

The term serpinopathies was introduced to describe a family of diseases caused by point mutations in serine protease inhibitors, or serpins. Serpins inhibit their cognate protease by an irreversible suicide mechanism starting with the attack of the active site serine on the reactive center loop of the inhibitor, followed by formation of a covalent complex between both proteins, insertion of the reactive center loop of the serpin into its own beta-sheet A, and culminating in distortion of the active site of the serine protease and thus irreversible inactivation. This inhibitory mechanism, reminiscent of the movement of a mousetrap, requires a structural flexibility that proves to be unfavorable when the folding of the serpin is altered by mutations responsible for conformational rearrangements, allowing an intermolecular domain exchange characterized by the insertion of the C-terminal domain of a molecule into a second one, thus forming a dimer. Expansion of this insertion reaction leads to the formation of serpin polymers that accumulate within the endoplasmic reticulum (ER) of the cell and consequently reduces the secretion of the wild-type serpin (Greene et al., 2016).
Alpha1-antitrypsin (AAT) is the best-characterized member of the serpin family. This serpin is synthesized prevalently by hepatocytes in the liver, secreted into the circulation and transported to the lung where it protects the tissue against the proteolytic activity of neutrophil elastase (Figure 1; left panel). AAT is also produced by other cell types including monocyte-derived macrophages and dendritic cells and alveolar macrophages, exerting a variety of inhibitory and noninhibitory functions across different tissues, including immunomodulatory roles. AAT deficiency is a genetic disease caused by mutations that lead to decreased AAT activity in plasma. Inactivating (dysfunctional) mutations as well as mutations that prevent correct production and secretion of AAT, either by promoting complete degradation of AAT (null mutations) or by causing its intracellular polymerization, have been described. The most common disease variant is the Z allele (E342K) with a high prevalence in northern and western Europe and the USA. This mutation is responsible for a structural destabilization and, although most of the protein is degraded within the cell, part of it accumulates within the ER of hepatocytes as ordered polymers, while a small fraction is secreted. In fact, circulating polymers have been detected in the plasma of patients carrying the Z and other AAT polymerogenic mutations. Clinically, these mutations in AAT are responsible for a gain-of-function phenotype in the liver, which in some cases manifests as neonatal hepatitis or, in adults, as cirrhosis and hepatocellular carcinoma, as well as a loss-of-function phenotype caused by reduced levels of circulating active AAT, resulting in uncontrolled neutrophil elastase activity and leading to early-onset lung emphysema in Z-AAT homozygotes (Greene et al., 2016).
Figure 1.
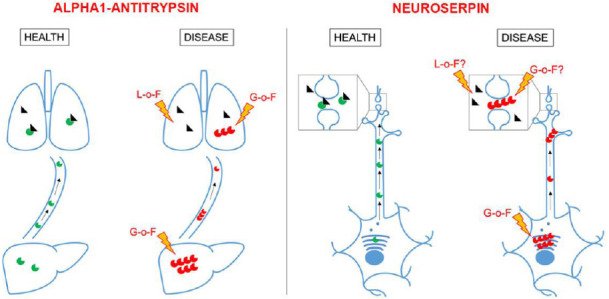
Physiological and pathological roles of alpha1-antitrypsin and neuroserpin.
Alpha1-antitrypsin is synthesized in the liver and transported to the lung where it inhibits neutrophil elastase. Mutations that destabilize its structure are responsible for polymer accumulation in hepatocytes and lung epithelial cells (G-o-F, gain-of-function) and reduced inhibition of neutrophil elastase in the lung (L-o-F, loss-of-function). Similarly, neuroserpin is synthesized in the endoplasmic reticulum of neurons, transported along axons and dendrites and secreted at synapses where it regulates the activity of tissue-plasminogen activator. Polymerogenic mutations lead to polymer accumulation in the endoplasmic reticulum (toxic G-o-F) and reduced neuroserpin secretion. If increased tissue-plasminogen activator activity (L-o-F) and/or extracellular polymer deposition (G-o-F) aggravate the pathomechanism of familial encephalopathy with neuroserpin inclusion bodies (FENIB) is still unknown. Green circle: Wild-type serpin; red circle: mutant serpin; black triangle: serine protease.
Familial encephalopathy with neuroserpin inclusion bodies (FENIB) is a serpinopathy affecting the nervous system. The disease, first described 1999 by Davis and colleagues (Davis et al., 1999), is an autosomal dominantly inherited neurodegenerative disease with cognitive decline, tremor and seizures, resulting in pre-senile dementia and death. Histological characterization of the patient’s brain revealed neuronal accumulation of inclusion bodies (IB), called Collins bodies, in several cortical and subcortical regions, whereas biochemical analysis showed that they consisted only of neuroserpin. Neuroserpin is an extracellular inhibitor of the serine protease tissue-plasminogen activator (tPA) mainly expressed in neurons. Since the first description of FENIB, six different point mutations have been identified as responsible for amino acid substitutions affecting the opening of the so-called shutter region, essential to the inhibitory mechanism and consequently to the conformational stability of this serpin. As for other serpinopathies, a correlation between conformational rearrangement triggered by the mutations, magnitude of intracellular protein deposition/reduction in secretion and severity of the disease has been observed (Lee et al., 2015). However, compared to AAT-deficiency, to attribute the pathophysiology of FENIB to gain-of-function or loss-of-function effects or both is complicated by the nature of neuroserpin expression and function. In the nervous system, neuroserpin is synthesized and secreted by neurons, and, unlike AAT, is mainly active at the site of synthesis. Although a toxic gain-of-function phenotype elicited by the accumulation of the mutant protein has been demonstrated in cell and animal models of FENIB, as described below, reduced secretion of wild-type neuroserpin has been hypothesized to disrupt the balance between serine protease and serpin, thus favoring an uncontrolled activity of tPA that could manifest in the seizure phenotype observed in FENIB patients (Figure 1; right panel). In fact, transgenic mice overexpressing human G392E-mutant neuroserpin were shown to be more susceptible to kainite-induced seizures (Takasawa et al., 2008). However, these mice have normal levels of murine neuroserpin, arguing against a possible loss-of-function phenotype. Furthermore, neither increased tPA activity, nor seizures or neuronal degeneration have been observed in neuroserpin-deficient mice so far. The possibility of a compensatory effect by another serpin is often discussed, the best candidate being plasminogen activator inhibitor-1, the serpin that regulates the activity of tPA in the circulation. However, plasminogen activator inhibitor-1 is present at very low levels in the brain under physiological conditions and analysis of the neocortex of neuroserpin-deficient mice did not reveal a compensatory upregulation of this serpin (Lee et al., 2015). The absence of neuroserpin in mice affects maturation and plasticity of hippocampal synapses, resulting in deficits in cognitive, emotional and social behaviour (Reumann et al., 2017), but a similar investigation has never been performed in a FENIB mouse model, therefore the contribution of the loss-of-function to FENIB remains to be investigated.
Many efforts have been conducted in the last twenty years to discover the pathomechanisms of FENIB using both cell culture and animal models. Overexpression of mutant neuroserpin has been performed in several cell lines of neuronal and non-neuronal origin, in which polymerization, IB formation within the ER and reduced protein secretion have always been observed (Miranda et al., 2008; Guadagno et al., 2017). FENIB mouse models recapitulating disease progression showed neuroserpin polymerization and ER-accumulation of intraneuronal IB in a time-, dose- and mutation-dependent fashion throughout the disease, as well as neuronal loss and a mild induction of apoptosis (Galliciotti et al., 2007; Takasawa et al., 2008). Other animal models of FENIB, generated in D. melanogaster (Miranda et al., 2008) and C. elegans (Schipanski et al., 2013), presented with IB accumulation without apparent effects on survival, but a reduction in locomotor activity that correlated with polymer load was found in flies. Intracellular polymers accumulate even if part of the monomeric mutant neuroserpin is specifically cleared by ER-associated degradation, as seen in cell lines and in a mouse model of FENIB, and basal autophagy cooperates by degrading both wild-type and mutant neuroserpin, as demostrated in COS7 and PC12 cell lines (Kroeger et al., 2009; Schipanski et al., 2014). Furthermore, although experiments with the mouse and worm models showed limited activation of the unfolded protein response at an early stage of the disease (Schipanski et al., 2013), contradictory results were reported with cell lines overexpressing polymerogenic variants of neuroserpin, in which unfolded protein response activation was not observed but neuroserpin accumulation triggered an ER overload response characterized by activation of NF-kB (Davies et al., 2009). This discrepancy could be explained by the different conditions present in the models tested, with polymers accumulating in post-mitotic cells over a long time period in the animal models and only in the short-term in dividing cultured cells. Similar findings have also been described for Z-AAT, with unfolded protein response activation depending on the particular cell type and activation of NF-kB as a characteristic consequence of polymer accumulation. A component of the toxicity elicited by the FENIB mutations was suggested in a study demonstrating that neural progenitor cells differentiated to neurons and expressing G392E neuroserpin upregulated genes involved in oxidative stress and were more susceptible to apoptosis upon inhibition of the antioxidant defense (Guadagno et al., 2017).
Despite having partially characterized the mechanism of polymerization, the fate of the mutant protein and some crucial mechanisms of toxicity, further questions are still unanswered. Is the presence of IB toxic to cells, or does the sequestration of the mutant protein within these structures confer protection, similar to Alzheimer disease, where insoluble beta-amyloid aggregating in plaques is now considered less toxic than soluble oligomeric species? A possible answer is offered by a study showing that hepatoma cells in culture, despite accumulating mutant AAT within IB, maintained ER function, whereas overexpression of calnexin inhibited IB formation and led to cell shrinkage and deficits in protein secretion (Granell et al., 2008). This would suggest that the soluble mutant form of the serpin that is not efficiently degraded by ER-associated degradation and/or autophagy could be the toxic species. Another open question is whether the intracellular or the secreted form is the most harmful. Since the severity of FENIB goes along with decreased secretion of the mutant protein, one could intuitively conclude that cell toxicity is caused by the intracellular form of the protein. Still, the possibility that the presence of mutant neuroserpin in the extracellular milieu aggravates the toxic phenotype had not been tested so far. Neuroserpin sorting has been investigated in primary neurons, where it is transported within large dense core vesicles along dendrites and axons, and is secreted perisynaptically (Lee et al., 2015). If the mutant protein shares the same fate, it might exert its toxicity at synapses. The idea of a synaptic phenotype being caused by extracellular deposition of pathogenic, misfolded proteins is not new. This concept has been described for several neurodegenerative diseases including Alzheimer’s disease, the tauopathies and prion diseases. Because synaptic pathology occurs at early stages of dementia, targeting synaptic loss might retard disease progression and ameliorate the symptomatology of the affected patients. Therefore, we have investigated the secretion pathway and the effects on the synapse of mutant neuroserpin in a recently published manuscript (Ingwersen et al., 2021). We observed that G392E-mutant neuroserpin, like its wild-type counterpart, was sorted along the ER-to-Golgi secretory pathway. Furthermore, using an in vitro model consisting of cultured hippocampal neurons, previously proven to reproduce synaptotoxicity in prion and Alzheimer’s diseases, we tested if exposure to the mutant serpin would lead to a synaptic phenotype, but we failed to observe this. This contradicts the dysregulated expression of the post-synaptic marker protein PSD-95 that we found in the hippocampus of aged FENIB mice, a feature also described in the hippocampus of neuroserpin-deficient mice, in which synaptic deficits have been observed at functional and behavioural levels (Reumann et al., 2017). The relevance of these findings for FENIB is still unknown, further research on the physiological functions of neuroserpin is needed to understand the contribution of gain-of-function and/or loss-of-function mechanisms to FENIB disease. The knowledge generated through these studies will also be informative for other serpinopathies and for other neurodegenerative diseases.
Footnotes
C-Editors: Zhao M, Liu WJ, Qiu Y; T-Editor: Jia Y
References
- 1.Davies MJ, Miranda E, Roussel BD, Kaufman RJ, Marciniak SJ, Lomas DA. Neuroserpin polymers activate NF-kappaB by a calcium signaling pathway that is independent of the unfolded protein response. J Biol Chem. 2009;284:18202–18209. doi: 10.1074/jbc.M109.010744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davis RL, Shrimpton AE, Holohan PD, Bradshaw C, Feiglin D, Collins GH, Sonderegger P, Kinter J, Becker LM, Lacbawan F, Krasnewich D, Muenke M, Lawrence DA, Yerby MS, Shaw CM, Gooptu B, Elliott PR, Finch JT, Carrell RW, Lomas DA. Familial dementia caused by polymerization of mutant neuroserpin. Nature. 1999;401:376–379. doi: 10.1038/43894. [DOI] [PubMed] [Google Scholar]
- 3.Galliciotti G, Glatzel M, Kinter J, Kozlov SV, Cinelli P, Rulicke T, Sonderegger P. Accumulation of mutant neuroserpin precedes development of clinical symptoms in familial encephalopathy with neuroserpin inclusion bodies. Am J Pathol. 2007;170:1305–1313. doi: 10.2353/ajpath.2007.060910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Granell S, Baldini G, Mohammad S, Nicolin V, Narducci P, Storrie B. Sequestration of mutated alpha1-antitrypsin into inclusion bodies is a cell-protective mechanism to main-tain endoplasmic reticulum function. Mol Biol Cell. 2008;19:572–586. doi: 10.1091/mbc.E07-06-0587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greene CM, Marciniak SJ, Teckman J, Ferrarotti I, Brantly ML, Lomas DA, Stoller JK, McEl-vaney NG. alpha1-Antitrypsin deficiency. Nat Rev Dis Primers. 2016;2:16051. doi: 10.1038/nrdp.2016.51. [DOI] [PubMed] [Google Scholar]
- 6.Guadagno NA, Moriconi C, Licursi V, D’Acunto E, Nisi PS, Carucci N, De Jaco A, Cacci E, Negri R, Lupo G, Miranda E. Neuroserpin polymers cause oxidative stress in a neuronal model of the dementia FENIB. Neurobiol Dis. 2017;103:32–44. doi: 10.1016/j.nbd.2017.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ingwersen T, Linnenberg C, D’Acunto E, Temori S, Paolucci I, Wasilewski D, Mohammadi B, Kirchmair J, Glen RC, Miranda E, Glatzel M, Galliciotti G. G392E neuroserpin causing the dementia FENIB is secreted from cells but is not synaptotoxic. Sci Rep. 2021;11:8766. doi: 10.1038/s41598-021-88090-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kroeger H, Miranda E, MacLeod I, Perez J, Crowther DC, Marciniak SJ, Lomas DA. Endoplasmic reticulum-associated degradation (ERAD) and autophagy cooperate to degrade polymerogenic mutant serpins. J Biol Chem. 2009;284:22793–22802. doi: 10.1074/jbc.M109.027102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee TW, Tsang VW, Birch NP. Physiological and pathological roles of tissue plasminogen activator and its inhibitor neuroserpin in the nervous system. Front Cell Neurosci. 2015;9:396. doi: 10.3389/fncel.2015.00396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miranda E, MacLeod I, Davies MJ, Perez J, Romisch K, Crowther DC, Lomas DA. The intracellular accumulation of polymeric neuroserpin explains the severity of the dementia FENIB. Hum Mol Genet. 2008;17:1527–1539. doi: 10.1093/hmg/ddn041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reumann R, Vierk R, Zhou L, Gries F, Kraus V, Mienert J, Romswinkel E, Morellini F, Ferrer I, Nicolini C, Fahnestock M, Rune G, Glatzel M, Galliciotti G. The serine protease inhibitor neuroserpin is required for normal synaptic plasticity and regulates learning and social behavior. Learn Mem. 2017;24:650–659. doi: 10.1101/lm.045864.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schipanski A, Lange S, Segref A, Gutschmidt A, Lomas DA, Miranda E, Schweizer M, Hoppe T, Glatzel M. A novel interaction between aging and ER overload in a protein conformational dementia. Genetics. 2013;193:865–876. doi: 10.1534/genetics.112.149088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schipanski A, Oberhauser F, Neumann M, Lange S, Szalay B, Krasemann S, van Leeuwen FW, Galliciotti G, Glatzel M. The lectin OS-9 delivers mutant neuroserpin to endoplasmic reticulum associated degradation in familial encephalopathy with neuroserpin inclusion bodies. Neurobiol Aging. 2014;35:2394–2403. doi: 10.1016/j.neurobiolaging.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 14.Takasawa A, Kato I, Takasawa K, Ishii Y, Yoshida T, Shehata MH, Kawaguchi H, Mohafez OM, Sasahara M, Hiraga K. Mutation-, aging-, and gene dosage-dependent accu-mulation of neuroserpin (G392E) in endoplasmic reticula and lysosomes of neurons in transgenic mice. J Biol Chem. 2008;283:35606–35613. doi: 10.1074/jbc.M804125200. [DOI] [PubMed] [Google Scholar]