

Abstract
Rotavirus (RV) replication occurs in cytoplasmic compartments, known as viroplasms, that are composed of viral and cellular proteins. Viroplasm formation requires RV nonstructural proteins NSP2 and NSP5 and cellular lipid droplets (LDs); however, the mechanisms required for viroplasm assembly remain largely unknown. We previously identified two conformationally-distinct forms of NSP2 (dNSP2, vNSP2) found in RV-infected cells that interact differentially with hypo- and hyper-phosphorylated NSP5, respectively, and indicate a coordinated phosphorylation-dependent mechanism regulating viroplasm assembly. We also reported that phosphorylation of dNSP2 on Serine 313 by the cellular kinase CK1α triggers the localization of vNSP2 to sites of viroplasm assembly and its association with hyper-phosphorylated NSP5. To directly evaluate the role of CK1α-mediated NSP2 phosphorylation on viroplasm formation, we used a recently published plasmid-based reverse genetics method to generate a recombinant rotavirus (rRV) with a phosphomimetic NSP2 mutation (rRV NSP2 S313D). The rRV NSP2 S313D virus is significantly delayed in viroplasm formation, virus replication, and interferes with wild type RV replication during co-infection. The rRV NSP2 S313A virus was not rescued. Taking advantage of the delay in viroplasm formation, the NSP2 S313D phosphomimetic mutant was used as a tool to observe very early events in viroplasm assembly. We show that (1) viroplasm assembly correlates with NSP5 hyper-phosphorylation, and (2) that vNSP2 S313D co-localizes with RV-induced LDs without NSP5, suggesting that vNSP2 phospho-S313 is sufficient for interacting with LDs and may be the virus factor required for RV-induced LD formation. Further studies with the rRV NSP2 S313D virus are expected to reveal new aspects of viroplasm and LD initiation and assembly.
1. Introduction
The advent of a long-awaited reverse genetics method for studying rotaviruses (RVs) was a very exciting development for the rotavirus field. The breakthrough development of an entirely plasmid-based reverse genetics method by Kanai, et al (Kanai et al., 2017) was welcomed as a method to finally begin targeted, mechanistic studies to investigate the roles of each of the RV proteins in rotavirus replication and pathogenesis. By the following year, Komoto et al published an optimized version of the protocol (Komoto et al., 2018).
One of the over-arching questions that has long intrigued our laboratory is how do rotavirus replication factories, or viroplasms, form inside the cytoplasm of rotavirus-infected cells? Viroplasms are sites of genome replication, genome packaging, and assembly of immature double-layered virus particles that interact and assemble on cellular lipid droplets (LDs) forming a viroplasm/LD replication platform (Crawford and Desselberger, 2016). Failure to assemble viroplasms or to induce LD formation results in loss of virus production (Cheung et al., 2010). An ongoing, basic cell biology challenge is determining the site of LD origin, as well as the mechanisms and machinery that are involved in LD formation (Chung et al., 2019). Understanding the molecular basis for initiation and formation of the viroplasm, including RV-induced lipid droplet biogenesis, is critical for understanding RV replication; but is also likely to provide clues to the replication processes of other RNA viruses (hepatitis C virus, Dengue virus) and bacterial pathogens (Chlamydia) that replicate in LD-dependent platforms or that require LDs for replication (Boulant et al., 2007; Elamin et al., 2012; Heaton and Randall, 2010; Kumar et al., 2006; Laufman et al., 2019; Miyanari et al., 2007; Samsa et al., 2009; Vallochi et al., 2018). This review summarizes the current knowledge of RV viroplasm formation and how we made and used genetically modified rotavirus to answers long-standing questions about the role of NSP2 in viroplasm assembly.
2. Viroplasm formation
2.1. RV nonstructural proteins NSP2 and NSP5 are required for viroplasm/LD formation
In 1999, Fabbretti et al. (Fabbretti et al., 1999) showed that in the absence of other viral proteins, transient co-expression of NSP2 with NSP5 results in the formation of spherical viroplasm-like structures (VLS), but when expressed alone each protein remains diffuse in the cytoplasm demonstrating the interdependent relationship between these two proteins. Viroplasm formation can be prevented using RNA interference (RNAi) or intrabodies that silence the expression of NSP2 and/or NSP5 in RV-infected cells (Campagna et al., 2005; Lopez et al., 2005; Silvestri et al., 2004; Vascotto et al., 2004). A rotavirus NSP2 isolate containing a temperature-sensitive (ts) mutation in genome segment 8 (A152V) is unable to form viroplasms at non-permissive temperatures (Ramig and Petrie, 1984; Taraporewala et al., 2002). Thus, both NSP2 and NSP5 are the minimum RV proteins that are absolutely required for viroplasm/LD formation. The molecular basis for how these two proteins interact with each other, and with LDs, to assemble into a structured, replication-competent platform has yet to be understood.
NSP2 is a 35 kDa protein that assembles into octamers in solution, formed by tail-to-tail interactions of two NSP2 tetramers, and is predicted to be the active form of the protein (Jayaram et al., 2002; Taraporewala et al., 2002). NSP2 is a multifunctional protein and may play a role in packaging the dsRNA genome. NSP2 specifically binds the 5’-end of ssRNA genome templates and catalyzes the removal of the 3’-phosphate required for replication of the second strand (Hu et al., 2012; Taraporewala et al., 1999). NSP2 also has been implicated as a chaperone for dsRNA genome segment assortment into assembling nascent particles (Borodavka et al., 2017). Additionally, phosphorylation of specific amino acids within NSP2, which has autokinase activity in addition to being phosphorylated by CK1α at serine 313 (S313) (Criglar et al., 2018), may regulate NSP2 activity, turning different functions on or off in specific contexts and regulating its interaction with other proteins and genomic RNA.
2.2. Phosphorylation plays a key role in viroplasm formation
Our research into viroplasm formation, and in particular the study of NSP2, led to the discovery that there are two distinct forms of NSP2 in a RV-infected cell; the cytoplasmically-dispersed dNSP2 that appears first, and the viroplasm-localized vNSP2 detected approximately one hour later, suggesting that dNSP2 transitions into vNSP2 by an unknown mechanism (Criglar et al., 2014). Each form of NSP2 is recognized by conformation-specific monoclonal antibodies raised against the full-length protein (Figure 1). NSP5, an O-linked glycosylated protein that contains a high number of serine residues, is post-translationally modified into various phosphorylated isoforms that migrate in SDS PAGE gels from 26 kDa to 34 kDa. Characterization of dNSP2 and vNSP2 showed that each form of NSP2 interacts differentially with NSP5 in a phosphorylation-dependent manner (Criglar et al., 2014). Co-immunoprecipitation of dNSP2 and vNSP2 from RV-infected cell lysates showed that dNSP2 only interacts with the least phosphorylated (hypophosphorylated) isoforms of NSP5, specifically the 26 and 28 kDa isoforms. In stark contrast, vNSP2 only interacts with NSP5 when it is hyperphosphorylated, specifically when the 32-34 kDa phosphorylated isoforms are present. These data indicated a phosphorylation-dependent interaction between NSP2 and NSP5 and suggested a phosphorylation-dependent mechanism for viroplasm assembly. This hypothesis was further supported when another group, utilized a trans-complementing wild type NSP5 cell line to evaluate replication deficient NSP5 phosphorylation mutant recombinant RVs, and showed that NSP5 hyperphosphorylation is required for viroplasm assembly (Papa et al., 2019).
Figure 1. Monoclonal antibody (mAb)-dependent detection of two forms of NSP2.

Panel A) viroplasm-specific vNSP2 (green) co-localizes with NSP5 (red) in viroplasms. Panel B) cytoplasmically-dispersed dNSP2 (green) does not colocalize with NSP5 (red) in viroplasms. C) Magnified view of orange boxed area in panel B shows a view of two viroplasms detected by mAb to dNSP2 (green) outside of viroplasms and mAb to vNSP2 (red) in viroplasms. Graph shows the path of the dotted white line in the merged image and the intensity of detection of, dNSP2 (green line) versus vNSP2 (red line). Adapted from Criglar et al, 2014.
2.3. NSP2 phosphorylation is critical for the formation of nascent viroplasms
Mass spectrometric analysis of dNSP2 and vNSP2 isolated from RV-infected cell lysates by mAb-specific immunoprecipitation showed that NSP2 is phosphorylated at serine 313 (S313) in the C-terminus of the protein (Criglar et al., 2014). Particularly intriguing, vNSP2 is 40 times more often phosphorylated at S313 than dNSP2, indicating that NSP2 phosphorylation correlates with the transition to vNSP2. Subsequently, we showed that the ubiquitous cellular protein kinase CK1α phosphorylates NSP2 on S313 in vitro, and that silencing CK1α in RV-infected cells results in an altered distribution of vNSP2 that is dispersed in the cytoplasm similar to dNSP2, and fails to assemble into viroplasms with NSP5 (Criglar et al., 2018). Thus, phosphorylation of NSP2 S313 by CK1α is the trigger that drives NSP2 to sites of viroplasm formation.
Further evidence points to NSP2 S313 phosphorylation as the potential key to the molecular assembly of the viroplasm structure. In solution, NSP2 monomers crystalize over the course of several days into octamers with enzymatic activity (Jayaram et al., 2002; Taraporewala et al., 2002). The recombinant phosphomimetic NSP2 protein, NSP2 S313D that structurally and chemically acts as a phosphorylated serine, also forms octamers in solution, but crystalizes in a matter of hours. The basis for the rapid crystallization is the formation of four interoctamer bonds in the NSP2 crystal lattice between D313 in one octamer and R287 in a neighboring octamer (Criglar et al., 2018). These data led to the hypothesis that during RV infection, phosphorylation of NSP2 on S313 catalyzes rapid viroplasm formation and enhances virus replication. To test this hypothesis, we attempted to generate a NSP2 S313D phosphomimetic recombinant RV, and a NSP2 S313A phospho-null recombinant RV that is incapable of being phosphorylated at S313, using the methods by Kanai, et al (Kanai et al., 2017).
3. Optimization and protocol for obtaining genetically engineered rotavirus
Practical execution of the published protocol was challenging and required quite a bit of trial and error for success in our laboratory. These difficulties included (i) defining the optimum cell density for transfection, (ii) determining specific incubation times for the various steps, (iii) defining the parameters of co-culture, and (iv) determining ways to maintain a balanced pH (7.2-7.4) for cell culture media throughout the various incubation periods. Below we discuss details incorporated into the protocol that allowed us to successfully obtain the rRV WT and rRV NSP2 S313D viruses.
3.1. Cells and media for transfection
Baby hamster kidney (BHK) cells that stably express phage T7 polymerase (BSR T7/5) were used for transfection and expression of the 14 plasmids engineered for reverse genetics for rotavirus (Buchholz et al., 1999). These cells are similar, but not identical, to the BHK T7-expressing cells used in the Kanai protocol. We chose to utilize these cells because we had used them previously in the lab and were familiar with their use and care. To our knowledge, the BSR T7/5 cells only differ genetically with regard to the additional phage T5 promotor and are generally hardy and readily transfectable to high efficiency. We determined that, for optimum health, the cells required additional supplementation. Therefore, the BSR T7/5 cells were maintained in high glucose DMEM media supplemented with 10% fetal bovine serum (FBS) (or without serum where noted), 2 mM L-glutamine, and 1 mg/mL Geneticin (G418) for continuous selection. The cells were most healthy, and we were most successful in recovering recombinants when the culture medium was additionally supplemented with 10% tryptose phosphate broth and 1% MEM non-essential amino acids. Additionally, we found that the extended incubation times required for the transfection and subsequent co-culture steps resulted in highly acidic culture media that could be toxic to the cells. Therefore, the cell culture media was further supplemented with 7.5% sodium bicarbonate up to 4% final concentration.
3.2. RV SA11 and helper protein plasmids
The eleven transcription plasmids carrying RV cDNA for each of the 11 SA11 genome segments, and the 3 additional expression plasmids that encode the fusion-associated small transmembrane (FAST) protein from Nelson Bay orthoreovirus and the vaccinia virus capping enzymes for enhanced replication, were supplied by Takeshi Kobayashi and acquired through the Addgene plasmid repository. Plasmids for generating recombinant RV NSP2 S313D and RV NSP2 S313A viruses were constructed by mutation of the pT7-NSP2SA11 plasmid, replacing the serine at position 313 with aspartic acid or alanine. Plasmids were purified from bacteria using the Marligen PowerPrep HP Plasmid DNA Maxiprep kit (OriGene), followed by an additional ethanol precipitation step.
3.3. Transfection and co-culture for recovery of recombinant viruses
For co-culture, we used MA104 cells maintained in high glucose DMEM supplemented with 10% FBS. The day prior to co-culture, the MA104 cell media was changed to DMEM without FBS. All incubations were carried out at 37°C, 5% CO2. After considerable trial and error, we finalized a transfection and co-culture protocol that was variably successful up to approximately 60% of the time for generating RV recombinants (Figure 2). Wild type recombinants were recovered most frequently, although not consistently, while recombinants with mutations were recovered far less often. The reason for the failure to at least recover wild type virus in every instance remains a mystery. Failure to recover many of our desired recombinants less frequently than wild type suggests that the introduced mutations are lethal, or less viable, and determining which of these is correct is discussed later.
Figure 2. Transfection and co-culture timeline for reverse genetics protocol.

The optimized timeline used to generate the recombinant RVs was strictly adhered to with little room for variation.
3.4. Seeding plates
We chose to perform our transfections in 6-well culture plates to increase the probability of virus recovery. BSR T7/5 cells were seeded in the late afternoon on the day prior to transfection at a density of 4 x 105 cells in the supplemented cell culture media. This number of cells consistently ensured a monolayer at 80% confluence at the time of transfection, approximately 18 hours later. As cells must be actively dividing to take up the lipofection reagent, the BSR T7/5 cells were well-dispersed around the well for transfection the following day. Plates were incubated overnight. Cells above 80% confluence were never used.
3.5. Preparing the transfection mixture
Transfections were carried out using the lipid-based Mirus TransIT-LT1 Transfection Reagent. All reagents and media, excluding the plasmid DNA, were warmed to room temperature (RT) for optimal formation of the DNA-liposome complexes. For each experimental reaction, 11 RV plasmids (0.8 μg each), the D12 and D1L plasmids (0.8 μg each), and the FAST plasmid (0.015 μg), were combined in OptiMEM Reduced Serum media per the Mirus transfection instructions and mixed well. Prior to use, the lipid transfection reagent was gently pulse vortexed to mix well. We found this to be a critical step since a homogenous liposome preparation is required for optimal liposome formation. Failure to do this also impacts future experiments using the same reagent tube because the ratios will be off and suboptimal DNA-lipid complexes will be formed. We found that adding 2 μL of the transfection reagent per μg of DNA worked well for our cells. The transfection reagent was added directly into the plasmid suspension and mixed gently but thoroughly. The DNA-lipid mixture was incubated 20-30 minutes at RT to form the DNA-containing liposomes.
3.6. Transfection of the BSR T7/5 cells
Per the manufacturer’s instructions, the BSR T7/5 cells were transfected without removing the culture media as serum is required for efficient transfection and reduced toxicity. Specifically, the DNA-liposome suspensions were mixed very gently after incubation and added dropwise in a circular pattern onto the cells. The plates were rocked gently and then incubated for 24 hours. We changed the transfection/culture media at 24 hours post transfection (hpt) to 2 mL serum-free supplemented DMEM so that the MA104 cells added for co-culture the following day (at 48 hpt) would be maximally susceptible for infection by any recombinant virus (culture cells are more susceptible to infection in the absence of serum (Smith et al., 1979)).
3.7. Co-culture with MA104 cells
At 48 hpt, the MA104 cells were trypsinized, resuspended in DMEM in 10% FBS. FBS is added at this step to deactivate the trypsin used for suspending the MA104 cells. This small amount of FBS in the MA104 cell suspension is diluted out once the small volume of MA104 cells is added to the larger media volume of BSR T7/5 cell media in the well (2 mL). The resuspended cells were added to the BSR T7/5 transfection plates (5 x 104 cells/well) in 0.2 mL volume and incubated for 4 hours, after which Worthington trypsin was added to each well for a final concentration of 0.5 μg/mL. The co-culture plates were incubated for an additional 2-4 days depending on development of cytopathic effect (CPE), but not longer than 4 days before collecting and freezing.
3.8. Preparing co-culture lysates and passage of potential recombinant virus
Co-culture lysates were prepared by freeze-thawing for three cycles before any potential recombinant virus was activated by adding 10 μg/mL Worthington trypsin and incubating at 37°C for 30 minutes, prior to inoculating the entire contents onto new MA104 cell monolayers, that were then further cultured in serum-free DMEM for 24 hours, in 6-well culture plates for 1 hour. After 60 minutes, the inoculum was removed and replaced with 2 ml serum-free DMEM. The Passage 1 (P1) plates were incubated for up to 7 days, or shorter depending on visualization of CPE. The plates were again collected, subjected to three cycles of freeze/thaw, activated (10 μg/mL Worthington trypsin for 30 minutes), and passaged again (P2) on new MA104 cells as before.
4. Generation and recovery of a recombinant phosphomimetic NSP2 rotavirus
To test our hypothesis that a NSP2 phospho-S313 protein enhances viroplasm formation and replication, we attempted to rescue three recombinant SA11 rotaviruses using the modified method described above of Kanai, et al. (Kanai et al., 2017); a wild type recombinant RV (rRV WT), a NSP2 S313D phosphomimetic recombinant RV (rRV NSP2 S313D), and a NSP2 S313A phospho-null recombinant RV that is incapable of being phosphorylated at S313 (Criglar et al., 2020).
To recover recombinant wild type and NSP2 S313D rotaviruses, P2 lysates from all our experiments were initially incubated up to 7 days. Recovery of rRV WT virus was achieved frequently using this modified protocol. However, no discernable CPE was ever observed for the NSP2 S313D or NSP2 S313A viruses. Subsequently, the NSP2 mutant plates were left in the incubator for 35 uninterrupted days. Once the plates were recovered, 1 ml of PBS was added to each well due to low media volume and subjected to three freeze/thaw cycles. The next passage (P3) showed clear CPE development for the NSP2 S313D virus after 48 hours. No visible CPE for NSP2 S313A virus was detected after three, 7-day passages. However, using primers surrounding the mutation site on gene 8, a RT-PCR product was recovered from the P3 lysates that was sequenced, and confirmed to contain the gene 8 mutation. Because no CPE was detected and virus could not be amplified, rescue of the NSP2 S313A virus was deferred for future investigation. Continued passage of both the rRV WT virus and rRV NSP2 S313D virus were carried out until sufficient virus was obtained for assays. Virus titer was determined by standard serial dilution plaque assay. Plates were overlayed with neutral red stain at 48 hpi and plaques were counted at 72 hpi. Ultimately, we generated high titer virus stock for the rRV WT virus by passage 4 (5.0 x 108 pfu/mL) and from the rRV NSP2 S313D virus by passage 5 (3.8 x 108 pfu/mL).The dsRNA from the recombinant viruses was extracted using a QIAamp Viral RNA Kit (Qiagen), and genome segment 8 was amplified by PCR, purified using a QIAquick PCR Purification Kit (Qiagen), and sequenced to confirm the WT NSP2 and the NSP2 S313D sequence.
5. The rRV NSP2 S313D mutant virus differs from rRV WT in many ways
Characterization of the phosphomimetic rRV NSP2 S313D mutant showed it was clearly different from wild type virus. We discovered that the phosphomimetic mutation, while rapidly catalyzing crystallization in vitro, actually significantly delayed viroplasm assembly for almost 24 hpi (Criglar et al., 2020), indicating that either the aspartic acid does not function exactly as a phospho-serine in infected cells, or the mutation has disrupted the temporal, ordered role of phosphorylation in viroplasm biogenesis. The latter is our favored hypothesis. We propose that by creating a virus with only the “phosphorylated” form of NSP2 at S313 (the D313 mimic),a post-translational modification more associated with vNSP2 than dNSP2, we have disrupted the functional activity associated exclusively with dNSP2. The few viroplasms detected in the NSP2 S313D mutant at 8 hpi were loosely assembled and disorganized (Figure 3).
Figure 3. The NSP2 S313D mutation results in loosely organized viroplasms.
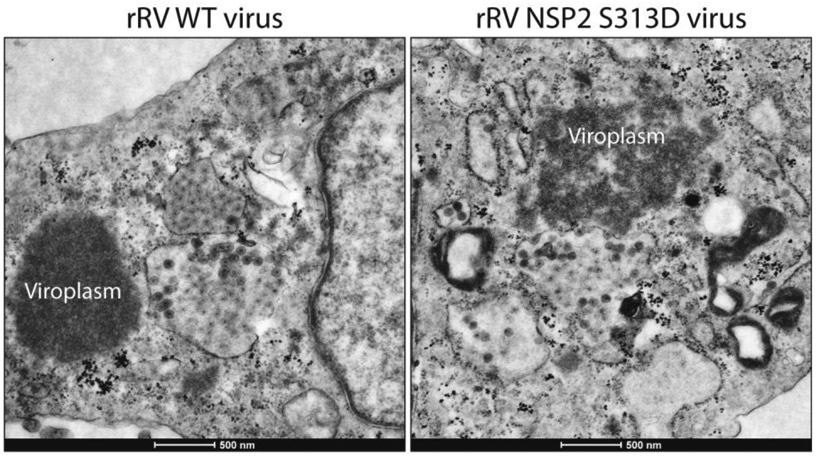
EM of cells infected with rWT rotavirus (left) compared to rNSP2 S313D rotavirus (right) shows the single amino acid mutation in NSP2 results in loosely assembled, less dense viroplasms. Adapted from Criglar et al, 2020.
Despite not being a gain-of-function mutant, the rRV NSP2 S313D virus has provided several important clues to aid our understanding of viroplasm formation. The phosphomimetic mutant is an excellent tool for studying the early events in nascent viroplasm assembly due to the delayed viroplasm assembly kinetics. By taking advantage of the delay in viroplasm formation in rRV NSP2 S313D virus-infected cells, we were able to determine that: (i) vNSP2 S313D is able to interact with nascent lipid droplets [identified by detection of the LD-specific protein phosphorylated perilipin 1 (PLIN1)] as early as 4 hpi, and that this interaction does not require NSP5; (ii) NSP5 hyperphosphorylation is significantly delayed in rRV NSP2 S313D virus-infected cells, indicating a crucial role for NSP2 in NSP5 phosphorylation; and (iii) NSP5 hyperphosphorylation correlates with the appearance of viroplasms (Criglar et al., 2020).
6. Reflections and ongoing challenges
Clearly, RV biology has been revitalized by the development of the plasmid-based reverse genetic system. Theoretically, all aspects of RV replication and pathogenesis can now be pursued. We have already learned unexpected information about viroplasm formation from one recombinant virus. Our original hypothesis, that a recombinant virus with a phosphomimetic NSP2 S313D mutation would be hyper-stimulated to make viroplasms was developed from the results of in vitro protein crystallization studies that showed that recombinant NSP2 S313D protein crystallizes in hours compared to days that are required for NSP2 S313 wild type crystals to form. However, our studies of the rRV NSP2 S313D virus clearly show that assumptions made from in vitro analyses can be incorrect despite our best, most educated suppositions. Reverse genetics for RVs will clearly push the field forward quickly with the vast array of possible mutations achievable by this advancement.
The method of Komoto, et al (Komoto et al., 2018) has been successfully used to study several human rotaviruses (Falkenhagen et al., 2020; Kawagishi et al., 2020; Komoto et al., 2020; Komoto et al., 2019; Song et al., 2020), which are the focus of many of our present studies. Human rotaviruses have unique biological characteristics, such as strain-specific binding to histoblood group antigens rather than to sialic acid used by most animal rotaviruses, as initial attachment factors. It is likely that many clinical isolates of human rotaviruses use unique receptors that remain to be identified. Furthermore, our pathophysiological studies using the biologically relevant human intestinal stem cell -derived organoids exhibit host range restriction and require infection with human rotaviruses.
The plasmid-based method, however, is not without its challenges. We found the system is sensitive to failure for many reasons. Having each step optimized and working perfectly together throughout the series of manipulations is a delicate balance. There are several crucial aspects of the protocol that require careful execution in order to recover recombinants including, cell health, optimum cell density and dispersion for maximum transfection efficiency, only transfecting monolayers at 80% cell confluence, using room temperature transfection reagents and media, ensuring that plasmids and the transfection reagent suspensions are well-mixed, and controlling the pH of cell culture by adding sodium bicarbonate, as previously mentioned, as needed throughout the many manipulations and lengthy incubation periods. We have used the method in the publication describing the optimized transfection protocol that does not require the addition of the helper plasmids (Komoto et al., 2018); however, we have yet to generate recombinants without the accessory helper plasmids. As we observed no detrimental effect of adding the additional plasmid DNA, we have maintained their use.
There is, of course, the inherent fallibility of introducing lethal mutations into any of the viral proteins. Not only must we wonder if a given mutation will prove lethal, but we must also now consider that any given mutation could dramatically slow the replication of a recombinant, simply because the generated viruses were not given sufficient time to amplify. This is well exemplified by our rRV NSP2 S313D mutant virus. Sheer frustration at the inability to recover the recombinant led to the exceptionally long incubation of the P2 cells and the seemingly miraculous recovery of the desired virus. Determining when to passage virus at each stage, how long to passage, and how many times to passage, adds complexity to an ostensibly simple system. Indeed, we are unsure if the failure to recover the rRV NSP2 S313A mutant is due to lethality or extremely slow replication kinetics. The fact that the NSP2 S313A passaged cell lysates are NSP2 PCR-positive suggests that at some point the mutant was generated, but the reasons for its failure to quickly propagate require further investigation. Added to these considerations, it should also be taken into account that lengthy incubations and passage times that result in recovery of a desired recombinant could also result in compensatory, and undesired, mutations elsewhere in the virus genome. Of course, compensatory mutations could be of great interest and yield valuable data about the interacting domains of RVs proteins in their own right. With these considerations, we recommend that the complete genome of genetically engineering recombinant viruses should be sequenced and are doing so now with our rescued viruses.
7. Ongoing questions
Our future plans include dissecting the many additional phosphorylations of NSP2, NSP5, and the mechanism of RV-mediated LD biogenesis. We previously reported that NSP2 has an autokinase function and is auto-phosphorylated on at least 22 residues prior to, and after, CK1α phosphorylation of S313 (Criglar et al., 2020). We hypothesize that the additional phosphorylations of NSP2 serve to regulate the different functions of both forms of NSP2, and one or more of these phosphorylations may be the modification that triggers the transition of dNSP2 to vNSP2. Many proteins and protein kinases with dual or multiple functions are regulated in this manner (Day et al., 2016; Narayanan and Jacobson, 2009).
We will further pursue the multiple phosphorylations of NSP5. Our recent report that the delay in viroplasm assembly in cells infected with recombinant RV NSP2 S313D correlates with the delayed appearance of the extra-phosphorylated isoforms of NSP5 (28 kDa and above) raises the question of the role of phosphorylated NSP5 in rotavirus biology. In particular we showed that the appearance of the 28 kDa isoform of NSP5, one of the least phosphorylated forms, was most delayed in the rRV NSP2 S313D virus infection. The appearance of higher order phosphorylated NSP5 isoforms correlated with formation of viroplasms detected by immunofluorescent microscopy (Criglar et al., 2020). Our findings were supported by work from Papa et al that characterized several RGs-derived NSP5 phosphorylation mutants suggesting that NSP5 hyperphosphorylation is required for viroplasm formation (Papa et al., 2019). Our earlier work clarified a role for the cellular kinase CK1α in RV-infected cells and showed that CK1α is required for vNSP2 localization to viroplasms, and for the interaction of vNSP2 with NSP5 (Criglar et al., 2018). CK1α is not required for the appearance of 26 kDa hypophosphorylated NSP5, as this isoform appears in CK1α-silenced cells (Criglar et al., 2018). Nor is CK1α the kinase required for phosphorylation of the 26 kDa NSP5 to the 28 kDa isoform as this is also present in cells infected with the rRV NSP2 S313D phosphomimetic virus. Thus, the kinase(s) responsible for the appearance of the 26 kDa and 28 kDa isoforms of NSP5 remains elusive. Given the work of Papa et al. (Papa et al., 2019), CK1α very likely plays a role in the subsequent NSP5 hyperphosphorylation events. What remains for us to decipher are the specific and temporal order of phosphorylations on both NSP2 and NSP5 that modulate their functions and interaction, and that ultimately lead to viroplasm formation.
Figure 4 represents our current model for viroplasm/LD formation and our current avenues of investigation are indicated in the numbered blue circles. (1) Do RVs induce LD formation or interact with preformed lipid droplets to assemble viroplasms? (2) Does dNSP2 interact with PLIN1 or other lipid droplet associated proteins? (3) Does NSP2, which has autokinase activity, also function as a protein kinase to phosphorylate NSP5 or cellular proteins required for viroplasm/LD formation such as PLIN1? (4) What phosphorylates unmodified NSP5 to become the 26 kDa NSP5 isoform? (5) Does NSP2 specifically phosphorylate the 26 kDa NSP5 to form the 28 kDa NSP5? (6) What triggers the transition of dNSP2 to vNSP2? (7) What kinase(s) is responsible for the rapid hyper-phosphorylation of NSP5? (8) How does the vNSP2/NSP5/LD nascent viroplasm complex associate with other cellular and viral proteins to form the macromolecular structure of the viroplasm/LD that is required for immature rotavirus particle assembly?
Figure 4. Model of viroplasm formation.

Questions under investigation are marked with blue circles marked 1-8 as discussed in the text. Ongoing questions seek to determine: 1) the mechanism of NSP2 and LD interaction; 2) the role of PLIN1 in LD interaction; 3) whether NSP2 phosphorylates PLIN1 and NSP5; 4) the kinase that phosphorylates the nascent NSP5 protein; 5) whether NSP2 is the kinase responsible for the 28 kDa form of NSP5; 6) the mechanism of dNSP2 to vNSP2 transition; 7) the kinase(s) responsible for NSP5 hyper-phosphorylation; and 8) the structural basis for viroplasm assembly.
The RV reverse genetics system will allow for our eventual understanding of the intricacies of the phosphorylation- and LD-dependent mechanisms of viroplasm formation and assembly of RVs, and hopefully, will also yield clues to the mechanism of viral factory formation in other important disease-causing pathogens. In addition, LDs are increasingly recognized for their role in disease (diabetes, steatohepatitis, obesity, heart disease, and lipodystrophies) and in the replication of other pathogenic viruses and intracellular bacteria. Using RV reverse genetics will reveal mechanisms that clarify the interplay between lipid accumulation, these pathogens, and disease.
Highlights.
RV reverse genetics was used to generate a recombinant phosphomimetic rotavirus
A single mutation in nonstructural protein 2 exhibits delayed viroplasm formation
NSP2 co-localizes with lipid droplets (LDs) and LD-specific protein PLIN-1
NSP5 hyper-phosphorylation correlates with viroplasm assembly
RGs allowed understanding of phosphorylation-dependent viroplasm assembly
Acknowledgements
We would like to thank the Texas Medical Center Digestive Diseases Consortium core facilities, particularly Jim Barrish at the Electron Microscopy Lab at Texas Children’s Hospital, Houston, TX.
Funding
The work described in this article was supported by National Institutes of Health (NIH) grants R01 AI080656 and R37 AI36040. This project was also supported by the Advanced Technology Core Laboratories (Baylor College of Medicine), specifically the Integrated Microscopy Core at Baylor College of Medicine with funding from NIH (DK56338, CA125123, ES030285), and CPRIT (RP150578, RP170719), the Dan L. Duncan Comprehensive Cancer Center, and the John S. Dunn Gulf Coast Consortium for Chemical Genomics. Additional support by the Advanced Technology Core Laboratories (Baylor College of Medicine) include the Protein and Monoclonal Antibody Production Core (NIH grant P30 CA125123) and the Pathway Discovery Proteomics Core (grants P30 CA125123, CPRIT RP120092).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Borodavka A, Dykeman EC, Schrimpf W, Lamb DC, 2017. Protein-mediated RNA folding governs sequence-specific interactions between rotavirus genome segments. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulant S, Targett-Adams P, McLauchlan J, 2007. Disrupting the association of hepatitis C virus core protein with lipid droplets correlates with a loss in production of infectious virus. J Gen Virol 88(Pt 8), 2204–2213. [DOI] [PubMed] [Google Scholar]
- Buchholz UJ, Finke S, Conzelmann KK, 1999. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J Virol 73(1), 251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campagna M, Eichwald C, Vascotto F, Burrone OR, 2005. RNA interference of rotavirus segment 11 mRNA reveals the essential role of NSP5 in the virus replicative cycle. The Journal of general virology 86(Pt 5), 1481–1487. [DOI] [PubMed] [Google Scholar]
- Cheung W, Gill M, Esposito A, Kaminski CF, Courousse N, Chwetzoff S, Trugnan GO, Keshavan N, Lever A, Desselberger U, 2010. Rotaviruses associate with cellular lipid droplet components to replicate in viroplasms, and compounds disrupting or blocking lipid droplets inhibit viroplasm formation and viral replication. Journal of virology 84(13), 6782–6798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung J, Wu X, Lambert TJ, Lai ZW, Walther TC, Farese RV Jr., 2019. LDAF1 and Seipin Form a Lipid Droplet Assembly Complex. Dev Cell 51(5), 551–563 e557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford SE, Desselberger U, 2016. Lipid droplets form complexes with viroplasms and are crucial for rotavirus replication. Curr Opin Virol 19, 11–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criglar JM, Anish R, Hu L, Crawford SE, Sankaran B, Prasad BVV, Estes MK, 2018. Phosphorylation cascade regulates the formation and maturation of rotaviral replication factories. Proc Natl Acad Sci U S A 115(51), E12015–E12023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criglar JM, Crawford SE, Zhao B, Smith HG, Stossi F, Estes MK, 2020. A Genetically Engineered Rotavirus NSP2 Phosphorylation Mutant Impaired in Viroplasm Formation and Replication Shows an Early Interaction between vNSP2 and Cellular Lipid Droplets. J Virol 94(15) : e00972–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criglar JM, Hu L, Crawford SE, Hyser JM, Broughman JR, Prasad BV, Estes MK, 2014. A novel form of rotavirus NSP2 and phosphorylation-dependent NSP2-NSP5 interactions are associated with viroplasm assembly. J Virol 88(2), 786–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day EK, Sosale NG, Lazzara MJ, 2016. Cell signaling regulation by protein phosphorylation: a multivariate, heterogeneous, and context-dependent process. Curr Opin Biotechnol 40, 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elamin AA, Stehr M, Singh M, 2012. Lipid Droplets and Mycobacterium leprae Infection. J Pathog 2012, 361374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbretti E, Afrikanova I, Vascotto F, Burrone OR, 1999. Two non-structural rotavirus proteins, NSP2 and NSP5, form viroplasm-like structures in vivo. The Journal of general virology 80 ( Pt 2), 333–339. [DOI] [PubMed] [Google Scholar]
- Falkenhagen A, Patzina-Mehling C, Gadicherla AK, Strydom A, O'Neill HG, Johne R, 2020. Generation of Simian Rotavirus Reassortants with VP4- and VP7-Encoding Genome Segments from Human Strains Circulating in Africa Using Reverse Genetics. Viruses 12(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaton NS, Randall G, 2010. Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe 8(5), 422–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L, Chow DC, Patton JT, Palzkill T, Estes MK, Prasad BV, 2012. Crystallographic analysis of rotavirus NSP2-RNA complex reveals specific recognition of 5' GG sequence for RTPase activity. Journal of virology 86(19), 10547–10557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaram H, Taraporewala Z, Patton JT, Prasad BV, 2002. Rotavirus protein involved in genome replication and packaging exhibits a HIT-like fold. Nature 417(6886), 311–315. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Komoto S, Kawagishi T, Nouda R, Nagasawa N, Onishi M, Matsuura Y, Taniguchi K, Kobayashi T, 2017. Entirely plasmid-based reverse genetics system for rotaviruses. Proc Natl Acad Sci U S A 114(9), 2349–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawagishi T, Nurdin JA, Onishi M, Nouda R, Kanai Y, Tajima T, Ushijima H, Kobayashi T, 2020. Reverse Genetics System for a Human Group A Rotavirus. J Virol 94(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komoto S, Fukuda S, Hatazawa R, Murata T, Taniguchi K, 2020. Generation of recombinant rotaviruses from just 11 cDNAs encoding a viral genome. Virus Res 286, 198075. [DOI] [PubMed] [Google Scholar]
- Komoto S, Fukuda S, Ide T, Ito N, Sugiyama M, Yoshikawa T, Murata T, Taniguchi K, 2018. Generation of Recombinant Rotaviruses Expressing Fluorescent Proteins by Using an Optimized Reverse Genetics System. J Virol 92(13) : e00588–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komoto S, Fukuda S, Kugita M, Hatazawa R, Koyama C, Katayama K, Murata T, Taniguchi K, 2019. Generation of Infectious Recombinant Human Rotaviruses from Just 11 Cloned cDNAs Encoding the Rotavirus Genome. J Virol 93(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar Y, Cocchiaro J, Valdivia RH, 2006. The obligate intracellular pathogen Chlamydia trachomatis targets host lipid droplets. Curr Biol 16(16), 1646–1651. [DOI] [PubMed] [Google Scholar]
- Laufman O, Perrino J, Andino R, 2019. Viral Generated Inter-Organelle Contacts Redirect Lipid Flux for Genome Replication. Cell 178(2), 275–289 e216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez T, Rojas M, Ayala-Breton C, Lopez S, Arias CF, 2005. Reduced expression of the rotavirus NSP5 gene has a pleiotropic effect on virus replication. The Journal of general virology 86(Pt 6), 1609–1617. [DOI] [PubMed] [Google Scholar]
- Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K, 2007. The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol 9(9), 1089–1097. [DOI] [PubMed] [Google Scholar]
- Narayanan A, Jacobson MP, 2009. Computational studies of protein regulation by post-translational phosphorylation. Curr Opin Struct Biol 19(2), 156–163. [DOI] [PubMed] [Google Scholar]
- Papa G, Venditti L, Arnoldi F, Schraner EM, Potgieter C, Borodavka A, Eichwald C, Burrone OR, 2019. Recombinant rotaviruses rescued by reverse genetics reveal the role of NSP5 hyperphosphorylation in the assembly of viral factories. J Virol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramig RF, Petrie BL, 1984. Characterization of temperature-sensitive mutants of simian rotavirus SA11: protein synthesis and morphogenesis. Journal of virology 49(3), 665–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samsa MM, Mondotte JA, Iglesias NG, Assuncao-Miranda I, Barbosa-Lima G, Da Poian AT, Bozza PT, Gamarnik AV, 2009. Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog 5(10), e1000632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestri LS, Taraporewala ZF, Patton JT, 2004. Rotavirus replication: plus-sense templates for double-stranded RNA synthesis are made in viroplasms. Journal of virology 78(14), 7763–7774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EM, Estes MK, Graham DY, Gerba CP, 1979. A plaque assay for the simian rotavirus SAII. The Journal of general virology 43(3), 513–519. [DOI] [PubMed] [Google Scholar]
- Song Y, Feng N, Sanchez-Tacuba L, Yasukawa LL, Ren L, Silverman RH, Ding S, Greenberg HB, 2020. Reverse Genetics Reveals a Role of Rotavirus VP3 Phosphodiesterase Activity in Inhibiting RNase L Signaling and Contributing to Intestinal Viral Replication In Vivo. J Virol 94(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taraporewala Z, Chen D, Patton JT, 1999. Multimers formed by the rotavirus nonstructural protein NSP2 bind to RNA and have nucleoside triphosphatase activity. Journal of virology 73(12), 9934–9943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taraporewala ZF, Schuck P, Ramig RF, Silvestri L, Patton JT, 2002. Analysis of a temperature-sensitive mutant rotavirus indicates that NSP2 octamers are the functional form of the protein. Journal of virology 76(14), 7082–7093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallochi AL, Teixeira L, Oliveira KDS, Maya-Monteiro CM, Bozza PT, 2018. Lipid Droplet, a Key Player in Host-Parasite Interactions. Front Immunol 9, 1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vascotto F, Campagna M, Visintin M, Cattaneo A, Burrone OR, 2004. Effects of intrabodies specific for rotavirus NSP5 during the virus replicative cycle. The Journal of general virology 85(Pt 11), 3285–3290. [DOI] [PubMed] [Google Scholar]