinoma and other solid tumors. Mol Carcinog 2001;32:44–53. [DOI] [PubMed] [Google Scholar]
- 38).Quetglas IM, Sia D, Jiao Y, Maeda M, Castro M, Esteller M, Llovet JM. TGF-β oncogenic pathway in HCC: PMEPA1 as a biomarker of treatment response. J Hepatol 2017;66:S466. [Google Scholar]
- 39).Anderson CM, Stahl A. SLC27 fatty acid transport proteins. Mol Aspects Med 2013;34:516–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40).Gao Q, Zhang G, Zheng Y, Yang YI, Chen C, Xia J, et al. SLC27A5 deficiency activates NRF2/TXNRD1 pathway by increased lipid peroxidation in HCC. Cell Death Differ 2020;27:1086–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.