

Abstract
Following treatment with androgen receptor (AR) pathway inhibitors, ≈20% of prostate cancer patients progress by shedding their AR-dependence. These tumors undergo epigenetic reprogramming turning castration-resistant prostate cancer adenocarcinoma (CRPC-Adeno) into neuroendocrine prostate cancer (CRPC-NEPC). No targeted therapies are available for CRPC-NEPCs, and there are minimal organoid models to discover new therapeutic targets against these aggressive tumors. Here, using a combination of patient tumor proteomics, RNA sequencing, spatial-omics, and a synthetic hydrogel-based organoid, putative extracellular matrix (ECM) cues that regulate the phenotypic, transcriptomic, and epigenetic underpinnings of CRPC-NEPCs are defined. Short-term culture in tumor-expressed ECM differentially regulated DNA methylation and mobilized genes in CRPC-NEPCs. The ECM type distinctly regulates the response to small-molecule inhibitors of epigenetic targets and Dopamine Receptor D2 (DRD2), the latter being an understudied target in neuroendocrine tumors. In vivo patient-derived xenograft in immunocompromised mice showed strong anti-tumor response when treated with a DRD2 inhibitor. Finally, we demonstrate that therapeutic response in CRPC-NEPCs under drug-resistant ECM conditions can be overcome by first cellular reprogramming with epigenetic inhibitors, followed by DRD2 treatment. The synthetic organoids suggest the regulatory role of ECM in therapeutic response to targeted therapies in CRPC-NEPCs and enable the discovery of therapies to overcome resistance.
Keywords: chemoresistance, dopamine receptors, epigenetics, neuroendocrine, tumor microenvironment
Graphical Abstract
TOC
Neuroendocrine prostate cancer is epigenetically transformed tumors for which no targeted therapies exist. Using a combination of transcriptomics, proteomics, spatial omics, and microscopy on patient biopsies, the extracellular matrix tumor microenvironment is defined. Informed by these findings, synthetic hydrogel-based prostate cancer organoids are developed to grow patient tumor cells under the defined microenvironment conditions, leading to the discovery of novel therapeutics.
1. Introduction
The acquired drug resistance in advanced prostate cancer to current Androgen Receptor (AR) pathway inhibitors, such as abiraterone acetate and enzalutamide, is driven, in part, by the ability of cancer cells to adopt AR-independent pathways for growth and survival.[1–3] A prolonged AR pathway inhibition can alter the archetypical course of the disease, leading to treatment-induced lineage transition where cellular dedifferentiation and alterations in the lineage of prostate cancer cells in the form of neuroendocrine differentiation.[1,4–6] This lineage plasticity is highly aggressive and acquires a lethal form when castration-resistant adenocarcinoma (CRPC-Adeno) tumors develop clinical transformation to a small cell neuroendocrine carcinoma-like presentation, termed neuroendocrine prostate cancer (NEPC).[7,8] CRPC-NEPCs evolve from CRPC-Adeno through genetic alterations and dysregulation of at least one epigenetic modifier, histone methyltransferase enhancer of zeste 2 (EZH2).[7,9–12] There are no targeted therapies currently available for CRPC-NEPCs, and the factors that induce tumor resistance to therapeutics remain poorly understood. A major limitation has been the lack of tumor models to grow CRPC-NEPC tumors from patients in the prostate cancer-specific tumor microenvironment (TME), regulating the tumor growth and drug resistance. Although patient-derived xenografts in mice are a viable platform to study these factors, one cannot precisely control the input signals to determine the role of TME on tumor outcomes. To model small cell NEPC, the only widely available cell line is the NCI-H660, initially thought to be small cell lung cancer but later reclassified as prostate.[13] To overcome these limitations, we have earlier developed Matrigel organoid models of patient-derived CRPC-NEPC[14] and discovered EZH2 as a new therapeutic target against these aggressive tumors. However, the success rate of generating these organoids remains low,[14] leading to the generation of two patient-derived Matrigel organoids, OWCM-155 and OWCM-154 by us (Puca et al.[14]). The EZH2 inhibitor (EZH2i) GSK343 decreases OWCM-155 and OWCM-154 tumor growth in Matrigel organoids.[14] However, the EZH2i alone requires a high dose[14] and may not eliminate the tumor in its entirety, as shown in other cancers.[15] Therefore, we can likely realize the full potential of EZH2i for CRPC-NEPC treatment if the tumors are grown in a more defined TME that recapitulates a controlled microenvironment of CRPC-NEPCs and combined with novel drug targets. A significant challenge is that Matrigel-based organoids[14] do not recapitulate patient- or disease-specific TME features of human CRPC-Adeno and CRPC-NEPC, such as the extracellular matrix (ECM), which is the focus of the current study.
Here, we performed transcriptomic analysis on 111 patients and a combination of specialized techniques such as proteomics, spatial omics, and microscopy analysis on patient biopsies to define ECM and related microenvironment in CRPC-Adeno and CRPC-NEPC. Informed by these findings, we developed a synthetic polymer-based, hydrogel platform of Maleimide-functionalized poly(ethylene glycol) (PEG-4MAL) to grow CRPC-Adeno and CRPC-NEPC patient tumor cells under the defined ECM microenvironment. We investigated the impact of biomimetic synthetic ECM on tumor morphology, actin cytoskeleton, and changes in transcriptomic and epigenetic signature associated with CRPC-NEPC progression. We demonstrate an ECM-dependent EZH2i response in CRPC-NEPCs, where the drug resistance could be overcome by cellular reprogramming with EZH2i, followed by treatment with neuroendocrine targeting experimental therapeutics. The synthetic hydrogel platform, which presents ECM-specific bioadhesive ligands, can provide a more defined microenvironment for CRPC-Adeno or CRPC-NEPCs, currently unavailable with Matrigel, subsequently regulating the transcriptome and epigenetics and enabling the discovery of novel single and combinatorial therapeutics.
2. Results and Discussion
2.1. CRPC-Adeno Patient Tumors Manifest a High Expression of Cell Adhesion Proteins
ECM-Integrin adhesion receptor interactions[16–22] provide critical signals that regulate the cellular and molecular fate of cancer cells, including survival, gene expression, and the ability to metastasize.[23–25] ECM-integrin interactions can also influence chromatin state through mechanotransduction. However, the ECM composition in TME in advanced prostate cancer and the expression of integrins on cells present at the tumor sites are not well defined. To address this gap, we performed a proteomic analysis of cell adhesion proteins using liquid chromatography-tandem mass spectrometry in de novo CRPC-Adeno tumors from 3 patients (primary tumors). We compared them to the adjacent nontumorous prostate tissue (benign). Across all three patients, we observed a higher expression of ECM proteins, including Collagen 1 (COL1A1), Fibronectin (FN1), Vitronectin (VTN), Laminin (LAMC2), and associated integrin (ITG) subunits α2, αV, α4, α5, α6, and several β subunits as compared to the benign tissue (Figure 1A). These differences motivated us to perform a more comprehensive analysis in CRPC-Adeno and CRPC-NEPC patient biopsies, to understand the change in integrin and ECM gene expression across disease subtypes.
Figure 1.
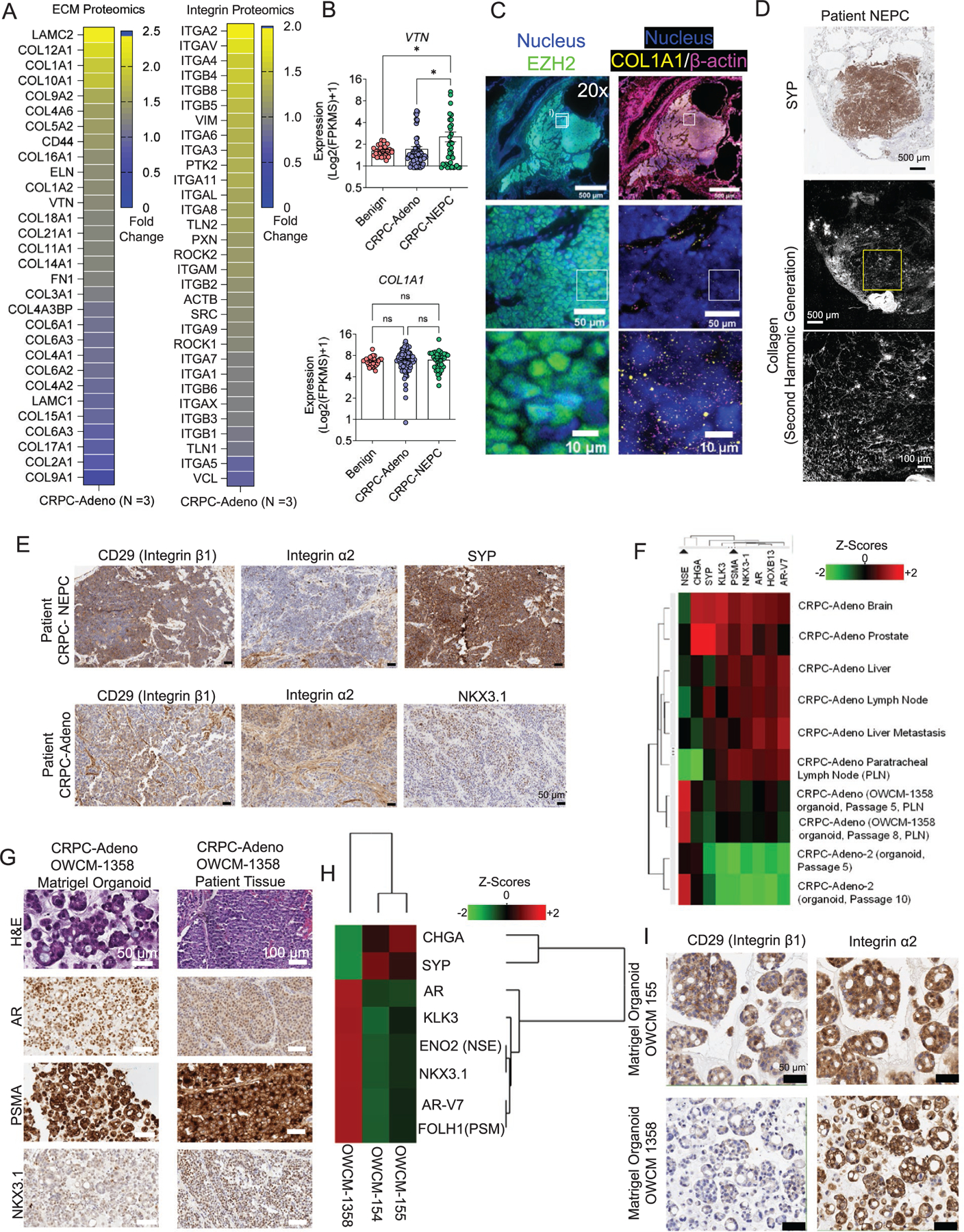
Proteomic, transcriptomic, and spatial omic analysis of patient tumors and establishment of CRPC-Adeno Matrigel organoids. A) Mass spectrometry analysis of ECM components (left) and integrin signaling-related components (right) in primary CRPC-Adeno tumors relative to adjacent normal tissue (average of n = 3). B) RNA-seq transcriptomic analysis of ECM components and integrins in CRPC-Adeno and CRPC-NEPC patient tumor biopsies, as compared to benign tissues (n = 74 CRPC-Adeno, n = 37 CRPC-NEPC, n = 31 benign samples). Data presented as mean ± SEM. C) Multiplexed single-cell spatial omics RNA analysis of prostate tumors. Left column: Immunostaining of EZH2 on CRPC-NEPC prostate tumors. Zoomed in images of single-cell EZH2 distributions (green) in the second and third rows. Right column: FISH based detection of single COL1A1 and β-actin RNA molecules in the same area of the CRPC-NEPC prostate tumors using a FISH signal amplification assay, HCR. Zoomed-in images of single-cell RNA distributions of COL1A1 (yellow) and β-actin (magenta) in the second and third rows. Data representative of two patient biopsies. D) SHG microscopy images of collagen fibers in CRPC-NEPC tissues. The tissue samples were embedded in paraffin and stained with Synaptophysin (SYP). The SHG of tissue samples were imaged on a deparaffinized unstained slide using a two-photon microscope. Data representative of two patient biopsies. E) IHC staining of CRPC-NEPC and CRPC-Adeno patient biopsies. SYP and NKX3.1 staining for CRPC-NEPC and CRPC-Adeno, respectively, and expression of integrin α2 and β1. Data representative of two patient biopsies. F) Development and characterization of a new CRPC-Adeno organoid (OWCM-1358) from a patient tumor biopsy. Nanostring analysis of hallmark gene expression in multiple metastatic sites and derived Matrigel organoids. G) H&E and IHC staining of CRPC-Adeno patient and OWCM-1358 Matrigel organoid that demonstrated CRPC-Adeno gene signature. H) Nanostring-based comparative analysis of prostate cancer-associated genes among CRPC-Adeno (OWCM-1358) and CRPC-NEPC Matrigel organoids (OWCM-154, OWCM-155). I) IHC staining of CRPC-NEPC and CRPC-Adeno Matrigel organoids. SYP and NKX3.1 staining for CRPC-NEPC and CRPC-Adeno, respectively, and expression of integrin α2 and β1. Data representative of two samples.
2.2. CRPC-Adeno and CRPC-NEPC Patient Tumors Manifest Similar Bioadhesive Microenvironment
We interrogated RNA sequencing results from 111 patient tumors, including 74 patients with clinical and histologic features of CRPC-Adeno and 37 with features of CRPC-NEPC, as confirmed by pathologic consensus criteria.[8] These tumors were compared to 31 benign prostate tissues. We confirmed the disease state of patient samples by the loss of AR gene in CRPC-NEPC tumors, relative to benign tissues and CRPC-Adeno tumors[7] (Figure S1A, Supporting Information). We did not perform further detailed characterization because we have already demonstrated previously, using whole-exome sequencing of metastatic tumor-normal tissue pairs, that the mutational landscape of CRPC-NEPC tumors is similar to that of CRPC-Adeno.[7] A comparative analysis of RNA sequencing readouts from CRPC-Adeno and CRPC-NEPC patients revealed an upregulated expression of VTN compared to benign prostate tissues and comparable, high expression levels of fibronectin (FN1), collagen 1 (COL1A1), similar to benign prostate tissues (Figure 1B, Figure S1B, Supporting Information). The patient cohort genes encoding for integrin α2 (ITGA2), β1 (ITGB1), α4 (ITGA4), and αV (ITGAV) were expressed at comparable levels to benign tissues. However, tumor tissues are expected to contain nontumorous stromal cells and the bulk RNA-sequencing could reflect the contributions from stroma, therefore we performed spatial-omics and protein imaging to confirm that tumor cells expressed the ECM and integrins.
To demonstrate that prostate tumors express collagen 1, we performed multiplexed single-cell imaging in CRPC-NEPC and CRPC-Adeno patient biopsies. To profile gene expression, the fluorescence in situ hybridization (FISH) method[26] was used to localize single RNA molecules in 5 μm thin formalin-fixed paraffin-embedded (FFPE) tissues. To amplify the FISH signal, a split-probe design of Hybridization Chain Reaction (HCR) assay[27] was utilized to efficiently detect RNA distributions in FFPE tissues. FISH-HCR measurements correspond to gene expression maps by visualizing RNA dots across the tissues, providing similar levels of COL1A1 RNA expression in tumor areas in distinct pathological cancer-rich regions of CRPC-NEPC and CPRC-Adeno tumors (Figure 1C, Figure S2A,B, Supporting Information). After the RNA visualization in prostate cancer biopsies, FISH labels were digested by a DNAse-I-based enzymatic treatment to remove the FISH signal on the tissues. To validate tumor-related protein expression on the same tissue sets, an immunofluorescence experiment was performed for visualization of EZH2 in CRPC-NEPC using antibodies (Figure 1C). Similarly, tissues for CPRC tumors were sequentially stained with NKX3.1, yielding positive NKX3.1 expression and confirming tumor regions (Figure S2B, Supporting Information). After chemically bleaching the fluorescence from the protein stains, the same tissue samples were subjected to hematoxylin-and-eosin (H&E) staining to further verify tumor regions (Figure S2B, Supporting Information).
To further confirm that collagen was present within the tumor, we performed protein localization analysis using second-harmonic generation (SHG). SHG microscopy illustrated that collagen was localized in both the tumor area and surrounding tissues. Figure 1D presents an immunohistochemistry stained biopsy slice of CRPC-NEPC cancer imaged by SHG under two-photon confocal microscopy. The collagen fibers are evident in the entire synaptophysin (SYP)+ tumor region (Figure 1D, Figure S2C, Supporting Information). Last, to determine that the tumor cells were expressing integrins identified in RNA-sequencing, such as integrin α2 and β1 that forms a heterodimer to bind collagen, we performed immunohistochemistry on patient tissue biopsies. Both CRPC-NEPC (SYP+) and CRPC-Adeno (NKX3.1+) were stained positive for integrin α2 and β1 (Figure 1E). The collective proteomic, RNA sequencing, spatial omics, SHG, and immunohistochemistry analysis highlights the presence of various ECM proteins and integrin ligands in a whole TME of prostate cancer. In contrast to primary prostate tumors, Matrigel organoids include mostly the tumor cells and ECM representing mouse sarcoma basement membrane, which consists primarily of collagen type IV, entactin, perlecan (heparan sulfate proteoglycan), and laminin.
2.3. CRPC-NEPC Matrigel Organoids Express Distinct Levels of Integrins than CRPC-Adeno Organoids
To understand whether these integrin proteins and genes found in patient tumor cells are expressed in Matrigel organoid models of patient-derived CRPC-NEPC and CRPC-Adeno, we first established a new CRPC-Adeno organoid in Matrigel and used CRPC-NEPC Matrigel organoids derived from CRPC-NEPC patients, reported earlier as OWCM-154 and OWCM-155.[14] The OWCM-155 patient cells were from a metastatic liver biopsy from a patient with prostatic adenocarcinoma Grade Group 5 (Gleason Score 4+5 = 9). We compared both tissue biopsy and Matrigel organoids of OWCM-155 using H&E and observed overlapping features between small cell carcinoma and adenocarcinoma (Figure S3A, Supporting Information). We have previously performed immunohistochemistry to confirm the loss of AR and the presence of SYP, chromogranin A (CHGA), and CD56 in OWCM-155 tumors in Matrigel organoids;[14] therefore we did not repeat these markers. CRPC-NEPC patients lose luminal morphology and transform into irregular shapes as compared to CRPC-Adeno.[28] However, in contrast to patient features, the Matrigel organoids adopted a near-spherical shape with semi-filled or hollow appearances (Figure S3A, Supporting Information), suggesting Matrigel may not capture morphological features of patient tumors.
To compare CRPC-NEPC organoids with CRPC-Adeno organoids, we derived a new CRPC-Adeno patient tumor organoid in Matrigel using procedures used earlier for CRPC-NEPCs.[14] The primary tumor tissues (Figure S1B, Supporting Information) were obtained from a rapid autopsy[29] and processed to establish multiple Matrigel organoid lines from various tumor sites. After five subsequent passages, we confirmed the identity of the CRPC-Adeno tumors in Matrigel organoids by comparing gene expression, using NanoString, against known molecular markers of CRPC-Adeno, including AR, KLK3, ENO2 (NSE), NKX3.1, AR-V7, CHGA, SYP, PSMA, and HOXB13 (Figure 1F). Among these organoids, the lymph node biopsy site expressed the markers associated with CRPC-Adeno, referred to as OWCM-1358. Immunohistochemistry analysis indicated that both patient’s tumor sample and OWCM-1358 tumor organoid manifested similar histological features and protein expression (AR, PSMA, and NKX3.1) (Figure 1G). In comparison to CRPC-NEPC organoids in Matrigel (OWCM-154 and OWCM-155) (Figure 1H), the CRPC-Adeno (OWCM-1358) organoids showed strikingly distinct genes, confirming that Matrigel-derivation did not induce transdifferentiation to neuroendocrine lineage. To determine whether the tumor cells in Matrigel organoids expressed integrins α2 and β1 similar to that seen in the patient biopsies, we performed immunohistochemistry and observed that while OWCM-155 Matrigel organoids were stained positive for integrin α2 and β1, the OWCM-1358 organoids only stained positive for integrin α2 (Figure 1I). Using mRNA analysis, we further observed that the OWCM-155 organoids upregulated a few integrin genes, including ITGA6 and ITGB1 that correspond to laminin-binding in Matrigel, however, unlike patient samples, both CRPC-Adeno and CRPC-NEPC Matrigel organoids downregulated ITGA4 (Figure S3C, Supporting Information).
2.4. Defined ECM in Synthetic Hydrogel-Based Prostate Cancer Organoids Regulates Phenotype and Morphology of Patient-Derived Cells
CRPC-NEPC and CRPC-Adeno tumors have ECM components not presented in Matrigel. Matrigel organoids also have batch-to-batch variability and are not conducive to controlled modifications[30,31] to present adhesion ligands of interest. Therefore, we engineered a synthetic four-arm PEG-4MAL-based hydrogel as an organoid platform to grow patient prostate tumor organoids initially derived in Matrigel and exposed to a week of defined ECM conditions in synthetic hydrogels. We were most interested in early changes in phenotype and transcriptome within the timeframe of the treatment with EZH2i, and therefore these studies focused on up to 7–10 days of cultures. We exploited the click-chemistry between PEG-4MAL and thiol moieties[31–33] to functionalize thiolated ECM mimic peptides and crosslink with di-thiolated materials (Figure 2A). To determine the hydrogel crosslinkers that would allow cell-mediated matrix remodeling through proteases,[34] we examined the metalloproteinases (MMPs) secreted by CRPC-Adeno and CRPC-NEPC patient tumors using RNA-sequencing. We observed high expression of MMP-3, 9, 14, and 16 in CRPC-NEPC and CRPC-Adeno tumors compared to benign prostate tissue (Figure 2B, Figure S4, Supporting Information). The patient tumors also expressed MMP1 and MMP2 at an average high levels, however, MMP-2 was expressed at lower levels than benign tissues (Figure S4, Supporting Information).
Figure 2.
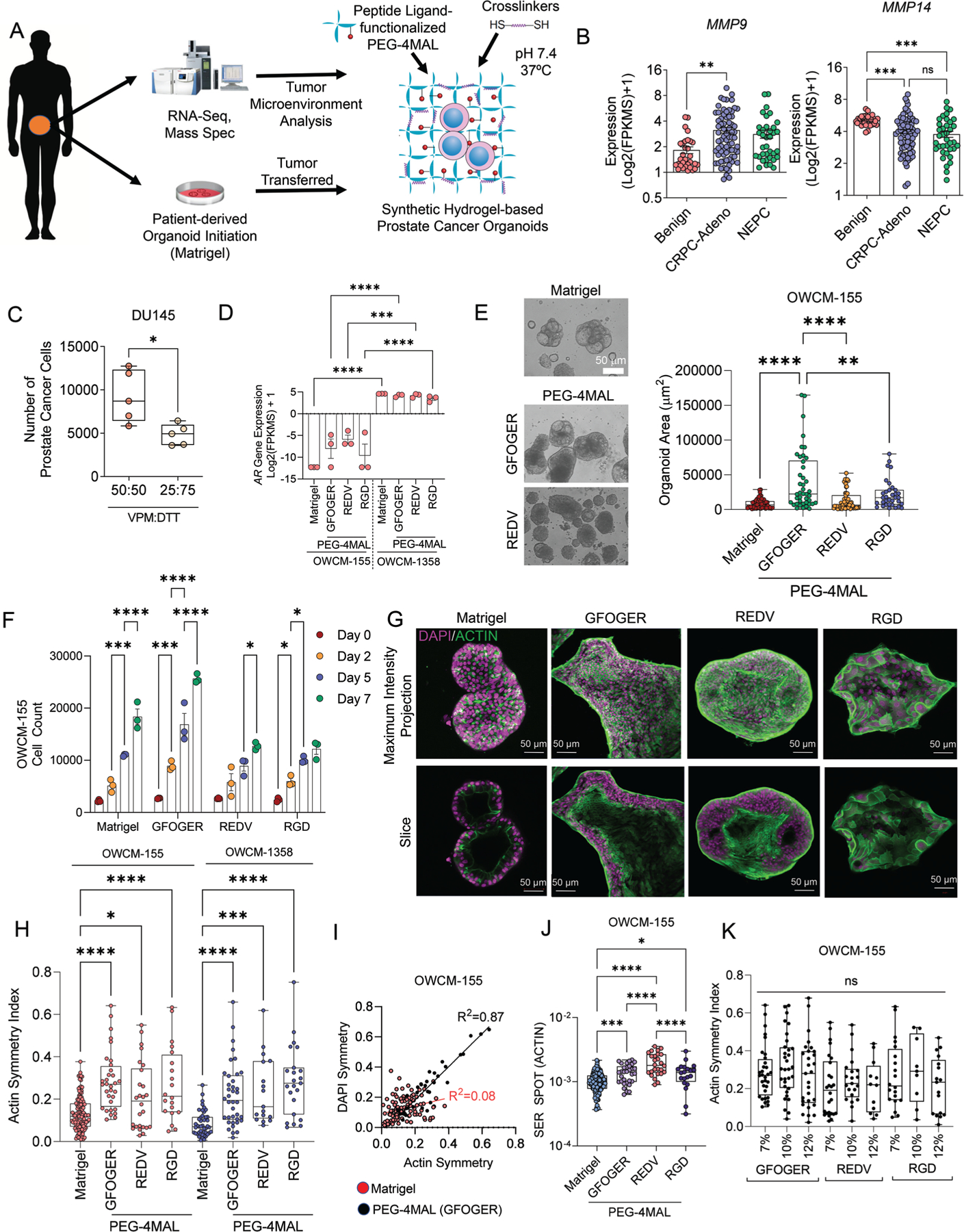
Development and characterization of PEG-4MAL-based synthetic hydrogels to grow prostate cancer organoids. A) Schematic of prostate tumor tissue processing, Matrigel organoid derivation of primary tumors, and serial implantation into ECM-specific PEG-4MAL hydrogels. B) RNA-seq transcriptomic analysis of the patient cohort for matrix MMPs (n = 74 CRPC-Adeno, n = 37 CRPC-NEPC, n = 31 benign samples). Data presented as mean ± S.E.M. C) Growth of DU145 prostate cells in PEG-4MAL organoids with varying VPM:DTT ratios (n = 5). Data presented as mean ± S.E.M. D) AR gene expression in CRPC-Adeno (OWCM-1358) and CRPC-NEPC (OWCM-155) organoids of Matrigel and PEG-4MAL (n = 3 per condition). Data presented as mean ± S.E.M. E) Phase-contrast image of Matrigel and PEG-4MAL organoids and quantified organoid cluster area from confocal microscopy. Each plot represents 3 hydrogels. Data presented as mean ± S.E.M. F) Growth of OWCM-155 cells under Matrigel and defined ECM conditions (n = 3). Data presented as mean ± S.E.M. G) Representative confocal imaging of organoid morphology across ECM conditions (DAPI: purple, actin: green). H) Actin symmetry index across organoid ECM conditions from high-content imaging (CRPC-NEPC: Matrigel n = 128, GFOGER n = 32, REDV n = 26, RGD n = 20; CRPC-Adeno: Matrigel n = 45, GFOGER n = 38, REDV n = 17, RGD n = 23 cell clusters). The box plots show mean (+), median, quartiles (boxes), and range (whiskers). I) Correlation between DAPI and Actin symmetry between Matrigel and GFOGER organoids (Matrigel n = 128, GFOGER n = 32, REDV n = 26, RGD n = 20 cell clusters). J) Texture analysis of actin morphology among organoids. The box plots show mean, median, quartiles (boxes), and range (whiskers). K) Actin symmetry index across organoid stiffness and ECM conditions from high-content imaging. The box plots show mean, median, quartiles (boxes), and range (whiskers). For (C), an unpaired t-test was performed. For all remaining comparisons, groups were compared by a one-way analysis of variance (ANOVA), with posthoc Tukey’s test. For *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
To present defined ECM, we functionalized one of the four Maleimide arms of PEG-4MAL with ECM-mimicking peptide ligands. We crosslinked the remaining three arms with di-thiolated MMP-2, 9, and −14-degradable peptide[35–39] (GCRDVPM↓SMRGGDRCG, referred to as VPM hereafter). However, hydrogels crosslinked with 100% VPM peptide biodegraded rapidly over 48 h (Figure S5A, Supporting Information), likely due to secreted proteases, therefore, we further optimized the stability of hydrogels by mixing VPM with nondegradable crosslinker dithiothreitol (DTT). To determine the effect of co-mixing DTT with VPM on tumor growth, we tested the growth of a model AR-negative prostate cancer cell line (DU145) in PEG-4MAL hydrogels crosslinked with a mixture of 50:50 or 25:75 ratio of VPM:DTT, functionalized with a 3 × 10−3 m concentration of fibronectin-mimicking tri-amino acid peptide sequence, arginine-glycine-aspartate (RGD). A 50:50 ratio of VPM to DTT supported the growth of prostate cancer cells over seven days without significantly degrading the gels, whereas 25% of VPM crosslinked hydrogels had relatively less growth (Figure 2C, Figure S5A, Supporting Information). AR-dependent cell line LNCaP responded similarly to the organoid degradation effect (Figure S5B, Supporting Information). Therefore, all further studies considered a 50:50 ratio of VPM:DTT.
Next, we determined the effect of PEG4MAL hydrogel-presented ECM peptide mimics (fibronectin/VTN mimicking RGD, fibronectin mimicking REDV, and collagen mimicking GFOGER peptide) and Matrigel on the growth of patient-derived CRPC-NEPC (OWCM-155) and CRPC-Adeno (OWCM-1358) tumor cells. GFOGER is a triple-helical synthetic peptide derived from type I collagen with high binding affinity for α1β1, α2β1, α10β1, and α11β1.[40,41] In contrast, REDV is a tetrapeptide Arg-Glu-Asp-Val that mimics Fibronectin in its ability to bind α4β1 integrins. RGD is a short linear peptide present in VTN, fibronectin, and other ECM proteins and binds several integrins, including αvβ3, αvβ1, and α5β1.[42–44] The mRNA analysis indicated that all PEG-4MAL ECM conditions retained AR gene expression in CRPC-Adeno (OWCM-1358) patient-derived lines. In contrast, the AR expression did not increase in CRPC-NEPC (OWCM-155) patient-derived cells suggesting that short-term ECM mimics do not revert to a CRPC-Adeno state from the NEPC state (Figure 2D).
We observed that the hydrogel ECM type influenced the organoid growth area of OWCM-155 patient cells, defined as the total 3D surface area of a cell cluster as measured by confocal imaging (Figure 2E). Cell grown in GFOGER conditions grew into the largest size organoids over a week compared to REDV and RGD functionalized matrices, which remained similar to Matrigel (Figure 2E). Along these lines, total cell count in organoids over seven days showed a significantly higher proliferation of OWCM-155 cells in GFOGER than in Matrigel, REDV, and RGD organoids (Figure 2F). Cell proliferation was confirmed by Ki67+ cell analysis using flow cytometry (Figure S5C, Supporting Information) and immunostaining (Figure S5D, Supporting Information).
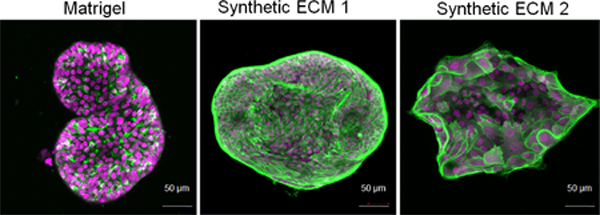
Confocal imaging suggested that tumors displayed spherical morphology in Matrigel organoids (Figure 2G). In contrast, the other three synthetic matrices induced distinct morphology. In GFOGER and RGD-functionalized PEG-4MAL organoids, we observed increased cell spreading and the expression pattern, distribution, and the amount of cytoskeletal protein, Actin (Figure 2G). High-content imaging using Operetta and cell cluster sphericity analysis indicated that GFOGER, REDV, and RGD matrices led to a wide range of nonspherical tumor clusters formed with both OWCM-155 and OWCM-1358 patient-derived cells, as compared to Matrigel induced spherical organoids (Figure S6, Supporting Information).
Remodeling of the actin cytoskeleton is necessary for epithelial–mesenchymal transition in cancer progression and metastasis.[45] To understand how hydrogels with specific ECM ligands regulated actin, we performed image analysis on organoids stained with phalloidin and DAPI, using the PhenoLOGIC machine-learning tool in Operetta high-content imaging system. The machine-learning tool quantifies texture parameters (Spots or Ridges) based on a characteristic intensity pattern within the image. In contrast, the symmetry morphology parameter quantifies the distribution of either texture or fluorescence intensities inside a region of interest. The cellular texture parameters can reflect the cytoskeletal status of the cells.[46] Compared to Matrigel, OWCM-155 tumors grown in all three PEG-4MAL-resented ECMs demonstrated a broader fluorescence distribution concerning cellular symmetry of actin filaments (Figure 2H), as well as nuclear stain DAPI (Figure S7A, Supporting Information).
We observed a high correlation between the actin symmetry and nuclear symmetry of CRPC-NEPCs in GFOGER (R2 = 0.87), REDV (R2 = 0.84), and RGD (R2 = 0.93) but a low correlation in Matrigel organoids (R2 = 0.08) (Figure 2I, Figure S7B, Supporting Information). In addition to cell morphology features, the texture parameters of cytoskeletal fibers at the 1px scale were determined to measure the cytoskeletal and nuclear structure differences. The synthetic hydrogel-based organoids with tumor-specific ECM resulted in significantly higher actin spots and ridges in CRPC-NEPC tumors than Matrigel (Figure 2J, Figure S7C, Supporting Information). However, in contrast to the pattern seen with actin symmetry, CRPC-Adeno (OWCM-1358) grown in synthetic ECMs did not show an increased texture than in Matrigel (Figure S7D, Supporting Information).
Finally, the physical properties of hydrogels (e.g., stiffness) could account for properties independent of ECM-mediated signaling. We changed the polymer weight % of PEG-4MAL from 7% to 10% and 12%. For context, the average storage modulus of 6% and 12% PEG-4MAL hydrogels are 250 and 430 Pa, respectively,[31] and average loss moduli are 15 and 20 Pa, respectively.[31] By keeping the adhesive ligand density the same, the change in storage modulus did not significantly change the actin symmetry index (Figure 2K), suggesting the differences are integrin ligand type-driven and not stiffness, at least in the tested conditions, however more detailed analysis is warranted.
2.5. ECM in PEG-4MAL Organoids Modulate the Expression of the Epigenetic Regulator EZH2 and Response to EZH2i
The transdifferentiation from CRPC-Adeno to CRPC-NEPC is strongly correlated with Polycomb group protein-mediated epigenetic silencing, due primarily to the upregulation of epigenetic modifiers EZH2, which methylates histone H3K27Me2 to H3K27Me3 to alter the expression of lineage specification genes, cell cycle checkpoint genes, and DNA repair genes.[1,7] Immunohistochemistry on patient biopsies confirmed that EZH2 protein was more abundant in CRPC-NEPC tumors than in CRPC-Adeno and absent in benign prostate tissue (Figure 3A). When grown in Matrigel, CRPC-NEPC (OWCM-155) organoids retained high levels and uniform expression of EZH2 (Figure 3A, Figure S8A, Supporting Information). In contrast, REDV peptide presentation significantly increased the expression of EZH2 and its target histone modification, H3k27Me3, compared to Matrigel (Figure 3B). Cells grown in RGD-functionalized PEG-4MAL had no significant increase over Matrigel, whereas those in GFOGER had increased expression of H3k27Me3. To determine whether the H3k27Me3 expression levels were consistent across multiple CRPC-NEPC organoids, we repeated the studies with another CRPC-NEPC patient-derived line, OWCM 154, derived initially in Matrigel from the sternum biopsy site.[14] Similar to OWCM 155, the metastatic tumor organoid from liver biopsy, the OWCM 154 patient-derived cells also demonstrated significantly increased expression of H3k27Me3 in REDV-functionalized hydrogels (Figure 3C), as compared to Matrigel. We observed a positive correlation between α4 and β1 integrins and EZH2 in OWCM-155 grown in PEG-4MAL REDV matrices and the CRPC-NEPC patient samples, but not in other matrices (Figure 3D). In contrast, integrins and EZH2 in PEG-4MAL grown CRPC-Adeno OWCM-1358 organoids showed a low correlation score, and therefore we continued with only CRPC-NEPC for EZH2 inhibition studies.
Figure 3.
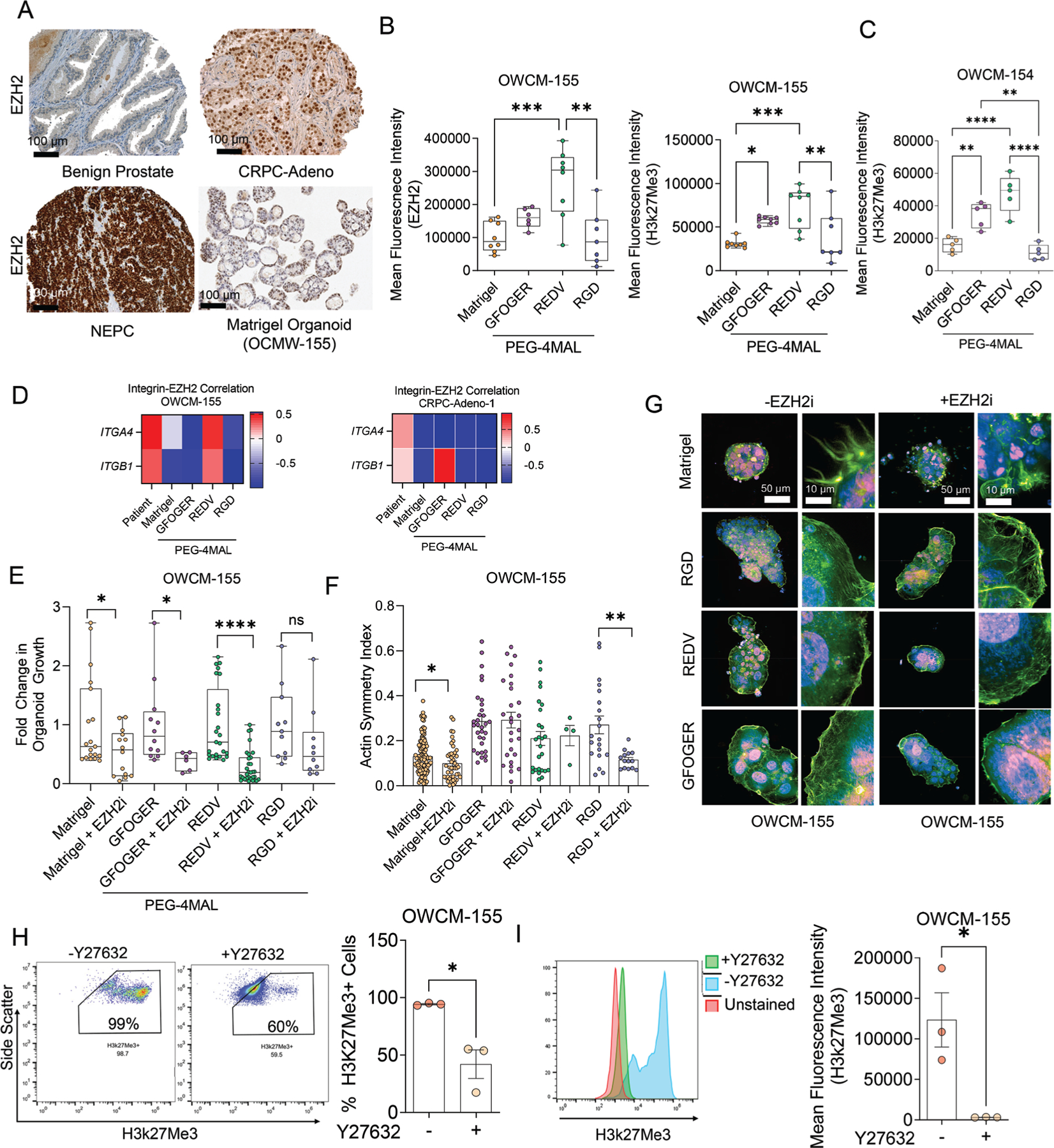
Synthetic ECM regulates EZH2 expression and therapeutic response to EZH2 inhibitor in CRPC-NEPC organoids. A) IHC staining demonstrating EZH2 expression in benign prostate, CRPC-Adeno, and CRPC-NEPC patient tumor biopsies. Comparative EZH2 levels in OWCM-155 (CRPC-NEPC) Matrigel organoids. B) Flow cytometry analysis of EZH2 and H3k27Me3 expression across OWCM-154 (CRPC-NEPC) organoid conditions (n = 5 per condition). Box plots show mean, median, quartiles (boxes), and range (whiskers). For *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. C) Flow cytometry analysis of H3k27Me3 expression across organoid conditions (n = 5 per condition). Box plots show mean, median, quartiles (boxes), and range (whiskers). For *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. D) Integrin and EZH2 gene expression correlation matrices for CRPC-NEPC and CRPC-Adeno organoids. Average of n = 3. E) Fold change in OWCM-155organoid growth area under treatment with an EZH2i GSK343 (Matrigel n = 19, Matrigel+EZH2i n = 14, GFOGER n = 12, GFOGER+EZH2i n = 6, REDV n = 25, REDV+EZH2i n = 31, RGD n = 11, RGD+EZH2i n = 10). A two-tailed t-test evaluated each treated and untreated comparison with *p < 0.05 and ****p < 0.0001. The box plots show mean, median, quartiles (boxes), and range (whiskers). F) Actin symmetry index from high-content imaging (Matrigel n = 128, Matrigel+EZH2i n = 49, GFOGER 7% n = 32, GFOGER 7%+EZH2i n = 27, GFOGER 10%+EZH2i n = 32, GFOGER 10%+EZH2i n = 11). The box plots show mean, median, quartiles (boxes), and range (whiskers). G) Representative images of organoids from high-content imaging under EZH2 inhibition and untreated conditions. (Purple: EZH2, blue: DAPI, green: actin). H,I) Percentage H3K27Me3+ cells (H) and expression level of H3k27Me3 (I) under ROCKi Y27632 conditions. The flow cytometry plots represent gating strategy (H) and histogram overlay represents a shift in the H3k27Me3 signal (I). Data are presented as mean ± S.E.M. An unpaired two-tailed t-test was performed for (H) and (I). For all other comparisons, a one-way ANOVA, with posthoc Tukey’s test was performed with *p < 0.05, **p < 0.01.
We hypothesized that interactions of prostate cancer cells with the ECM ligands regulate EZH2 and its activity. We treated PEG-4MAL grown CRPC-NEPC organoids with EZH2i GSK343 and evaluated growth using a luminescence assay (Figure 3E). Compared to untreated controls, cells in GFOGER- and REDV-functionalized hydrogels demonstrated reduced tumor growth when treated with GSK343. In contrast, RGD matrices did not show any significant reduction in tumor growth. The reduction in tumor growth in GFOGER and REDV matrices correlated with high H3k27Me3 expression, suggesting an increased sensitivity to the drug in these synthetic matrices. Similar results were seen with CRPC-NEPC OWCM-154 patient-derived cells (Figure S8B, Supporting Information).
Next, we characterized the role of EZH2 inhibition on actin localization in organoids using high-content imaging of organoids under EZH2 inhibition (Figure 3F,G). In comparison to untreated organoids, when treated with EZH2i GSK343, we observed a loss of actin cytoskeleton structure in OWCM-155 tumors grown in Matrigel. In contrast, PEG-4MAL hydrogel-based organoids did not show the same actin loss. Notably, Matrigel organoids had uniform EZH2 expression throughout the nucleus. Contrary to that, the PEG-4MAL hydrogel-based organoids showed heterogeneous expression of EZH2 across the nucleus, with differences being especially abundant among REDV and GFOGER matrices. We did not observe a significant difference in actin symmetry in GFOGER and REDV matrices with inhibition of EZH2 (Figure 3F,G). In contrast, RGD-functionalized hydrogels demonstrated a significant reduction in actin symmetry with inhibition of EZH2 (Figure 3F). Although RGD conditions did not reduce cell growth under EZH2i conditions, they seem to disrupt the actin symmetry. These results support the context of ECM ligand dependence of CRPC-NEPC biology and drug response. We next determined whether the integrins were regulating EZH2 activity, that is, trimethylation of histone 3, through actin cytoskeleton. We added Rho-associated protein kinase (ROCK) inhibitor[47] Y-27632 to the REDV organoid culture and observed that ROCK inhibition reduced the percentage of H3k27Me3+ cells and expression level of H3k27Me3 (Figure 3H,I, Figure S8C, Supporting Information), determined via flow cytometry. Our findings provide evidence that ECM may regulate the activity of epigenetic markers, such as EZH2.
2.6. The ECM Composition of PEG-4MAL Hydrogel-Based Organoids Regulates the DNA Methylation Profile of CRPC-NEPCs
To further explore the effect of ECM components on epigenetics, we performed Reduced-representation bisulfite sequencing (RRBS-Seq) to determine DNA methylation programming in cells grown in synthetic ECM-functionalized PEG-4MAL hydrogel organoids and Matrigel organoids and compared to patient RRBS-Seq. Promoter methylation was calculated by averaging the methylation levels of inside CpGs. Genes were ranked based on the standard deviation of promoter methylation across all the samples. Top 100 genes were selected to generate the plots of Pearson correlation matrix and hierarchical clustering. The CRPC-NEPC (OWCM-155) cells grown in PEG-4MAL hydrogels functionalized with REDV and RGD ECMs shared higher similarities of promoter methylation compared to GFOGER ECM and the patient sample (Figure 4A, Figure S9A, Supporting Information). Of note, the patient sample was more similar to RGD and REDV compared to GFOGER based on the correlation of promoter methylation (Figure 4B). These findings suggest that RGD and REDV-functionalized PEG-4MAL hydrigels can better recapitulate the epigenetic signatures of CRPC-NEPC
Figure 4.
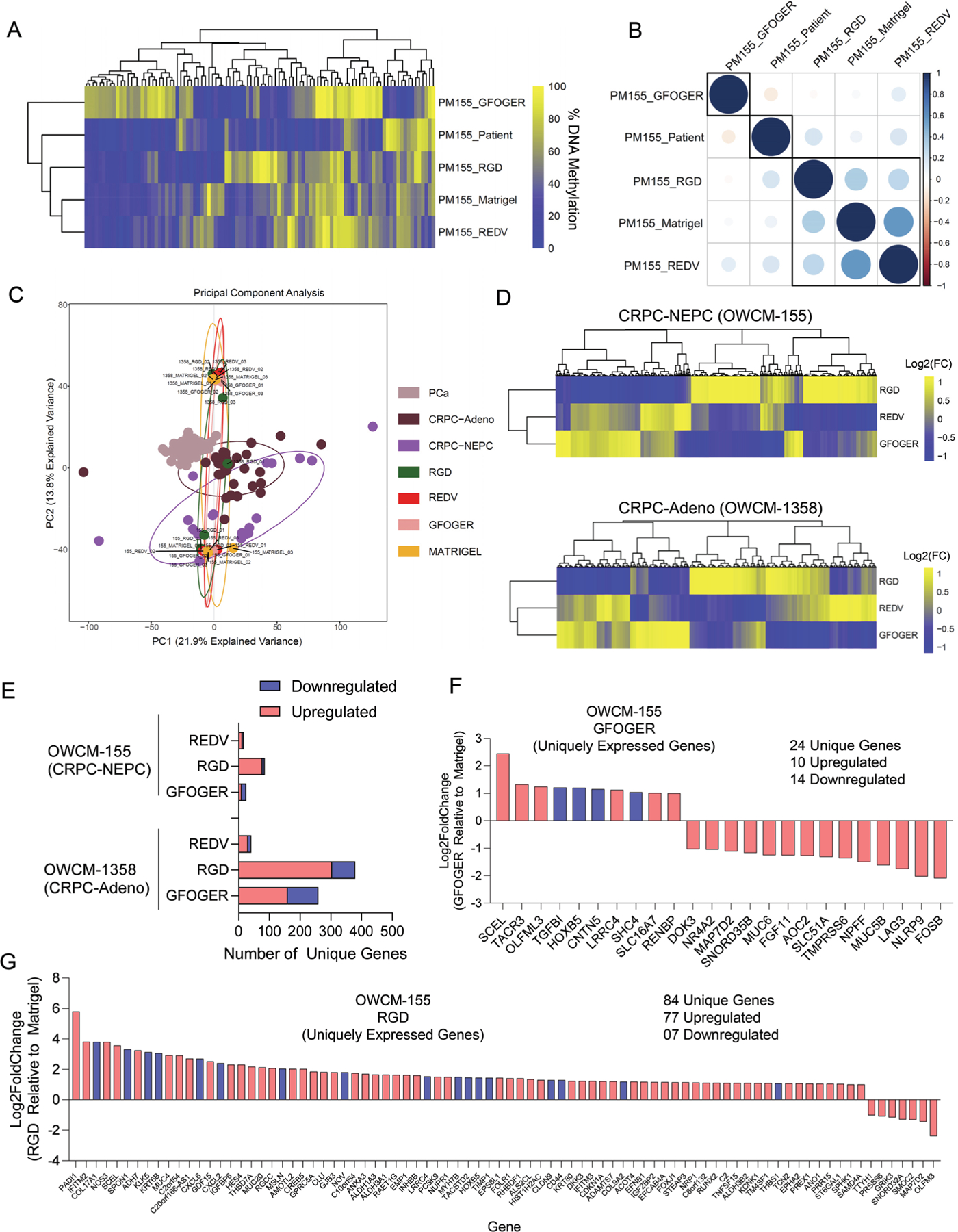
ECM-dependent differential DNA methylation and gene mobilization in Peg-4MAL hydrogel-based prostate cancer organoids. A) Heirchial clustering heatmap of the DNA methylation levels of the top 100 most variable promoters of methylation. Genes were ranked based on the standard deviation of promoter methylation across all the samples. Data represent pooled cells from 24 PEG-4MAL hydrogel-based organoids and 30 Matrigel organoids. B) Pearson correlation of methylation profiles between patient samples and organoids. The numbers represent pair-wise Pearson correlation coefficients between each pair. Promoter methylation was calculated by averaging the methylation levels of inside CpGs. C) PCA for gene expression in organoids compared to primary patient tumors CRPC-NEPC, CRPC-Adeno, and localized prostate adenocarcinoma PCa. D) Heatmap of differentially expressed genes in CRPC-NEPC OWCM-155 and CRPC-Adeno OWCM-1358 cultured across GFOGER, RGD, and REDV-functionalized PEG-4MAL hydrogel and normalized to Matrigel. Data represent an average of 3 organoids per condition. Yellow (high), blue (low). E) Number of differentially expressed unique genes in OWCM-155 and OWCM-1358 tumors grown in PEG-4MAL organoids relative to Matrigel. Data represent an average of 3 organoids per condition. F) Unique genes expressed by OWCM-155 tumors grown in GFOGER hydrogels. Blue indicates genes that are related to cell adhesion or cytoskeletal pathways. G) Unique genes expressed by OWCM-155 tumors grown in RGD hydrogels. The blue bars indicate genes that are related to cell adhesion or cytoskeletal pathways. The data represent an average of 3 organoids per condition.
2.7. The ECM Composition of PEG-4MAL Hydrogel-Based Organoids Regulates the Gene Mobilization of CRPC-Adeno and CRPC-NEPCs
To further explore the effect of ECM components on transcriptional programming, we performed bulk RNA sequencing on cells grown in defined ECM-functionalized PEG-4MAL hydrogel-based organoids and Matrigel organoids. PCA analysis of batch normalized gene expression suggested that individual CRPC-NEPC organoids were closer to their corresponding gene expressions of patient tumor type (Figure 4C), reported earlier.[7,14] We next normalized genes from tumor cells grown in each PEG-4MAL hydrogel condition to genes expressed by tumor cells grown in Matrigel. Using −log2fc > 1 and padj < 0.05 or log2fc < −1 and padj < 0.05 as thresholds, we found that no major changes were seen in hallmark NEPC genes in PEG-4MAL conditions, relative to Matrigel (Figure S9B, Supporting Information). Unsupervised analyses for OWCM-155 and OWCM-1358 cells suggested that most differentially expressed genes clustered together in GFOGER and REDV but were distinct from RGD organoids (Figure 4D). These findings suggest that while overall the CRPC-NEPC cells grown in PEG-4MAL conditions are close to patient samples in gene expression, the ECM still distinctly upregulates genes and therefore can be used as modular systems to investigate the impact of individual ECMs.
RNA sequencing further revealed distinct gene mobilization in CRPC-NEPC OWCM 155 and CRPC-Adeno OCMW-1358 cells grown in various ECM conditions (Figure 4E). The gene mobilization was markedly higher in CRPC-Adeno OWCM-1358 grown in all PEG-4MAL hydrogels than seen in CRPC-NEPCs grown in the same type of ECM-functionalized hydrogels (379 genes in CRPC-Adeno, 84 in CRPC-NEPC in RGD-functionalized hydrogels) (Figure 4E). The GFOGER matrix upregulated 10 unique genes and downregulated 14 genes in OWCM-155 cells (Figure 4F). RGD organoids induced changes to 84 uniquely expressed genes in OWCM-155 cells (77 upregulated and 7 downregulated) (Figure 4G), not found to be different in GFOGER- and REDV-functionalized hydrogels (Figure S9C, Supporting Information). Several of these genes were associated with cell adhesion or cytoskeletal pathways (highlighted in blue). The REDV-functionalized hydrogels induced 12 uniquely upregulated genes and four downregulated genes in OWCM-155 cells.
In contrast, CRPC-Adeno tumor cells grown in the GFOGER-functionalized PEG-4MAL showed an upregulation of 160 genes and downregulation of 99 genes (Figure S10, Supporting Information), whereas REDV-functionalized PEG-4MAL induced 30 unique upregulated genes and 11 downregulated genes (Figure S10, Supporting Information). None of the upregulated genes in OWCM-1358 grown in REDV-functionalized hydrogels could be traced to cell adhesion or cytoskeletal pathways. These results highlight that short-term exposure to different ECM types can mobilize distinct genes in CRPC-Adeno and CRPC-NEPC, which could be traced to cell-cell adhesion or cytoskeletal pathways, as well as correlate with differences in therapeutic drug responses. These findings further suggest that distinct ECM components can differentially regulate gene expression and DNA methylation.
2.8. ECM Differentially Regulates Gene Expression Pathways in CRPC-NEPCs
Next, we performed gene set enrichment analysis (GSEA) to assess gene expression pathways associated with each synthetic ECM (Figure 5, Figures S11 and S12, Supporting Information). The enriched pathways and hallmarks were identified by pre-ranked GSEA using the gene list ranked by log-transformed p values with signs set to positive/negative for a fold change of > 1 or <1. GSEA on multiple pathways associated with CRPC-NEPCs revealed that PEG-4MAL hydrogel-derived organoids upregulated ECM, embryonic morphogenesis, sequence-specific DNA binding, Pattern specification process, cell adhesion, neuroendocrine cell differentiation, ECM receptor, EMT transition, epigenetic regulator, and EZH2 target pathways as compared to Matrigel organoids (Figure 5A,B, Figure S11, Supporting Information). These pathways are distinctly altered in CRPC-NEPCs, as identified previously by Beltran et al.[7] Interestingly, the results identify that while synthetic ECMs enriched for all pathways relative to Matrigel, no single ECM can enrich all pathways to the same extent. The ECM pathway was more enriched in GFOGER, whereas the epigenetics pathway was more enriched in RGD (Figure 5B).
Figure 5.
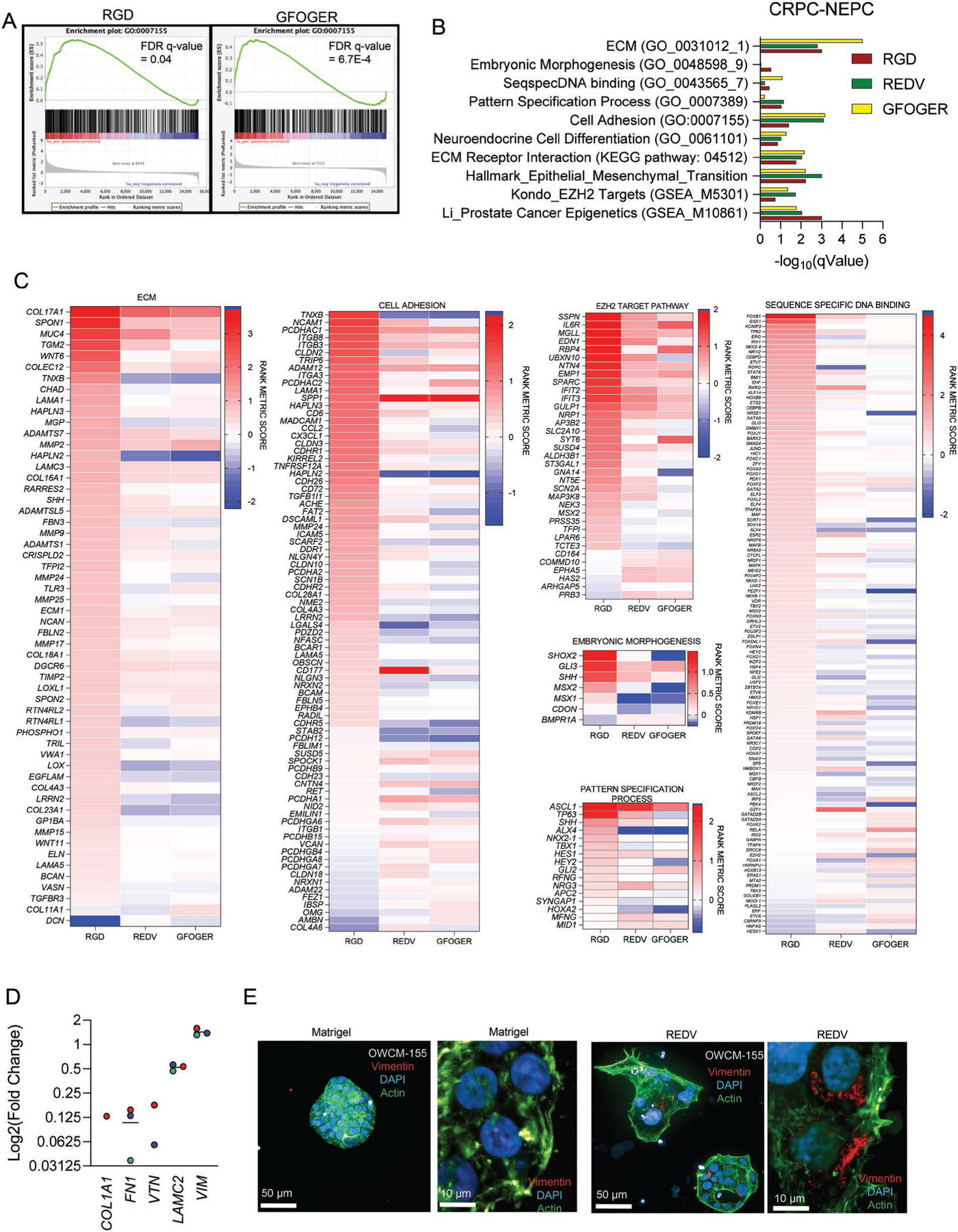
ECM in synthetic hydrogels drives a transcriptionally distinct phenotype. A) Cumulative enrichment plot of CRPC-NEPC OWCM-155 grown in PEG-4MAL organoids. B) False discovery rate q-values of GSEA pathways. All samples were compared to Matrigel, and a q-value below 0.25 is considered significant. C) Heat maps of pathway-specific enriched genes for OWCM-155 tumors grown in RGD, REDV, and GFOGER-functionalized PEG-4MAL hydrogels. Heat map values are relative to Matrigel. All sequencing data presented here were generated from whole transcriptome sequencing of n = 3 organoids per condition. D) Expression of Vimentin in CRPC-NEPC tumors grown in PEG-4MAL hydrogel-based organoids. E) Representative confocal imaging of Matrigel and REDV organoids for Vimentin (red), DAPI (blue), and Actin (green).
We observed an upregulated ECM pathway and cell adhesion pathway among CRPC-NEPCs (Figure 5C), indicating that the cells differentially responded to the distinct microenvironment through integrin-ECM interactions. For instance, the RGD-functionalized PEG-4MAL matrix enriched for the ITGB3 gene, which corroborates with RGD’s ability to bind to integrin αvβ3. Cells grown in RGD-functionalized PEG-4MAL also upregulated integrin α3, which was one of the integrins expressed in CRPC-NEPC patient RNA sequencing (Figure S1B, Supporting Information). The REDV and GFOGER matrices were mostly distinct in the rank metric scores, and some ECM-associated genes were downregulated in these two matrices as compared to RGD (e.g., tenascin-X TNXB, Hyaluronan, and Proteoglycan Link Protein 2 HAPLN2).
As shown in Figure S9A, Supporting Information, EZH2 is expressed at twofold higher in CRPC-NEPC compared to CRPC-Adeno patient tumors. Therefore, we performed an epigenetic pathway analysis and EZH2 target gene analysis (Figure 5C, Figure S12A, Supporting Information) to identify the role of ECM ligand type in modulating EZH2 target gene expression. Several genes were enriched in organoids grown in synthetic ECMs, compared to Matrigel. Among these, a few genes were differentially regulated within GFOGER, REDV, and RGD functionalized PEG-4MAL hydrogels. Specifically, we observed differences in the enrichment of HIC1 (Figure S12B, Supporting Information), which plays a role in chromatin condensation, with lower enrichment in REDV and GFOGER. Among other EZH2 target genes, G protein α subunit 14 (GNA14) was differentially enriched, with negative scores in REDV and GFOGER (Figure 5C). GNA14 depletion inhibits the proliferation of cells and potentially slows down growth genes or cell cycle genes.[48] EZH2 facilitates cell proliferation by repressing cyclin-dependent kinase inhibitors, especially CDKN2A (p16Ink4a p14Arf), a canonical Polycomb target gene and tumor suppressor, and inhibits progressions from G1 into S phase.[49] We observed a modest enrichment of CDKN2A in all three PEG-4MAL ECM organoids relative to Matrigel organoids (Figure S12, Supporting Information). RGD also had a markedly large number of enriched genes in Embryonic Morphogenesis and Sequence-Specific DNA binding pathways, with RGD and REDV being more aligned with each other than GFOGER (Figure 5C). In contrast. RGD was largely distinct from REDV and GFOGER in Pattern Specification process pathway analysis.
A hallmark of CRPC-NEPCs is that they exhibit reactivation of epithelial–mesenchymal transition plasticity. Therefore, we suspected that, among neuroendocrine tumors, distinct integrin ligands would further upregulate genes to drive an epithelial–mesenchymal transition. While we observed an enrichment of the epithelial–mesenchymal transition pathway in all three matrices, OCWM-155 tumors grown in REDV- and GFOGER-conditions had a distinct signature than those grown in RGD-functionalized hydrogels (Figure S12B, Supporting Information). These findings further suggest that while overall gene expression is close to patient samples for all PEG-4MAL organoids, different ECM ligands induce distinct changes to pathways in CRPC-NEPCs. This distinction cannot be captured in Matrigel alone, therefore emphasizing the need for a synthetic, modular ECM. Among genes near equally enriched in all three ECMs, as compared to Matrigel, cytoskeletal gene vimentin (VIM) was significantly upregulated for all 3 ECM ligands (Figure 5D). Vimentin is an intermediate filament protein that forms networks extending from a juxtanuclear cage to the cell periphery. Using confocal microscopy, we observed that Vimentin was upregulated at the protein level in REDV-functionalized hydrogels than in Matrigel and localized near the nucleus (Figure 5E).
2.9. Dopamine Receptor D2 (DRD2) as a Novel Therapeutic for CRPC-NEPCs
To identify a potential new targeted therapy, we hypothesized that differentiation to a neuroendocrine phenotype would result in a greater expression of genes associated with a neural lineage. RNA sequencing analysis in patient tumors revealed that Dopamine Receptor D2 (DRD2) was significantly upregulated in CRPC-NEPC patients than CRPC-Adeno patients and benign tissues (Figure 6A). Neuroendocrine cancers upregulate DRD2, and its targeting has emerged as a promising therapy in the treatment of glioma.[50] However, DRD2’s comparative presence in CRPC-NEPC and CRPC-Adeno is not well understood. Before testing the ECM effect, we confirmed a high expression of DRD2 among the OWCM-155 cells grown in PEG-4MAL hydrogel-based organoids and Matrigel organoids (Figure 6B). In contrast, OWCM-1358 cells did not express DRD2 among any synthetic ECM conditions or Matrigel (Figure 6B).
Figure 6.
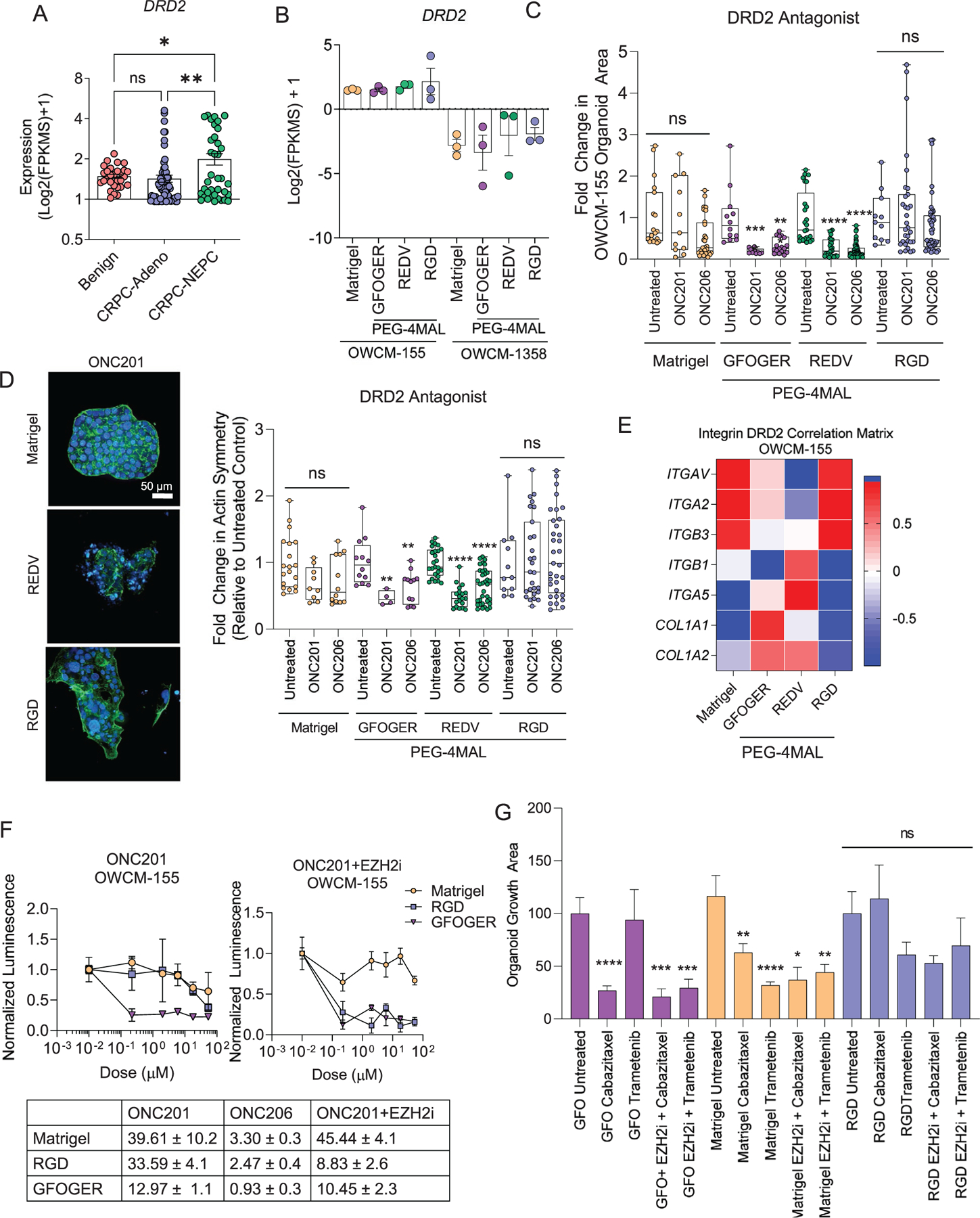
Dopamine Receptor 2 as a single agent and combinatorial therapeutic target in CRPC-NEPC. A) Transcriptomic expression of Dopamine Receptor 2 (DRD2) gene across disease progression from the patient cohort (n = 31 Benign, n = 74 CRPC-Adeno, n = 37 CRPC-NEPC). A one-way ANOVA compared all groups with a posthoc Tukey’s test with ****p < 0.0001. B) DRD2 gene expression in PEG-4MAL and Matrigel prostate organoids (n = 3 per condition). C) Fold change in the organoid growth area, determined by high-content imaging of cultures grown under DRD2 inhibition conditions. A one-way ANOVA, with posthoc Tukey’s test with **p < 0.01, ***p < 0.001, and ****p < 0.0001 was used for comparison. D) Left, representative high-content imaging of Matrigel and PEG-4MAL organoid architecture, cultured with ONC201. (DAPI: blue, Actin: green). Right, quantification of actin symmetry of organoids under DRD2 antagonist treatment. Treated groups were compared to untreated controls by a one-way ANOVA, with posthoc Tukey’s test with **p < 0.01, ***p < 0.001, and ***p < 0.0001. E) Correlation heatmap of transcriptomic expression of DRD2 signal with genes differentially expressed by synthetic organoids (n = 3 per condition). F) Drug response curves for ONC201 treatment with and without EZH2 inhibitor, GSK343 (n = 5 per condition). Each growth condition was normalized to untreated conditions for that group. G) Drug response for Cabazitaxel and Trametinib single-agent treatment and with the EZH2 inhibitor, GSK343 (n = 5 per condition). A one-way ANOVA, with posthoc Tukey’s test with **p < 0.01, ***p < 0.001, and ****p < 0.0001 was used for comparison.
As a potential therapeutic strategy targeting DRD2, we tested emerging therapeutics, imipridone, anti-cancer compounds that possess a three-ring heterocyclic core structure with two substitutable amines. We specifically tested two emerging small molecule inhibitors ONC201 and ONC206. ONC201 (benzyl-benzylmethyl-imipridone or 1,2,6,7,8,9-hexahydroimidazo[1,2-a] pyrido[3,4-e]pyrimidin-5(4H)-one) is a small molecule discovered through a phenotypic screen for p53-independent inducers of TRAIL-mediated apoptosis—currently in Phase II and Phase I clinical trials for high-grade glioma, with an emphasis in diffuse midline glioma. Using a Bayesian machine-learning approach, we had recently identified that the ONC201 binding target is DRD2,[51] and the drug is in Phase I clinical trial with solid glioma tumors, which express high DRD2.[50] ONC201 has also shown that it is effective against certain subtypes of glioma. It is currently in two Phase II clinical trials[50] and being tested in metastatic neuroendocrine tumors [ClinicalTrials.gov Identifier: NCT03034200]. Similarly, ONC206 (benzyl-flurobenzyl impridone) is an imipridone with highly potent activity in preclinical models of neuroendocrine tumors. Imipridones have shown high bioavailability and an excellent clinical safety profile and, therefore, a promising neuroendocrine tumor approach. Although ONC201 has been tested with hormone-sensitive prostate cancers,[52] the effect of ECM on the efficacy of ONC201 and ONC206 in CRPC-NEPC remains to be explored.
We first performed high-content imaging to characterize the morphological response of organoids to the DRD2 antagonist (Figure 6C). Among OWCM-155 tumors grown in Matrigel and RGD-functionalized PEG-4MAL hydrogels, we observed no response to ONC201 and ONC206 drug treatment. However, organoids in both REDV and GFOGER-functionalized hydrogels had a significantly smaller cluster area after treatment with either ONC201 or ONC206. Intriguingly, since DRD2 can act on cytoskeleton inhibition through microtubules,[51] we examined the changes in actin symmetry after the addition of the DRD2 antagonist. A comparison of maximum intensity projections yielded that among organoids grown in Matrigel and RGD-based hydrogels, cell clusters maintained their actin cytoskeletal structure (Figure 6D). On the other hand, organoids grown in REDV matrices treated with DRD2 antagonists were notably less symmetrical, with a partial loss of underlying actin structure (Figure 6D). We observed a strong correlation between ECM-binding integrins and DRD2 expression among PEG-4MAL hydrogel-based organoids in RNA sequencing results (Figure 6E), suggesting that the ECM mimicking peptides may have impacted the interactions between DRD2 and the corresponding ECM genes.
We tested whether the synthetic ECM matrices could impact the anti-tumor response to ONC201 and ONC206. Using a metabolism-based luminescent assay, we determined that drug treatment up to supraphysiological doses failed to kill cells in Matrigel or RGD-functionalized PEG-4MAL hydrogels. In contrast, cells grown in GFOGER-functionalized PEG-4MAL hydrogels showed susceptibility to treatment at sub-micromolar doses, among both ONC201 and ONC206 treatment (Figure 6F, Figure S14, Supporting Information). We next hypothesized that EZH2 mediates the link between ECM sensing and response to therapeutics. We disrupted EZH2 by pre-treatment with EZH2i GSK343 for five days and observed that PEG-4MAL-based RGD-functionalized organoids became susceptible to DRD2 inhibition, while cells grown in Matrigel organoids remained less responsive to ONC201. A comparison of area under the curve suggested low tumor growth under GFOGER and drug conditions (Figure 6F), and a synergistic effect of EZH2i and DRD2 in both GFOGER and RGD conditions, but not Matrigel. We tested whether the EZH2i mediated reprogramming was also observed with other drugs reported in prostate cancer treatment, Cabazitaxel and Trametinib. We found that the single-agent treatment with Cabazitaxel and Trametinib was ECM-type dependent (Figure 6G). The combinatorial effect with EZH2i was more significantly effective in GFOGER conditions than RGD or Matrigel. These findings underscore the importance of engaging defined ECM with tumor cells and synergistic epigenetic targeting in the treatment of CRPC-NEPCs.
Finally, to establish the in vivo efficacy of DRD2 as a novel therapeutic target for CRPC-NEPCs, we performed in vivo studies by subcutaneously engrafting the CRPC-NEPC patient tumors in immunodeficient mice. For xenograft propagation, tumors were harvested from an established PDX developed from OWCM-154 tumors. Subsequently, 4 tumor fragments, each measuring 2 mm × 2 mm × 2 mm, were transplanted subcutaneously into the flank of seven-week old male nude mice. Once the subcutaneous tumors reached a volume of ≈150 mm3, mice were randomized into 2 treatment groups, ONC201 and vehicle (n = 5 each). ONC201 was administered at 125 mg kg−1 diluted in PBS once a week by oral gavage for a total of 5 weeks after which the mice were humanly euthanized. Mouse weight was monitored throughout the experiment. Mice receiving the PBS control had markedly large tumors and significantly larger tumor volume as compared to mice receiving ONC-201 treatment (Figure 7A–C). The area under the curve was significantly smaller for the ONC-201 treatment group (9272 ± 670) as compared to the control PBS group (15 632 ± 1500) (Figure 7C).
Figure 7.
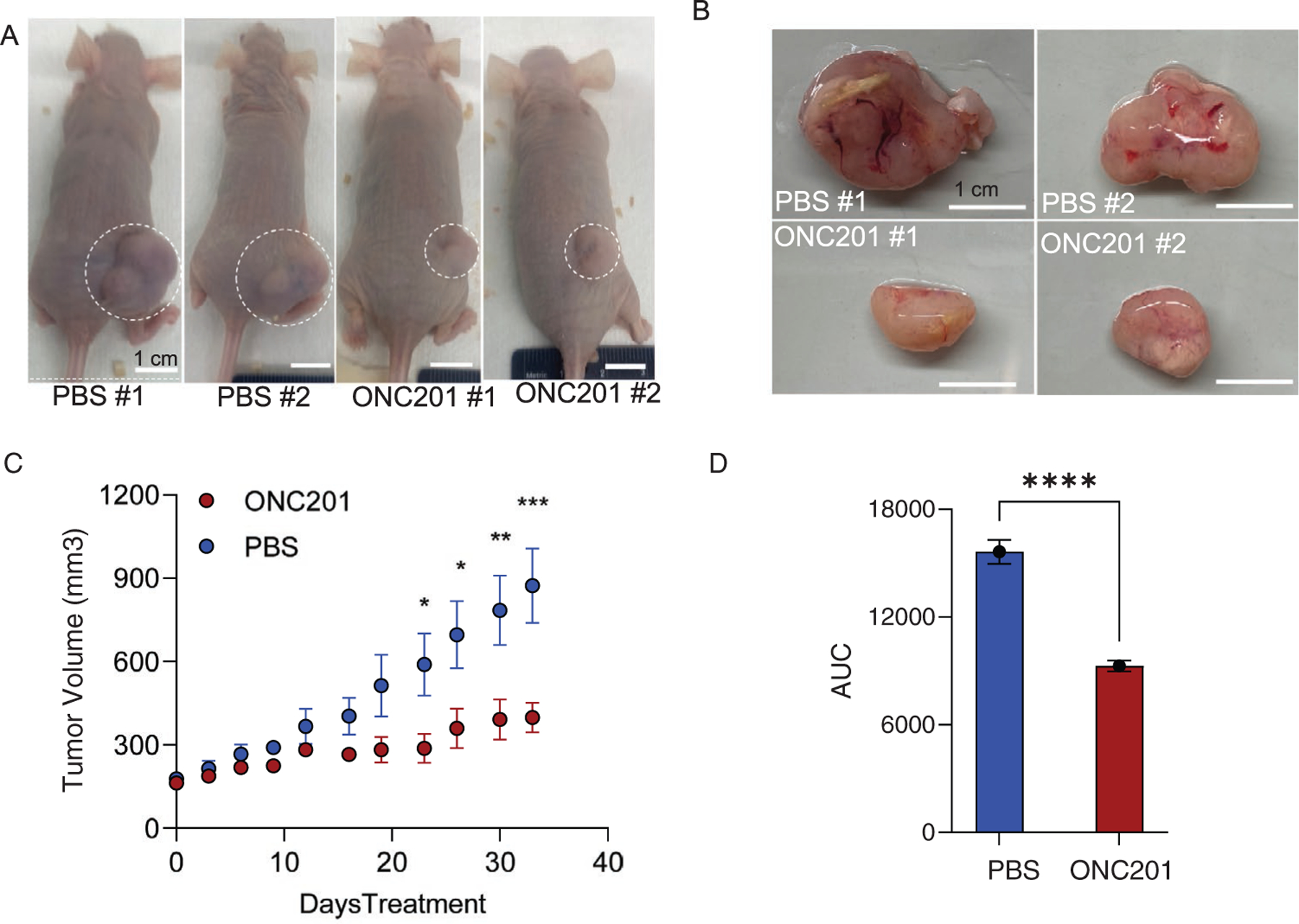
DRD2 inhibitor ONC-201 reduces tumor volume in the PDX-engrafted nude mouse model of CRPC-NEPC. A) Representative images of mice bearing tumors obtained at week 5. PDX line developed from OWCM-154 Matrigel organoids were transplanted subcutaneously into the flank of male nude mice and once the tumors reached a volume of ≈150 mm3, mice were randomized into treatment groups (n = 5 each), with ONC-201 administered at 125 mg kg−1 once a week by oral gavage for a total of 5 weeks. B) Representative images of tumors harvested from mice at week 5. C) Tumor volume curves for nude mice implanted with CRPC-NEPC OWCM-154 PDX tumors and weekly treated with Saline or ONC-201 (125 mg kg−1, oral) continuously until they reached the study endpoint (n = 5). D) Average area under the curves for the CRPC-NEPC OWCM-154 PDX-engrafted nude mice in the different treatment groups (n = 5).
3. Conclusion
A major hurdle in the study of CRPC-NEPC tumors is a lack of existing preclinical models and very few CRPC-NEPC organoids exist. In this work, we have engineered synthetic hydrogel-based ECM to grow CRPC-NEPC and CRPC-Adeno organoids. The synthetic ECM was bioinspired from patient tumor tissue proteomics, spatial omics, immunohistochemistry, and transcriptomics data. In contrast to Matrigel, we demonstrated that bioengineered ECM regulated the growth, EZH2 expression, and its activity, DNA methylation status and mobilized distinct genes in CRPC-NEPC, which could correlate with differences in therapeutic drug responses. These findings suggest that distinct ECM components can differentially regulate gene expression and DNA methylation. Using these synthetic PEG-4MAL hydrogel-grown organoids, we have identified a potential therapeutic target, DRD2 inhibitor ONC-201, for CRPC-NEPC and elucidated how ECM-integrin interactions can render these tumors susceptible to EZH2 and DRD2 antagonist activity. We anticipate that this work will motivate future efforts to study long-term responses in the ECM-specific TME and incorporate other cell-based factors, including cancer-associated fibroblasts, immune cells, and stromal cells. We further anticipate that the PEG-4MAL organoids will improve the success rate of deriving patient organoids from patients’ primary tumors, which currently remain at 16% (4/25) with Matrigel.[14]
4. Experimental Section
Proteomics Analysis:
Prostate cancer tissue processing and mass spectrometric analysis were performed as reported earlier with minor modifications.[53] Briefly, tissues were homogenized, and protein was extracted using 9M urea. After extraction, protein samples were subjected to reduction with DTT (Sigma) and alkylation with iodoacetamide (Sigma) before overnight digestion with Trypsin (Gibco) at 37 °C. The resulting peptides were desalted and labeled with the tandem mass tag (TMT) reagents according to the manufacturer’s protocol (catalog no. 90 110, Thermo Fisher Scientific). Labeled peptides were mixed and desalted before peptide fractionation by high-pH reverse phase chromatography to obtain 12 fractions. About 5% of each fraction was desalted and used for protein expression profiling, and 95% was used for phosphopeptide enrichment by titanium dioxide (TiO2). These fractions were analyzed by Liquid Chromatography with tandem mass spectrometry (LC-MS). For the LC-MS data acquisition, a Thermo Fisher Scientific EASY-nLC 1200 coupled online to a Fusion Lumos mass spectrometer was used. Buffer A (0.1% formic acid in water) and buffer B (0.1% formic acid in 80% acetonitrile) were used as mobile phases for gradient separation. A 75 μm I.D. column (ReproSil-Pur C18-AQ, 3 μm, Dr. Maisch GmbH, German) was packed in-house for peptides separation. Peptides were separated with a gradient of 5–10% buffer B over 1 min, 10%−35% buffer B over 110 min, and 35%−100% B over 10 min at a flow rate of 300 nL min−1. The Fusion Lumos mass spectrometer was operated in data-dependent mode. Full MS scans were acquired in the Orbitrap mass analyzer over a range of 400–1500 m/z with a resolution of 120 000 at m/z 200. The top 15 most abundant precursors were selected with an isolation window of 0.7 Thomson and fragmented by higher-energy collisional dissociation with a normalized collision energy of 40. MS/MS scans were acquired in the Orbitrap mass analyzer. The automatic gain control target value was 1 × 106 for full scans, and 5 × 104 for MS/MS scans, respectively, and the maximum ion injection time was 54 ms for both.
The raw data files were processed for protein identification using the MaxQuant[54] computational proteomics platform version 1.6.1.0 (Max Planck Institute, Munich, Germany). The fragmentation spectra were used to search for the UniProt human protein database (downloaded September 21, 2017). Oxidation of methionine and protein N-terminal acetylation were used as variable modifications for database searching. Phosphorylation on serine, threonine, and tyrosine was also used as a variable modification for the phosphopeptide analysis. The precursor and fragment mass tolerances were set to 7 and 20 ppm, respectively. Both peptide and protein identifications were filtered at a 1% false discovery rate based on a decoy search using a database with the protein sequences reversed.
Patient Cohort Description, Pathology Classification, and Organoid Development:
Fresh tumor biopsy specimens were obtained and processed into organoids, as outlined in the previous work.[14] Briefly, discarded biopsy specimens were obtained through a next-generation sequencing-based clinical study[55,56] approved by the Institutional Review Board at Weill Cornell Medicine (IRB #1305013903). Pathologists reviewed all hematoxylin and eosin-stained and immunohistochemistry slides. Histologic criteria were from the proposed classification of prostate cancer with neuroendocrine differentiation.[8]
For tissue processing and development, the pipeline outlined in the previously established work was followed again.[14] Fresh biopsy samples were isolated and placed in DMEM (Invitrogen) supplemented with GlutaMAX (1×, Invitrogen), 100 U Lml−1, 100 μg mL−1 streptomycin (Gibco), Primocin 100 μg mL−1 (InvivoGen), and 10 μmol L−1 ROCK inhibitor (Selleck Chemical Inc.). Tissue samples were washed 2×, and the tissue was then enzymatically digested in 250 U mL−1 collagenase IV (Life Technologies) and TrypLE Express (Gibco) in a 1:2 ratio. Incubation time was dependent on the amount of tissue, ranging from 30–90 min.
After digestion, tissue fragments were washed in Advanced DMEM/ F12 and centrifuged at 300 rcf for 3 min. The pellet was resuspended with prostate-specific culture media composed of Advanced DMEM (Invitrogen) with GlutaMAX (1×, Invitrogen), 100 U mL−1 penicillin, 100 μg mL−1 streptomycin (Gibco), Primocin 100 μg mL−1 (Invitrogen), B27 (Gibco), N-acetylcysteine 1.25 × 10−3 m (Sigma-Aldrich), Mouse Recombinant EGF 50 ng mL−1 (Invitrogen), Human Recombinant FGF-10 20 ng mL−1 (Peprotech), Recombinant Human FGF-basic 1 ng mL−1 (Peprotech), A-83–01 500 × 10−9 m (Tocris), SB202190 10 × 10−6 m (Sigma-Aldrich), Nicotinamide 10 × 10−3 m (Sigma-Aldrich), (DiHydro) Testosterone 1 × 10−9 m (Sigma-Aldrich), PGE2 1 × 10−6 m (R&D Systems), Noggin conditioned media (5%), and R-spondin conditioned media (5%). The final resuspended pellet was combined with Matrigel (Corning) in a 1:2 volume Matrigel, with 6 50 μL droplets pipetted onto each well of a six-well suspension culture plate (Sarstedt LTD). The plate was then incubated for 30 min at 37 °C to crosslink Matrigel before 3 mL of media was added to each well. The culture was replenished with fresh media changed twice a week. Throughout prostate organoid development, cultures were screened for various Mycoplasma strains using the MycoAlert Kit (Lonza) and confirmed negative before being used for experimental assays.
Immunohistochemistry and Scoring:
Immunohistochemistry (IHC) for Integrin Alpha-2 (ITGA-2), CD29, Synaptophysin (SYN), and NKX3.1 was performed on 4 μm-thick unstained slides using the following primary antibodies: monoclonal rabbit anti-ITGA-1 EPR5788 (Abcam, Cambridge, MA, USA), monoclonal rabbit anti-CD29 (EP1041Y Epitomics, Cambridge, MA), monoclonal mouse anti-Synaptophysin 27G12 (Leica, Buffalo Grove, IL, USA), and polyclonal rabbit anti-NKX3.1 (Biocare Medical, Pacheco, CA, USA). IHC was performed on the BenchMark ULTRA automated staining system (Leica Bond Systems, Inc.). IHC scores were given based on intensity: weak (1+), moderate (2+), or intense (3+), and extent: local, multifocal, or diffuse. At least 5% of positive tumor cells were considered positive. Positive and negative controls were included in each run.
Development of PEG-4MAL Hydrogel Organoids of Prostate Cancer:
To engineer synthetic hydrogel-based organoids using a four-arm PEG-4MAL (MW 22 000, Laysan Bio, Inc., >90% purity), the PEG-4MAL macromers were functionalized with adhesive peptides and crosslinked using di-thiolated crosslinkers as reported earlier.[33] The following single thiol adhesive peptides were used: RGD (GRGDSPC), GFOGER (GYGGGP(GPP)5GFOGER(GPP)5GPC), REDV (GREDVSPC), all purchased from Apptec Peptides. Dilithiolated crosslinker peptide VPM: GCRDVPMSMRGGDRCG was purchased from Apptec Peptides. PEG-4MAL was dissolved in HEPES buffer at a 20 × 10−3 m concentration and functionalized with bioadhesive peptides RGD, REDV, or GFOGER (in HEPES at a 10.0 × 10−3 m concentration to achieve a 5× final ligand density) with at a 4:1 macromer-to-peptide ratio. The PEG-4MAL macromers were crosslinked with di-thiolated MMP-degradable VPM (GCRDVPMSMRGGDRCG) peptide and a mixture of nondegradable DTT at a defined ratio and a 4:1.5 macromer-to-crosslinker molar ratio at 37 °C and 7.4 pH. The final peptide concentration of adhesive peptides in PEG hydrogels was 3 × 10−3 m. OWCM-155, OWCM-154, and OWCM-1358 cells grown in Matrigel were isolated at passage 25–27, as described earlier,[14] and re-encapsulated in PEG-4MAL hydrogels, followed by culture in the prostate cancer media conditions. The OWCM-155 and OWCM-154 were previously established[14] and OWCM-1358 was the newly established Matrigel organoid in the current study.
RNA Sequencing and Analysis of Synthetic Hydrogel-Based Organoids:
RNA-sequencing and data processing of synthetic organoids was performed according to the protocol described earlier.[7,57] Briefly, RNA was extracted from frozen material for RNA-sequencing (RNA-seq) using the Promega Maxwell 16 MDx instrument, (Maxwell 16 LEV simplyRNA Tissue Kit (cat. # AS1280)). Specimens were prepared for RNA sequencing using TruSeq RNA Library Preparation Kit v2. RNA integrity was verified using the Agilent Bioanalyzer 2100 (Agilent Technologies). cDNA was synthesized from total RNA using Superscript III (Invitrogen). Sequencing was then performed on GAII, HiSeq 2000, or HiSeq 2500.[7,57] All reads were independently aligned with STAR_2.4.0f1[58] for sequence alignment against the human genome sequence build hg19, downloaded via the UCSC genome browser (http://hgdownload.soe.ucsc.edu/goldenPath/hg19/bigZips/), and SAMTOOLS v0.1.19[59] for sorting and indexing reads. Cufflinks (2.0.2)[60] was used to estimate the expression values (FPKMS), and GENCODE v19[61] GTF file for annotation. Rstudio (1.0.136) with R (v3.3.2) and ggplot2 (2.2.1) was used for the statistical analysis. The gene counts from htseq-count[62] and DESeq2 Bioconductor package[63] were used to identify differentially expressed genes. The hypergeometric test and GSEA[64] were used to identify enriched signatures using the different pathways collection in the MSigDB database.[65,66] The GSEA pre-ranked method from GSEA was used for the purpose.
Nanostring Analysis:
Molecular characterization was completed on both patient samples and organoids. A targeted gene panel described previously was utilized.[67] Briefly, a targeted gene panel was utilized for this study using the NanoString nCounter that was applied to FFPE, biopsy, and RP tissues with limited RNA input requirement (<300 ng). The gene panel included 163 genes based on known or potential roles in prostate cancer progression. Raw data counts were normalized using the nSolver analysis software version 2.0, which normalizes samples according to positive and negative control probes. The edgeR package was used to determine genes that were differentially expressed when comparing the treated and untreated cases. The differentially expressed genes were identified by fitting a generalized linear cluster model, comparing specimens between disease subtypes. Hierarchical clustering in edgeR library package distance was used.
Flow Cytometry, Immunohistochemistry, and Microscopy:
Tissue sections of patient samples were obtained from the Translational Research Program at Weill Cornell Medicine, previously collected under IRB#1305013903. Immunohistochemistry was performed on deparaffinized FFPE sections (organoid, xenograft, or patient tissue) using a Bond III automated immunostainer (Leica Microsystems, IL, USA). Heat-mediated antigen retrieval was performed using the Bond Epitope Retrieval solution 1 (ER1) at pH 6 or 2 (ER2) at pH9. EZH2 antibody was used at a 1:20 dilution (clone 11/EZH2, BD Biosciences, CA, USA; ER1, 1:20 dilution.)
For flow cytometry analysis, organoids were digested in TrypLE (Gibco) for Matrigel or collagenase (Worthington Biomedical) for PEG-4MAL. After digestion, cells were spun down at 400× to attain single-cell suspensions. Cells were subsequently blocked for half an hour in PBS with 1% BSA (Sigma). Cells were subsequently washed (400×) and resuspended in FACS buffer. After an hour of incubation on ice, cells were washed 3× before being analyzed on an Accuri C6 flow cytometer (Becton Dickinson). Flow cytometry analysis was performed using FlowJo (Treestar). All antibodies or their fluorophore variations were used in these studies at a 1:250 dilution. The list of antibodies, clones, and vendors is available in Figure S15, Supporting Information.
For confocal imaging, cells were seeded onto a glass-bottom coverslip (Matsunami). After culture for seven days to allow organoids to grow, dishes were carefully washed 3× with PBS. Cells were fixed with 4% PFA for 30 min, and organoids were then stained with Phalloidin (40× dilution) for 40 min on ice. Organoids were then permeabilized by incubation with 0.5% Triton x-100 in PBS for 30 min, with a subsequent 3× wash in PBS. Organoids were then incubated with primary antibody for 60 min, washed 3×, and then incubated with secondary antibody for a subsequent 60 min. Finally, organoids were stained with DAPI for ten min. Cells were imaged on an LSM 710 microscope (Zeiss).
For high-content imaging, cells were stained as indicated above except on a 96-well glass-bottom plate (X) and imaged using Operetta CLS High-Content Analysis System. Organoid clusters were identified and analysis was performed using Harmony High-Content Imaging and Analysis Software v4.9. Downstream analysis of organoid readouts was performed in Excel and Graphpad Prism 8 (Graphpad).
Human Prostate Cancer Tissue and Second-Harmonic Generation (SHG) in Collagen:
NEPC and CRPC tissues from patients were screened for this study. The tissue samples were embedded in paraffin, sectioned at a thickness of 5 μm, and stained with Synaptophysin (SYP). Optical images of the tissue immunostaining were acquired by scanning the slides in the Leica Aperio AT2 brightfield slide scanner at 40× magnification.
The SHG of tissue samples were imaged on a deparaffinized unstained slide using a two-photon microscope (Leica SP8 with DIVE optics, Wetzlar, Germany). The sample was excited at 780 nm using a Mai Tai DeepSee femtosecond pulsed laser tunable between 690–1040 nm (Spectra-Physics, Milpitas, CA). The SHG signal from collagen was detected at a wavelength range of 380–407 nm. Images were acquired with either a 5×/0.15 numerical aperture (NA) or 10×/0.4 NA dry objective. Autofluorescence images were acquired for reference between 443–561 nm.
Drug Treatment Studies:
For drug treatment studies, organoids were seeded into either Matrigel or a PEG-4MAL hydrogel. Study timelines were defined as per the drug mechanism of action and previous work.[14] For EZH2 studies, the organoids were allowed to grow for two days, followed by treatment with EZH2 inhibitor GSK343 at a concentration of 3 × 10−6 m, according to the previously established EC50 in Matrigel organoids. The organoids were grown further for five days, and proliferation was quantified using a CellTiterGlo-3D assay (Promega). A more extended incubation period (5 days) with an EZH2 inhibitor was necessary to facilitate the cells’ epigenetic reprogramming. CellTiterGlo-3D was added to the organoid culture at a 1:1 ratio with media, and after 30 min, luminescence was evaluated using a Biotek Synergy H4 plate reader.
For DRD2 antagonist studies, cells were grown for five days and added DRD2 antagonists pre-determined concentrations according to a sixfold dilution, and incubated for two more days. For luminescence studies, a similar protocol was used as described for the EZH2 studies. For high-content imaging, cells were stained with phalloidin and DAPI as described above and imaged them on Perkin Elmer Operetta CLS High-Content Analysis System (PerkinElmer). For synergistic studies, organoids were grown for two days, without any drug, followed by the addition of EZH2i (GSK343) for five days, and finally, the DRD2 antagonist was added for two days and quantified luminescence.
Reduced-Representation Bisulfite Sequencing (RRBS-Seq):
For DNA Methylation studies, 24 PEG-4MAL hydrogels of 125 μL each with 1000 cell μL−1 were fabricated. For Matrigel organoids, 30 droplets of 100 μL each with 1000 cell μL−1 were fabricated. The organoids were allowed to grow for 10 days and the cells were then extracted from the Matrigel using Gibco TryplE Express (Cat #12 605 028) and from the PEG-4MAL gels using 125 U mL−1 Collagenase IV (Cat # 17104–019) solution in media. The organoid cell pellets were processed using the Omega Mag-Bind Blood & Tissue DNA HDQ kit (M6399–01), using the standard cell pellet protocol. Briefly, cells were resuspended in 180 μL ice-cold PBS and the standard protocol was followed as per the manufacturer.
RRBS data were aligned to the bisulfite-converted hg19 reference genome using Bismark. Promoters were defined as the regions encompassing 2 kb upstream and downstream of the transcription start site (TSS) of UCSC genes. Only promoters with a minimum of 5 CpGs covered by at least 10 reads were included in the analysis. Promoter methylation was calculated by averaging the methylation levels of inside CpGs. Genes were ranked based on the standard deviation of promoter methylation across all the samples. Top 500 genes were selected to generate the plots of principal component analysis (PCA), Pearson correlation matrix, and hierarchical clustering.
In Vivo PDX Studies:
For xenograft propagation, established PDX developed from OWCM-154 tumors were prepared as 4 tumor fragments, each measuring 2 mm × 2 mm × 2 mm, and transplanted subcutaneously into the flank of seven-week old male nude mice. Once the subcutaneous tumors reached a volume of ≈150 mm3, mice were randomized into 2 treatment groups, ONC201 and vehicle. Five mice per group were used. DRD2 inhibitor ONC201 was administered at 125 mg kg−1 diluted in PBS once a week by oral gavage for a total of 5 weeks after which the mice were humanly euthanized. Mouse weight was monitored throughout the experiment. Animal care and experiments were carried out following Weill Cornell Medicine’s IACUC guidelines.
Statistical Analysis:
Graphpad Prism 8.0 (Graphpad) was used to perform all statistical analyses, and experimental conditions and analysis are detailed in figure captions. All groups were compared by a one-way ANOVA or a two-way ANOVA, with a posthoc Tukey’s test. In select studies, a t-test was performed. In all studies, significance is defined as *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Supplementary Material
Acknowledgements
The authors acknowledge funding support from the National Cancer Institute of the US National Institutes of Health (5P50CA211024 awarded to O.E., H.B., J.M.M., M.R., D.S.R., and A.S.; 1R01CA238745–01A1 awarded to A.S.), the National Institute of Allergy and Infectious Diseases (NIAID) of the US National Institutes of Health (5R01AI132738–04 awarded to A.S.), the Wellcome Leap HOPE Program (awarded to A.S.), and a US National Science Foundation CAREER award (DMR-1554275 awarded to A.S.). The authors further acknowledge funding support from the NIH Immunoengineering T32 training grant to M.J.M. (NIBIB, 1T32EB023860–01A1) and the National Science Foundation (EEC-1648035, seed funding awarded to A.S). A.F.C holds a Career Award at the Scientific Interface from Burroughs Wellcome Fund. The authors thank Dr. Ari Melnick at Weill Cornell Medicine for insights on epigenetics. Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the funding agency. Animal care and experiments were carried out following Weill Cornell Medicine’s IACUC guidelines and all animal experiments were approved by the IACUC. For the experiments involving human samples, experiments involving samples such as PM154, which were used to form organoids, were approved by the Institutional Review Board at Weill Cornell Medicine (IRB #1305013903) and had informed patient consent. Experiments involving discarded, deidentified samples that were obtained by pathology for immunostaining were also approved by the Institutional Review Board at Weill Cornell Medicine (IRB #1305013903) and had waived consent.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
V.P. and J.E.A. are employees and shareholders of Chimerix, Inc. M.J.M is an employee at Teva Pharmaceutical and this work was completed prior to his employment at Teva. L.P is an employee at Loxo Oncology at Lilly, this work was completed prior to her employment at Loxo, she is acting on her own, and these endeavors are not in any manner affiliated with Loxo Oncology at Lilly.
Contributor Information
Matthew J. Mosquera, Sibley School of Mechanical Engineering, Cornell University, Ithaca, NY 14850, USA
Sungwoong Kim, Woodruff School of Mechanical Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA; Coulter Department of Biomedical Engineering, Georgia Institute of Technology and Emory University School of Medicine, Atlanta, GA 30332, USA.
Rohan Bareja, Englander Institute for Precision Medicine, Weill Cornell Medicine-New York-Presbyterian Hospital, New York, NY 10021, USA; Institute for Computational Biomedicine, Weill Cornell Medicine, New York, NY 10021, USA.
Zhou Fang, Coulter Department of Biomedical Engineering, Georgia Institute of Technology and Emory University School of Medicine, Atlanta, GA 30332, USA.
Shuangyi Cai, Coulter Department of Biomedical Engineering, Georgia Institute of Technology and Emory University School of Medicine, Atlanta, GA 30332, USA.
Heng Pan, Englander Institute for Precision Medicine, Weill Cornell Medicine-New York-Presbyterian Hospital, New York, NY 10021, USA; Institute for Computational Biomedicine, Weill Cornell Medicine, New York, NY 10021, USA.
Muhammad Asad, Department of Pathology and Laboratory Medicine, Weill Cornell Medicine, New York, NY 10021, USA.
Maria Laura Martin, Englander Institute for Precision Medicine, Weill Cornell Medicine-New York-Presbyterian Hospital, New York, NY 10021, USA.
Michael Sigouros, Englander Institute for Precision Medicine, Weill Cornell Medicine-New York-Presbyterian Hospital, New York, NY 10021, USA.
Florencia M. Rowdo, Englander Institute for Precision Medicine, Weill Cornell Medicine-New York-Presbyterian Hospital, New York, NY 10021, USA
Sarah Ackermann, Englander Institute for Precision Medicine, Weill Cornell Medicine-New York-Presbyterian Hospital, New York, NY 10021, USA.
Jared Capuano, Englander Institute for Precision Medicine, Weill Cornell Medicine-New York-Presbyterian Hospital, New York, NY 10021, USA.
Jacob Bernheim, Englander Institute for Precision Medicine, Weill Cornell Medicine-New York-Presbyterian Hospital, New York, NY 10021, USA.
Cynthia Cheung, Englander Institute for Precision Medicine, Weill Cornell Medicine-New York-Presbyterian Hospital, New York, NY 10021, USA.
Ashley Doane, Englander Institute for Precision Medicine, Weill Cornell Medicine-New York-Presbyterian Hospital, New York, NY 10021, USA.
Nicholas Brady, Department of Pathology and Laboratory Medicine, Weill Cornell Medicine, New York, NY 10021, USA.
Richa Singh, Department of Pathology and Laboratory Medicine, Weill Cornell Medicine, New York, NY 10021, USA.
David S. Rickman, Department of Pathology and Laboratory Medicine, Weill Cornell Medicine, New York, NY 10021, USA
Varun Prabhu, Chimerix, Inc., Durham, NC 27713, USA.
Joshua E. Allen, Chimerix, Inc., Durham, NC 27713, USA
Loredana Puca, Englander Institute for Precision Medicine, Weill Cornell Medicine-New York-Presbyterian Hospital, New York, NY 10021, USA.
Ahmet F. Coskun, Coulter Department of Biomedical Engineering, Georgia Institute of Technology and Emory University School of Medicine, Atlanta, GA 30332, USA
Mark A. Rubin, Department for BioMedical Research, University of Bern, Bern 3012, Switzerland
Himisha Beltran, Department of Medical Oncology, Dana Farber Cancer Institute, Harvard Medical School, Boston, MA 02215, USA.
Juan Miguel Mosquera, Englander Institute for Precision Medicine, Weill Cornell Medicine-New York-Presbyterian Hospital, New York, NY 10021, USA; Department of Pathology and Laboratory Medicine, Weill Cornell Medicine, New York, NY 10021, USA.
Olivier Elemento, Englander Institute for Precision Medicine, Weill Cornell Medicine-New York-Presbyterian Hospital, New York, NY 10021, USA; Institute for Computational Biomedicine, Weill Cornell Medicine, New York, NY 10021, USA.
Ankur Singh, Sibley School of Mechanical Engineering, Cornell University, Ithaca, NY 14850, USA; Woodruff School of Mechanical Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA; Coulter Department of Biomedical Engineering, Georgia Institute of Technology and Emory University School of Medicine, Atlanta, GA 30332, USA.
Data Availability Statement
The RNA-seq data for organoids generated during the current study are available through Gene Expression Omnibus (GEO) accession number: GSE181462 with the following sub-series: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE181462. The published human data are available through dbGap:phs000909.v.p1 (http://www.cbioportal.org/study?id=nepc_wcm_2016)(7).
References
- [1].Davies AH, Beltran H, Zoubeidi A, Nat. Rev. Urol 2018, 15, 271. [DOI] [PubMed] [Google Scholar]
- [2].Siegel RL, Miller KD, Jemal A, Ca-Cancer J. Clin 2018, 68, 7. [DOI] [PubMed] [Google Scholar]
- [3].Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A, Ca-Cancer J. Clin 2018, 68, 394. [DOI] [PubMed] [Google Scholar]
- [4].Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, de Wit R, Mulders P, Chi KN, Shore ND, Armstrong AJ, Flaig TW, Flechon A, Mainwaring P, Fleming M, Hainsworth JD, Hirmand M, Selby B, Seely L, de Bono JS, N. Engl. J. Med 2012, 367, 1187. [DOI] [PubMed] [Google Scholar]
- [5].de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, Chi KN, Jones RJ, Goodman OB Jr., Saad F, Staffurth JN, Mainwaring P, Harland S, Flaig TW, Hutson TE, Cheng T, Patterson H, Hainsworth JD, Ryan CJ, Sternberg CN, Ellard SL, Flechon A, Saleh M, Scholz M, Efstathiou E, Zivi A, Bianchini D, Loriot Y, Chieffo N, Kheoh T, Haqq CM, Scher HI, N. Engl. J. Med 2011, 364, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, Wasielewska T, Welsbie D, Chen CD, Higano CS, Beer TM, Hung DT, Scher HI, Jung ME, Sawyers CL, Science 2009, 324, 787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, Marotz C, Giannopoulou E, Chakravarthi BV, Varambally S, Tomlins SA, Nanus DM, Tagawa ST, Van Allen EM, Elemento O, Sboner A, Garraway LA, Rubin MA, Nat. Med 2016, 22, 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Epstein JI, Amin MB, Beltran H, Lotan TL, Mosquera JM, Reuter VE, Robinson BD, Troncoso P, Rubin MA, Am. J. Surg. Pathol 2014, 38, 756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Clermont PL, Lin D, Crea F, Wu R, Xue H, Wang Y, Thu KL, Lam WL, Collins CC, Wang Y, Helgason CD, Clin. Epigenet 2015, 7, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dardenne E, Beltran H, Benelli M, Gayvert K, Berger A, Puca L, Cyrta J, Sboner A, Noorzad Z, MacDonald T, Cheung C, Yuen KS, Gao D, Chen Y, Eilers M, Mosquera JM, Robinson BD, Elemento O, Rubin MA, Demichelis F, Rickman DS, Cancer Cell 2016, 30, 563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kleb B, Estecio MR, Zhang J, Tzelepi V, Chung W, Jelinek J, Navone NM, Tahir S, Marquez VE, Issa JP, Maity S, Aparicio A, Epigenetics 2016, 11, 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, Goodrich MM, Labbe DP, Gomez EC, Wang J, Long HW, Xu B, Brown M, Loda M, Sawyers CL, Ellis L, Goodrich DW, Science 2017, 355, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mertz KD, Setlur SR, Dhanasekaran SM, Demichelis F, Perner S, Tomlins S, Tchinda J, Laxman B, Vessella RL, Beroukhim R, Lee C, Chinnaiyan AM, Rubin MA, Neoplasia 2007, 9, 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Puca L, Bareja R, Prandi D, Shaw R, Benelli M, Karthaus WR, Hess J, Sigouros M, Donoghue A, Kossai M, Gao D, Cyrta J, Sailer V, Vosoughi A, Pauli C, Churakova Y, Cheung C, Deonarine LD, McNary TJ, Rosati R, Tagawa ST, Nanus DM, Mosquera JM, Sawyers CL, Chen Y, Inghirami G, Rao RA, Grandori C, Elemento O, Sboner A, Demichelis F, Rubin MA, Beltran H, Nat. Commun 2018, 9, 2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Scholze H, Stephenson RE, Reynolds R, Shah S, Puri R, Butler S, Trujillo-Alonso V, Teater M, van Besien H, Gibbs-Curtis D, Ueno H, Parvin S, Letai AG, Mathew S, Singh A, Cesarman E, Melnick A, Giulino-Roth L, Blood Adv 2020, 4, 5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Barnes JM, Kaushik S, Bainer RO, Sa JK, Woods EC, Kai F, Przybyla L, Lee M, Lee HW, Tung JC, Maller O, Barrett AS, Lu KV, Lakins JN, Hansen KC, Obernier K, Alvarez-Buylla A, Bergers G, Phillips JJ, Nam DH, Bertozzi CR, Weaver VM, Nat. Cell Biol 2018, 20, 1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Paszek MJ, DuFort CC, Rossier O, Bainer R, Mouw JK, Godula K, Hudak JE, Lakins JN, Wijekoon AC, Cassereau L, Rubashkin MG, Magbanua MJ, Thorn KS, Davidson MW, Rugo HS, Park JW, Hammer DA, Giannone G, Bertozzi CR, Weaver VM, Nature 2014, 511, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Weaver VM, Lelievre S, Lakins JN, Chrenek MA, Jones JC, Giancotti F, Werb Z, Bissell MJ, Cancer Cell 2002, 2, 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, Hammer DA, Weaver VM, Cancer Cell 2005, 8, 241. [DOI] [PubMed] [Google Scholar]
- [20].Apoorva F, Loiben AM, Shah SB, Purwada A, Fontan L, Goldstein R, Kirby BJ, Melnick AM, Cosgrove BD, Singh A, Cell Rep 2018, 23, 499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Singh A, Suri S, Lee T, Chilton JM, Cooke MT, Chen W, Fu J, Stice SL, Lu H, McDevitt TC, Garcia AJ, Nat. Methods 2013, 10, 438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Dumbauld DW, Lee T, Singh A, Scrimgeour J, Gersback CA, Zamir EA, Fu J, Chen C, Curtis JE, Craig SW, Garcia AJ, Proc. Natl. Acad. Sci. USA 2013. [DOI] [PMC free article] [PubMed]
- [23].Goel HL, Li J, Kogan S, Languino LR, Endocr.-Relat. Cancer 2008, 15, 657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Cress AE, Rabinovitz I, Zhu W, Nagle RB, Cancer Metastasis Rev 1995, 14, 219. [DOI] [PubMed] [Google Scholar]
- [25].Fischbach C, Kong HJ, Hsiong SX, Evangelista MB, Yuen W, Mooney DJ, Proc. Natl. Acad. Sci. USA 2009, 106, 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Coskun AF, Cai L, Nat. Methods 2016, 13, 657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Choi HMT, Schwarzkopf M, Fornace ME, Acharya A, Artavanis G, Stegmaier J, Cunha A, Pierce NA, Development 2018, 145, dev165753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Vlachostergios PJ, Puca L, Beltran H, Curr. Oncol. Rep 2017, 19, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Pisapia DJ, Salvatore S, Pauli C, Hissong E, Eng K, Prandi D, Sailer VW, Robinson BD, Park K, Cyrta J, Tagawa ST, Kossai M, Fontugne J, Kim R, Sigaras A, Rao R, Pancirer D, Faltas B, Bareja R, Molina AM, Nanus DM, Rajappa P, Souweidane MM, Greenfield J, Emde AK, Robine N, Elemento O, Sboner A, Demichelis F, Beltran H, Rubin MA, Mosquera JM, JCO Precis. Oncol 2017, 1, 10.1200/PO.16.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Gjorevski N, Sachs N, Manfrin A, Giger S, Bragina ME, Ordonez-Moran P, Clevers H, Lutolf MP, Nature 2016, 539, 560. [DOI] [PubMed] [Google Scholar]
- [31].Cruz-Acuna R, Quiros M, Farkas AE, Dedhia PH, Huang S, Siuda D, Garcia-Hernandez V, Miller AJ, Spence JR, Nusrat A, Garcia AJ, Nat. Cell Biol 2017, 19, 1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lee TT, Garcia JR, Paez JI, Singh A, Phelps EA, Weis S, Shafiq Z, Shekaran A, Del Campo A, Garcia AJ, Nat. Mater 2015, 14, 352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Graney PL, Lai K, Post S, Brito I, Cyster J, Singh A, Adv. Funct. Mater 2020, 30, 2001232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kessenbrock K, Plaks V, Werb Z, Cell 2010, 141, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Phelps EA, Enemchukwu NO, Fiore VF, Sy JC, Murthy N, Sulchek TA, Barker TH, García AJ, Adv. Mater 2012, 24, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Patterson J, Hubbell JA, Biomaterials 2011, 32, 1301. [DOI] [PubMed] [Google Scholar]
- [37].Matera DL, DiLillo KM, Smith MR, Davidson CD, Parikh R, Said M, Wilke CA, Lombaert IM, Arnold KB, Moore BB, Baker BM, Sci. Adv 2020, 6, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Gutowski SM, Shoemaker JT, Templeman KL, Wei Y, Latour RA Jr., Bellamkonda RV, LaPlaca MC, Garcia AJ, Biomaterials 2015, 44, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Purwada A, Shah SB, Beguelin W, August A, Melnick AM, Singh A, Biomaterials 2019, 198, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Knight CG, Morton LF, Onley DJ, Peachey AR, Messent AJ, Smethurst PA, Tuckwell DS, Farndale RW, Barnes MJ, J. Biol. Chem 1998, 273, 33287. [DOI] [PubMed] [Google Scholar]
- [41].Zeltz C, Gullberg D, J. Cell Sci 2016, 129, 653. [DOI] [PubMed] [Google Scholar]
- [42].Clark AY, Martin KE, Garcia JR, Johnson CT, Theriault HS, Han WM, Zhou DW, Botchwey EA, Garcia AJ, Nat. Commun 2020, 11, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Arnaout MA, Mahalingam B, Xiong JP, Annu. Rev. Cell Dev. Biol 2005, 21, 381. [DOI] [PubMed] [Google Scholar]
- [44].Bellis SL, Biomaterials 2011, 32, 4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Haynes J, Srivastava J, Madson N, Wittmann T, Barber DL, Mol. Biol. Cell 2011, 22, 4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kowal JM, Schmal H, Halekoh U, Hjelmborg JB, Kassem M, Stem Cells Transl. Med 2020, 9, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Sahai E, Marshall CJ, Nat. Cell Biol 2003, 5, 711. [DOI] [PubMed] [Google Scholar]
- [48].Wang J, Lv X, Xu F, Wei M, Liu C, Yang Y, Biosci. Rep 2018, 38, BSR20180574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kim KH, Roberts CW, Nat. Med 2016, 22, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Arrillaga-Romany I, Chi AS, Allen JE, Oster W, Wen PY, Batchelor TT, Oncotarget 2017, 8, 79298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Madhukar NS, Khade PK, Huang L, Gayvert K, Galletti G, Stogniew M, Allen JE, Giannakakou P, Elemento O, Nat. Commun 2019, 10, 5221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lev A, Lulla AR, Ross BC, Ralff MD, Makhov PB, Dicker DT, El-Deiry WS, Mol. Cancer Res 2018, 16, 754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Huang FK, Zhang G, Lawlor K, Nazarian A, Philip J, Tempst P, Dephoure N, Neubert TA, J. Proteome Res 2017, 16, 1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Cox J, Mann M, Nat. Biotechnol 2008, 26, 1367. [DOI] [PubMed] [Google Scholar]
- [55].Beltran H, Eng K, Mosquera JM, Sigaras A, Romanel A, Rennert H, Kossai M, Pauli C, Faltas B, Fontugne J, Park K, Banfelder J, Prandi D, Madhukar N, Zhang T, Padilla J, Greco N, McNary TJ, Herrscher E, Wilkes D, MacDonald TY, Xue H, Vacic V, Emde AK, Oschwald D, Tan AY, Chen Z, Collins C, Gleave ME, Wang Y, Chakravarty D, Schiffman M, Kim R, Campagne F, Robinson BD, Nanus DM, Tagawa ST, Xiang JZ, Smogorzewska A, Demichelis F, Rickman DS, Sboner A, Elemento O, Rubin MA, JAMA Oncol 2015, 1, 466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Sailer V, Eng KW, Zhang T, Bareja R, Pisapia DJ, Sigaras A, Bhinder A, Romanel A, Wilkes D, Sticca E, Cyrta J, Rao R, Sahota S, Pauli C, Beg S, Motanagh S, Kossai M, Fontunge J, Puca L, Rennert H, Zhaoying Xiang J, Greco N, Kim R, MacDonald TY, McNary T, Blattner-Johnson M, Schiffman MH, Faltas BM, Greenfield JP, Rickman D, Andreopoulou E, Holcomb K, Vahdat LT, Scherr DS, van Besien K, Barbieri CE, Robinson BD, Fine HA, Ocean AJ, Molina A, Shah MA, Nanus DM, Pan Q, Demichelis F, Tagawa ST, Song W, Mosquera JM, Sboner A, Rubin MA, Elemento O, Beltran H, JCO Precis. Oncol 2019, 3, 10.1200/PO.19.00047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Chakravarty D, Sboner A, Nair SS, Giannopoulou E, Li R, Hennig S, Mosquera JM, Pauwels J, Park K, Kossai M, MacDonald TY, Fontugne J, Erho N, Vergara IA, Ghadessi M, Davicioni E, Jenkins RB, Palanisamy N, Chen Z, Nakagawa S, Hirose T, Bander NH, Beltran H, Fox AH, Elemento O, Rubin MA, Nat. Commun 2014, 5, 5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, Bioinformatics 2013, 29, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Bioinformatics 2009, 25, 2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L, Nat. Protoc 2012, 7, 562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, Lagarde J, Veeravalli L, Ruan X, Ruan Y, Lassmann T, Carninci P, Brown JB, Lipovich L, Gonzalez JM, Thomas M, Davis CA, Shiekhattar R, Gingeras TR, Hubbard TJ, Notredame C, Harrow J, Guigo R, Genome Res 2012, 22, 1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Anders S, Pyl PT, Huber W, Bioinformatics 2015, 31, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Love MI, Huber W, Anders S, Genome Biol 2014, 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, Proc. Natl. Acad. Sci. USA 2005, 102, 15545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P, Cell Syst 2015, 1, 417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP, Bioinformatics 2011, 27, 1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Beltran H, Wyatt AW, Chedgy EC, Donoghue A, Annala M, Warner EW, Beja K, Sigouros M, Mo F, Fazli L, Collins CC, Eastham J, Morris M, Taplin ME, Sboner A, Halabi S, Gleave ME, Clin. Cancer Res 2017, 23, 6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq data for organoids generated during the current study are available through Gene Expression Omnibus (GEO) accession number: GSE181462 with the following sub-series: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE181462. The published human data are available through dbGap:phs000909.v.p1 (http://www.cbioportal.org/study?id=nepc_wcm_2016)(7).