

Epigenetic mechanisms regulate the chromatin structure and gene expression levels without changing the primary DNA sequence, and include DNA methylation and hydroxymethylation, post-translational modifications of histone tails and nucleosome positioning, as well as mechanisms mediated by long and short non-coding RNA molecules. These mechanisms are required for human development and cell differentiation, and are pivotal in differentiated cells for cellular functions, allowing a tight regulation of gene expression levels in response to environmental stimuli and cellular metabolic demands (Coppedè, 2021b). Particularly, the neuronal epigenome is highly sensitive to external stimuli and its function is required for learning and memory processes (Creighton et al., 2020). Several enzymes, collectively referred to as the “epigenetic machinery”, add, remove and read epigenetic marks, allowing chromatin remodeling to promote or repress gene transcription. The activity of these enzymes allows the reversibility of epigenetic marks, which is pivotal in neurons for memory formation and consolidation (Creighton et al., 2020). The epigenome is altered in the aging brain, although the factors driving these changes, their contribution to age-related memory decline, and their potential modulation with environmental interventions are still a matter of debate (Creighton et al., 2020). Furthermore, both global and gene-specific epigenetic changes are observed in blood and brain tissues of individuals with major neurodegenerative diseases, including among others Alzheimer’s disease (AD) (Coppedè, 2021a), Parkinson’s disease (PD) (Rathore et al., 2020), and amyotrophic lateral sclerosis (ALS) (Coppedè, 2020). An open question is which of these changes result from gene-environment interactions that lead to age-related declines in cognitive and motor functions, thus contributing to the onset of the disease, and which ones are consequences of the degenerative processes occurring in neurons. However, due to their reversibility, several epigenetic marks have been proposed as potential therapeutic targets to treat or delay neurodegeneration (Coppedè, 2021a). In the present article, the author provides a brief overview of the literature and its own perspective opinion on targeting the epigenome to treat neurodegeneration.
DNA methylation is catalyzed by DNA methyltransferases (DNMTs), a family of enzymes that transfer the methyl group from S-adenosylmethionine (SAM) to cytosine, forming 5-methylcytosine. In neurons, both CpG methylation, i.e., the methylation of a cytosine that is followed by a guanine, and non-CpG methylation (CpA, CpT and CpC) occur. These marks are recognised and bound by methyl-binding proteins, such as MeCP2, which in turn recruit other factors to regulate the chromatin structure and orchestrate gene expression levels during brain development and function. Members of the ten-eleven translocation protein family convert 5-methylcytosine to 5-hydroxymethylcytosine, another stable epigenetic mark in mammalian brain regions where it binds to proteins important for neuronal function and development (Coppedè, 2021b). Two therapeutic options can be used to regulate DNA methylation, the first based on the use of compounds that inhibit DNMTs, known as DNMT inhibitors, and the second based on the provision of methyl donor compounds such as SAM or folates and other B-group vitamins required for SAM production (Coppedè, 2021a). DNMT inhibitors include azacitidine and decitabine, both approved by the Food and Drug Administration for the treatment of hematological malignancies, and other compounds that are being tested in hematological and solid tumors. Unfortunately, these compounds are often toxic and relatively unstable, so that they seem more useful in rapidly proliferative cancer cells than in non-proliferative neurons. For example, it was shown that decitabine exacerbates neurotoxicity and upregulates PD-related genes in cultured dopaminergic neurons, including demethylation of the SNCA gene coding for α-synuclein (Wang et al., 2013). SAM is the universal intracellular methyl donor compound generated through one-carbon metabolism, a key metabolic pathway that couples the folate and methionine cycles. Dietary folates and related B-group vitamins are therefore required for SAM production, and their restriction can impair the cellular methylation potential (Coppedè, 2021b). The PSEN1 gene is one of the causative genes of familial AD forms and codes for the presenilin 1 protein, a component of the ϒ-secretase complex required for the production of the amyloid-beta peptide, the neurotoxic peptide that accumulates in AD brains. We recently observed reduced PSEN1 methylation levels, resulting in increased gene expression, in both blood of living AD patients and post-mortem AD brain regions, suggesting that it could represent an epigenetic biomarker of the disease (Monti et al., 2020). Previous research in AD epigenetics revealed that a diet poor in methyl donor compounds, such as folates and other B-group vitamins, induced PSNE1 hypomethylation followed by increased amyloid-beta peptide production and AD-like symptoms in rodents, whilst dietary supplementation of SAM restored PSEN1 methylation levels and ameliorated cognitive functions in the animals, suggesting that SAM supplementation could represent a therapeutic option for AD (Fuso et al., 2012). I recently reviewed cell culture studies, animal studies and human trials investigating SAM as a potential compound to reverse AD-related epigenetic changes and attenuate disease symptoms (Coppedè, 2021a). Overall, studies performed in animal or cell culture models of AD demonstrated that SAM possesses epigenetic and antioxidant properties, reducing the burden of amyloid-beta peptide accumulation and enhancing antioxidant defense mechanisms. Similarly, formulations including SAM, folic acid, vitamin B12, and nutraceuticals such as alpha-tocopherol, N-acetyl cysteine, and acetyl-L-carnitine, have been investigated in AD patients and showed some improvement of cognitive symptoms. Unfortunately, changes in DNA methylation levels were not evaluated in human trials, making not possible to evaluate the epigenetic properties of these formulations (Coppedè, 2021a). Concerning PD, it was shown that intracranial injection of SAM induces PD-like changes in rodents, including striatal dopamine depletion, tremors, and hypokinesia, likely because it impairs the methylation levels and function of several enzymes involved in dopamine metabolism, including dopamine receptors and transporters (Lee et al., 2004). Therefore, epigenetic interventions in PD animal models were mainly focused on targeting histone tail modifications than DNA methylation (Coppedè, 2014).
Post-translational modifications occurring on histone tails, collectively known as the “histone code”, regulate the chromatin structure and the accessibility of gene regulatory regions. Among them, histone tail acetylation is linked to a relaxed chromatin structure allowing transcription, whilst histone tail deacetylation, mediated by histone deacetylases (HDACs), results in chromatin compaction and transcriptional repression. Targeting histone tail acetylation with inhibitors of HDACs (HDACi) is a promising strategy to treat human cancers, and four of these drugs, namely vorinostat, belinostat, panobinostat, and romidepsin, received Food and Drug Administration approval for the treatment of T-cell lymphomas or multiple myeloma, whilst additional HDACi are in clinical trials for human cancers and other complex diseases. In recent years I reviewed the literature related to the use of HDACi for the treatment of AD (Coppedè, 2021a), ALS (Coppedè, 2020), PD and other neurodegenerative diseases (Coppedè, 2014), observing that collectively, the investigations performed in animal models of these conditions have shown that treatment with HDACi was able to improve synaptic plasticity as well as cognitive and motor functions in the animals. However, most of these compounds are scarcely selective and neurotoxic, so that there is concern for long-term treatment in humans. For example, the HDACi valproic acid (VPA) is an anti-epileptic drug used as a first-choice agent for most forms of epilepsy and, despite that VPA treatment was neuroprotective in PD animal models, chronic usage of VPA in humans induced Parkinsonism as an adverse effect in some individuals (Muralidharan et al., 2020). Moreover, there is still no consensus from animal studies about which HDAC isoforms should be selectively inhibited to achieve the therapeutic effect and an adequate safety profile in neurons, so that some clinical trials with HDACi are ongoing to determine safety and tolerability in patients with neurodegenerative disorders (Cummings et al., 2021).
Both short and long non-coding RNA molecules are dysregulated in tissues from patients with neurodegenerative diseases, so that they are increasingly proposed as disease biomarkers as well as possible targets of therapeutic interventions (Huaying et al., 2021). Indeed, in my opinion, non-coding RNA molecules will be further explored as targets of small molecules designed to regulate the expression of their target genes to treat individuals with neurodegenerative diseases and, although further efforts are required to improve the delivery and stability of these molecules, their development is an attractive and promising field of research (Coppedè, 2020).
Overall, based on studies in cell culture and animal models of neurodegeneration, targeting epigenetic marks could represent a promising approach to improve cognition, synaptic plasticity and motor functions in an attempt to delay disease progression in individuals affected by neurodegenerative disorders. Anyway, the observed neurotoxicity, scarce selectivity and relative instability of most of the available drugs prompt the design of novel, more selective and safer compounds. In cancer research both approved and experimental epigenetic drugs are undergoing clinical trials in combination with standard therapeutic agents in order to benefit from synergistic effects of multi-target approaches, and something similar is likely to be expected in non-cancer diseases, including neurodegeneration. However, there is a substantial difference between cancer and neurodegeneration, which is represented by the fact that cancer cells are characterized by rapid divisions that require a continuous re-writing of epigenetic marks in daughter cells, so that targeting enzymes of the epigenetic machinery with DNMT inhibitors, HDACi or similar compounds can re-activate genes whose silencing is responsible of cancer aggressiveness and resistance to conventional therapies. By contrast, neurodegenerative disorders are characterized by neuronal death, and most of their signs and symptoms manifest when a considerable number of neurons are already died. Therefore, treating patients already suffering from the disease might be too late, and enhancing the plasticity of the surviving neurons with epigenetic molecules or compounds could only slightly delay disease progression, without significantly improving the quality of life. For example, despite encouraging results in transgenic ALS mice, prolonged VPA treatment did not show beneficial effects on survival or disease progression in ALS patients (Piepers et al., 2009). Most of AD, PD, and ALS forms are sporadic, resulting from complex gene-environment interactions within the course of life, and according to the Developmental Origin of Health and Disease theory, the origins of complex diseases is formed at the time of fertilization, embryonic, fetal, and neonatal stages by the interrelation between genes and the environment. Indeed, there is a growing body of evidence from animal studies suggesting that early life environmental stressors including chemical exposure, dietary restrictions, maternal stress, etc., can perturb the neuronal epigenome predisposing to neurocognitive, neurobehavioral and/or neurodegenerative disorders later in life (Lahiri et al., 2009). Therefore, neurodegeneration and other complex diseases can be viewed as the result of gene-environment interactions occurring throughout life, with epigenetic modifications as the cellular response to these interactions. With this in mind, our challenging opportunity to prevent or delay neurodegeneration could be making use of recent genome-wide technologies to detect as early as possible individuals at risk to develop neurodegeneration later in life (Figure 1), coupled to the design of prospective studies to see if natural compounds with epigenetic properties, exercise, brain stimulation and/or epigenetic drugs can restore the synaptic plasticity preventing or reducing neuronal death in those subjects. Several natural compounds with epigenetic properties are available in foods, including folate, vitamin B12, epigallocatechin-3-gallate, curcumin, genistein, resveratrol, quercetin, and many others. Furthermore, exercise and brain stimulation can modify the neuronal epigenome, enhancing synaptic plasticity. Therefore, questions that we should be able to answer are the following: (a) Can we detect in easily accessible biological fluids such as blood or saliva epigenetic marks of an early life adverse environment potentially leading to neuronal impairment? (b) Can we reverse those marks with natural compounds, exercise, brain stimulation or psychological therapies before adolescence? (c) Can we detect middle-aged individuals at risk to develop age-related neurodegenerative diseases, maybe using the polygenic risk score or identifying genome-wide epigenetic biomarkers of disease risk? (d) Can we potentiate the epigenetic neuronal plasticity in those subjects with dietary interventions, exercise and brain stimulation in order to prevent or delay neurodegeneration? (e) Can we identify in elderly individuals epigenetic biomarkers of preclinical neurodegenerative conditions and see if dietary interventions, environmental manipulations and/or epigenetic drugs are able to delay the conversion into clinical stages?
Figure 1.
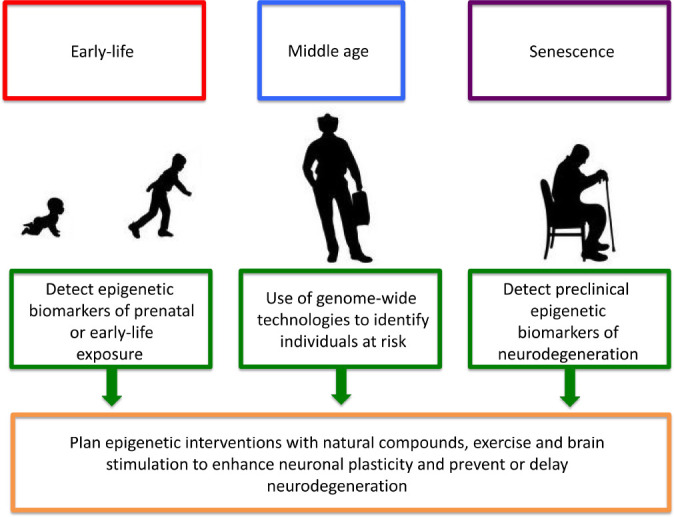
Potential strategies to prevent or delay neurodegeneration.
Epigenetic modifications leading to impaired neuronal function and plasticity can be triggered by early-life environmental factors such as pre- and post-natal dietary restrictions or overfeeding, parental stressful events, and exposure to neurotoxicants. Subsequent harmful environmental exposures, dietary habits and lifestyles within the course of life can further contribute to the neuronal damage, ultimately leading to the onset of neurodegenerative diseases later in life. Our ability to detect as early as possible epigenetic marks of early-life exposure as well as epigenetic biomarkers of increased risk to develop neurodegeneration later in life could represent a challenging opportunity to plan epigenetic interventions with natural compounds, exercise and brain stimulation to enhance neuronal plasticity and prevent or delay neurodegeneration. Particular effort should be put in the search of preclinical epigenetic biomarkers of neurodegeneration, in order to identify older adult subjects that could particularly benefit from those interventions.
Overall, the design of novel, more stable, selective and less toxic epigenetic drugs is a promising strategy to improve neuronal function and delay disease progression in individuals already affected by neurodegenerative diseases. However, we need to shed light on the complex gene-environment interactions occurring within the course of life and make advantage of recent genome-wide technologies to identify epigenetic biomarkers able to detect as early as possible individuals at risk to develop neurodegeneration later in life, in order to plan epigenetic interventions with natural compounds, exercise, brain stimulation, etc. that could enhance neuronal plasticity to prevent or delay the onset of neurodegeneration.
Footnotes
C-Editors: Zhao M, Liu WJ, Qiu Y; T-Editor: Jia Y
References
- 1.Coppedè F. The potential of epigenetic therapies in neurodegenerative diseases. Front Genet. 2014;5:220. doi: 10.3389/fgene.2014.00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coppedè F. Epigenetics of neuromuscular disorders. Epigenomics. 2020;12:2125–2139. doi: 10.2217/epi-2020-0282. [DOI] [PubMed] [Google Scholar]
- 3.Coppedè F. Epigenetic regulation in Alzheimer’s disease: is it a potential therapeutic target. Expert Opin Ther Targets. 2021a;25:283–298. doi: 10.1080/14728222.2021.1916469. [DOI] [PubMed] [Google Scholar]
- 4.Coppedè F. One-carbon epigenetics and redox biology of neurodegeneration. Free Radic Biol Med. 2021b;170:19–33. doi: 10.1016/j.freeradbiomed.2020.12.002. [DOI] [PubMed] [Google Scholar]
- 5.Creighton SD, Stefanelli G, Reda A, Zovkic IB. Epigenetic mechanisms of learning and memory: implications for aging. Int J Mol Sci. 2020;21:6918. doi: 10.3390/ijms21186918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cummings J, Lee G, Zhong K, Fonseca J, Taghva K. Alzheimer’s disease drug development pipeline: 2021. Alzheimers Dement (N Y) 2021;7:e12179. doi: 10.1002/trc2.12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fuso A, Nicolia V, Ricceri L, Cavallaro RA, Isopi E, Mangia F, Fiorenza MT, Scarpa S. S-adenosylmethionine reduces the progress of the Alzheimer-like features induced by B-vitamin deficiency in mice. Neurobiol Aging. 2012;33:1482. doi: 10.1016/j.neurobiolaging.2011.12.013. [DOI] [PubMed] [Google Scholar]
- 8.Huaying C, Xing J, Luya J, Linhui N, Di S, Xianjun D. A signature of five long non-coding RNAs for predicting the prognosis of Alzheimer’s disease based on competing endogenous RNA networks. Front Aging Neurosci. 2021;12:598606. doi: 10.3389/fnagi.2020.598606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lahiri DK, Maloney B, Zawia NH. The LEARn model: an epigenetic explanation for idiopathic neurobiological diseases. Mol Psychiatry. 2009;14:992–1003. doi: 10.1038/mp.2009.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee ES, Chen H, Shepherd KR, Lamango NS, Soliman KF, Charlton CG. The inhibitory role of methylation on the binding characteristics of dopamine receptors and transporter. Neurosci Res. 2004;48:335–344. doi: 10.1016/j.neures.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 11.Monti N, Cavallaro RA, Stoccoro A, Nicolia V, Scarpa S, Kovacs GG, Fiorenza MT, Lucarelli M, Aronica E, Ferrer I, Coppedè F, Troen AM, Fuso A. CpG and non-CpG Presenilin1 methylation pattern in course of neurodevelopment and neurodegeneration is associated with gene expression in human and murine brain. Epigenetics. 2020;15:781–799. doi: 10.1080/15592294.2020.1722917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muralidharan A, Rahman J, Banerjee D, Hakim Mohammed AR, Malik BH. Parkinsonism: a rare adverse effect of valproic acid. Cureus. 2020;12:e8782. doi: 10.7759/cureus.8782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Piepers S, Veldink JH, de Jong SW, van der Tweel I, van der Pol WL, Uijtendaal EV, Schelhaas HJ, Scheffer H, de Visser M, de Jong JM, Wokke JH, Groeneveld GJ, van den Berg LH. Randomized sequential trial of valproic acid in amyotrophic lateral sclerosis. Ann Neurol. 2009;66:227–234. doi: 10.1002/ana.21620. [DOI] [PubMed] [Google Scholar]
- 14.Rathore AS, Birla H, Singh SS, Zahra W, Dilnashin H, Singh R, Keshri PK, Singh SP. Epigenetic modulation in Parkinson’s disease and potential treatment therapies. Neurochem Res. 2021;46:1618–1626. doi: 10.1007/s11064-021-03334-w. [DOI] [PubMed] [Google Scholar]
- 15.Wang Y, Wang X, Li R, Yang ZF, Wang YZ, Gong XL, Wang XM. A DNA methyltransferase inhibitor, 5-aza-2’-deoxycytidine, exacerbates neurotoxicity and upregulates Parkinson’s disease-related genes in dopaminergic neurons. CNS Neurosci Ther. 2013;19:183–190. doi: 10.1111/cns.12059. [DOI] [PMC free article] [PubMed] [Google Scholar]