

SUMMARY
Dopamine degeneration in Parkinson’s disease (PD) dysregulates the striatal neural network and causes motor deficits. However, it is unclear how altered striatal circuits relate to dopamine-acetylcholine chemical imbalance and abnormal local field potential (LFP) oscillations observed in PD. We perform a multimodal analysis of the dorsal striatum using cell-type-specific calcium imaging and LFP recording. We reveal that dopamine depletion selectively enhances LFP beta oscillations during impaired locomotion, supporting beta oscillations as a biomarker for PD. We further demonstrate that dynamic cholinergic interneuron activity during locomotion remains unaltered, even though cholinergic tone is implicated in PD. Instead, dysfunctional striatal output arises from elevated coordination within striatal output neurons, which is accompanied by reduced locomotor encoding of parvalbumin interneurons and transient pathological LFP high-gamma oscillations. These results identify a pathological striatal circuit state following dopamine depletion where distinct striatal neuron subtypes are selectively coordinated with LFP oscillations during locomotion.
Graphical Abstract
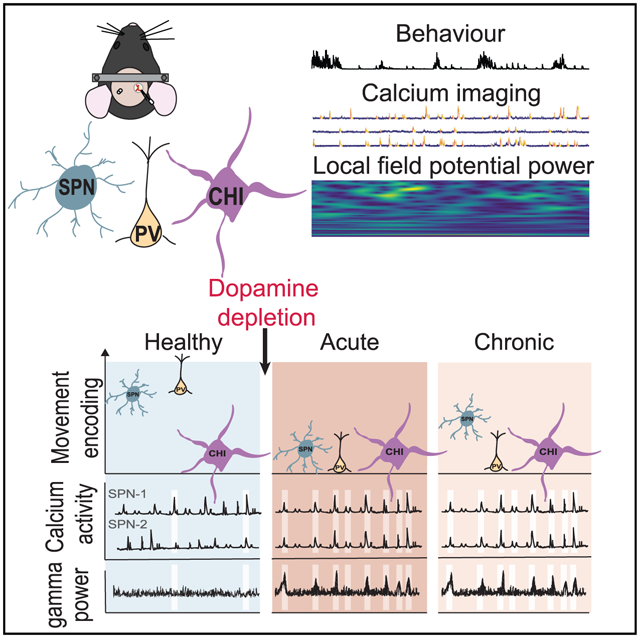
In brief
Zemel et al. demonstrate that dopamine loss disrupts striatal neural network and enhances local field potential beta oscillations during impaired locomotion. Specifically, striatal projecting neuron activation is abnormally coordinated and accompanied by pathological high-gamma oscillations. While parvalbumin interneurons reduce locomotor encoding, cholinergic interneurons strengthen their interactions with projecting neurons.
INTRODUCTION
Parkinson’s disease (PD) is characterized by the degeneration of substantia nigra dopamine neurons that project to the dorsal striatum, a major basal ganglia structure important for motor function (DeLong, 1990; Bolam et al., 2000). The dorsal striatum is composed of mainly medium-sized GABAergic spiny projecting neurons (SPNs) and a sparse population of various interneuron subtypes (Tepper et al., 2010). The classical spike-rate-based two-pathway model of PD states that an imbalance between the activity of SPNs expressing D1- (D1-SPNs) versus D2-dopamine receptors (D2-SPNs) contributes to PD motor deficits (DeLong, 1990; Calabresi et al., 2014; Albin et al., 1989; Bolam et al., 2000). Acute dopamine depletion increases D2-SPN activity, but decreases D1-SPN activity, leading to an overall reduction in striatal activity (Parker et al., 2018; Costa et al., 2006). Chronic depletion leads to persistent D2-SPN activation (Sharott et al., 2017; Parker et al., 2018) and impairs D2-SPN locomotion encoding ability (Parker et al., 2018), although conflicting results have been reported on the overall striatal neuron spiking rates in PD animal models and PD patients (Liang et al., 2008; Chen et al., 2001; Singh et al., 2016; Valsky et al., 2020; Parker et al., 2018).
Striatal cholinergic interneurons (CHIs) provide the striatum with a high cholinergic tone through extensive arborization and tonic firing (Bonsi et al., 2011; Kreitzer, 2009). Dopamine inhibits CHIs via D2 receptors (Calabresi et al., 2014), and loss of striatal dopamine is thought to elevate striatal cholinergic tone in PD. In addition, dopamine loss strengthens CHI to D2-SPN connections but weakens CHI to D1-SPN connections (Salin et al., 2009), suggesting a role for CHIs in promoting the indirect pathway function in PD. These findings, along with the therapeutic benefit of anti-cholinergic drugs in PD, support a dopamine-acetylcholine chemical imbalance in PD (Smith et al., 2012; Goetz, 2011). Recent calcium imaging studies demonstrated that CHI activity paralleled striatal dopamine levels, and transient elevation of CHI activity facilitated movement transitions (Howe et al., 2019; Gritton et al., 2019; Kondabolu et al., 2016), highlighting a dynamic role of CHIs in locomotion. Parvalbumin-positive interneurons (PVs) are another major striatal interneuron subtype. PVs receive strong cortical excitation, provide powerful feedforward inhibition to SPNs (Tepper et al., 2010; Owen et al., 2018), and closely track locomotion (Gritton et al., 2019). PVs mainly express D1/D5 receptors and are activated by dopamine (Calabresi et al., 2014; Bonsi et al., 2011; Bracci et al., 2002). However, dopamine loss does not alter the spontaneous activity of PVs in anesthetized rats or in brain slices, but does acutely enhance feedforward inhibition to D2-SPNs. This is followed by a chronic reduction of PV connectivity to both D1- and D2-SPNs (Mallet et al., 2006; Gittis et al., 2011; Salin et al., 2009). These studies suggest a dynamic remodeling of PV-SPN circuits following loss of dopamine.
PD also involves prominent changes in the local field potentials (LFPs) across the cortical-basal ganglia circuit. Intracranial recordings in PD patients consistently reveal pathological LFP oscillations at beta frequencies (~10–30 Hz) in the basal ganglia, including the striatum (Little and Brown, 2014; Singh and Papa, 2020). Pathological beta oscillations closely parallel key PD motor deficits and are largely suppressed by effective dopamine replacement treatment and deep brain stimulation therapies (Oswal et al., 2013; Boraud et al., 2006; Little and Brown, 2014). Recent studies in PD patients have found that high-gamma oscillations (~60–90 Hz) in the subthalamic nucleus (STN) and the motor cortex are associated with dyskinesia (Swann et al., 2016), and STN low-gamma oscillations (35–55 Hz) are augmented during strong tremor (Weinberger et al., 2009). While the cellular and network mechanisms of these LFP features remain largely unknown, animal studies support alteration of striatal LFP dynamics following dopamine loss (Lemaire et al., 2012).
To understand the effect of dopamine loss on different striatal neuron subtypes and their interactions with striatal LFPs, we performed simultaneous Ca2+ imaging from SPNs in conjunction with either PV or CHI interneurons, while monitoring striatal LFPs and locomotion. We evaluated the acute dopamine depletion effects within 2 weeks of 6-OHDA injury, and the chronic effects in the 2–5 weeks after 6-OHDA injury. We found that dopamine loss reduces the locomotor encoding ability of individual SPNs and PVs, but not CHIs. Furthermore, dysfunctional striatal output arose from abnormally coordinated SPNs, providing evidence for pathological network interactions beyond the classical basal ganglia spike rate model of PD. Finally, we found that SPN and PV activation was uniquely associated with a transient exaggeration of LFP high-gamma oscillations, which was rescued by dopamine replacement, suggesting a specific abnormal striatal circuit state and providing a basis for future LFP-based clinical functional biomarker studies.
RESULTS
Localized unilateral dopamine depletion selectively impairs movement initiation and rotational behavior
To examine how dopamine depletion affects dorsal striatal activity during locomotion, we performed simultaneous Ca2+ imaging of striatal neurons and striatal LFP recordings, while mice voluntarily ran on a spherical treadmill (Figure 1). Dopamine depletion was induced by a focal unilateral 6-OHDA infusion into the striatum close to the recording site on day 0. Histological quantification with tyrosine hydroxylase immunofluorescence confirmed that dopamine depletion was confined to a few hundred micrometers of the imaging site (Figures S1A and S1B). To capture dynamic striatal changes following dopamine depletion, we examined three conditions: healthy (over the 2-week period prior to 6-OHDA infusion), acute depletion (days 1–13 post-lesion), and chronic depletion (days 14–35 post-lesion).
Figure 1. Experimental setup and timeline.
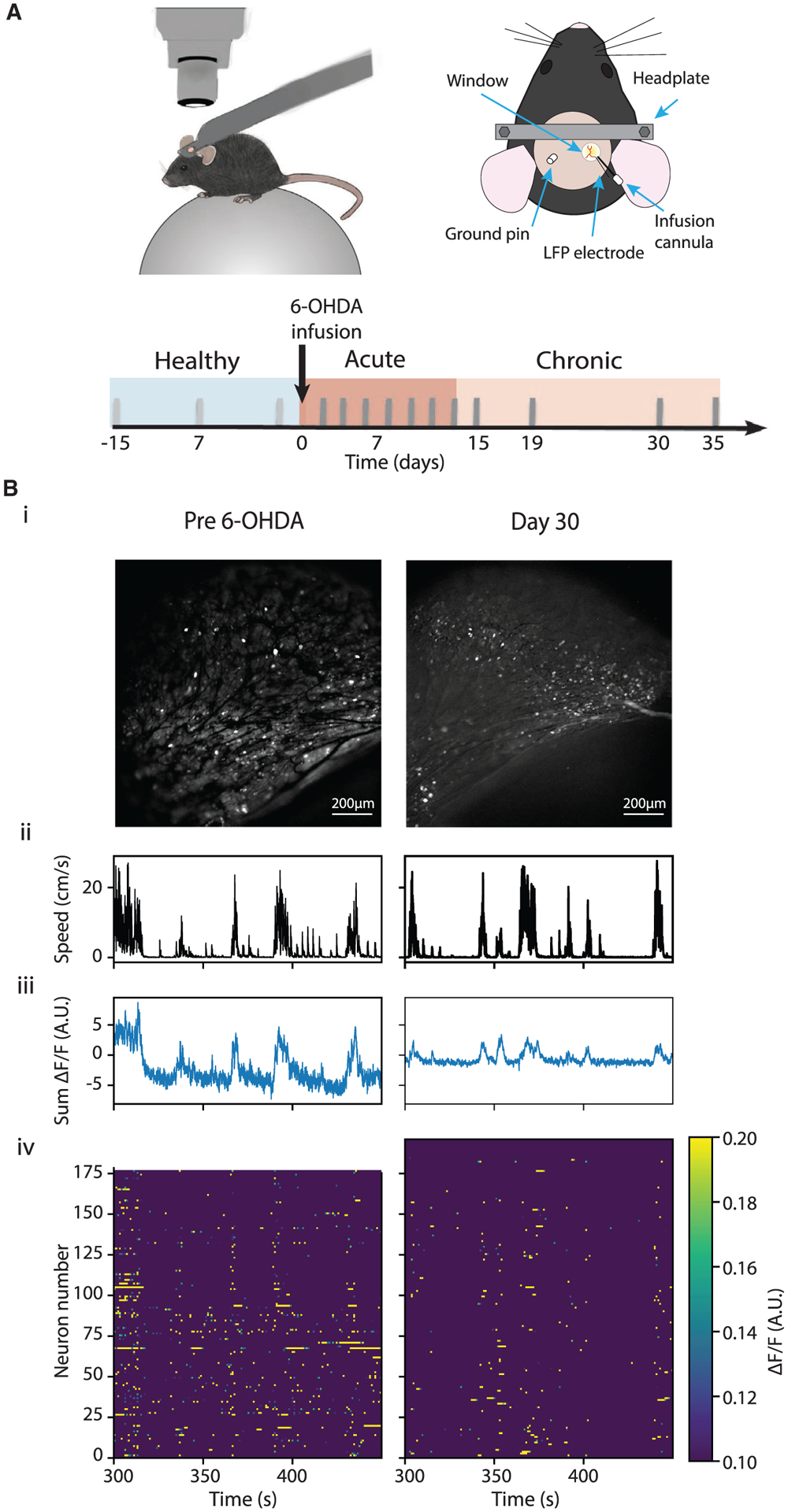
(A) Top: illustration of the experimental setup where a mouse is positioned under a microscope on the spherical treadmill (left), and the imaging window coupled with a LFP electrode and an infusion cannula (right). Bottom: experimental timeline. Three recording sessions, 5–7 days apart, were performed before 6-OHDA infusion in each animal, and multiple recording sessions were performed under the acute and chronic conditions.
(B) Example recording sessions from the same animal pre-6-OHDA infusion (left) and on day30 after 6-OHDA infusion (right). (i) A maximum-minimum GCaMP6 fluorescence intensity (ΔF/F) projection image, (ii-iv) movement speed, sum of ΔF/F of all neurons, and a heatmap of ΔF/F over an example session.
We found that dopamine depletion did not alter the frequency or duration of high-speed or low-speed movement bouts, or the overall speed distribution across the three conditions (Figures 2A and 2Bii). However, there was a significant decrease in the frequency of speed-onset transitions under both acute and chronic conditions (Figure 2Div). Furthermore, animals spent significantly less time in contralateral rotational movement under both depletion conditions (Figure 2C), consistent with the imbalance created with unilateral dopamine lesion. Similarly, there was a significant reduction in contralateral rotational onset transitions under the acute and chronic conditions (Figure 2Dii), whereas the reduction in ipsilateral rotational onset transitions was restricted to the acute condition (Figure 2Diii). The behavioral effect of 6-OHDA lesion developed gradually and stabilized after ~5 days (Figure S2E), and acute saline infusion produced no behavioral changes (Figures S2F–S2K). Finally, the number of recorded cells remained constant throughout our recording sessions (Figures 1 and 3), and we did not notice any detrimental tissue alterations around the injection site upon histological analysis (Figures S1C and S1D). Thus, the behavioral deficits observed were specific to the 6-OHDA lesion. Additionally, we measured locomotor behavior on spherical treadmills following systemic amphetamine injection. Under healthy conditions, amphetamine administration increased high-speed movement (Figure S2A), but not rotational movement in either direction (Figures S2B and S2C). Under both depletion conditions, mice spent more time in high-speed movement and ipsilateral rotational movement (Figures S2A–S2C), further confirming the effects of unilateral lesions in these mice. Together, the selective reduction in onset transitions, both speed and rotation transitions, without overall effects on movement duration or speed suggests that movement initiation is more sensitive to focal dopamine depletion.
Figure 2. Localized unilateral striatal dopamine depletion selectively impaired movement initiation and rotational behavior.
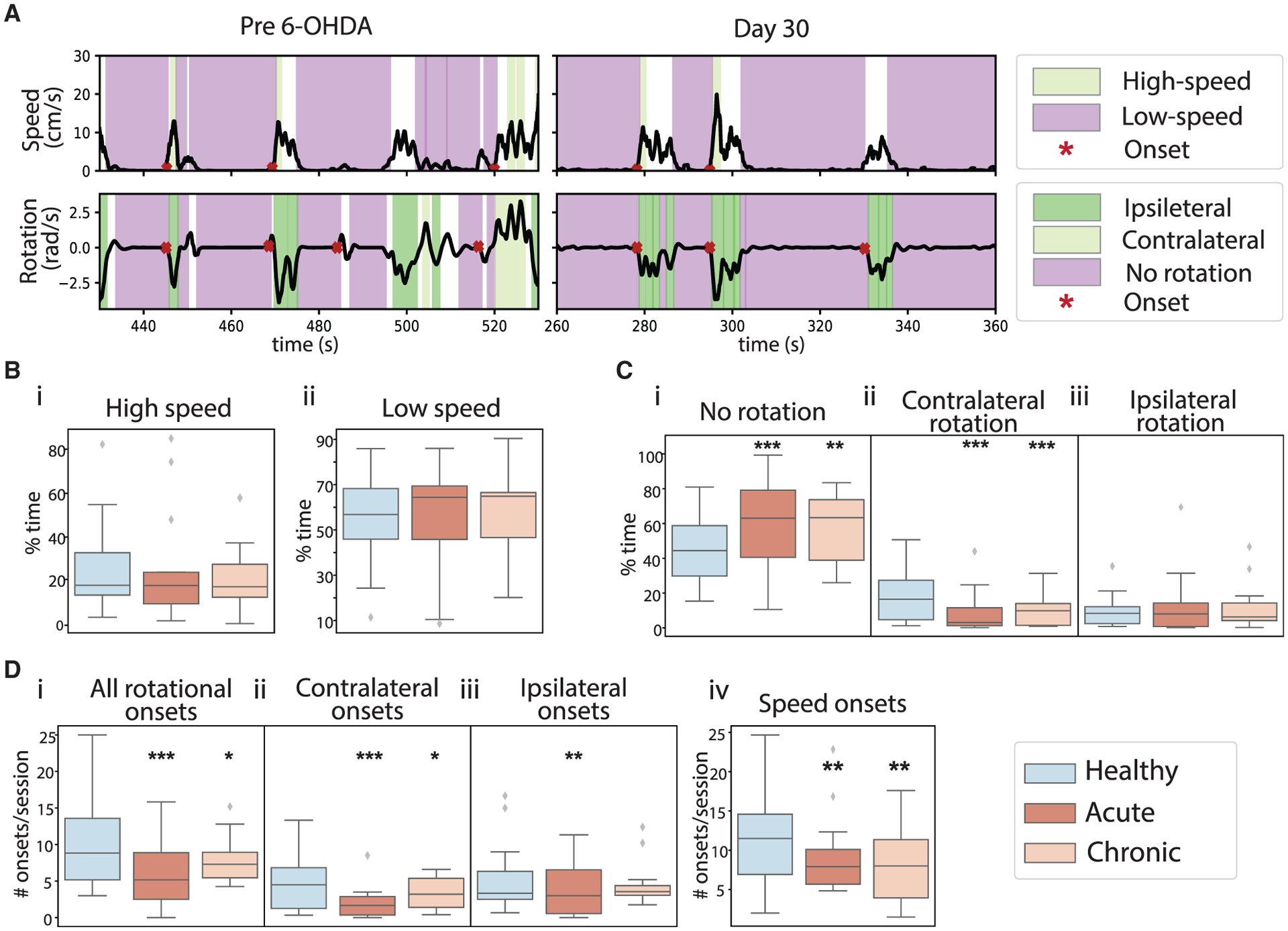
(A) Top: example movement speed traces (black) from the same mouse pre 6-OHDA infusion (left) and from day 30 after 6-OHDA infusion (right). Color shadings mark high-speed (green) and low-speed bouts (purple). Red stars mark onset. Bottom: same as top but for rotational movement. Color shading marks ipsilateral (dark green), contralateral (light green), and no rotation bouts (purple).
(B) The percentage of time mice spent in high-speed (i) and low-speed bouts (ii) under healthy (blue), acute (dark orange), and chronic (light orange) conditions. No differences across the three conditions for high-speed bouts (ANOVA, F(2,211) = 0.072, p = 0.931, 214 sessions from 14 mice) or low-speed bouts (ANOVA, F(2,211) = 0.397, p = 0.673, 214 sessions from 14 mice).
(C) The percentage of time mice spent in no rotation (i), contralateral rotation (ii), and ipsilateral rotation bouts (iii). The time spent in no rotation bouts was significantly increased under both depletion conditions (ANOVA, F(2,212) = 9.889, p = 8.03 × 10−5; mixed-effect model, acute versus healthy, t = 4.446, p = 1.44 × 10−5; chronic versus healthy, t = 2.745, p = 0.007). The time in contralateral rotation bouts was significantly reduced under both depletion conditions (ANOVA, F(2,211) = 9.873, p = 8.17 × 10−5; mixed-effect model, acute versus healthy, t = −4.374, p = 1.96 × 10−5; chronic versus healthy, t = −3.376, p = 8.82 × 10−4). There was no difference in ipsilateral rotation bouts across conditions (ANOVA, F(2,211) = 0.389, p = 0.678).
(D) The number of rotational onsets (i), contralateral rational onsets (ii), ipsilateral rotational onsets (iii), and speed onsets (iv). The number of rotational onsets decreased after dopamine depletion (ANOVA for interaction of total rotational onsets and conditions, F(2,211) = 14.42, p = 1.39 × 10−6; mixed-effect model, acute versus healthy, t = −5.221, p = 4.32 × 10−7; chronic versus healthy, t = −2.285, p = 0.023. ANOVA for contralateral onsets and conditions, F(2,211) = 10.92, p = 3.14 × 10−5; mixed-effect model, acute versus healthy, t = −4.577, p = 8.12 × 10−6; chronic versus healthy, t = −2.112, p = 0.036. ANOVA for ipsilateral onsets and conditions, F(2,211) = 4.38, p = 0.014; mixed-effect model, acute versus healthy, t = −2.859, p = 0.005; chronic versus healthy, t = −1.195, p = 0.233). The number of speed onsets decreased after dopamine depletion (ANOVA, F(2,211) = 5.838, p = 0.003; mixed-effect model, acute versus healthy, t = −2.676, p = 0.008; chronic versus healthy, t = −3.334, p = 0.001). All boxplots are plotted by subject (n = 14), with the box representing the median (middle line) and the 25th (Q1, bottom line) and 75th (Q3, top line) percentiles, and the whiskers are Q1 − 1.5 * (Q3 − Q1) and Q3 + 1.5 * (Q3 − Q1). Outliers that exceed these values are shown as diamonds. **p < .01, ***p < 0.001.
Figure 3. Dopamine depletion reduced locomotor encoding ability of SPNs and PVs, but not CHIs.
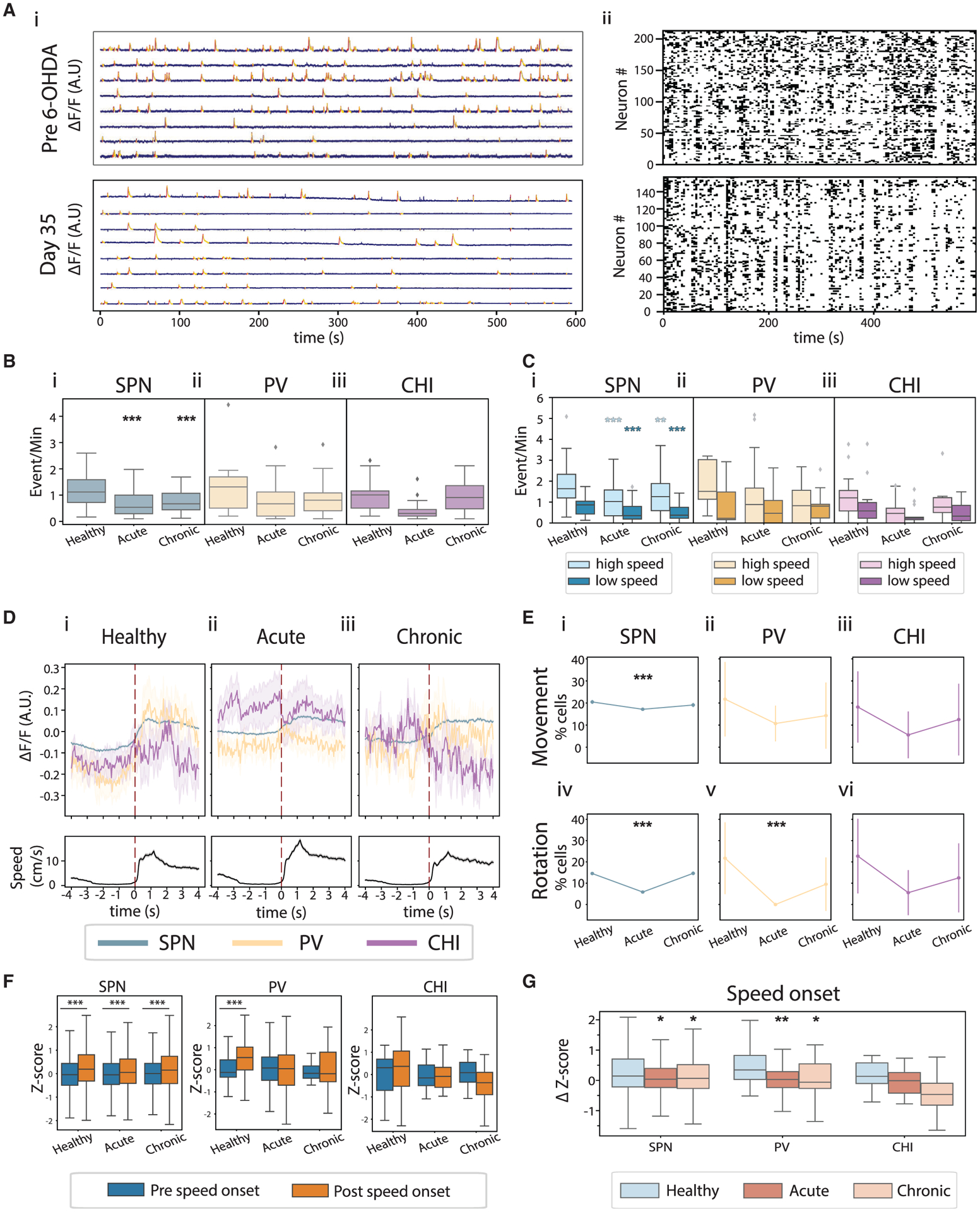
(A) Example GCaMP6 recordings before 6-OHDA infusion (top) and on day 30 after 6-OHDA infusion (bottom), from the same animal. (i) Example single-neuron GCaMP6 fluorescence traces (blue) with identified Ca2+ event rising (red) and falling phases (yellow) marked. (ii) Raster plots of binarized Ca2+ events from all neurons recorded in these sessions.
(B) Ca2+ event rates of SPNs (i), PVs (ii), and CHIs (iii) across conditions. SPN rates were significantly reduced in acute and chronic conditions (ANOVA, F(2,211) = 20.74, p = 6.05 × 10−9; mixed-effect model, acute versus healthy, t = −8.654, p = 1.69 × 10−15; chronic versus healthy, t = −5.767, p = 3.05 × 10−8). PV and CHI rates were not different across conditions (ANOVA for interaction of PV rate and conditions, F(2,80) = 2.035, p = 0.137; CHI rate and conditions, F(2,36) = 2.275, p = 0.117).
(C) Ca2+ event rates in high-speed and low-speed bouts for SPNs (i), PVs (ii), and CHIs (iii) across conditions. SPN rate was significantly decreased during high-speed and low-speed bouts (ANOVA for interaction of SPN rates during high-speed bouts and conditions, F(2,206) = 10.53, p = 4.41 × 10−5; mixed-effect model, acute versus healthy, t = −6.028, p = 7.92 × 10−9; chronic versus healthy, t = −3.149, p = 0.002. ANOVA for interaction of SPN rates during low-speed bouts and conditions, F(2,209) = 13.43 p = 3.25 × 10−6; mixed-effect model, acute versus healthy, t = −6.146, p = 4.26 × 10−9; chronic versus healthy, t = −4.664, p = 5.70 × 10−6).
(D) Population GCaMP6 fluorescence of SPNs (blue), PVs (yellow), and CHIs (purple) aligned with speed onset (top), and the corresponding movement speed (bottom).
(E) Percentage of speed- (top) and rotation- (bottom) responsive SPNs, PVs, and CHIs. Significantly fewer SPNs were movement responsive under the acute condition (Fisher’s exact test with Bonferroni correction for multiple comparisons: movement-responsive SPNs, 1,690/9,850 in acute versus 1,604/7,837 in healthy, p = 2.13 × 10−8; 1,248/6,539 in chronic versus 1,604/7,837 in healthy, p = 0.04; rotation-responsive SPNs, 577/9,850 in acute versus 1,140/7,837 in healthy, p = 1.39 × 10−83; 957/6,539 in chronic versus 1,140/7,837 in healthy, p = 0.887). Only rotation-responsive PVs were reduced after dopamine depletion (movement-responsive PVs, 6/56 in acute versus 5/23 in healthy, p = 0.282; 3/21 in chronic versus 5/23 in healthy, p = 0.701; rotation-responsive PVs, 0/56 in acute versus 5/23 in healthy, p = 0.001; 2/21 in chronic versus 5/23 in healthy, p = 0.416). There was no change in responsive CHIs (movement-responsive CHIs, 1/18 in acute versus 4/22 in healthy, p = 0.36; 2/16 in chronic versus 4/22 in healthy, p = 1; rotation-responsive CHIs, 1/18 in acute versus 5/22 in healthy, p = 0.197; 2/16 in chronic versus 5/22 in healthy, p = 0.675).
(F) Z scores of population responses during the 4-s period before versus after movement onset for SPNs (left), PVs (middle), and CHIs (right) (two-tailed paired t tests: SPN, healthy, t = 27.55, p = 1.08 × 10−159; acute, t = 18.19, p = 3.82 × 10−73; chronic, t = 18.94, p = 1.89 × 10−78; PV, healthy, t = 3.76, p = 6.80 × 10−4; acute, t = 1.39, p = 0.166; chronic, t = 1.35, p = 0.187; CHI, healthy, t = 1.015, p = 0.32; acute, t = 0.346, p = 0.732; chronic, t = 1.28, p = 0.212).
(G) Change in Z scores of population fluorescence of SPNs, PVs, and CHIs at speed onset (ANOVA: SPN, F(30,539,2) = 68.05, p = 3.25 × 10−30; PV, F(181,2) = 4.24, p = 4.89 × 10−4; CHI, F(83,2) = 1.55, p = 0.21. Tukey’s honest post hoc test: SPN, acute versus healthy, p = 0.001; chronic versus healthy, p = 0.001; PV, acute versus healthy, p = 0.001; chronic versus healthy, p = 0.043). Boxplot details are as in Figure 2. *p < 0.05, **p < 0.01, ***p < 0.001.
Dopamine depletion selectively reduces the locomotor encoding ability of SPNs and PVs, but not CHIs
To examine how dopamine depletion alters the activity of individual neurons, we expressed the fluorescent Ca2+ indicator GCaMP6 nonspecifically across SPNs and interneurons using AAV9-syn-GCaMP6f (n = 14 mice). PVs or CHIs were co-labeled with tdTomato using AAV9-CAG-flex-tdTomato in PV-cre (n = 6) or Chat-cre (n = 5) mice, respectively. Since ~95% of striatal neurons are SPNs (Tepper et al., 2010), all tdTomato-negative cells were considered putative SPNs. We simultaneously imaged SPNs and labeled interneurons throughout the study duration without observing substantial changes in the number of recorded neurons per session (healthy: SPN, 179.3 ± 22.8; CHI, 1.5 ± 0.4; PV, 1.8 ± 0.3; chronic: SPN, 152.1 ± 20.3; CHI, 1.1 ± 0.2; PV, 1.0 ± 0.3; mean ± standard deviation).
Under the healthy condition, all three cell types had Ca2+ event rates of 1–1.4 events/min, with no difference between cell types (ANOVA, F(2,76) = 0.276, p = 0.759), similar to previous findings (Gritton et al., 2019; Tran et al., 2020; Rendón-Ochoa et al., 2018). Acute and chronic depletion decreased SPN event rates regardless of movement bouts (Figures 3B and 3C). This decrease in SPN activity occurred without movement speed changes (Figure 2B), suggesting that unaffected SPNs can compensate for the hypoactivity of the affected SPNs to maintain normal locomotion. In contrast, event rates for CHIs and PVs remained unaltered by dopamine loss across all movement conditions analyzed (Figures 3B and 3C).
Next, we quantified locomotor encoding ability of individual cells by comparing the event rate of each neuron during high- versus low-speed or rotation bouts. We found a significant fraction of movement- and rotation-responsive SPNs, PVs, and CHIs under the healthy condition (Figure 3E). Dopamine depletion significantly reduced the fraction of speed- and rotation-responsive SPNs (Figures 3Ei, and 3Eiv). In contrast, the only change in interneuron populations was a reduction in rotation-responsive PVs under the acute condition (Figures 3Eii, 3Eiii, 3Ev, and 3Evi). Thus, dynamic CHI locomotor encoding ability remains intact at the individual neuron level following dopamine loss, even though CHIs are critical in maintaining high striatal cholinergic tone and dopamine-acetylcholine imbalance is implicated in PD. Furthermore, we found that at the population level, PVs and SPNs were activated at movement and rotation onsets under the healthy condition (Figures 3D, 3F, and S3). While the SPN population remained responsive at onset transitions, the PV population lost its responses after dopamine loss (Figure 3F). In contrast, the CHI population showed little change around onset transitions due to the heterogeneity among individual CHIs, even though many individual CHIs were movement responsive (Figures 3E, 3F, and S3). Together, our results demonstrate that dopamine depletion led to a profound reduction in SPN locomotor encoding ability for both sustained locomotor bouts and onset transitions. In contrast, PVs exhibited some transient impairment under the acute condition only, whereas CHIs remained largely unaltered (Table S1).
Striatal LFP beta, low-gamma, and high-gamma oscillations support distinct locomotor activity, and dopamine loss selectively promotes beta oscillations during impaired movement
Basal ganglia LFP oscillations are broadly associated with locomotion. Thus, we analyzed striatal LFPs recorded from electrodes near the imaging site. We found that beta (10–15 Hz) power was significantly higher during low-speed bouts compared with high-speed bouts and during no-rotation bouts compared with rotational bouts across all conditions (Figures 4B–4D and S4A). In contrast, low-gamma (40–60 Hz) and high-gamma (60–100 Hz) oscillations were significantly stronger during active movement states, exhibiting higher power in high-speed bouts than in low-speed bouts (Figures 4E and 4F) and in rotational bouts than in no-rotation bouts (Figures S4B and S4C). These results confirm the distinct roles of striatal oscillations during locomotion, where beta oscillations promote immobility and gamma oscillations support heightened movement, as in previous studies (Miller et al., 2007; Lemaire et al., 2012). Since localized dopamine depletion produced specific behavioral impairment on movement initiation and rotation (Figures 2C and 2D), we further characterized beta oscillations during different movement bouts. We found that beta oscillations were elevated only during the impaired contralateral-rotational bouts, but not during other unimpaired-movement bouts (Figure 4C). Thus, dopamine depletion selectively augments striatal beta oscillations during sustained locomotor bouts that were impaired, highlighting that pathological beta oscillations are present in the striatum even after mild focal dopamine depletion.
Figure 4. Locomotion differentially modulated striatal LFP oscillations, and dopamine depletion selectively augmented beta oscillations during impaired locomotion bouts.
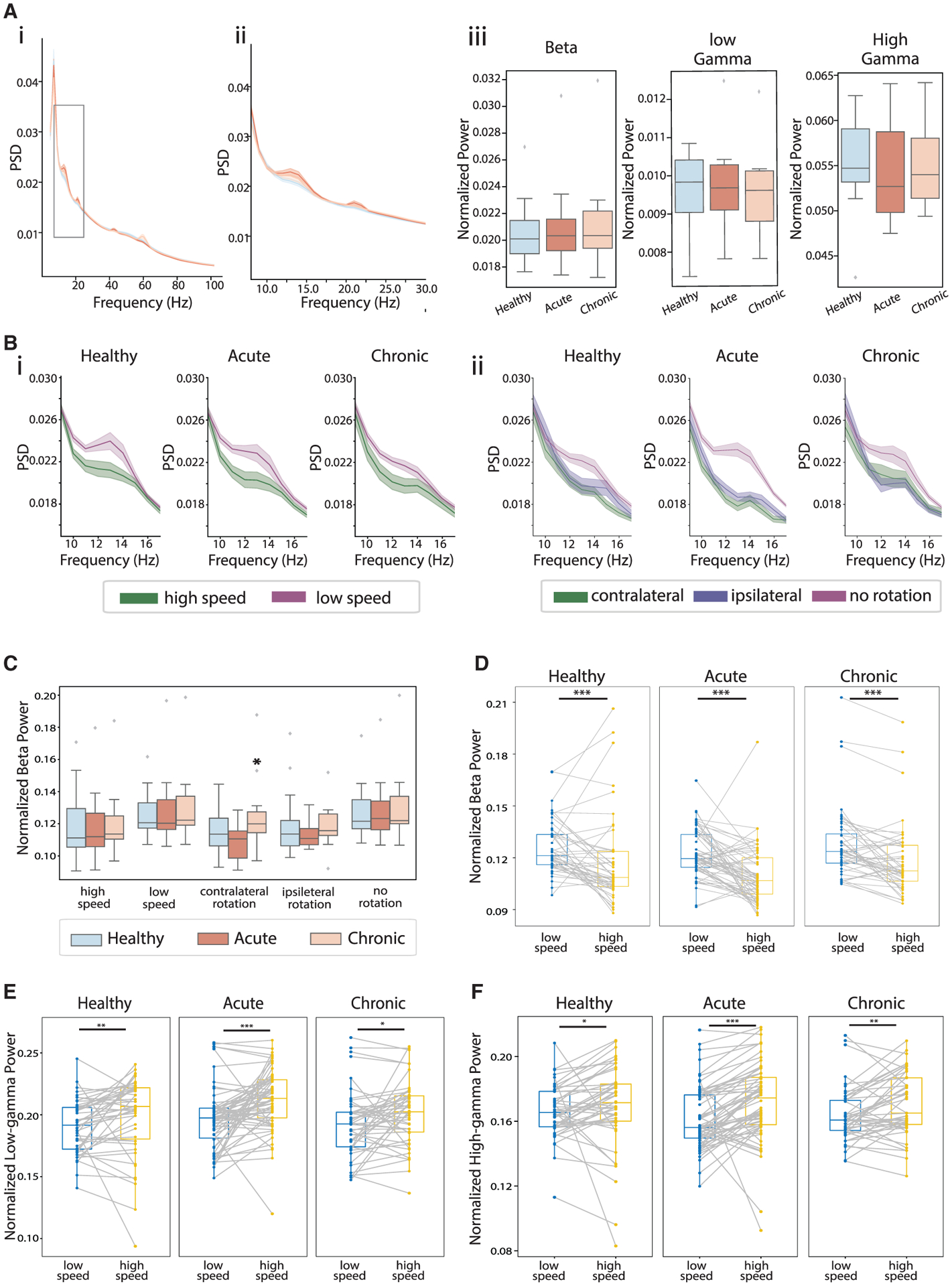
(A) Mean LFP power spectrum density (PSD) across sessions in healthy (blue), acute (dark orange), and chronic (light orange) conditions (i) and zoom-in view around beta (10–15 Hz) frequencies (ii). LFP power at beta, low-gamma (40–60Hz), and high-gamma (60–100) frequencies (iii). There was no significant differences across conditions (ANOVA for interaction of beta power and conditions, F(2,210), p = 0.360; low-gamma power and conditions, F(2,210), p = 0.419; high-gamma power and conditions, F(2,210), p = 0.222).
(B) LFP PSD at the beta frequencies in high-speed (green) and low-speed bouts (purple), across conditions (i). Same as (i), but during contralateral-rotation bouts (green), ipsilateral-rotation bouts (blue), and no-rotation bouts (purple).
(C) Beta power during different movement bouts. The only significant difference was the increase in beta power during contralateral rotation under the chronic condition (ANOVA, F(2,159) = 3.698, p = 0.027; mixed-effect model, acute versus healthy, t = −0.344, p = 0.732; chronic versus healthy, t = 2.014, p = 0.045).
(D) Beta power during low-speed (blue) versus high-speed bouts (yellow) across sessions in healthy (left), acute (middle), and chronic conditions (right) (Wilcoxon signed-rank test: high-speed versus low-speed bouts, healthy, p = 7.6 × 10−4; acute, p = 5.7 × 10−6; chronic, p = 2.3 × 10−5).
(E) Same as (D) but for low-gamma power (healthy, p = 0.006; acute, p = 2.8 × 10−5; chronic, p = 0.028).
(F) Same as (D) but for high-gamma power (healthy, p = 0.021; acute, p = 5.7 × 10−6; chronic, p = 0.006). *p < 0.05, **p < 0.01, ***p < 0.001.
Dopamine loss enhances correlated activity within the SPN network, and between SPN and CHI networks, but not between SPN and PV networks
To understand how dopamine depletion alters SPN, PV, and CHI circuit interactions, we quantified SPN-SPN and SPN-interneuron pairwise co-activity by calculating the Pearson correlation coefficient (PCC). To determine whether an observed PCC between a neuron pair is significantly higher than that expected from random overlap of Ca2+ events in that neuron pair, we estimated the shuffled distribution of PCC by assigning random delays between the two Ca2+ event traces. Neuron pairs with PCC significantly greater than expected from their corresponding shuffled distributions were deemed significantly correlated (“correlated pairs”), and those otherwise were considered randomly correlated (“random pairs”).
We found a gradual increase in correlated SPN-SPN pairs after dopamine loss, which was significant under the chronic condition (Figures 5A and 5Bi). This increase in correlated SPN-SPN pairs was accompanied by augmented PCC values between correlated pairs (Figure 5Di) and an overall increase in PCC across all pairs (Figure 5Ci). In contrast, under the acute condition, even though the overall PCC across all SPN-SPN pairs was unaltered, the PCC between random pairs decreased (Figure 5Dii), and that between correlated pairs increased (Figure 5Di). In theory, PCC may increase with increasing event rates, since more events lead to an increased probability of event overlap. The reduction in individual SPN event rates (Figures 3Bi and 3Ci) under the acute condition thus may contribute to the drop in PCC between random SPN pairs but not the increase in PCC between correlated pairs. Further examination of correlation changes during different movement bouts revealed that the increase in correlated SPN-SPN pairs and the increase in PCC between correlated pairs occurred during immobility, in either low-speed or no-rotation bouts (Figures S5C, S5E, and S5G). Thus, SPN populations exhibit abnormally elevated correlated activity specific to immobility, which may relate to the movement initiation deficits observed (Figures 2Ci and 2Di).
Figure 5. Dopamine loss enhanced correlated activity within the SPN circuit and between SPN and CHI circuits, but not between SPN and PV circuits.
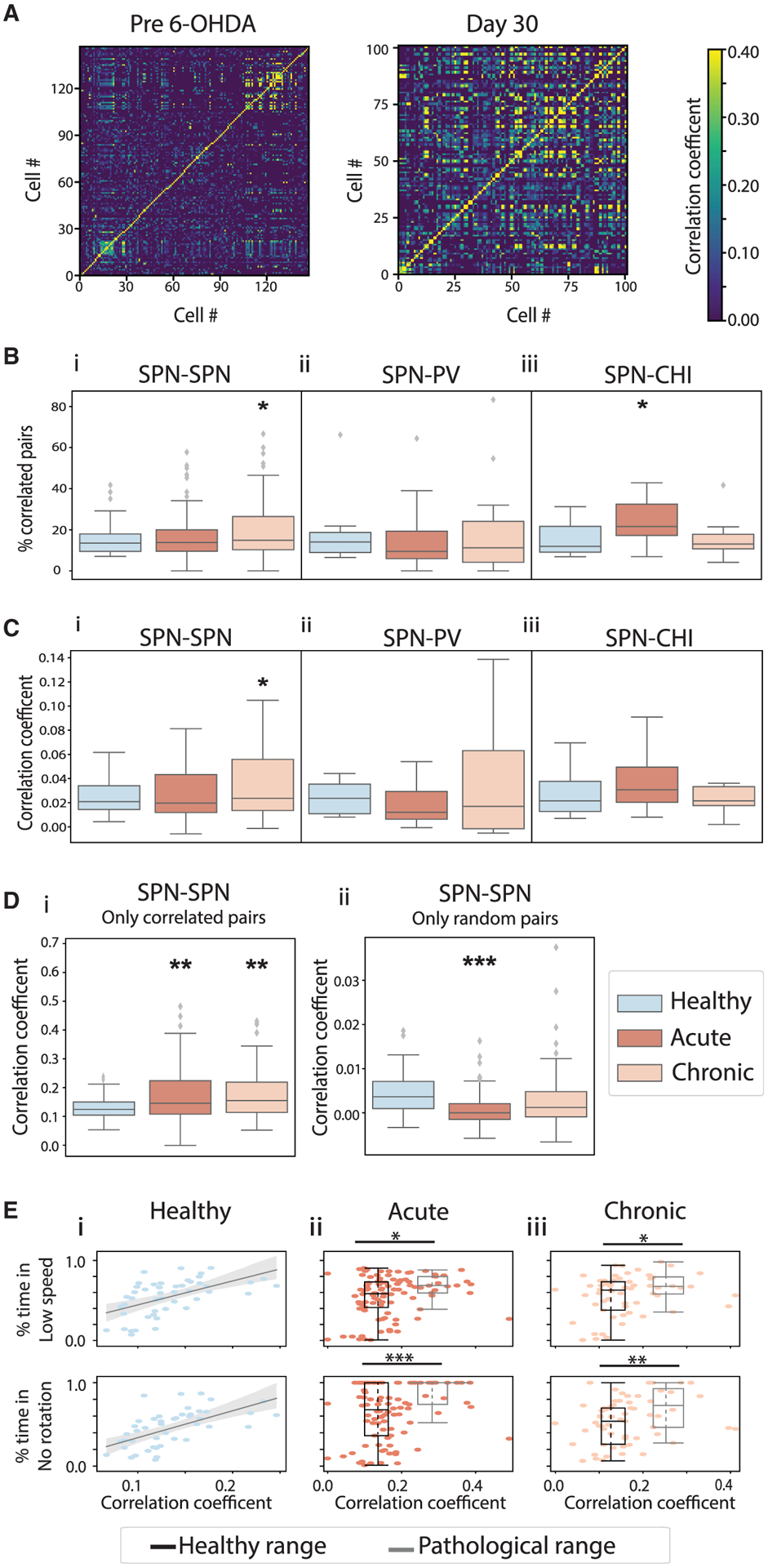
(A) An example SPN-SPN pairwise correlation heatmap from recording sessions before 6-OHDA lesion (left) and on day 30 (right), from the same animal.
(B) Percentage of correlated SPN-SPN pairs (i), SPN-PV pairs (ii), and SPN-CHI pairs (iii) across conditions. Correlated SPN-SPN pairs increased under the chronic condition (ANOVA, F(2,208) = 3.033, p = 0.050; mixed-effect model, acute versus healthy, t = 0.410, p = 0.683; chronic versus healthy, t = 2.156, p = 0.032). Correlated SPN-CHI pairs increased under the acute condition (ANOVA, F(2,38) = 3.532, p = 0.039; mixed-effect model, acute versus healthy, t = 2.325, p = 0.026; chronic versus healthy, t = −0.045, p = 0.964).
(C) Pearson correlation coefficient (PCC) of all SPN-SPN pairs (i), SPN-PV pairs (ii), and SPN-CHI pairs (iii) across conditions. PCC of all SPN-SPN pairs increased under the chronic condition (ANOVA, F(2,208) = 3.283, p = 0.040; mixed-effect model, acute versus healthy, t = 1.130, p = 0.260; chronic versus healthy, t = 2.505, p = 0.013).
(D) PCC of correlated SPN-SPN pairs (i) and random SPN-SPN pairs (ii). PCC of correlated SPN-SPN pairs increased for both acute and chronic conditions (ANOVA, F(2,204) = 5.395, p = 0.005; mixed-effect model, acute versus healthy, t = 2.995, p = 0.003; chronic versus healthy, t = 3.097, p = 0.002). PCC of random SPN-SPN pairs decreased under the acute condition (ANOVA, F(2,208) = 9.245, p = 1.42 × 10−4; mixed-effect model, acute versus healthy, t = −4.097, p = 6.03 × 10−5; chronic versus healthy, t = −1.099, p = 0.273).
(E) The fraction of time animals spent in low-speed (top) or low-rotation bouts (bottom) versus mean PCC of correlated SPN-SPN pairs across sessions in healthy (i), acute (ii), and chronic (iii) conditions. The gray lines in (i) correspond to linear regression. Boxplots in (ii) and (iii) correspond to sessions with PCC within that observed under healthy conditions (healthy range, black) or higher (pathological range, gray). Individual session results (dots) are plotted on top of the corresponding boxplots representing the median (middle dotted lines) and the 25th (Q1, bottom line) and 75th (Q3, top line) percentiles of PCC (x axis) and the fraction of time (y axis). Animals spent significantly more time in low-speed and no-rotation bouts in sessions with SPN-SPN PCC in the pathological range (Mann-Whitney U test: acute, low-speed, U = 630.0, p = 0.013; no rotation, U = 519.0, p = 8.1 × 10−4; chronic, low-speed, U = 259.0, p = 0.049; no rotation, U = 213.5.0, p = 0.008). Boxplots are as detailed for Figure 2. *p < 0.05, **p < 0.01, ***p < 0.001.
To further confirm the role of SPN-SPN correlation in immobility, we plotted the fraction of time animals spent in low-speed and no-rotation bouts in a recording session versus the session-wise mean PCC across correlated SPN-SPN pairs (Figure 5E). Under the healthy condition, SPN-SPN PCCs (0.13 ± 0.04, mean ± standard deviation, n = 48 sessions) are significantly correlated with the fraction of time animals spent in low-speed bouts (correlation, C = −0.50, p = 0.0002). After dopamine loss, we noticed that sessions with abnormally high SPN-SPN correlations often exhibited profound immobility. Thus, we separated sessions after dopamine depletion into those with PCC within the 2 standard deviations of that observed under the healthy condition (healthy range <0.21, n = 72 sessions under acute and n = 45 under chronic condition) and those with abnormally higher PCC (pathological range >0.21, n = 25 sessions under acute and n = 16 under chronic condition). We found that in sessions with abnormally high SPN-SPN PCC, animals spent significantly more time in immobility, confirming that abnormal increase in SPN-SPN correlation accompanies immobility following dopamine depletion (Figure 5Eii and 5Eiii).
When we considered SPN-interneuron co-activity, we found a significant increase in the proportion of correlated SPN-CHI pairs under the acute condition, but not the chronic condition (Figure 5Biii). Even though the overall PCC for SPN-CHI populations remained constant (Figure S5A), the SPN-CHI correlation increased during high-speed bouts under the acute condition (Figure S5D). Since individual CHIs did not exhibit any change in locomotor encoding after dopamine loss (Figures 3D–3F, Table S1), the strengthened SPN-CHI coordination under the acute condition likely reflects compensatory effects. In contrast, SPN-PV co-activity remained constant, showing no change in the fraction of correlated SPN-PV pairs (Figure 5Bii) or their PCC across conditions (Figures S5A and S5B) or movement bouts (Figure S5F). It is interesting that individual PVs reduced their movement encoding ability (Figures 3D–3F, Table S1), but their coordination with SPNs remained unaltered after dopamine depletion. Together, these results demonstrate that following dopamine loss, the SPN network exhibits abnormally high coordination that parallels the specific movement impairment observed, whereas changes in SPN-interneuron network coordination are independent of the interneurons’ movement encoding ability.
Dopamine loss leads to transient pathological LFP high-gamma oscillations during SPN and PV activation, but not CHI activation
Exaggerated cortical-basal ganglia beta oscillations are a functional biomarker for PD (Wingeier et al., 2006), and recent studies also suggested a role for cortical high-gamma oscillations in PD (Swann et al., 2016). To examine how striatal LFPs relate to striatal neural activities, we aligned the LFP power spectrum to movement onset or Ca2+ event onsets (Figure 6A). Transient increases in beta oscillations have been suggested to be important for movement transitions (Leventhal et al., 2012). However, dopamine depletion did not affect beta power around movement onset even though onset transitions were impaired (Figures 6Ai, S6A, and S6B). It is possible that pathological changes in beta power exist around intended transitions, which were missed, as we can detect only successful transitions. It is also possible that transient beta oscillations around transitions are less sensitive to localized dopamine depletion, or beta oscillations may not be as tightly time locked to onset transitions.
Figure 6. Dopamine loss led to a transient increase in LFP high-gamma oscillations during SPN and PV activation, but not CHI activation.
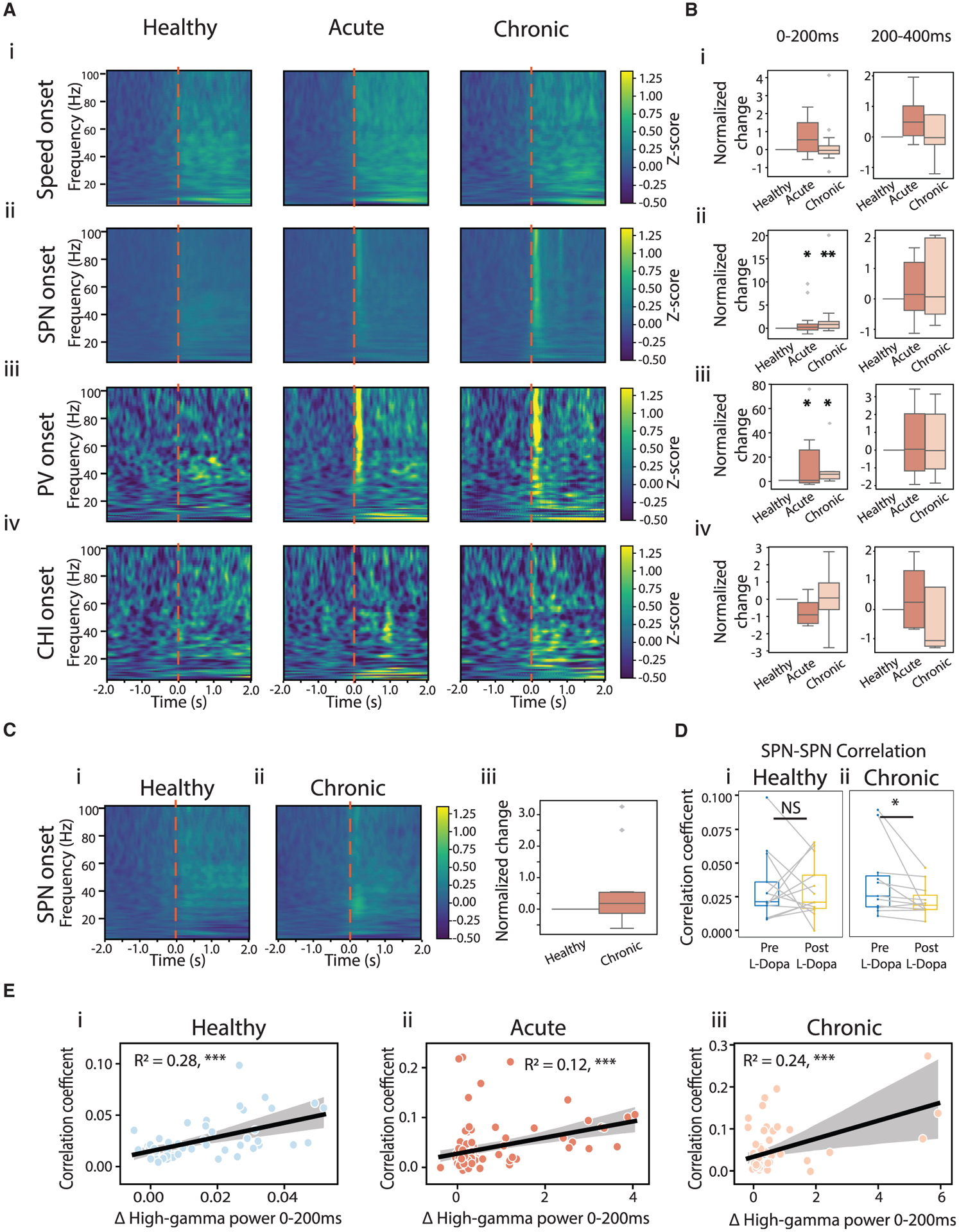
(A) LFP power spectrum aligned to movement onset (i), SPN Ca2+ event onset (ii), PV event onset (iii), and CHI event onset (iv) across conditions. Shown are Z scored power spectra, normalized across all events in all mice.
(B) Changes in high-gamma power (60–100 Hz) in the 0–200 ms (left) and 200–400 ms (right) windows following the corresponding event onsets, normalized to the change observed under the healthy condition (ANOVA for interaction of high-gamma power following SPN onset and conditions: F(2,206) = 4.961, p = 0.007; mixed-effect model, acute versus healthy, t = 2.168, p = 0.031; chronic versus healthy, t = 3.139, p = 0.002. ANOVA for high-gamma power following PV onset and conditions: F(2,53) = 4.021, p = 0.024; mixed-effect model, acute versus healthy, t = 2.547, p = 0.014; chronic versus healthy, t = 2.58, p = 0.013).
(C) LFP power spectrum aligned to SPN event onset post L-Dopa administration under healthy (i) and chronic conditions (ii), and changes in high-gamma power within 0–200 ms of SPN event onset (iii). No difference was observed (ANOVA, F(1,88), p = 0.085).
(D) PCC across all SPN-SPN pairs before (blue) and after (yellow) L-Dopa administration, under healthy (left) and chronic conditions (right). L-Dopa significantly decreased PCC of SPN-SPN pairs under the chronic condition (Wilcoxon signed-rank, p = 0.014), but not under the healthy condition (p = 1).
(E) High-gamma oscillation power within 0–200 ms of SPN event onsets versus PCC of correlated SPN-SPN pairs across sessions. Black lines indicate linear regression. *p < 0.05, **p < 0.01, ***p < 0.001.
We found that after dopamine loss, SPN Ca2+ events were associated with a prominent increase in high-gamma power, but not beta power or low-gamma power (Figures 6Aii and S6B). This increase in high-gamma power was transient, restricted to the 200 ms after SPN event onset, but not the subsequent 200–400 ms time window (Figures 6Aii, Bii, and S6C). Increase in neural activity is generally associated with increased higher frequency oscillations, and indeed we detected a small increase in high-gamma oscillations at SPN event onset under healthy conditions (Figure S6Di). Thus, to test whether the transient exaggeration of high-gamma oscillations during SPN activation relates to abnormal coordination between SPNs (Figure 5), we performed a linear regression analysis, and found that high-gamma power during SPN activation was significantly correlated with the PCC between correlated SPN-SPN pairs across sessions under all conditions (Figure 6E). While high-gamma power also increased within 200 ms of speed onset, this movement-related high-gamma oscillation was insensitive to 6-OHDA injury (Figure 6Bi), and thus reflects a physiological circuit state. Furthermore, since the movement-related high-gamma oscillations also paralleled the general increase in striatal neural activities at onset transitions (Figures 3C–3F), this observation is also consistent with the 6-OHDA insensitive elevation of physiological high-gamma oscillations during high-speed and high-rotation bouts (Figures 4F and S4C). In contrast, the exaggerated transient high-gamma oscillations during SPN activation capture a pathological circuit state following dopamine depletion.
To further confirm that exaggerated SPN coordination and the related LFP transient high-gamma oscillations are due to dopamine deletion, we administered levodopa (L-Dopa) systemically during one healthy session and two chronic sessions (on days 19 and 35) to elevate striatal dopamine levels. Consistent with the focal lesion, we did not detect significant changes in the animals’ overall locomotor behavior after L-Dopa administration. Animals spent similar amount of time in high-speed and high-rotation bouts during healthy and chronic conditions (Wilcoxon signed-rank test: high-speed, healthy, p = 0.146; chronic, p = 0.074; high-rotation, healthy, p = 0.068; chronic, p = 0.176). Despite a lack of effect on behavior, we found that L-Dopa replacement significantly reduced SPN-SPN correlation under the chronic condition (Figure 6Dii) but not the healthy condition (Figure 6Di). In addition, L-Dopa eliminated the exaggerated high-gamma oscillation transients associated with SPN activation under the chronic condition (Figure 6Cii), without affecting high-gamma power under the healthy condition (Figure 6Ci). These results further confirm that dopamine loss promotes abnormal correlation within the SPN network and leads to transient pathological high-gamma oscillations during SPN activation (Table S1).
When we examined LFP power around interneuron activation, we detected a transient increase in high-gamma oscillations around PV activation like that around SPN activation, but not around CHI activation (Figures 6Aiii–6Aiv and 6Biii–6Biv). The selective association of pathological high-gamma oscillation transients with PV activation is consistent with the general role of PVs in contributing to high-frequency oscillations in the basal ganglia (van der Meer, 2010; van der Meer and Redish, 2009). It is interesting that we did not detect an increase in PV-SPN coordination following dopamine loss (Figures 5Bii and 5Cii), even though Ca2+ events in both cell types are accompanied by abnormal high-gamma oscillation transients. This suggests that although both SPNs and PVs contribute to the pathological high-gamma oscillations, their contribution is independent of their coordination. While CHI and SPN circuits exhibit increased coordination under the acute condition (Figure 5Biii), the absence of high-gamma power change around CHI activation suggests that CHIs have minimal involvement in the mechanisms of pathological high-gamma oscillation transients. Together, these results demonstrate that interneurons are differentially related to high-gamma oscillation transients, and the exaggeration of high-gamma oscillations during SPN activation captures a pathological striatal circuit state that accompanies an abnormal increase in SPN coordination following dopamine loss.
DISCUSSION
Dopamine degeneration in PD alters the neural circuit function of the dorsal striatum, leading to dysregulated striatal outputs, dopamine-acetylcholine chemical imbalance, and pathological LFP oscillations (Obeso et al., 2000; DeLong, 1990). To understand how chemical imbalance and LFP dynamics relate to dysregulated striatal outputs, we performed simultaneous Ca2+ imaging from individual SPNs in conjunction with PV or CHI interneurons, while monitoring striatal LFPs and locomotor behavior. We found that both acute and chronic dopamine loss reduces the locomotor encoding ability of SPNs and PVs, but not CHIs. At the circuit level, we detected a pathological elevation of correlated activity within SPN circuits under both acute and chronic conditions, and a transient increase in SPN-CHI coordination under the acute condition only. Interestingly, SPN-PV coordination remains largely unaltered, although both SPN and PV activation is accompanied by exaggerated LFP high-gamma oscillation transients. These results provided direct evidence of the differential roles of PVs and CHIs in regulating striatal circuit dynamics following dopamine loss and identified a pathological striatal circuit state involving abnormal coordination of SPNs and pathological LFP high-gamma oscillations.
We induced localized dopamine depletion through a focal striatal 6-OHDA infusion, which selectively impaired movement initiation and rotational behavior, without altering movement duration or speed. Animals showed significantly fewer voluntary onset transitions for both linear and rotational movement, suggesting that transitions are more sensitive to dopamine depletion than sustained movement. This observation is consistent with the more prominent transition difficulty noted in PD patients, where PD patients struggle with movement initiation, but once initiated they have less difficulty maintaining ongoing movement (Müller et al., 1997). Our previous work revealed that under healthy conditions, SPN, PV, and CHI population activity is modulated by movement, although individual cells exhibit heterogeneity in locomotor encoding (Gritton et al., 2019). Here, we extended such analysis to dopamine-depletion conditions, and discovered that SPNs, but not PVs or CHIs, exhibit a profound reduction in locomotor encoding ability following dopamine loss, despite animals not showing significant movement deficits.
At the circuit level, we observed elevated correlations within the SPN network following dopamine loss, which is accompanied by exaggerated LFP high-gamma oscillations that occur within 200 ms of SPN activation. Dopamine replacement with systemic L-Dopa administration reversed both the abnormal coordination of SPNs and the pathological high-gamma oscillation transients. Interestingly, SPN-SPN correlations occurred primarily during immobility and were negatively correlated with movement speed and rotation, during both healthy and dopamine-depleted conditions, suggesting a role for synchronized SPN-SPN activation in movement inhibition. The role of SPN-SPN correlations in movement inhibition observed here is distinct from the movement-promoting effect of individual SPN activation that is not accompanied by extensive synchronization with other SPNs. It is possible that dopamine depletion exaggerates the synchronization of the SPN network that naturally occurs during immobility, leading to more frequent occurrence of movement-inhibiting network states that result in akinesia. While high-gamma power also increased after movement onset, this movement-related high-gamma oscillation was insensitive to dopamine depletion, similar to the 6-OHDA-insensitive high-gamma oscillations during high-speed and high-rotation bouts. Thus, the movement-related high-gamma oscillations reflect a physiological circuit state where individual SPN neuron activity is high, but synchronization between SPNs is low. In contrast, the pathological high-gamma oscillations during SPN activation following dopamine depletion represent a circuit state where SPNs are pathologically coordinated, biasing the striatal network toward movement inhibition.
A previous study showed that dopamine depletion increased D1-SPN co-activity, but reduced D2-SPN co-activity during heightened movement states (Parker et al., 2018). We did not detect a significant change in SPN-SPN co-activity during high-speed or rotational movement (Figure S5), likely because of the opposing changes in D1- versus D2-SPNs. Future studies that selectively examine D1- versus D2-SPNs will provide insights on how D1- and D2-SPN networks are differentially engaged in different aspects of movement. Previous computational studies also linked D2-SPN activation to exaggerated striatal beta oscillations (McCarthy et al., 2011). It will be interesting for future studies to probe the relationship between instantaneous SPN-SPN co-activity and LFP oscillations using techniques with better temporal resolution, such as voltage imaging.
We detected a significant increase in CHI-SPN coordination during high-speed bouts under the acute condition, a movement aspect that was not altered by 6-OHDA injury. This suggests that CHI and SPN network remodeling following dopamine loss helps maintain movement, consistent with the role of CHIs in promoting movement completion through coordinating SPN networks (Gritton et al., 2019). In contrast, we did not detect any change in SPN-PV correlation, suggesting that dopamine depletion does not broadly alter coordination between SPN and PV networks, although we cannot rule out differential changes in PV interactions with D1- versus D2-SPNs as previously noted (Gittis et al., 2011; Salin et al., 2009). Since our correlation measure is biased toward positive correlations between Ca2+ events, it is also possible that our analysis is not sensitive enough to detect changes in feedforward inhibition of PVs to SPNs (Tepper et al., 2008).
Sustained elevation of beta oscillations is increasingly considered a biomarker for PD (Wingeier et al., 2006). Since our 6-OHDA lesions were focal, leaving most of the striatal dopamine signaling intact, we did not detect an overall increase in beta oscillations, consistent with the idea that global pathological beta oscillations likely emerge after widespread dopamine degeneration. However, we detected a selective increase in beta power during contralateral rotations, the only sustained locomotor behavior that was impaired. Our focal 6-OHDA injury method also impaired movement transitions. But beta oscillations are known to temporarily increase during transitions (Howe et al., 2011), making it difficult to assess beta power changes around transitions. Thus, our data provide direct experimental support that mild focal dopamine depletion results in pathological striatal beta oscillations that are specific to impaired movement. While low-gamma and high-gamma power increased during movement, dopamine depletion did not alter movement-related gamma oscillations, again likely due to the focal injury method used. However, we found transiently exaggerated high-gamma oscillations that were selectively associated with SPN and PV activation, but not CHI activation following 6-OHDA injury, which was eliminated upon dopamine replacement with L-Dopa. These pathologically high gamma oscillations are correlated with SPN-SPN co-activity, suggesting that pathological high-gamma oscillation transients are associated with disrupted striatal output network that is independent of changes in dynamic cholinergic signaling.
Limitations of the study
Ca2+ imaging measures changes in intracellular Ca2+, and Ca2+ events most likely capture spike bursting (Huang et al., 2021). This may contribute to the failure in detecting changes in CHI locomotor encoding ability following dopamine loss. However, previous electrophysiology studies in primates also reported that CHI firing rates were unaltered or decreased after dopamine depletion (Aosaki et al. 1994; Raz et al., 1996). Further studies with more sensitive techniques will be necessary to further differentiate CHI’s tonic versus dynamic influence on striatal circuit function and how tonic versus dynamic cholinergic signaling is altered in PD. In addition, due to the poor temporal resolution of Ca2+ imaging, we were unable to capture instantaneous changes in cellular dynamics associated with the transient pathological LFP high-gamma oscillations. Finally, our study does not distinguish D1- and D2-SPNs, nor can we simultaneously measure from many individual PV or CHI. Future studies are necessary to examine specific changes within PV or CHI networks, or between interneuron and D1- versus D2-SPN networks.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Xue Han (xuehan@bu.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
All original code has been deposited at GitHub https://github.com/HanLabBU/Dopamine-Depletion-CellReports/releases/tag/v1.0 and Zenodo https://doi.org/10.5281/zenodo.5786809 and is publicly available as of the date of publication.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Adult GM24Gsat (Chat-cre, MMRRC Stock Number: 017269-UCD), B6; 129P2-Pvalbtm1 (cre)Arbr/J (PV-cre, Jax Stock Number: 017320) and C57B6 mice (Charles River Laboratories, Cat# C57BL/6) were purchased from Charles River Laboratories, MMRRC, Jackson Laboratories or bred in-house. Animals were house in a temperature and humidity-controlled environment with a 12-h light-dark cycle (lights on at 7 am to 7 pm). Both female and male mice were used, and all mice were 8–12 weeks old at the start of the study. All animal procedures used in this study were approved by Boston University Institutional Animal Care and Use Committee (IACUC).
METHOD DETAILS
Surgical preparation
All animal procedures used in this study were approved by Boston University Institutional Animal Care and Use Committee (IACUC). Adult GM24Gsat (Chat-cre, MMRRC Stock Number: 017269-UCD, n = 7) were used to examine CHIs and their interactions with SPNs before 6-OHDA lesion, and 5 of the 7 mice that survived 6-OHDA lesions were used to analyze the effect post 6-OHDA injection. B6; 129P2-Pvalbtm1 (cre)Arbr/J (PV-cre, Jax Stock Number: 017320, n = 6) transgenic mice were used to examine PVs and their interactions with SPNs. These 13 transgenic mice, and another 3 C57BL/6 mice were used to examine SPNs. All mice were 8–12 weeks old at the start of the study. Under isoflurane anesthesia, mice were surgically implanted with a custom imaging window attached with a drug infusion cannula to permit AAV and 6-OHDA delivery, and a stainless steel LFP electrode (P1 Technologies, 7N003736501F) with the tip positioned at about 200um below the imaging window, and above the virus injection site, as described previously (Gritton et al., 2019). Imaging windows were positioned above the dorsal striatum, centered at (AP: +0.5 mm, ML: 1.8 mm, and DV: 1.6 mm from brain surface), with the overlying cortical tissue removed. In the same surgery, a metal pin serving as LFP ground was implanted on the skull of the contralateral hemisphere above the occipital cortex, and a head fixation bar was also implanted. Buprenorphine was administered as post-operative analgesic. About 2 weeks after the surgery, 1 μL cocktail containing 500nL AAV9-Syn-GCaMP6f.WPRE.SV40 (Addgene, titer 6.6 × 1012 GC ml−1) and 500nL AAV9-CAG-flex-tdTomato.WPRE.SV40 virus (UNC vector core, 5.9 × 1012GC ml−1) was infused into the dorsal striatum through the infusion cannula.
Habituation
Upon recovery from surgery, animals were first habituated, for at least 10 sessions over a period of 2–3 weeks, before the first imaging session. During habituation, mice were head-fixed on the spherical treadmill under the microscope with the LED on, up to 5 days a week for increasingly longer periods (5 min–1 h per day).
Imaging set-up and data acquisition
Animals were head-fixed under the microscope and imaged on a spherical treadmill that permits voluntary movement using a setup as descried in Gritton et al.(Gritton et al., 2019). Briefly, we used a custom wide field microscope equipped with ORCA-Flash4.0 LT Digital sCMOS camera (No. C11440-42U, Hamamatsu). 5W light-emitting diode (No. LZ1-00B200, 460 nm; LedEngin) was used for GCaMP6f excitation, and 1,000 mA light-emitting diode (No. LXML-PX02-0000, 567 nm, Lumileds) was used for tdTomato excitation. Prior to each GCaMP6f imaging session, we recorded a tdTomato fluorescence video for 10s, 200ms per frame exposure time. This tdtomato fluorescence video was used to identify CHI or PV interneurons during data analysis. GCaMP6 and tdTomato fluorescence was imaged at approximately 20Hz with a filter set containing a dichroic mirror (Semrock, FF493/574-Di01-25×36), a dual-band emission filter (Semrock, FF01-512/630-25), and a dual-band excitation filter (Semrock, FF01-468/553-25).
LFPs were recorded via a Tucker Davis Technologies (TDT) multichannel recording system (RZ5D). LFPs were recorded at 3051.75 Hz, with a band-pass filter of 1–1000Hz. The spherical treadmill was composed of a custom plastic housing, a Styrofoam ball supported by air, and two motion sensors. The motion sensors were mounted 3–4mm away from the ball and 78°C apart. Motion sensor data was collected via a custom MATLAB ViRMEen environment on a separate computer used for calcium imaging data acquisition. A custom multi-threaded Python script was used to send packaged < dx,dy > motion sensor data at 100 Hz to the image acquisition computer via a RS232 serial link. A custom MATLAB script was used to extract the time stamps of the calcium imaging frame captured with HC image Live (HC Image Live; Hamamatsu), the motion sensor data captured via the MATLAB ViRMEen environment, and the LFP data recorded via the TDT system, to allow for offline alignment of these different signals.
Study timeline and 6-OHDA injection
Each calcium imaging session was 10 min long. We first performed three healthy control imaging sessions, 5–7 days apart, starting 15 days prior to 6-OHDA infusion (day −15). One day after the last healthy session (day 0), mice received intracranial infusion of 5ug 6-OHDA (1ul, 5ug/ul) dissolved in 0.9% sterile saline containing 0.02% ascorbic acid through the infusion cannula, 30 min after systemic desipramine injection (I.P 25mg/kg) to limit 6-OHDA effects on non-dopaminergic neurons. Mice were then imaged every 1–2 days for two weeks after 6-OHDA infusion, and then again on days 15,19, 30, and 35. 14 of the 16 mice survived the 6-OHDA injection (Chat-Cre: n = 5; PV-Cre: n = 6; Wild Type: n = 3), resulting in a total of 214 imaging sessions for this study. As a control for acute injection procedures, we analyzed the effect of intracranial saline infusion in a group of 9 additional animals. Specifically, these mice were imaged as detailed above for 1–2 healthy baseline sessions, then infused with 1ul of saline instead of 6-OHDA, and then recorded for another 2–5 sessions over the 3–10 days period after saline infusion.
Systemic L-Dopa and amphetamine testing procedure
During one healthy session before 6-OHDA infusion, and on days 15 and 30 post 6-OHDA infusion, amphetamine induced rotational behavioral assays were performed on the spherical treadmill and in an open field to verify dopamine depletion. On these testing days, baseline locomotion was first recorded in the open field for 10 min without amphetamine administration, and then mice were placed on the spherical treadmill for a regular 10-minute-long calcium imaging recording session. After the calcium imaging session, animals received systemic amphetamine injection (IP, 5mg/kg) while on the spherical treadmill. Animals’ locomotor behavior on the spherical treadmill was then recorded for another 35 min. After the head fixed treadmill behavior assay, animals were placed back into the open field for an additional 10 min of locomotion recording. To observe the amphetamine effect, we analyzed the recording period starting 5 min post amphetamine administration. Locomotor recording on the spherical treadmill was analyzed as detailed below. Locomotor recording in the open field was analyzed with custom Python scripts, which identified the location of the mouse head and body and quantified the frequency of rotations.
During one healthy session before 6-OHDA infusion, and on days 19 and 35 after 6-OHDA infusion, we examined the effect of L-Dopa on striatal dynamics. The long intervals between the three L-Dopa injection sessions (16 days or longer between sessions) help avoid potential L-Dopa induced dyskinesia. In these recording sessions, we first recorded LFPs and calcium dynamics for 10 min, and then administered L-Dopa systematically (30mg/kg IP) while the animals were on the spherical treadmill. We then resumed recording of LFPs and calcium dynamics for another 35 min. To observe dopamine replacement effect on the striatum, we analyzed the recording period starting 5 min post L-Dopa administration.
6-OHDA lesion quantification and histology
Animals were perfused at the completion of the study, their brains extracted, post fixed in 4% paraformaldehyde, and sliced into 40μm horizontal slices. Dopamine depletion was assessed by analyzing immunofluorescence of tyrosine hydroxylase (TH) on 6–8 dorsal striatum slices within ~500 μm of the imaging sites from each animal. Striatal brain slices were stained using a Rabbit anti-TH antibody (Abcam, No. AB112, 1:250) followed by either Alexa Fluor 633 goat anti-rabbit secondary antibody (Invitrogen, No. A21070, 1:500), or Biotinylated goat anti-rabbit antibody (Vector PK-6101, 1:400). The stained slices were imaged using Olympus VS120 slide scanner microscope at 4× magnification to visualize tdTomato fluorescence or Alex Fluor 633 fluorescence, or DAB intensity in white field. Each image was then tiled, and the lesioned side was confirmed using the GCaMP6f fluorescence. Striatal TH staining intensity was calculated for both the lesioned hemisphere (THlesion) and the intact hemisphere (THhealthy). The percent reduction of dopamine innervation was defined as: (THlesion – THhelathy)/THhealthy. To quantify the specificity of tdTomato expression in PVs and CHIs, we used antibodies against PV (rabbit anti-PV, SWANT PV25, 1:1,000) or Chat (goat anti-Chat antibody, Abcam, No. ab254118 1:500), followed by either Alexa Fluor 633 goat anti-rabbit secondary antibody (Invitrogen, No. A21070, 1:1000) for PV staining or Alexa Fluor 633 donkey anti-goat secondary antibody (Thermo Fisher, No. A21082, 1:200) for Chat staining. Images were taken on an Olympus FV1,000 scanning confocal microscope using a 60 × water immersion lens (Figure S1).
QUANTIFICATION AND STATISTICAL ANALYSIS
Movement data pre-processing
Movement data was first pre-processed as detailed in Gritton et al. (Gritton et al., 2019). Briefly, we calculated liner and rotational velocity from the two motion sensors displacement readings. Linear velocity was calculated using Equation (1):
| (Equation 1) |
Where L is the left sensor’s reading, R is the right sensor’s reading, θ is the angle between the sensors (78°). V is the linear velocity and S is speed. Rotational velocity was calculated by converting the horizontal component of both sensors to radians/second using a calibration factor obtained by manually moving the Styrofoam ball, and then averaged between the two sensors. We then interpolated both the linear and rotational velocity traces to 0.05 s intervals to align with calcium imaging and LFP recordings.
Movement bouts identification
We first smoothed the movement speed trace by applying a 1 s (20 data points) moving average filter with a step size of 1 data point. We then identified high-speed bouts and low-speed bouts, based on movement speed using the following steps. To account for variability in movement speed between different mice, we first identified the mean maximum movement speed of each mouse during the three recording sessions before 6-OHDA infusion. High-speed bouts were defined as continuous time intervals when mice ran faster than 20% of their maximum speed during baseline sessions. The speed thresholds ranged from 1.96cm/s to 16.04cm/s across mice, with an average threshold of 7.15cm/s across all mice (7.15 ± 4.54, mean ± standard deviation, n = 16 mice). The average speed during the identified high-speed bouts was 16.97cm/s (16.97 ± 11.05, mean ± standard deviation, n = 16 mice). Low-speed bouts were defined as time intervals when mouse speed was continuously below 10% of their maximum speed during baseline sessions. The average speed for all mice during these low-speed bouts was 0.87 cm/s (0.87 ± 0.75, mean ± standard deviation, n = 16 mice).
We also defined three rotational bouts based on rotational velocity: ipsilateral rotational bouts, contralateral rotational bouts, and no rotation bouts. This was done in two steps. First, we defined high rotation and no rotation velocity bouts using the same criteria as described above but applied to the absolute values of the rotational velocity vectors. Then we further divided the rotational bouts into ipsilateral versus contralateral bouts based on rotational direction.
Movement onset identification
To locate the movement onset transition time point, we first determined the high-speed threshold for each mouse as 25% of the mean maximum speed for that mouse during the three baseline sessions. Movement onsets were then defined as the time points that satisfied all three following conditions:
The mouse speed at the onset point must be greater than 1cm/s
The mouse speed in the two seconds prior to the onset point must not exceed 3.3cm/s, or 40% of the high-speed threshold, whichever is lower.
The mouse speed must exceed the mouse’s high-speed threshold within 1s after the onset point.
Rotational onsets were defined using the same procedure and criteria but applied to the absolute value of the smooth rotational velocity trace. The thresholds used to define high rotational velocity were set to 5% of the mean maximum rotational velocity during the three recording sessions before 6-OHDA injection. After rotational onsets were identified, they were assigned to ipsilateral versus contralateral directions based on the direction at the onset point.
Calcium data pre-processing
GCaMP6f videos were first processed with homomorphic filtering, motion correction and background subtraction similar to that described in Gritton et al. (Gritton et al., 2019). Briefly, motion correction was done using cross correlation against reference frames. The reference frame was initialized with the first frame in the video and updated by adding each newly corrected frame to the reference. Background subtraction was applied in two steps. First, we calculated the minimum projection image by finding the minimum fluorescence value of each pixel over the entire video. The minimum projection image was then spatially smooth and subtracted from each frame. In the second step, we identified image areas with no cells by finding pixels whose intensity range was smaller than the mean intensity range over for all pixels, and then subtracted the mean of these pixels from all pixels in all frames.
Region of interest (ROI) identification and ΔF/F trace extraction for each ROI
After pre-processing of the calcium videos, ROIs were manually identified on a maximum minus minimum projection image of the entire video. Each ROI was defined as a circle with a radius of 6 pixels (7.8um), corresponding to a neuron. The mean fluorescence value of all pixels within each ROI in each frame was extracted and interpolated to 0.05s intervals between points. We then calculated ΔF/F for each ROI as (F-Fmean)/Fmean, where F is the fluorescence at each time point and Fmean is the mean fluorescence of the entire trace for that ROI.
To identify PV or CHI interneurons, we first motion corrected the tdTomato videos using the same algorithm as with the GCaMP6f videos, and then manually identified ROIs on the tdTomato maximum projection image of the entire video. Next, we automatically aligned each GCaMP6f ROI map with the corresponding tdTomato ROI map, using the transformation matrix determined by the MATLAB built in functions “estimateGeometricTransform” and “imwrap” that considered blood vessels as landmarks. We used a threshold of 70% overlapping in these algorithms to identify interneurons in GCaMP6f map based on the interneuron tdTomato ROI map. All automatically identified interneurons in GCaMP6f recordings were further manually inspected and confirmed.
Calcium event identification
To identify calcium events, we first applied a Butterworth band-pass filter between 0.2Hz and 9.9Hz to the ΔF/F traces, and then we calculated the spectrogram for each calcium trace using a multi-taper method with 3 tapers and a time band-with of 2. For the multi-taper calculation, we converted the “mtspecgramc” function and its dependency functions from the MATLAB “chronux” package (“Chronux Home”, n.d.) into Python. Through manual inspection we noted that calcium events are associated with increased low frequency (<2Hz) power, therefore, we calculated the “power trace” as the mean Z scoreZ-score of the power across frequencies below 2Hz. Then, we identified potential event rising locations as points where the derivative of the “power trace” is greater than its scaled median defined by Equation (2):
| (Equation 2) |
If two potential event rising locations were within 2 data points, the later one was discarded to avoid detecting the same event twice. We removed any data point where Δpower<mean (Δpower), which corresponds to decreases in low frequency power. We then processed all potential event rising locations to identify the final calcium events using the following steps. First, we identified potential event falling locations as the time point after each potential event rising location where the “power trace” decreased. Then, we identified the calcium event peak as the time points when the ΔF/F amplitude peaked between the potential rising and falling locations. Finally, we examined each data point backward in time from the peak to identify the calcium event onset as the first data point, where the ΔF/F amplitude exceeded 7 standard deviations of that during the two seconds prior. Calcium event rise time was defined as the time interval between the onset and the calcium event peak. To identify calcium event offset, we analyzed the time window between the data point after the calcium event peak, until the onset of the following calcium event onset or for 4 times of the calcium event’s rise time, whichever is shorter. We then determined the calcium event offset as the time point with the lowest ΔF/F amplitude within the examined window. Calcium event fall time was defined as the interval from the first data point after calcium event peak to the calcium event offset, including the event offset.
This event detection method does not work well when there are multiple calcium events in close proximity that resulted in increased standard deviation leading to the failure of finding event rising locations. Therefore, after going through the trace for the first round, we remove all identified calcium events, from the onset until the offset, by replacing them with NaNs. We then repeated the process to find additional calcium events using the same procedure as described above in this new iteration. The algorithm stops when an iteration does not identify any new calcium events. On the first iteration, we started with a threshold of 7 standard deviations, and then on every new iteration, we reduced the threshold by 10%. If more than 3 iterations were performed, we increased the window for calculating the standard deviation by 75% for each extra iteration to account for the increasing number of NaNs. Manual inspection confirmed that these criteria were able to detect most calcium events in our dataset.
Binarized calcium event vector construction and event rate analysis
Using the identified calcium events, we generated binarized calcium event vector by assigning ones to the rising phase of all calcium events and zeros elsewhere. Calcium event rates were calculated as the density of onset events in the binarized calcium event vector, either across individual sessions, or for different periods of interest.
Calcium event Pearson correlation calculation
Correlation coefficient between a neuron pair was estimated as the Pearson correlation of the two binarized calcium event vectors of the neuron pair. To determine whether a correlation coefficient value observed between a neuron pair was significantly higher than that expected from random overlap of calcium events in that neuron pair, we used a shuffling procedure to estimate the baseline distribution of correlation values. Specifically, we assigned random delays between the two calcium event vectors, and computed the corresponding correlation values during each shuffle. In each shuffle, we applied a circular shift to one of the event vectors by a random number of time points, and calculated the correlation coefficients. This procedure was repeated 1000 times to build the distribution of the shuffled baseline distribution. Neuron pairs with correlation values greater than 95% of the shuffled distribution were deemed significantly correlated (“correlated pairs”). Neuron pairs with correlation values within 95% of the shuffled distributions were deemed randomly correlated (“random pairs”).
To compare changes in Pearson correlation coefficient values between different time periods before and after 6-OHDA injury, we first computed the average correlation coefficient of all neuron pairs in a session, and then performed ANOVA and mixed effect model as detailed below. To compare the correlation coefficients pre- and post- L-Dopa injection, we used a Wilcoxon signed rank test.
Individual neuron’s response to movement analysis
To determine if a neuron responded to movement, we examined whether a neuron significantly changed its activity during high-speed bouts relative to during low-speed bouts using binarized calcium event vectors. Specifically, we calculated the observed difference in the activity rate, defined as the density of ones in the binarized calcium event vectors, between high-speed and low speed bouts for each neuron. Then, we created a baseline shuffled distribution of such differences by shuffling the location of high-speed bouts 1000 times randomly throughout the entire trace duration and calculating the difference in activity rate between shuffled high-speed and low-speed locations in every shuffle. If the observed activity rate difference for a given cell was higher than the 97.5% or lower than the 2.5% of the shuffled distribution, that cell was deemed responsive to movement. Similar procedure was used to identify cells responsive to rotation.
Neuron population response to onset transition analysis
To quantify how SPN, PV or CHI cell populations responded to movement or rotation onset transitions, we first normalized the mean GCaMP6 fluorescence of each cell during the 2 s period after all onsets in each session to the mean fluorescence during the 2 s before all onsets in each session. We then computed the Z score of the normalized GCaMP6 fluorescence across all cells in each session. To determine if a cell population responded to movement or rotation onset, we performed two-sided paired t test, comparing the Z-scores of the normalized fluorescence across all sessions of a given cell type. To determine whether population responses of each cell type were altered by 6-OHDA injury, we computed the difference in the Z-scores of each cell type across sessions under each condition, and compared across healthy, acute and chronic conditions.
LFP power spectrum analysis
LFP motion artifacts were first automatically identified and removed (replaced with NaN) by finding points in which the LFP power was more than 10 times larger than the mean power in the recording. In general, these outliers were transient, with durations of a few milliseconds. In two LFP recording sessions, we detected a significant number of motion artifacts, over a period of 2–4 min, and thus we omitted those periods from further analysis. We used a wavelet transformation for all frequency domain analysis. Wavelet transformation was performed with the PyWavelets package (Lee et al., 2019), using Complex Morlet Wavelet with bandwidth of 14, center frequency of 1.5 and scales between 45 and 950, resulting in frequency bins of approximately 1Hz, between 5Hz and 100Hz. To examine the relative distribution of power at specific frequency domains, we scaled the wavelet power spectrogram at each time point by the total power across all frequencies at that time point.
LFPs spectrum aligned to calcium event onset analysis
LFPs were acquired at 3051.75 Hz, whereas calcium imaging was performed at 20Hz. To align these two signals, we first up-sampled the calcium event onset vector to match the sampling rate of the LFP power spectrum vector, by setting the first point of the calcium event onset equal to 1, and the subsequent up-sampled points as zeros.
We first calculated the mean LFP power spectrogram across all event onsets for each neuron, and then Z-scored the power spectrogram to the mean power during the 2s period prior to the event onsets. We then obtained session averaged LFP power spectrogram by averaging across Z-scored power spectrograms of all neurons in each session. LFP power at different frequency bands, high-gamma (60–100), low-gamma (40–60 Hz), and beta (10–15Hz) were calculated by averaging the power across the frequency bins within these frequency bands. LFP power changes in different frequency bands were compared across sessions, using ANOVA and mixed effect model as detailed below.
Linear regression between Pearson correlation coefficients of SPNs and movement across sessions
Linear regression was plotted using seaborn plotting package, and R2 values computed using the Statsmodel python package (Seabold and Perktold, 2010). For each session, we calculated Pearson correlation coefficients among correlated SPN-SPN pairs, and the fraction of time animals spend in low-speed bouts or no rotation bouts. Linear regressions were performed separately for sessions under healthy condition, acute condition, and chronic condition.
Statistics
The main statistical test used was ANOVA to test whether condition was a significant parameter followed with linear mixed effects models. All linear mixed effect models contained one fixed effect – time period (healthy, versus acute and chronic depletion conditions), and one mixed effect – mouse, which resulted in the model structure described in Equation (3).
| (Equation 3) |
The models were fitted using the R package “lme4” and p values were calculated using the R package “lmerTest”. ANOVA test was preformed using the R basic stats package. Additionally, we used Fisher’s exact test, and Wilcoxon signed rank test as specified in each Figure. All statistics were done on recording session level.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-TH antibody | Abcam | Cat# ab112; RRID:AB_297840 |
| Goat Anti-Rabbit IgG (H+L) Antibody, Alexa Fluor 633 Conjugated | Invitrogen | Cat# A-21070; RRID:AB_2535731 |
| Donkey anti-Goat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 633 | Thermo Fisher Scientific | Cat#A21082; RRID: AB_2535739 |
| Anti-Choline Acetyltransferase antibody | Abcam | Cat#ab254118, RRID: N/A |
| Rabbit anti-parvalbumin antibody | SWant | Cat# PV 25; RRID:AB_10000344 |
| Bacterial and virus strains | ||
| AAV9-Syn-GCaMP6f.WPRE.SV40 6.6 × 1012 GC ml−1 | Addgene | Cat# 100837-AAV9, RRID:Addgene_100837 |
| AAV9-CAG-flex-tdTomato.WPRE.SV40 5.9 × 1012GC ml−1 | UNC vector core | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| 6-Hydroxydopamine hydrobromide | Sigma | Cat#H116-5MG |
| Desipramine hydrochloride | Sigma | Cat#D3900-1G |
| L-Ascorbic acid,BioXtra, ≥99.0%, crystalline | Sigma | Cat#A5960-25G |
| D-Amphetamine hemisulfate salt | Sigma | Cat#A5880-5G |
| Levodopa | Sigma | Cat#1361009-200MG |
| Sterile Solutions for Injection, 0.9% Sodium (Saline) | Baxter | Cat#68000-314 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6 | Charles River Laboratories | Cat#: C57BL/6 |
| Mouse: GM24Gsat (Chat-cre) | MMRRC | Cat#017269-UCD, RRID:MMRRC_017269-UC |
| Mouse: 129P2-Pvalbtm1(cre)Arbr/J | Jackson Laboratories | Cat#JAX:008069, RRID:IMSR_JAX:008069 |
| Software and algorithms | ||
| Matlab2019a | Mathworks | RRID:SCR_001622 |
| HCImage | Hamamatsu Photonics | RRID:SCR_015041 |
| RStudio | RStudio | RRID:SCR_000432 |
| Python Programming Language | NA | RRID:SCR_008394 |
| Code archived | N/A | https://doi.org/10.5281/zenodo.5786809 |
Highlights.
Dopamine loss selectively enhances LFP beta oscillations during impaired locomotion
6-OHDA injury reduces locomotor encoding ability of PV interneurons and SPNs
Elevated coordination within SPN network after dopamine loss inhibits movement
Dopamine loss leads to pathological LFP high-gamma oscillations during SPN activation
ACKNOWLEDGMENTS
We thank members of the Han Lab for suggestions on the manuscript. X.H. acknowledges funding from the NIH (1R01NS115797) and NSF (CBET-1848029, CIF-1955981).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.110265.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Albin RL, Young AB, and Penney JB (1989). The functional anatomy of basal ganglia disorders. Trends Neurosci. 12, 366–375. 10.1016/0166-2236(89)90074-X. [DOI] [PubMed] [Google Scholar]
- Aosaki T, Graybiel AM, and Kimura M (1994). Effect of the nigrostriatal dopamine system on acquired neural responses in the striatum of behaving monkeys. Science 265, 412–415. 10.1126/science.8023166. [DOI] [PubMed] [Google Scholar]
- “Chronux Home”.http://chronux.org/.
- Bolam JP, Hanley JJ, Booth PAC, and Bevan MD (2000). Synaptic organisation of the basal ganglia. J. Anat 196, 527–542. 10.1046/j.1469-7580.2000.19640527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonsi P, Cuomo D, Martella G, Madeo G, Schirinzi T, Puglisi F, Ponterio G, and Pisani A (2011). Centrality of striatal cholinergic transmission in basal ganglia function. Front. Neuroanat 5, 1–9. 10.3389/fnana.2011.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boraud T, Brown P, Goldberg JA, Graybiel AM, and Magill PJ (2006). Oscillations in the basal ganglia: the good, the bad, and the unexpected.” in. The Basal Ganglia VIII, 1–24. 10.1007/0-387-28066-9_1. [DOI] [Google Scholar]
- Bracci E, Centonze D, Bernardi G, and Calabresi P (2002). Dopamine excites fast-spiking interneurons in the striatum. J. Neurophysiol 87, 2190–2194. 10.1152/jn.00754.2001. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Picconi B, Tozzi A, Ghiglieri V, and Di Filippo M (2014). Direct and indirect pathways of basal ganglia: a critical reappraisal. Nat. Neurosci 17, 1022–1030. 10.1038/nn.3743. [DOI] [PubMed] [Google Scholar]
- Chen MT, Morales M, Woodward DJ, Hoffer BJ, and Janak PH (2001). In Vivo extracellular recording of striatal neurons in the awake rat following unilateral 6-hydroxydopamine lesions. Exp. Neurol 171, 72–83. 10.1006/exnr.2001.7730. [DOI] [PubMed] [Google Scholar]
- Costa RM, Lin SC, Sotnikova TDD, Cyr M, Gainetdinov RRR, Caron MGG, and Nicolelis MAAL (2006). Rapid alterations in corticostriatal ensemble coordination during acute dopamine-dependent motor dysfunction. Neuron 52, 359–369. 10.1016/j.neuron.2006.07.030. [DOI] [PubMed] [Google Scholar]
- DeLong MR (1990). Primate models of movement disorders of basal ganglia origin. Trends in Neurosci. 10.1016/0166-2236(90)90110-V. [DOI] [PubMed] [Google Scholar]
- Gittis AH, Hang GB, LaDow ES, Shoenfeld LR, Atallah BV, Fink-beiner S, and Kreitzer AC (2011). Rapid target-specific remodeling of fast-spiking inhibitory circuits after loss of dopamine. Neuron 71, 858–868. 10.1016/j.neuron.2011.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz CG (2011). The history of Parkinson’s disease: early clinical descriptions and neurological therapies. Cold Spring Harb. Perspect. Med 1. 10.1101/cshperspect.a008862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritton HJ, Howe WM, Romano MF, DiFeliceantonio AG, Kramer MA, Saligrama V, Bucklin ME, Zemel D, and Han X (2019). Unique contributions of parvalbumin and cholinergic interneurons in organizing striatal networks during movement. Nat. Neurosci 22, 586–597. 10.1038/s41593-019-0341-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe MW, Atallah HE, McCool A, Gibson DJ, and Graybiel AM (2011). Habit learning is associated with major shifts in frequencies of oscillatory activity and synchronized spike firing in striatum. Proc. Natl. Acad. Sci. U S A 108, 16801–16806. 10.1073/pnas.1113158108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe M, Ridouh I, Letizia A, Mascaro A, Larios A, Azcorra M, and Dombeck DA (2019). Coordination of rapid cholinergic and dopaminergic signaling in striatum during spontaneous movement. ELife. 10.7554/eLife.44903.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Ledochowitsch P, Knoblich U, Lecoq J, Murphy GJ, Reid RC, de Vries SEJ, Koch C, Zeng H, Buice MA, et al. (2021). Relationship between simultaneously recorded spiking activity and fluorescence signal in Gcamp6 transgenic mice. ELife 10. 10.7554/ELIFE.51675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondabolu K, Roberts EA, Bucklin M, McCarthy MM, Kopell N, and Han X (2016). Striatal cholinergic interneurons generate beta and gamma oscillations in the corticostriatal circuit and produce motor deficits. Proc. Natl. Acad. Sci. U S A 113, E3159–E3168. 10.1073/pnas.1605658113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer Anatol C. (2009). Physiology and pharmacology of striatal neurons. Annu. Rev. Neurosci 32, 127–147. 10.1146/annurev.neuro.051508.135422. [DOI] [PubMed] [Google Scholar]
- Lee G, Gommers R, Waselewski F, Wohlfahrt K, and O’Leary A (2019). PyWavelets: a Python package for wavelet analysis. J. Open Source Softw 4, 1237. 10.21105/joss.01237. [DOI] [Google Scholar]
- Lemaire N, Hernandez LF, Hu D, Kubota Y, Howe MW, and Graybiel AM (2012). Effects of dopamine depletion on LFP oscillations in striatum are task- and learning-dependent and selectively reversed by L-DOPA. Proc. Natl. Acad. Sci. U S A 109, 18126–18131. 10.1073/pnas.1216403109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leventhal DK, Gage GJ, Schmidt R, Pettibone JR, Case AC, and Berke JD (2012). Basal ganglia beta oscillations accompany cue utilization. Neuron 73, 523–536. 10.1016/j.neuron.2011.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang L, DeLong MR, and Papa SM (2008). Inversion of dopamine responses in striatal medium spiny neurons and involuntary movements. J. Neurosci 28, 7537–7547. 10.1523/JNEUROSCI.1176-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little S, and Brown P (2014). The functional role of beta oscillations in Parkinson’s disease. Parkinsonism Relat. Disord 20, S44–S48. 10.1016/S1353-8020(13)70013-0. [DOI] [PubMed] [Google Scholar]
- Mallet N, Ballion B, Le Moine C, and Gonon F (2006). Cortical inputs and GABA interneurons imbalance projection neurons in the striatum of parkinsonian rats. J. Neurosci 26, 3875–3884. 10.1523/JNEUROSCI.4439-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meer M (2010). Integrating early results on ventral striatal gamma oscillations in the rat. Front. Neurosci 4, 300. 10.3389/fnins.2010.00300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meer MAA, and Redish AD (2009). Low and high gamma oscillations in rat ventral striatum have distinct relationships to behavior, reward, and spiking activity on a learned spatial decision task. Front. Integr. Neurosci 3, 9. 10.3389/neuro.07.009.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy MM, Moore-Kochlacs C, Gu X, Boyden ES, Han X, and Kopell N (2011). Striatal origin of the pathologic beta oscillations in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 108, 11620–11625. 10.1073/pnas.1107748108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KJ, Leuthardt EC, Schalk G, Rao RPN, Anderson NR, Moran DW, Miller JW, and Ojemann JG (2007). Spectral changes in cortical surface potentials during motor movement. J. Neurosci 27, 2424–2432. 10.1523/JNEUROSCI.3886-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller V, Mohr B, Rosin R, Pulvermüller F, Müller F, and Birbaumer N (1997). Short-term effects of behavioral treatment on movement initiation and postural control in Parkinson’s disease: a controlled clinical study. Movement Disord. 12, 306–314. 10.1002/mds.870120308. [DOI] [PubMed] [Google Scholar]
- Obeso JA, Rodriguez-Oroz MC, Rodriguez M, Lanciego JL, Artieda J, Gonzalo N, and Warren Olanow C (2000). Pathophysiology of the basal ganglia in Parkinson’s disease. Trends Neurosci. 23, S8–S19. 10.1016/S1471-1931(00)00028-8. [DOI] [PubMed] [Google Scholar]
- Oswal A, Brown P, and Litvak V (2013). Synchronized neural oscillations and the pathophysiology of Parkinson. Curr. Opin. Neurol 26, 662–670. 10.1097/wco.0000000000000034. [DOI] [PubMed] [Google Scholar]
- Owen SF, Berke JD, and Kreitzer AC (2018). Fast-spiking interneurons supply feedforward control of bursting, calcium, and plasticity for efficient learning. Cell 172, 683–695.e15. 10.1016/j.cell.2018.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JG, Marshall JD, Ahanonu B, Wu Y-W, Kim TH, Grewe BF, Zhang Y, Li JZ, Ding JB, Ehlers MD, et al. (2018). Diametric neural ensemble dynamics in parkinsonian and dyskinetic states. Nature 557, 177–182. 10.1038/s41586-018-0090-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz A, Feingold A, Zelanskaya V, Vaadia E, and Bergman H (1996). Neuronal synchronization of tonically active neurons in the striatum of normal and parkinsonian primates. J. Neurophysiol 76, 2083–2088. 10.1152/jn.1996.76.3.2083. [DOI] [PubMed] [Google Scholar]
- Rendón-Ochoa EA, Hernández-Flores T, Avilés-Rosas VH, Cáceres-Chávez VA, Duhne M, Laville A, Tapia D, Galarraga E, and Bargas J (2018). Calcium currents in striatal fast-spiking interneurons: dopaminergic modulation of CaV1 channels. BMC Neurosci. 19, 1–14. 10.1186/S12868-018-0441-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salin P, López IP, Kachidian P, Barroso-Chinea P, Rico AJ, Gómez-Bautista V, Coulon P, Kerkerian-Le Goff L, and Lanciego JL (2009). Changes to interneuron-driven striatal microcircuits in a rat model of Parkinson’s disease. Neurobiol. Dis 34, 545–552. 10.1016/j.nbd.2009.03.006. [DOI] [PubMed] [Google Scholar]
- Seabold S, and Perktold J (2010). Statsmodels: econometric and statistical modeling with Python. In 9th Python in Science Conference. [Google Scholar]
- Sharott A, Vinciati F, Nakamura KC, and Magill PJ (2017). A population of indirect pathway striatal projection neurons is selectively entrained to parkinsonian beta oscillations. J. Neurosci 37, 9977–9998. 10.1523/JNEUROSCI.0658-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, and Papa SM (2020). Striatal oscillations in parkinsonian non-human primates. Neuroscience 449, 116–122. 10.1016/j.neuro-science.2020.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Mewes K, Gross RE, DeLong MR, Obeso JA, and Papa SM (2016). Human striatal recordings reveal abnormal discharge of projection neurons in Parkinson’s disease. Proc. Natl. Acad. Sci. U S A 113, 9629–9634. 10.1073/pnas.1606792113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith Y, Wichmann T, Factor SA, and Delong MR (2012). Parkinson’s disease therapeutics: new developments and challenges since the introduction of levodopa. Neuropsychopharmacology. 10.1038/npp.2011.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swann NC, De Hemptinne C, Miocinovic S, Qasim S, Wang SS, Ziman N, Ostrem JL, San Luciano M, Galifianakis NB, and Starr PA (2016). Gamma oscillations in the hyperkinetic state detected with chronic human brain recordings in Parkinson’s disease. J. Neurosci 36, 6445–6458. 10.1523/JNEUROSCI.1128-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepper JM, Wilson CJ, and Koós T (2008). Feedforward and feedback inhibition in neostriatal GABAergic spiny neurons. Brain Res. Rev 58, 272–281. 10.1016/j.brainresrev.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepper JM, Tecuapetla F, Koós T, and Ibáñez-Sandoval O (2010). Heterogeneity and diversity of striatal GABAergic interneurons. Front. Neuroanat 10.3389/fnana.2010.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran CH, Vaiana M, Nakuci J, Somarowthu A, Goff KM, Goldstein N, Murthy P, Muldoon SF, and Goldberg EM (2020). Interneuron desynchronization precedes seizures in a mouse model of dravet syndrome. J. Neurosci 40, 2764–2775. 10.1523/JNEUROSCI.2370-19.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valsky D, Grosberg SH, Israel Z, Boraud T, Bergman H, and Deffains M (2020). What is the true discharge rate and pattern of the striatal projection neurons in Parkinson’s disease and dystonia? ELife 9, 1–27. 10.7554/ELIFE.57445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger M, Hutchison WD, Lozano AM, Hodaie M, and Dostrovsky JO (2009). Increased gamma oscillatory activity in the subthalamic nucleus during tremor in Parkinson’s disease patients. J. Neurophysiol 101, 789–802. 10.1152/jn.90837.2008. [DOI] [PubMed] [Google Scholar]
- Wingeier B, Tom T, Koop MM, Hill BC, Heit G, and Bronte-Stewart HM (2006). Intra-operative STN DBS attenuates the prominent beta rhythm in the STN in Parkinson’s disease. Exp. Neurol 197, 244–251. 10.1016/j.expneurol.2005.09.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
All original code has been deposited at GitHub https://github.com/HanLabBU/Dopamine-Depletion-CellReports/releases/tag/v1.0 and Zenodo https://doi.org/10.5281/zenodo.5786809 and is publicly available as of the date of publication.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request