

Abstract
PURPOSE
DNA polymerase epsilon is critical to DNA proofreading and replication. Mutations in POLE have been associated with hypermutated tumors and antitumor response to immune checkpoint inhibitor (ICI) therapy. We present a clinicopathologic analysis of patients with advanced cancers harboring POLE mutations, the pattern of co-occurring mutations, and their response to ICI therapy within the context of mutation pathogenicity.
METHODS
We conducted a retrospective analysis of next-generation sequencing data at MD Anderson Cancer Center to identify patient tumors with POLE mutations and their co-occurring mutations. The pathogenicity of each mutation was annotated using InterVar and ClinVar. Differences in therapeutic response to ICI, survival, and co-occurring mutations were reported by POLE pathogenicity status.
RESULTS
Four hundred fifty-eight patient tumors with POLE mutations were identified from 14,229 next-generation sequencing reports; 15.0% of POLE mutations were pathogenic, 15.9% benign, and 69.1% variant of unknown significance. Eighty-two patients received either programmed death 1 or programmed death ligand-1 inhibitors as monotherapy or in combination with cytotoxic T-cell lymphocyte-4 inhibitors. Patients with pathogenic POLE mutations had improved clinical benefit rate (82.4% v 30.0%; P = .013), median progression-free survival (15.1 v 2.2 months; P < .001), overall survival (29.5 v 6.8 months; P < .001), and longer treatment duration (median 15.5 v 2.5 months; P < .001) compared to those with benign variants. Progression-free survival and overall survival remained superior when adjusting for number of co-occurring mutations (≥ 10 v < 10) and/or microsatellite instability status (proficient mismatch repair v deficient mismatch repair). The number of comutations was not associated with response to ICI (clinical benefit v progressive disease: median 13 v 11 comutations; P = .18).
CONCLUSION
Pathogenic POLE mutations were associated with clinical benefit to ICI therapy. Further studies are warranted to validate POLE mutation as a predictive biomarker of ICI therapy.
INTRODUCTION
DNA polymerase epsilon, encoded by the POLE gene, is a critical protein involved in DNA proofreading and replication.1 POLE synthesizes the leading strand of DNA in the replication fork and has a 3′-5′ exonuclease domain that increases replication accuracy by approximately 100-fold through recognition and excision of mismatched base pairs.2,3 Somatic and germline POLE proofreading defects, particularly mutations occurring in the exonuclease domain representing codons 268-471, are more often found in mismatch repair proficient tumors and associated with hypermutagenesis.4-8
CONTEXT
Key Objective
Determining appropriate predictive biomarkers of response to optimize patient selection to immune checkpoint inhibitor (ICI) therapy remains a challenge. This retrospective clinicopathologic analysis of patients with POLE mutations examined the correlation between POLE pathogenicity and patient outcomes to ICI therapy. To our knowledge, this is the first and largest report of patient data in the context of POLE pathogenicity.
Knowledge Generated
Patients with pathogenic POLE mutations, compared to those with benign variants, had improved clinical benefit rate, median progression-free survival, median overall survival, and a longer duration on ICI treatment. Survival analyses remained superior when adjusting for number of co-occurring mutations within the tumor and/or microsatellite instability status.
Relevance
These findings support further study in patients with advanced solid tumors harboring POLE variants to further clarify the utility of POLE mutation location and pathogenicity as a predictive biomarker for ICI therapy.
Determining appropriate predictive biomarkers of response to optimize patient selection for immune checkpoint inhibitor (ICI) therapy remains a challenge.9 The programmed death 1 (PD-1) inhibitor pembrolizumab is US Food and Drug Administration (FDA)-approved in multiple tumor-specific indications and also for histology-agnostic use in tumors that are microsatellite instability high (MSI-H), mismatch repair deficient (dMMR), and/or those with tumor mutation burden (TMB) ≥ 10 mutations/megabase.10-13 Other ICI therapies have varied programmed death ligand-1 (PD-L1) or combined positive score cutoffs.14 However, PD-L1 expression is often not predictive of ICI response.15
Wang et al16 evaluated the prevalence of mutations in POLE and POLD1, another proofreading protein, in 47,721 patients with different cancer types via the cBioPortal database. They found variants in POLE and POLD1 at mutational frequencies of 2.8% and 1.4%, respectively. Patients with one of these mutations had improved overall survival (OS; 34 v 18 months; P = .004) and were more likely to benefit from ICI therapy.16 However, this study did not examine whether mutation location or pathogenicity had an effect on therapeutic response, a key consideration in the development of any clinical biomarker.
Here, we present a clinicopathologic analysis of patients with advanced cancers harboring POLE mutations at The University of Texas MD Anderson Cancer Center (MDACC). The primary aim of this study was to determine the correlation between POLE mutation pathogenicity and patient outcomes to ICI therapy. Secondary aims included determining the relationship of POLE mutations to patient prognosis, other ICI biomarkers, and co-occurring mutation patterns. To our knowledge, this is the first and largest report of patient data of this magnitude in the context of POLE pathogenicity.
METHODS
A retrospective electronic database search of Clinical Laboratory Improvement Amendments–certified next-generation sequencing data was conducted to identify MDACC patient tumors with POLE mutations and co-occurring mutations, as this previously has been shown to be a surrogate for TMB.17,18 Clinical data through April 1, 2020, were collected from the electronic medical record. MDACC institutional review board approval was obtained before study initiation, and all data were collected and stored according to best practices, protecting patient confidentiality and data integrity.
The pathogenicity of each POLE mutation was annotated via InterVar19 and ClinVar.20 If one of these two sources indicated a variant of unknown significance (VUS), but the other provided a non-VUS annotation, then the non-VUS status was used. All mutations were then reviewed using peer-reviewed publications to further update pathogenicity21-25 (Appendix 1). All POLE mutation annotations were checked independently by a second reviewer. Benign and likely benign as well as pathogenic and likely pathogenic were grouped together for analysis.
Descriptive statistics were used to summarize patient's characteristics. Chi-squared or Fisher exact tests were used to evaluate differences of category variables. The distributions of progression-free survival (PFS), OS, and time on first immunotherapy treatment were estimated using the Kaplan-Meier method.26 Log-rank test27 was performed to test the difference in survival between groups. The Cox proportional hazards model28 were used for the multivariate analyses of survival, adjusting for MSI status and/or number of comutations (≥ 10 and ≥ 20).
See Appendix 1 for additional information.
RESULTS
Of 14,229 patients with solid tumors and available next-generation sequencing data, we identified 486 (3.4%) patients with a POLE-aberrant tumor. This percentage is comparable to that identified on The Cancer Genome Atlas database (4.0%, accessed on September 24, 2020). Of these 486 patients, 458 had comutation data and 453 had available clinical data in the electronic medical record.
POLE Mutation Pathogenicity
POLE mutations were annotated as the following (n = 453): pathogenic (n = 68, 15.0%), benign (n = 72, 15.9%), or variant of unknown significance (n = 313, 69.1%) mutations. Sixty-eight patients had a tumor with a mutation in the POLE exonuclease domain: 47.1% pathogenic, 8.8% benign, and 50% VUS (Fig 1).
FIG 1.
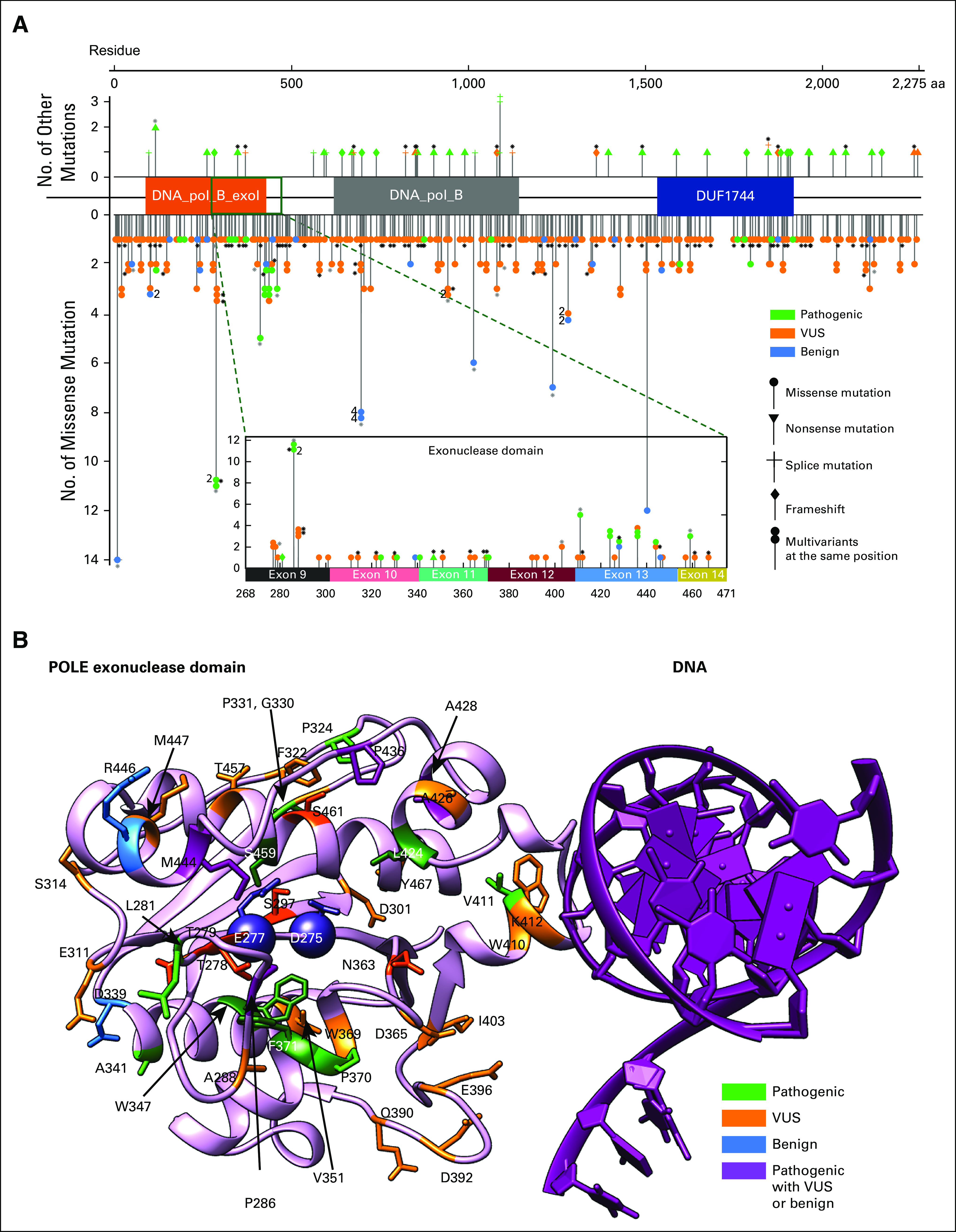
(A) Distribution of POLE mutations within POLE whole-length sequence. Among 450 evaluable patients, one had a POLE amplification and 449 had a POLE mutation, contributing to 424 unique POLE variants, as plotted against the mutational sites. Each data point with certain symbol represents a unique variant. All 375 unique missense mutations are shown downward with solid circles. Thirteen frameshift mutations, 13 splice variants, 23 nonsense mutations are plotted upwards in respective symbols. Stacked symbols indicate different mutations have been found at the same position. For example, POLE_P286 has 11 missense mutations occurrences among 449 patients in total, with one pathogenic mutation (P286L) in two patients and another VUS mutation found in nine patients. Black asterisks next to some data points indicate that those variants are in patients containing multiple POLE mutations, while gray asterisks represent a mixture of patients with a single POLE mutation and multiple POLE mutations. Fifty unique variants (47 missense mutations) are found within the exonuclease domain (268-471). (B) POLE exonuclease domain mutations mapped to structure. The exonuclease domain of human POLE modeled by AlphaFold229 (pink) is aligned to Saccharomyces cerevisiae POLE-DNA complex30 (DNA in purple) and then the POLE chain from Saccharomyces cerevisiae is removed. The two catalytic residues (D275 and E277) are shown as spheres. Mutations found in this domain are displayed in the structure with different colors indicating pathogenic status. Residues that are physically adjacent to the catalytic sites (all atom distance < 6 Å) are highlighted in darker colors, with VUS mutations S297, T278, T279, N363, and S461 in dark orange and pathogenic mutations M444, L424, S459, F371, W347, and P286 in dark green. These pathogenic mutations surround the two catalytic residues (D275 and E277)31 and likely affect the catalytic pocket, whereas pathogenic mutations at V411 might affect DNA binding, although its location is distal to the catalysis center. All benign mutations occur at residues far from catalytic sites. Other residues W410, I403, D365, D396, and D392 may also contribute to DNA binding, although the functional annotation of those mutations remains unknown. Different mutations at A428 (A428T and A428S) lead to conflicting pathogenic status in available databases; no patient treated with anti–PD-1/L1-based therapy in this cohort had an A428 mutation. PD-1, programmed death 1; PD-L1, programmed death ligand-1; VUS, variant of unknown significance.
Response to Immune Checkpoint Inhibition
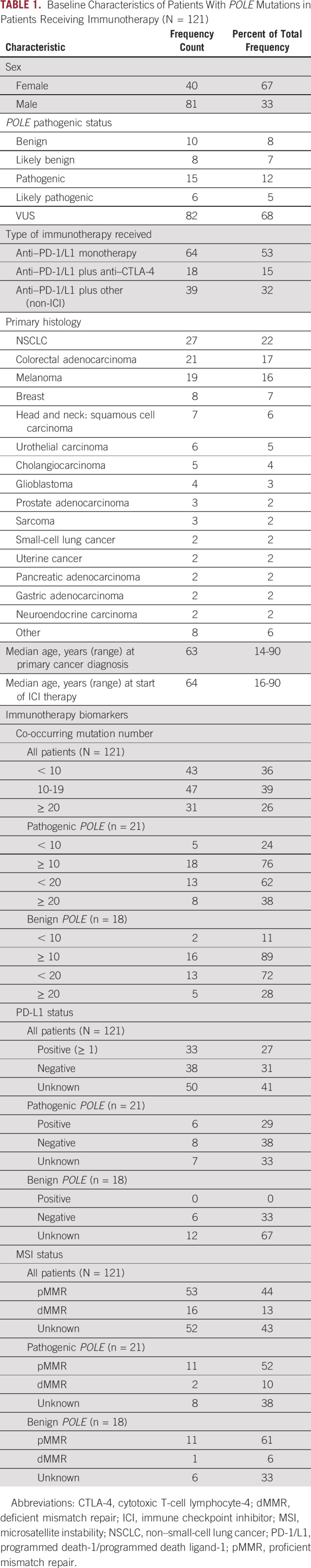
Of the 453 patients with available clinical data, 172 had received treatment with either a PD-1 or PD-L1 (PD-1/L1) inhibitor. One hundred twenty-one patients were considered suitable for response analysis after excluding those with limited-stage disease, insufficient follow-up time for response evaluation (ie, had no restaging scans performed), and/or received their treatment as neoadjuvant, adjuvant, or maintenance therapy. Ninety-six of 121 (79.3%) had a tumor or molecular subtype with an FDA-approved indication for ICI therapy. Overall, 64 patients received PD-1/L1 inhibitors as monotherapy, 18 as combination therapy with a cytotoxic T-cell lymphocyte-4 (CTLA-4) inhibitor, and 39 in combination with either chemotherapy, a molecular targeted agent, or a vaccine (Table 1). See additional clinical information in Appendix Table A1.
TABLE 1.
Baseline Characteristics of Patients With POLE Mutations in Patients Receiving Immunotherapy (N = 121)
Patients who achieved radiologic complete response (CR), partial response (PR), or stable disease (SD) were considered to have derived clinical benefit. Response data are shown in Table 2 and Appendix Table A1. Clinical benefit rate (CBR) of all 121 patients to PD-1/L1 inhibitor–based therapy was 55.4% (95% CI, 46.5 to 64.2). CBR was greater in patients with pathogenic POLE mutations when compared to patients with benign variants: 81.0% (pathogenic), 38.0% (benign); pathogenic versus benign, P = .01. We then grouped patients with benign or VUS mutations together as nonactionable variants, as this would be a meaningful distinction when selecting patients for therapy in clinic. CBR was also greater in patients with pathogenic mutations with tumors harboring pathogenic POLE mutations compared with nonactionable variants; 81.0% versus 50.0%, P = .014. The overall response rate (ORR, CR, and PR) was also higher in patients with pathogenic versus benign mutations (52.4% v 11.1%; P = .008) and trended toward significance with pathogenic versus nonactionable variants (52.4% v 31.0%; P = .061).
TABLE 2.
Best Response to Immune Checkpoint Inhibitor Therapy in Patients Receiving Immunotherapy
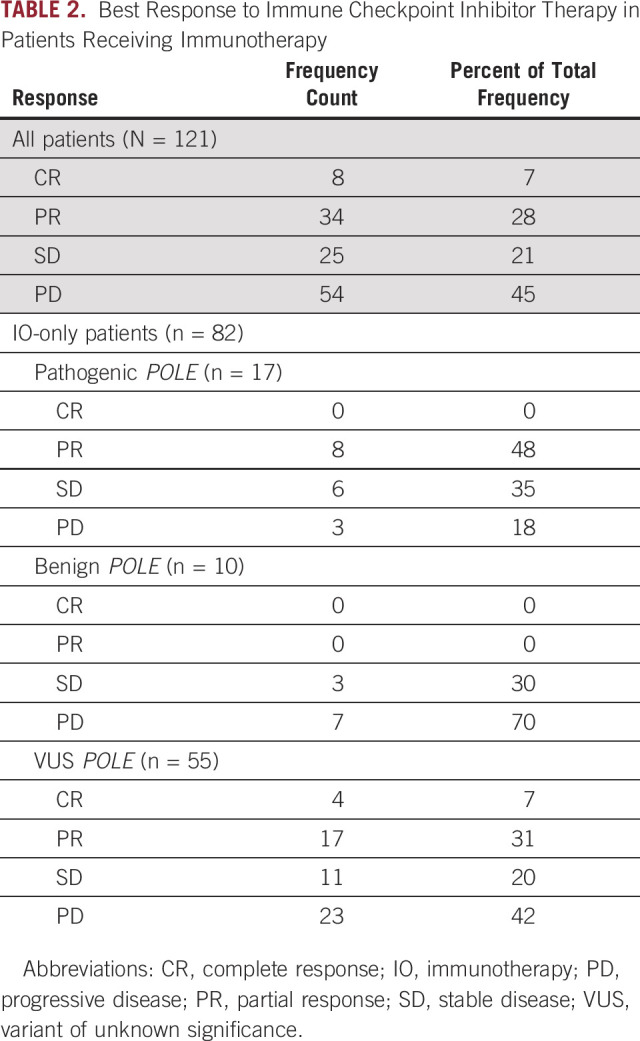
As 32.2% of patients received PD-1/L1 inhibitors in combination with another agent that could influence response, we next analyzed the 82 patients who received immunotherapy-only (IO-only) regimens, either PD-1/L1 inhibitor as monotherapy or dual therapy in combination with a CTLA-4 inhibitor. CBR was again higher in patients with pathogenic POLE mutations; pathogenic versus benign, 82.4% versus 30.0%, P = .013; 82% versus 53.8%, P = .50. There were no CR or PR in patients with benign mutations (ORR pathogenic v benign: 47.1% v 0%; P = .019).
Among patients who received IO-only therapy (n = 82), eight had pathogenic POLE mutations in the exonuclease domain (all had missense and two had additional nonsense mutations; Fig 1B and Appendix Table A1); response rate (RR) was 37.5% (3 of 8 patients). No patient had a benign variant in the exonuclease domain and one patient had a VUS in the exonuclease domain (missense, RR 0%). Nine patients had pathogenic mutations outside of the exonuclease domain: two had single missense mutations (RR 100%; 2 of 2 patients), three had single frameshift (RR 66.7%; 2 of 3 patients), one had splice (RR 100%), one had nonsense (RR 0%), and two had both frameshift and missense mutations (RR 0%). No responses were observed in the 10 patients with benign POLE mutations outside the exonuclease domain (missense, RR 0%; 0 of 10 patients). Fifty-four patients had VUSs outside the exonuclease domain (all missense, RR 31.5%; 17 of 54 patients).
Survival Analysis
Median PFS for the 121 patients that received therapy with an anti–PD-1/L1-based regimen was 5.4 (95% CI, 3.5 to 7.9) months and PFS at 12 months was 35% (95% CI, 26 to 44). Median PFS was greater in patients with pathogenic mutations compared with benign mutations: 15.1 versus 2.8 months; P < .001. Median PFS was 4.9 (95% CI, 3.1 to 11.4) months in patients with VUS (Fig 2A). Median PFS improved by > 10 months in patients with pathogenic mutations compared with nonactionable variants, although this did not reach statistical significance (15.1 v 4.2 months; P = .075).
FIG 2.

PFS for patients treated with an anti–PD-1/L1-based regimen. Among all 121 patients treated, two patients did not have data evaluable for survival analysis. Median PFS was as follows: pathogenic POLE mutation 15.1 months, VUS 4.9 months, and benign 2.8 months; pathogenic versus benign P < .001 (A). Among the 82 patients who received an immunotherapy-only regimen: pathogenic 15.1 months, VUS 6.2 months, benign 2.2 months; pathogenic versus benign P < .001. With adjustment for comutation number (≥ 10 v < 10) and MSI/MMR status (MSS/pMMR v MSI-H/dMMR), patients with pathogenic mutations had a superior PFS than those with benign mutations (HR, 0.07; 95% CI, 0.02-0.31; P < .001). When adjusting for number of comutations (≥ 20 v < 20) and MSI status, patients with pathogenic mutations continued to have superior PFS than those with benign mutations (HR, 0.07; 95% CI: 0.02-0.31; P < .001) (B). dMMR, deficient mismatch repair; MSI-H, microsatellite instability high; MSS, microsatellite stable; PD-1, programmed death 1; PD-L1, programmed death ligand-1; PFS, progression-free survival; VUS, variant of unknown significance.
Median OS from time of anti–PD-1/L1-based therapy for these patients was 29.5 (95% CI, 26.0 to not reached [NR]) and OS at 12 months was 77% (95% CI, 67 to 84); median follow-up was 15.6 months. Median OS was greater for patients with pathogenic mutations than benign variants: 29.5 versus 11.6 months; P < .001. Patients with VUS had median OS that was NR (Fig 3A).
FIG 3.

OS for patients treated with an anti–PD-1/L1-based regimen. Among all 121 patients treated, median OS was as follows: pathogenic POLE mutation 29.5 months, VUS NR, and benign 11.6 months; pathogenic versus benign P < .001 (A). Among the 82 patients who received an immunotherapy-only regimen: pathogenic 29.5 months, VUS NR, benign 6.8 months; pathogenic versus benign P < .001. Patients with pathogenic POLE mutations had greater median OS compared to patients with benign mutations when adjusting for number of comutations (≥ 10 v < 10: HR, 0.10 [95% CI, 0.03 to 0.40], P = .001; ≥ 20 v < 20: HR, 0.10 [95% CI, 0.03 to 0.41], P = .001) or MSI status (HR, 0.14; 95% CI, 0.03 to 0.81; P = .028) (B). (C) OS from time of diagnosis for all 453 patients with POLE mutations including patients who did not receive anti–PD-1/L1 therapy. Median OS was as follows: pathogenic POLE mutation NR, VUS 8.0 years, and benign 3.4 years; pathogenic versus benign P < .001. NR, not reached; OS, overall survival; PD-1, programmed death 1; PD-L1, programmed death ligand-1; VUS, variant of unknown significance.
Patients Who Received IO-Only Treatment Regimens (n = 82)
Among patients who received IO-only regimens, median PFS was 6.0 (95% CI, 4.1 to 13.3) months with a 12-month PFS of 41% (95% CI, 29 to 51). Median PFS was longer in patients with pathogenic compared with benign POLE mutations: 15.1 versus 2.2 months; P < .001. Patients with VUS had a median PFS of 6.2 (95% CI, 3.9 to 20.6) months (Fig 2B). With adjustment for comutation number (≥ 10 v < 10) and MSI/MMR status (microsatellite stable [MSS]/proficient mismatch repair v MSI-H/dMMR), patients with pathogenic mutations had a superior PFS than those with benign mutations (HR, 0.07; 95% CI, 0.02 to 0.31; P < .001), as did patients with VUS mutations compared with benign mutations (HR, 0.16; 95% CI, 0.05 to 0.53; P = .003). When adjusting for number of comutations (≥ 20 v < 20) and MSI status, patients with pathogenic mutations continued to have superior PFS than those with benign mutations (HR, 0.07; 95% CI, 0.02 to 0.31; P < .001), as did those with VUS mutations compared with benign mutations (HR, 0.12; 95% CI, 0.04 to 0.43; P = .001).
Among patients who received IO-only regimens, median OS from time of therapy start was 29.5 (95% CI, 26.0 to NR) months and OS at 12 months was 80% (95% CI, 68 to 87); median follow-up was 17.5 months. Median OS was greater in patients with pathogenic compared with benign POLE mutations: 29.5 months versus 6.8 months, P < .001. Median OS was NR in patients with VUS mutations (Fig 3B). Differences in median OS between patients with pathogenic and nonactionable variants were not statistically significant (29.5 months v NR; P = .265). Patients with pathogenic POLE mutations had greater median OS compared to patients with benign mutations when adjusting for number of comutations (≥ 10 v < 10: HR, 0.10 [95% CI, 0.03 to 0.40], P = .001; ≥ 20 v < 20: HR, 0.10 [95% CI, 0.03 to 0.41], P = .001) or MSI status (HR, 0.14; 95% CI, 0.03 to 0.81], P = .028).
Of note, patients with pathogenic compared with benign POLE mutations had longer median IO-only treatment duration of first IO-only therapy (15.5 v 2.5 months; P < .001). Patients with VUS had a median duration of 6.2 (95% CI, 3.6 to 14.2) months.
Total Population of Patients With POLE Mutations (N = 453)
Among the total population of patients with POLE mutations and available clinical information, median follow-up was 2.5 years and median OS was 6.8 (95% CI, 5.0 to 9.4) years. Median OS from time of diagnosis was greater for those with pathogenic POLE mutations compared to those with benign mutations (NR v 3.4 years; P < .001) and those with VUS (NR v 8.0 years; P = .012; Fig 3C). Patients with pathogenic mutations had a superior median OS compared to those with nonactionable variants (NR v 6.4 years; P = .003). Patients with pathogenic mutations continued to have superior OS with adjustment for TMB (≥ 10 v < 10) or MSI status (HR, 0.23; 95% CI, 0.07 to 0.75; P = .014).
Relationship of POLE Pathogenicity and Immunotherapy Biomarkers
Compared to patients with benign POLE mutations, those with pathogenic variants were significantly not more likely to have number of comutations ≥ 10 (76.2% v 88.9%; P = .417) or ≥ 20 (38.1% v 27.8%; P = .734), PD-L1–positive on pathology examination (42.9% v 0%; P = .115), or MSI-high status (13.3% v 8.3%; P > .99; Table 1).
Co-occurring Mutations
Figure 4A shows the landscape of comutations in all patients with POLE mutations (n = 450). The most common co-occurring mutations included TP53 (52%), ARID1A (22%), BRCA2 (21%), KRAS (21%), NF1 (19%), NOTCH3 (18%), NOTCH1 (18%), ATM (18%), PIK3CA (17%), SETD2 (17%), and SMARCA4 (17%).
FIG 4.
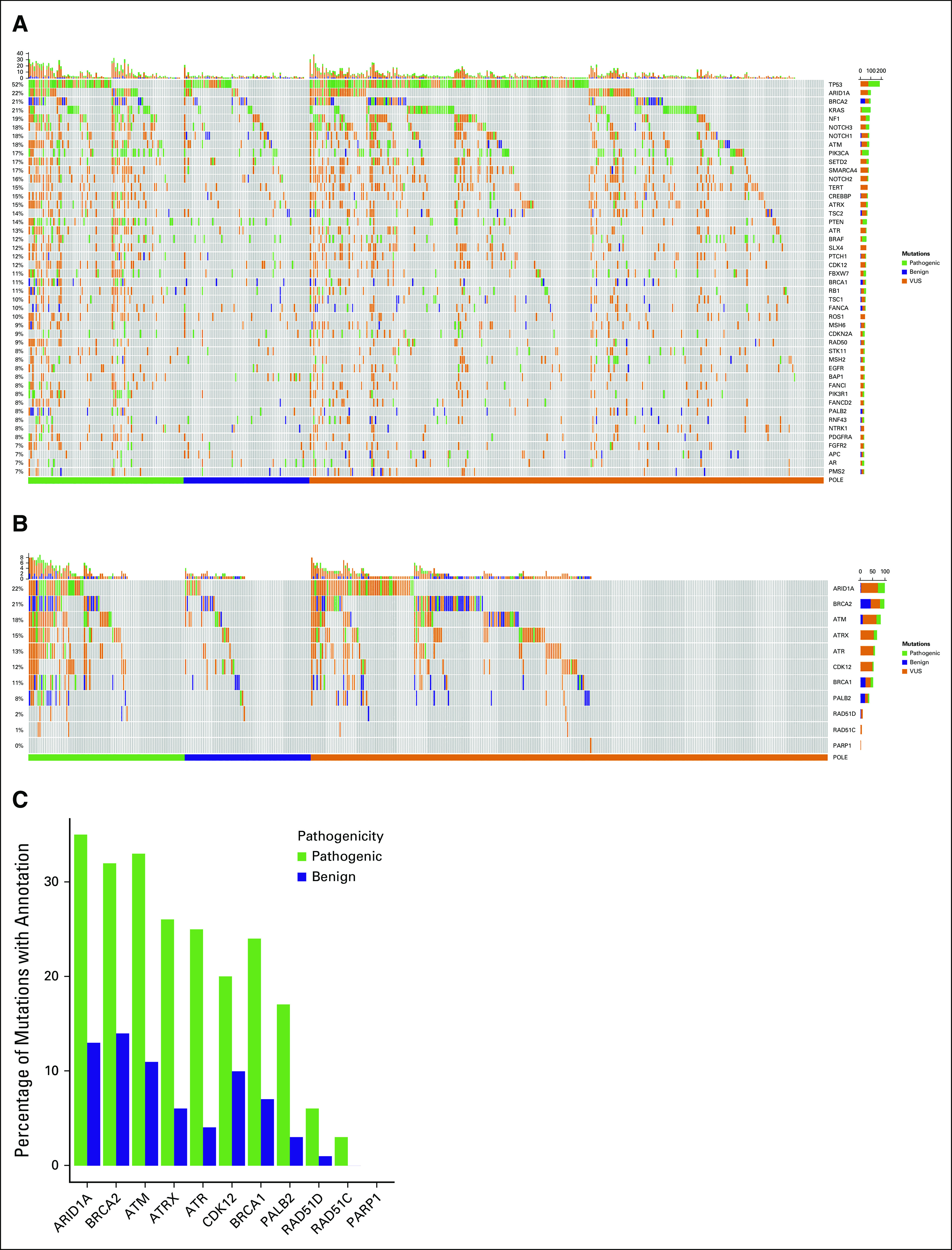
(A) The landscape of co-occurring mutations in all patients with evaluable POLE mutations (n = 450). The most common co-occurring mutations included TP53 (52%), ARID1A (22%), BRCA2 (21%), KRAS (21%), NF1 (19%), NOTCH3 (18%), NOTCH1 (18%), ATM (18%), PIK3CA (17%), SETD2 (17%), and SMARCA4 (17%). Mutations are classified as pathogenic (green), benign (blue), and VUS (orange). Patients with pathogenic POLE mutations had more comutations in DDR genes as shown in the (B) heat map and (C) bar graphs above. DDR, DNA damage response; VUS, variant of unknown significance.
Patients with pathogenic POLE mutations had more comutations in DNA damage response (DDR) pathway genes (pathogenic v benign POLE mutation): ARID1A (30.0% v 14.1%; P = .032), ATM (26.3% v 12.7%; P = .059), ATR (18.8% v 4.2%; P = .012), ATRX (20.0% v 7.0%; P = .039), BRCA1 (20.0% v 9.9%; P = .132), BRCA2 (26.3% v 14.1%; P = .100), CDK12 (20.0% v 12.7%; P = .323), and PALB2 (13.8% v 2.8%; P = .036; Figs 4B and 4C).
The number of mutations was not significantly associated with antitumor response to an IO-only regimen (clinical benefit v progressive disease [PD]: median 13 v 11; P = .18). However, there was a trend toward increased CBR in patients with ≥ 20 co-occurring mutations (odds ratio, 2.6; P = .086); there was no trend when a lower threshold of ≥ 10 co-occurring mutations was used (odds ratio, 1.3; P = .65). There was also no difference in OS in patients with low or high numbers of comutations. Overall, among the 29 patients with responses (CR or PR) to IO-only regimens, 10 (34%) had < 10 co-occurring mutations. Notably, the number of unique co-occurring DDR gene mutations was associated with clinical benefit to therapy (median 1 v 0; P = .009). Additionally, among patients with nonactionable POLE variants (n = 65), the number of comutations was not predictive of antitumor response (CR/PR v SD/PD, median 16 v 11; P = .838) or clinical benefit (CR/PR/SD v PD, 13 v 11; P = .522).
DISCUSSION
Current research studies have focused on the association between POLE mutations and tumors with a hypermutation phenotype,5-7,21,32 providing the rationale for targeting patients with these tumors with immunotherapeutic agents. To the best of our knowledge, this study is the first to highlight the importance of the pathogenic status of POLE mutations. CBR in patients who received an IO-only regimen (PD-1/L1 inhibitor monotherapy or in combination with CTLA-4 inhibitor) was greater in patients with pathogenic POLE mutations compared to those with nonactionable variants (82.4% v 30.0%; P = .013) and there were no radiologic responses in patients with benign variants.
Mutations in the POLE exonuclease domain are associated with hypermutated cancers, antitumor responses to immunotherapy agents, and a trend toward improved prognosis.3,4,7,33-37 Only 15 (12.4%) patients treated with a PD-1/L1 inhibitor had a mutation in the exonuclease domain. Comparative analysis relating POLE pathogenicity, location and type of mutation, and IO response was limited because of a small sample size. Although mutations in the exonuclease domain are often pathogenic, our study highlights that there are pathogenic alterations outside of this domain that may be successfully targeted by ICI therapy. Our cohort includes IO-only responses in patients with POLE missense, splice, and frameshift alterations outside of the exonuclease domain.
Patients with pathogenic mutations (n = 17) who received IO-only regimens had improved median PFS (15.1 v 2.2 months; P < .001), OS (29.5 v 6.8 months; P < .001), and duration on therapy (15.5 v 2.5 months; P < .001) than patients with benign mutations (n = 10). PFS differences remained significant when adjusting for MSI and number of comutations at either cutoffs of ≥ 10 or ≥ 20. Additionally, including all patients with POLE mutations treated with IO-only regimens, 10 of 29 (34%) responses were found in tumors with < 10 co-occurring mutations. This provides preliminary evidence that these POLE-mutated tumors may be more immunogenic and/or responsive to ICI, irrespective of the number of comutations.
A major limitation of this study was that there was not a sufficient patient population to comprehensively evaluate the effect of POLE pathogenicity in historically immune-resistant tumors. Seventy-nine percent of patients included in our series had FDA-approved ICI indications based upon tumor or molecular subtype, limiting our ability to draw meaningful conclusions from the remaining immune-resistant patients. Nevertheless, responses observed across both immune-sensitive and immune-resistant patients indicate that POLE pathogenicity may indicate benefit to ICI therapy irrespective of traditional immunosensitivity of tumor histology or hypermutagenic status. These data highlight the need for further analysis in larger patient populations without FDA-approved ICI indications, including tumor types that have not previously been linked to POLE proofreading-defect tumorigenesis,1,8 and prospective clinical testing. Another major limitation was that the majority (68%) of patients had a POLE VUS. In this series, 38% of patients with a VUS treated with an IO-only regimen had a response. Further work is necessary to better annotate these mutations to clarify potential immune sensitization differences within current VUSs within the context of intrinsic tumor immunosensitivity.
Data from an early phase trial of 16 patients with POLE mutations treated with nivolumab were recently presented.38 At 84 days after treatment, ORR was 50% for patients with pathogenic mutations (n = 8) compared to 0% in patients with benign mutations (n = 5), consistent with our findings. A phase II trial is randomizing patients with POLE- or POLD1-mutated solid tumors to treatment with nivolumab plus ipilimumab versus nivolumab monotherapy (NCT03461952).39 Another phase II trial is assessing treatment with the PD-L1 inhibitor durvalumab for patients with previously treated, metastatic, and MSI-high or POLE-mutated colorectal cancer (NCT03461952).40 Additionally, the phase III POLEM study is randomly assigning patients with stage III MMR-deficient or POLE exonuclease domain mutant colon cancer to further adjuvant treatment with avelumab (v no intervention) after standard-of-care fluoropyrimidine-based chemotherapy (NCT03827044).41 To improve the utility of POLE as a robust predictive biomarker of response to select patients for ICI therapy, on the basis of our data, these trials should also include assessments of the pathogenicity of POLE mutations, location within the gene, number of comutations, and associated tumor immune infiltration. Of note, a recent study found that nine of 11 tumor samples with POLE or POLD1 mutations had high levels of tumor-infiltrating lymphocytes, although this must be placed into the context that these patients had an average TMB of 158 mutations/megabase.37
Among the total population, median OS was NR in patients with pathogenic mutations and was 3.4 years in patients with benign variants (P < .001). Previous data have shown that patients with either POLE or POLD1 mutations have significantly longer OS compared with a wild-type population (2.8 v 1.5 years, respectively).16 Our study is consistent with these data and adds to these findings by demonstrating the impact of POLE pathogenicity on survival outcomes.
To the best of our knowledge, this study is the first to provide a detailed description of the landscape of comutations in patients with POLE-mutated tumors. Interestingly, of patients who received an IO-only regimen, the number of comutations was not significantly associated with antitumor response. We also examined whether the number of comutations could predict for antitumor response in patients with POLE benign variants or VUS, but this analysis did not reveal significant results, indicating again that the number of mutations a tumor had was not the primary driver of response to ICI therapy. We also noted that tumors with pathogenic mutations were more likely to have comutations in DDR pathway genes and the number of unique co-occurring DDR mutations was associated with benefit to an IO-only regimen, but the sample size analyzed was small, limiting any formal conclusions to be made.
In summary, patients with pathogenic POLE mutations had improved antitumor responses and greater median PFS and OS with ICI therapy. These data should be interpreted within the context of a limited sample size and intrinsic tumor immune-sensitivity in most patients. Our findings nevertheless support further study in patients with advanced solid tumors harboring POLE pathogenic or VUS variants to further clarify the utility of POLE mutation location and pathogenicity as a predictive biomarker for ICI therapy.
APPENDIX 1. SUPPLEMENTAL MATERIAL
Methods
Clinical data were collected from the MD Anderson Cancer Center electronic medical record between June 18, 2019, and April 1, 2020, and included basic demographic parameters, tumor stage, pathology, and treatment response, as well as survival data. Next-generation sequencing data were collected on April 29, 2019, and again on January 14, 2020, from five panels: FoundationOne (Foundation Medicine, Cambridge, MA), FoundationOne CDx (Foundation Medicine, Cambridge, MA), STGA-DNA 2018 (MD Anderson Cancer Center, Houston, TX), Tempus xT (Tempus, Chicago, IL), and MI Profile (Caris Life Sciences, Irving, TX). The five panels included the following numbers of genes: FoundationOne (416), FoundationOne CDx (324), STGA-DNA (147), Tempus xT (648), and MI Profile (260). Next-generation sequencing data collected included gene symbols/names, protein changes, Human Genome Variation Society (HGVS) expressions for transcripts and corresponding proteins,42 and complementary DNA changes. Initial annotation of POLE mutation pathogenicity was conducted via InterVar19 and ClinVar.20 Review of peer-reviewed published literature through August 15, 2021, further identified known pathogenic POLE hotspot mutations, including P286R, V411L, V411R, V411M, S297F, A456P, and S459F.21-25 Patients with these mutations that had previously been labeled as having a variant of unknown significance were updated to pathogenic.
In the analyses of progression-free survival (PFS), overall survival (OS) and time on immunotherapy of the patients receiving immunotherapy (IO), PFS was defined as the time from treatment initiation to the time of progression or death, whichever occurred first; OS was defined as the time from treatment initiation to death; and time on immunotherapy was defined as the time from first IO initiation to the end of first IO treatment or last follow-up. For the analysis of OS with both the patients receiving IO and those who did not receive IO included, OS was defined as the time of diagnosis to death. For events that have not occurred by the time of data analysis, times were censored at the last contact at which the patient was known to be progression-free for PFS or the last time the patient was known to be alive for OS.
Additional statistical methods included the following: Wilcoxon rank-sum or Kruskal-Wallis tests were used to detect differences for continuous variables between groups.43 A two-proportion Z-test was used to compare the percentage of patients with a DNA damage response comutation among patients with pathogenic versus benign POLE mutations. The Fisher's exact test was used to determine the association between response and comutation load. Odds ratio estimation, confidence intervals, and P value were calculated in R package Epitools.26 The P value was adjusted for multiple hypotheses testing using the Bonferroni-Holm method.
TABLE A1.
Additional Data for Patients Receiving Immunotherapy

Benjamin Garmezy
Uncompensated Relationships: AVEO (Inst)
Xianli Jiang
Patents, Royalties, Other Intellectual Property: I am in the process of a patent application, which is nonrelevant to this study. The application is for United States Letters Patent, Serial No. 63/015,317
Patrick G. Pilié
Consulting or Advisory Role: Novartis
Patents, Royalties, Other Intellectual Property: Patent pending-biomarker
Kenna R. Shaw
Consulting or Advisory Role: Guidepoint Global
Research Funding: Guardant Health (Inst), Tempus (Inst), Philips Healthcare (Inst)
Jordi Rodon
Consulting or Advisory Role: Peptomyc, Kelun, Merck Sharp & Dohme, Spectrum Pharmaceuticals, Pfizer, Roche/Genentech, Ellipses Pharma, Ionctura, Novartis, Lilly, Orion, Servier, Molecular Partners, NovellusDx, Certara, Bayer, KisoJi Biotechnology
Research Funding: Novartis, Bayer, Spectrum Pharmaceuticals, Tocagen, Symphogen, BioAtla, Pfizer, Genmab, CytomX Therapeutics, Kelun, Takeda/Millennium, GlaxoSmithKline, Ipsen, Blueprint Medicines
Travel, Accommodations, Expenses: ESMO, Department of Defense, Louisiana State University, Huntsman Cancer Institute, Cancer Core Europe, Karolinska Cancer Institute, King Abdullah International Medical Research Center, Molecular Partners, Merck Sharp & Dohme, Kelun Pharmaceuticals/Klus Pharma, Bayer, WIN Consortium, Janssen
Other Relationship: European Journal of Cancer, Chinese University of Hong Kong, SOLTI, Elsevier, GlaxoSmithKline, Vall d'Hebron University Hospital Institute of Oncology, VHIO/Ministero De Empleo Y Seguridad Social
John-Paul Shen
Stock and Other Ownership Interests: Agios, Syndax
Consulting or Advisory Role: Engine Biosciences
Research Funding: Celsius Therapeutics
Ying Yuan
Honoraria: Ono Pharmaceutical
Consulting or Advisory Role: Boehringer Ingelheim, Amgen, AbbVie, Servier, Starpax Medical, Vertex, MicuRx Pharmaceuticals, BeyondSpring Pharmaceuticals, Bristol Myers Squibb/Celgene/Juno
Funda Meric-Bernstam
Employment: MD Anderson Cancer Center
Honoraria: Rutgers Cancer Institute of New Jersey
Consulting or Advisory Role: Samsung Bioepis, Xencor, Debiopharm Group, Silverback Therapeutics, IBM Watson Health, Roche, PACT Pharma, eFFECTOR Therapeutics, Kolon Life Sciences, Tyra Biosciences, Zymeworks, Puma Biotechnology, Zentalis, Alkermes, Infinity Pharmaceuticals, AbbVie, Black Diamond Therapeutics, Eisai, OnCusp Therapeutics, Lengo Therapeutics, Tallac Therapeutics, Karyopharm Therapeutics, Biovica
Speakers' Bureau: Chugai Pharma
Research Funding: Novartis (Inst), AstraZeneca (Inst), Taiho Pharmaceutical (Inst), Genentech (Inst), Calithera Biosciences (Inst), Debiopharm Group (Inst), Bayer (Inst), Aileron Therapeutics (Inst), PUMA Biotechnology (Inst), CytomX Therapeutics (Inst), Jounce Therapeutics (Inst), Zymeworks (Inst), Curis (Inst), Pfizer (Inst), eFFECTOR Therapeutics (Inst), AbbVie (Inst), Boehringer Ingelheim, Guardant Health (Inst), Daiichi Sankyo (Inst), GlaxoSmithKline (Inst), Seattle Genetics (Inst), Klus Pharma (Inst), Takeda (Inst)
Travel, Accommodations, Expenses: Beth Israel Deaconess Medical Center
Timothy A. Yap
Consulting or Advisory Role: Pfizer, EMD Serono, Clovis Oncology, Ignyta, AstraZeneca, Atrin Pharmaceuticals, Aduro Biotech, Merck, Almac Diagnstics, Bayer, Bristol Myers Squibb, Calithera Biosciences, Cybrexa Therapeutics, Janssen, Roche, Seattle Genetics, Axiom Biotechnologies, F-Star, Guidepoint Global, I-Mab, Repare Therapeutics, Rubius Therapeutics, Schrodinger, Varian Medical Systems, Zai Lab
Research Funding: AstraZeneca (Inst), Vertex (Inst), Pfizer (Inst), Bayer (Inst), Tesaro (Inst), Jounce Therapeutics (Inst), Seattle Genetics (Inst), Kyowa Hakko Kirin (Inst), Constellation Pharmaceuticals (Inst), Lilly (Inst), Artios (Inst), Clovis Oncology (Inst), Cyteir (Inst), EMD Serono (Inst), Forbius (Inst), F-Star (Inst), GlaxoSmithKline (Inst), Genentech (Inst), ImmuneSensor Therapeutics (Inst), Ipsen (Inst), Karyopharm Therapeutics (Inst), Merck (Inst), Novartis (Inst), Ribon Therapeutics (Inst), Regeneron (Inst), Repare Therapeutics (Inst), Sanofi (Inst), Scholar Rock (Inst)
No other potential conflicts of interest were reported.
PRIOR PRESENTATION
Some data included in this manuscript were presented as oral presentation at the 2020 Virtual ASCO Annual Meeting, May 29-31, 2020. Associated abstract: DOI: 10.1200/JCO.2020.38.15_suppl.3008 Journal of Clinical Oncology 38, 2020 (suppl 15; abstr 3008).
SUPPORT
Supported in part by The Cancer Prevention and Research Institute of Texas (award no. RP150535), the Sheikh Khalifa Bin Zayed Al Nahyan Institute for Personalized Cancer Therapy, National Center for Advancing Translational Sciences Grant No. UL1 TR000371 (Center for Clinical and Translational Sciences), and the MD Anderson Cancer Center Support Grant No. P30 CA016672. This work was supported by the National Cancer Institute (K22 CA234406 to J.P.S., P50CA221707 and P50CA127001 to Y.Y., and U01CA247760 to K.C.) and the Cancer Prevention & Research Institute of Texas (RP180248 to K.C. and RR180035 to J.P.S.; J.P.S. is a CPRIT Scholar in Cancer Research).
DATA SHARING STATEMENT
A data sharing statement provided by the authors is available with this article at DOI https://doi.org/10.1200/PO.21.00267.
AUTHOR CONTRIBUTIONS
Conception and design: Benjamin Garmezy, Ying Yuan, Timothy A. Yap
Financial support: Kenna R. Shaw, Ken Chen
Administrative support: Kenna R. Shaw, Funda Meric-Bernstam, Ken Chen, Timothy A. Yap
Provision of study materials or patients: Kyaw Z. Thein, Wanlin Wang, Kenna R. Shaw, Jordi Rodon, Funda Meric-Bernstam, Timothy A. Yap
Collection and assembly of data: Benjamin Garmezy, Jinesh Gheeya, Kyaw Z. Thein, Wanlin Wang, Kenna R. Shaw, Funda Meric-Bernstam, Timothy A. Yap
Data analysis and interpretation: Benjamin Garmezy, Jinesh Gheeya, Heather Y. Lin, Yuefan Huang, Taebeom Kim, Xianli Jiang, Kyaw Z. Thein, Patrick G. Pilié, Fadl Zeineddine, Jordi Rodon, John-Paul Shen, Ying Yuan, Funda Meric-Bernstam, Ken Chen, Timothy A. Yap
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Benjamin Garmezy
Uncompensated Relationships: AVEO (Inst)
Xianli Jiang
Patents, Royalties, Other Intellectual Property: I am in the process of a patent application, which is nonrelevant to this study. The application is for United States Letters Patent, Serial No. 63/015,317
Patrick G. Pilié
Consulting or Advisory Role: Novartis
Patents, Royalties, Other Intellectual Property: Patent pending-biomarker
Kenna R. Shaw
Consulting or Advisory Role: Guidepoint Global
Research Funding: Guardant Health (Inst), Tempus (Inst), Philips Healthcare (Inst)
Jordi Rodon
Consulting or Advisory Role: Peptomyc, Kelun, Merck Sharp & Dohme, Spectrum Pharmaceuticals, Pfizer, Roche/Genentech, Ellipses Pharma, Ionctura, Novartis, Lilly, Orion, Servier, Molecular Partners, NovellusDx, Certara, Bayer, KisoJi Biotechnology
Research Funding: Novartis, Bayer, Spectrum Pharmaceuticals, Tocagen, Symphogen, BioAtla, Pfizer, Genmab, CytomX Therapeutics, Kelun, Takeda/Millennium, GlaxoSmithKline, Ipsen, Blueprint Medicines
Travel, Accommodations, Expenses: ESMO, Department of Defense, Louisiana State University, Huntsman Cancer Institute, Cancer Core Europe, Karolinska Cancer Institute, King Abdullah International Medical Research Center, Molecular Partners, Merck Sharp & Dohme, Kelun Pharmaceuticals/Klus Pharma, Bayer, WIN Consortium, Janssen
Other Relationship: European Journal of Cancer, Chinese University of Hong Kong, SOLTI, Elsevier, GlaxoSmithKline, Vall d'Hebron University Hospital Institute of Oncology, VHIO/Ministero De Empleo Y Seguridad Social
John-Paul Shen
Stock and Other Ownership Interests: Agios, Syndax
Consulting or Advisory Role: Engine Biosciences
Research Funding: Celsius Therapeutics
Ying Yuan
Honoraria: Ono Pharmaceutical
Consulting or Advisory Role: Boehringer Ingelheim, Amgen, AbbVie, Servier, Starpax Medical, Vertex, MicuRx Pharmaceuticals, BeyondSpring Pharmaceuticals, Bristol Myers Squibb/Celgene/Juno
Funda Meric-Bernstam
Employment: MD Anderson Cancer Center
Honoraria: Rutgers Cancer Institute of New Jersey
Consulting or Advisory Role: Samsung Bioepis, Xencor, Debiopharm Group, Silverback Therapeutics, IBM Watson Health, Roche, PACT Pharma, eFFECTOR Therapeutics, Kolon Life Sciences, Tyra Biosciences, Zymeworks, Puma Biotechnology, Zentalis, Alkermes, Infinity Pharmaceuticals, AbbVie, Black Diamond Therapeutics, Eisai, OnCusp Therapeutics, Lengo Therapeutics, Tallac Therapeutics, Karyopharm Therapeutics, Biovica
Speakers' Bureau: Chugai Pharma
Research Funding: Novartis (Inst), AstraZeneca (Inst), Taiho Pharmaceutical (Inst), Genentech (Inst), Calithera Biosciences (Inst), Debiopharm Group (Inst), Bayer (Inst), Aileron Therapeutics (Inst), PUMA Biotechnology (Inst), CytomX Therapeutics (Inst), Jounce Therapeutics (Inst), Zymeworks (Inst), Curis (Inst), Pfizer (Inst), eFFECTOR Therapeutics (Inst), AbbVie (Inst), Boehringer Ingelheim, Guardant Health (Inst), Daiichi Sankyo (Inst), GlaxoSmithKline (Inst), Seattle Genetics (Inst), Klus Pharma (Inst), Takeda (Inst)
Travel, Accommodations, Expenses: Beth Israel Deaconess Medical Center
Timothy A. Yap
Consulting or Advisory Role: Pfizer, EMD Serono, Clovis Oncology, Ignyta, AstraZeneca, Atrin Pharmaceuticals, Aduro Biotech, Merck, Almac Diagnstics, Bayer, Bristol Myers Squibb, Calithera Biosciences, Cybrexa Therapeutics, Janssen, Roche, Seattle Genetics, Axiom Biotechnologies, F-Star, Guidepoint Global, I-Mab, Repare Therapeutics, Rubius Therapeutics, Schrodinger, Varian Medical Systems, Zai Lab
Research Funding: AstraZeneca (Inst), Vertex (Inst), Pfizer (Inst), Bayer (Inst), Tesaro (Inst), Jounce Therapeutics (Inst), Seattle Genetics (Inst), Kyowa Hakko Kirin (Inst), Constellation Pharmaceuticals (Inst), Lilly (Inst), Artios (Inst), Clovis Oncology (Inst), Cyteir (Inst), EMD Serono (Inst), Forbius (Inst), F-Star (Inst), GlaxoSmithKline (Inst), Genentech (Inst), ImmuneSensor Therapeutics (Inst), Ipsen (Inst), Karyopharm Therapeutics (Inst), Merck (Inst), Novartis (Inst), Ribon Therapeutics (Inst), Regeneron (Inst), Repare Therapeutics (Inst), Sanofi (Inst), Scholar Rock (Inst)
No other potential conflicts of interest were reported.
REFERENCES
- 1.Rayner E, van Gool IC, Palles C, et al. A panoply of errors: Polymerase proofreading domain mutations in cancer Nat Rev Cancer 1671–812016 [DOI] [PubMed] [Google Scholar]
- 2.Albertson TM, Ogawa M, Bugni JM, et al. DNA polymerase epsilon and delta proofreading suppress discrete mutator and cancer phenotypes in mice Proc Natl Acad Sci USA 10617101–171042009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Church DN, Briggs SE, Palles C, et al. DNA polymerase ε and δ exonuclease domain mutations in endometrial cancer Hum Mol Genet 222820–28282013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palles C, Cazier JB, Howarth KM, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas Nat Genet 45136–1442013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shlien A, Campbell BB, de Borja R, et al. Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers Nat Genet 47257–2622015 [DOI] [PubMed] [Google Scholar]
- 6.Campbell BB, Light N, Fabrizio D, et al. Comprehensive analysis of hypermutation in human cancer Cell 1711042–1056.e102017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hatakeyama K, Ohshima K, Nagashima T, et al. Molecular profiling and sequential somatic mutation shift in hypermutator tumours harbouring POLE mutations. Sci Rep. 2018;8:8700. doi: 10.1038/s41598-018-26967-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park VS, Pursell ZF.POLE proofreading defects: Contributions to mutagenesis and cancer DNA Repair (Amst) 7650–592019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Havel JJ, Chowell D, Chan TA.The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy Nat Rev Cancer 19133–1502019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer Science 348124–1282015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goodman AM, Kato S, Bazhenova L, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers Mol Cancer Ther 162598–26082017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade Science 357409–4132017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency N Engl J Med 3722509–25202015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guo L, Wei R, Lin Y, et al. Clinical and recent patents applications of PD-1/PD-L1 targeting immunotherapy in cancer treatment-current progress, strategy, and future perspective. Front Immunol. 2020;11:1508. doi: 10.3389/fimmu.2020.01508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Davis AA, Patel VG. The role of PD-L1 expression as a predictive biomarker: An analysis of all US Food and Drug Administration (FDA) approvals of immune checkpoint inhibitors. J Immunother Cancer. 2019;7:278. doi: 10.1186/s40425-019-0768-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang F, Zhao Q, Wang Y-N, et al. Evaluation of POLE and POLD1 mutations as biomarkers for immunotherapy outcomes across multiple cancer types JAMA Oncol 51504–15062019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Roszik J, Haydu LE, Hess KR, et al. Novel algorithmic approach predicts tumor mutation load and correlates with immunotherapy clinical outcomes using a defined gene mutation set. BMC Med. 2016;14:168. doi: 10.1186/s12916-016-0705-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chalmers ZR, Connelly CF, Fabrizio D, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9:34. doi: 10.1186/s13073-017-0424-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Q, Wang K.InterVar: Clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines Am J Hum Genet 100267–2802017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Landrum MJ, Lee JM, Benson M, et al. ClinVar: Improving access to variant interpretations and supporting evidence Nucleic Acids Res 46D1062–D10672018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahn SM, Ansari AA, Kim J, et al. The somatic POLE P286R mutation defines a unique subclass of colorectal cancer featuring hypermutation, representing a potential genomic biomarker for immunotherapy Oncotarget 768638–686492016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xing X, Kane DP, Bulock CR, et al. A recurrent cancer-associated substitution in DNA polymerase epsilon produces a hyperactive enzyme. Nat Commun. 2019;10:374. doi: 10.1038/s41467-018-08145-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guenther M, Veninga V, Kumbrink J, et al. POLE gene hotspot mutations in advanced pancreatic cancer J Cancer Res Clin Oncol 1442161–21662018 [DOI] [PubMed] [Google Scholar]
- 24. Imboden S, Nastic D, Ghaderi M, et al. Phenotype of POLE-mutated endometrial cancer. PLoS One. 2019;14:e0214318. doi: 10.1371/journal.pone.0214318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Davila JI, Chanana P, Sarangi V, et al. Frequent POLE-driven hypermutation in ovarian endometrioid cancer revealed by mutational signatures in RNA sequencing. BMC Med Genomics. 2021;14:165. doi: 10.1186/s12920-021-01017-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaplan EL, Meier P.Nonparametric estimation from incomplete observations J Am Stat Assoc 53457–4811958 [Google Scholar]
- 27.Mantel N.Evaluation of survival data and two new rank order statistics arising in its consideration Cancer Chemother Rep 50163–1701966 [PubMed] [Google Scholar]
- 28.Cox DR.Regression models and life-tables J R Stat Soc Series B Methodol 34187–2021972 [Google Scholar]
- 29.Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold Nature 596583–5892021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hogg M, Osterman P, Bylund GO, et al. Structural basis for processive DNA synthesis by yeast DNA polymerase varepsilon Nat Struct Mol Biol 2149–552014 [DOI] [PubMed] [Google Scholar]
- 31.Henninger EE, Pursell ZF.DNA polymerase epsilon and its roles in genome stability IUBMB Life 66339–3512014 [DOI] [PubMed] [Google Scholar]
- 32.Yuza K, Nagahashi M, Watanabe S, et al. Hypermutation and microsatellite instability in gastrointestinal cancers Oncotarget 8112103–1121152017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shinbrot E, Henninger EE, Weinhold N, et al. Exonuclease mutations in DNA polymerase epsilon reveal replication strand specific mutation patterns and human origins of replication Genome Res 241740–17502014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fang H, Barbour JA, Poulos RC, et al. Mutational processes of distinct POLE exonuclease domain mutants drive an enrichment of a specific TP53 mutation in colorectal cancer. PLoS Genet. 2020;16:e1008572. doi: 10.1371/journal.pgen.1008572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Temko D, Van Gool IC, Rayner E, et al. Somatic POLE exonuclease domain mutations are early events in sporadic endometrial and colorectal carcinogenesis, determining driver mutational landscape, clonal neoantigen burden and immune response J Pathol 245283–2962018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Billingsley CC, Cohn DE, Mutch DG, et al. Prognostic significance of POLE exonuclease domain mutations in high-grade endometrioid endometrial cancer on survival and recurrence: A subanalysis Int J Gynecol Cancer 26933–9382016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keshinro A, Vanderbilt C, Kim JK, et al. Tumor-infiltrating lymphocytes, tumor mutational burden, and genetic alterations in microsatellite unstable, microsatellite stable, or mutant POLE/POLD1 colon cancer JCO Precis Oncol 5817–8262021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rousseau B.High activity of nivolumab in patients with pathogenic exonucleasic domain POLE (edPOLE) mutated Mismatch Repair proficient (MMRp) advanced tumours Ann Oncol 31S462–S5042020suppl 4; abstr 5260 [Google Scholar]
- 39. Rizvi N, Tang P, Bhardwaj N, et al. Nivolumab +/- ipilimumab in patients with hypermutated cancers detected in blood: NIMBLE. Cancer Immunol Res. 2019;7:B207. abstr B207. [Google Scholar]
- 40.Durvalumab for MSI-H or POLE Mutated Metastatic Colorectal Cancer (NCT03435107) 2020, https://clinicaltrials.gov/ct2/show/NCT03435107 [Google Scholar]
- 41. Lau D, Kalaitzaki E, Church DN, et al. Rationale and design of the POLEM trial: Avelumab plus fluoropyrimidine-based chemotherapy as adjuvant treatment for stage III mismatch repair deficient or POLE exonuclease domain mutant colon cancer: A phase III randomised study. ESMO Open. 2020;5:e000638. doi: 10.1136/esmoopen-2019-000638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.den Dunnen JT, Antonarakis SE.Mutation nomenclature extensions and suggestions to describe complex mutations: A discussion Hum Mutat 157–122000 [DOI] [PubMed] [Google Scholar]
- 43.Woolson RFC, William R. Statistical Methods for the Analysis of Biomedical Data. ed 2. New York, NY: John Wiley & Sons; 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
A data sharing statement provided by the authors is available with this article at DOI https://doi.org/10.1200/PO.21.00267.