

ABSTRACT
Ulcerative colitis (UC) is a complex immune-mediated disease in which the gut microbiota plays a central role, and may determine prognosis and disease progression. We aimed to assess whether a specific microbiota profile, as measured by a machine learning approach, can be associated with disease severity in patients with UC. In this prospective pilot study, consecutive patients with active or inactive UC and healthy controls (HCs) were enrolled. Stool samples were collected for fecal microbiota assessment analysis by 16S rRNA gene sequencing approach. A machine learning approach was used to predict the groups’ separation. Thirty-six HCs and forty-six patients with UC (20 active and 26 inactive) were enrolled. Alpha diversity was significantly different between the three groups (Shannon index: p-values: active UC vs HCs = 0.0005; active UC vs inactive UC = 0.0273; HCs vs inactive UC = 0.0260). In particular, patients with active UC showed the lowest values, followed by patients with inactive UC, and HCs. At species level, we found high levels of Bifidobacterium adolescentis and Haemophilus parainfluenzae in inactive UC and active UC, respectively. A specific microbiota profile was found for each group and was confirmed with sparse partial least squares discriminant analysis, a machine learning-supervised approach. The latter allowed us to observe a perfect class prediction and group separation using the complete information (full Operational Taxonomic Unit table), with a minimal loss in performance when using only 5% of features. A machine learning approach to 16S rRNA data identifies a bacterial signature characterizing different degrees of disease activity in UC. Follow-up studies will clarify whether such microbiota profiling are useful for diagnosis and management.
KEYWORDS: Inflammatory bowel disease, microbiota, ulcerative colitis, machine learning
Introduction
Ulcerative colitis (UC) is a chronic disorder characterized by inflammation of the gastrointestinal tract, with relapsing and remitting phases, and is associated with a reduced quality of life.1–3 The pathogenesis is partially understood, but it has been hypothesized that it arises from dysregulation of the innate and adaptive immune systems,4 leading to an abnormal inflammatory response to commensal bacteria in a genetically susceptible individual.5 Therefore, a perturbation of the structure of the gut microbiota seems to play a key role in determining intestinal inflammation.
Recent investigations based on 16S rRNA gene sequencing showed significant differences between the microbiota of patients with inflammatory bowel disease (IBD) and healthy controls, suggesting a potential role of gut microbiota not only in the development, but also in determining prognosis and disease progression.6 In particular, the dysbiosis associated with UC is characterized by reduced bacterial diversity, a decline in Firmicutes such as Faecalibacterium prausnitzii and other short chain fatty acid (SCFA)-producing bacteria, and an increase in Proteobacteria.7–9 Despite technological advancements in microbiota analysis, such as next-generation sequencing, high-throughput omics data generation, and molecular networks opening up new horizons in microbial research, the complex relationship between the gut microbiota and IBD is poorly understood.10 Indeed, although a potential role of gut microbiota dysbiosis has been widely recognized in IBD, causality is yet to be established. In addition, data on the composition of the gut microbiota in patients with IBD vary widely among studies.11–13 Thus, most have failed to observe a specific microbiota signature in association with either IBD type or severity.14
Recently, machine learning models have grown in popularity among microbiome researchers because they can effectively account for the interpersonal microbiome variations and the ecology of disease.15 However, data in the field of IBD remain limited. A machine learning approach could assist in both the diagnosis and prediction of disease course of patients with IBD. Also, it could also be used to predict response to therapy and drug-related adverse effects.16 In addition, machine learning could be applied to profile stool samples revealing specific microbiota signatures to further study a disease specific causal link.16
Therefore, we decided to perform a pilot, monocentric, prospective study to assess whether a specific microbiota profile was associated with disease severity in patients with UC. Thus, also with the application of the available machine learning approaches, we investigated how the microbiota profile could be used as a noninvasive marker for monitoring and predicting outcomes in IBD.
Methods
Study populations, sample and data collection
We recruited consecutive patients with a histologically confirmed diagnosis of UC for at least 6 months, both with inactive and active disease, from the IBD Unit of Padova Hospital (Italy) from April 2019 to February 2020. Moreover, data about an historical control group of healthy controls (HCs) was used for analysis comparison.9 Inclusion and exclusion criteria are reported in Box 1.
Box 1.
Inclusion and exclusion criteria
|
Healthy Controls Inclusion Criteria: The Healthy controls enrolled were subjects of both sexes, aged ≥18 years, of Italian nationality, with no relatives with UC, and who did not present evidence of illness on the basis of the anamnestic data collected. |
Patients with Inactive Disease Inclusion Criteria
|
Patients with Active Disease Inclusion Criteria
|
UC Patients (Both Active and Inactive) Exclusion Criteria
|
For each patient, a stool sample was collected for fecal microbiota assessment analysis and for fecal calprotectin (FC) analysis. For patients with inactive UC, fecal samples were collected at home on the evening before or the morning of each visit and stored at 4°C. Upon arrival at the hospital, samples were frozen at −80°C, in all cases within 24 hours of defecation, for the analysis of microbiota. Similarly, fecal samples of HCs were collected at home on the evening before, or the morning of, its delivery to our laboratory, again within 24 hours of defecation. For patients with UC with moderately-to–severely active disease, fecal samples were collected at home if they were not hospitalized, or were collected within 24 hours of hospitalization in the case of hospitalized patients.
The following data were recorded for each patient with UC at baseline: age, gender, age at diagnosis, disease duration, disease location and extent, previous biological treatments, and presence of extraintestinal manifestations. Clinical activity was measured using a total and partial Mayo (p-Mayo) score, while the Mayo endoscopic subscore was used to assess endoscopic activity. For the purpose of the study, all endoscopic examinations were performed within 3 to 5 days of stool collection.
Sample processing and sequencing
For the microbiota analysis, the stool samples were solubilized and stabilized by degradation in Xpedition Buffer (Zymo Research) and stored at −80°C until analysis. Sequencing protocol was performed at BMR Genomics srl. Briefly: V3–V4 regions of 16S rRNA gene were amplified using the primers Pro341F: 5′-CCTACGGGNBGCASCAG-3′ and Pro805R: Rev 5′-GACTACNVGGGTATCTAATCC-3′.17 Primers were modified with forward overhang: 5′-TCGTCGGCAGC GTCAGATGTGTATAAGAGACAG [locus-specific sequence]-3′ and with reverse overhang: 5′-GTCTCGTGGGCTCGGAGATGTGTA TAAGAGACAG [locus-specific sequence]-3′ necessary for dual-index library preparation, following Illumina protocol https://web.uri.edu/gsc/files/16s-metagenomic-library-prep-guide-15044223-b.pdf. Samples were normalized, pooled, and run on Illumina MiSeq with 2 × 300 bp approach.
Fecal calprotectin levels were determined using the ELISA Buhlmann fCAL Turbo (Buhlmann Laboratories AG, Schonenbuch, Switzerland), known to perform with high sensitivity and specificity.18,19
Bioinformatic analysis and statistics
The raw reads underwent a filtering procedure performed within QIIME2 analysis framework (version 2020.2).20 The primer removal was done via cutadapt plugin, while the quality filtering, denoising and chimera checking steps were performed using DADA2 plugin. Alpha diversity was evaluated on rarefied counts (Richness, Shannon, and Pielou indices; rarefaction level: 28,366), while beta diversity was calculated on normalized counts (Bray-Curtis, Jaccard, Canberra, Weighted and Unweighted Unifrac; counts normalized with GMPR).21 The diversity analysis was conducted in R (version 3.6.3) using DiversitySeq package, and the statistical tests (Kruskal-Wallis) on differences in alpha diversity indices distributions between groups were performed using base R functions.
A permutational analysis of variance (PERMANOVA) test on Bay-Curtis dissimilarity was used to test for differences in the microbiota composition between disease status groups (vegan package). The ANCOM2 package was then used to perform differential abundance analysis at all taxonomic levels (a conservative detection threshold for differentially abundant taxa of 0.8 was chosen).22 Supervised and unsupervised machine learning algorithms were applied to normalized data to explore the possibility of grouping and classifying samples, and to identify the most important taxa for class discrimination. To this aim, we performed the following analyses: hierarchical clustering using Ward algorithm on Canberra distance, non-metric multidimensional scaling (NMDS) on Bray-Curtis distance, random forests (training set: 62 patients; test set: 20 patients) and sparse partial least squares discriminant analysis (sPLS-DA).
Machine learning approaches were also run in R, using stats, phyloseq,23 randomForest24 and mixOmics25 packages. With the SPLS-DA, a supervised machine learning approach, it is possible to discriminate Amplicon Sequence Variants (ASVs) that best characterize each group. sPLS-DA analysis identified a subset of discriminant ASVs: for each ASV, a loading value that represents the discriminant power of that ASV in explaining differences among the three different examined conditions (active UC, inactive UC, and HCs) was obtained.
Using a one-way analysis of variance (ANOVA) test we tested data for a possible association between disease status and FC values in patients to see whether FC levels could be considered as an identifier for disease groups and their associated microbiota. A post-hoc test (Tukey) was then performed to attribute the observed difference to sub-comparisons between disease statuses.
Finally, we compared demographic, clinical, and biochemical data between inactive and active UC using SPSS for Windows (version 24.0 SPSS Inc., Chicago, IL, USA). In particular, continuous variables were reported as medians with ranges, and categorical variables as frequencies and percentages. Comparison between the two groups was carried out using Mann-Whitney tests for numerical data and χ2 test for categorical data. A p-value ≤0.05 was considered statistically significant.
Ethical statement
The study was approved by University of Padova’s Ethics Committee as part of a larger study aimed to evaluate disease course and characteristics of patients with IBD from the introduction of biologics in clinical practice (N.3312/AO/14). Written informed consent was obtained from all eligible participants, or their legal representatives, before participation.
Results
Stool samples were collected from 46 patients with UC (20 active UC and 26 inactive UC) and 36 HCs at IBD Unit of Padua University (Italy). Detailed demographic and clinical characteristics of patients with UC are reported in Table 1. Included HCs had a median age of 37 years, with a male to female ratio of 1:1.
Table 1.
Characteristics of study population
| Patients with active UC (N = 20) | Patients with inactive UC (N = 26) | P value * | |
|---|---|---|---|
| Male, n (%) | 14 (70.0) | 13 (50.0) | 0.21 |
| Age (median and range) | 40 (20–77) | 56.5 (28–75) | 0.01 |
| Age at diagnosis (median and range) | 26 (17–71) | 38 (13–62) | 0.01 |
| Smoker, n (%) | 4 (20.0) | 6 (23.1) | 0.90 |
| BMI (median and range) | 23.6 (15.8–27.8) | 23.7 (16.3–39.2) | 0.65 |
| Disease localization -Proctitis or proctosigmoiditis -Left-sided colitis -Extensive colitis |
1 (5.0) 7 (35.0) 12 (60.0) |
2 (7.7) 12 (46.1) 12 (46.1) |
0.64 |
| Fecal calprotectin µg/g (median and range) | 1456.5 (204–3800) | 60.0 (2–744) | <0.001 |
| Disease activity (p-Mayo), n (%) -Remission -Mild -Moderate -Severe |
--1 (5.0) 19 (95.0) |
22 (84.6) 4 (15.4)-- |
<0.001 |
| Endoscopic Mayo -Remission -Mild -Moderate -Severe |
--2 (5.0) 18 (95.0) |
24 (92.3) 2 (7.7)-- |
<0.001 |
| Previous abdominal surgery, n (%) | 1 (5.0) | 2 (7.7) | 0.74 |
| Previous steroids, n (%) | 17 (85.0) | 22 (84.6) | 0.92 |
| Naïve to biological drugs, n (%) | 5 (25.0) | 3 (11.5) | 0.21 |
* We used Mann-Whitney tests for numerical data and χ2 test for categorical data.
Pre-processing: from reads to Annotated Amplicon Sequence Variant (ASV) table
A total of 6.724.392 reads (mean: 82,442.02; SD: 31,275.40) were obtained from the sequencing procedure. After the filtering, denoising and chimera checking steps, a total of 3.647.949 (mean: 44,831.48; SD: 11,765.93) non-chimeric reads were retained. Details on read loss at each step can be found in Supplementary Table 1. The resulting ASV table collected 3754 ASVs belonging to 14 phyla, 25 classes, 42 orders, 81 families, 189 genera, and 302 species.
Metataxonomics results
At phylum level, we found that Tenericutes, Verrucomicrobia, Euryarchaeota (Archaea) and Cyanobacteria characterized the HCs. In particular, Tenericutes phylum was increasingly reduced with more active disease, while Verrucomicrobia phylum was absent in active UC. Conversely, Actinobacteria was significantly more abundant in patients with both active and inactive UC, and higher levels of Proteobacteria were detected consistently in a subset of patients with active UC. Interestingly, in the active UC group there were five patients with a dissimilar microbiota profile compared with the other patients in the same group; demonstrating very high levels of Proteobacteria (Figure 1). A more detailed species barplot is provided as Supplementary Figure 1.
Figure 1.
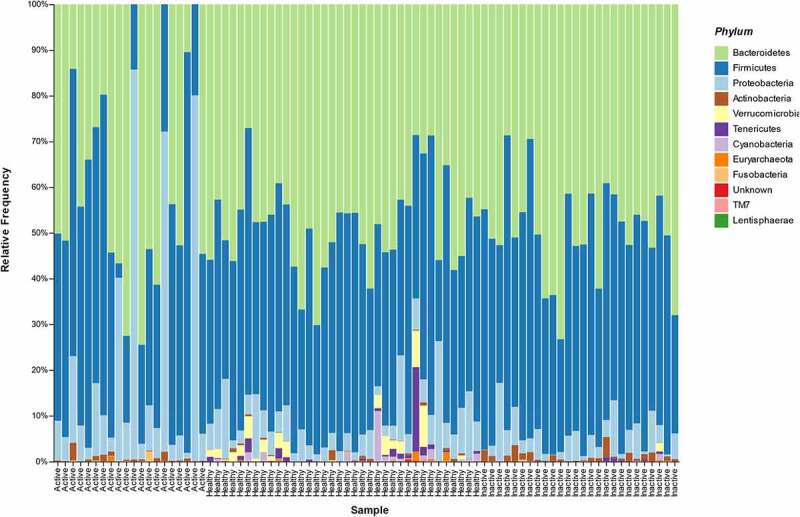
Microbiota composition (Phylum level) in the three different groups of patients (active UC, inactive UC, HCs).
At class, order, genus, and species levels we observed differences in microbiota composition between the three groups, summarized in Table 2.
Table 2.
Bacteria at phylum, class, order, genus, and species level increased and decreased in stool samples of healthy controls, and patients with inactive or active UC
| Healthy Controls | Inactive UC | Active UC | |
|---|---|---|---|
| Increased | p__Cyanobacteria p__Tenericutes c_Alphaproteobacteria o__Clostridiales g__Butyricimonas s__Akkermansia muciniphila s__Coprococcus eutactus |
g__Holdemania s__Eubacterium dolichum s__Blautia producta s__Ruminococcus gnavus s__Bifidobacterium adolescentis |
c_Gammaproteobacteria g__Granulicatella s__Haemophilus parainfluenzae s__Streptococcus anginosus s__Clostridium symbiosum |
| Decreased | g__Blautia g__Dorea s__Clostridium celatum s__Eubacterium dolichum |
p__Tenericutes p__Verrucomicrobia g__Lachnospira g__Oscillospira |
UC: ulcerative colitis; p_: phylum level; c_: class; o_: order level; g_: genus level; s_: species level.
Alpha diversity analysis
Alpha diversity results (ASV richness, Shannon index, and Pielou index) showed a marked difference among the three groups (Figure 2a, 2b and 2c). In particular, patients with active UC had the lowest values for all the indexes, followed by inactive UC, and HCs. The detected differences were statistically significant for all the pairwise comparisons, both for richness (p-values: active UC vs HCs = 0.0009; active UC vs inactive UC = 0.05; HCs vs inactive UC = 0.008) and for Shannon index (p-values: active UC vs HCs = 0.0005; active UC vs inactive UC = 0.03; HCs vs inactive UC = 0.03). Regarding the Pielou index, the pairwise comparisons were statistically significant between active UC and HCs (p = .004) and between active and inactive UC (p = .05), although the comparison between HCs and patients with inactive UC was not statistically significant (Table 3).
Figure 2.
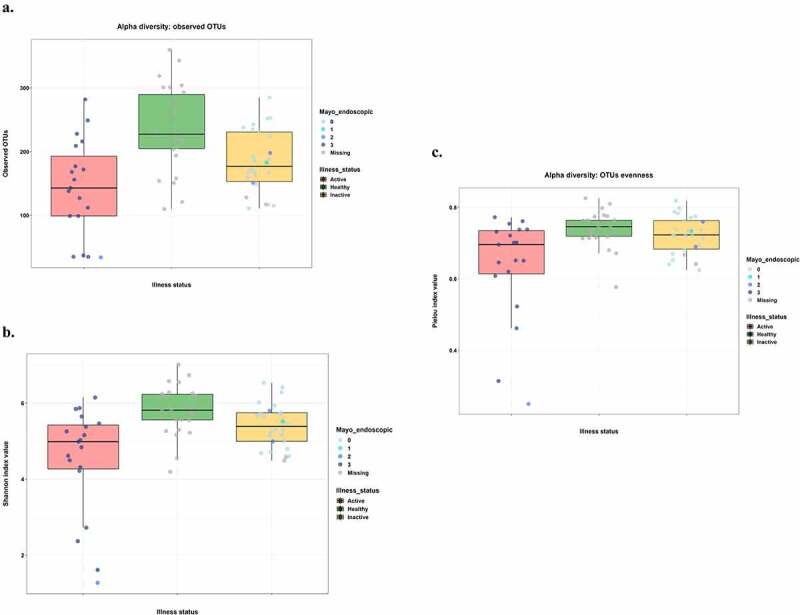
Alpha diversity analysis results for richness (a), Shannon (b), and Pielou (c) indices.
Points were colored corresponding to Mayo endoscopic subscores. Thus, we combined each metric quantitative information with endoscopic activity.
Table 3.
ASV Richness, Shannon index, Pielou index comparisons
| Richness |
Shannon index |
Pielou index |
|||
|---|---|---|---|---|---|
| Comparison | p-value* | Comparison | p-value* | Comparison | p-value* |
| Active UC-Healthy | 0.0009 | Active UC-Healthy | 0.0005 | Active UC-Healthy | 0.004 |
| Active UC-Inactive | 0.05 | Active UC-Inactive | 0.03 | Active-Inactive UC | 0.05 |
| Healthy–Inactive UC | 0.008 | Healthy-Inactive UC | 0.03 | Healthy-Inactive UC | 0.14 |
*Kruskal Wallis pairwise test
Figure 2 and Supplementary Figure 2 show diversity plots for microbiota patient samples (plots of Richness, Shannon index, and Pielou index, respectively) according to disease activity status (based on Mayo Endoscopic score) and fecal calprotectin values, respectively.
After performing the ANOVA test, FC values were statistically significantly different among the three groups (p < .001). Post-hoc, with the Tukey test, we demonstrated that there were statistically significant differences between HCs and active UC (p < .001) and between patients with inactive and active UC (p < .001). Conversely, no differences were found between HCs and patients with inactive UC (p = .86).
Beta diversity analysis
The PERMANOVA analysis based on Bray-Curtis dissimilarity showed that the composition of the microbiota at the ASV level was statistically significantly different among the three groups (p < .001). A hierarchical clustering using Ward algorithm on Canberra distance was performed (Supplementary Figure 3). As shown in the figure, cutting the tree at level 1.15, four different clusters were visible: the first one consisted mainly of HCs; the second consisted of five patients with active UC; the third mainly of patients with inactive UC; and the final cluster consisting of stool samples belonging to all groups (active and inactive UC and HCs) demonstrating similar microbial composition.
Non-metric multidimensional scaling
After the ASV table construction, an ordination graphical analysis was performed to represent the highest possible fraction of the complete information into a bidimensional plot (NMDS) based on the Bray-Curtis distance between samples (Figure 3). Beta-diversity based on Bray-Curtis distances showed a marked disease-associated pattern, with samples from patients with active UC located furthest from HCs and patients with inactive UC. These last two groups, again, were slightly separated from each other in the graph. Interestingly, the aforementioned five patients with active UC, and with a very dissimilar microbiota composition, were placed furthest from other patients in the same group.
Figure 3.
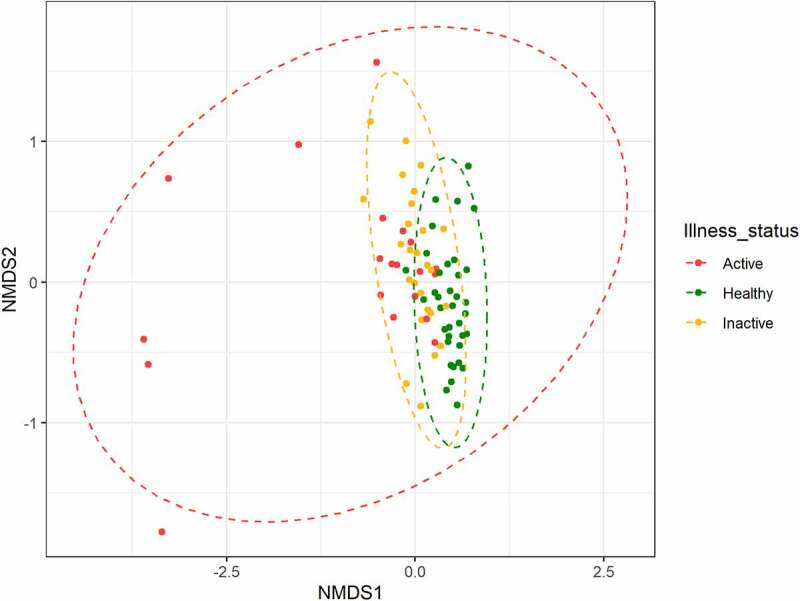
Non metric multidimensional scaling (NMDS) plot of Beta diversity (Bray-Curtis distance matrix).
Each point represented the gut microbiota of a patient while colors represent each clinical status (healthy, inactive UC, active UC). Patients with similar microbiota composition tended to be in the same area of the graph, while points far apart from each other represent patients with dissimilar microbiota.
sPLS-DA (Sparse partial least squares discriminant analysis) and random forest analyses
The sPLS-DA analysis allowed us to observe a perfect class prediction and group separation using the complete information (full ASV table) (Figure 4a), with a minimal loss in performance when using only the 5% of most important features (Figure 4b). Interestingly, the majority of the features selected by the sPLS-DA algorithm were the ones marked as differentially abundant among groups. The host trait predictive potential of the microbiota was also confirmed by a Random Forest analysis (Supplementary Table 2).
Figure 4.
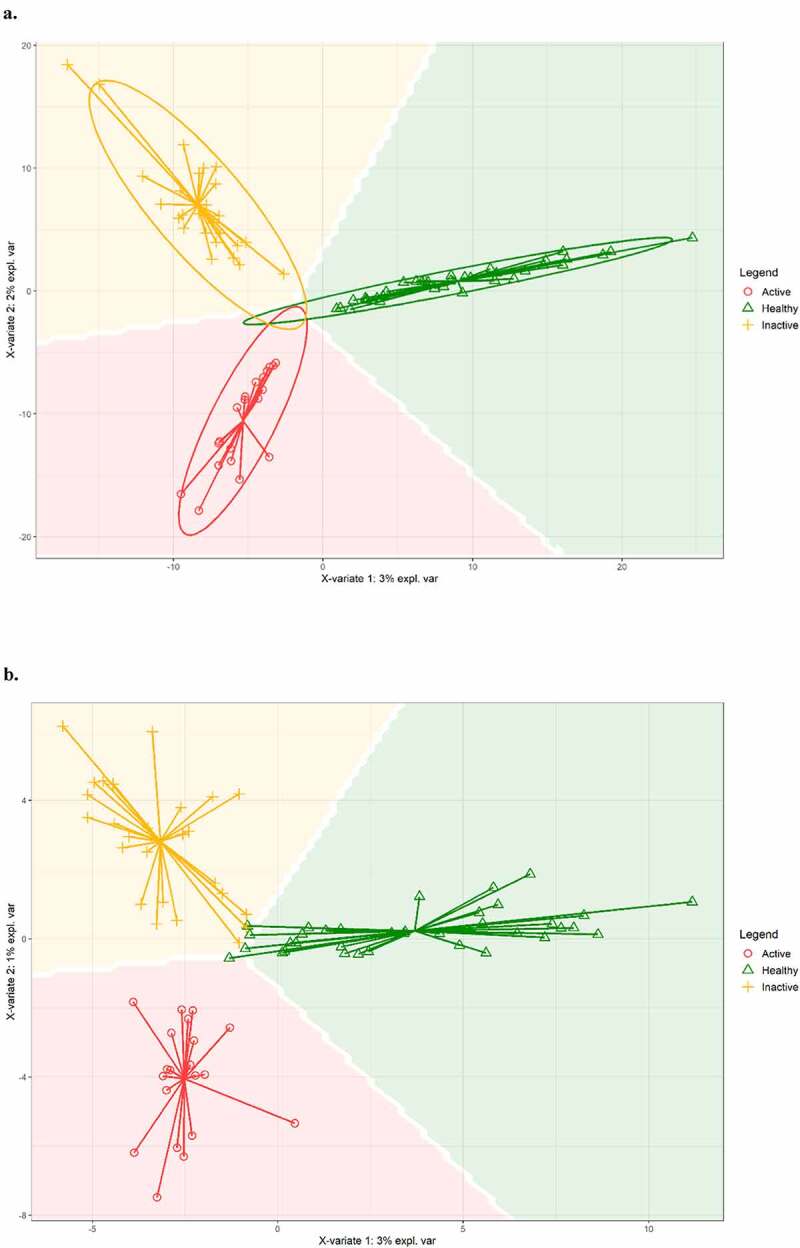
Sparse Partial Least Squares Discriminant Analysis (SPLS-DA), a machine learning-supervised approach using all ASVs (a) and 5% of all ASVs (b).
Discussion
Bacterial dysbiosis is one of the most widely proposed etiological factors in IBD,26 with variations affecting the α-diversity and abundance of phyla, families and genera.26 However, it remains unclear whether alterations in the intestinal microbiota are a cause or an effect of inflammation in IBD, as well as how the microbiota changes based on intestinal inflammation. In our study, we aimed to assess whether a specific microbiota profile, as measured by a machine learning approach, was associated with disease severity in patients with UC. We also aimed to verify if it could be used as a noninvasive marker for IBD monitoring, as well as a predictor of outcome before it become clinically manifested.
Overall, in our study, fecal microbiota alpha diversity (ASV richness, Shannon index, and Pielou index) was significantly reduced in patients with UC compared with HCs, and this difference remained statistically significant between patients with active and inactive disease. The gut microbiota of the HCs, at phylum level, was characterized by high levels of Tenericutes, Verrucomicrobia, Euryarchaeota, and Cyanobacteria. While, Actinobacteria were significantly more abundant in patients with both active and inactive UC and higher levels of Proteobacteria were detected in a consistent subset of patients with active UC. Of the microbial species found to be significantly increased in HCs, there were Akkermansia muchiniphila and Coprococcus eutactus, which were practically absent in active UC. On the other hand, among the microbial species found to be significantly higher in active UC, there were Haemophilus parainfluenzae, Clostridium symbiosum, Clostridium perfringens, and Streptococcus anginosus. In addition, at genus level, we found high levels of Granulicatella. Regarding inactive UC, we observed higher levels of Ruminococcus gnavus, Blautia producta, Eubacterium dolichum, and Bifidobacterium adolescentis. Moreover, at genus level, in inactive UC Holdemania was significantly increased. The application of sPLS-DA, a machine learning-supervised approach, allowed us to observe a perfect class prediction and group separation using the complete information, with a minimal loss in performance when using only 5% of features. Overall, machine learning approaches highlighted a disease-state signature that agreed well with differential abundance analysis results.
Numerous studies have supported evidence for intestinal dysbiosis in IBD patients compared with healthy controls,11,12 and our results confirmed this finding. As our results also show, current literature reports that the gut microbiota of healthy individuals is dominated at phylum level by the bacterial phyla Firmicutes and Bacteroidetes, and to a lesser extent by Proteobacteria, Actinobacteria, and Verrucomicrobia.13 In fact, a relevant abundance of Akkermansia muchiniphila, belonging to Verrucomicrobia phylum, was found in our controls, whereas it was very low in our patients with UC, especially those with active disease. Interestingly, we did not find a statistically significant difference in Faecalibacterium prausnitzii in patients with UC compared with healthy controls, or between active and inactive UC, in contrast with current literature.27 Fecal samples of patients with active UC had a lower abundance of F. prausnitzii, but this difference was not statistically significant.
Several studies using meta-genomics analysis have demonstrated that members of the phylum Firmicutes are less abundant in patients with UC or CD.11,28,29 Among Firmicutes, Clostridium clusters XIVa and IV are largely underrepresented in the gut of patients with IBD. Clostridium cluster XIVa comprises species belonging to the Clostridium, Ruminococcus, Lachnospira, Roseburia, Eubacterium, Coprococcus, Dorea, and Butyrivibrio genera.30–32 We found that g_Ruminococcus was lower in UC, mostly in those with active disease, compared with controls. Likewise, Coprococcus eutactus was statistically higher in HCs compared with patients with UC. Previous studies have evaluated microbial differences between patients with active and inactive IBD.27 A decrease in the Clostridium family was found in active UC compared with inactive UC and healthy controls.33 Furthermore, a decrease of Clostridium coccoides and Clostridium leptum was reported in the feces of patients with active compared with inactive UC.34 Interestingly, in patients with active UC we found high levels of Clostridum perfringens and symbiosum compared with patients with inactive UC and controls. Several microbiome analyses have revealed there is an expansion of the Proteobacteria phylum in patients with IBD.11,28 In keeping with these findings, we demonstrated a relevant and significant increase of Proteobacteria in patients with active UC. In particular, we observed high levels of Hemophilus parainfluenzae, a Gammaproteobacteria, at class level. It is worth of noting that our samples demonstrated a specific signature even in patients with inactive UC, who demonstrated higher levels of Ruminococcus gnavus, Blautia producta, Eubacterium dolichum, and g_Holdemania, all belonging to Firmicutes phylum, and Bifidobacterium adolescentis, belonging to Actinobacteria phylum. Another recent study by Clooney et al. applied a machine learning approach to stool samples of patients with both active and inactive IBD. They recruited 303 patients with CD, 228 with UC, and 161 controls, and demonstrated that machine learning separated IBD from controls, and active from inactive IBD, when consecutive time points were modeled.16
Our study had some limitations. First, the sample size is relatively small. However, this drawback is balanced by the fact that we enrolled a study population as homogeneous as possible. In particular, we tried to limit the influence of ongoing treatments on microbiota characterization and we divided our patients based on their disease activity. Indeed, we included patients who were taking only mesalazine, and not biological drugs or immunosuppressants. In addition, although our cohort is homogeneous because it belongs to a single center, a validation cohort from multiple sites will be required in future studies to determine the quality of the machine learning. However, we are aware that also dietary factors, which we did not consider, could lead to microbiota variability in patients with IBD and controls. Another limitation is the cross-sectional design of the study, with a lack of longitudinal data to further support our findings. Moreover, we only assessed the fecal microbiota profile, without examining the microbiota adherent to the colonic mucosa. Finally, we did not match the microbiota of both active and inactive patients based on characteristics such as diet or smoking or alcohol consumption. On the other hand, we considered as exclusion criteria the medications (antibiotics and probiotics) known to influence the microbiota assessment in such patients.
However, despite these limitations, the distinct signatures observed for the gut microbiota in active and inactive UC could have several implications. Firstly, these results further strengthen the association between dysbiosis and IBD and have given a proof-of-concept for a potential correlation between disease severity and degree of dysbiosis. Secondly, since the accuracy of diagnosis in IBD is key to commencing prompt and effective treatment, there is an urgent need to develop a novel classification technique that can expedite IBD diagnosis, before intestinal damage ensues. In our study, we used supervised and unsupervised machine learning algorithms on an IBD associated metagenomics data, which might improve diagnostic accuracy and elucidate which subsets of microbiota are most informative to identify these patients. Potentially, all of this could enable the identification of individuals who will develop IBD before symptoms, using their microbiota. Finally, finding a specific microbiota signature will represent an opportunity for personalized prognostics or therapeutics based on microbiota manipulation.
In conclusion, in our study, the application of sPLS-DA, a machine learning-supervised approach, allowed us to observe a perfect class prediction and group separation using the complete information (full OTU table), with a minimal loss in performance when using only 5% of features. In addition, the majority of the features selected by the sPLS-DA algorithm were the ones marked as differentially abundant between groups. Our data support the concept that implementing current 16S rRNA data will be helpful to improve management of IBD patients. Further follow-up studies will aim to clarify whether such microbiota profiling also predicts disease outcomes.
List of abbreviations
ASVs: Amplicon Sequence VariantsCD: Crohn’s diseaseHBI: Harvey-Bradshaw indexHCs: Healthy controlsIBD: inflammatory bowel diseaseFC: fecal calprotectinNMDS: non-metric multidimensional scalingp-MAYO: partial Mayo ScoresPLS-DA: sparse partial least squares discriminant analysisUC: ulcerative colitis
Supplementary Material
Acknowledgments
We thank our patients for helping us to perform this study.
Funding Statement
The author(s) reported there is no funding associated with the work featured in this article.
Disclosure statement
Brigida Barberio has received lecture or consultancy fees from Janssen, Sofar, Alfasigma, Procise, Doxapharma.
Ilaria Patuzzi: none; Alexander Ford: none; Davide Massimi: none; Giorgio Valle: none; Eleonora Sattin: none; Barbara Simionati: none; Elena Bertazzo: none; Fabiana Zingone: none.
Sonia Facchin has received lecture or consultancy fees from Grifols and Unifarco.
Edoardo Savarino has received lecture or consultancy fees from Abbvie, Amgen, Bristol-Myers Squibb, Fresenius Kabi, Grifols, Janssen, Johnson&Johnson, Innovamedica, Merck & Co, Novartis, Sandoz, Shire, SILA, Sofar, Takeda, Unifarco.
Author contributions
BB, SF, EVS conceived and drafted the study. BB, ACF, and EVS drafted the manuscript. BB, IP, SF, GV, ES, BS analyzed and interpreted all data. BB, DM, EB collected all data. All authors have approved the final draft of the manuscript.
Ethics committee approval
The study was approved by University of Padova’s Ethics Committee as part of a larger study aimed to evaluate disease course and characteristics of IBD patients from the introduction of biologics in clinical practice (N. 3312/AO/14).
Supplementary material
Supplemental data for this article for this article can be accessed on the publisher’s website.
Data availability statement
The data underlying this study is available within the manuscript and supplementary materials
References
- 1.Marinelli C, Savarino E, Inferrera M, Lorenzon G, Rigo A, Ghisa M, Facchin S, D’Incà R, Zingone F.. Factors influencing disability and quality of life during treatment: a cross-sectional study on IBD patients. Gastroenterol Res Pract. 2019;2019:5354320. doi: 10.1155/2019/5354320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barberio B, Zingone F, Savarino EV. Inflammatory bowel disease and sleep disturbance: as usual, quality matters. Dig Dis Sci. 2021;66(1):3–12. doi: 10.1007/s10620-020-06268-5. [DOI] [PubMed] [Google Scholar]
- 3.Barberio B, Zamani M, Black CJ, Savarino, EV, Ford , AC. Prevalence of anxiety and depression in inflammatory bowel disease: systematic review and meta-analysis. The Lancet Gastroenterology & Hepatology. 2021;6(5):359-370. [DOI] [PubMed] [Google Scholar]
- 4.Choy MC, Visvanathan K, Cruz PD. An overview of the innate and adaptive immune system in inflammatory bowel disease. Inflamm Bowel Dis. 2017;23(1):2–13. doi: 10.1097/MIB.0000000000000955. [DOI] [PubMed] [Google Scholar]
- 5.Guo XY, Liu XJ, Hao JY. Gut microbiota in ulcerative colitis: insights on pathogenesis and treatment. J Dig Dis. 2020;21(3):147–159. doi: 10.1111/1751-2980.12849. [DOI] [PubMed] [Google Scholar]
- 6.Varela E, Manichanh C, Gallart M, Torrejón A, Borruel N, Casellas F, Guarner F, Antolin M. Colonisation by Faecalibacterium prausnitzii and maintenance of clinical remission in patients with ulcerative colitis. Aliment Pharmacol Ther. 2013;38(2):151–161. doi: 10.1111/apt.12365. [DOI] [PubMed] [Google Scholar]
- 7.Machiels K, Joossens M, Sabino J, De Preter V, Arijs I, Eeckhaut V, Ballet V, Claes K, Van Immerseel F, Verbeke K, et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut. 2014;63(8):1275–1283. doi: 10.1136/gutjnl-2013-304833. [DOI] [PubMed] [Google Scholar]
- 8.Nagalingam NA, Lynch SV. Role of the microbiota in inflammatory bowel diseases. Inflamm Bowel Dis. 2012;18(5):968–984. doi: 10.1002/ibd.21866. [DOI] [PubMed] [Google Scholar]
- 9.Facchin S, Vitulo N, Calgaro M, Buda A, Romualdi C, Pohl D, Perini B, Lorenzon G, Marinelli C, D’Incà R, et al. Microbiota changes induced by microencapsulated sodium butyrate in patients with inflammatory bowel disease. Neurogastroenterol Motil. 2020;32(10):e13914. doi: 10.1111/nmo.13914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Segal JP, Mullish BH, Quraishi MN, Acharjee, A, Williams, HRT, Iqbal, T, Hart, AL, Marchesi, JR, et al. The application of omics techniques to understand the role of the gut microbiota in inflammatory bowel disease. Therap Adv Gastroenterol. 2019;12:1756284818822250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frank DN, Amand ALS, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A. 2007;104(34):13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Swidsinski A, Loening-Baucke V, Vaneechoutte M, Doerffel Y. Active Crohn’s disease and ulcerative colitis can be specifically diagnosed and monitored based on the biostructure of the fecal flora. Inflamm Bowel Dis. 2008;14(2):147–161. doi: 10.1002/ibd.20330. [DOI] [PubMed] [Google Scholar]
- 13.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Microbiology: diversity of the human intestinal microbial flora. Science (80-). 2005;308(5728):1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsuoka K, Kanai T. The gut microbiota and inflammatory bowel disease. Semin Immunopathol. 2015;37(1):47–55. doi: 10.1007/s00281-014-0454-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seyed Tabib NS, Madgwick M, Sudhakar P, Verstockt B, Korcsmaros T, Vermeire S. Big data in IBD: big progress for clinical practice. Gut. 2020;69(8):1520–1532. doi: 10.1136/gutjnl-2019-320065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clooney AG, Eckenberger J, Laserna-Mendieta E, Sexton, KA, Bernstein, MT, Vagianos, K, Sargent, M, Ryan, FJ, Moran, C, Sheehan, D, Sleator, RD, Targownik, LE, Bernstein, CN, Shanahan, F, Claesson, MJ, et al. Ranking microbiome variance in inflammatory bowel disease: a large longitudinal intercontinental study. Gut. 2021;70.3: 499-510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takahashi S, Tomita J, Nishioka K, Hisada T, Nishijima M. Development of a prokaryotic universal primer for simultaneous analysis of bacteria and archaea using next-generation sequencing Bourtzis K, ed. PLoS One. 2014;9(8):e105592. doi: 10.1371/journal.pone.0105592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mumolo MG, Bertani L, Ceccarelli L, Laino G, Fluri GD, Albano E, Tapete G, Costa F. From bench to bedside: fecal calprotectin in inflammatory bowel diseases clinical setting. World J Gastroenterol. 2018;24(33):3681–3694. doi: 10.3748/wjg.v24.i33.3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barberio B, D’Incà R, Facchin S, Dalla Gasperina M, Fohom Tagne CA, Cardin R, Ghisa M, Lorenzon G, Marinelli C, Savarino EV, et al. Matrix metalloproteinase 3 predicts therapeutic response in inflammatory bowel disease patients treated with infliximab. Inflamm Bowel Dis. 2020;26(5):756–763. doi: 10.1093/ibd/izz195. [DOI] [PubMed] [Google Scholar]
- 20.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37(8):852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen L, Reeve J, Zhang L, Huang S, Wang X, Chen J. GMPR: a robust normalization method for zero-inflated count data with application to microbiome sequencing data. PeerJ. 2018;6:e4600. doi: 10.7717/peerj.4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mandal S, Treuren WV, White RA, Eggesbo, M, Knight, R, Peddada, SD. Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb Ecol Heal Dis. 2015;26(1):27663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McMurdie PJ, Holmes S, Watson M. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data Watson M, ed. PLoS One. 2013;8(4):e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liaw A, Wiener M. Classification and regression by randomForest. undefined 2007.
- 25.Rohart F, Gautier B, Singh A, Lê Cao K-A. mixOmics: an R package for ‘omics feature selection and multiple data integration Schneidman D, ed. PLOS Comput Biol. 2017;13(11):e1005752. doi: 10.1371/journal.pcbi.1005752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scharl M, Rogler G. Inflammatory bowel disease pathogenesis: what is new? Curr Opin Gastroenterol. 2012;28(4):301–309. doi: 10.1097/MOG.0b013e328353e61e. [DOI] [PubMed] [Google Scholar]
- 27.Sokol H, Seksik P, Furet JP, Firmesse O, Nion-Larmurier I, Beaugerie L, Cosnes J, Corthier G, Marteau P, Doré J, et al. Low counts of faecalibacterium prausnitzii in colitis microbiota. Inflamm Bowel Dis. 2009;15(8):1183–1189. doi: 10.1002/ibd.20903. [DOI] [PubMed] [Google Scholar]
- 28.Gophna U, Sommerfeld K, Gophna S, Doolittle WF, Veldhuyzen van Zanten SJO. Differences between tissue-associated intestinal microfloras of patients with Crohn’s disease and ulcerative colitis. J Clin Microbiol. 2006;44(11):4136–4141. doi: 10.1128/JCM.01004-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma HQ, Yu TT, Zhao XJ, Zhang Y, Zhang H-J. Fecal microbial dysbiosis in Chinese patients with inflammatory bowel disease. World J Gastroenterol. 2018;24(13):1464–1477. doi: 10.3748/wjg.v24.i13.1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mondot S, Kang S, Furet JP, Aguirre de Carcer D, McSweeney C, Morrison M, Marteau P, Doré J, Leclerc M. Highlighting new phylogenetic specificities of Crohn’s disease microbiota. Inflamm Bowel Dis. 2011;17(1):185–192. doi: 10.1002/ibd.21436. [DOI] [PubMed] [Google Scholar]
- 31.Joossens M, Huys G, Cnockaert M, De Preter V, Verbeke K, Rutgeerts P, Vandamme P, Vermeire S. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut. 2011;60(5):631–637. doi: 10.1136/gut.2010.223263. [DOI] [PubMed] [Google Scholar]
- 32.Vigsnaes LK, Van Den AP, Sulek K, Frandsen HL, Steenholdt C, Brynskov J, Vermeiren J, van de Wiele T, Licht TR. Microbiotas from UC patients display altered metabolism and reduced ability of LAB to colonize mucus. Sci Rep. 2013;3(1):1–10. doi: 10.1038/srep01110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andoh A, Kuzuoka H, Tsujikawa T, Nakamura S, Hirai F, Suzuki Y, Matsui T, Fujiyama Y, Matsumoto T. Multicenter analysis of fecal microbiota profiles in Japanese patients with Crohn’s disease. J Gastroenterol. 2012;47(12):1298–1307. doi: 10.1007/s00535-012-0605-0. [DOI] [PubMed] [Google Scholar]
- 34.Takaishi H, Matsuki T, Nakazawa A, Takada T, Kado S, Asahara T, Kamada N, Sakuraba A, Yajima T, Higuchi H, et al. Imbalance in intestinal microflora constitution could be involved in the pathogenesis of inflammatory bowel disease. Int J Med Microbiol. 2008;298(5–6):463–472. doi: 10.1016/j.ijmm.2007.07.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study is available within the manuscript and supplementary materials