

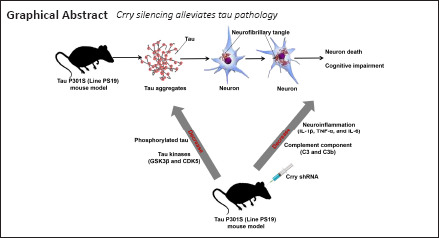
Key Words: Alzheimer's disease, cognitive function, complement system, CR1, Crry, neurodegeneration, neuroinflammation, tauopathy
Abstract
Complement component (3b/4b) receptor 1 (CR1) expression is positively related to the abundance of phosphorylated microtubule-associated protein tau (tau), and CR1 expression is associated with susceptibility to Alzheimer’s disease. However, the exact role of CR1 in tau protein-associated neurodegenerative diseases is unknown. In this study, we show that the mouse Cr1-related protein Y (Crry) gene, Crry, is localized to microglia. We also found that Crry protein expression in the hippocampus and cortex was significantly elevated in P301S mice (a mouse model widely used for investigating tau pathology) compared with that in wild-type mice. Tau protein phosphorylation (at serine 202, threonine 205, threonine 231, and serine 262) and expression of the major tau kinases glycogen synthase kinase-3 beta and cyclin-dependent-like kinase 5 were greater in P301S mice than in wild-type mice. Crry silencing by lentivirus-transfected short hairpin RNA led to greatly reduced tau phosphorylation and glycogen synthase kinase-3 beta and cyclin-dependent-like kinase 5 activity. Crry silencing reduced neuronal apoptosis and rescued cognitive impairment of P301S mice. Crry silencing also reduced the levels of the neuroinflammatory factors interleukin-1 beta, tumor necrosis factor alpha, and interleukin-6 and the complement components complement 3 and complement component 3b. Our results suggest that Crry silencing in the P301S mouse model reduces tau protein phosphorylation by reducing the levels of neuroinflammation and complement components, thereby improving cognitive function.
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disease characterized by cognitive function deficits and progressive memory loss; it was first discovered in 1906 by Alois Alzheimer (Alzheimer, 1906). Two histopathological hallmarks of AD are intraneuronal neurofibrillary tangles, caused by abnormal aggregation of hyperphosphorylated microtubule-associated protein tau (tau), and extracellular amyloid plaques, composed of amyloid beta (Aβ) peptides (Kidd, 1963; Hardy and Higgins, 1992). As a microtubule-associated protein, tau is usually expressed in neuronal axons and is implicated in the dissociation of microtubules by binding to the tubulin dimers that constitute microtubules (Cleveland et al., 1977). Truncation and phosphorylation of tau protein cause tau to aggregate as neurofibrillary tangles in the proximal axoplasm. A study based on the P301S mouse model (widely used for investigating tau pathology) previously indicated that filamentous tau lesions were associated with loss of neurons and atrophy of the hippocampus and entorhinal cortex; microgliosis and impaired hippocampal synaptic function occurred prior to the formation of filamentous tau tangles (Yoshiyama et al., 2007).
Complement cascades are a vital part of the immune system that function to protect the central nervous system (CNS) from infection; dysregulation of the complement system in the CNS results in neurodevelopmental damage and disease (Lee et al., 2019; Lo and Lee, 2021; Yang et al., 2021). Several potential genes and loci, including the complement component (3b/4b) receptor 1 (CR1) gene (CR1), are associated with susceptibility to AD (Corneveaux et al., 2010; Kucukkilic et al., 2018; Kunkle et al., 2019). The CR1 protein affects the deposition of Aβ in brain tissue (Biffi et al., 2012; Thambisetty et al., 2013). Furthermore, Zhu et al. (2017) implicated CR1 in the progression of AD because of its effect on Aβ deposition, glucose metabolism, and brain structure. However, the role of CR1 in AD-related tauopathy is not clear. Killick et al. (2013) showed that the levels of complement factor H and the phosphorylation of tau protein at serine 235 were greatly decreased in Cr1-related protein Y (Crry)–deficient (–/–) mice in comparison with controls, implicating Crry protein in AD-associated tauopathy. However, these observations illustrate only that Crry affects tau phosphorylation and does not explain the mechanisms of action of Crry in AD-related tauopathy. The expression and distribution of CR1 proteins differ between rodents and humans: the murine complement receptor type 2 (CR2) gene (Cr2) encodes both CR1 and CR2 proteins, whereas human CR1 and CR2 proteins are encoded by separate genes (Jacobson and Weis, 2008). Crry, a rodent-specific gene also known as Cr1-like (Cr1l), is more highly expressed in microglia than astrocytes and is expressed only to the RNA level in mouse neurons (Davoust et al., 1999). The Crry gene in rodents is the closest ortholog of the human CR1 gene, and it is widely accepted that Crry in murine models acts as the equivalent of CR1 in humans (Molina et al., 1992; Jacobson and Weis, 2008; Killick et al., 2013). Therefore, the Crry gene in murine models can be used to investigate the possible role of CR1 in AD-related tauopathy, which may contribute to further understanding the neuropathological processes in AD.
The complement system is a crucial driver of inflammation (Lee et al., 2019). Overactivation of complement signals can result in neuron injury and neuroinflammation in many neurological diseases (Krance et al., 2019; Parker et al., 2019). Synapse loss in AD is mediated by the complement pathway and microglia and leads to cognitive decline (Stevens et al., 2007). Complement C1q levels were elevated in postsynaptic densities of P301S mice and patients with AD, and the accumulation of C1q was correlated with phosphorylated tau levels and synapse loss (Dejanovic et al., 2018). Wu et al. (2019) showed that P301S mice had elevated levels of classical complement components and the central complement component complement 3 (C3), and C3 expression was associated with tau phosphorylation. Litvinchuk et al. (2018) demonstrated positive correlations between cognitive decline and the expression levels of C3 and C3a anaphylatoxin chemotactic receptor in patients with AD.
The tau P301S (PS19) mouse model is one of the most widely used models for investigating AD-related tauopathies (Yoshiyama et al., 2007; Iba et al., 2013). In this study, the role of CR1 protein in AD-related tauopathy was investigated by examining the role of Crry in wild-type (WT) and P301S tauopathy mouse models. Our study aimed to discover the role that the CR1 protein plays in the progression of AD and to identify potential therapeutic targets for AD.
Materials and Methods
Animals
We acquired the mice used in our current study from the Model Animal Research Center, Nanjing University. We used heterozygous transgenic mice (Tg(Prnp-MAPT*P301S)PS19Vle/JNju, specific-pathogen-free grade, 3–9 months old, 12–25 g) that had the murine prion protein promoter on a B6C3 background and expressed the P301S human 1N4R tau isoform on chromosome 3 (Yoshiyama et al., 2007). For WT controls, we selected littermates with matched genetic background and age. We selected only male mice to minimize the interference of estrogen on cognitive function and neuroinflammation (Wei et al., 2014). Mice were housed at 22°C and 50% relative humidity in a 12-hour light/dark cycle. The Medical Ethics Committee of the Wuxi No. 2 People’s Hospital affiliated to Nanjing Medical University, Wuxi, China, approved the study protocol with approval No. 2020-Y-45 on October 21, 2020. All operations were carried out as per the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines (Kilkenny et al., 2010). Sodium pentobarbital (50 mg/kg; MilliporeSigma, Burlington, MA, USA) was used as anesthesia in the study and was administered by intraperitoneal injection. The detailed experimental procedure is shown in Additional Figure 1 (1.5MB, tif) . The mice were divided into groups randomly.
Double immunofluorescence staining
In order to explore the localization of Crry in the brains of P301S mice, double immunofluorescence staining was performed. Paraffin sections (8-μm thick) of mouse brain tissues were dewaxed and rehydrated with serial dilutions of ethanol in water, followed by antigen retrieval in citrate buffer. The samples were treated with bovine serum albumin (50 g/L) and Triton X-100 (1 mg/mL) diluted in 1× phosphate buffer saline and then incubated with primary antibodies at 4°C overnight. The samples were then treated with secondary antibodies at 37°C for 1 hour. All antibodies used are listed in Additional Table 1. The sections were photographed with a cooled CCD camera (ORCA-ER; Hamamatsu Photonics K.K., Hamamatsu, Japan) on a fluorescence microscope (BX63; Olympus, Tokyo, Japan). We counted cells in ten random visual fields across each tissue section for each mouse and calculated the proportion of positive cells.
Addtional Table 1.
Antibodies used in western blot and double immunofluorescence staining
| Antibody | Concentration | RRID No. | Species and clonality | Catalog No. | Supplier |
|---|---|---|---|---|---|
| Crry | 1:200 | AB_2085152 | Rabbit monoclonal | sc-30214 | Santa Cruz Biotechnology, Santa Cruz, CA, USA |
| Iba-1 | 1:500 | AB_2636859 | Rabbit monoclonal | ab178846 | Abcam, Cambridge,UK |
| GFAP | 1:500 | AB_305808 | Rabbit polyclonal | ab7260 | Abcam, Cambridge,UK |
| NeuN | 1:1000 | AB_2532109 | Rabbit monoclonal | ab177487 | Abcam, Cambridge,UK |
| OSP | 1:500 | AB_305935 | Rabbit polyclonal | ab7474 | Abcam, Cambridge,UK |
| Hyperphosphorylated tau at Ser202:Thr205 (AT8) | 1:1000 | AB_223647 | Mouse monoclonal | mn1020 | ThermoFisher Scientific, Sunnyvale, CA, USA |
| Hyperphosphorylated tau at Thr231 | 1:1000 | AB_2533742 | Rabbit polyclonal | 44-746G | ThermoFisher Scientific, Sunnyvale, CA, USA |
| Hyperphosphorylated tau at Ser262 | 1:1000 | AB_2533743 | Rabbit polyclonal | 44-750G | ThermoFisher Scientific, Sunnyvale, CA, USA |
| Total tau | 1:1000 | AB_628327 | Mouse monoclonal | sc-32274 | Santa Cruz Biotechnology, Santa Cruz, CA, USA |
| Phospho-GSK-3β (Ser9) | 1:1000 | AB_2798546 | Rabbit monoclonal | 5558T | Cell Signaling Technology, Danvers, MA, USA |
| GSK-3α:β | 1:1000 | AB_2636978 | Rabbit monoclonal | 12456T | Cell Signaling Technology, Danvers, MA, USA |
| AMPK | 1:1000 | AB_1118940 | Mouse monoclonal | sc-74461 | Santa Cruz Biotechnology, Santa Cruz, CA, USA |
| Phospho-AMPKα (Thr172) |
1:1000 | AB_2799368 | Rabbit monoclonal | 50081s | Cell Signaling Technology, Danvers, Massachusetts, USA |
| Phospho-p38 MAPK (Thr180:Tyr182) |
1:1000 | AB_2139682 | Rabbit monoclonal | 4511T | Cell Signaling Technology, Danvers, MA, USA |
| p38 MAPK | 1:1000 | AB_2533144 | Mouse monoclonal | 33-8700 | ThermoFisher Scientific, Sunnyvale, CA, USA |
| JNK | 1:1000 | AB_2140722 | Mouse monoclonal | sc-137019 | Santa Cruz Biotechnology, Santa Cruz, CA, USA |
| Phospho-JNK (Thr183:Tyr185) |
1:1000 | AB_2574777 | Mouse monoclonal | sc-293136 | Santa Cruz Biotechnology, Santa Cruz, CA, USA |
| CDK5 | 1:1000 | AB_627241 | Mouse monoclonal | sc-6247 | Santa Cruz Biotechnology, Santa Cruz, CA, USA |
| Synaptophysin | 1:1000 | AB_2286949 | Rabbit monoclonal | ab32127 | Abcam, Cambridge,UK |
| PP2Ac | 1:1000 | AB_2170107 | Mouse monoclonal | sc-81603 | Santa Cruz Biotechnology, Santa Cruz, CA, USA |
| C3 | 1:500 | AB_2066623 | Rat monoclonal | ab11862 | Abcam, Cambridge,UK |
| C3b | 1:2000 | AB_627277 | Mouse monoclonal | sc-28294 | Santa Cruz Biotechnology, Santa Cruz, CA, USA |
| Phospho-CaMKII-α (Tyr231) |
1:1000 | AB_2070310 | Rabbit polyclonal | 3356s | Cell Signaling Technology, Danvers, MA, USA |
| CaMKII-α | 1:1000 | AB_2721906 | Mouse monoclonal | 50049s | Cell Signaling Technology, Danvers, MA, USA |
| Cleaved caspase-3 | 1:500 | AB_2341188 | Rabbit polyclonal | 9661 | Cell Signaling Technology, Danvers, MA, USA |
| β-Actin | 1:1000 | AB_2714189 | Mouse monoclonal | sc-47778 | Santa Cruz Biotechnology, Santa Cruz, CA, USA |
| Goat anti-Mouse IgG (H+L) Poly-HRP Secondary Antibody |
1:1000 | AB_1965958 | Polyclonal | 32230 | ThermoFisher Scientific, Sunnyvale, CA, USA |
| Fluorescein-conjugated affinipure Goat anti-Rabbit IgG antibody | 1:200 | AB_2571576 | Polyclonal | ZF-0311 | Zhongshan Goldenbridge Inc., Beijing, China |
| FITC labeled Goat anti-mouse IgG antibody | 1:200 | AB_2716306 | Polyclonal | ZF-0312 | Zhongshan Goldenbridge Inc., Beijing, China |
AMPK: AMP-activated protein kinase; C3: complement 3; C3b: complement component 3b; CaMKII-α: CaM-dependent protein kinase II-α; CDK5: cyclin-dependent kinase 5; Crry: Cr1-related protein Y; GFAP: glial fibrillary acidic protein; GSK: glycogen synthase kinase; Iba-1: ionized calcium binding adaptor molecule 1; JNK: c-Jun N-terminal kinase; MAPK: mitogen-activated protein kinases; NeuN: neuronal nuclei; OSP: oligodendrocyte specific protein; PP2Ac: protein phosphatase 2A catalytic subunit.
Western blot assay
Brain tissues were lysed using 50 mg protein lysate:100 μL radioimmunoprecipitation assay buffer (Beyotime Biotechnology Co. Ltd., Shanghai, China). Extracted proteins were quantified using the bicinchoninic acid kit (Beyotime Biotechnology Co. Ltd.), in accordance with the manufacturer’s instructions. Total protein was separated using 10% polyacrylamide gels and sodium dodecyl sulfate-polyacrylamide electrophoresis, and separated proteins were transferred to a nitrocellulose membrane. After treatment with 50 mg/mL skim milk/bovine serum albumin in phosphate buffer saline, membranes were incubated with primary antibodies at 4°C overnight. Then, membranes were treated with horseradish peroxidase-labeled anti-mouse secondary antibodies at 37°C for 1 hour, followed by chemiluminescent horseradish peroxidase substrate (Thermo Fisher Scientific Inc., Waltham, MA, USA) at 37°C for 1 hour. All antibodies used are listed in Additional Table 1. Finally, protein bands and signal intensities were analyzed using X-ray film (Fujifilm Corporation, Tokyo, Japan) and Quantity One 1-D analysis software version 4.6.2 (Bio-Rad Laboratories Inc., Hercules, CA, USA), respectively.
Lentiviral vector preparation and intracerebral lentiviral particle injection
Crry protein expression was suppressed by intracerebral injection of lentiviral vectors because direct Crry knockout in embryos would be lethal (Xu et al., 2000). Briefly, short hairpin RNA (shRNA) sequences corresponding to mouse Crry and the matching negative control were synthesized (Santa Cruz Biotechnology Inc., Dallas, TX, USA) and cloned into the lentiviral vector under the myeloid-specific CD11b promoter. The lentiviral vectors were transfected into HEK293-FT cells (ATCC Cat# PTA-5077, RRID:CVCL_6911; Cell Bank of the Chinese Academy of Sciences, Shanghai, China), and we collected the lentiviral particles after 48 hours using an ultracentrifuge (titer: 1 × 106 –1 ×107 viral particles/mL) and suspended them in phosphate-buffered saline. Mice were administered 150 mg/kg imidazole sodium (MilliporeSigma) to prevent postoperative pain. Lentiviral particles (3 μL) were injected into the cerebral cortex (anteroposterior: –0.3 mm, mediolateral: 2 mm, dorsoventral: –1.5 mm; anteroposterior: –2 mm, mediolateral: 1.2 mm, dorsoventral: –1.2 mm) and the hippocampus (anteroposterior: –2 mm, mediolateral: 1.2 mm, dorsoventral: –2 mm) of the brains of 6-month-old P301S mice (each mouse received injections in all regions) via a micropipette attached to a 10-μL Hamilton syringe (MilliporeSigma), as previously described (Jiang et al., 2016). Because of the long-term neurotoxicity of lentiviruses (Dodart et al., 2005), the influence of the lentiviral vector on Crry expression was observed 2 months following intervention using western blot assays.
Quantitative real-time polymerase chain reaction
Total RNA from brain tissue samples from each group was extracted using TRIzol reagent (Thermo Fisher Scientific Inc.). Then, using PrimeScript RT Master Mix (Takara Bio Inc., Kusatsu, Japan), RNA (10 μg) was reverse-transcribed in accordance with the manufacturer’s protocol on a 7500 Real-Time PCR system (Thermo Fisher Scientific Inc.). Reverse transcription was carried out at 37°C for 15 minutes, 85°C for 5 seconds, and then samples were kept at 4°C. The complementary DNA was then amplified under the following reaction conditions: 95°C for 30 seconds, 40 cycles at 95°C for 5 seconds, and 60°C for 34 seconds. The primer sequences used are listed in Additional Table 2. Signals were collected at the endpoint of each cycle extension, and amplification curve data were evaluated. The 2–ΔΔCT method (Livak and Schmittgen, 2001) was used for analyzing expression data.
Additional Table 2.
Primer sequences used in this study
| Name | Primer forward (5’-3’) | Primer reverse (5’-3’) | GenBank accession number* | Amplic on size (bp) |
|---|---|---|---|---|
| Crry | CTTCCCTCTCGCATCAGTG | AAGGATACCGCCTCATTGG | NM_01349 | 209 |
| TTGCA | TTCCTC | 92.2 | ||
| TNF-α | GTCTACTGAACTTCGGGG | ATGATCTGAGTGTGAGGGT | NM_01369 | 102 |
| TGAT | CTG | 3.3 | ||
| IL-1β | GAAGAGCCCATCCTCTGT | TTCATCTCGGAGCCTGTAGT | NM_00836 | 96 |
| GA | G | 1.3 | ||
| IL-6 | ACAAAGCCAGAGTCCTTC | CATTGGAAATTGGGGTAGG | NM_03116 | 105 |
| AGAG | A | 8.1 | ||
| GAPD | CAACAGCAACTCCCACTC | GGTCCAGGGTTTCTTACTCC | NM_00808 | 164 |
| H | TTC | TT | 4.3 |
* The GenBank accession numbers were obtained from the NCBI (https://www.ncbi.nlm.nih.gov/). Crry: Cr1-related protein Y; GAPDH: reduced glyceraldehyde-phosphate dehydrogenase; IL-1β: interleukin-1beta; IL-6: interleukin-6; TNF-α: tumour necrosis factor alpha.
Cresyl violet staining
Briefly, all tissue samples were paraffin embedded, cut, dewaxed, rehydrated, and stained in cresyl violet (10 mg/mL, MilliporeSigma). The sections were rinsed with water, dehydrated in increasing concentrations of ethanol, and viewed with a light microscope. Neurons in the hippocampus and cortex of WT and P301S mice were counted. Only neurons with visible nuclei and whose entire edges were well-defined were counted. These neurons were defined as survival neurons. We evaluated six sections of cortex and hippocampus from each mouse, and the mean survival neuron count was used for statistical analysis. We calculated the hippocampal and ventricular volumes of mice using cresyl violet staining and ImageJ software (National Institutes of Health, Bethesda, MD, USA) (Schneider et al., 2012). Two independent, experienced experimenters performed the neuron counts.
Morris water maze test
The Morris water maze test was carried out 2 months following lentiviral particle injection as per the methods provided in a previous study (Jiang et al., 2014), with 6 mice in each group. Briefly, for the hidden-platform training, a circular platform with a diameter of 10 cm was submerged 0.5 cm below the water surface in the southeast quadrant of a large, water-filled container. A video camera (SMART Video Tracking System; Panlab, Harvard Apparatus, Barcelona, Spain) was used to track the swimming paths for 60 seconds, and then a computer-controlled system (SMART Video Tracking System; Panlab, Harvard Apparatus) was used to analyze the swimming paths. All mice were trained to complete 4 trials per day for 5 consecutive days. The length of time taken to reach the submerged platform over four trials was averaged. When a mouse failed to find the platform within 60 seconds, we picked it up and placed it on the platform for 15 seconds. The platform was removed 24 hours after the last trial, and this was followed by a probe test for 60 seconds. In a probe test, the platform was removed, and the swimming path of the mouse was recorded.
Passive avoidance step-through task
The passive avoidance step-through task was carried out 2 months following lentiviral particle injection, as previously described (Kumaran et al., 2008), with six mice in each group. The test cage (TopScan; CleverSys Inc., Reston, VA, USA) had two equally sized compartments, with a guillotine door separating light and dark compartments. For the acquisition trial, mice were kept in the test cage for 5 minutes and had free movement between the light and dark compartments. For the training test, mice were placed in the light compartment. The door between the compartments was opened after 10 seconds, and we recorded the latency period, i.e., the time it took the mouse to step all four paws into the dark compartment. We kept track of the number of mice that entered the dark compartment after more than 5 minutes, and we reported these as errors. When a mouse entered the dark compartment with all four paws, we closed the door and administered a 0.5 mA foot shock for 10 seconds. We excluded animals that took more than 100 seconds to cross to the dark compartment. When a mouse did not enter the dark compartment, we recorded the latency as 300 seconds.
Statistical analysis
The sample sizes in this study were not predetermined using statistical methods, but the sample sizes we used are similar to those in previous studies (Ruan et al., 2020; Izzy et al., 2021). All experiments were evaluated using blind methods. Statistical analyses were performed with SPSS Statistics for Windows software, version 17.0 (SPSS Inc., Chicago, IL, USA). Data are expressed as the mean ± standard error of the mean (SEM). The Kolmogorov-Smirnov test was employed for analysis of variables with normal distribution. Independent sample t-tests were applied for pairwise group comparisons. For comparisons among multiple groups, we performed one-way analysis of variance followed by Tukey’s post hoc test for data from western blots, quantitative real-time polymerase chain reaction, double immunofluorescence staining, cresyl violet staining, and the passive avoidance step-through task. Two-way repeated-measures analysis of variance followed by Tukey’s post hoc test was applied to analyze the escape latency of each group in the hidden-platform training of the Morris water maze test. P < 0.05 was selected as the cutoff value for significant results.
Results
Crry protein expression increases with age in the context of AD
The localization of Crry in the brain was explored via double immunofluorescence staining in 6-month-old P301S mice. Crry and the microglial marker ionized calcium-binding adapter molecule 1 (Iba-1) (Ohsawa et al., 2004) were colocalized, suggesting Crry is expressed in microglia (Figure 1A). There was relatively weak colocalization between Crry and glial fibrillary acidic protein, an astrocyte marker (Takizawa et al., 2008), suggesting lower expression of Crry in astrocytes (Figure 1B). However, we observed no association between Crry and neurons labeled with neuronal nuclei antigen (NeuN) (Duan et al., 2016) or oligodendrocytes labeled with oligodendrocyte-specific protein (OSP) (Bronstein et al., 1996), suggesting that Crry is not expressed in neurons and oligodendrocytes of P301S mice (Figure 1C and D).
Figure 1.
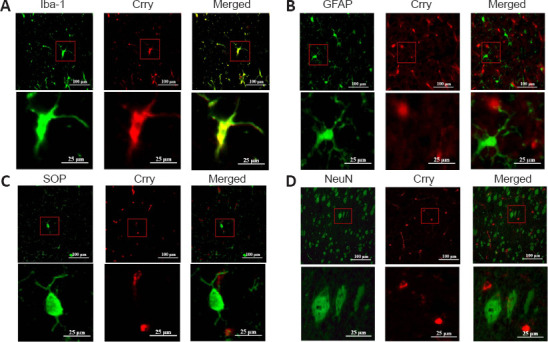
Double immunofluorescence staining to identify the localization of Crry in the brains of 6-month-old P301S mice (original magnification 400×).
(A) Crry (FITC, green fluorescence) and the microglial marker Iba-1 (rhodamine, red fluorescence) colocalize in the brains. (B) Weak colocalization between Crry and the astrocyte marker GFAP (rhodamine, red fluorescence). (C) No colocalization between the oligodendrocyte marker OSP (rhodamine, red fluorescence) and Crry. (D) No colocalization between the neuron marker NeuN (rhodamine, red fluorescence) and Crry. Scale bars: 100 μm (upper) and 25 μm (lower). Crry: Cr1-related protein Y; FITC: fluorescein isothiocyanate; GFAP: glial fibrillary acidic protein; Iba-1: ionized calcium binding adaptor molecule 1; NeuN: neuronal nuclei; OSP: oligodendrocyte specific protein.
The change in Crry protein levels in the brain during the aging process in the AD context was revealed by western blot assay. Our results show that Crry protein levels in the hippocampus and cortex were markedly upregulated in 6-month-old and 9-month-old P301S mice in comparison with 3-month-old P301S mice (n = 6, both P < 0.05), but no statistical difference was found between 6-month-old and 9-month-old P301S mice (n = 6, P > 0.05; Figure 2A–D). Moreover, Crry protein levels in the hippocampus and cortex were also markedly increased in 6-month-old and 9-month-old P301S mice compared with those in age-matched WT mice (n = 6, both P < 0.05; Figure 2A–D). Immunofluorescence staining of Crry confirmed that Crry protein levels in the hippocampus and cortex were increased in 6-month-old P301S mice compared with those in age-matched WT mice (Additional Figure 2 (2MB, tif) ). Furthermore, analysis based on the microglia marker Iba-1 demonstrated increasing microglia activation in the hippocampus and cortex of 3-, 6-, and 9-month-old P301S mice compared with that in age-matched WT mice (n = 6, all P < 0.05), with no statistical difference among 3-, 6-, and 9-month-old P301S mice (n = 6, all P > 0.05; 2A, C, E, and F). However, statistically significant differences were observed when Crry levels were normalized to Iba-1 expression in microglia in 6-month-old P301S mice (n = 6, both P < 0.05; Figure 2G and H), which indicates that Crry upregulation is not a result of the increased number of microglia in this AD mouse model.
Figure 2.

Crry protein expression increases with age in an Alzheimer’s disease context.
(A, C) Crry, Iba-1, and β-actin protein expression in the cortex and hippocampus of P301S mice and age-matched WT mice at different ages, as visualized using western blot. (B, D–F) Quantification of Crry and Iba-1 protein levels in the cortex and hippocampus of P301S mice and age-matched WT mice. (G, H) Crry expression was significantly upregulated in the cortex and hippocampus of 6-month-old P301S mice after normalizing Crry expression to microglial Crry expression. Protein expression levels were normalized to β-actin. Data are represented as the mean ± SEM (n = 6 for each group) and were analyzed by one-way analysis of variance followed by Tukey’s multiple comparison test (B, D–F) and by independent sample t-test (G, H). †P < 0.05. Crry: Cr1-related protein Y; Iba-1: ionized calcium binding adaptor molecule 1; WT: wild type.
Crry expression in brain tissues is inhibited by Crry-specific shRNA
We investigated Crry gene and protein expression and the silencing efficiency of Crry shRNA infusion. Crry gene and protein expression in the cortex of 6-month-old mice that received Crry shRNA decreased by 41.0% and 42.6%, respectively, compared with those in 6-month-old mice that received control shRNA (n = 6, both P < 0.05; Figure 3A–C). Similarly, Crry shRNA infusion decreased Crry gene and protein expression in the hippocampus of 6-month-old mice by 37.2% and 35.3%, respectively, compared with those in 6-month-old mice that received control shRNA (n = 6, both P < 0.05; Figure 3D–F). These results indicate that Crry expression is successfully inhibited by shRNA infusion.
Figure 3.

Crry expression in the brains of P301S mice Is significantly decreased by Crry shRNA.
(A–C) mRNA and protein expression of Crry in the cortex of 6-month-old WT and P301S mice after treatment with control shRNA or Crry shRNA. (D–F) mRNA and protein expression of Crry in the hippocampus of 6-month-old WT and P301S mice after treatment with control shRNA or Crry shRNA. mRNA and protein expression levels were normalized to β-actin. Data are represented as the mean ± SEM (n = 6 for each group) and were analyzed by one-way analysis of variance followed by Tukey’s multiple comparison test. †P < 0.05. Crry: Cr1-related protein Y; shRNA: short hairpin RNA; WT: wild type.
Decrease in hyperphosphorylated tau levels after Crry silencing
Western blot analyses showed changes at four phosphorylation sites (serine 202, threonine 205, threonine 231, and serine 262) in tau protein in 6-month-old WT and P301S mice that received Crry shRNA or control shRNA. AT8/total tau, threonine 231/total tau, and serine 262/total tau were significantly elevated in whole brains of 6-month-old P301S mice compared with those in WT mice (n = 6, all P < 0.05; Figure 4A–D). Following Crry shRNA infusion, AT8/total tau, threonine 231/total tau, and serine 262/total tau decreased in whole brains of 6-month-old WT and P301S mice by 59.0% (n = 6, P < 0.05; Figure 4A and B), 59.85% (n = 6, P < 0.05; Figure 4A and C), and 52.4% (n = 6, P < 0.05; Figure 4A and D), respectively, compared with those in 6-month-old WT and P301S mice that received control shRNA. Total tau levels did not change in any of the mice tested.
Figure 4.

Hyperphosphorylated tau is decreased after infusion with Crry shRNA in the brains of 6-month-old P301S mice.
(A) Levels of AT8 (which recognizes serine 202 and threonine 205 hyperphosphorylated tau), threonine (thr) 231 hyperphosphorylated tau, serine (ser) 262 hyperphosphorylated tau, and total tau were measured by western blot. (B–D) Quantification of the proportion of AT8/total tau (B), threonine 231 hyperphosphorylated tau/total tau (C), and serine 262 hyperphosphorylated tau/total tau (D). Protein expression levels were normalized to β-actin. Data are presented as the mean ± SEM (n = 6 for each group) and were analyzed by one-way analysis of variance followed by Tukey’s multiple comparison test. †P < 0.05. Crry: Cr1-related protein Y; shRNA: short hairpin RNA; WT: wild type.
Crry silencing influences the balance of tau kinases and tau phosphatase
The activation of major tau kinases, including glycogen synthase kinase-3 beta (GSK3β), AMP-activated protein kinase (AMPK), mitogen-activated protein kinase p38 beta (p38 MAPK), c-Jun N-terminal kinase 1 (JNK), cyclin-dependent-like kinase 5 (CDK5), and calcium/calmodulin-dependent protein kinase type II subunit alpha (CaMKII-α), was studied to determine the effect of Crry shRNA infusion on tau kinases (Figure 5A–G). The expression levels of p-GSK3β, p-AMPK, p-JNK, CDK, and p-CaMKII-α were elevated in 6-month-old P301S mice compared with those in WT mice. In addition, GSK3β phosphorylation and CDK5 levels were significantly decreased in P301S mice after treatment with Crry shRNA (all P < 0.05). However, there were no significant differences in phosphorylation for the other tau kinases tested (all P > 0.05). Furthermore, P301S mice had lower levels of protein phosphatase 2A (PP2A) catalytic subunit (PP2Ac) expression than WT mice, and the activity of the main tau phosphatase, PP2A, remained unchanged in P301S mice after treatment with Crry shRNA (P < 0.05; Figure 5H and I). These results indicate that infusion of Crry shRNA decreased hyperphosphorylated tau by repressing the activity of the tau kinases GSK3β and CDK5, without influencing tau phosphatase levels.
Figure 5.

Effect of Crry shRNA on the tau kinase/phosphatase balance in the brains of 6-month-old P301S mice.
(A) Expression levels of p-GSK3b (serine 9), total GSK3β, phospho-AMPKα (threonine 172), total AMPK, phospho-p38 MAPK (threonine 180 and tyrosine 182), p38 MAPK, phospho-JNK (threonine 183 and tyrosine 185), JNK, CDK5, phospho-CaMKII-α (tyrosine 231), and total CaMKII-α, as detected by western blot. (B–G) Quantification of p-GSK3β/GSK3β, p-P38/P38, p-AMPK/AMPK, p-JNK/JNK, CDK5/β-actin, and p-CaMKII-α/CaMKII-α. (H) PP2Ac levels were detected by western blot. (I) Quantification of PP2Ac levels. Protein expression levels were normalized to the WT + control shRNA group. Data are presented as the mean ± SEM (n = 6 for each group) and were analyzed by one-way analysis of variance followed by Tukey’s multiple comparison test. †P < 0.05. AMPK: AMP-activated protein kinase; CaMKII-α: CaM-dependent protein kinase II-α; CDK5: cyclin-dependent kinase 5; Crry: Cr1-related protein Y; GSK3β: glycogen synthase kinase-3beta; JNK: c-Jun N-terminal kinase; MAPK: mitogen-activated protein kinases; PP2Ac: protein phosphatase 2A catalytic subunit; shRNA: short hairpin RNA; WT: wild type.
Crry silencing reverses neurodegenerative changes in the brains of P301S mice
P301S mice had a lower neuron count in the hippocampus and cortex compared with WT mice. In addition, the neuron count in these two brain regions of P301S mice injected with control shRNA was significantly lower than that of P301S mice injected with Crry shRNA (both P < 0.05; Figure 6A–C). We found higher cleaved caspase-3 levels in the hippocampus and cortex of P301S mice compared with those in WT mice, as measured by western blot. Moreover, cleaved caspase-3 expression was lower in P301S mice that received Crry shRNA compared with that in P301S mice that received control shRNA (Figure 6D and E), which suggests that Crry shRNA may have reduced apoptosis. Furthermore, we analyzed hippocampal atrophy and ventricle volume based on cresyl violet staining. We observed hippocampal atrophy in P301S mice, and Crry silencing significantly alleviated hippocampal atrophy (P < 0.05; Figure 6F). Ventricle volume was increased in P301S mice, and Crry silencing decreased this change in ventricle volume (P < 0.05; Figure 6G). Synaptophysin expression was lower in P301S mice compared with that in WT mice, and synaptophysin levels in P301S mice were upregulated after Crry shRNA infusion compared with that in P301S mice injected with control shRNA (P < 0.05; Figure 7). These results indicate that there were significant neurodegenerative changes in the brains of P301S mice. When Crry protein expression in the brain was decreased, the neuron count and synaptophysin expression levels increased in the cerebral cortex and hippocampus.
Figure 6.

Decreasing Crry expression in the brain can delay neuron loss in P301S transgenic mice.
(A) Cresyl violet staining shows the survival of neurons in the cortex and hippocampus. Mice in the WT + control shRNA and WT + Crry shRNA groups have more neurons than P301S mice, and mice in the P301S + Crry shRNA group have more neurons compared with mice in the P301S + control shRNA group. Arrows indicate the neurons stained by cresyl violet. Scale bar: 100 μm. (B, C) Quantification of surviving neurons stained by cresyl violet in the cortex and hippocampus of mice in each group. (D, E) Cleaved caspase-3 expression in neurons of the cortex and hippocampus of mice in each group visualized by western blot. Protein expression levels were normalized to the WT + control shRNA group. (F, G) Quantitative analysis of hippocampal volume (F) and ventricle volume (G). Data are presented as the mean ± SEM (n = 6 for each group) and were analyzed by one-way analysis of variance followed by Tukey’s multiple comparison test. †P < 0.05. Crry: Cr1-related protein Y; shRNA: short hairpin RNA; WT: wild type.
Figure 7.

Expression of synaptophysin is elevated following Crry silencing in P301S transgenic mice.
(A) Protein expression of synaptophysin visualized by western blot. (B) Quantification of synaptophysin protein expression. Protein expression levels were normalized to β-actin. Data are presented as the mean ± SEM (n = 6 for each group) and were analyzed by one-way analysis of variance followed by Tukey’s multiple comparison test. †P < 0.05. Crry: Cr1-related protein Y; shRNA: short hairpin RNA; WT: wild type.
Crry silencing restores the cognitive function of P301S mice
The Morris water maze test was used to determine the influence of Crry shRNA infusion in 6-month-old P301S mice. We found that the difference in swimming speed between individual 6-month-old P301S mice was not significant, which eliminates any potential for interference of motivation effects and sensorimotor input on measurements of cognitive performance (P > 0.05; Figure 8A). The hidden-platform task, which assesses cognitive deficiency, showed that P301S mice had higher latencies compared with WT mice when finding the submerged platform (P < 0.05; Figure 8B). After infusion with Crry shRNA, the average time it took to reach the hidden platform was markedly decreased in P301S mice in comparison with P301S mice infused with control shRNA or WT mice on days 4 and 5. No significant differences were found within the group of P301S mice infused with control shRNA or within the group of WT mice during this test (P > 0.05; Figure 8B).
Figure 8.

Cognitive function deficiency is rescued in P301S mice after infusion with Crry shRNA.
(A) Swimming speed during the 5-day training in the Morris water maze. (B) Escape latency in the hidden-platform training of the Morris water maze. WT mice had significantly lower escape latencies compared with P301S mice during the 5-day training. P301S mice injected with Crry shRNA had shorter escape latencies than P301S mice injected with control shRNA on days 4 and 5. (C) Results of the probe trial of the Morris water maze. Mice in the P301S + control shRNA group spent similar amounts of time in all quadrants, whereas mice in the other three groups spent significantly more time in the target quadrant (Q1) than in the other three quadrants. (D, E) Latency (D) and number of errors (E) in the passive avoidance step-through task in 6-month-old P301S mice. Data are presented as the mean ± SEM (n = 6 for each group). Data from the passive avoidance step-through task and the swimming speed test were analyzed by one-way analysis of variance followed by Tukey’s post hoc test; the escape latency data of each group from the hidden-platform training of the Morris water maze were analyzed by two-way repeated-measures analysis of variance followed by Tukey’s post hoc test. *P < 0.05, vs. WT + control shRNA and WT + Crry shRNA; #P < 0.05, vs. P301S + control shRNA. †P < 0.05. Crry: Cr1-related protein Y; shRNA: short hairpin RNA; WT: wild type.
For the probe test, we removed the platform in the water and performed probe trials to evaluate the effects of infusion with Crry shRNA on 6-month-old P301S mice. As shown in Figure 8C, three groups of mice (6-month-old WT mice with control shRNA, 6-month-old WT mice with Crry shRNA, and 6-month-old P301S mice with Crry shRNA) spent markedly more time in the target quadrant than in the other three quadrants (all P < 0.05), while 6-month-old P301S mice with control shRNA spent similar amounts of time in all quadrants (P > 0.05). Our results indicate that infusion with Crry shRNA significantly alleviated spatial memory impairment in P301S mice. Our results also indicate that infusion with Crry shRNA in 6-month-old P301S mice reduced the number of errors and improved latency times (both P < 0.05; Figure 8D and E).
Crry silencing modulates neuroinflammation and the complement system in the brains of P301S mice
We studied the effects of Crry shRNA treatment on neuroinflammation by analyzing the expression of interleukin-1 beta (IL-1β), tumor necrosis factor alpha (TNF-α), and interleukin-6 (IL-6). Downregulation of Crry significantly suppressed the expression of IL-1β (by 47.3%, n = 6, P < 0.05), TNF-α (by 53.4%, n = 6, P < 0.05), and IL-6 (by 35.4%, n = 6, P < 0.05) compared with those in P301S mice infused with control shRNA (Figure 9A–C). Furthermore, we observed a decrease in Crry levels and proinflammatory cytokines after infusion with Crry shRNA. We also found that the levels of C3 and complement component 3b (C3b) proteins decreased by 48.6% and increased by 51.9%, respectively, after infusion with Crry shRNA compared with those in P301S mice that received control shRNA (both P < 0.05; Figure 10A–C).
Figure 9.
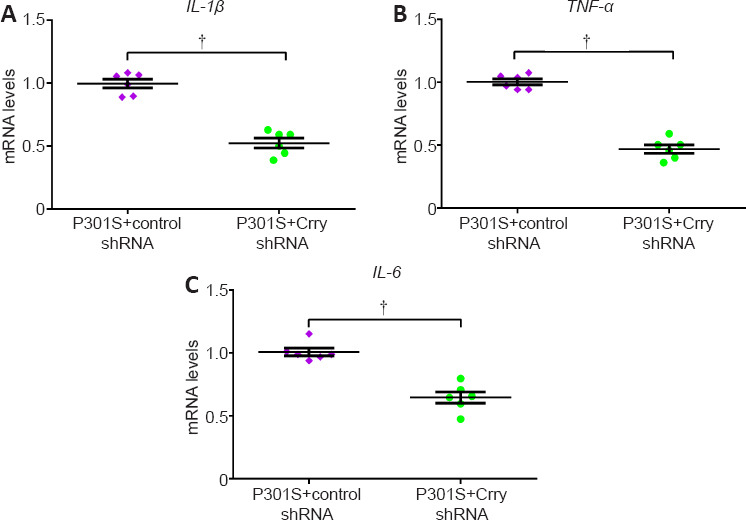
Infusion with Crry shRNA modulates neuroinflammation in P301S mice.
(A–C) mRNA levels of IL-1β, TNF-α, and IL-6 in brains measured by quantitative real-time polymerase chain reaction (normalized to GAPDH). Data represent the mean ± SEM (n = 6 for each group) and were analyzed by independent sample t-test. †P < 0.05. Crry: Cr1-related protein Y; GAPDH: reduced glyceraldehyde-phosphate dehydrogenase; IL-1β: interleukin-1beta; IL-6: interleukin-6; shRNA: short hairpin RNA; TNF-α: tumour necrosis factor alpha.
Figure 10.
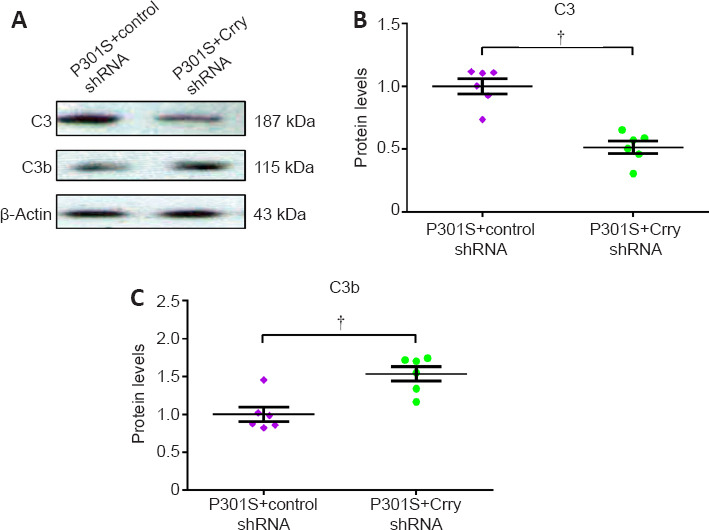
Infusion with Crry shRNA modulates the complement system in P301S mice.
(A) Expression levels of C3 and C3b proteins in the brains of P301S mice determined by western blot. (B, C) Quantification of C3 and C3b protein levels. Protein expression levels were normalized to β-actin. Data are presented as mean ± SEM (n = 6 for each group) and were analyzed by independent sample t-test. †P < 0.05. C3: complement 3; C3b: complement component 3b; Crry: Cr1-related protein Y; shRNA: short hairpin RNA.
Discussion
For this study, we intended to explore the potential role of the CR1 gene in AD-related tauopathy by investigating the rodent-specific Crry gene (the ortholog of CR1 in humans) in the P301S tauopathy mouse model. Double immunofluorescence staining showed that Crry was expressed in microglia and had lower expression in astrocytes (weak colocalization with the astrocyte marker glial fibrillary acidic protein) but was not expressed in neurons. In mice, Davoust et al. (1999) were the first to detect mRNA and protein expression of Crry in astrocytes and microglia, and they showed that microglia had higher Crry expression levels than astrocytes. Only Crry mRNA was observed in neurons. Our results are consistent with the findings of Davoust et al. (1999). Microglia-mediated neuroinflammation is a hallmark of a variety of neurodegenerative diseases, including AD (Tang and Le, 2016). CR1, SPI1, and CD33 are highly expressed in microglia and contribute to the progression of AD in a non-Aβ-dependent manner (Villegas-Llerena et al., 2016; Efthymiou and Goate, 2017). Therefore, we speculated that CR1 expressed in microglia might contribute to AD progression by influencing tau pathology.
We detected tau phosphorylation levels and the activity of tau kinases by silencing Crry expression with a Crry shRNA-encoding lentiviral vector in P301S mice. P301S mice had significantly elevated tau protein phosphorylation at the AT8, threonine 231, and serine 262 sites, and had increased activity of the major tau kinases GSK3β and CDK5 compared with WT mice. Crry silencing decreased tau protein phosphorylation and the activity of these two tau kinases. GSK3β is a serine/threonine protein kinase that regulates the phosphorylation of many cellular substrates and has a crucial role in the phosphorylation of tau protein (Hernández et al., 2009). GSK3β expression was significantly elevated in P301S mice compared with that in WT mice, and aged P301S mice had increased levels of hyperphosphorylated tau coupled with increased GSK3β expression (Dumont et al., 2011; Hou et al., 2020). CDK5 is a cyclin-dependent kinase and also a tau kinase that phosphorylates tau at serine/threonine residues (primarily serine 202, serine 235, serine 404, and threonine 205) in serine/threonine-proline sequences (Kimura et al., 2014; Cortés et al., 2019). CDK5 is abnormally activated in AD and its deregulation contributes to various histopathological changes in patients with AD (Liu et al., 2016). Saito et al. (2019) showed that CDK5 could elevate the phosphorylation and accumulation of tau by regulating tau phosphorylation at serine 262. Hyperphosphorylation of tau protein is one of the many histopathological changes in AD. Our results suggest that Crry silencing might partly improve AD by alleviating tau pathology. Killick et al. (2013) found that Crry–/– mice had decreased levels of complement factor H (a serum biomarker for AD progression) and tau phosphorylation at the serine 235 site. To a certain extent, the study of Killick et al. (2013) confirmed our speculation that Crry silencing might partly restore AD by alleviating tau pathology. In addition, we found that Crry silencing decreased neuron death and hippocampal atrophy and improved the deficiency in cognitive function in P301S mice, as expected. Therefore, we confirm that Crry silencing is beneficial in the P301S tauopathy mouse model.
The complement system represents a crucial part of the innate immune system. Overactivation of the complement system will directly or indirectly lead to damage of the body’s own tissues (Bajic et al., 2015). Crry is an important regulator of the complement pathway that has the same effect as other complement regulatory proteins, such as complement decay-accelerating factor and membrane cofactor protein. Crry can repress the activity of C3/C5 convertase to block complement activation (Kim et al., 1995). Crry–/– mice are embryo lethal, but they can be rescued by C3 deletion as complement activation in the mother cannot be inhibited when Crry is absent, which results in complement attacks on the placenta (Xu et al., 2000). The double knockout of Crry in kidneys can cause unrestricted complement activation, marked inflammation, and progressive renal failure (Bao et al., 2007). Ruseva et al. (2009) showed that significantly decreased C3 levels were observed in Crry–/– mice. In our study, we used lentivirus transfection to reduce Crry expression, and we observed decreased C3 and increased C3b levels. We speculate that C3 convertases persisted for longer as a result of Crry shRNA activity; therefore, more C3b was produced, and that C3b could not be efficiently degraded. Our results suggest that there was more complement activation in Crry shRNA-treated mice, which is inconsistent with the claim that Crry silencing has beneficial effects. In fact, Crry levels were clearly elevated in P301S mice compared with those in WT mice, and we observed beneficial effects in P301S mice after receiving Crry shRNA. Our study preliminarily investigated the effect of Crry silencing on complement C3 and C3b levels. However, many complement components and receptors are involved in complement activation and its subsequent effects. For example, it has been reported that accumulation of inactive C3b (iC3b) causes microglial priming via interaction of iC3b with CR3; microglial priming exacerbates neurodegenerative disease in the context of experimental autoimmune encephalomyelitis (Ramaglia et al., 2012). Ramaglia et al. (2012) suggested that elevated local expression of C3 and fB were observed in Crry–/– mice, and both C3 and fB were dependent on and required for microglial priming, indicating microglial priming was caused by uncontrolled complement activation through an alternative pathway. In Crry–/– mice, fH catalyzes the first cleavage of C3b, resulting in iC3b accumulation (Kim et al., 1995), and Ramaglia et al. (2012) suggested that local accumulation of iC3b engaged its receptor CR3 and triggered microglial priming in the Crry–/– CNS. However, complement mediates microglial phagocytosis and clearance of fibrillar Aβ by the interaction of iC3b with CR3 on the microglia surface (Fu et al., 2012). Shi et al. (2017) suggested that inhibition of complement and downstream iC3b/CR3 signals could protect synapses against Aβ aggregation-related loss in an APP/PS1 mouse model before amyloid plaque aggregation. C3b was also implicated in the clearance of Aβ in peripheral blood by interacting with CR1 on erythrocytes (Rogers et al., 2006). Increasing evidence indicates that Aβ plays roles in the pathways upstream of tau and induces neurotoxicity mediated by tau (Rapoport et al., 2002; Roberson et al., 2007; Choi et al., 2014). Therefore, we speculate that C3b or iC3b might contribute to the clearance of Aβ by interacting with their receptors and decreasing tau-mediated neurotoxicity. Studies have also suggested that microglia can degrade and clear pathological tau (Luo et al., 2015; Bolós et al., 2016). For example, Luo et al. (2015) found that hyperphosphorylated pathological tau derived from brain tissue from patients with AD was rapidly internalized and degraded by microglia. We speculate that complement might also mediate microglial phagocytosis and clearance of pathological tau by the interaction of C3b/iC3b with the CR3 receptor on the microglia surface.
Our findings emphasize the complexity of the complement system. Therefore, more work is required to understand the dual nature of the complement system (namely, that it can exacerbate or attenuate neurodegenerative disease), especially for Crry and human CR1 in the CNS. Moreover, more experiments should be carried out to determine the activities of iC3b, CR3, C3a, C5a, and the terminal complement complex, as well as their associations with microglial priming and activation. Additionally, studies have reported that Crry is involved in regulation of CD4+ T cell activation and acts as a costimulatory molecule to elevate T cell receptor-dependent activation signals (such as extracellular signal-regulated kinase and protein kinase B) and costimulation-dependent kinases (such as MAPK and JNK) (Fernández-Centeno et al., 2000; Jiménez-Periañez et al., 2005). MAPK and JNK were investigated in this study because of their roles in tau phosphorylation. To comprehensively understand the role of Crry, the effects of Crry on immunobiology or other biological processes should also be further investigated.
Our study’s design and methods had some limitations. We performed all experiments in vivo, and no in vitro experiments were designed. We used quantitative real-time polymerase chain reaction for the detection of inflammatory cytokines, but we did not use western blot on tissue lysates or enzyme-linked immunosorbent assays on tissue homogenates to investigate inflammatory cytokines.
In conclusion, Crry might play an important role in the P301S tauopathy mouse model, and Crry downregulation might be associated with modulation of microglial activation, neuroinflammation, and the complement system. Our study indicates that complement dysregulation is likely to be important in neurodegenerative diseases. Drugs that modulate complement activation may be repurposed, or brain-penetrant complement therapeutics could be developed for the treatment of neurodegenerative diseases.
Additional files:
Addtional Table 1: Antibodies used in western blot and double immunofluorescence staining.
Additional Table 2: Primer sequences used in this study.
Additional Figure 1 (1.5MB, tif) : The detailed experimental protocol.
The detailed experimental protocol.
CR1: Complement component 3b/4b receptor 1; Crry: Cr1-related protein Y; shRNA: short hairpin RNA; WT: wild type.
Additional Figure 2 (2MB, tif) : Immunofluorescence staining to determine the expression of Crry in cortex and hippocampus of 6-month-old WT and P301S mice.
Immunofluorescence staining to determine the expression of Crry in cortex and hippocampus of 6-month-old WT and P301S mice.
(A) Representative images of immunofluorescence staining (original magnification, 200×) and quantified for Crry immunopositivity in cortex of WT and P301S mice. (B) Representative images of immunofluorescence staining (original magnification, 400×) and quantified for Crry immunopositivity in hippocampus of WT and P301S mice. The arrows indicate the merged fluorescent signal from green and red fluorescent signals. Immunofluorescence staining of Crry in these two brain regions were obvioulsly increased in P301S mice versus to that of age-matched WT mice. Data are presented as the mean ± SEM (n = 6 for each group), and analyzed by independent sample t-test. Crry: Cr1-related protein Y; DAPI: 4‘,6-diamidino-2-phenylindole; Iba-1: ionized calcium binding adaptor molecule 1; WT: wild type.
Additional file 1: Open peer review reports 1 (103.9KB, pdf) and 2 (112.5KB, pdf) .
Footnotes
Conflicts of interest: The authors declare that they have no competing interests.
Availability of data and materials: All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open peer reviewers: Tim Hughes, Siemens plc, UK; Megan Torvell, Cardiff University, UK; Claudia Bregonzio, Universidad Nacional de Cordoba, Argentina.
Funding: This study was supported by the National Natural Science Foundation of China (No. 81801054), the Natural Science Foundation of Jiangsu Province of China (No. BK20180166), the Wuxi Municipal Health and Family Planning Commission Fund of China (No. Q201722), Wuxi Top Talent Support Program for Young and Middle-aged People of Wuxi Health Committee of China (No. HB2020023), and China Postdoctoral Funding (all to XCZ).
P-Reviewers: Hughes T, P Torvell M, Bregonzio C; C-Editor: Zhao M; S-Editors: Yu J, Li CH; L-Editors: Yu J, Song LP; T-Editor: Jia Y
References
- 1.Alzheimer A. Über einen eigenartigen Schweren Erkrankungsprozeβ der Hirnrinde. Neurolog Centralbl. 1906;23:1129–1136. [Google Scholar]
- 2.Bajic G, Degn SE, Thiel S, Andersen GR. Complement activation, regulation, and molecular basis for complement-related diseases. EMBO J. 2015;34:2735–2757. doi: 10.15252/embj.201591881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bao L, Wang Y, Chang A, Minto AW, Zhou J, Kang H, Haas M, Quigg RJ. Unrestricted C3 activation occurs in Crry-deficient kidneys and rapidly leads to chronic renal failure. J Am Soc Nephrol. 2007;18:811–822. doi: 10.1681/ASN.2006101176. [DOI] [PubMed] [Google Scholar]
- 4.Biffi A, Shulman JM, Jagiella JM, Cortellini L, Ayres AM, Schwab K, Brown DL, Silliman SL, Selim M, Worrall BB, Meschia JF, Slowik A, De Jager PL, Greenberg SM, Schneider JA, Bennett DA, Rosand J. Genetic variation at CR1 increases risk of cerebral amyloid angiopathy. Neurology. 2012;78:334–341. doi: 10.1212/WNL.0b013e3182452b40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bolós M, Llorens-Martín M, Jurado-Arjona J, Hernández F, Rábano A, Avila J. Direct evidence of internalization of tau by microglia in vitro and in vivo. J Alzheimers Dis. 2016;50:77–87. doi: 10.3233/JAD-150704. [DOI] [PubMed] [Google Scholar]
- 6.Bronstein JM, Kozak CA, Chen XN, Wu S, Danciger M, Korenberg JR, Farber DB. Chromosomal localization of murine and human oligodendrocyte-specific protein genes. Genomics. 1996;34:255–257. doi: 10.1006/geno.1996.0278. [DOI] [PubMed] [Google Scholar]
- 7.Choi SH, Kim YH, Hebisch M, Sliwinski C, Lee S, D’Avanzo C, Chen H, Hooli B, Asselin C, Muffat J, Klee JB, Zhang C, Wainger BJ, Peitz M, Kovacs DM, Woolf CJ, Wagner SL, Tanzi RE, Kim DY. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature. 2014;515:274–278. doi: 10.1038/nature13800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cleveland DW, Hwo SY, Kirschner MW. Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J Mol Biol. 1977;116:207–225. doi: 10.1016/0022-2836(77)90213-3. [DOI] [PubMed] [Google Scholar]
- 9.Corneveaux JJ, Myers AJ, Allen AN, Pruzin JJ, Ramirez M, Engel A, Nalls MA, Chen K, Lee W, Chewning K, Villa SE, Meechoovet HB, Gerber JD, Frost D, Benson HL, O’Reilly S, Chibnik LB, Shulman JM, Singleton AB, Craig DW, et al. Association of CR1, CLU and PICALM with Alzheimer’s disease in a cohort of clinically characterized and neuropathologically verified individuals. Hum Mol Genet. 2010;19:3295–3301. doi: 10.1093/hmg/ddq221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cortés N, Guzmán-Martínez L, Andrade V, González A, Maccioni RB. CDK5: a unique CDK and its multiple roles in the nervous system. J Alzheimers Dis. 2019;68:843–855. doi: 10.3233/JAD-180792. [DOI] [PubMed] [Google Scholar]
- 11.Davoust N, Nataf S, Holers VM, Barnum SR. Expression of the murine complement regulatory protein crry by glial cells and neurons. Glia. 1999;27:162–170. doi: 10.1002/(sici)1098-1136(199908)27:2<162::aid-glia6>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 12.Dejanovic B, Huntley MA, De Mazière A, Meilandt WJ, Wu T, Srinivasan K, Jiang Z, Gandham V, Friedman BA, Ngu H, Foreman O, Carano RAD, Chih B, Klumperman J, Bakalarski C, Hanson JE, Sheng M. Changes in the synaptic proteome in tauopathy and rescue of tau-induced synapse loss by C1q antibodies. Neuron. 2018;100:1322–1336.e7. doi: 10.1016/j.neuron.2018.10.014. [DOI] [PubMed] [Google Scholar]
- 13.Dodart JC, Marr RA, Koistinaho M, Gregersen BM, Malkani S, Verma IM, Paul SM. Gene delivery of human apolipoprotein E alters brain Abeta burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2005;102:1211–1216. doi: 10.1073/pnas.0409072102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duan W, Zhang YP, Hou Z, Huang C, Zhu H, Zhang CQ, Yin Q. Novel insights into neun: from neuronal marker to splicing regulator. Mol Neurobiol. 2016;53:1637–1647. doi: 10.1007/s12035-015-9122-5. [DOI] [PubMed] [Google Scholar]
- 15.Dumont M, Stack C, Elipenahli C, Jainuddin S, Gerges M, Starkova NN, Yang L, Starkov AA, Beal F. Behavioral deficit, oxidative stress, and mitochondrial dysfunction precede tau pathology in P301S transgenic mice. FASEB J. 2011;25:4063–4072. doi: 10.1096/fj.11-186650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Efthymiou AG, Goate AM. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol Neurodegener. 2017;12:43. doi: 10.1186/s13024-017-0184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fernández-Centeno E, de Ojeda G, Rojo JM, Portolés P. Crry/p65, a membrane complement regulatory protein, has costimulatory properties on mouse T cells. J Immunol. 2000;164:4533–4542. doi: 10.4049/jimmunol.164.9.4533. [DOI] [PubMed] [Google Scholar]
- 18.Fu H, Liu B, Frost JL, Hong S, Jin M, Ostaszewski B, Shankar GM, Costantino IM, Carroll MC, Mayadas TN, Lemere CA. Complement component C3 and complement receptor type 3 contribute to the phagocytosis and clearance of fibrillar Aβ by microglia. Glia. 2012;60:993–1003. doi: 10.1002/glia.22331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 20.Hernández F, de Barreda EG, Fuster-Matanzo A, Goñi-Oliver P, Lucas JJ, Avila J. The role of GSK3 in Alzheimer disease. Brain Res Bull. 2009;80:248–250. doi: 10.1016/j.brainresbull.2009.05.017. [DOI] [PubMed] [Google Scholar]
- 21.Hou TY, Zhou Y, Zhu LS, Wang X, Pang P, Wang DQ, Liuyang ZY, Man H, Lu Y, Zhu LQ, Liu D. Correcting abnormalities in miR-124/PTPN1 signaling rescues tau pathology in Alzheimer’s disease. J Neurochem. 2020;154:441–457. doi: 10.1111/jnc.14961. [DOI] [PubMed] [Google Scholar]
- 22.Iba M, Guo JL, McBride JD, Zhang B, Trojanowski JQ, Lee VM. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J Neurosci. 2013;33:1024–1037. doi: 10.1523/JNEUROSCI.2642-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Izzy S, Brown-Whalen A, Yahya T, Sarro-Schwartz A, Jin G, Chung JY, Lule S, Morsett LM, Alquraini A, Wu L. Repetitive traumatic brain injury causes neuroinflammation before tau pathology in adolescent P301S mice. Int J Mol Sci. 2021;22:907. doi: 10.3390/ijms22020907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jacobson AC, Weis JH. Comparative functional evolution of human and mouse CR1 and CR2. J Immunol. 2008;181:2953–2959. doi: 10.4049/jimmunol.181.5.2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang T, Yu JT, Zhu XC, Tan MS, Gu LZ, Zhang YD, Tan L. Triggering receptor expressed on myeloid cells 2 knockdown exacerbates aging-related neuroinflammation and cognitive deficiency in senescence-accelerated mouse prone 8 mice. Neurobiol Aging. 2014;35:1243–1251. doi: 10.1016/j.neurobiolaging.2013.11.026. [DOI] [PubMed] [Google Scholar]
- 26.Jiang T, Zhang YD, Chen Q, Gao Q, Zhu XC, Zhou JS, Shi JQ, Lu H, Tan L, Yu JT. TREM2 modifies microglial phenotype and provides neuroprotection in P301S tau transgenic mice. Neuropharmacology. 2016;105:196–206. doi: 10.1016/j.neuropharm.2016.01.028. [DOI] [PubMed] [Google Scholar]
- 27.Jiménez-Periañez A, Ojeda G, Criado G, Sánchez A, Pini E, Madrenas J, Rojo JM, Portolés P. Complement regulatory protein Crry/p65-mediated signaling in T lymphocytes: role of its cytoplasmic domain and partitioning into lipid rafts. J Leukoc Biol. 2005;78:1386–1396. doi: 10.1189/jlb.1104642. [DOI] [PubMed] [Google Scholar]
- 28.Kidd M. Paired helical filaments in electron microscopy of Alzheimer’s disease. Nature. 1963;197:192–193. doi: 10.1038/197192b0. [DOI] [PubMed] [Google Scholar]
- 29.Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Killick R, Hughes TR, Morgan BP, Lovestone S. Deletion of Crry, the murine ortholog of the sporadic Alzheimer’s disease risk gene CR1, impacts tau phosphorylation and brain CFH. Neurosci Lett. 2013;533:96–99. doi: 10.1016/j.neulet.2012.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim YU, Kinoshita T, Molina H, Hourcade D, Seya T, Wagner LM, Holers VM. Mouse complement regulatory protein Crry/p65 uses the specific mechanisms of both human decay-accelerating factor and membrane cofactor protein. J Exp Med. 1995;181:151–159. doi: 10.1084/jem.181.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kimura T, Ishiguro K, Hisanaga S. Physiological and pathological phosphorylation of tau by Cdk5. Front Mol Neurosci. 2014;7:65. doi: 10.3389/fnmol.2014.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krance SH, Wu CY, Zou Y, Mao H, Toufighi S, He X, Pakosh M, Swardfager W. The complement cascade in Alzheimer’s disease: a systematic review and meta-analysis. Mol Psychiatry. 2019 doi: 10.1038/s41380-019-0536-8. doi: 101038/s41380-019-0536-8. [DOI] [PubMed] [Google Scholar]
- 34.Kucukkilic E, Brookes K, Barber I, Guetta-Baranes T, Morgan K, Hollox EJ. Complement receptor 1 gene (CR1) intragenic duplication and risk of Alzheimer’s disease. Hum Genet. 2018;137:305–314. doi: 10.1007/s00439-018-1883-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kumaran D, Udayabanu M, Kumar M, Aneja R, Katyal A. Involvement of angiotensin converting enzyme in cerebral hypoperfusion induced anterograde memory impairment and cholinergic dysfunction in rats. Neuroscience. 2008;155:626–639. doi: 10.1016/j.neuroscience.2008.06.023. [DOI] [PubMed] [Google Scholar]
- 36.Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, Boland A, Vronskaya M, van der Lee SJ, Amlie-Wolf A, Bellenguez C, Frizatti A, Chouraki V, Martin ER, Sleegers K, Badarinarayan N, Jakobsdottir J, Hamilton-Nelson KL, Moreno-Grau S, Olaso R, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet. 2019;51:414–430. doi: 10.1038/s41588-019-0358-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee JD, Coulthard LG, Woodruff TM. Complement dysregulation in the central nervous system during development and disease. Semin Immunol. 2019;45:101340. doi: 10.1016/j.smim.2019.101340. [DOI] [PubMed] [Google Scholar]
- 38.Litvinchuk A, Wan YW, Swartzlander DB, Chen F, Cole A, Propson NE, Wang Q, Zhang B, Liu Z, Zheng H. Complement C3aR inactivation attenuates tau pathology and reverses an immune network deregulated in tauopathy models and Alzheimer’s disease. Neuron. 2018;100:1337–1353.e5. doi: 10.1016/j.neuron.2018.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu SL, Wang C, Jiang T, Tan L, Xing A, Yu JT. The role of Cdk5 in Alzheimer’s disease. Mol Neurobiol. 2016;53:4328–4342. doi: 10.1007/s12035-015-9369-x. [DOI] [PubMed] [Google Scholar]
- 40.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 41.Lo MW, Lee JD. Complement: a global immunometabolic regulator in amyotrophic lateral sclerosis. Neural Regen Res. 2021;16:1210–1211. doi: 10.4103/1673-5374.300441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luo W, Liu W, Hu X, Hanna M, Caravaca A, Paul SM. Microglial internalization and degradation of pathological tau is enhanced by an anti-tau monoclonal antibody. Sci Rep. 2015;5:11161. doi: 10.1038/srep11161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Molina H, Wong W, Kinoshita T, Brenner C, Foley S, Holers VM. Distinct receptor and regulatory properties of recombinant mouse complement receptor 1 (CR1) and Crry, the two genetic homologues of human CR1. J Exp Med. 1992;175:121–129. doi: 10.1084/jem.175.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ohsawa K, Imai Y, Sasaki Y, Kohsaka S. Microglia/macrophage-specific protein Iba1 binds to fimbrin and enhances its actin-bundling activity. J Neurochem. 2004;88:844–856. doi: 10.1046/j.1471-4159.2003.02213.x. [DOI] [PubMed] [Google Scholar]
- 45.Parker SE, Hanton AM, Stefanou SN, Noakes PG, Woodruff TM, Lee JD. Revisiting the role of the innate immune complement system in ALS. Neurobiol Dis. 2019;127:223–232. doi: 10.1016/j.nbd.2019.03.003. [DOI] [PubMed] [Google Scholar]
- 46.Ramaglia V, Hughes TR, Donev RM, Ruseva MM, Wu X, Huitinga I, Baas F, Neal JW, Morgan BP. C3-dependent mechanism of microglial priming relevant to multiple sclerosis. Proc Natl Acad Sci U S A. 2012;109:965–970. doi: 10.1073/pnas.1111924109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to beta -amyloid-induced neurotoxicity. Proc Natl Acad Sci U S A. 2002;99:6364–6369. doi: 10.1073/pnas.092136199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 49.Rogers J, Li R, Mastroeni D, Grover A, Leonard B, Ahern G, Cao P, Kolody H, Vedders L, Kolb WP, Sabbagh M. Peripheral clearance of amyloid beta peptide by complement C3-dependent adherence to erythrocytes. Neurobiol Aging. 2006;27:1733–1739. doi: 10.1016/j.neurobiolaging.2005.09.043. [DOI] [PubMed] [Google Scholar]
- 50.Ruan Z, Delpech JC, Venkatesan Kalavai S, Van Enoo AA, Hu J, Ikezu S, Ikezu T. P2RX7 inhibitor suppresses exosome secretion and disease phenotype in P301S tau transgenic mice. Mol Neurodegener. 2020;15:47. doi: 10.1186/s13024-020-00396-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ruseva MM, Hughes TR, Donev RM, Sivasankar B, Pickering MC, Wu X, Harris CL, Morgan BP. Crry deficiency in complement sufficient mice: C3 consumption occurs without associated renal injury. Mol Immunol. 2009;46:803–811. doi: 10.1016/j.molimm.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 52.Saito T, Oba T, Shimizu S, Asada A, Iijima KM, Ando K. Cdk5 increases MARK4 activity and augments pathological tau accumulation and toxicity through tau phosphorylation at Ser262. Hum Mol Genet. 2019;28:3062–3071. doi: 10.1093/hmg/ddz120. [DOI] [PubMed] [Google Scholar]
- 53.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shi Q, Chowdhury S, Ma R, Le KX, Hong S, Caldarone BJ, Stevens B, Lemere CA. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci Transl Med. 2017;9 doi: 10.1126/scitranslmed.aaf6295. eaaf6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 56.Takizawa T, Gudla PR, Guo L, Lockett S, Misteli T. Allele-specific nuclear positioning of the monoallelically expressed astrocyte marker GFAP. Genes Dev. 2008;22:489–498. doi: 10.1101/gad.1634608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tang Y, Le W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol Neurobiol. 2016;53:1181–1194. doi: 10.1007/s12035-014-9070-5. [DOI] [PubMed] [Google Scholar]
- 58.Thambisetty M, An Y, Nalls M, Sojkova J, Swaminathan S, Zhou Y, Singleton AB, Wong DF, Ferrucci L, Saykin AJ, Resnick SM Baltimore Longitudinal Study of Aging and the Alzheimer’s Disease Neuroimaging Initiative. Effect of complement CR1 on brain amyloid burden during aging and its modification by APOE genotype. Biol Psychiatry. 2013;73:422–428. doi: 10.1016/j.biopsych.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Villegas-Llerena C, Phillips A, Garcia-Reitboeck P, Hardy J, Pocock JM. Microglial genes regulating neuroinflammation in the progression of Alzheimer’s disease. Curr Opin Neurobiol. 2016;36:74–81. doi: 10.1016/j.conb.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 60.Wei J, Yuen EY, Liu W, Li X, Zhong P, Karatsoreos IN, McEwen BS, Yan Z. Estrogen protects against the detrimental effects of repeated stress on glutamatergic transmission and cognition. Mol Psychiatry. 2014;19:588–598. doi: 10.1038/mp.2013.83. [DOI] [PubMed] [Google Scholar]
- 61.Wu T, Dejanovic B, Gandham VD, Gogineni A, Edmonds R, Schauer S, Srinivasan K, Huntley MA, Wang Y, Wang TM, Hedehus M, Barck KH, Stark M, Ngu H, Foreman O, Meilandt WJ, Elstrott J, Chang MC, Hansen DV, Carano RAD, et al. Complement C3 is activated in human AD brain and is required for neurodegeneration in mouse models of amyloidosis and tauopathy. Cell Rep. 2019;28:2111–2123e6. doi: 10.1016/j.celrep.2019.07.060. [DOI] [PubMed] [Google Scholar]
- 62.Xu C, Mao D, Holers VM, Palanca B, Cheng AM, Molina H. A critical role for murine complement regulator crry in fetomaternal tolerance. Science. 2000;287:498–501. doi: 10.1126/science.287.5452.498. [DOI] [PubMed] [Google Scholar]
- 63.Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 64.Yang MS, Xu XJ, Zhang B, Niu F, Liu BY. Comparative transcriptomic analysis of rat versus mouse cerebral cortex after traumatic brain injury. Neural Regen Res. 2021;16:1235–1243. doi: 10.4103/1673-5374.301028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhu XC, Wang HF, Jiang T, Lu H, Tan MS, Tan CC, Tan L, Tan L, Yu JT Alzheimer’s Disease Neuroimaging Initiative. Effect of CR1 genetic variants on cerebrospinal fluid and neuroimaging biomarkers in healthy, mild cognitive impairment and Alzheimer’s disease cohorts. Mol Neurobiol. 2017;54:551–562. doi: 10.1007/s12035-015-9638-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The detailed experimental protocol.
CR1: Complement component 3b/4b receptor 1; Crry: Cr1-related protein Y; shRNA: short hairpin RNA; WT: wild type.
Immunofluorescence staining to determine the expression of Crry in cortex and hippocampus of 6-month-old WT and P301S mice.
(A) Representative images of immunofluorescence staining (original magnification, 200×) and quantified for Crry immunopositivity in cortex of WT and P301S mice. (B) Representative images of immunofluorescence staining (original magnification, 400×) and quantified for Crry immunopositivity in hippocampus of WT and P301S mice. The arrows indicate the merged fluorescent signal from green and red fluorescent signals. Immunofluorescence staining of Crry in these two brain regions were obvioulsly increased in P301S mice versus to that of age-matched WT mice. Data are presented as the mean ± SEM (n = 6 for each group), and analyzed by independent sample t-test. Crry: Cr1-related protein Y; DAPI: 4‘,6-diamidino-2-phenylindole; Iba-1: ionized calcium binding adaptor molecule 1; WT: wild type.