

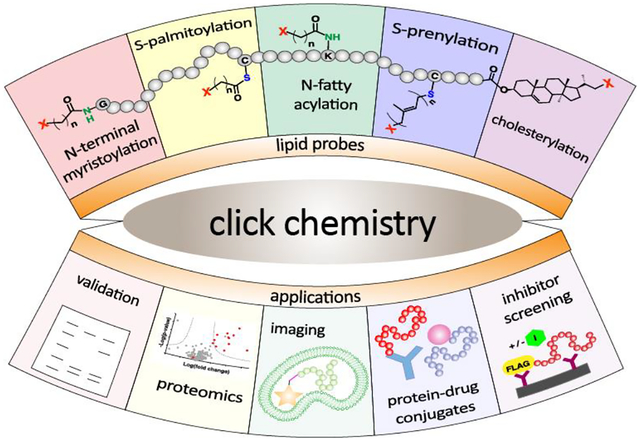
Abstract
Protein lipid modification involves the attachment of hydrophobic groups to proteins via ester, thioester, amide or thioether linkages. In this review, the specific click chemical reactions that have been employed to study protein lipid modification and their use for specific labeling applications are first described. That is followed by an introduction to the different types of protein lipid modifications that occur in biology. Next, the roles of click chemistry in elucidating specific biological features including the identification of lipid-modified proteins, studies of their regulation, and their role in diseases are presented. A description of the use of protein-lipid modifying enzymes for specific labeling applications including protein immobilization, fluorescent labeling, nanostructure assembly and the construction of protein drug conjugates is presented next. Concluding remarks and future directions are presented in the final section.
Graphical abstract
1. Introduction
Protein lipid modification involves the attachment of hydrophobic groups to proteins via ester, thioester, amide or thioether linkages; this process occurs in eukaryotes but not prokaryotes.1,2 The most common purpose of such modifications is to cause the association of the resulting proteins to various membranes where they can interact with other proteins involved in signal transduction pathways. Proteins containing such modifications are involved in a diverse range of cellular functions including cell division, subcellular organization, secretion and differentiation.3–7 Due to their critical role in such processes, these lipid-modified proteins are often mutated in various diseases or targets for therapeutic intervention.8–11 In addition, the consensus sequences that mark proteins for lipidation are often simple and can be incorporated into other proteins to render them membrane-bound or provide a new site for selective modification. That feature has been exploited for a myriad of applications ranging from protein immobilization to the creation of protein-drug conjugates for therapeutic applications.12,13 Click chemistry has played a key role in both illuminating features of protein lipid modification as well as exploiting it for the aforementioned alternative applications.14–18
In this review, the specific click chemical reactions that have been employed to study protein lipid modification and their use for specific labeling applications are first described. That is followed by an introduction to the different types of protein lipid modifications that occur in biology. Next, the roles of click chemistry in elucidating specific biological features including the identification of lipid-modified proteins, studies of their regulation, and their role in diseases are presented. That is followed by a description of the use of protein-lipid modifying enzymes for specific labeling applications including protein immobilization, fluorescent labeling, assembly of nanostructures and the construction of protein drug conjugates. Concluding remarks and future directions are presented in the final section.
2. Click chemistry
A term coined by Sharpless and coworkers, click chemistry is defined as reactions that are modular, wide in scope, give very high yields, generate only inoffensive byproducts, require simple reaction conditions, and use benign solvents such as water.19 Click chemistry closely overlaps with bio-orthogonal chemistry, which is defined as reactions that do not interfere with biological processes that proceed rapidly and selectively under physiological conditions.20 Tools developed for click chemistry enable bio-orthogonal reactions and developments in chemical biology stimulate the conception of novel click chemistry. Before the birth of these two concepts, chemistry and biology were somewhat disconnected as many advances in chemistry were inapplicable to living systems. The development of click chemistry revolutionized chemical biology, especially when performed in tandem with enzymatic labeling due to the synergetic effects generated by the unique advantages that the two components each provide. To be used in cellular environments, the participating click chemistry functionalities require a few additional characteristics. The molecular reaction components must possess cell penetrating capabilities and click-induced modifications must introduce minimal perturbation, which often necessitates the reaction components to be hydrophobic, neutrally charged, and small in size. Fortunately, many types of click reactions meet these criteria. Therefore, the bio-orthogonality of click chemistry has allowed selective labeling and therefore real-time investigation of enzymatic processes of interest both in vitro and in vivo, providing insights into many biological questions and biomedical challenges including cancer, Alzheimer’s disease, and coronavirus, among others. The site-specificity and rapid rates of both click chemistry and enzymatic labeling have also been purposed to tackle those biomedical challenges by efficiently functionalizing therapeutic molecules of interest. The combination of the two principles has created a powerful and multifunctional “Swiss Army knife-like” tool. In this section, click reactions that are most commonly used specifically in tandem with lipid modifying enzymes are described (Fig. 1). For a broader scope of click chemistry or applications to other specific fields, readers are directed to other reviews including those included in the current thematic issue.21–25
Figure 1. Reaction scheme of biorthogonal click reactions for modifying proteins of interest.
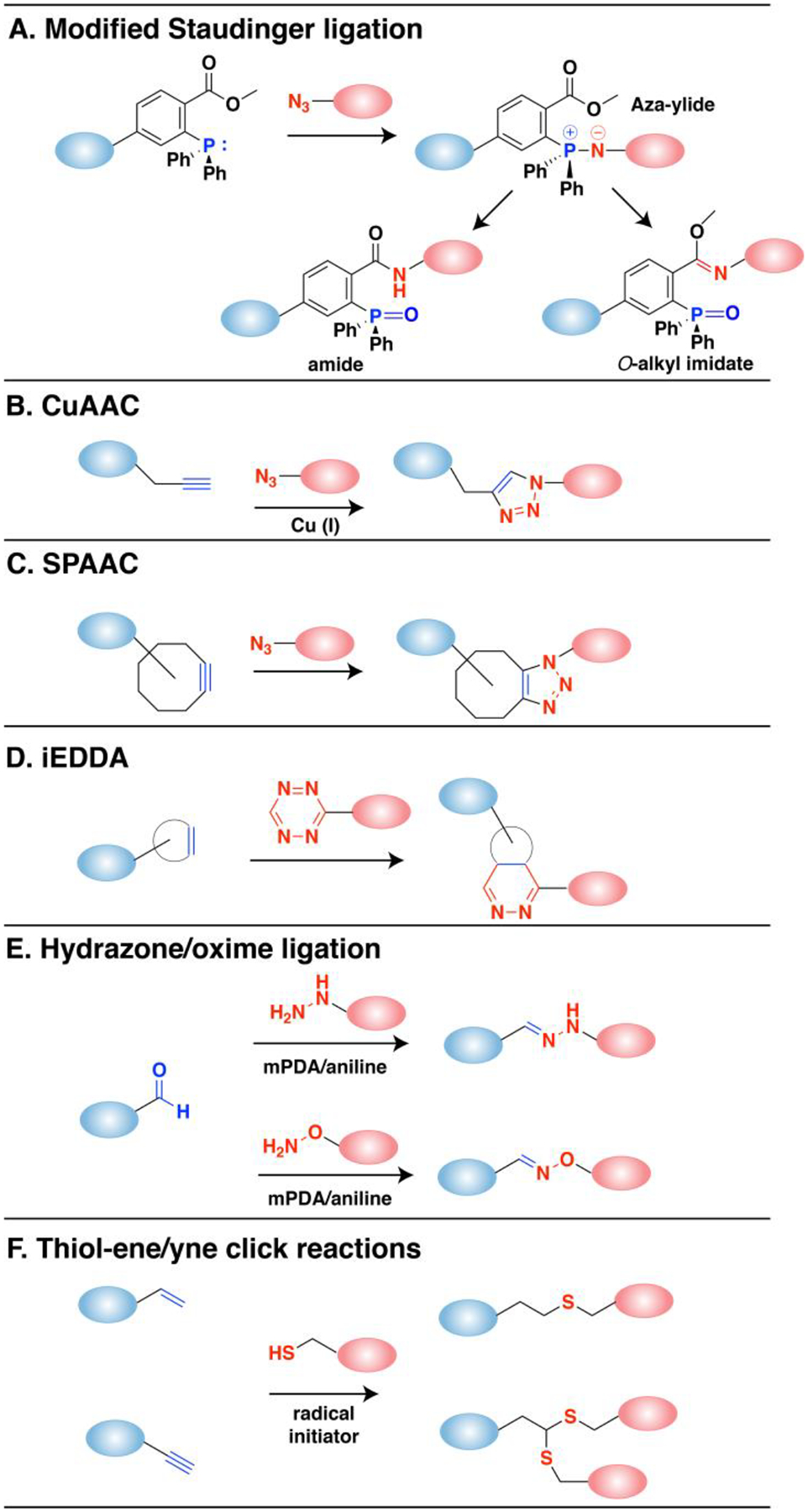
(A) Modified Staudinger ligation between an azide and phosphine reagent. An aza-ylide intermediate is formed followed by a transfer to the ester forming an amide bond. The O-alkyl imidate product can occur especially with aryl azides. (B) The copper-catalyzed azide-alkyne cycloaddition reaction (CuAAC) is a 1,3-dipolar cycloaddition between alkynes and azides that results in a 1,2,3-triazole product. (C) Strain-promoted azide-alkyne cycloaddition (SPAAC) reaction that employs strained octynes. (D) Inverse-electron demand Diels-Alder (iEDDA) reaction between strained alkenes (trans-cyclooctene or norbornene) with tetrazine. (E) Reaction between aldehyde and hydrazines or aminooxy compounds to form hydrazone or oxime bonds, respectively. These reactions can be catalyzed by aniline and related reagents (mPDA). (F) Radical reactions between thiols and alkenes or alkynes initiated by UV light.
One type of click reactions involve azides which are a versatile functionality due to their small size, stability and inertness. Azides are known to be truly bio-orthogonal as they are essentially absent in biological systems.22 The first bio-orthogonal click reaction reported was a Staudinger ligation between an azide and a functionalized triphenylphosphine (Fig. 1A). Bertozzi and coworkers exploited a modified version of the classic Staudinger reaction where they appended an ester to the phosphine component to prevent the hydrolysis of the aza-ylide intermediate, and instead via intramolecular trapping pushed the reaction towards a stable amide-linked product.26 This classic reaction has been and is still widely used for many applications both in cellular environments and in live animals due to its selectivity and biocompatibility.27 However, slow reaction kinetics (k ~ 10−3 M−1s−1) and the oxidation-prone phosphine reagents have driven investigations into alternative click chemistries,28 which has led to the emergence of the copper-catalyzed azide-alkyne cycloaddition (CuAAC, Fig. 1B), the most widely used click chemistry to date. CuAAC takes advantage of a Cu(I) catalyst which lowers the activation barrier for the formation of the triazole ring, resulting in significantly enhanced kinetics (k ~ 10–100 M−1s−1).29–32 Cu(I) is typically prepared in situ from Cu(II) salts by reducing agents, and is stabilized by various ligands that allows the reaction to proceed in aqueous conditions.33–35 The use of copper however is a double-edged sword, as its cytotoxicity is a significant liability. Cu(I) is involved in protein oxidation via reactive oxygen species (ROS) that also causes DNA strand breaks,32 which restricts the application of CuAAC with proteins or living cells. Efforts have been made to overcome the toxicity issue by utilizing novel ligands36,37 or improved azide structures38–40 which allow reduced copper concentrations while maintaining reaction efficiency, or reducing the generation of reactive oxygen species (ROS).34,41 Strain-promoted azide-alkyne cycloaddition (SPAAC, Fig. 1C) is an alternative to CuAAC which exploits activated alkynes incorporating ring strain to eliminate the requirement for metal catalysts.42 The non-toxic nature of copper-free click chemistry has allowed SPAAC to be employed in various in vivo applications.25 The use of SPAAC has significantly grown in the field of protein conjugation, often used in tandem with enzymatic labeling to maximize its selectivity and site-specificity. The growth is expected to continue as many pharmaceutical companies have established platforms for synthesizing site-specific antibody drug conjugates (ADC) based on site-specific installation of an azide and use of strained-alkyne payloads.43 In terms of accelerating the rate of SPAAC, progress has been made through various strategies including adding electron-withdrawing fluorines adjacent to the alkyne,44 adding a ketone to distort the alkyne into a more reactive geometry,45 adding a nitrogen in the ring for increased hydrophilicity,46 or forming a fused ring system for additional ring strain.47 However, the rate of SPAAC (k ~ 0.1–1 M−1s−1) still falls far behind CuAAC, and there are concerns that the large size and hydrophobicity of the ring-fused strained alkynes are prone to modulate the properties of target molecules or introduce off-target binding.48,49
Strained alkenes such as trans-cyclooctenes (TCO) or norbornenes undergo inverse electron demand Diels-Alder (iEDDA, Fig. 1D) reactions with tetrazines, which is the fastest click reaction identified to date (k > 103 M−1s−1). Although tetrazine ligation exploits ring strain to achieve accelerated rates under copper free conditions, it has been shown to be orthogonal to SPAAC which therefore allows the use of both reactions simultaneously for selective labeling of multiple targets.50,51 The fast rate even at low concentrations makes it especially useful in radiolabeling studies where the short half-life of isotopes requires rapid labeling,52 or tracking and imaging of fast dynamic biological processes in living cells.23 In imaging studies, tetrazine-conjugated fluorophores have been used as turn-on probes due to the dual functionality of the tetrazine as both a bio-orthogonal moiety and a fluorescence quencher.25 Similar to SPAAC, the biggest drawback in tetrazine ligation is the hydrophobicity and large size of the ring-containing alkenes. However, smaller dienophiles such as cyclopropenes or azetines have been shown to retain reactivity and aqueous stability which introduces minimal perturbation at the target site comparable to products of the CuAAC reaction.53–55
Aldehydes and ketones undergo transamination reactions with amine nucleophiles that exhibit the α-effect, namely, increased nucleophilicity from the presence of lone pair electrons on the atom adjacent to the amine.24 to form oximes or hydrazones. Although endogenous intracellular aldehydes and ketones do exist, oxime and hydrazone ligation (Fig. 1E) are considered bio-orthogonal as the α-effect nucleophile counterpart is rare and the reaction can be useful in other biological environments where aldehydes and ketones are absent. Oxime and hydrazone ligation products are among the smallest possible products in size among other click reaction products which therefore results in minimal structural perturbation. Rapid rate (k ~ 1–103 M−1s−1) at physiological conditions has been achieved by careful structural design of the substrates56 and use of various catalysts.57–60 While an oxime linkage is more stable than a hydrazone,61 both ligation products are susceptible to hydrolysis, and especially for hydrazones that have a low equilibrium constant (Keq) which at low concentration reactions causes incomplete product conversion and dissociation of the products.59 However, the instability of oximes and hydrazones has actually provided them with a unique reversible capability which has been exploited for various applications.62–64 For studies that require stable products, modified ligation reactions have been developed that utilize the reaction of aldehydes with alternative nucleophiles that yield stable C-C bonds instead of the labile C=N bonds.65–67 Due to their unique advantages, the use of oxime and hydrazone ligation has been well established for site-specific protein labeling and in vitro cell surface labelling especially in combination with enzymatic labelling methods which allows controlled installment of an aldehyde.68,69 However, in vivo applications employing aldehyde and ketones are considerably more limited.
Thiol-ene (and thiol-yne) reactions (Fig. 1F) are click reactions that occur between thiols and alkenes or alkynes. Thiol-ene click chemistry has been heavily utilized for the functionalization of polymeric materials or nanoparticles.70,71 While the use of thiol-ene chemistry in biochemical applications has held challenges in retaining its bio-orthogonality and site-specificity due to cellular thiols and other nucleophilic and electrophilic groups prone to side reactions,72–74 it has been shown that exploiting photochemistry, in which the reaction proceeds via a single electron radical chemistry, provides enhanced selectivity.75 Additionally, using light as the external stimulus allows applications in studies that require spatiotemporal control. These properties of thiol-ene click chemistry have prompted its use for protein modification such as surface immobilization, extracellular matrix (ECM) generation, and protein patterning in hydrogels.76,77
3. Protein lipid modifications
A large number of proteins synthesized within cells undergo post-translational modifications (PTMs) essential for their biological function, cellular localization and activity. More than 200 PTMs are currently known that diversify protein function and dynamically synchronize complex signaling networks.78 Among these PTMs, protein lipidation involves the covalent attachment of small, hydrophobic molecules that promote stable membrane association of proteins, regulate protein trafficking, and mediate protein-protein interactions.3 Lipid PTMs can be categorized into two types—those that occur on the cytosolic side of membranes and those that take place in the lumen of secretory organelles. There are three major protein lipidation processes known to occur in the cytoplasm or cytoplasmic face of membranes. The first two modify proteins with fatty acyl groups, palmitoyl and myristoyl, generally on thiols present in cysteine (S-acylation) and N-terminal amines of glycine (N-myristoylation), respectively. The thioester bonds formed in S-acylated proteins are labile and are therefore reversible, as opposed to the amide bonds in N-myristoylated proteins that are thought to be stable and irreversible. The third major type of protein lipidation is S-prenylation where the thiols of cysteines present near the C-terminus of proteins are linked to an isoprenoid through a thioether bond. In contrast, lipidation of secreted proteins includes cholesterylation and the attachment of glycosylphosphatidylinositol (GPI) anchors. Cholesterylation involves the esterification of the carboxyterminus of proteins with a cholesteryl functional group, while modification with a GPI anchor appends a fatty acylated phosphatidyl inositol containing a glycan core and linked to the carboxyterminus of proteins through an amide bond with ethanolamine. An overview of the major cytoplasmic protein lipid modifications is presented below.
3.1. S-acylation (S-palmitoylation)
Protein S-acylation is the addition of fatty acids of varying carbon chain lengths (C14-C20) to a cysteine residue forming a thioester bond. In particular, protein is S-palmitoylation is the most common type of this modification (Fig. 2A). This lipid PTM is essential for stable anchoring, trafficking and localization of a plethora of membrane-associated proteins.79 The discovery of a protein modified with a palmitoyl moiety was initially made via radiolabeling in virus-infected cells with [3H] palmitate.80 Soon thereafter, a number of other S-acylated proteins were reported including rhodopsin and p21 Ras protein.81,82 The protein acyltransferase (PAT) enzymes that catalyze this thioester linkage formation were initially discovered in yeast,83 and later found to bear a conserved aspartate-histidine-histidine-cysteine (DHHC) motif in its cysteine-rich domain (CRD).84 The first mammalian homolog identified contained a zinc finger domain,85 which is a salient feature of all mammalian PATs reported to date. Hence, they are typically referred to as zDHHC-PATs. These enzymes are relatively ubiquitous as they localize in the golgi, endoplasmic reticulum (ER), and plasma membrane,86 although recent findings indicate that they mainly reside in golgi membranes.87 In both human and mouse genomes, bioinformatic analyses revealed that there are 23 proteins known to possess the DHHC-CRD motifs, raising the possibility of a large family of zDHHC-PATs.88 Indeed, these 23 enzymes are recognized as fatty acyl transferases particularly for protein S-palmitoylation. However, the catalytic motif is not strictly constrained to the DHHC amino acid residues, as other motifs such as DHYC in yeast Akr1 and DQHC in mammalian zDHHC13 display efficient S-palmitoylation activity with their substrates.89,90 Furthermore, zDHHC-PATs generally catalyze the transfer of fatty acids with varying lengths or containing unsaturated bonds, although palmitate-CoA is their preferred substrate.
Figure 2. Fatty acylation on proteins.
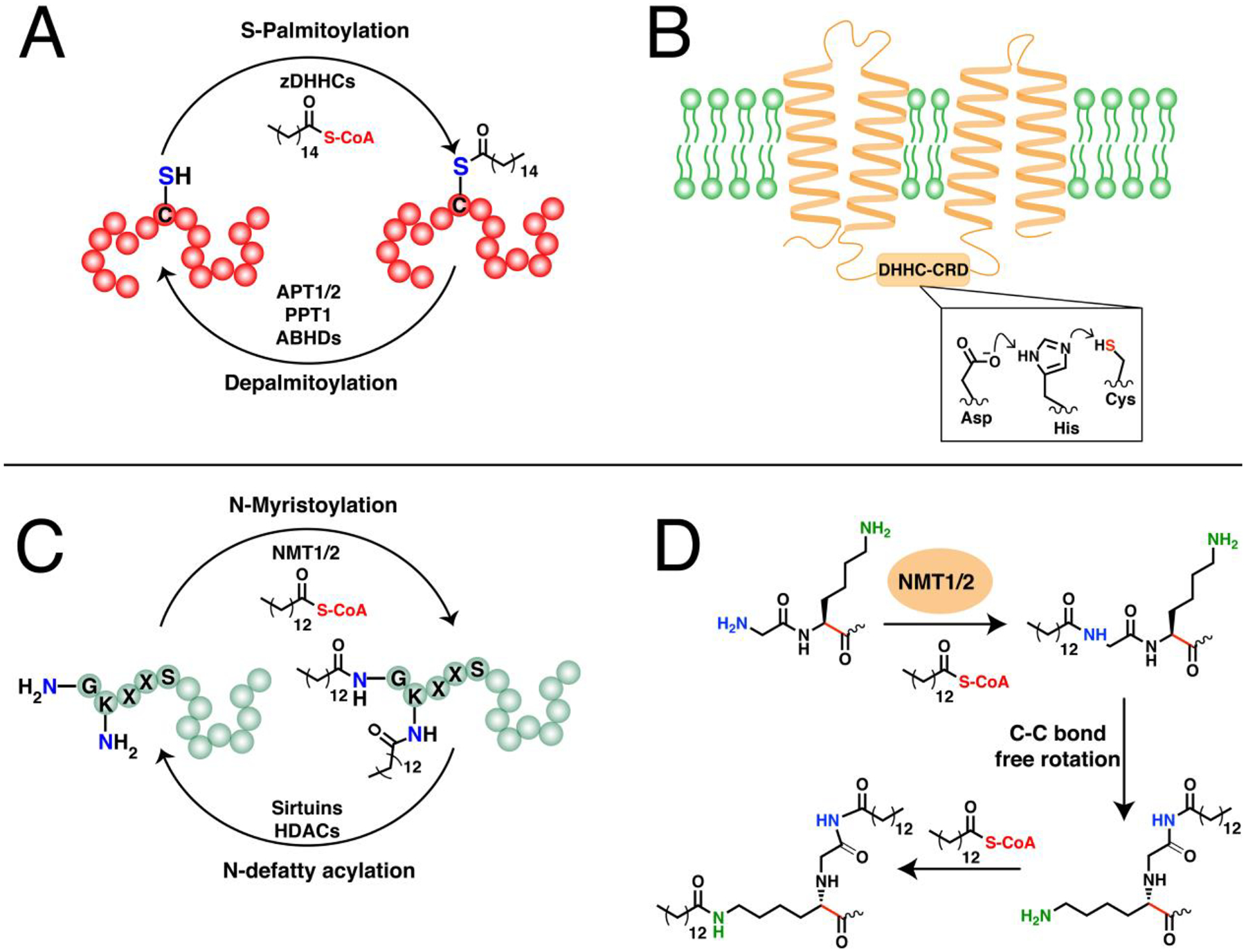
(A) The dynamic cycling of protein S-palmitoylation and depalmitoylation on cysteine residues. zDHHCs catalyze S-acylation on proteins particularly S-palmitoylation while APTs, PPT1, and ABHDs can remove these modifications. (B) A representative structure of a zDHHC-PAT embedded in a membrane through its transmembrane domains (TMDs).(Ref. 93) The DHHC active site is oriented towards the cytoplasm and resembles a catalytic triad that generates a thiolate nucleophile for activation of fatty acyl-CoAs. (C) The N-myristoylation and N-defatty acylation cycling of N-lipidated proteins. NMTs can catalyze both N-terminal and side-chain fatty acylation, although the lysine is typically adjacent to the N-terminal glycine. Sirtuins and HDACs can reverse this modification (D) The mechanism for the dual myristoylation on the N-terminus of myristoylated AFR6 GTPase. The free rotation of the C-C bond shown in red allows for dual myristoylation on the N-terminal glycine and the side-chain of the adjacent lysine.(Ref 116)
The DHHC motif in PATs is highly conserved and usually necessary for catalysis, while the CRD is speculated to be important for zinc ion binding.91,92 Although PATs have been studied for several years, until recently, no atomic structure was available and most studies relied mainly on predictive models. The recent breakthrough of successfully solving the crystal structures for human zDHHC20 and zebrafish zDHHC15 illuminated and supported the observed and predicted features of some zDHHCs crucial to understanding their structure and mechanism.93,94 The crystal structures of these two zDHHCs revealed the projection of transmembrane domains (TMDs) and other key intramolecular contacts that rationalize the exposure of the DHHC-CRD to the cytoplasm, as well as the placement of the active site at the cytoplasm-membrane interface (Fig. 2B). Their active sites also resemble a catalytic triad wherein the first His in the DHHC motif is polarized by the Asp to deprotonate the catalytic Cys, generating a thiolate for efficient nucleophilic attack on palmitoyl-CoA, and resulting in an auto-palmitoylated zDHHC enzyme with a concomitant release of CoA. The covalently attached palmitoyl moiety is then transferred to a bound protein substrate.95 The auto-acylation mechanism is common among zDHHCs, however, not all seem to require such an intermediate. Depending on the mutations introduced into the positions in the active site, some zDHHCs remain functional and can still S-palmitoylate their protein substrates regardless of whether the machinery for auto-palmitoylation is present or not.89,96,97 It is important to note that these crystal structures are for only two zDHHCs and may not necessarily be representative of other zDHHC family members. Factors such as structural and chemical requirements of individual zDHHCs, the mechanism of fatty acyl transfer, or the reactivity of the protein substrate’s cysteine may play key roles into determining the precise mechanistic details of each zDHHC enzyme’s reaction with their cognate substrates.94
Deducing substrate specificities across zDHHCs is non-trivial due to the lack of a consensus sequence within the substrates that these enzymes recognize. The functional redundancy observed among them coupled with the fact that they share the same substrates makes the generality of their substrate recognition mechanism implausible. Multiple factors may dictate the specificity of particular zDHHCs for their substrates such as the presence of specific binding domains and substrate proximity. For example, zDHHC13 and zDHHC17 contain ankyrin-repeat domains that interact with a consensus sequence present in SNAP23, SNAP25, and huntingtin.96 On the other hand, zDHHC5 engages with its substrate GRIP1b through its PDZ motif, yet interacts with another substrate PLM through the enzyme’s C-terminal 120-amino acid domain.98,99 Clearly, these varying modes of interaction between zDHHCs and their substrates requires further investigations to elucidate the distinctive substrate recognition features of individual enzyme-substrate pairs. Despite this lack of consensus, a powerful predictive tool, based on a clustering and scoring algorithm (CSS-Palm), was developed for global in silico screening of S-palmitoylation sites in proteomes.100
One characteristic feature of protein S-palmitoylation is its dynamic nature (Fig. 2A). Acyl protein thioesterases (APTs) and lysosomal protein palmitoylthioesterase-1 (PPT1) serve as deacylases/depalmitoylases that catalyze the cleavage of palmitate groups for protein cycling and degradation, respectively.101 Members of the α/β-Hydrolase domain-containing (ABHDs) family of thioesterases were also found to catalyze depalmitoylation of mammalian proteins.102 These depalmitoylases themselves require palmitoyl modification for proper localization and function. The plasma membrane localization of small GTPases and G proteins, for example, is controlled by a continuous S-palmitoylation-depalmitoylation cycle, manifesting the requirement of both zDHHCs and depalmitoylases for regulating localization, and in effect, the function of palmitoylated proteins.103 However, the stability of the palmitate modification varies over a range of substrates, with most proteins remaining stably S-palmitoylated with others undergoing rapid dynamic cycling.104
The broad substrate scope of protein S-acylation/deacylation highlights its major role in mediating numerous and diverse biological processes. This includes regulation of intracellular signaling and trafficking, shaping of neuronal synaptic plasticity, and immunity against bacterial and viral infections, among others.105–107 Dysregulation and perturbations in this lipidation pathway have been linked to a myriad of diseases including neurological disorders and cancer.108,109 In later sections, this review describes the impact of click chemistry as a valuable tool to characterize the role of S-acylation particularly of S-palmitoylation in regulating the biological functions of known and novel S-palmitoylated proteins, as well as the development of technological innovations based on the clickable analogues of palmitic acid.
3.2. N-Myristoylation
A second major type of fatty acylation is the covalent attachment of a myristoyl group, a saturated 14-carbon chain, onto the amine of an N-terminal glycine residue (Fig. 2C). Similar to S-acylation, the activated myristoyl-CoA form is used as a substrate to acylate proteins and form an amide bond catalyzed by N-myristoyltransferases (NMTs).110 This lipidation is often critical for proper localization of modified proteins. However, N-myristoylation alone is generally insufficient to confer stable anchoring and is therefore usually present in tandem with a second membrane-targeting signal such as a polybasic region (PBR) or other lipid modifications including S-palmitoylation.111,112 N-myristoylation can proceed either through co- or post-translational mechanisms. The co-translational mechanism occurs on nascent proteins with initial cleavage of Met catalyzed by MetAPs and subsequent glycine N-myristoylation on substrates with consensus N-terminal sequence of MG[^DEFRWY]X[^DEKR][*ACGST][*KR]X, where ^ denotes exclusion of the amino acids listed in that specific position, * indicates preferred residues, and X is any amino acid.113 The transfer of the myristoyl moiety then takes place during translation. In contrast, post-translational modification has been observed in pro-apoptotic proteins that are cleaved by caspases during apoptosis, revealing a new N-terminal glycine fated for myristoyl modification.112 The NMTs themselves are truncated during apoptosis, which alters their cellular localization and influences their rates of activity and to some extent, their specificities towards their fragment substrates.114
NMT-mediated N-myristoylation follows a sequential ordered bi-bi mechanism where the initial binding of myristoyl-CoA elicits a conformational change that enables subsequent protein substrate binding.115,116 Unlike S-palmitoylation where the palmitoyl group is transiently attached to zDHHCs prior to transfer, NMTs directly append the myristoyl group onto their substrates without participation of a covalent intermediate. A recent analysis of high-resolution structures of human NMT1 co-crystalized with myristoyl-CoA and substrate peptides allowed for atomic-level dissection of the molecular mechanism of NMT catalysis.116 In these structures, an oxy-anion hole is present that activates the acyl group for efficient attack of the N-terminal amine from Gly1 of the substrate. The Ab-loop of NMT represents a fluid structure that promotes pre-organization of the substrates, which then triggers water-mediated deprotonation of the amino group by a carboxy catalytic base from Gln496, followed by nucleophilic attack of myristoyl-CoA. This concerted reaction is promoted by Thr282 that serves as a key residue in this mechanism. Importantly, the absence of a side chain on Gly permits free rotation of the N-terminal amine that is necessary for this mechanism, making it preferentially modified by NMTs over other amino acids. In addition, a hydrophilic pocket near the enzyme’s active site interacts with the fifth amino acid of the substrate, favoring the polar residues Ser, Thr, or Cys that occupy this position. These features serve as the basis for the specificity of NMTs toward N-terminal glycine-containing substrates.117,118
There is no evidence of innate expression of NMTs in prokaryotes and hence, they usually exploit host machinery to perform myristoyl modification required during infection.119 In some cases, pathogenic eukaryotes require intracellular N-myristoylation for their survival and virulence, prompting efforts to design pathogen-specific NMT inhibitors as therapeutic agents.111 Lower eukaryotes typically encode a single NMT gene while higher organisms express two characterized enzymes NMT1 and NMT2, which share approximately 77% sequence identity in the human homologs.120 Although both NMTs generally have overlapping substrates, biochemical and kinetic assays suggest that their substrate affinities differ and are not functionally redundant.121 Separate knockdowns of each enzyme resulted in differential effects in cell proliferation, embryonic development, and T-cell receptor signaling.3 Furthermore, they behave differently during apoptosis—NMT1 translocates from the plasma membrane to the cytosol while NMT2 does the contrary.114 These changes in their localization cause subtle differences in their substrate scope reflected by the downstream effects that ensue from the aberrations on the biological functions of their substrates.
While N-myristoylation is frequently ascribed to covalent attachment on an N-terminal glycine, Nε-side chain fatty acylation on Lys residues was recognized as early as 1992.122 The growing number of side-chain fatty acylated proteins identified has gained more attention over recent years and highlights their more frequent occurrence than was previously thought. Members of the Ras family of small GTPases such as KRAS, RRAS2, and RalB are Nε-fatty acylated on a Lys in their PBR, in addition to S-palmitoylation or S-prenylation on Cys residues.123–125 NMTs were recently discovered as the first enzymes to catalyze such Lys modification, at least for ARF6 GTPase.126 After the initial N-terminal N-myristoylation on Gly1 of ARF6, the lipid moiety shifts to a conformation that positions the side-chain amine of the adjacent Lys2 for efficient Nε-myristoylation (Fig. 2D). High-resolution structures of Gly1-Lys2-containing peptides in NMTs reveal that the Lys2 amino side-chain directly interacts with the carboxy catalytic base for efficient deprotonation, as opposed to the observed water-mediated deprotonation of Gly1.116 This possible dimyristoylation of ARF6 adds another layer to the intricacies of lipid modification and provides a rationale for its potentially unique regulation compared to other glycine-myristoylated members of the ARF family.126 While this demonstrates the capability of NMTs to modify side-chain amines, the mechanism for the fatty acylation on Lys residues positioned within the protein sequence or in the C-terminal PBRs remains unknown.
For many years, dynamic lipid modification has been focused on S-palmitoylation until recently, when the unexpected hydrolytic removal of fatty acyl groups from Nε-modified Lys residues was reported. The landmark discovery of sirtuins and histone deacetylases (HDACs) as lysine defatty-acylases broadened the paradigm to include the idea that Lys fatty acylation may also be dynamic.127 This family of proteins were initially described as “erasers” of short acyl modifications (e.g. acetyl) that is essential mainly in regulating epigenetic processes.128,129 However, recent studies provide evidence of their ability to remove long-chain fatty acids from modified substrates associated with other biological processes. For example, SIRT6 and HDAC11 can efficiently cleave long-chain lipids from RRAS2 and serine methylhydroxytransferase (SHMT), respectively.124,130 ARF6 itself is regulated by both NMTs and SIRT2, where NMT may myristoylate Lys2 in its GTP-bound active form while SIRT2 demyristoylates ARF6 while in its GDP-bound inactive state, thereby regulating another aspect of the GTPase cycle.126 Other members of these family of hydrolyzing enzymes exhibit defatty-acylase activity but no current bonafide substrates have been identified to date.127
Being an important lipid modification, N-myristoylation is a key regulator of protein stability, activity, localization, and protein-protein interactions that are linked to multiple aspects of immunity, autophagy, infection, and cancer.3,111,131–133 The use of click chemistry has enabled significant progress towards addressing numerous biological questions with substantial efforts focused on parasitic infections. A comprehensive description of those studies are discussed in Section 6. Furthermore, the convenient placement of N-myristoylation on the N-terminus of proteins has enabled site-specific N-terminal modification of proteins, offering strategies to decorate proteins of interest. These accounts are detailed in Section 9.2.
3.3. S-Prenylation
While the previous two major types of lipidation are based on acylation, S-prenylation is the attachment of isoprenoids onto cysteine residues forming a thioether bond. It was first observed in fungi and much later found to be present in mammals.134,135 There are now three recognized classes of protein S-prenylation. The first two involve a single addition of an isoprenoid either as a shorter-chain farnesyl or a longer-chain geranylgeranyl group from the native substrates farnesyldiphosphate (FPP) or geranylgeranyldiphosphate (GGPP), respectively, catalyzed by farnesyltransferase (FTase) or geranylgeranyltransferase type I (GGTase-I) as shown in Fig. 3A.136 The protein substrates for these enzymes are recognized through a C-terminal motif with a canonical sequence generalized as the Ca1a2X box. C is the cysteine that is modified with the prenyl group, a1 and a2 are generally hydrophobic amino acids, and X determines the fate of the protein substrate, either farnesylation or geranylgeranylation. Farnesylation is usually observed in proteins with alanine, methionine, serine, or glutamate at the X position, whereas hydrophobic residues leucine, isoleucine, and phenylalanine promote geranylgeranylation.136 Although these general rules are widely acknowledged, some proteins are capable of bearing either type of S-prenylation. For example, the Rho GTPase RhoB terminating in CKVL exhibits a prenylation-dependent function, wherein it promotes cell growth when farnesylated but its geranylgeranylated form induces apoptosis.137 Furthermore, the a1 and a2 positions are not strictly limited to hydrophobic amino acids as other polar or charged residues occupying these positions display efficient farnesylation activities.138,139 Another feature of these protein substrates is a linker region containing small or flexible hydrophilic residues that often precedes the Ca1a2X box. This facilitates the accessibility of the C-terminus for prenyltransferases and may impact substrate reactivity.140,141 Recent efforts to expand the existing accepted paradigm of protein S-prenylation have shown that proteins terminating in longer (CXXXX) or shorter (CXX) C-terminal motifs can be efficiently modified by the prenyltransferase enzymes.142,143 Although these sequences have been shown to be substrates in vitro and within cells, there is no reported evidence of these types of modifications on endogenous proteins to date.
Figure 3. Protein prenylation.
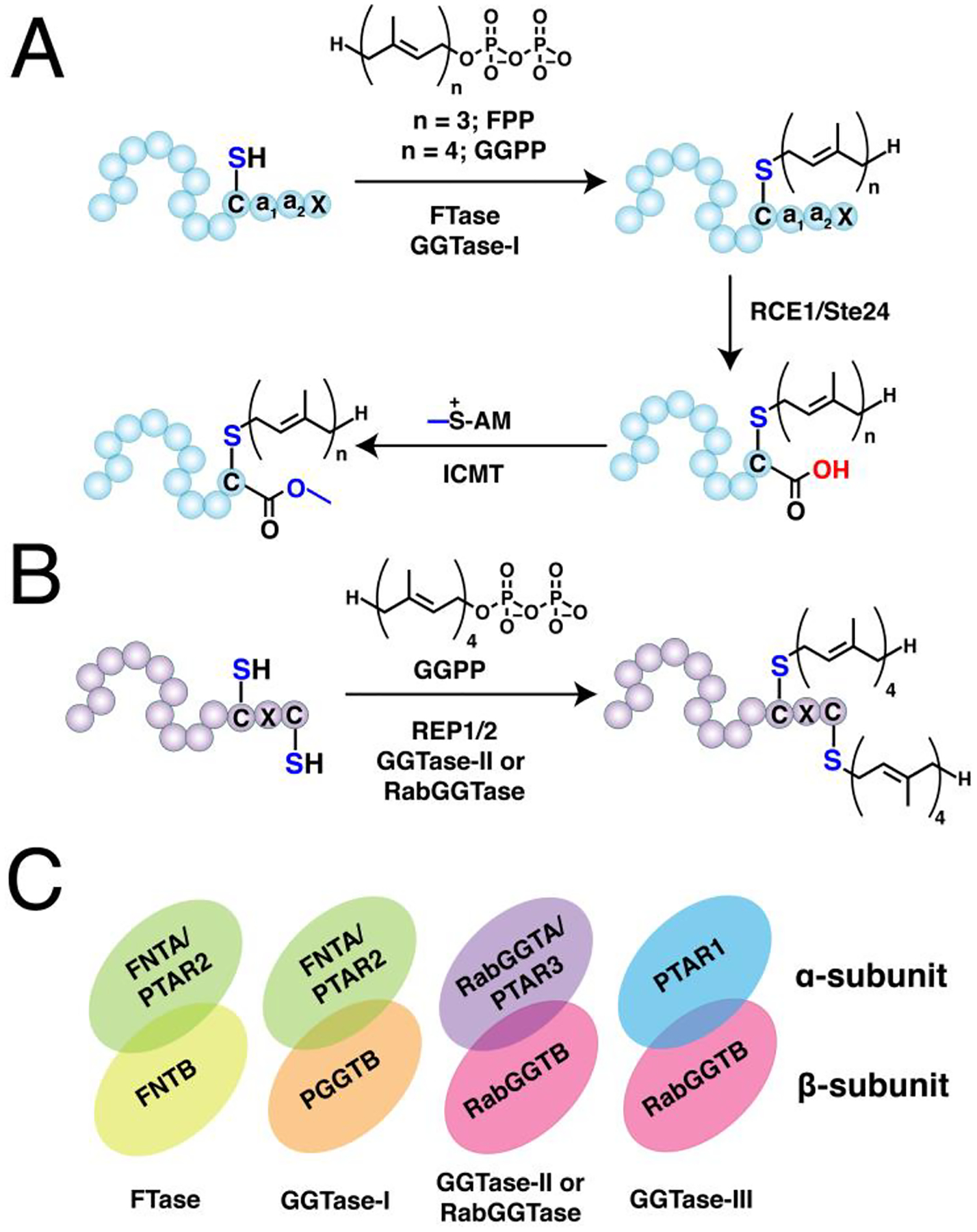
(A) Single prenylation on Ca1a2X box-containing proteins with farnesyl diphosphate (FPP) or geranylgeranyl diphosphate (GGPP) catalyzed by farnesyltransferase (FTase) or geranylgeranyltransferase type I (GGTase-I), respectively. The prenylated protein undergoes a maturation process involving cleavage of -a1a2X tripeptide catalyzed by ras converting enzyme (RCE1) or sterile 24 (Ste24), followed by methylation of the exposed carboxy terminus by isoprenylcysteine methyl transferase (ICMT). (B) Dual geranylgeranylation on two proximal C-terminal cysteine residues in Rab proteins. The Rab substrate is initially recruited by the rab escort protein 1 or 2 (REP1/2) and subsequently geranylgeranylated by the geranylgeranyltransferase type II (GGTase-II) or RabGGTase. (C) Representation of the heterodimeric structures of prenyltransferases showing their α and β subunits. FTase and GGTase-I share a common α subunit but differs in their β that determines their reactivity and substrate specificity (e. g. FPP vs GGPP). GGTase-II and GGTase-III share a common β subunit for geranylgeranylation but differ in their α subunit that influences their substrate recognition.
Singly prenylated proteins often undergo additional processing steps to increase their hydrophobicity. The -a1a2X tripeptide is cleaved off by the endoproteases Ras converting enzyme (RCE1) or Ste24 (Fig. 3A), followed by methylation with S-adenosyl methionine (SAM) on the exposed carboxyterminus catalyzed by isoprenylcysteine carboxyl methyltransferase (ICMT).144,145 These mature forms of S-prenylated proteins are then usually directed to the plasma membrane tethered through the prenyl anchor. However, in several instances, the prenyl group is not sufficient for stable anchoring, similar to N-myristoylation. Thus, prenylated proteins may also possess a PBR or undergo S-palmitoylation.146 In fewer cases, Ca1a2X-containing proteins circumvent the maturation process and are rather directed to a shunt pathway, thereby retaining their C-terminal Ca1a2X intact that is essential to their subsequent function.147
The third class of S-prenylation usually confers dual geranylgeranylation on two cysteine residues positioned near the C-terminus catalyzed by geranylgeranyltransferase type II (GGTase-II), also conveniently referred to as RabGGTase, since its currently known substrates are members of the Rab family of proteins (Fig. 3B). The Rab protein substrates are initially recruited by the Rab escort protein 1 or 2 (REP1 or REP2) recognized through their C-terminal interacting motif (CIM).148 This pre-formed complex then associates with RabGGTase to install the prenyl modification. While most of these proteins terminate in -CC, -CXC, or CCXX motifs poised for dual cysteine S-prenylation, some of these Rabs terminate with Ca1a2X box motifs that are singly geranylgeranylated and are also subjected to the maturation process described above.149 The dual geranylgeranylation on many of these Rabs might be required for proper localization function as monoprenylated variants of yeast Rab homologs failed to function both in vitro and in vivo.150
All three prenyltransferase enzymes are heterodimers consisting of α and β subunits, with the active site situated at the interface but mainly comprised of residues from the latter (Fig. 3C).151 The β subunits also influence the preference of each enzyme for FPP versus GGPP. Both FTase and GGTase-I share a common α- subunit, FNTA (or prenyltransferase alpha subunit repeat containing 2, PTAR2), but differ in their β-subunits—FNTB and PGGTB, respectively—that manifest 25% sequence identity in mammalian homologs.152 Both farnesylation and geranylgeranylation type I prefer an ordered sequential kinetic mechanism.153–155 The isoprenoid substrate initially binds to the enzyme, followed by the Ca1a2X-containg protein substrate binding and thioether bond formation.153 A second isoprenoid binds to the ternary complex that triggers product release either prior to or simultaneously with the binding of a second Ca1a2X protein substrate (rate determining step). In the case of geranylgeranylation, the GGPP in the GGTase-I active site adopts a bent conformation and the terminal isoprene unit is accommodated in a deeper pocket, which is not present in the corresponding region in the FTase active site.156 This serves as the critical determinant for the isoprenoid specificity of each enzyme. On the other hand, RabGGTase is comprised of RabGGTA (or PTAR3 in some organisms) and RabGGTB as its α and β subunits, respectively. It sequentially adds geranylgeranyl groups to the Rab protein substrate in the Rab:REP complex with the second isoprenoid addition and product release being slow steps.148,157 Although the substrate specificity features of REPs and RabGGTase towards the Rab substrates are not clear, variations in the extent of S-prenylation across these proteins are apparent.158 These observed differences in the extent of S-prenylation should be interpreted with caution as these were conducted under physiologically perturbed conditions, i.e. in the presence of statins. Regardless, further studies are needed to define the substrate specificity of Rab prenylation under native conditions and across cellular systems. These differences are of particular interest as altered levels of S-prenylation of a few key Rab proteins may contribute to diseases such as choroideremia as discussed in later sections.
Being a ubiquitous post-translation modification, protein S-prenylation modulates protein localization and a plethora of protein functions, which can be targeted for therapeutic interventions in diseases and infections where it is implicated.11,159 In particular, inhibition of the farnesylation of oncogenic Ras proteins was initially the main motivation for the development of prenyltransferase inhibitors (PTIs).160 However, candidate PTIs often failed in clinical trials partially owing to the ability of these protein targets to be alternatively prenylated typically by geranylgeranylation in lieu of farnesylation, and none of these potential drugs has been approved for clinical use to date in treating cancer. Despite these limitations, several lines of evidence show that targeting protein S-prenylation remains a promising strategy in treating neurogenerative diseases and progeria, as well as bacterial, protozoal, and viral infections.159,161–163 For example, the farnesyltransferase inhibitor (FTI) lonafarnib has been recently approved by the FDA as the first drug to treat Hutchinson-Gilford progeria syndrome (HGPS).164 Specifically, it inhibits the farnesylation of a genetic variant of nuclear Lamin A that is incapable of the normal Lamin A processing—a mechanism that involves cleavage of a portion of the protein containing the farnesyl modification. Clinical studies showed that patients with HGPS treated with lonafarnib, manifested improved vascular function and bone structure.165 In addition, oral administration of lonafarnib demonstrated success in a Phase II clinical trial of chronic hepatitis D infected patients by suppressing the prenylation of large hepatitis delta antigen (LHDAg) in human delta virus (HDV).166 These results clearly emphasize that targeting specific prenylated proteins offers an avenue for the development of therapeutic agents but requires a clearer understanding of both the specificity and dynamics between PTIs and protein S-prenylation in an actual biological milieu.
Clickable analogues of isoprenoids paved the way for profiling the S-prenylated proteomes in different species and delineating the effects of potential therapeutic agents that target protein S-prenylation. Click chemistry also aided the discovery of a fourth class of prenyltransferase enzyme GGTase-III, which consists of an orphaned protein PTAR1 (α-subunit) and RabGGTB (β-subunit).167 Additionally, the short Ca1a2X sequence has found significant applications in in vitro C-terminal modification of proteins of interest that can be further functionalized with fluorescent reporters or drug cargos. Details on the utility of click chemistry in uncovering the biological significance of S-prenylated proteins, as well as its application in enzymatic protein C-terminal modification are described in this review.
4. Click chemistry in studying the biological significance of protein lipidation
Studies of protein lipid modifications have faced significant challenges. Classical methods to detect and identify lipidated proteins involve radioactive techniques using isotopic analogues including [3H]palmitic acid, [3H]myristate and [I125]myristate.122,168,169 However, these techniques are expensive, labor-intensive, generate radioactive waste and require special permitting, and suffer from low sensitivity that often entails lengthy film exposures to acquire sufficient and quantifiable signals. Proteins bearing canonical sequences for lipidation motifs determined by available prediction tools such as MyrBase170 or PrenBase140 may be validated using this approach. For those with new or undefined motifs or those for proteins with inherently low expression levels, such methods may not be suitable. Mass spectrometry-based approaches for intact and fragmented lipidated proteins may also be exploited to confirm and map the modification sites in individual proteins. Although useful, the hydrophobic character of these lipid PTMs hampers the separation and isolation of proteins and peptides, which often results in lower signal-to-noise intensities and false identification of PTM sites.171
In the past decade, the development of a repertoire of bio-orthogonal, clickable analogues of these hydrophobic molecules has made a major impact in the arena of lipidation science. This chemical toolbox consists primarily of alkyne- and azide-modified lipids that allow conjugation of fluorescent reporters, affinity handles, and drug cargos.3,13,14,18,172,173 For most biological investigations, these chemical probes are typically metabolically incorporated into proteins using cells of interest—from here on referred to as click chemistry-based metabolic labeling (CCML) in this review (Fig. 4A). Within cells, the host machinery converts these probes into bonafide substrates that can be incorporated by host enzymes into the lipid-modified proteins, in lieu of the native lipid substrates. The convenient introduction of the small alkyne or azide tags causes minimal interference on protein processing, subcellular localization, and crucial protein-protein interactions. After a certain period of probe incubation, the labeled cells are fixed and reacted with fluorophores for cellular imaging and flow cytometric analysis, or harvested and lysed, followed by ligation in lysates with the corresponding click reagent partner of the probes used. Conjugation via click reaction with fluorescent dyes allows visualization of tagged proteins typically separated via gel electrophoresis. Immunoprecipitation and antibody-based detection such as western and streptavidin blots can be combined with CCML to verify the PTMs on proteins of interest.
Figure 4. Click chemistry-based metabolic labeling (CCML) for the analysis of protein lipid modifications.
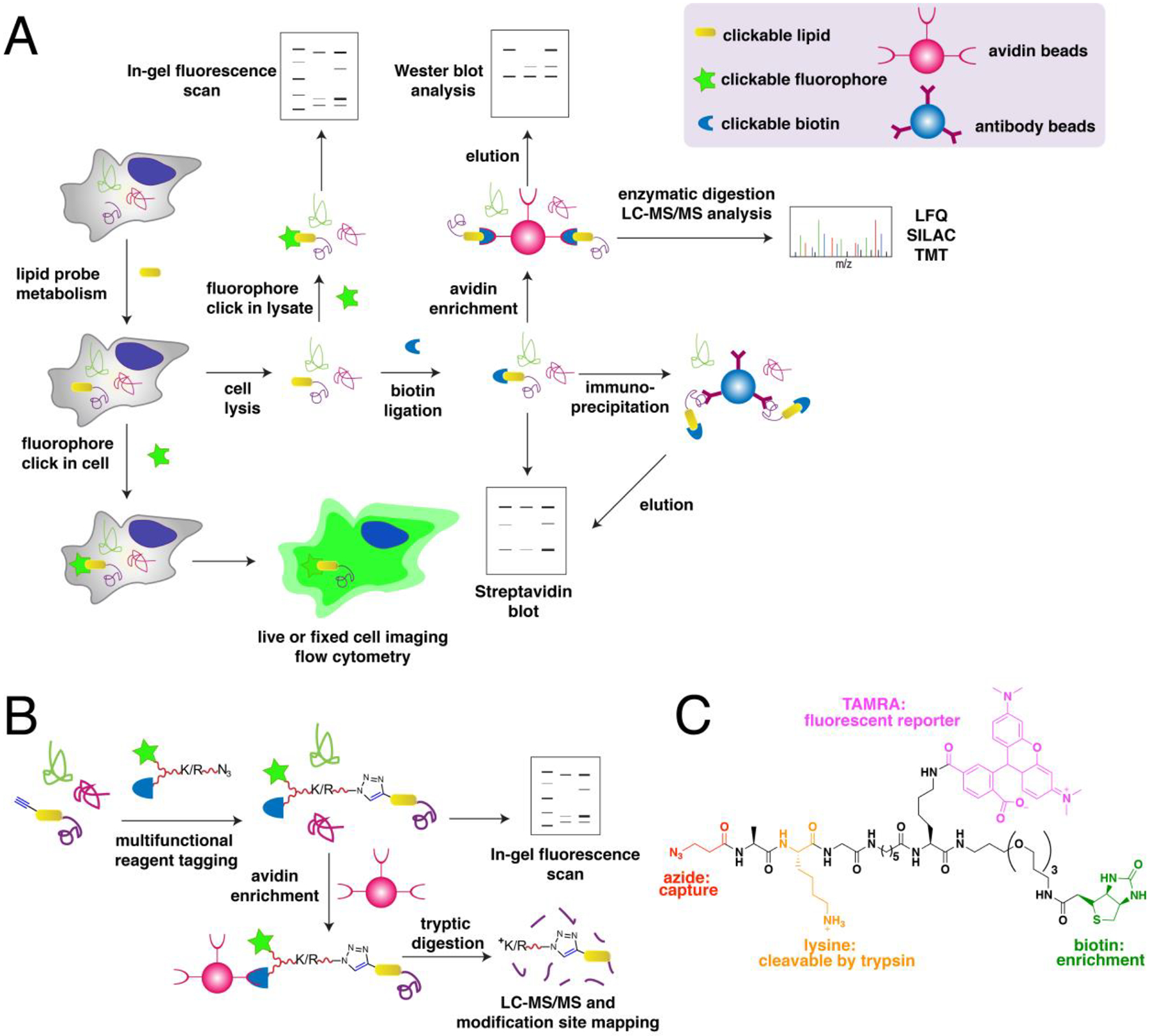
(A) A general scheme for metabolic labeling of cells with clickable analogues of small lipid molecules and subsequent detection and identification. Lysates from cells metabolically labeled with clickable probes are subjected to click reaction with their cognate bio-orthogonal tag linked to a fluorophore for fluorescence imaging or biotin for enrichment and/or streptavidin blotting. Enriched proteins from biotinylated samples can be detected through western blot or identified through label-free or isotopic tag-based quantitative LC-MS/MS proteomic analysis. Metabolic probe labeling and subsequent fluorophore tagging also allows for imaging of whole live or fixed cells and flow cytometric analysis.(B) The use of cleavable multifunctional reagents allows inclusion of the lipid-modified peptide for mapping of the site of lipid modification. Tryptic digestion cleaves the lysine or arginine in the linker and releases the lipidated peptide in solution. (C) An example of a cleavable multifunctional reagent containing a reactive group, fluorescent reporter, biotin handle, and cleavable linker. (Ref. 177)
More importantly, the use of affinity reagents for click reaction with the labeled lysates enables enrichment of the probe-modified proteins. The ability to enrich these labeled proteins is a pivotal step in chemical proteomics, a powerful tool that affords a catalogue of multiple PTM-proteins in a single experiment (Fig. 4A).14,18 In this scheme, the probe-labeled lysates are often ligated with biotin and subjected to protein enrichment using avidin-functionalized resins, which serve to isolate the labeled proteins. The lipid-modified proteins immobilized on the solid support are digested either on-bead or eluted for subsequent enzymatic digestion in solution. The resulting peptides are then chromatographically separated and introduced into a mass spectrometer (MS) for qualitative or label-free quantitative (LFQ) analysis. Alternatively, improved quantitation can be achieved using more sophisticated approaches including Stable Isotope Labeling with Amino acids in Cell culture (SILAC) or Tandem Mass Tag (TMT) labeling.174,175 In SILAC, isotopically labeled amino acids (typically Lys and Arg) are incorporated into all proteins via prolonged cell culture prior to metabolic labeling. These isotopic tags provide distinct mass signatures that permit calculation of fold-changes by computing the ratios of modified peptides from enriched proteins across samples. This approach ensures consistent downstream processing of proteins but is typically performed with two or three samples at a time. In contrast, TMT introduces isotopic tags on the digested peptides after enrichment which offers a rapid labeling strategy, as well as an improved multiplexing scheme (up to 16 samples per experiment). However, inconsistencies in the upstream processes can introduce additional variability among samples and may diminish the accuracy of the results.
Early efforts to identify the site of lipid modifications were impeded by the low ionization of these hydrophobic peptides during MS analysis. For this reason, cleavable multifunctional reagents were designed to contain a fluorescent reporter, a biotin handle, and a cleavable linker (typically basic residues Arg or Lys for trypsin digestion).176,177 This allows fluorescent detection, enrichment, and site identification using a single reagent (Fig. 4B and 4C). The cleavage of the linker releases the modified peptides for inclusion in the LC-MS/MS analysis, with an additional positive charge from Arg or Lys that enhances peptide ionization.
Indeed, the development of an array of chemical probes with small clickable tags has permitted the detection, validation, and large-scale chemical proteomic analysis of lipidated proteins. Accounts in the literature where click chemistry has aided in the discovery of novel regulation of protein function and localization through protein lipidation are reviewed in this section. The technologies based on these clickable lipid probes that provided platforms for rapid evaluation of enzymatic lipidation activities are also included.
5. Biological applications in S-acylation (S-palmitoylation)
Nonradioactive click chemistry-based labeling of fatty acylated proteins generally employs alkyne- or azide-modified analogues of the fatty acid. Although the azido analogues were first developed,178 the alkyne-modified versions are typically preferred owing to enhanced sensitivity and lower background signal compared to azide-modified fatty acids.179,180 Several analogues with various chain lengths have been synthesized for metabolic or in vitro labeling of fatty acylation substrates (Fig. 5).15 These fatty acid probes are converted into their acyl-CoA forms for in vitro experiments. For metabolic labeling studies, the free or saponified forms of the fatty acid analogues are metabolically converted inside cells by acyl-CoA synthetases into the bonafide fatty acyl-CoA substrates.181 Among the fatty acid analogues developed, Alk-C16 (15-HDYA) and Alk-C18 (17-ODYA) are preferentially incorporated by palmitoyl acyl transferases (PATs) onto the S-palmitoylation sites within protein substrates.172,180,182 Similar to their native counterparts, these probes can also undergo β-oxidation at the C2 and C3 positions to yield α,β-unsaturated esters that may result in aberrant palmitoyl-protein adducts via Michael addition. To address this issue, a different version of the probe modified at C3 (15-HDYOA, Fig. 5) was explored and found to largely label the same proteins (70%) identified with Alk-C18.183 The disparity in the sets of proteins identified between treatments suggests that a fraction of the Alk-C18-labeled proteins may be a result of adduct formation with the oxidized probe. Regardless, Alk-C16 and Alk-C18 have been widely used in click chemistry-based S-acylation studies reported up to the present. While Alk-C16 better mimics a palmitoyl moiety and Alk-C18 more likely resembles a stearoyl group in terms of chain length, both of these probes have been interchangeably used in investigations of protein S-palmitoylation. The preference for the incorporation of each analogue is dictated by the specific protein substrates and/or fatty acyltransferase.184,185 Furthermore, these probes are not only metabolized to label fatty acylated proteins, but they can also be incorporated into biosynthetically more complex phospholipids and neutral lipids.186,187
Figure 5. Structures of clickable probes for protein S-palmitoylation.
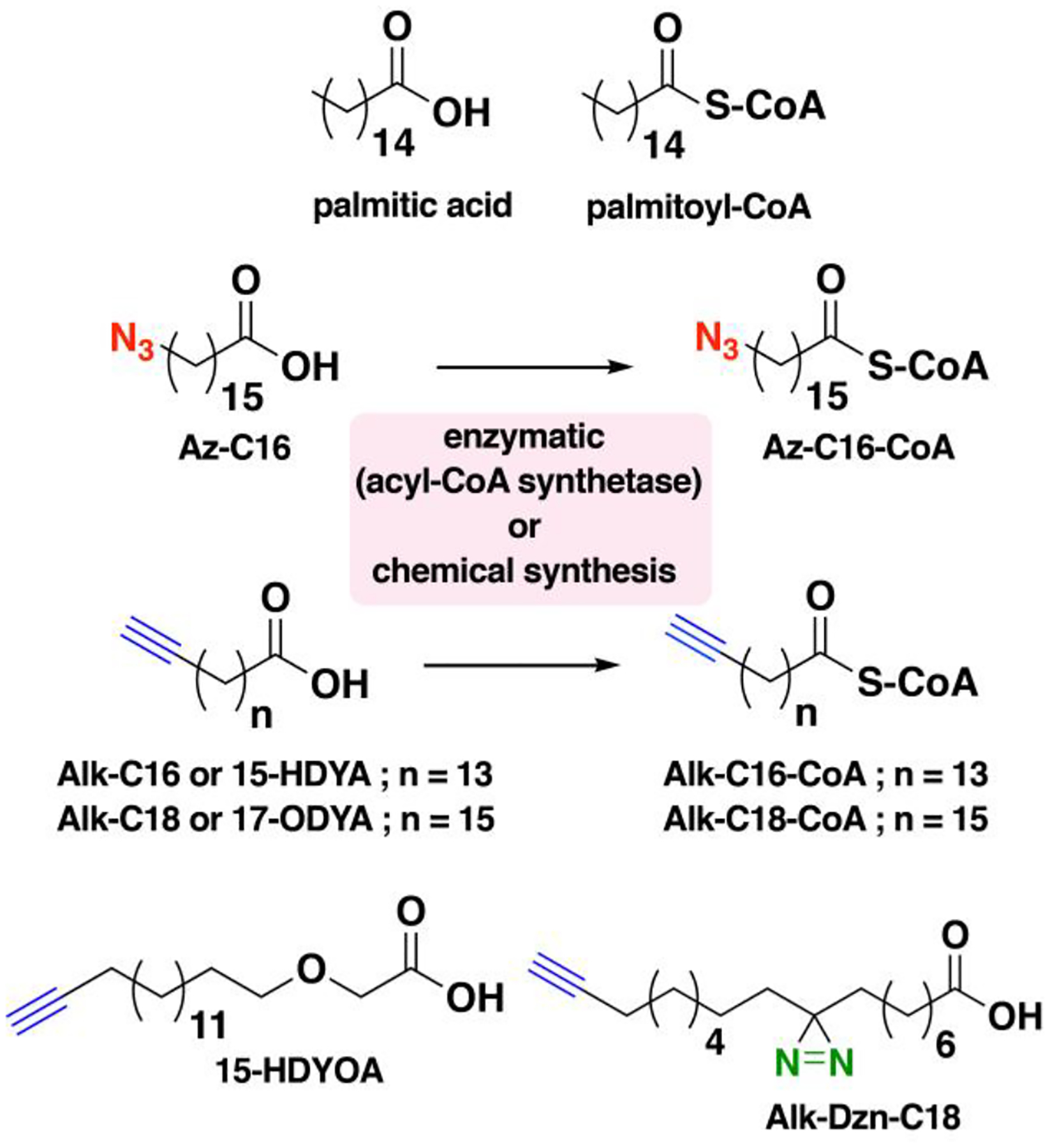
The native structures of palmitic acid and palmitoyl-CoA along with their azide- and alkyne-modified analogues are shown. Varying chain lengths can serve as surrogate for this lipid modification and hence can be generalized as S-acylation. The presence of a diazirine moiety in Alk-Dzn-C18 allows for a photocrosslinking strategy that enables identification of proteins that interact with fatty acylated proteins through the lipid modification.
The use of bio-orthogonal analogues of fatty acids for click chemistry tremendously impacted and revolutionized the field of protein S-palmitoylation.15,17,173,188 These tools permitted rapid and robust fluorescence-based detection and large-scale proteomic analysis of fatty acylated proteins, thereby facilitating the discovery of novel S-acylated proteins and contributing to the understanding of S-palmitoylation dynamics. The development of a bifunctional fatty acid reporter with both clickable and photoaffinity tags (Alk-Dzn-C16) enabled the discovery of new membrane protein-protein interactions as well.189–191 In this section, several strategies using clickable fatty acid analogues for proteomic profiling of S-palmitoylated proteins, as well as their utility in biochemical assays and enabling platforms to validate such lipid modification and screen for potent PAT inhibitors are described. It is important to note that in this review, the term S-palmitoylated proteins is used according to the claims made by the following studies in the literature, although this type of modification is more precisely described as protein S-acylation.
5.1. Proteomic studies for profiling S-palmitoylated proteins
Advances in proteomics strategies to identify S-acylated proteins have contributed to unveiling regulatory mechanisms and critical functions of protein S-palmitoylation. Early proteomic methods to detect and identify S-acylated proteins took advantage of the lability of thioesters, via procedures described as acyl-biotin exchange (ABE) or acyl-resin-assisted capture (acyl-RAC).192,193 In these multistep procedures, free cysteines in protein lysates are blocked with N-ethylmaleimide (NEM) and subsequently treated with hydroxylamine to selectively cleave the thioesters (Fig. 6A). S-acylated proteins are enriched by either biotinylation with a thiol-reactive biotin (biotin-HPDP) and streptavidin pulldown for ABE or immobilization using a thiol-reactive resin for acyl-RAC. Although these typically provide extensive lists of identified proteins, both methods are prone to false positives since neither are exclusive to S-acylated proteins and are not suitable for dynamic profiling studies. CCML with fatty acid analogues incorporating small azide or alkyne tags allows for more specific labeling and enrichment of S-acylated proteins (Fig. 1A). Employing such a strategy afforded several lists of S-acylated proteins from multiple research groups obtained from different cellular systems and provided insights into the dynamic nature of protein S-palmitoylation. These catalogues of S-acylated proteins have been consolidated into a comprehensive database, SwissPalm©, which is accessible online for free.194
Figure 6. Strategies for profiling of S-acylated proteins.
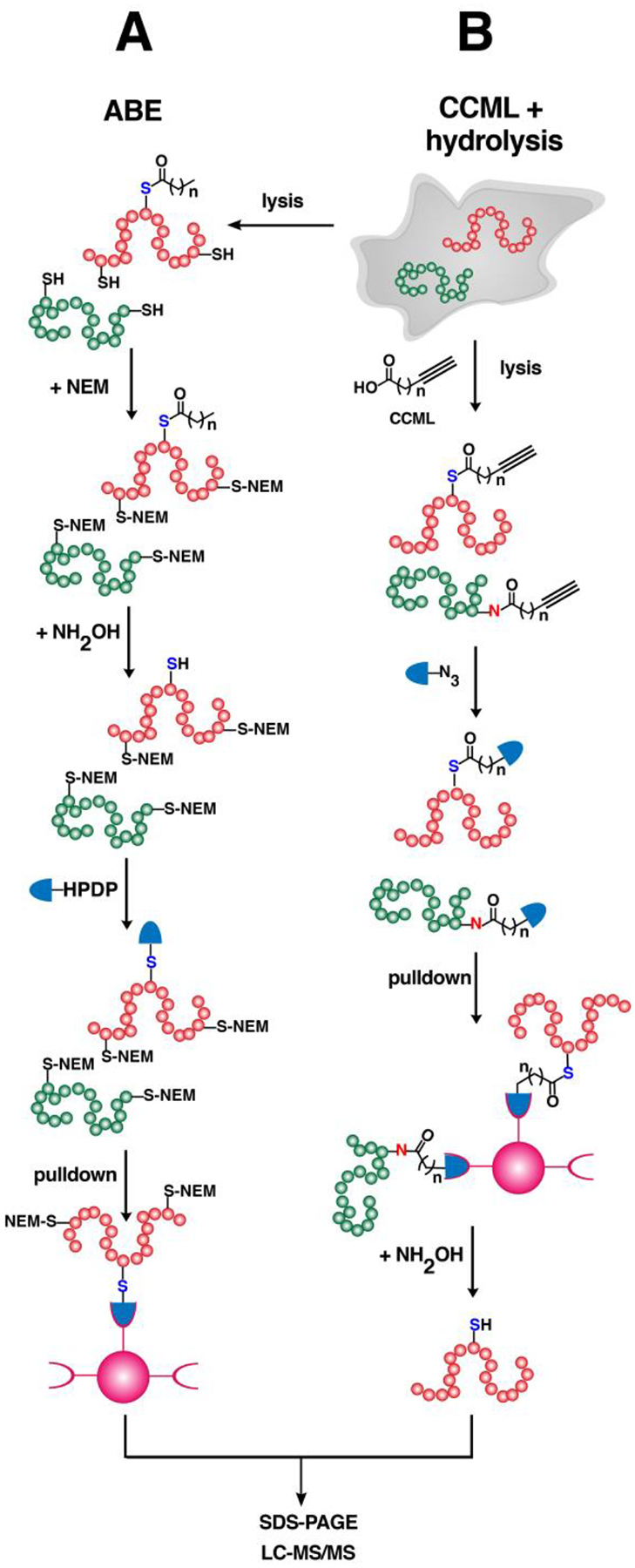
(A) An acyl-biotin exchange (ABE) method that does not require metabolic labeling with clickable analogues. Lysates are treated with N-ethyl maleimide to block free cysteines followed by hydrolysis using NH2OH to expose the thiols of S-acylated proteins. The newly unmasked free cysteines are reacted with thiol-reactive reagents such as biotin-HPDP (see Abbreviations section) and enriched. Immobilized proteins are eluted for SDS-PAGE or processed for proteomic analysis. (B) Cells can be metabolically labeled with clickable fatty acid analogues followed by click reaction with biotin-based reagents for enrichment of labeled proteins. Immobilized proteins that are probe-labeled are selectively released from the beads through hydrolysis with NH2OH, improving the confidence of the identities of the profiled S-acylated proteins.
5.1.1. S-acylated (S-palmitoylated) proteins in mammals
The tagging strategy for S-palmitoylated proteins using biorthogonal analogues combined with mass spectrometric analysis facilitated the identification of multiple S-palmitoylated proteins in a single experiment. The first report using bio-orthogonal labeling for proteomic identification of S-palmitoylated proteins in vitro used Az-C16-CoA (Fig. 5), an azide analogue of palmitate-CoA, ligated with a phosphine-biotin reagent via Staudinger ligation.195 A total of 21 putative S-palmitoylation substrates were identified from lysates of rat liver mitochondria matrix, which included 19 novel S-palmitoylated proteins. Following this chemical proteomic approach, large-scale proteomic profiling of S-palmitoylated proteins relied on metabolic labeling with palmitic acid analogues introduced into cultured cells of interest. The commercially available Alk-C18 or 17-ODYA (Fig. 5) has been the probe of choice owing to its better specificity and minimal background in tagging S-acylated proteins amenable to labeling with shorter fatty acyl chains.179 This CCML strategy combined with proteomic analysis affords identification of hundreds of S-palmitoylated proteins involved in multiple cellular mechanisms and processes. Cravatt and coworkers first reported the metabolic labeling of Alk-C18 in mammalian cells followed by click reaction with biotin-azide for CuAAC and subsequent enrichment.196 A total of 125 candidate S-palmitoylated proteins were identified at high confidence including G proteins, receptors, and Fam108 serine hydrolases.
Similarly, Hang and coworkers identified 178 S-acylated proteins at high confidence from Jurkat cells using alkyne-modified fatty acyl probes of various chain lengths (myristic, palmitic and stearic acid analogues).197 This list of proteins contained less than 30% overlap with a previous study performed by Cravatt and coworkers. The differences in the identities of profiled S-acylated proteins may be attributed to variations in identity of the probes and their incubation periods, proteomic strategies (gel-based vs MudPIT) and subtle differences in MS analysis.197 Moreover, other proteomic studies reported by the Hang and coworkers in dendritic cell lines profiled more than 150 S-palmitoylated proteins, which included those involved in innate immunity.182,198 Another group analyzed the differential S-palmitoylation of proteins in macrophages upon treatment with bacterial surface lipopolysaccharides (LPS), a recapitulation of the initial engagement of bacteria that activates a cascade of cellular events inside the host.199 A total of 154 and 186 upregulated and downregulated S-palmitoylated proteins, respectively, responded throughout the immune stimulation. In particular, LPS activates S-palmitoylation on phosphatidylinositol kinase II (PI4KII) that results in increased levels of the phosphorylated forms of phosphatidylinositol, leading to enhanced production of cytokines that support inflammatory responses.
S-palmitoylated proteome profiling has also provided a tool to hunt for biomarkers of diseases. For example, androgen-dependent malignant tumors in prostate cancer were investigated for potential S-palmitoylated protein biomarkers using CCML with Alk-C18 in a human prostate adenocarcinoma cell line.200 By comparing the set of S-palmitoylated proteins in the presence or absence of androgen, the eIF3L subunit of the initiation factor eIF3 exhibited a remarkable extent of enrichment. Further assays confirmed the androgen-induced S-palmitoylation of eIF3L that may be implicated in cancer progression and may also serve as a novel prostate cancer biomarker.
5.1.2. S-acylated proteins in lower class organisms and viruses
Protein S-palmitoylation is a ubiquitous PTM across kingdoms and can be a key regulator for the pathogenicity of lower organisms. Bacterial lipoproteins (LPPs) are tethered to membranes often by diacylglyceryl moieties modified with fatty acids.201 This modification is essential for proper LPP localization and function for the virulence of pathogenic bacteria, as well as for the recognition of host cell receptors. Large-scale profiling in the Gram-negative Escherichia coli using Alk-C16 identified more than 90 high- and medium-confidence LPPs involved in diverse biological processes.202 However, half of these proteins are not annotated and therefore these studies should stimulate further investigation into the role of lipidation in the biological functions of these LPPs.
S-palmitoylation also plays key roles in the development and virulence of protozoans and fungi. Using Alk-C18 for labeling S-palmitoylated proteins and identification in the asexual stage of the causal agent of malaria, Plasmodium falciparum, resulted in 176 statistically enriched S-palmitoylated proteins essential for cytoadherence, drug resistance, and host-cell invasion mechanisms, among others.203 Analysis of the S-palmitoylated proteome in a related parasite, Toxoplasma gondii, identified 501 proteins, revealing the essential role of S-palmitoylation in all stages of its life cycle.204 Furthermore, it was shown that blocking the S-palmitoylation of apical membrane antigen 1 (TgAMA1) can trigger its release along with other proteins implicated in host-cell invasion. This also highlights the parasite-specific nature of S-palmitoylation; TgAMA1 is S-palmitoylated while the P. falciparum homolog PfAMA1 is not.203 In Cryptococcus neoformans, an opportunistic fungus that causes lethal meningitis, Alk-C18 labeling identified 72 S-palmitoylation substrates essential for fungal integrity and virulence.205 S-palmitoylation in this pathogen is catalyzed by a single PAT, Pfa4, and its deletion causes morphological defects and virulence attenuation. Developing an inhibitor of S-palmitoylation in C. neoformans could be of great advantage since specificity can be better achieved for a single PAT compared to mammalian zDHHCs, which have functional redundancies and broader substrate scope to which the current inhibitors developed lack specificity.
Viral proteins may also require palmitoylation for their function. In epithelial cells infected with herpes simplex virus (HSV), click chemistry-based S-palmitoylated proteome analysis using Alk-C17 afforded a novel set of virus-encoded proteins S-palmitoylated by the host machinery.206 Selective repression of the global fatty acylation of host proteins was also observed, suggesting that the virus hijacks the S-palmitoylation pathway to promote its virulence. HIV-1 infection was also found to alter cellular acyltransferase activity.207 As a consequence, differential modification of proteins such as phosphatases lead to altered cellular phosphorylation levels, potentially favoring viral pathogenicity. Identifying the key zDHHCs implicated in the virulence of these viruses is essential to delineating which of these enzymes could serve as potential therapeutic targets to treat infections caused by these viral pathogens.
5.2. Quantitative proteomics unravels S-palmitoylation dynamics
Combining CCML with palmitic acid analogues and quantitative proteomic approaches such as SILAC and TMT not only provides robust and accurate quantitation of S-palmitoylated protein levels but also enables S-palmitoylated proteome-wide analysis of dynamic S-palmitoylation events.203,206 In a study reported by Cravatt and coworkers, a pulse-chase technique combined with SILAC in mouse hybridoma T-cells was employed to uncover the role of dynamic cycling controlled by depalmitoylases.208 Alk-C18 was added to the cells (pulse) and subsequently competed by the addition of natural palmitic acid at different time points (chase), showing a decrease in total S-palmitoylation with time. Through the addition of a serine lipase (depalmitoylase) inhibitor hexadecylfluorophosphonate (HDFP), stably S-palmitoylated proteins were distinguished from those that undergo rapid cycling, allowing the important regulatory mechanism of depalmitoylation in dynamic S-palmitoylation to be examined. The phosphonofluoridate warhead is particularly reactive towards the hydroxyl group of the catalytic serine in serine hydrolases as evidenced by other proteomic studies.209,210 A recent study combining CCML with TMT in mouse endothelial cells with an APT1 (depalmitoylase) deletion identified proteins involved in focal adhesion potentially regulated by depalmitoylation.211 Pulse-chase analysis on the candidate RRas revealed the importance of dynamic regulation by APT1 on its role in vascular function. Despite its utility in profiling a number of dynamically modified proteins, the pulse-chase technique is often compromised by low sensitivity since membrane-localized labeled proteins are often stable with slow turnover rates104 and the methods employed may require additional optimization (e.g., appropriate detergents) for improved detection. Furthermore, fatty acyl analogues are widely incorporated across various phospholipid species and may result in reduced labeling of the actual fatty acylation substrates.186
As an alternative to the pulse-chase method, a temporal profiling strategy of CCML combined with TMT-based multiplexing was used to streamline the evaluation of S-palmitoylation kinetics.104 Cells were treated with Alk-C18 or natural palmitic acid at various time points, multiplexed using TMT tags, and analyzed as a single experiment in a highly accurate multinotch SPS-MS3 LC-MS approach. This revealed conserved S-palmitoylation kinetic profiles in different cell lines and that the previously observed effect of HDFP only represents a small fraction of the bulk S-palmitoylated form of various proteins. An orthogonal proteomic analysis using acyl-RAC revealed that inhibition of depalmitoylation with HDFP affects the rate of S-palmitoylation but steady-state S-palmitoylation levels remain unchanged.
5.3. Combining CCML with hydrolysis-based methods for robust profiling
Results from CCML labeling for S-palmitoylated proteome analysis have similarities and differences when compared with biotin-switch approaches (ABE and acyl-RAC). Initial reports on the direct quantitative comparison between Alk-C18 labeling and ABE showed large complementarity in the identities of the S-palmitoylated proteins profiled.203 In Trypanosoma brucei, analysis of the S-palmitoylated proteins using CCML identified more than 100 potentially S-palmitoylated proteins, 70 of which were not found in a previous report that employed the ABE method.212,213 While both approaches may be useful, the differences in the life stages of the parasite studied in each method may contribute to the observed variations in the S-palmitoylated proteome. ABE was also used to validate proteins with altered S-palmitoylation levels identified through Alk-C18 labeling in breast cancer cells after Snail-induction, an event associated with chemoresistance and metastasis.214 While some proteins showed consistent enrichment levels with metabolic labeling, other candidate proteins were not detected using ABE potentially due to their lack of a hydroxylamine-dependent linkage (such as an amide or ester modifications) or high background enrichment. While it was thought that these methods should give similar results, a more recent study found that hydroxylamine-based switch methods are significantly more sensitive than metabolic labeling, with only 10% overlap of profiled S-palmitoylated proteomes.104 One of the disadvantages of using CCML is that stably S-palmitoylated proteins containing endogenously produced palmitate are unresponsive, and therefore clickable analogues only label those proteins that reveal free cysteines available for labeling during the course of probe treatment. It was therefore suggested that this disparity between methods warrants reevaluation of mass spectrometry-based S-palmitoylated proteome analyses reported in the literature.
Selective release of labeled proteins is desirable for robust identification of S-palmitoylated proteins. Improved detection of lipidated proteins labeled with alkyne-modified analogues of fatty acids can be achieved using a biotin-azide reagent containing a linker such as azobenzene cleavable by sodium dithionite.197,202 However, this may not discriminate S-palmitoylated proteins from those that are irreversibly N- or O-palmitoylated with the bio-orthogonal probes.184,215 Combining the principles of CCML and acyl-switch methods provides a robust method for the delineation of S- versus O- and N-palmitoylated proteins. In this method, cells are metabolically labeled with Alk-C18 (or Alk-C16), clicked with biotin-N3 and enriched on beads (Fig. 6B). Treatment with NH2OH cleaves thioester linkages and selectively releases S-palmitoylated proteins from the resin. Employing such method afforded a robust list of 282 NH2OH-sensitive S-palmitoylated proteins out of the total 501 labeled with Alk-C18 in T. gondii.204 Extending this approach to Raw264.7 murine macrophages identified <150 NH2OH-sensitive proteins out of > 400 total Alk-C18-labeled proteins.216 Furthermore, this provides a more accurate determination of S-palmitoylated sites. Comparison with the acyl-RAC approach revealed overlapping but distinct sets of protein substrates.216 Since acyl-RAC captures other thioester-linked proteins including ubiquitin processing enzymes, the Alk-C18 labeling combined with NH2OH treatment is more specific for S-palmitoylated proteins.
Indeed, chemical proteomics presents a powerful tool not only to rapidly identify multiple fatty acylated proteins in a single experiment but also to delineate dynamics of protein S-palmitoylation. It is noteworthy that optimization of click chemistry conditions is critical to maximize the labeling and number of identified S-palmitoylated proteins in these types of proteomic approaches.216–218 The choice of buffer and the amount of detergent for solubilization may significantly impact the extent of labeling particularly for proteome-wide analyses. Details on the click chemistry conditions and the buffer systems used in several studies are summarized in Table 1. In gel-based analyses, sample preparation must be carefully designed since these reporter tags are labile to hydrolysis. Inhibitors of depalmitoylases may be included in the lysis buffer to block depalmitoylation activities and retain the incorporated probes onto proteins.208,214 The use of dithiothreitol (DTT) and sample heating may hydrolyze thioester linkages, leading to decreased detection of labeled proteins.219 Furthermore, MS-compatible detergents are recommended to avoid S- to N-palmitoyl transfer, which may complicate the MS data analysis in discriminating S- vs N-palmitoylated proteins or peptides.220
Table 1.
Click chemistry conditions from studies using clickable analogues of lipids for biological investigations.
| probe | cell line/organism | applicationa | validated protein | click reaction conditionsb | buffer | proteins identifiedc | ref |
|---|---|---|---|---|---|---|---|
| S-Palmitoylation/depalmitoylation | |||||||
| Alk-C16 (100 μM), Alk-C18 (100 μM) | Raw 264.7 (macrophages) | imaging | 0.1 mM biotin-N3 or rhodamine- N3, 1 mM TCEP, 0.2 mM TBTA, 1 mM CuSO4 | PBS | 180 | ||
| Az-C16-CoA (100 μM) | rat liver matrix (in vitro) | GBP | 0.2 mM phosphine-biotin | HEPES + 1% SDS | 21 | 195 | |
| Alk-C18 (25 μM) | Jurkat (T cells) | EP, V | FAM108 family | 0.1 mM biotin-N3* | PBS | > 325 (125 HC) | 196 |
| Az-C12, Alk-C14, Alk-C16, Az-C15, Alk-C18 (100 μM) | Jurkat | EP | histones H3.1, H3.2, H3.3 | 0.1 mM biotin-N3 or rhodamine- N3* | 50 mM TEA (pH 7.4) + 4% SDS | 361 (178 HC) | 197 |
| Alk-C18 (50 μM) | DC2.4 (dendritic cells) | EP, V | IFITM3, TLR2 | 0.1 mM biotin-N3 or rhodamine- N3* | 0.1 mM TEA + 1% Brij-97, PBS + 1% Brij-97 | 157, 247 | 182,198 |
| Alk-C18 (50 μM) | Raw 264.7 | EP, V | PI4KII | 0.1 mM biotin-N3 or 10 μM IRDye 800CW- N3* | 50 mM TEA (pH 7.4) + 4% SDS | 646 (167 HC) | 199 |
| Alk-C18 (100 μM) | LNCaP (prostate adenocarcinoma) | EP, V | eIF3L | 1 mM azide-agarose beads, 1 mM TCEP, 0.2 mM TBTA, 1 mM CuSO4 | PBS (pH 7.5) + 1% Nonidet | 835 | 200 |
| Alk-C16 (20 μM) | E.coli | EP | 0.1 mM biotin-N3 or rhodamine- N3* | 50 mM TEA + 2% SDS | > 90 | 202 | |
| Alk-C18 (25 μM) | P. falciparum | EP (SILAC), V | GAP45, MTIP | 0.1 mM biotin-N3* | PBS + 2% SDS | > 400 (176 HC) | 203 |
| Alk-C18 (25 μM) | T. gondii | EP, V | MLC1, MyoA, PhIL1, AMA1 | 20 μM rhodamine-N3* | PBS | 501 (282 HC) | 204 |
| Alk-C18 | C. neoformans | EP, V | Chs3 | 0.2 mM biotin-N3, 1 mM TCEP, 0.5 mM TBTA, 1 mM CuSO4 | 50 mM TEA (pH 7.4) + 4% SDS | 427 (72 HC) | 205 |
| Alk-C17 (25 μM) | HSV-infected RPE-1 | EP (SILAC) | 0.1 mM AzTB* | 100 mM Tris-HCl (pH 7.6) + 4% SDS | 1292 | 206 | |
| Alk-C18 (20 μM) | BW5147 (T-cell hybridoma) | EP (SILAC) | 0.5 mM biotin-N3* | PBS | 338 (118 HC) | 208 | |
| Alk-C18 (20 μM) | HEK 293T (kidney) | EP (TMT) | 0.5 mM biotin-N3* | DPBS | 195 | 104 | |
| Alk-C17 (100 μM) | T. brucei | EP, inhibition | 0.1 mM AzTB* | PBS + 1% SDS | 134 | 212 | |
| Alk-C18 (50 μM) | Raw 264.7 | EP, V | CANX, RAS, IFITM3 | 0.1 mM biotin-N3* | 50 mM HEPES (pH 7.4) + 0.25% SDS | > 400 (103 HC) | 216 |
| Alk-C18 (50 μM) | MC3T3 (osteoblasts) | V | IFITM5 | 0.1 mM TAMRA- N3* | 227 | ||
| Alk-C18 (100 μM) | HEK 293T | V | DLK | 10 μM IRDye 800CW- N3, 5 mM ascorbate, 5 mM THPTA, 1 mM CuSO4 | PBS | 240 | |
| Alk-C18 | hTLR4–293T (kidney) | V | MYD88 | 20 μM biotin-N3* | 5 mM HEPES (pH 7.4) + 1% Triton X-100 | 241 | |
| Alk-C18 (25 μM) | HEK 293 | V | Nav1.5 | IRDye 680RD- N3* | 20 mM HEPES + 1% Triton X-100 | 244 | |
| Alk-C18 (50 μM) | S. typhimurium-infected HEK 293 | V | SspH2, SseI | 0.1 mM rhodamine- N3* | RIPA | 229 | |
| Alk-C18 (10 μM) | Schizosaccharomyces pombe (yeast) | V | Rho3 GTPase | 0.1 mM rhodamine- N3* | 50 mM TEA (pH 7.4) + 1% Brij-97 | 231 | |
| Alk-C16 (50 μM) | HEK 293T | V | JAM-C | 0.15 mM BODIPY- N3, 2.5 mM TCEP, 0.3 mM TBTA, 2.5 mM CuSO4 | 50 mM Tris-HCl (pH 7.4) + 1% Nonidet | 234 | |
| Alk-C18 (50 μM) | MDCK (kidney) | V | PMP22 | 0.25 mM biotin-N3, 1 mM TCEP, 0.25 mM TBTA, 1 mM CuSO4 | 50 mM HEPES (pH 7) + 2% SDS | 245 | |
| Alk-C18 (100 μM) | HEK 293T | V | TEAD | 0.1 mM biotin-N3, 1 mM TCEP, 0.2 mM TBTA, 1 mM CuSO4 | PBS + 0.1% Tween-20 | 249 | |
| Alk-C16 (100 μM) | COS-7 (kidney) or myocytes | V, imaging (Palm-PLA) | JPH-2 | 1 mM biotin-PEG3- N3, 1 mM ascorbate, 1 mM TCEP, 0.1 mM TBTA, 1 mM CuSO4 | PBS | 252 | |
| Alk-C18 (25 μM) | LpdA-transfected HeLa | V | LpdA | 0.1 mM AzTB* | PBS + 1% Triton X-100, 0.1% SDS | 255 | |
| Alk-C16 (100 μM) or Alk-C18 (100 μM) | HEK 293 | V | ASBT | 50 μM biotin-PEG3- N3, 1.5 mM ascorbate, 0.5 mM THPTA, 0.1 mM CuSO4 | 50 mM HEPES, 0.1% Triton X-100, 1% SDS | 257 | |
| Alk-C18 (100 μM) | U937 (myeloid leukemia) | V | TNF-R1 | 0.5mM biotin-N3, 4 mM ascorbate, 0.2 mM TBTA, 2 mM CuSO4 | 25 mM HEPES (pH 7.4) + 0.5% Triton X-100 | 266 | |
| Alk-C16 (100 μM) | L. donovani | imaging, flow cytometry | 0.1 mM Oregon Green 488- N3, 1 mM TCEP, 1 mM CuSO4 | water | 274 | ||
| Alk-C18 (100 μM) | P. falciparum | imaging | 0.1 mM Oregon Green 488- N3, 1 mM TCEP, 1 mM CuSO4 | PBS | 275 | ||
| Alk-C16 (50 μM) | cortical neurons | imaging | 0.1 mM Oregon Green 488- N3, 1 mM TCEP, 1 mM CuSO4 | PBS | 277 | ||
| Alk-C18 (100 μM) | HEK 293T | ratiometric quantification | STX-19 | 2.5 μM IR-800- N3, 4 mM ascorbate, 0.2 mM TBTA, 2 mM CuSO4 | 50 mM Tris-HCl (pH 8.0) + 0.5% SDS | 236 | |
| Alk-C16 (100 μM) | L cells (fibroblast) | imaging (Palm-PLA) | Wnt3a | 0.1 mM biotin- N3, 1 mM TCEP, 1 mM CuSO4 | PBS | 184 | |
| Alk-C17 (20 μM) | HEK 293 | inhibitor screen | Shh | 0.1 mM AzTB* | PBS + 1% Triton X-100, 0.1% SDS | 286 | |
| Alk-C16 (100 μM) | E.coli | inhibitor screen | P. falciparum DHHCs | 0.1 mM Oregon Green 488- N3, 1 mM TCEP, 1 mM CuSO4 | PBS | 291 | |
| Alk-cer (10 μM) | 501Mel (melanoma) | ABPP | 0.1 mM biotin-N3* | PBST | >200 | 295 | |
| Az-2-BP (50 μM) | HEK 293T | ABPP | 20 μM biotin-N3* | PBS | 450 | 294 | |
| Alk-HDFP (5 μM) | BW5147 | ABPP | PPT1 | 0.5 mM biotin-N3* | PBS | 50 (21 HC) | 208 |
| Alk-C18-CoA (15 μM) | inhibitor screen | 45 μM CalFluor 488- N3, 10 mM ascorbate, 0.5 mM THPTA, 0.1 mM CuSO4 | 100 mM MES (pH 7.2) + 0.2% Triton X | 296 | |||
| Alk-C17-CoA (1 μM) | inhibitor screen | 10 μM FLAG- N3, 1 mM TCEP, 1 mM TBTA, 1 mM CuSO4 | PBST + 1% BSA | 297,298 | |||
| Alk-C16-CoA (40 μM) | inhibitor screen | 20 μM biotin-N3, 1 mM TCEP, 1 mM TBTA, 1 mM CuSO4 | 50 mM NaH2PO4 (pH 6.5) + 1% Triton X-100 | 301 | |||
| Az-C16 (100 μM) | U2OS (osteosarcoma) | V | Click-IT® pre-mix | Click-IT® buffer | 269 | ||
| Az-C16 (100 μM) | HEK 293T | mPEG-click | SNAP25 | 0.2 mM mPEG-5k-alkyne, 8 mM ascorbate, 0.4 mM TBTA, 4 mM CuSO4 | 25 mM Tris (pH 8.0) + 0.25% SDS | 302 | |
| N- Myristoylation/defatty-acylation | |||||||
| Alk-C14 (50 μM) | P. falciparum | EP, V | GAP45, MTIP, CDPK1, ARO | 0.1 mM AzKTB* | 10 mM Na2PO4 (pH 8.2) + 1% Triton X-100 | > 30 HC | 177 |
| Alk-C14 (20 μM) | HeLa | EP, V | FLOT2, MARC2, HCCS | 0.1 mM AzTB* | PBS + 1% Triton X-100, 0.1% SDS | 108 HC | 315 |
| Alk-C14 (20 μM) | HeLa, HEK 293T, MCF7 | EP | 0.1 mM AzKTB* | PBS + 1% Triton X-100, 0.1% SDS | 69, 65, 50 (all HC) | 176 | |
| Alk-C16 (50 μM) | mouse embryonic fibroblasts | EP (SILAC), V | RRAS2 | 0.1 mM biotin-PEG3- N3, 1 mM TCEP, 0.5 mM TBTA, 1 mM CuSO4 | 25 mM Tris-HCl (pH 7.4) + 1% Nonidet, 10% glycerol | > 800 (5 HC) | 124 |
| Alk-C16 (50 μM) | HAP 1 (haploid leukemia) | EP (SILAC), V | SHMT2 | 0.1 mM biotin-N3, 0.5 mM TCEP, 40 μM TBTA, 1 mM CuSO4 | 50 mM TEA (pH 7.4) + 4% (w/v) SD | 80 (2 HC) | 130 |
| Alk-C14 (100 μM) | HEK 293T | V | ARF6 | 80 μM TAMRA- N3, 1.6 mM TCEP, 0.4 mM TBTA, 1.6 mM CuSO4 | 25 mM Tris-HCl (pH 7.4) + 0.2% Nonidet | 126 | |
| Alk-C14 (50 μM) | P. berghei | V | ISP1, ISP3 | 0.1 mM AzTB* | 10 mM Na2PO4 (pH 8.2) + 1% Triton X-100 | 344 | |
| Alk-C14 (100 μM) | T. brucei | V | ARL6 | 50 μM AzTB, 0.5 mM TCEP, 50 μM TBTA, 0.5 mM CuSO4 | PBS | 346 | |
| Az-C12 (50 μM) | T. cruzi | EP (SILAC) | Click-IT® pre-mix | Click-IT® buffer | 56 | 349 | |
| Alk-C14 (50 μM) | L. donovani | EP, V | HASPB | 0.1 mM AzTB* | PBS + 1% SDS | 30 HC | 350 |
| Alk-C14 (25 μM) | T. gondii | EP, V | CDPK1, MIC7 | 0.1 mM capture reagent* | PBS + 1% Triton X-100, 0.1% SDS | 76 HC | 354 |
| Alk-C14 (30 μM) | Z. tritici | EP, inhibition | 0.1 mM AzRB* | PBS + 0.2% SDS | 20 HC | 361 | |
| Alk-C14 (20 μM) | S. flexneri-infected HeLa | EP, V | ARF1 | 0.2 mM biotin-N3, 1 mM TCEP, 0.2 mM TBTA, 1 mM CuSO4 | 50 mM TEA (pH 7.4) + 4% SDS | 34 HC | 368 |
| Alk-C14 (20 μM) | Rhinovirus-infected HeLa | EP, V | RV-A16 | 0.1 mM AzTB* | PBS + 1% Triton X-100, 0.1% SDS | 376 | |
| Az-C12 (20 μM) | Jurkat, HEK 293T | V | Lck | 0.2 mM phosphine-biotin | 50 mM Tris (pH 7.4) + 0.5% NP-40 | 178 | |
| Az-C12 | COS-7 | V | DCNL3 | 0.25 mM phosphine-biotin | 100 mM NaH2PO4 (pH 7.2) + 1% SDS | 323 | |
| Az-C12 (40 μM) | HASMC, MASMC (smooth muscles) | V | LMCD1 | 0.25 mM phosphine-biotin | 100 mM NaH2PO4 (pH 7.2) + 1% SDS | 324 | |
| Az-C12-CoA (10 – 200 μM) | V, inhibitor screen | Lck, Nef, Arf | 12.5 μM phosphine-biotin | 12.5 mM Tris (pH 7.2) + 0.05% BSA, 0.025% Tween | 310 | ||
| Alk-C14 (50 μM) | HaCaT (keratinocyte) | V | PPM1A | 0.1 mM AzTB* | PBS + 1% SDS | 332 | |
| Az-C12 (40 μM) | zebrafish | imaging | 1 μM AlexaFluor 488-alkyne, 0.5 mM TCEP, 0.2 mM THPTA, 0.5 mM CuSO4 | 284 | |||
| S-Prenylation | |||||||
| C15-Az-OH (20 μM) | COS-1 (kidney) | EP | 1 mM phosphine-biotin | PBS + 2% SDS | 17 | 394 | |
| C10-Alk-OH (50 μM) | HeLa | GBP | 25 μM TAMRA- N3* | PBS + 0.2% SDS | 6 | 405 | |
| C15-Alk-OH (50 μM) | Raw 264.7 | EP, V | ZAP | biotin-N3 * | 50 mM TEA (pH 7.4) + 4% SDS | 37 | 412 |
| C15-Alk-OPP (10 μM) | P. falciparum | EP | 0.1 mM biotin-N3* | PBS + 1% SDS | 15 | 415 | |
| C15-Alk-OH (1 μM) | P. falciparum | EP, V | FCP, Rab5b | 0.1 mM biotin-N3* | 50 mM TEA (pH 7.4) + 4% SDS | 19 | 417 |
| YnF (10 μM), YnGG (10 μM) | EA.hy926 (endothelial) | EP (SILAC), V, inhibition | RHOA, HRAS, ULK3, CEP85, DCAF8 | 0.1 mM biotin-N3* | PBS + 1% Triton X-100, 0.2% SDS | 80 | 418 |
| C15-Alk-OH (50 μM) | HeLa | GBP, V | GNAI-1, GNAI-2 | 25 μM Cy3-N3 or Cy5-N3* | PBS + 1% SDS | 19 | 422 |
| C20-Az-OH (30 μM) | primary mouse macrophages | V, inhibition | RAC1 | 10 μM AlexaFluor 488-N3* | PBS | 427 | |
| C15-Alk-OH (10 μM) | HeLa, A549 (lung epithelial), COS-7, MCF10A | imaging, flow cytometry | 0.1 mM 5-FAM- N3, 1 mM TCEP, 0.2 mM TBTA, 1 mM CuSO4 | PBS | 431 | ||
| C15-Alk-OH (25 μM) | primary sympathetic neurons | imaging, V | RAC1 | 0.25 mM biotin-N3, 1 mM TCEP, 1 mM CuSO4 | PBS (pH 7.4) | 434 | |
| C20-Az-OH (30 μM) | RPE1-HTERT (retinal epithelium) | V, inhibition | FBXL2 | 20 μM DIBO-biotin | PBS | 167 | |
| C15-Alk-OH (50 μM) | HeLa | V, inhibition | Spindly | 0.1 mM rhodamine-N3* | PBS | 436 | |
| Other lipids | |||||||
| Az-C17 | HeLa | V | TFR1 | Click-IT (R) pre-mix | Click-IT (R) buffer | 447 | |
| Alk-C18 (50 μM) | S. flexneri-infected HeLa | EP (SILAC), V | RHOA, VAMP8, RAB13 | 0.1 mM biotin-N3* | 50 mM TEA (pH 7.4) + 4% SDS | 60 | 450 |
| Alk-C20 (50 μM), Alk-C22 (50 μM) | HeLa | imaging, in-gel fluorescence | 0.25 mM biotin-N3, 1 mM TCEP, 0.25 mM TBTA, 1 mM CuSO4 | PBS + 4% SDS | 453 | ||
| Alk-C5 (50 mM) | S. typhimurium | EP, V | HilA | 0.1 mM biotin-N3* | PBS + 0.5% NP-40 | 56 | 455 |
| Alk-C8-CoA (1 μM) | inhibitor screen | 1 μM HRP- N3, 5 mM ascorbate, 0.5 mM THPTA, 0.1 mM CuSO4 | PBS (pH 7.4) | 464 | |||
| Alk-C18:1 (200 μM), Alk-C19:1 (200 μM), Alk-C20:1 (200 μM) | Raw 264.7 | EP, imaging, V | IFITM3 | 0.1 mM biotin-N3, 1 mM TCEP, 0.5 mM TBTA, 1 mM CuSO4 | 50 mM TEA (pH 7.4) + SDS | > 100 | 477 |
| Alk-C20:4 (10 μM) | peritoneal macrophages | EP, V | ACADL, GAPDH | 0.25 mM biotin-N3, 15 mM ascorbate, 5 mM THPTA, 1 mM CuSO4 | RIPA | 221 | 484 |
| Az-Chol-2 (5 μM) | HEK293a, zebrafish | EP (SILAC), imaging, V | Shh | 0.1 AzTB* | PBS + 0.2% SDS | 501 | |
| Az-Chol (2 μg /mL) | HEK 293T, CHO-7 (ovarian) | EP, V | 0.1 mM biotin-N3, 2.5 mM ascorbate, 1 mM TBTA, 1 mM CuSO4 | 20 | 504 | ||
The clickable probes used in gel-based proteomics (GBP), enrichment proteomics (EP), activity-based protein profiling (ABPP), validation (V), imaging, flow cytometry, or inhibition to validate response to lipidation inhibitors.
Concentrations of reagents used. *Standard conditions employ 1 mM TCEP, 0.1 mM TBTA, and 1 mM CuSO4. AzTB, AzRB and AzKTB are trifunctional capture reagents containing azide, biotin and TAMRA (AzTB) or rhodamine (AzRB). AzKTB contains a lysine for trypsin cleavage. Click-IT® is a commercial click reaction kit.
Total number of putative lipidated proteins identified. Proteins identified at high confidence (HC) are indicated.
5.4. Discovery of S-palmitoylation-regulated proteins
Proteomic studies using palmitic acid analogues containing bio-orthogonal functionality has indeed led to the discovery of several novel S-palmitoylation substrates. Click-chemistry based analysis can also be exploited to explore and validate the S-palmitoylation status of individual proteins of interest. This subsection describes the use of several strategies to detect and validate the S-acylation or S-palmitoylation of a given protein of interest
5.4.1. Direct fluorescence-based detection
Assessment of successful labeling of proteins treated with clickable probes can be conveniently performed through click reaction with a fluorophore and visualization through in-gel fluorescence analysis. Using this method, Hang and coworkers first discovered the S-palmitoylation of interferon-induced transmembrane protein 3 (IFITM3) in murine models using a chemical proteomic approach, which was later confirmed to have three S-palmitoylation sites on membrane-proximal cysteines (Cys 71, 72, and 105) essential for its antiviral activity.182,221 The palmitate modification was validated by immunoprecipitating the metabolically labeled IFITM3 and subjecting it to click reaction with rhodamine-azide.222 More than half of the known zDHHC-PATs can S-palmitoylate IFITM3, with a preference for zDHHC20 that uniquely increases its antiviral activity upon co-expression.223 Later studies employing transfection of wild-type and mutant forms of human and bat IFITM3 followed by CCML revealed discrepancies and contradictions in the S-palmitoylation of the three conserved cysteine residues.224,225 Thus, the regulatory mechanism of S-palmitoylation might differ among these homologs. Furthermore, IFITM3 also requires an amphipathic helix that impacts its antiviral activity upon mutation that does not influence S-palmitoylation as shown through a CCML assay.226 After the discovery of IFITM3 S-palmitoylation, other members of the IFITM family were also examined for potential S-palmitoylation. This includes IFITM5 whose S-palmitoylation was validated using Alk-C18 and found to mediate its interaction with FKBP11 that is essential for bone formation.227 In the same vein, S-palmitoylation was also confirmed at a non-conserved position as well as on three conserved cysteine residues in IFITM1, which are crucial for its protection from proteasomal degradation and antiviral activity against influenza A.228
Non-mammalian S-palmitoylated proteins were also validated through CCML and fluorescence labeling. In bacteria, S-palmitoylation on the two bacterial effector proteins SspH2 and SseI is indispensable for their localization in the host-cell plasma membrane, highlighting a mechanism for how pathogens exploit the host-cell PTM machinery.229 Interestingly, mycobacterial IFITM is S-palmitoylated when expressed in human cells and confers antiviral activity against influenza.230 This demonstrates that a plausible IFITM gene transfer between bacteria and eukaryotes may be beneficial to eukaryotic cells to combat viral infections. In yeast cells undergoing cell division, differential S-palmitoylation on Rho3 GTPase was visualized, implicating its role during meiosis under the control of Erf2 palmitoylase.231 Beyond validation, CCML is also useful for mapping essential residues that influence S-palmitoylation such as in yeast protein Yck2.232 Through site-directed mutagenesis, a dipeptide Phe-Phe near the C-terminus of Yck2 was found to be essential for its S-palmitoylation and surmised to be involved in enzyme recognition.
Labeling with the shorter fatty acid analogues Alk-C16 and Alk-C17 combined with fluorescent detection further illuminated the role of S-palmitoylation on novel and known S-palmitoylated proteins. Dual S-palmitoylation drives the amyloid precursor protein (APP) to concentrate in lipid rafts where it undergoes amyloidogenic processing, thereby enhancing the production of Aβ peptides, a hallmark of Alzheimer’s disease pathology.233 Along the same lines, zDHHC7-catalyzed dual S-palmitoylation on junction adhesion molecule C (JAM-C) mediates its localization in the tight junction regions in cells, which may serve as a target to attenuate cancer metastasis.234 Alk-C17 was used to confirm that knockout of hedgehog acyltransferase (Hhat) results in diminished sonic hedgehog (Shh) S-palmitoylation, leading to attenuation of the proliferation and invasiveness of human carcinoma cells.235
In vitro fluorescence labeling may also provide a platform for quantitative assessment of the levels of S-palmitoylation of specific proteins within cells. For example, GFP-fused STX-19 was co-transfected with the 23 mammalian zDHHCs and labeled with Alk-C18, followed by click reaction with an azide-dye, IR-800.236 Ratiometric quantitation of the IR-800/GFP fluorescence after electrophoretic separation in gels provided information on the extent of labeling promoted by each zDHHC of STX-19, revealing zDHHCs 2,3,7, 11, and 12 as efficient enzymes that modify STX-19. S-palmitoylation of this Q-SNARE protein by these zDHHCs is essential for its targeting to tubular recycling and may also mediate Rab8 trafficking to the plasma membrane.
5.4.2. Antibody-based methods and streptavidin blotting
Detection methods based on CCML and fluorescent-azide or -alkyne conjugation combined with antibody-based recognition or immunoprecipitation approaches were carried out to validate the S-palmitoylation of proteins. The click reaction can be carried out in solution after elution of immunoprecipitated proteins or directly on proteins immobilized on beads. Pulse-chase labeling with Alk-C18 on the tyrosine kinase Lck demonstrated its highly dynamic S-palmitoylation kinetics that plays key roles in transduction downstream of Fas receptor activation, a process that mediates T-cell responses during inflammation.237–239 Novel S-palmitoylation on another kinase, dual leucine-zipper kinase (DLK), was found to control its localization and activity that is critical in axonal injury signaling.240 Another example is the zDHHC6-catalyzed S-palmitoylation on MYD88 that was found to influence toll-like receptor inflammatory signaling via regulation by fatty acid synthase (FASN).241
Alk-C18 labeling also demonstrated that S-palmitoylation reduces the depalmitoylase activity of PPT1, an enzyme linked to the neurodegenerative disease neuronal ceroid lipofuscinosis (NCL).242 PPT1-deficient mouse models displayed higher levels of S-palmitoylated proteins as well as mislocalization of S-palmitoylated proteins that regulate neuronal ciliogenesis including Rab3IP.243 Results of these studies implicated PPT1 S-palmitoylation as a key player in the progression of NCL and that this disease is a form of ciliopathy. Similarly, Alk-C18 labeling in cardiac myocytes showed that S-palmitoylation of the voltage-gated sodium channel Nav1.5 can markedly modulate cardiac sodium currents, leading to alterations in cardiac excitability.244
Although in-gel fluorescence scans can be used to directly visualize labeled proteins, indirect approaches by means of click reaction with biotin have been widely applied. The labeled proteins in these lysates can be probed for biotin-modification via streptavidin blotting. Through this strategy, the novel S-palmitoylation of peripheral myelin protein 22 (PMP22) was discovered and shown to be critical for its role in cell shape and motility.245 Likewise, the S-palmitoylation site near the N-terminus of the transframe (TF) protein of Sindbis virus was mapped and its role in trafficking TF to the host plasma membrane to promote virus budding was established.246
S-palmitoylation on proteins of interest from biotinylated samples can be more specifically probed or validated by isolating the protein via immunoprecipitation prior to streptavidin blot detection. For example, probing the S-palmitoylation state of calnexin showed that ER stress promotes its depalmitoylation.247 This approach validated the autopalmitoylation on a conserved residue in TEA domain (TEAD) transcription factors that is required for their proper folding, stability, and interaction with coactivators.248,249 Novel S-palmitoylation on plakophilin was discovered and found to be indispensable for the assembly of desmosomes that are specialized structures that serve a key role in cell-cell adhesion of epithelial and cardiac tissues.250 Similarly, SOD1 was confirmed to be S-palmitoylated and the enhanced S-palmitoylation of its disease-relevant mutants may contribute to the pathogenesis of familial amyotrophic lateral sclerosis (FALS).251 S-palmitoylation was also discovered for junctophilin-2 (JPH-2), a modification essential for its function in tethering the sarcoplasmic reticulum to the plasma membrane.252 Moreover, S-palmitoylation of the tumor suppressor Scribble (SCRIB) by zDHHC7 was evaluated to be essential for its plasma membrane targeting, and impairment of this modification activates pathways that promote tumorigenesis.253 A follow up study combining CCML and fluorescent labeling revealed that Snail overexpression, implicated in cancer, stimulates depalmitoylation of SCRIB, resulting in enhanced growth signaling and malignancy.254
5.4.3. Enrichment approaches
Alternatively, metabolically labeled samples can be reacted with biotin-azide or -alkyne, enriched using avidin beads, eluted and detected via western blot using an antibody against the protein of interest or fused tags such as FLAG or GFP. Using this method, the S-palmitoylation by the host-cell machinery on the effector protein LpdA in the pneumonia-causing bacteria Legionella pneumophilia was validated, a mechanism that allows spatial control of LpdA’s activity inside the host.255 The novel S-palmitoylation on Glut4 and IRAP was confirmed and altered levels of their S-palmitoylation may play a role in obesity.256 Similarly, S-palmitoylation was validated and found to modulate the function of the apical sodium-dependent bile acid transporter (ASBT) crucial for the circulation of bile acids in human cells.257 Moreover, the S-palmitoylation on the C-terminal region of mucolipin 3 whose activation induces autophagy in a S-palmitoylation-dependent manner was verified.258 This approach also aided in the discovery of the regulatory function of dickkopf1 (DKK1) in the dynamic S-palmitoylation of its receptors CKAP4 and LRP6, which may play pivotal roles in cancer proliferation.259 Recently, both the programmed cell death ligand 1 (PD-L1) and its cognate receptor PD-1—two interacting proteins that are key therapeutic targets in cancer treatment—were discovered to be S-palmitoylated.260,261 Inhibition of S-palmitoylation in PD-L1 enhances the immune response against tumors while blocking PD-1 S-palmitoylation manifested anti-tumor effects. Therefore, designing selective inhibitors capable of suppressing the S-palmitoylation of both cancer targets should yield new weapons in the battle against cancer.
5.4.4. Validating S-palmitoylated receptors
The methods described above were useful to validate or identify sites of modification on protein receptors and provided insights into the regulatory role of S-palmitoylation for proteins including the D2 dopamine receptor (D2R), c-Met receptor tyrosine kinase (RTK) epidermal growth factor receptor (EGFR), bone morphogenic protein receptor 1a (BMPR1a), and tumor necrosis factor alpha receptor 1 (TNF-R1).262–266 Multiple PATs were found to mediate S-palmitoylation of the μ-opioid receptor D2R on Cys433 that is essential for its stability and membrane trafficking.262 The localization of the frequently overexpressed or mutated protein c-Met RTK in cancer was found to be controlled by S-palmitoylation by modulating transport from the Golgi to the plasma membrane.263 zDHHC20 was discovered to S-palmitoylate EGFR and disruption of its S-palmitoylation combined with inhibition of EGFR phosphorylation may serve as an effective therapeutic strategy to treat EGFR-driven cancers.264 S-palmitoylation on Cys173/175/180 of BMPR1a may affect BMP signal transduction and proper embryonic development in neural stem cells (NSC), therefore regulating the fate of NSC.265An earlier study using azido palmitate to investigate S-palmitoylation on the ligand TNF-α provided the first evidence for the role of S-palmitoylation in TNF-R1 signaling.267 This most recent study of the S-palmitoylation of the TNF-R1 receptor itself exemplifies how S-palmitoylation may regulate multiple aspects of a single signaling pathway.
5.4.5. Validating depalmitoylation substrates
Validation and mapping of S-palmitoylation sites in proteins using CCML has also served to unveil information regarding their regulation through depalmitoylation as well as well as contributed to the discovery of novel depalmitoylases. For example, Wnt5a signaling was shown to drive the APT1-mediated depalmitoylation of melanoma cell adhesion molecule (MCAM), a protein upregulated in melanoma, that influences it localization and ability to promote cell invasion—a mechanism often linked to metastasis.268 CCML with Az-C16 in melanoma cells treated with Wnt5a displayed reduced MCAM S-palmitoylation. Wnt5a was speculated to control depalmitoylation through the Wnt adaptor protein Dvl2, which interacts with APT1. Moreover, oncogenic mutants of zDHHC20 in melanoma were also assessed for their inability to S-palmitoylate MCAM through CCML, which delineates the importance of these mutations to support the dominance of unpalmitoylated MCAM for melanoma cell invasion. In another study, APT1 was found to regulate the differential localization of Numb and β-catenin—two proteins that determine cell fate.269 The S-palmitoylation status of these proteins, probed through CCML with Az-C16, including the three conserved residues in Numb predicted to be S-palmitoylation sites was verified through site-directed mutagenesis. The tight control of protein localization conferred by APT1 on Numb and β-catenin affects the downstream transcriptional activity of Notch and Wnt signaling during cell division.
Both palmitoylation and depalmitoylation can proceed simultaneously to promote protein function.270 For example, Alk-C16 labeling showed that the T helper 17 (TH17) cell differentiation stimulator, STAT3, undergoes a cooperative palmitoylation and depalmitoylation by zDHHC7 and APT2, respectively. zDHHC7 palmitoylates STAT3 at Cys108 for membrane recruitment and subsequent phosphorylation. APT2 then selectively depalmitoylates phosphorylated STAT3 (p-STAT3) over unphosphorylated species, which is a critical step for the translocation of p-STAT3 in nuclear membranes and its delivery to the nucleus to promote cell differentiation. However, overactivation of p-STAT3 leads to accelerated differentiation that is often associated with inflammatory diseases such as inflammatory bowel disease (IBD). Impairing the function of zDHHC7 or APT2 through knockout or chemical inhibition relieved symptoms in mouse models of colitis—a form of IBD characterized by colon inflammation. Moreover, zDHHC7 and APT2 were also found to be upregulated in IBD patients. Therefore, designing strategies to specifically inhibit zDHHC7- or APT2-catalyzed palmitoylation/depalmitoylation of STAT3 offers a novel mechanism for therapeutic intervention to treat IBD.270
CCML also facilitated the discovery of α/β-hydrolase domain-containing thioesterases (ABHDs) as novel classes of depalmitoylases. These enzymes are directed to the plasma membrane through S-palmitoylation on their N-termini.102,196 Pulse-chase labeling using Alk-C18 revealed the role of ABHD17 proteins in controlling palmitate turnover on PSD95 and NRAS. All three ABHD17 isoforms (ABHD17A, ABHD17B, and ABHD17C) are required for dynamic cycling of NRAS in vivo.102 Similarly, ABHD10 governs the anti-oxidant activity of PRDX5 (an S-palmitoylated protein validated via Alk-C18 labeling) in mitochondria through the depalmitoylation of a nucleophilic cysteine in its active site.271
In summary, these various approaches for validating the lipid modification on proteins offer a range of options for researchers to achieve their goals. Although fluorescence-based methods offer direct confirmation of the PTM on the protein, several other labeled proteins in the lysate that co-migrate with the protein may overwhelm the signal and therefore complementary validation through western blotting may be required. The use of streptavidin enrichment of biotinylated samples may enhance the confidence on the labeling of proteins, although it generally suffers with issues of high background. Efforts to improve this background problem through optimizing the blocking step have been reported.272
5.5. Imaging methods to visualize dynamic PAT activity and localization
Earlier methods to tag S-acylated proteins using isotopically labeled fatty acyl analogues were limited to detection in gels. The advent of chemical probes to tag S-palmitoylated proteins enabled the visualization and monitoring of changes in S-palmitoylation events in fixed cells.273 Cellular imaging of S-acylated proteins using clickable fatty analogues allows for global visualization of the modified proteins, as well as the detection of a specific protein of interest using antibodies.
5.5.1. Global S-acylated proteins
The first data that provided a cellular view of global fatty acylation was conducted on PC3 cells, which were metabolically labeled with various alkyne-modified fatty acyl analogues and clicked with rhodamine-azide after cell fixation.180 Fatty-acylated proteins were distributed as punctate patterns across the cytoplasm and plasma membrane, with subtle variations observed among varying fatty acyl analogues employed. Changes in S-palmitoylation events were also monitored at different stages of cell division, revealing that S-palmitoylation is spatiotemporally regulated during the cell cycle.
Imaging of protein S-acylation in protozoans illuminated its multiple roles in the invasion mechanism of these parasites. Flow cytometric analysis and cellular imaging on Leishmania donovani cells labeled with Alk-C16 and treated with 2-BP showed a drastic decrease in global S-palmitoylation with concomitant morphological defects.274 Inhibition of S-palmitoylation further led to a severe loss of cell movement and disconcerted invasion pattern. Cellular imaging using Alk-C18 allowed for quantification of the total S-palmitoylated proteome in multiple life stages of P. falciparum.275 The PAT activity in free merozoites were imaged for the first time, with the schizont stage showing the highest number of S-palmitoylation events compared to the ring and trophozoite stages. S-palmitoylation in merozoites was found to be dynamic and regulated in response to natural signals that trigger microneme secretion, an important mechanism for red blood cell (RBC) invasion. Similarly, visualization of the S-palmitoylated proteome shed some light on the relationship between S-palmitoylation and other regulatory mechanisms involving cysteine residues. A recent study employing Alk-C18 observed similar results, showing that the schizont stage harbors the most S-acylated proteins followed by trophozoite and ring stages, respectively.276 Higher resolution images were acquired by using single-molecule switching super-resolution microscopy (SMS) on samples directly labeled with a fluorophore-azide, or through electron microscopy on biotinylated samples tagged with streptavidin-gold nanoparticles. These methods provide rapid and high-throughput strategies for microscopic examination of protein S-acylation without the need for transfection, which is a difficult endeavor to achieve in many cellular systems such as P. falciparum.
CCML and fluorescent labeling with primary mouse cortical neurons revealed that alterations in S-nitrosylation result in decreased global S-palmitoylation.277 Perturbations of S-nitrosylation/S-palmitoylation crosstalk of proteins associated with synaptic transmission and plasticity might lead to the destabilization of synaptic systems implicated in chronic stress-related diseases. In dorsal root ganglion (DRG) neurons, cellular labeling and imaging revealed that cGKI, a kinase involved in axon branching, co-localizes in the DRG growth cone along with the bulk S-palmitoylated proteome.278 This evidence, together with other biochemical data, suggests a potential role for cGKI-mediated transduction in modulating S-palmitoylation, which contributes to cone enlargement and neurite extension of DRG neurons.
The dynamic feature of S-palmitoylation is central to its regulatory role in protein function, stability, and localization. Pulse labeling techniques in imaging PAT activity may provide insights into the rate of turnover in cells similar to chemical proteomics. CCML using palmitate analogues as a pulse can be a valuable tool to determine protein turnover. Pulse-washout experiments using Alk-C16 revealed rapid turnover of global S-palmitoylation in PC3 cells.180 Pulsing with Alk-C16 and subsequent chasing with palmitic acid enabled tracking of dynamic S-palmitoylation of specific proteins.184 The O-palmitoylated Wnt3a appeared to have the slowest turnover rate compared to the S-palmitoylated proteins investigated. Pulse-chase experiments in cellular imaging also allowed for monitoring of protein transport. Anterograde transport proteins were found to be S-palmitoylated by zDHHC3 and zDHHC7 in the cis-golgi and this modification accelerates their intragolgi (cis to trans) transport.279 The intrinsic physical properties of the fatty acyl side chain promotes localization in the highly curved regions of the cis-golgi, resulting in the observed enhanced rate of transport.
5.5.2. Specific S-acylated protein
Another strategy to assess the localization of specific S-acylated/S-palmitoylated proteins is through subcellular fractionation of labeled protein lysates, followed by immunoprecipitation, click reaction and western blot detection.280 However, imperfections in the cell lysis may affect the integrity of the generated fractions and could complicate subsequent interpretation concerning the true localization of S-acylated/S-palmitoylated proteins of interest.15 To this end, Hannoush and coworkers developed a robust fluorescence imaging platform by combining CCML using Alk-C16 with proximity-ligation assay (Palm-PLA) to track specific S-acylated/S-palmitoylated proteins in intact cells (Fig. 7A).281,282 In this method, cells expressing the protein of interest are metabolically labeled with Alk-C16 (or Alk-C18), fixed and permeabilized, then clicked with an azide-modified tag. Two primary antibodies that recognize the protein and the tag are introduced, followed by treatment with two secondary antibodies specifically binding to each primary antibody. These secondary antibodies are conjugated to complementary oligonucleotides, which hybridize to form a closed circle when both are in close proximity. A rolling-circle amplification is then carried out with a fluorophore-labeled oligonucleotide complementary to the closed circle, generating a fluorescence signal to visualize localization of the tagged protein. This approach enabled the visualization of various proteins in multiple intact cell lines,281 as well as successful tracking of Wnt3a through the secretion pathway.184
Figure 7. Imaging of proteins inspired by proximity ligation assay (PLA).
(A) Palm-PLA visualizes localization of specific palmitoylated proteins. Intact cells metabolically labeled with clickable fatty acids are tagged with a biotinylated click reaction partner. Two primary antibodies that recognize the target protein and biotin tag are added, followed by treatment with a pair of oligonucleotide-labeled secondary antibodies that bind to the primary antibodies separately. When they are in close proximity, the oligonucleotides complement and form a closed circle that is required for rolling-circle amplification (RCA). (B) UnPalm-PLA detects the unpalmitoylated species of the protein of interest (POI). Cells are treated with biotin-maleimide to directly tag the free form of the POI followed by treatment with antibodies as described in Palm-PLA. An orthogonal fluorophore is used to distinguish localization of the unpalmitoylated POI from the palmitoylated species.
Palm-PLA was also used with Alk-C18 to examine the distribution pattern of S-palmitoylated JPH2 (palm-JPH2) in rat ventricular myocytes, while the total JPH2 was concurrently visualized by immunofluorescence (IF-JPH2).252 In order to authenticate the observed palm-PLA signals, unpalmitoylated forms were also imaged by reaction with a maleimide derivative of biotin in lieu of the palmitate-alkyne/biotin-azide tag (Unpalm-PLA, Fig. 7B). Through the complementary methods Palm-PLA, Unpalm-PLA and IF, palm-JPH2 was found to exhibit slow turnover rates of S-palmitoylation along the z-axis of the cell periphery. Using structured illumination microscopy on the myocyte surface, palm-JPH2 and IF-JPH2 were observed to localize at distinctly different plasma membrane subdomains with little overlap. These results indicated that JPH2 S-palmitoylation is stable in the cell periphery while dynamic along the lateral cell surface.
Indeed, imaging S-acylated/S-palmitoylated proteins in intact cells has been made possible through the use of CCML. In labeling with azide-modified probes, significant background arises from non-specific reaction of the alkyne reagent with free thiols. Activation of terminal alkynes by copper (I) can result in reactions with other nucleophiles (beyond azides), leading to strong background labeling when the alkyne reagent is present in excess. In fact, this copper (I)-catalyzed reaction between terminal alkynes and thiols has been repurposed for the construction of alkylsulfides in the presence of molecular oxygen.283 Blocking these functional groups via alkylation with iodoacetamide may improve visualization284 and excluding the complexing ligands (e.g. TBTA and THPTA) may also provide enhanced quality of cellular imaging of cells treated with clickable analogues.184,275,277 Furthermore, these methods have generally been limited to fixed cells since the requirement for toxic Cu(I) in CuAAC restricts its application to live-cells. Imaging of S-palmitoylation activity in living cells and in vivo may provide real-time monitoring of S-palmitoylation dynamics and should be of great interest in future studies.
5.6. Evaluation of fatty acyltransferase activity and inhibition
Inhibitors of PATs are essential to study protein S-palmitoylation and have potential to serve as therapeutic agents for relevant diseases. Inhibition of PATs can be evaluated through examining the S-palmitoylation states of proteins of interest metabolically labeled with Alk-C16, Alk-C17, or Alk-C18 and detected via western blotting. Inhibition of zDHHC3-mediated S-palmitoylation of integrin β4 (ITG β4) by curcumin occluded autoacylation of zDHHC3 and effectively reduced levels of S-palmitoylated ITG β4 in invasive breast cancer cells.285 Inhibition of N-palmitoylation of Shh by Ruski-201 was visualized via a gel mobility shift assay by using Alk-C17 labeling followed by click reaction with a multifunctional reagent.286 Likewise, several small molecules including IWPs, C59, and ETC-159 were shown to effectively inhibit the O-palmitoylation of Wnt proteins by blocking the acyltransferase activity of porcupine (PORCN) using Alk-C16 labeling experiments.280,287–289 Alterations in Wnt signaling including overexpression of Wnts and their receptors are implicated in various human cancers.290 Using these inhibitors may offer an effective strategy to prevent such Wnt-driven diseases.
Cell-based in vitro assays were developed to evaluate PAT inhibition on S-palmitoylation of specific proteins through qualitative or quantitative analysis using fluorescence microscopy as previously described.184,236,281,282 These techniques can be extended to high-throughput analysis in a 96-well format that may facilitate rapid screening of PAT inhibitors.184,282 Recently, a cell-based approach was reported utilizing bacterial expression systems to visualize PAT inhibition by 2-BP.291 P. falciparum PATs ectopically expressed in an engineered PTM-null E. coli expression system can incorporate Alk-C18 into bacterial proteins. Effects on the global S-palmitoylation enforced by PfDHHCs can be evaluated to screen for effective inhibitors of specific zDHHCs. This technique provides potential for chemotherapeutic targeting of pathogenic zDHHCs via a high-throughput screening platform.
The identification of PAT inhibitor targets using CCML can also be accomplished via mass spectrometry activity-based protein profiling (ABPP) and fluorescence-based assays. For example, the palmitic acid analogue 2-bromopalmitatic acid (2-BP, Fig. 8) has been commonly used as an irreversible inhibitor and thought to be selective for the S-palmitoylation activities of PATs.292 However, profiling for its protein targets using its corresponding azide (Az-2-BP) and alkyne (Alk-2-BP) analogues (Fig. 8) revealed its poor selectivity, reacting with many targets beyond PATs such as transporters and chaperones.293,294 Therefore, studies employing 2-BP to validate S-acylated proteins should be interpreted carefully and take into account any phenotypic changes or off-target effects this reagent induces.203,274 The natural product cerulenin (Fig. 8), a known PAT inhibitor, was derivatized into an alkyne-containing clickable form (Alk-cer, Fig. 8) to evaluate its protein targets.295 Metabolic labeling of zDHHC4-expressing HEK293 cells with Alk-C16 or Alk-cer followed by biotinylation and western blot detection showed that Alk-C16 adducts are sensitive to hydroxylamine while Alk-cer derived modifications are resistant. This confirms that cerulenin directly and irreversibly alkylates zDHHCs similar to 2-BP. While Alk-cer displayed improved potency and could label all zDHHCs tested in this study, it also reacts with some zDHHCs bearing mutant active sites, suggesting it is not truly on-target and “activity-based”. This may be attributed to the design of the probe which contains a linear 12-carbon chain and differs from the native cerulenin structure. The loss of the double bonds may affect the conformation of this probe and may explain its difference in target specificity with the native cerulenin.
Figure 8. Structures of PAT and APT inhibitors and their clickable analogues for activity-based protein profiling (ABPP).
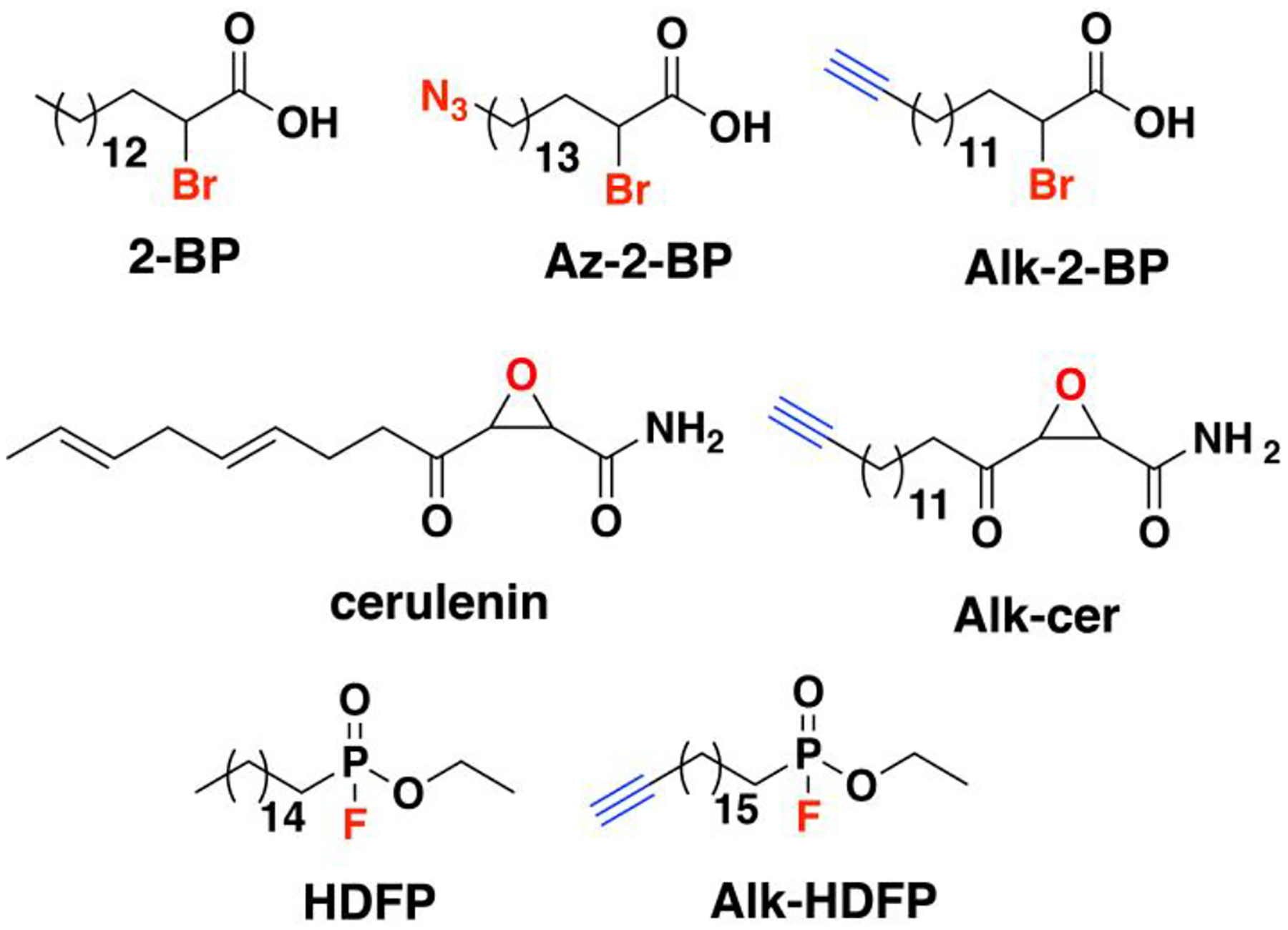
2-bromopalmitic acid and the natural product cerulenin are pan inhibitors of zDHHCs. Hexadecyl fluorophosphonate (HFDP) is a serine lipase inhibitor that targets many hydrolase enzymes.
As mentioned earlier, fatty acyl modification of proteins can be reversed by acylthioesterases such as APT1/2, which are known to be targets of the serine lipase inhibitor HDFP (Fig. 8). Activity-based protein profiling of depalmitoylases/deacylases using Alk-HDFP showed reactivity with a specific subset of serine hydrolases with a strong preference for lipid substrates, particularly lipases.208 HDFP-inhibition of depalmitoylases increases Alk-C18 probe incorporation but only represents a small fraction of the bulk S-palmitoylated form of the proteins.104 The targets of selective inhibitors for specific fatty acyl transferases can be also identified through the use of Alk-C18 labeling. A SILAC-based quantitative proteomic approach was employed to demonstrate that the inhibitor RUSKI-201 can selectively reduce the palmitoylation levels of Hhat palmitoylation substrates at submicromolar concentrations.286
The development of cell-based in vitro assays and the use of clickable analogues of PAT inhibitors can indeed allow for evaluation of their inhibition capacity. More importantly, employing chemical proteomics with inhibitor analogues can help delineate side reactions with proteins and may offer information on how to improve their design to avoid reactivity with these off-target proteins. These current inhibitors for zDHHCs are largely based on fatty acid structures and other scaffolds may be required to achieve greater target specificity. With the recent success in obtaining crystal structures for zDHHC20, the development of inhibitors tailored to be specific to this enzyme should be possible. It is likely that additional crystal structures for other zDHHCs will be acquired in the future, which should aid in the rational design of inhibitors specific to a particular zDHHC of interest.
5.7. Enabling technologies based on clickable palmitic acid
Although cell-based and chemical proteomic approaches presented above may be useful in evaluating inhibition of fatty acyltransferase activity, more rapid and higher throughput methods are desirable. In an effort to innovate non-radioactive detection methods for acyltransferase activity suitable for high-throughput screening of inhibitors, click chemistry-based screening platforms were developed. A high-throughput method designed for screening inhibitors of zDHHC9-catalyzed N-Ras S-palmitoylation employed immobilized N-Ras peptide and Alk-C18-CoA adaptable with a 384-well format (Fig. 9A).296 Through a turn-on fluorescence assay conferred by successful N-Ras modification, the inhibition efficiency of inhibitors were quantified and IC50 values were calculated from dose-response curves. Counter-screening with an unrelated S-palmitoylated protein such as Fyn aided in the validation of the specificity of candidate antagonists. Such a method is suitable for pursuing potent inhibitors with high specificity for the protein substrates of interest.
Figure 9. Enabling technologies based on click chemistry.
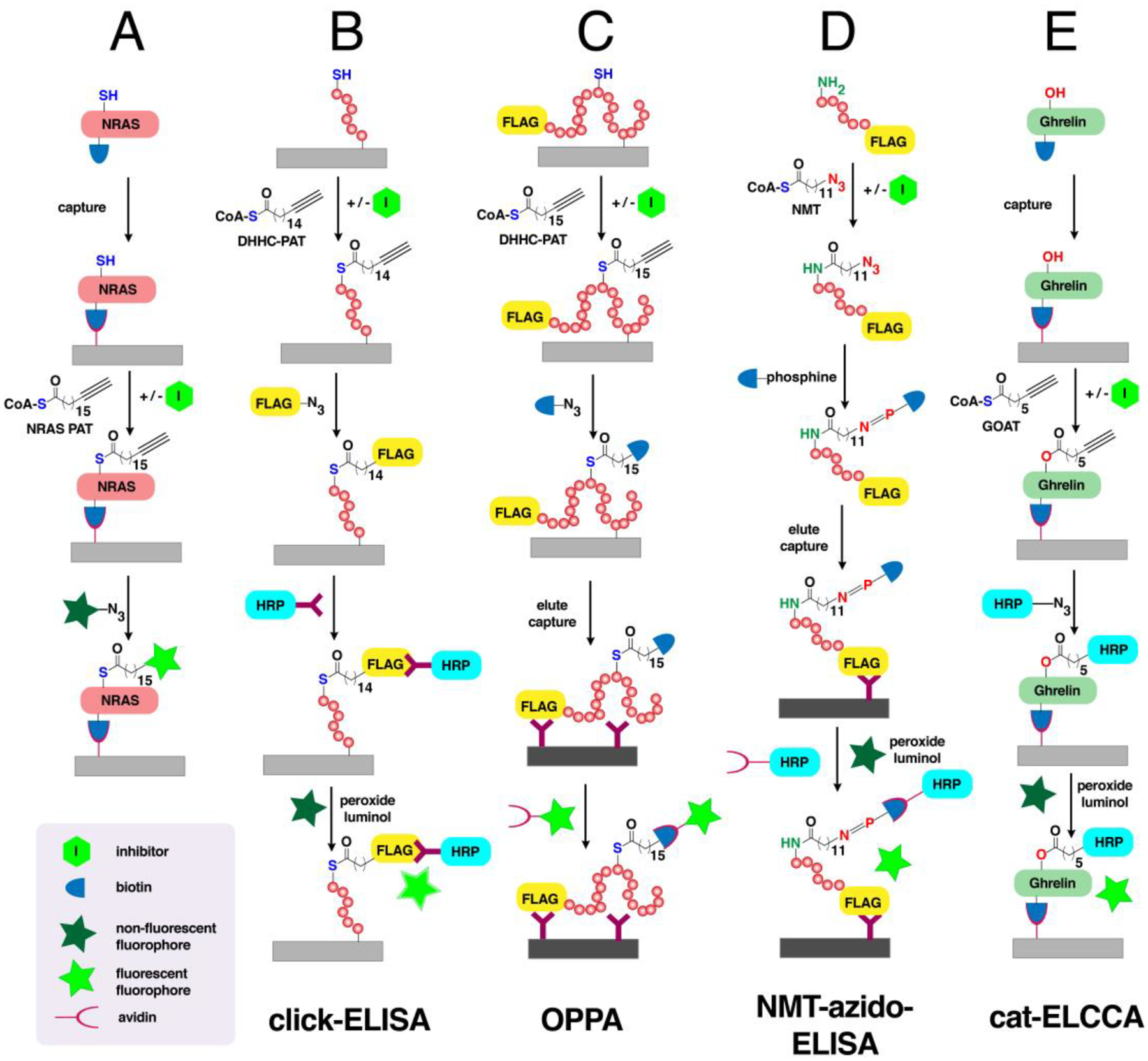
(A) A high-throughput method for screening inhibitors of NRAS palmitoylation. Immobilized NRAS is labeled with a clickable palmitate analogue and the extent of inhibition is evaluated based on turn on fluorescence after click reaction. (B) An ELISA-inspired click chemistry-based technology (click-ELISA) for inhibitor screening. Immobilized substrates are labeled with a clickable fatty acyl-CoA analogue and tagged with FLAG-N3. Addition of anti-FLAG conjugated to HRP in the presence of luminol and peroxide gives of chemiluminescence inversely proportional to extent of inhibition. (C) On-plate palmitoylation assay (OPPA) that involves labeling of a FLAG-tagged substrate a fatty acid analogue and biotin and subsequent immobilization on plates coated with anti-FLAG. Detection is based on recognition of the biotin-fatty acid label on proteins using fluorophore-conjugated anti-biotin or streptavidin. (D) NMT-azido-ELISA especially designed for evaluating NMT activity. FLAG-tagged substrates modified with azide are ligated with phosphine-biotin through Staudinger ligation. Labeled-substrates are then trapped on anti-FLAG-coated plates and NMT activity is quantified through chemiluminescence. (E) Catalytic enzyme-linked click chemistry assay (cat-ELCCA) to detect GOAT activity. Ghrelin substrate captured on a plate is labeled with a clickable octanoyl-CoA analogue and clicked with HRP. Detection is based on chemiluminescence.
Tate and coworkers established ELISA-inspired methods for high-throughput analysis of fatty acylation. They initially introduced click chemistry ELISA (click-ELISA) to detect and measure PAT activity of Hhat on Shh (Fig. 9B).297,298 In this approach, an immobilized Shh peptide is acylated by Hhat with Alk-C17-CoA and functionalized with azido-FLAG peptide, followed by probing with anti-FLAG fused to horseradish peroxidase (anti-FLAG-HRP) to evaluate levels of Hhat labeling. Such a method was able to quantify the potencies of inhibitors and calculate kinetic parameters for the Hhat-catalyzed Shh N-palmitoylation. The same group developed a fluorescence-based microfluidic mobility shift and acylation-coupled lipophilic induction of polarization (acyl-cLIP) assays that obviate the need for the multiple handling steps involved in click-ELISA.299,300 In acyl-cLIP, a fluorescently labeled Shh peptide is N-palmitoylated and the resulting hydrophobicity increase drives binding to BSA, providing fluorescence polarization readouts. Alk-C18-CoA was used to counter-screen candidate inhibitors for Hhat. On the basis of lipid binding to BSA, this assay is broadly applicable to other protein lipid transferases and hydrolase enzymes.
Albeit high-throughput, the aforementioned assays are limited to truncated peptides of S-palmitoylation substrates. A rapid click chemistry-based On-Plate Palmitoylation Assay (OPPA) was demonstrated to be efficient for quantifying time-dependent S-palmitoylation on intact proteins (Fig. 9C).301 His- and FLAG-tagged protein substrates were bound to Ni-NTA plates, S-palmitoylated with Alk-C16-CoA and clicked with azido-biotin. Products were then eluted and captured onto anti-FLAG coated plates and detected using fluorescently labeled streptavidin. S-palmitoylation on VAMP7 and LAT by full-length and truncated zDHHC18 was confirmed using this method. Transforming this method into a high-throughput scheme has yet to be accomplished.
While well-suited for determining the S-palmitoylation state of proteins of interest or evaluating PAT inhibition, none of the methods described above directly provide details regarding the number of fatty acylated sites nor the differential S-palmitoylation on multiple sites in a given protein. PEG-switch or acyl-PEG exchange (APE) assays based on thioester hydrolysis strategy were initially developed (Fig. 10A).99,221 These methods use maleimide-functionalized heavy PEG groups reactive with putative S-palmitoylation sites on free cysteines that can be used to delineate several S-palmitoylated species of a protein based on mass shifts in electrophoretic gels. An orthogonal approach based on click chemistry was recently innovated by the Chamberlain group referred to as mPEG-click (Fig. 10B).302 In this strategy, cells expressing the proteins of interest were treated with a clickable probe, e.g. Az-C16, followed by click reaction with a heavy PEG linker and detected via western blot. Similar to PEG-switch assays, the number of S-acylated sites can be visualized as the number of differentially migrated bands, as well as used to distinguish between mutations that abrogate either site-specific or global protein S-acylation. It also facilitates the study of dynamic acylation cycling within a single protein when combined with pulse-chase and time-course labeling techniques. Using this strategy, the extents of modification of multiple S-palmitoylated cysteine residues in SNAP25 were determined, as influenced by mutations in its linker region that alters its interaction with zDHHCs. Indeed, mPEG-click complements PEG-switch assays in that it provides a broader scope for investigating S-acylation dynamics in active living cells, while the latter is suitable for analyzing the extent of S-acylation in samples not amenable to metabolic labeling such as tissues.302
Figure 10. Methods to determine number of fatty acyl modifications in a protein.
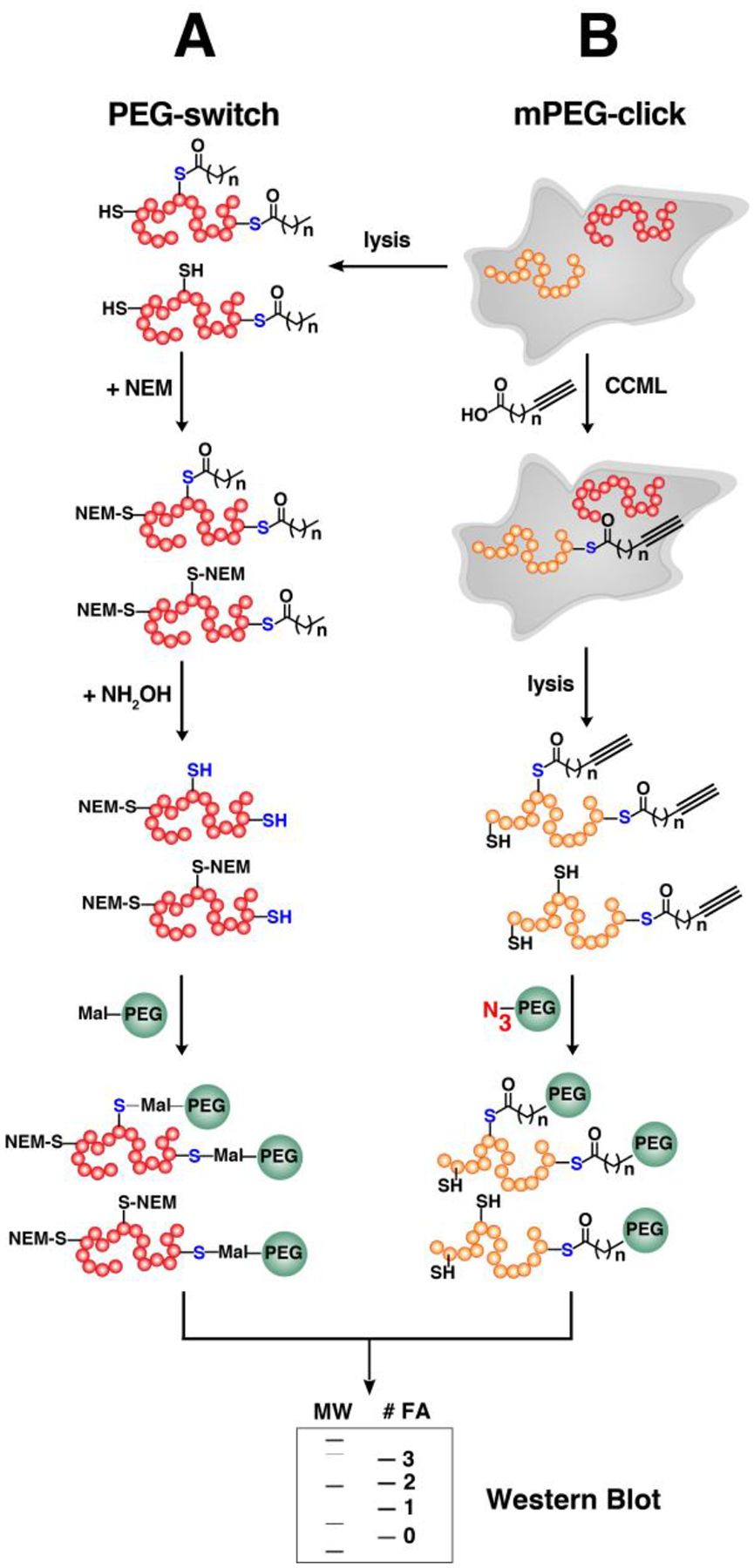
(A) PEG-switch assay similar to ABE where free cysteines are blocked with NEM and the thiols of modified residues are liberated through hydrolysis. These free residues are subsequently reacted with maleimide-functionalized heavy PEG reagents and resolved in gels for western blot analysis. PEG-switch assay is suitable for non-active samples (e.g. tissues) but may not be specific to detect fatty S-acylated proteins. (B) mPEG-click assay involves metabolic labeling of clickable fatty acids in cultured cells and lysates are subsequently reacted with a heavy PEG linker via click reaction. Samples are resolved in an SDS-PAGE and proteins of interest are detected through western blotting. The number of fatty acyl modifications reflect the number of PEG conjugates displayed as protein bands with retarded migration.
In summary, these enabling methods developed for detecting fatty acyltransferase activity have indeed expanded the utility of the clickable analogues of palmitic acid. However, technologies based on in vitro enzymatic activities of zDHHCs have been limited to the screening platforms noted above and not generally for selective modification and bioconjugation of proteins. Unlike N-myristoylation and S-prenylation where the site modification and substrate recognition is well-defined, S-acylation lacks these features and hence may not be useful for site-specific protein labeling applications discussed in Section 9.
6. Biological applications in N-Myristoylation
The recent efforts in defining the N-terminal canonical sequence, as well as the development of bioinformatic tools has aided our ability to predict N-myristoylated proteins.113,303 Although these tools are useful, their less than perfect predictive power necessitates experimental validation of putative N-myristoylated proteins of interest. Click chemistry-based analogues of myristic acid including Az-12 and Alk-C14 (Fig. 11) have been used to simplify the validation of N-myristoylated proteins, assessment of NMT inhibitor efficacy, and proteome-wide profiling of N-myristoylated proteins.18,179,304–306 Although selective, these probes can also be recognized as substrates that modify S-palmitoylated and GPI-anchored proteins.177,180 Owing to the stability of the amide bond in N-acylation versus S-acylation (S-palmitoylation) and ester (GPI anchor) bonds, selective monitoring of N-myristoylated proteins can be achieved through releasing S-palmitoylated and GPI-APs aided by basic hydrolysis prior to detection (Fig. 6A).192
Figure 11. Clickable analogues of myristic acid and myristate-S-CoA for metabolic and in vitro labeling of myristoylated proteins.
These probes are more selective for labeling N-myristoylated proteins.
The use of clickable myristic acid analogues has also facilitated cellular imaging,284 when used in concert with additional technologies involving transfection vectors,307,308 cell-free expression systems,309 and ELISA-based techniques.310 These methods have allowed the characterization of N-myristoylation substrates and probing for NMT activity. Furthermore, methods complementary to bio-orthogonal labeling such as photo-crosslinking, and Sort-A and Phospho tagging strategies have expanded the available tools to study N-myristoylation.311–313 Although N-myristoylation plays major roles in cancer and immunity,314 significant efforts have been invested into understanding and targeting this lipidation process in pathogens via CCML-based methods. This section focuses on those studies enabled by CCML with clickable myristic acid probes that highlight the importance of N-myristoylation in regulating protein functions.
6.1. Proteome-wide analysis on N-myristoylation and defatty-acylation
Although the probes developed for fatty acylation of proteins are often amenable for incorporation into both N-myristoylated and S-acylated proteins, the analogues Az-C12 or Alk-C14 more efficiently label N-myristoylated proteins.305 The preference for these probes corroborates observations made regarding the substrate specificity of NMTs which include the importance of chain length over hydrophobicity and the polarity of functional groups present in the fatty acyl-CoA analogue.111 Their active site architecture also allows for bending of the fatty acyl substrate with exquisite control of acceptable chain length and steric bulk in the chain end. Moreover, comparison of the extent of labeling of biorthogonal fatty acid probes with varying chain lengths revealed that metabolic labeling with Az-C12 or Alk-C14 provides robust and irreversible labeling compared to analogues 2–4 carbons longer.178,179 Their observed resistance to base hydrolysis in streptavidin blots and in-gel fluorescence analysis is a strong indication of amide bond formation, a characteristic feature of N-myristoylated proteins. Their labeling profiles also exhibit distinct labeled proteins compared to the longer probes across varying cell lines, suggesting that the substrate scope of Az-C12 or Alk-C14 differs from the longer probes that are potentially S-palmitoylation substrates. More importantly, Alk-C14 is preferentially and effectively blocked by inclusion of the native myristic acid or the weak NMT inhibitor 2-hydromyristic acid, further validating this probe as a suitable surrogate for labeling N-myristoylated proteins.
6.1.1. Profiling N-terminal myristoylation
Tate and coworkers first described the use of Alk-C14 combined with CCML and quantitative proteomics in P. falciparum.177 In this protozoan, both N-myristoylated proteins and GPI anchors were identified. To distinguish NMT substrates from GPI-anchored proteins, basic hydrolysis before enrichment enabled identification of more than 30 N-myristoylated proteins, with some showing evidence of intact modification on their N-terminal peptide determined through the use of a trypsin-cleavable reagent, AzKTB. These proteins participate in a diverse range of functions including motility, transport, development and phosphorylation pathways.
When quantitative mass spectrometric methods described above were applied to mammalian cells, more than 100 co- and post-translationally N-glycine myristoylated proteins were detected in HeLa cells.315 To validate those candidates, both NMT knockdown and inhibition experiments using a small molecule were conducted, revealing 70 proteins responsive to inhibition at high confidence. This NMT inhibition perturbs the function of myristoylated proteins, leading to ER stress, cell cycle arrest, and induced apoptotic cancer cell death.316 Furthermore, 87 candidate N-myristoylated proteins were identified across HeLa, MCF7, and HEK293 cells, with 36 found in common among these cancer cell lines.317 This suggests that the levels of NMTs and expression of their substrates are cell-type specific. The same group also presented the first quantitative analysis of the dynamic changes in N-myristoylation in a developing organism through pulse-chase labeling with Alk-C14.176 N-myristoylation events were found to be more prominent during the early development of zebrafish, particularly with those involved in maturation, melanogenesis, meiosis, and hedgehog and Wnt pathways.
6.1.2. Lysine fatty and defatty-acylation
As discussed previously, fatty acylation on the Nε-side chain of lysine was previously thought to occur on a limited number of proteins with TNFα described as the first Nε-lysine myristoylated protein.122 There is burgeoning evidence that this modification is regulated by members of the histone deacetylase (HDAC) and sirtuin families of hydrolases.130,318 Although early studies suggested that this lysine modification occurs through N-myristoylation, metabolic labeling with the palmitate analogue Alk-C16 results in better incorporation compared with Alk-C14.179,319 In contrast, recent findings suggest that Nε-lysine fatty acylation is catalyzed by NMTs in mammalian cells, at least for the potentially Nε-lysine myristoylation of ARF6 GTPase.126 Due to this ambiguity, this modification is more rigorously referred to as Nε-lysine fatty/defatty-acylation. Proteome-wide analysis of probe-labeled Nε-lysine fatty acylated proteins can be accomplished through combining CCML and quantitative proteomics with genetic manipulation, chemical inhibition of the hydrolases, or selective hydrolysis of N-glycine myristoylated proteins. Lin and coworkers utilized both Alk-C14 and Alk-C16 in a SILAC-based quantitative proteomic analysis to profile the targets of the defatty-acylase SIRT6 in mouse embryonic fibroblasts (MEFs) with wild-type and SIRT-6 knockout phenotypes.124 Out of 865 and 1285 proteins enriched from Alk-C14 and Alk-C16 labeling, respectively, the list was narrowed down to 5 proteins as targets of SIRT6 after strict data filtering. One of those proteins was RRas2, where Nε-lysine fatty acylation is essential for the activation of P13K/Akt pathway and promotion of cell proliferation. Owing to the stringent criteria used in this study, the data should be interpreted with caution as many actual protein targets may have not qualified to be included in this extremely reduced list of SIRT6 substrates. Following the same approach using Alk-C16, a proteome-wide analysis of MCF-7 cells identified SHMT2 as an HDAC11-mediated defatty-acylated protein.130 The Nε-lysine fatty/defatty-acylation of SHMT2 was shown to play a key role in regulating the type I interferon (IFN) signaling during immune response.
Several other hydrolases including SIRT1, SIRT2, and SIRT3 and HDAC8 display appreciable lysine defatty-acylation activity in vitro.127,320 SIRT2 efficiently hydrolyzes Alk-C16 in metabolically labeled KRas-4a, RalB, and ARF6, but no validated fatty acyl-modified substrates were identified for the other hydrolases.123,125,126,321 Proteins with fatty acylation regulated by these hydrolases are anticipated and their discovery should be facilitated by the established CCML and chemical proteomic approaches described here. Importantly, while these relatively recent studies presented strongly suggest that lysine fatty/defatty-acylation is plausible based on clickable probe labeling, they have been conducted through exogenous treatment with fatty acid analogues. It is possible that this approach may result in induced hypermodification of proteins that may not necessarily reflect physiological conditions. In order to further hone in on the role this type of modification and its potential biological implications, direct evidence of Nε-lysine modification under more native (unperturbed) conditions may be required. Future investigations should strive towards providing concrete evidence for the existence of this type of modification and deconstruct the precise molecular mechanisms that rationalize its occurrence. It is noteworthy that while NMTs can facilitate Nε-fatty acylation in Gly1Lys2-containing substrates, the enzymes responsible and mechanisms involved in this type of modification on Lys residues near the C-terminus of a protein such as in KRas-4a and RalB remain unknown. It is possible that an S-to-N type of lipid transfer may occur from initially formed S-acylated proteins but further studies are needed to investigate this possibility.
6.2. Mammalian N-myristoylated proteins
The use of Az-C12 was pioneered by Hang and co-workers for detecting the N-myristoylation of the kinase Lck at endogenous levels via Staudinger ligation.178 This marked the birth of using clickable myristic acid probes to validate N-myristoylation substrates. While intact proteins are viable for post-translational labeling in vitro,322 the co-translational nature of N-myristoylation on nascent proteins can be examined by tagging proteins in one pot using cell-free transcription/translation systems (Fig. 12A).309 In this strategy, genes for proteins of interest are inserted in plasmids and transcribed and translated in lysates obtained from rabbit reticulocytes or insects. These lysates contain the essential elements to express proteins and in the presence of a clickable myristic acid analogue, co-translational labeling takes place. Importantly, the free acid form of the lipid analogues can be used since they are converted to their -CoA forms in situ.
Figure 12. Mechanisms for labeling the N-myristoylated proteins.
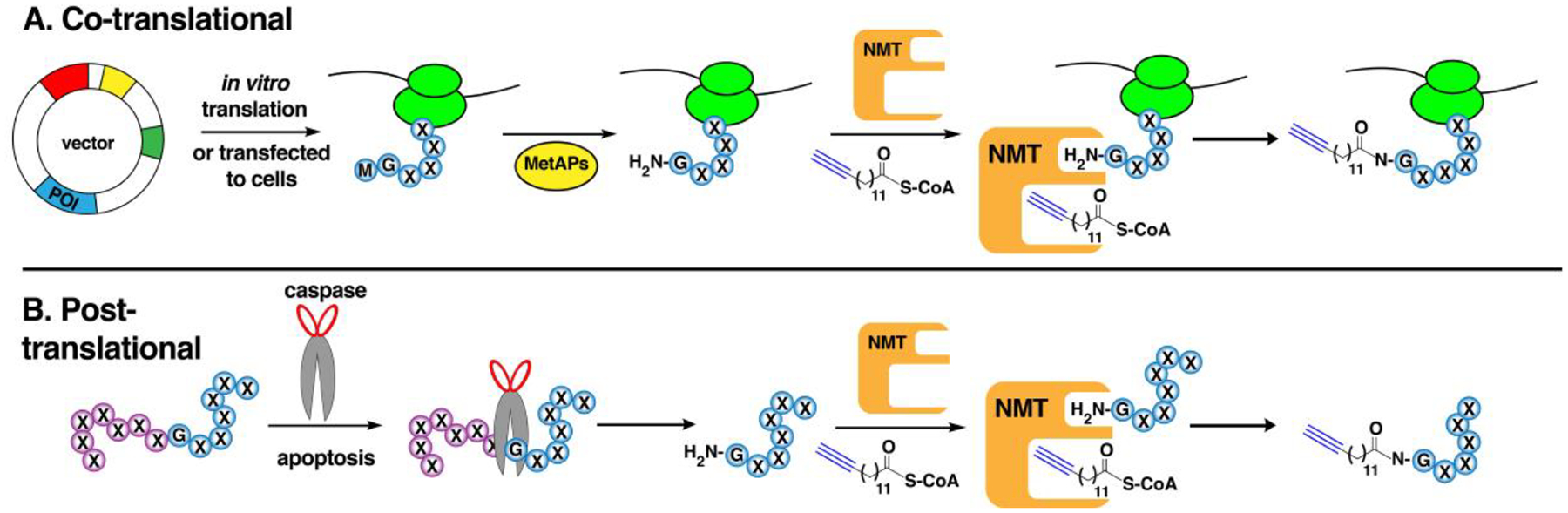
(A) Co-translational modification where probes are added to either cultured cells or in vitro in lysates containing plasmids that express proteins of interest (POI) for cell-free expression. The N-terminal methionine is cleaved off by MetAPs and the clickable myristoyl analogue is appended by NMTs to the N-terminal amine on the exposed glycine of the nascent protein. (B) Post-translational N-myristoylation during apoptosis on protein substrates generated through cleavage by caspases that expose a new N-terminal glycine.
Prior to the development of Alk-C14, the complementarity between azides and phosphines for Staudinger ligation enabled the use of Az-C12 to rapidly detect and identify post-translationally N-myristoylated proteins during apoptosis.304 The Staudinger reaction was also useful in confirming the N-myristoylation and S-palmitoylation of DCNL3 in cells treated with Az-C12 and Az-C14, respectively.323 While related proteins DCNL1 and DCNL2 share the same function by mediating Cul neddylation, only DCNL3 is directed to the plasma membrane through a myristoyl anchor, facilitating neddylation of the membrane-localized Cul protein. This exemplifies the role of lipid modification in modulating localization and subsequent substrate specificity of related proteins sharing the same function. Likewise, this method aids in understanding how species-specific function is regulated by N-myristoylation. Human LMCD1 activates transcription factors that stimulate cell proliferation and migration while the mouse homolog conversely represses this pathway. The NMT2-mediated N-myristoylation of mouse LMCD1, as evidenced by Az-C12 labeling, reversed its repressor activity, underscoring the differential roles of LMCD1 transactivation activity in humans versus mice.324 Furthermore, N-myristoylation regulates the cell type-specific sorting of proteins such as the spermatozoan variant Hexokinase 1 (HK1S).325 Cell-free translation combined with co-translational labeling with Az-C12 and subsequent Staudinger detection confirmed HK1S N-myristoylation; and the presence of myristate directs it to the plasma membrane, which is not the case in other cell-types.
Labeling with Az-C12 was also the basis for the development of an ELISA-based method called NMT-azido-ELISA (Fig. 9D).310 This approach differs from those designed for palmitoylation in that it does not involve immobilization in the first step. Instead, peptide substrates bearing a FLAG tag are enzymatically labeled with Az-C12-CoA in solution followed by Staudinger coupling with a biotin-phosphine reagent. The biotinylated substrates are then captured with immobilized anti-FLAG antibodies and detected through peroxidation-driven chemiluminescence. This versatile technology offers the ability to measure enzymatic activities of wild-type and mutant NMTs, as well as measure the efficacy of candidate NMT inhibitors. Using this method, the loss of NMT activity in NMT-deficient knockouts was confirmed, serving as a framework for the succeeding biochemical assays that pinpointed that NMT deficiency results in aberrant T cell development; this was later ascribed to changes in calcineurin activity during IFN signaling.326,327 Although this method uses Staudinger reaction in its current form, it could be adapted to function with other bio-orthogonal reactions that manifest faster kinetics and use more stable reagents.
The Alk-C14 probe has found wider applications in discovering and characterizing N-myristoylated proteins owing to its lower background, similar to the clickable palmitic acid probes.179,180 CCML with Alk-C14 aided the discovery of regulatory mechanisms of proteins or their binding partners through their subcellular localization dictated by N-myristoylation. The N-myristoylation on Fragile X-related protein 2 (FXR2P) was shown to control its distribution in neuronal axons and its interaction with Fragile X mental retardation protein (FMRP).328 Since FMRP plays important roles in axonal and presynaptic functions and relies on FXR2P for proper localization, N-myristoylation on FXR2P may be implicated in Fragile X syndrome linked to intellectual disability and autism. N-myristoylation also influences the subcellular localization of proteins associated with mitochondrial morphology and degradation (mitophagy). Myristate modification directs the autophagy receptor mouse Stbd1 to ER-mitochondria junctions, and impairment of this process induced significant mitochondrial fragmentation and clustering, possibly associated with autophagy.329 Similarly, the N-myristoylation-dependent localization of AMP-activated proteins kinase (AMPK) to damaged mitochondria was also found to mediate mitophagy.330 Treatment with a 2-hydroxymyristic acid, a weak inhibitor of NMTs,331 validated its N-myristoylation through mislocalization upon treatment with effective concentrations of this inhibitor. Furthermore, N-myristoylation can be controlled by proteins that modulate the interaction between NMTs and their substrates. The CSIG protein facilitates N-myristoylation of PPM1A by mediating the NMT-PPM1A interaction.332 Evidence for a CSIG-PPM1A-NMT complex was obtained by co-immunoprecipitation and reduced Alk-C14 labeling of PPM1A was observed upon CSIG knockdown. Indeed, the use of clickable analogues of myristic acid not only directly validates the myristoyl modification on proteins but also can indirectly confirm the role of this lipid modification in the assembly of protein complexes.
Although most N-myristoylated proteins are co-translationally modified, a number of substrates are post-translationally modified during apoptosis.315 Apoptotic caspase activation leads to N-myristoylation of a newly exposed N-terminal glycine (Fig. 12B).333 Thus, CCML with Az-C12 or Alk-C14 was used to show the upregulation and post-translational N-myristoylation of protein kinases A and C during chemically induced apoptosis.238,307 Alk-C14 labeling readily detected drastic changes in the N-myristoylated proteome during apoptosis, with concomitant caspase-mediated cleavage of NMTs resulting in impaired NMT activity.114 The interplay between NMTs and caspases may explain the high expression levels of NMTs in cancer cells subverting apoptosis. Moreover, the caspase-cleaved huntingtin protein (HTT) liberates a myristoylated C-terminal product detected using Alk-C14.334 The lipidated product localizes to the ER and promotes accumulation of autophagosomes, suggesting a connection between HTT and autophagy with possible implications in Huntingtin’s disease (HD). A later study showed that an HTT variant with G533E mutation blocked caspase cleavage and subsequent N-myristoylation, leading to cytotoxicity.335 It was noted that this HTT variant was an important disease marker that may lead to earlier onset of HD.
Several proteins associated with oncogenesis such as Src and various kinases are dependent on N-myristoylation for their proper cellular function.336 Alk-C14 labeling revealed that excessive exogenous levels of myristic acid elevates levels of N-myristoylated Src, implicating high-fat diets as drivers of prostate cancer tumorigenesis.337 In a separate study, Az-C12 labeling illustrated that NMT1 regulates cancer cell proliferation through Src N-myristoylation and blocking the lipid-modification suppresses Src kinase activity.338 A recent study has shown that treatment with the NMT pan-inhibitor PCLX-001 along with Alk-C14 labeling in B-cell lymphoma induced global impacts on the N-myristoylated proteome and particularly abrogated Src N-myristoylation.339 This protein along with non-myristoylated proteins involved in B-cell receptor signaling were degraded upon NMT inhibition, leading to cancer cell death both in vitro and in vivo. In addition to Src, the N-myristoylated protein fibroblast growth factor receptor substrate 2 (FRS2) also contributes to carcinogenesis through FGF/FGFR-mediated oncogenic signaling and FGF10-induced tumorigenesis.340 Pharmacologically targeting its N-myristoylation, as verified through diminished Az-C12 incorporation, inhibits cancer cell proliferation and migration as well. These studies provide evidence that designing effective NMT inhibitors can lead to promising drug candidates for cancer treatment. Clinical applications for such compounds including PCLX-001 are anticipated. However, exploring their on-target selectivity through enzymology and crystallographic studies will be required to validate these inhibitors, especially in the case with PCLX-001 treatment where many non-myristoylated proteins were also degraded.
6.3. Parasitic protozoans and fungi
Targeting NMTs is a viable strategy for the development of therapeutic agents owing to the requirement of N-myristoylation for the survival of human pathogens.111,341–343 Integrated chemical biology methods combining CCML and quantitative proteomics with selective NMT inhibition allowed for the robust identification of the parasite N-myristoylated proteome in P. falciparum.177 From that proteome-wide analysis, the proteins ISP1 and ISP3 required for RBC invasion emerged as attractive targets; additionally, their N-myristoylation status was previously validated using Alk-C14 in a related malaria parasite, P. berghei.344 Increasing resistance of these parasites to current antimalarial drugs underscores the need to generate new classes of parasite-specific chemotherapeutics. In an effort to preempt potential resistance to lead candidate inhibitors of P. falciparum NMTs, a mutant PfNMT with a single amino acid substitution (G386E) was generated that exhibited resistance to current known parasitic NMT inhibitors.345 Such an approach enabled the generation of a new class of NMT inhibitors capable of inhibiting the growth of predicted resistant strains. IMP-1002 emerged as a lead candidate with on-target inhibition of NMT as shown by decreased labeling by Alk-C14 of myristoylated proteins. Future studies can be directed towards designing pharmacological inhibitors against G386E-mediated resistance without compromising target selectivity.
Trypanosomes are the causative agents for fatal diseases including African sleeping sickness and Chagas disease caused by Trypanosoma brucei and Trypanosoma cruzi, respectively. N-myristoylation is essential for the cell viability and development of these parasites and therefore offers an attractive target for treating those diseases. For example, N-myristoylation of ARL6 was demonstrated using CCML with Alk-C14 and is important for its membrane localization to promote flagellum extension in T. brucei.346 Global profiling robustly identified 101 enriched proteins with 46 possessing the N-terminal glycine motif and 53 responded to TbNMT inhibition.212 On the other hand, the azide analogue Az-C12 was employed to validate NMT inhibition as a potential therapeutic strategy to target T. cruzi.347,348 Subsequent CCML with Az-C12 and quantitative proteomic analysis profiled more than 50 N-myristoylated proteins sensitive to NMT inhibition.349 Only half of that list of proteins overlaps with those identified from T. brucei.212 This observed disparity in the sets of myristoylated proteins between two species could be attributed to the type of myristic acid analogues used or differences in metabolism such as the rate of plasma turnover, as evidenced by their differential responses to NMT inhibition.212
Other neglected tropical diseases such as leishmaniasis and toxoplasmosis are caused by protozoans that also require N-myristoylation for proper protein functions.343 Label-free quantitative analysis with Alk-C14 labeling in L. donovani revealed 67% overlap between the parasite and host N-myristoylated proteomes, of which 30 parasitic N-myristoylated proteins displayed sensitivity to LdNMT inhibition.350 Although this indicates that LdNMT is a druggable target, some bonafide N-myristoylated proteins including CAP5.5 and phosphatase PPEF did not respond to chemical inhibition. This prompted the development of more potent pharmacological inhibitors based on benzothiophene and pyrazolyl scaffolds that exhibit high selectivity for LdNMT.351,352 However, these effective inhibitors suffer from problems of chemical stability and display poor cellular uptake. A recent study reported a comprehensive development of a series of thienopyrimidine-based small molecules that showed high selectivity towards LdNMT versus human NMT.353 Through the use of Alk-C14 labeling, a dose-dependent decrease in signals from N-myristoylated proteins was observed, signifying the on-target intracellular activity of this novel set of LdNMT inhibitors. Furthermore, these promising therapeutic agents are effective against the amastigote form of L. donovani, which is an excellent model to study owing to its clinical relevance.
Global profiling of N-myristoylation in T. gondii identified 76 myristoylated proteins including 31 N-myristoylated peptides using azide-modified, trypsin- and TEV-cleavable multifunctional reagents.354 Validation of N-myristoylation through Alk-C14 labeling on two proteins, CDPK1 and MIC7, was accomplished, followed by biochemical assays resulting in defining their key roles in egress and host-cell invasion, respectively. Interestingly, a recent study validated the N-myristoylation of the previously uncharacterized active serine hydrolase 4 (ASH4) in T. gondii through Alk-C14 labeling.355 N-myristoylation on ASH4 does not influence membrane localization but is involved in a mechanism important for parasite organization in the parasitorous vacuole. Prior evidence suggested that this enzyme is a protein depalmitoylase.356 However, biochemical and cell-based assays in this more recent work demonstrated its esterase activity on small molecules with short chain acyl esters.
N-myristoylation is also of functional importance in opportunistic fungal pathogenic genera including Candida, Cryptococcus, and Aspergillus, indicating that NMTs could serve as attractive targets for antifungal agents.357–360 Despite the fact that this lipid modification has been established in these pathogens for decades, information on their NMT substrates is limited. Advances in chemical proteomic strategies should pave the way for defining their sets of N-myristoylated proteins. Recently, Tate and coworkers profiled the N-myristoylated proteome using Alk-C14 in the fungal pathogen of wheat, Zymoseptoria tritici.361 Out of the 25 predicted N-myristoylated proteins from the fungal proteome, 20 were detected through LC-MS analysis, providing a robust list of N-myristoylated proteins in Z. tritici. Furthermore, a ZtNMT-selective inhibitor IMP-1088 showed an NMT-dependent mode-of-action, in contrast to a previously reported NMT inhibitor IMP-162 that displayed off-target effects.
It is noteworthy that there are no Nε-lysine fatty/defatty-acylation substrates amongst these protozoan and fungal pathogens reported to date. However, the previously characterized P. falciparum SIRT2 can catalyze hydrolysis of medium and long chain fatty acyl groups on model peptides in vitro.362 Hence, lipid modifications on the Nε-side-chain may yet be discovered in protozoans and chemical biology tools such as CCML should aid in the characterization of its potential functional importance in these parasites. Similar to those experiments conducted in mammalian cells, detection of this type of modification under native conditions is necessary to demonstrate its true existence in lower eukaryotes.
6.4. Bacteria and viruses
As mentioned above, bacterial LPPs can be modified with diacylglyceryl bearing fatty acids on an exposed N-terminal cysteine (Fig. 13A). Compared with S-palmitoylation, only a modest number of myristoyl-containing LPPs are present in Gram-negative E. coli.202 However, clickable fatty acid labeling in the Gram-positive Clostridium difficile, a bacteria associated with severe gastrointestinal diseases, revealed that Alk-C14 can efficiently label its LPPs.363 Inactivation of lipoprotein diacylglyceryltransferase (Lgt) led to almost complete loss of probe labeling, validating the observed N-myristoyl modification that occurs on LPPs. Quantitative proteomic analysis then yielded 65 candidate LPPs with 56 responsive to competition with the native myristate, comprising 74% of the predicted LPP proteome in C. difficile. The transcription factor Spo0A which regulates sporulation, a self-preservation mechanism of bacteria, was further shown to be modulated by LPP expression. These results highlight the involvement of LPPs in C. difficile sporulation and lays the groundwork for future studies on LPP lipidation in other Gram-positive bacteria.363
Figure 13. Bacteria-mediated lipidation/delipidation.
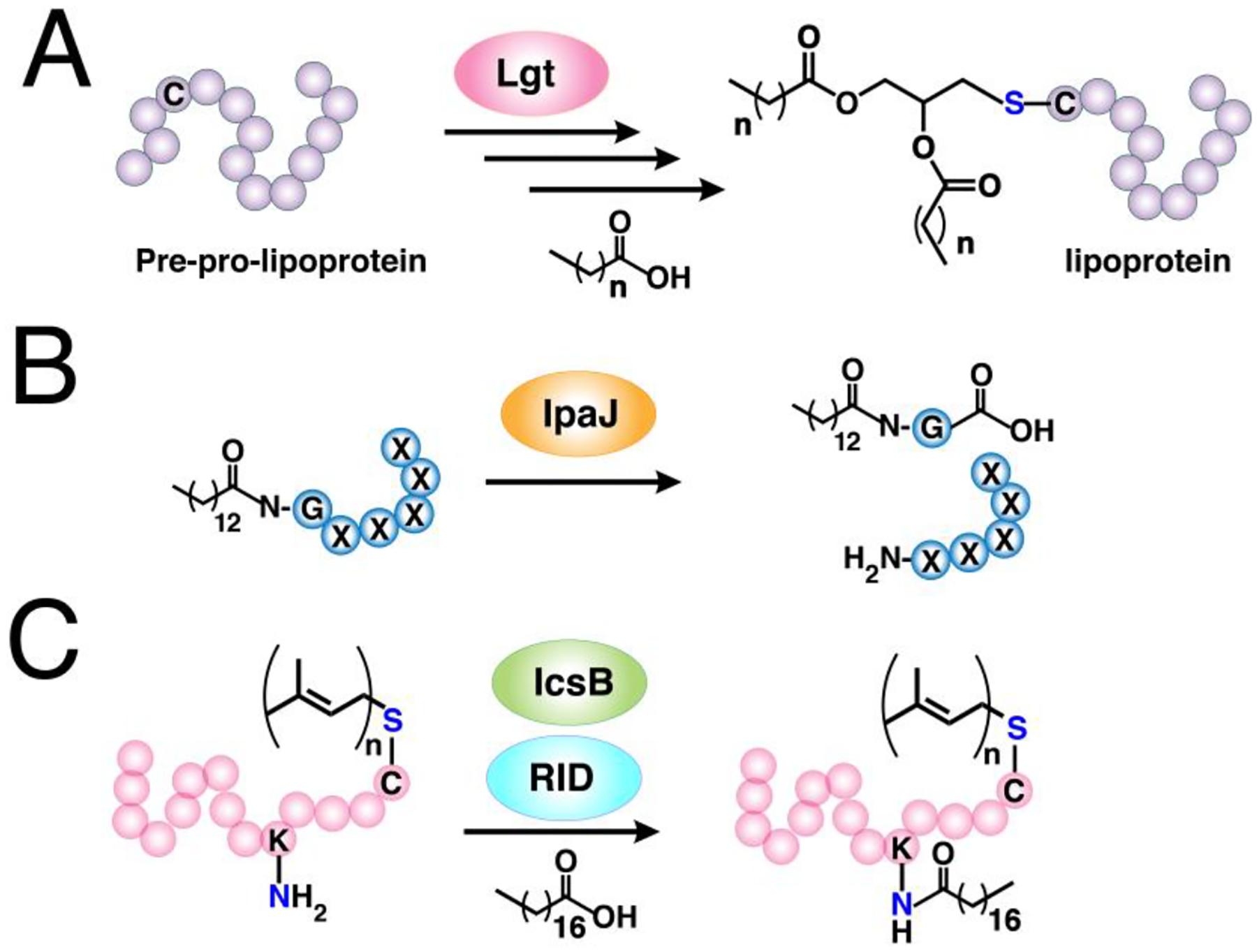
(A) Incorporation of saturated fatty acids into the S-linked diacylglycerol unit on bacterial lipoproteins (LPPs). The pre-pro-lipoprotein undergoes processing steps that exposes a new N-terminal cysteine and its thiol side-chain is modified by lipoprotein diacylglyceryltransferase (Lgt) with a glyceryl group containing two fatty acyl moieties. (B) Demyristoylation on host proteins catalyzed by the bacterial effector protein IpaJ. The N-myristoylated N-terminal glycine is cleaved by this peptidase. (C) N-fatty acylation on prenylated small GTPases by bacterial effector proteins IcsB or RID. The prenyl modification on their substrates appears to be essential for their recognition.
Bacteria also leverage protein N-myristoylation to direct their secreted effector proteins to appropriate subcellular locations critical for their virulence.119,364,365 Concurrently, these pathogens may also alter PTMs on host proteins to redirect host pathways that promote their replication and escape from the host immune system.366 A bacterial T3SS effector protein IpaJ peptidase, a cysteine protease from Shigella flexneri, was discovered to efficiently cleave the N-myristoylated glycine of host proteins (Fig. 13B).367 Lysates from mammalian cells metabolically labeled with Alk-C14 showed diminished global N-myristoylation after infection with S. flexneri.368 Global N-myristoylated proteome profiling revealed a number of substrates cleaved by IpaJ with high specificity to N-myristoylated ARF/ARL GTPases. The myristoyl group on these proteins serve as the binding site of IpaJ, leading to hydrolysis of the substrate glycine peptide bond catalyzed by its active site cysteine. Interestingly, ARF6 was the only protein in ARF family found to be an unsuitable substrate for IpaJ. It is possible that the observed regulation on ARF6 dimyristoylation by NMTs for its localization in the plasma membrane is implicated in the poor cleavage by IpaJ.126 In a subsequent study, BT3 from Vibrio vulnificus, which belongs to a family of multifunctional auto-processing repeats-in-toxin (MARTX) effector proteins, was observed to bind myristoylated ARF1.369 Its domain X (DmX) is structurally homologous to a cysteine protease AvrPphB and it was therefore speculated to hydrolyze N-myristoylated glycine. However, Alk-C14 labeling experiments did not reveal diminished N-myristoylation of ARF1 upon incubation with DmX unlike IpaJ.
N-myristoylation of viral proteins is critical for their host cell entry, replication, assembly, and infectivity.370–372 Due to the small proteomes of viruses, N-myristoylated proteins can be conveniently assigned based on predicted N-terminal glycine motifs unlike S-palmitoylation. However, viruses not only require lipid-modification of their proteins but also modulate host-cell PTM machinery.366 Therefore, CCML allows the detection and identification of not only N-myristoylated viral proteins, but also host proteins impacted due to infection. Az-C12 labeling in infected cells validated the N-myristoylation of Pr55gag HIV-1 protein, and that NMT1 promotes HIV-1 replication by modulating the expression of viral proteins.373 In HSV, quantitative profiling of N-myristoylated proteins using Alk-C14 in HSV-infected cells afforded 5 putative myristoylated viral proteins and revealed a dramatic reduction in host global N-myristoylation.206 Although novel myristoylated viral proteins were identified, known N-myristoylated viral proteins were not enriched, potentially owing to the small number of tryptic peptides from these short proteins.
Among the diverse roles of N-myristoylation in promoting viral virulence, the co-translational N-myristoylation of viral structural capsid proteins (VPs) is critical for viral capsid assembly.374 Therefore, inhibiting their N-myristoylation may attenuate viral infectivity. Effective inhibition by a non-selective human NMT inhibitor DDD85646 on the capsid protein VP0 was verified using Alk-C14 and in-gel fluorescence assays.375 Cells infected with the picornavirus CVB3 and treated with DDD85646 diminished viral RNA encapsidation and viral genome transfer, rendering the virus less infectious. At the same time, Tate and coworkers screened a series of HsNMT inhibitors on cells infected with the common cold virus, rhinovirus (RV).376 The most potent and HsNMT-selective inhibitor IMP-1088 reduced N-myristoylation of host proteins and capsid protein VP0 in a chemical proteomic profiling experiment. This picomolar inhibitor is a derivative of the P. vivax NMT-specific IMP-72,377 refined through a fragment reconstruction and linking approach. Moreover, it displayed effective inhibition of replication of poliovirus and foot-and-mouth disease virus with no observed toxicity to host cells.
Given the key role of N-myristoylation in oncogenesis and infections, designing human and parasitic NMT-selective inhibitors as potential chemotherapeutic agents against cancer and infectious diseases remains an active area of research.336,343 Several inhibitors have emerged and been claimed to be selective for targeting HsNMT. However, a recent study investigated the potency of five commonly used inhibitors against HsNMT through a series of biochemical assays including metabolic labeling with Az-C12.331 Three of those compounds failed to arrest NMT activity while two high-affinity inhibitors displayed on-target NMT inhibition with low cytotoxicity. Transforming these potent inhibitors into formulations viable for clinical trials is therefore anticipated.
7. Biological applications in S-Prenylation
The existing paradigm for protein S-prenylation is that it occurs on proteins that terminate with Ca1a2X box motifs for farnesylation and geranylgeranylation, and CCXX, XXCC, XCXC motifs for dual geranylgeranylation of the Rab family of proteins.378–380 Basing on those criteria, previously validated prenylated proteins have been used as models to generate computational tools that allow in silico screening for protein S-prenylation substrates.140,381 However, despite the existence of canonical S-prenylation motifs and prediction tools, several approaches have been put forth to experimentally validate the true S-prenylation status of proteins. These methods involve in vitro reactions on Ca1a2X box-containing substrates in peptide libraries or genetic screening in yeast thermotolerance assays.138,139,382,383 Although these methods are generally high throughput, they are often limited to Ca1a2X box peptide substrates mimicking the C-termini of proteins. The sequences upstream of the Ca1a2X box on proteins contribute to substrate reactivity and therefore these peptides may not completely reflect substrate recognition on intact proteins.141
Bio-orthogonal labeling of prenylated proteins rapidly emerged and a number of isoprenoid analogues were reported.16 The promiscuity of prenyltransferase enzymes offers the advantage of introducing a variety of functional groups onto the isoprenoid probe.384,385 For example, light-activatable and antigenic groups enabled photoaffinity labeling and prenyl-targeted antibody-based detection of prenylated proteins.386–390 A biotin-conjugated geranyldiphosphate analogue (BGPP, Fig. 14) was also utilized to directly detect and enrich prenylated proteins in both metabolic labeling and in vitro S-prenylation experiments, obviating the need of secondary tagging unlike click reaction-based approaches.158,391–393 Although that probe is an efficient substrate for RabGGTase owing to this enzyme’s larger binding site, it however requires an engineered FTase and GGTase-I to effectively label singly prenylated proteins.391 Therefore, the scope of BGPP in probing prenylated proteins is restricted.
Figure 14. Bio-orthogonal isoprenoid analogues for probing protein prenylation.
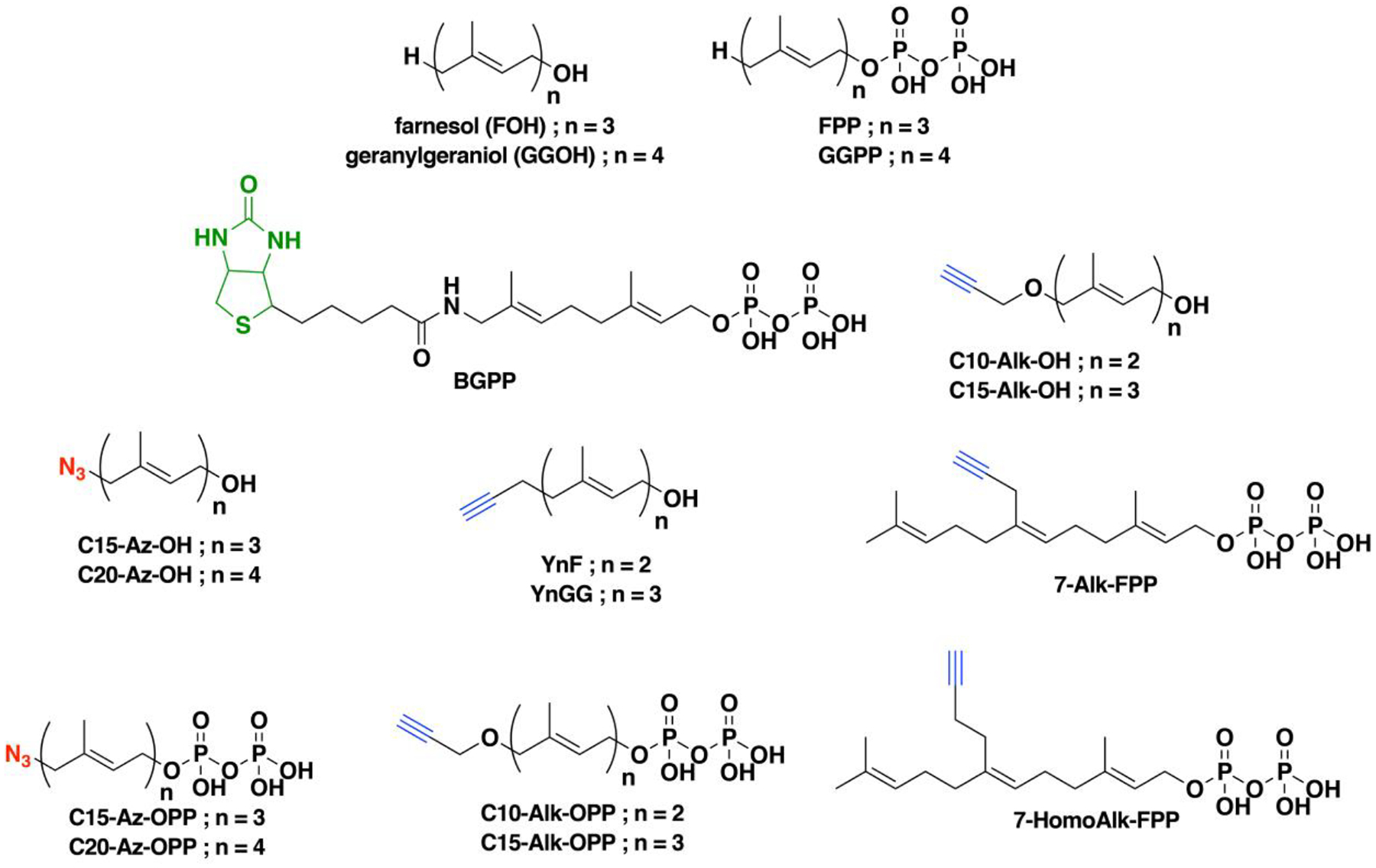
Native substrates farnesol (FOH) and geranylgeraniol (GGOH) or their diphosphate forms FPP and GGPP modified with azide or alkyne functionalities suitable for CCML. A biotinylated isoprenoid can also be used to directly label Rab substrates but not of those for FTase and GGTase-I.
Clickable analogues of isoprenoids containing azides and alkynes were developed in different versions of alcohol and diphosphate forms (Fig. 14).394–397 Although isoprenoid diphosphates are the bonafide substrates for prenyltransferases, the alcohol versions of the probes have been extensively used in metabolic labeling experiments since they are readily incorporated into proteins through intracellular conversion by host-cell kinases.398,399 This additional pathway (distinct from the well characterized mevalonate pathway) for converting the alcohol forms to their diphosphate counterparts is not well understood and warrants further investigation concerning what enzymes are involved and how this process occurs. However, it is important to note that the exogenous treatment of isoprenols (e.g. farnesol) induces potential toxicity that may impact cell physiology. This may complicate the biological interpretation of experiments using alcohol analogues of isoprenoid probes.400,401 The azide functionalized farnesol (C15-Az-OH) and the corresponding diphosphate form, C15-Az-OPP, were first reported by two independent groups to detect prenylated proteins via Staudinger ligation.394,395,402 Later studies demonstrated that the longer C20-Az-OH and C20-Az-OPP are more suitable for detecting geranylgeranylated proteins.403,404 However, employing alkyne-modified isoprenoids results in lower noise signal compared to the azide versions, consistent with the observations in using fatty acylation probes.179,180,405 Therefore, alkyne-modified isoprenoids were exploited to identify and characterize prenylated proteins. The position of the alkyne was also varied along the isoprenoid chain such as in the second isoprene unit of FPP (7-Alk-FPP and 7-HomoAlk-FPP).406 Kinetic analysis comparing C15-Alk-OPP and 7-substituted FPP indicates that alkyne-modification at the terminus of FPP presents the best mimic for the native FPP.407 In this section, studies that utilized these isoprenoid analogues to profile and validate known and novel prenylated proteins are reviewed.
7.1. S-prenylated proteome profiling
Proteomic profiling of prenylated proteins has been reported using various isoprenoid analogues via metabolic labeling in a number of studies. These methods often involve the use of statins that impede isoprenoid biosynthesis by inhibiting HMG-CoA reductase in the mevalonate pathway.408 This reduction of the pool of endogenously occurring isoprenoids generally improves labeling efficiency by forcing the prenyltransferase enzymes to incorporate the analogues in lieu of the native isoprenoids. Methods to profile a set of S-prenylated proteins from cells of interest can be performed through metabolic labeling or in vitro S-prenylation approaches as described below.
7.1.1. Metabolic labeling with isoprenoid analogues
Early pioneering work utilized the alcohol probe C15-az-OH to metabolically label farnesylated proteins in statin-treated cells.394 Tagging via Staudinger ligation with biotin-phosphine for affinity enrichment, followed by proteomic analysis resulted in the identification of 17 enriched proteins containing Ca1a2X motifs. A longer analogue C20-az-OH was used in a subsequent study to tag geranylgeranylated proteins, which were detected using an alkyne-containing fluorophore.403 Separation of the labeled proteins via two-dimensional SDS-PAGE and excision of the labeled spots subjected to LC-MS/MS afforded a list of 10 geranylgeranylated proteins dominated by Rabs.
The alkyne containing analogues C10-Alk-OH and C15-Alk-OH along with their diphosphate counterparts C10-Alk-OPP and C15-Alk-OPP, respectively, were evaluated for their labeling efficiencies in HeLa cells.405 Replacing the alcohol probes with the diphosphate analogues enabled effective labeling although no direct comparison was performed. However, other cell lines may have behave differently such as COS-7, which displayed intense labeling of prenylated proteins using the diphosphate analogue versus the alcohol form. Treatment with lovastatin displayed modest enhancement of labeling in HeLa, indicating that this cell line maybe less responsive to statin treatment, or isoprenoid labeling in this cell line is less efficient such that small differences cannot be observed. Following a two-dimensional SDS-PAGE approach for proteomic analysis, only 6 Ca1a2X -containing proteins were identified, reflecting a low efficiency of labeling in HeLa cells treated with C10-Alk-OH. Optimal concentrations of lovastatin may have not been attained to furnish enhanced probe labeling.409 Furthermore, HeLa cells may not respond to isoprenoid labeling as efficiently as other cell lines do such as COS-7, which displayed excellent labeling with C15-Alk-OPP but not with C15-Alk-OH.410 This disparity in labeling between cell lines using different forms of the isoprenoid probe can be rationalized with the observation that different cell-types respond with varying degrees of probe labeling with C15-Alk-OH.411 Profiling of S-prenylated proteome in the macrophage cell line RAW264.7 yielded 23 Ca1a2X-containing and 12 Rab proteins using C15-Alk-OH.412 Furthermore, a novel prenylated zinc antiviral protein (ZAP) was discovered and further characterized to show that its farnesylation enhances its membrane-targeting and inhibitory activity against Sindbis virus.412
Like other protein lipid-modifications, S-prenylation mediates diverse pathways across organisms and is therefore implicated in numerous human diseases, as well as viral, bacterial, and protozoal infections.11,413 For example, prenyltransferase inhibitors (PTIs) directed towards the development of antimalarial drugs indicate that protein S-prenylation is indispensable for parasite development.414 Two independent studies sought to define the S-prenylated proteome in that organism. First, Distefano and co-workers employed bioinformatic analysis and proteomic profiling using C15-Alk-OPP in blood-stage P. falciparum.415 A total of 15 putative S-prenylated proteins were identified comprising 78% of the bioinformatically predicted protein S-prenylation substrates in the parasite. One of the proteins identified, heat shock protein 40 (Hsp40), was later shown to require farnesylation to promote parasite thermotolerance and facilitate vesicular trafficking.416 A subsequent study provided a similar list of S-prenylated proteins in the parasite using C15-Alk-OH.417 It was shown that farnesylation of the protein FCP directs it to the parasitorous vacuole and treatment with an antimalarial farnesyltransferase inhibitor (FTI) disrupts its localization. Furthermore, an unusual S-prenylation on PfFRab5b was observed, which does not contain the canonical S-prenylation motifs. These studies demonstrated that the malaria parasite possesses a restricted set of S-prenylated proteins, making prenyltransferases attractive targets to develop antimalarial drugs.
More than 200 proteins bearing S-prenylation motifs in mammalian cells are predicted to be S-prenylation substrates based on genomic, structural, biochemical, and functional analyses.2 Although the C15-Alk-OH and C15-Alk-OPP can be incorporated into the three classes of S-prenylation substrates, early reports on profiling of S-prenylated proteins in diverse mammalian cell lines provided less than a quarter of the predicted number.397,405,412 Recently, Tate and coworkers applied a dual chemical probe labeling strategy for CCML, where both alkyne-modified farnesyl (YnF) and geranylgeranyl (YnGG) isoprenoid analogues were used in tandem to identify protein substrates of the three classes of S-prenylation in a human endothelial cell line.418 These probes are shorter by one atom compared to the previously reported S-prenylation probes C10-Alk-OH and C15-Alk-OH. A total of 80 known and novel S-prenylated proteins were identified with 64 detected at endogenous levels in the absence of statins. The assignment of the S-prenylation status of proteins (farnesylated or geranylgeranylated) were deduced through their ability to be prenylated by YnF or YnGG and their corresponding response to FTI or GGTI treatment, as well as to competition with FPP or GGPP. In addition, a cleavable multifunctional reagent, originally developed for the detection of N-myristoylated proteins, was used in the enrichment step,176 which allowed for the detection of prenylated peptides in the LC-MS/MS analysis. A total of 26 intact S-prenylated peptides from 18 distinct proteins were successfully detected. Furthermore, a choroideremia disease model with knocked out Rep-1, the escort protein that recruits Rabs for S-prenylation, showed 10 out of 29 quantified Rab proteins as having a significant reduction in S-prenylation. In particular, Rab12 and Rab27b displayed the most significant reduction in prenylation in this model. Previous in vitro S-prenylation studies indicated that Rab27b along with Rab27a, Rab38, and Rab42 are the least efficient substrates of RabGGTase, suggesting that these proteins are the key contributors to the disease.158 The latter three Rab proteins were not quantified nor identified in the MEFs with Rep-1 knockouts, perhaps owing to the limitations in the detectability of these proteins in the chemical proteomics workflow. It is also worth noting that these results in Rep-1 knockout models indicate that under conditions where Rep-1 is dysfunctional, rescue by the other escort protein Rep-2 is not efficient. This further supports the previous findings that suppression of Rab S-prenylation occurs in retinal degeneration in the choroideremia disease.419
While the study above demonstrates that the use of more than one isoprenoid probe widened the scope of S-prenylated proteins profiled through quantitative chemical proteomics, a recent report revealed that C15AlkOPP acts as a surrogate for both FPP and GGPP, leading to the identification of a comparable number of prenylated proteins in COS-7.420 Using this single isoprenoid analogue also led to the identification of novel prenylated proteins that were not reported in the dual isoprenoid probe labeling strategy (although the converse of this was also true). In an effort to rationalize what factors affect the detectability of S-prenylated proteins in this chemical proteomics scheme, predicted scores and reported in vitro activity assays for S-prenylation substrates were investigated but none appeared to correlate with the degree of enrichment of each substrate. Rather, the best correlation was observed when the fold-enhancement from the proteomic analysis was compared with the native abundances of the S-prenylation substrates. Furthermore, COS-7 displayed superior labeling compared with HeLa and the brain-derived cell lines for neurons, microglia, and astrocytes, with variations in the extent of labeling across cell lines. This indicates that the cell line of choice also greatly affects the number of detectable prenylated proteins that can be identified. These differences may be attributed to the innate expression levels of the prenylation substrates in each cell line, their levels of the prenyltransferases, and their respective efficiencies for probe uptake.420 It is noteworthy that these experiments were performed using both stable cell lines and primary cells suggesting that these isoprenoid analogues should be useful for a broad range of experiments with other primary cells and perhaps even tissue samples or whole animals, thereby greatly increasing their utility.
7.1.2. In vitro labeling
The chemical proteomic approaches discussed so far for profiling of S-prenylated proteins involve metabolic labeling with isoprenoid probes. In some instances, samples of interest may not be metabolically active and hence not suitable for metabolic labeling. These experiments may require labeling of prenylated proteins outside of the biological matrix. In vitro S-prenylation on protein substrates was pioneered by Alexandrov and co-workers using the biotinylated isoprenoid analogue BGPP (Fig. 14).391 Lysates from cultured COS-7 cells treated with statin were prenylated with BGPP in the presence of the prenyltransferases in vitro and subjected to streptavidin blot detection and enrichment for proteomic analysis. Chemical proteomic analysis was only performed for Rab S-prenylation substrates, leading to a total of 42 Rab proteins identified. Although this method is excellent for detecting Rab proteins—perhaps owing to the flexibility of RabGGTase active site to accommodate the bulky analogue—engineered mutants of FTase and GGTase-I are required to label Ca1a2X box-containing protein models. These mutants were only shown to modify lysates in conjunction with Rab S-prenylation through streptavidin blotting and no proteomic analysis were performed. Following the same approach, lysates from a macrophage cell line identified 18 Rabs and 3 GGTase-I substrates, supporting the previous observation of the sensitivity of this method to Rab proteins.393 Treatment with zoledronic acid (ZOL), an N-bisphosphonate inhibitor of RabGGTase, allowed for the identification of a few Rab proteins responsive to inhibition by this inhibitor.
While previous attempts for in vitro labeling of FTase and GGTase-I substrates showed limited detectability, a recent study combined in vitro S-prenylation and click chemistry to identify compromised prenylated proteins in mouse brain tissues.421 Previous studies have shown that genetic deletion of FTase and GGTase-I mitigates neuropathology in mouse models, suggesting a possible role of S-prenylation in Alzheimer’s disease.163 Neuron-specific deletion of FTase in mouse forebrain can result in diminished synaptic plasticity and memory retention while haplodeficient GGTase-I knockout is sufficient for synaptic and cognitive impairment.421 In order to identify these compromised proteins upon prenyltransferase deletion, lysates from mouse brain with wild-type and FTase or GGTase-I knockout phenotypes were subjected to in vitro labeling with C15AlkOPP followed by biotinylation, enrichment and proteomic analysis. Putative S-prenylated proteins remain unprenylated in knockout models and therefore achieve higher levels of C15AlkOPP incorporation upon in vitro labeling compared to the wild-type controls. A total of 11 FTase and 5 GGTase-I substrates were enriched compared to their wild-type controls. Among these farnesylated proteins, Rheb was identified which is known to regulate synaptic plasticity, neuronal morphology, and memory functions. FTase deletion may have impaired Rheb function and contributed to the observed reduction of synaptic and cognitive functions in FTase knockout mice. A geranylgeranylated protein RhoA was also identified which is involved in regulating the structure and function of dendritic spine in the brain. This novel in vitro labeling strategy offers significant improvements from the previous approaches using BGPP since it allows the detection of FTase and GGTase-I substrates. This method should also be suitable in future studies that involve samples less amenable to metabolic labeling techniques such as tissues.
Collectively, the above metabolic and in vitro labeling techniques using isoprenoid analogues that aimed to define the set of S-prenylated proteins indeed provided the scope of prenylated proteomes detectable in various systems. However, it should be noted that while isoprenoids modified at the terminal isoprene unit are the best substrates for farnesylation, no single analogue recapitulates the reactivity and specificity of FPP in vitro and in vivo.407 Different alkyne-containing isoprenoid analogues manifest intrinsic bias concerning which substrates are efficiently modified. Therefore, in order to broaden the list of farnesylated proteins identified from proteomic profiling strategies, a variety of these isoprenoid analogues may be used to overcome analogue bias.407 Additionally, the development of new biorthogonal approaches may be necessary to improve the detection and to validate the substrate scopes of the prenylated proteomes in various systems.
7.2. Profiling proteins responsive to perturbations
Proteomic strategies enable quantitation of the differential responses of modified proteins upon small molecule-induced disruption of the normal lipidation process.104 Since a large number of PTIs have been developed, efforts to understand their selectivity and impact on individual proteins have been undertaken using clickable isoprenoid probes. The effects of the FTI L-744,832 on S-prenylated proteins labeled with C15-Alk-OH were visualized using a dual color approach in a two-dimensional gel electrophoresis analysis.422 Proteomic analysis of these labeled proteins not only revealed that known farnesylated proteins displayed reduction in labeling but also that GGTase-I and RabGGTase substrates showed increased S-prenylation levels. Furthermore, FTI treatment facilitated the discovery of potentially novel farnesylated proteins GNAI-1 and GNAI-2. Enzymatic farnesylation on their C-terminal Ca1a2X box confirmed that these proteins are farnesylated in vitro.422 CCML and quantitative proteomics employing YnF and YnGG also enabled proteome-wide quantification of individual S-prenylated proteins in response to PTIs, revealing a wide range of estimated individual IC50.418 The identified novel S-prenylated proteins also displayed significant sensitivity to PTI treatment, further validating their true S-prenylation status inside the cell.
Inhibition of protein S-prenylation using PTIs offers pharmacological benefits, especially as anticancer agents targeting oncogenic isoforms of proteins involved in signal transduction.160 However, the potency of FTIs are sometimes offset by the ability of some targets to switch to geranylgeranylation catalyzed by GGTase-I. To identify these proteins capable of this “rescue prenylation”, the proteome-wide YnF/YnGG incorporation was compared to inhibition of YnF labeling in response to FTI treatment.418 The small GTPase proteins KRAS, NRAS, and RRAS2 exhibited robust dose-dependent increases in labeling, confirming switch-like behavior for these proteins; this effect is partially responsible for the failure of FTIs in clinical trials. FTI treatment in primary astrocytes labeled with C15AlkOPP also revealed that RRAS2 can switch from farnesylation to geranylgeranylation.420 Being a surrogate for both FPP and GGPP, C15AlkOPP is able to label RRAS2 even in the presence of an FTI. This further supports the switch-like behavior of RRAS2, potentially making it an interesting target for developing inhibitors that block both farnesylation and geranylgeranylation.
While extensive efforts were focused on targeting the S-prenylation of oncogenic small GTPases through inhibition of FTase using FTIs, a more specific strategy directed towards this subset of S-prenylation substrates could be conceived. Some small GTPases are regulated and trafficked through GGTase-I-catalyzed S-prenylation by the splice variants SmgGDS607 and SmgGDS558 chaperone proteins, with SmgGDS607 bearing an extra exon (Ex5).423,424 SmgGDS607 delivers the protein substrate to the prenyltransferase while SmgGDS shuttles the prenylated protein to the ER for maturation (Fig. 15A). Since a high SmgGDS607:SmgGDS558 ratio is implicated in some cancers, a splice-switch oligonucleotide (SSO) therapeutic strategy was developed to reprogram the ratio of the SmgGDS isoforms.425 A small oligonucleotide SSOEx5 site-specifically targets Ex5, which reduces and increases the levels of SmgGDS607 and SmgGDS558, respectively, resulting in a reduced SmgGDS607:SmgGDS558 ratio. Through the use of C15-Alk-OPP labeling, the SSO was found to suppress global protein S-prenylation, inducing apoptotic cell death in cancer cell lines and diminishing tumorigenesis in vivo. Since all three classes of S-prenylation substrates respond to alterations in SmgGDS607:SmgGDS558, there may be unexplored interactions between SmgGDS proteins and the other prenyltransferases FTase and RabGGTase.
Figure 15. Other mechanisms for regulating protein prenylation.
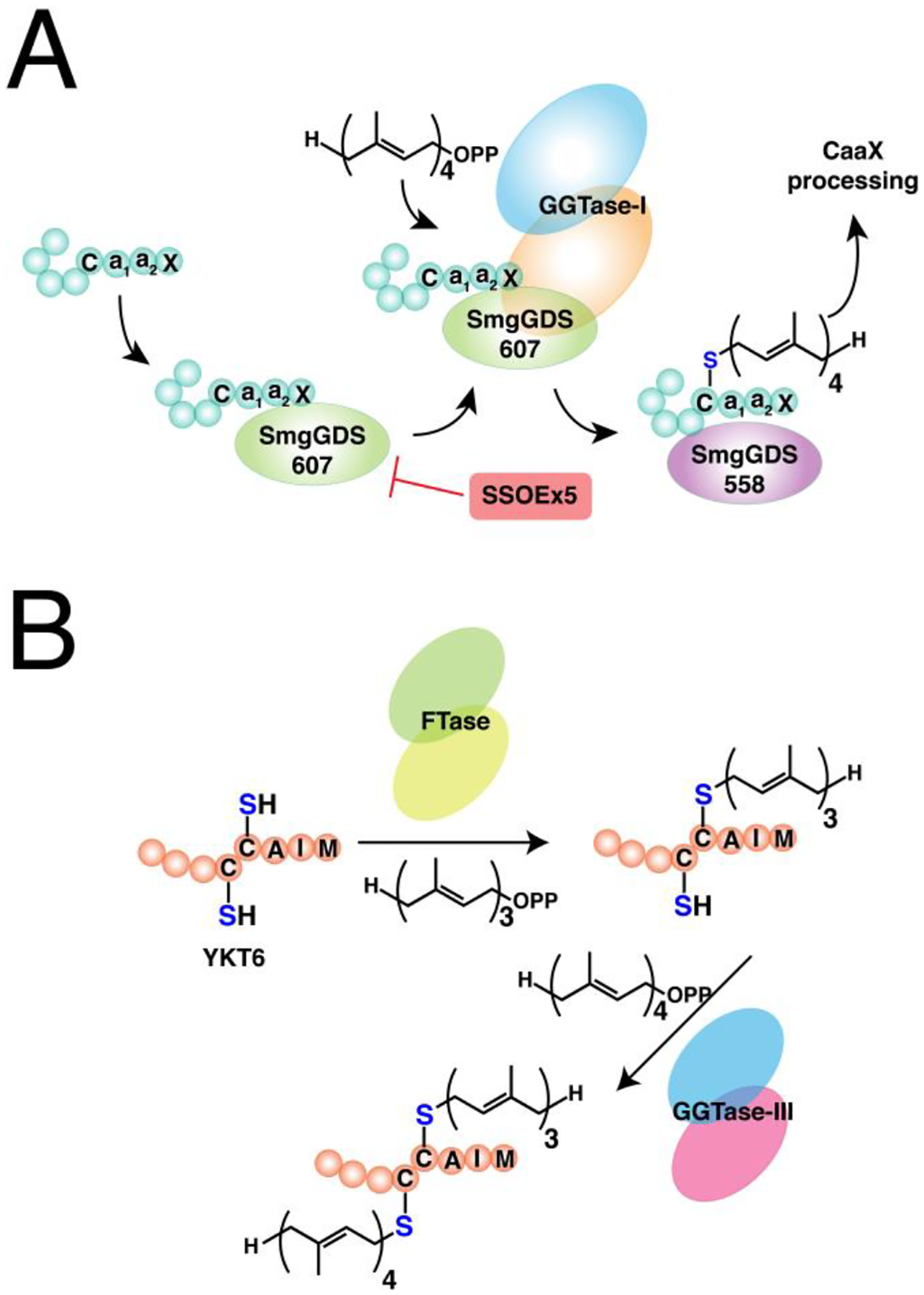
(A) Trafficking of prenylation substrates mediated by SmgGDS chaperone proteins. SmgGDS607 recruits the substrate for prenylation by GGTase-I and the prenylated protein is sequestered and delivered by SmgGDS558 the endoplasmic reticulum for subsequent processing steps. Splice-switch oligo (SSOEx5) can reduce the ratio of SmgGDS607:SmgGDS558 for potential treatment of cancer. (Ref. 425) (B) Dual prenylation on Ykt6 catalyzed by FTase and GGTase-III. Ykt6 is first farnesylated by FTase on the Ca1a2X box cysteine followed by geranylgeranylation by GGTase-III on the adjacent cysteine upstream from the farnesylated residue.
7.3. S-Prenylation of Rho GTPases
S-prenylation of small GTPases is essential for their membrane localization and interaction with effector proteins that regulate their activity.426 The Rho family of small GTPases are implicated in tumor growth and metastasis, as well as in regulating phagocytosis and signaling in inflammatory cells. Protein S-prenylation was found to control innate immunity by restricting the interaction between the Rho protein Rac1 with its effector proteins.427 Through C20-Az-OH labeling, Rac1 was shown to be unprenylated in GGTase-I-deficient mouse macrophages, stimulating proinflammatory signaling and severe rheumatoid arthritis. The inflammation in the GGTase-I-deficient model was reversed upon Rac1 deletion, but not when other geranylgeranylated Rho proteins RhoA and Cdc42 were knocked down. The reduction in S-prenylation of Rac1 enhanced its affinity with its interacting partner proteins Tiam1 and Iqgap1, resulting in enhanced GTP-loading and its ubiquitin-mediated degradation, respectively. Inhibition of these interactions is a key component for understanding the molecular basis of how protein S-prenylation normally restrains innate immunity.427
The brain specific splice variant of the Cdc42 (bCdc42) which contains a C-terminal Ca1a2X box188CCIF191 was initially shown to be S-palmitoylated rather than exclusively S-prenylated, in contrast to its canonical splice variant with a C-terminal CVLL sequence.428 However, a subsequent study showed that bCdc42 is sequentially geranylgeranylated and S-palmitoylated using C20-Az-OH and Alk-C18 labeling at C188 and C189, respectively.429 Thus, bCdc42 may not undergo the typical Ca1a2X processing (Fig. 3A) and instead become S-palmitoylated, which stabilizes the association of this protein bearing two different lipid modifications with membranes. CCML with isoprenoid and palmitoyl probes on a few other proteins terminating in CCa2X demonstrated that some may mature through the normal Ca1a2X processing pathway while others can be dually lipidated with prenyl and palmitoyl modifications.429 The dual lipidation in bCdc42 prevents its binding with RhoGDI which usually facilitates the trafficking of Rho proteins including the canonical archetype Cdc42. Approximately 80–95% of the total bCdc42 population is left singly prenylated which is subsequently processed in Ca1a2X maturation steps. A recent study conducted on mouse brain tissues identified the post-prenylation processed C-terminal geranylgeranylated peptide of the canonical splice variant of Cdc42 but not the brain specific isoform.430 Therefore, modifications on their analysis should have been applied to discern the concurrent geranylgeranylation and S-palmitoylation on the brain-specific variant bCdc42 since their targeted database only contained only singly lipidated peptides. Moreover, the archetype Cdc42 is responsive to both competition by farnesol or geranylgeraniol, suggesting that it may be both farnesylated and geranylgeranylated.418 The bCdc42 protein may also be a substrate of farnesylation and therefore warrants further investigation. Regardless, this aberrant CCa2X S-palmitoylation exemplifies the complexity of lipidation and may not be limited only to bCdc42.
7.4. Imaging S-prenylated proteins
The versatility engendered by clickable isoprenoid probes allows conjugation with fluorophores and subsequent quantitative flow cytometric analysis and cellular imaging on fixed cells.431 C15-Alk-OH labeling on various cell types analyzed via flow cytometry revealed variations in the levels of expressed S-prenylated proteins, consistent with in-gel fluorescence analysis from other studies.411,418 In cellular models of autophagy and aging, significantly higher levels of labeled proteins were observed compared to normal cells, highlighting the potential involvement of protein S-prenylation in compromised autophagy.431 In addition, CCML enabled cellular imaging of fixed cells to visualize the S-prenylated proteins localizing in the ER. Despite their established localization in plasma membrane, S-prenylated proteins were not detected on cell surfaces. It should be noted that a limited number of proteins modified with alkyne-functionalized isoprenoids were shown to proceed through Ca1a2X processing and should therefore be directed to the plasma membrane.418,432 It is possible that the alkyne moiety on plasma membrane-localized S-prenylated proteins may be buried in the lipid bilayer, making it inaccessible by the bulky azide-fluorophores for effective visualization.433
Confocal imaging of labeled S-prenylated proteins also provides insights into changes in protein S-prenylation in response to stimuli. Nerve growth factor (NGF) was discovered to trigger S-prenylation of proteins in neuronal axons with a dependence on axonal protein synthesis.434 C15-Alk-OH labeling revealed that S-prenylation does not take place throughout the cytoplasm but rather appeared as puncta in subcellular sites after NGF stimulation. Among several S-prenylated proteins, Rac1 was shown to be geranylgeranylated in axons and serve as a key effector of NGF signaling. The overall results of this study imply that aberrant localization, activity, or stability of S-prenylated proteins may contribute to the pathology of neurodegenerative diseases.434
7.5. Discovery of both a novel prenyltransferase and new S-prenylation substrates
Since its discovery over the past decades, the mechanism of protein S-prenylation by the three types of prenyltransferases have been extensively studied in efforts to develop selective and specific PTIs.11 Recently, a novel prenyltransferase has emerged whose discovery was facilitated through the use of bio-orthogonal isoprenoid analogues. A previously unknown human prenyltransferase consisting of PTAR1 and the β-subunit of RabGGTase was recently reported as GGTase-III (Fig. 3C).167 CCML with C20-Az-OH and streptavidin blotting showed that FBXL2 is specifically geranylgeranylated by GGTase-III. The leucine-rich repeat domain of FBXL2 interacts with PTAR1, serving as the structural basis for GGTase-III specificity.167 Shortly after, another group discovered the same enzyme but presented contradicting results, indicating that GGTase-III could not prenylate FBXL2 under conditions where GGTase-I robustly prenylated the substrate.435 Instead, they showed through BGPP-labeling that the SNARE protein Ykt6 is dually S-prenylated; it is first farnesylated by FTase and subsequently geranylgeranylated by GGTase-III on the upstream cysteine adjacent to the farnesylated residue (Fig. 15B). The use of BGPP in this study was suitable as previous reports showed excellent incorporation of this probe into Rab proteins by RabGGTase,391 which shares the same catalytic β-subunit with GGTase-III. It was suggested that since FBXL2 also bears an adjacent cysteine upstream of its Ca1a2X box, it is possible that GGTase-III appends a second geranylgeranyl moiety to this substrate after initial geranylgeranylation by GGTase-I.
The current paradigm for protein S-prenylation has been restricted to the Ca1a2X box motif on singly modified proteins. Recent efforts to probe for a broader substrate scope for S-prenylation demonstrated that extended CXXXX sequences including CVAGP and CMIIM can be farnesylated.142 CCML with C15-Alk-OPP on model proteins terminating in such motifs were found to be prenylated in vitro and within cells, as detected through fluorescence labeling and chemical proteomic approaches. In addition, the same group reported that peptide models with shortened CXX sequences can also be recognized as S-prenylation substrates in vitro.143 These studies highlight the promiscuity of prenyltransferase enzymes, and may expand our current understanding of their substrate recognition and potentially enlarge the scope of S-prenylation substrates. However, it has been noted that no endogenous proteins terminating in such unconventional prenylation motifs have been identified. If some of these sequences are indeed recognized as substrates under native conditions, several factors may hamper their detectability in click-chemistry based approaches including low expression levels.420
Beyond identification, clickable isoprenoid probes can be utilized to validate the S-prenylation status of novel S-prenylated proteins. For example, farnesylation of the human mitotic check point protein Spindly was found to be essential for its targeting to kinetochores.436 Disruption of its farnesylation abrogates its localization, resulting in prolonged prometaphase and delayed anaphase with concurrent defects in chromosome alignment. FTI treatment, however, does not affect other farnesylated proteins including CENP-E and CENP-F that are also involved in mitosis. These results indicate that HsSpindly is a novel mitotic FTI target in specific cancers that manifest high levels of its expression such as in oral cancer.437
Bacterial effector proteins not only rely on fatty acylation, as previously described, but also on S-prenylation. Several Legionella pneumophila effector proteins that modulate host functions in host cell membrane compartments depend on prenylation.438 By individually validating the S-prenylation of proteins using C15-Alk-OH labeling with subsequent immunoprecipitation and fluorescence detection, at least eight L. pneumophila effector proteins were shown to require S-prenylation for proper localization. Farnesylation does not seem to direct localization of two S-prenylated bacterial proteins but may play key roles in regulating their function. Similarly, the Salmonella T3SS effector protein SifA was shown to be prenylated with C15-Alk-OH.411 It was previously reported that GGTase-I modifies its C-terminal sequence,331CLCCFL336, possibly at C333.439 However, mutational studies on the C-terminal cysteines and C15-Alk-OH labeling suggest that SifA may be heterogeneously prenylated.411 With the recent discovery of GGTase-III adding a second prenyl moiety on proteins, it is tempting to speculate that SifA maybe a substrate of GGTase-III.
As a final comment, it is important to note that in the studies described above, the use of isoprenoids bearing small clickable handles in metabolic labeling studies does not appear to compromise the function of prenylated proteins or cause toxicity. Studies with a-factor, a farnesylated pheromone involved in yeast mating indicate that azide and alkyne substitutions have minimal effects on the activity of the mature pheromone; additionally, no significant effects on the subsequent proteolysis or methylation steps were observed either.432 While high concentrations of the alcohol forms of the analogues may exhibit some level of toxicity as was observed in the malaria parasite,417 the use of the diphosphate analogues may circumvent this issue, particularly when working with sensitive cell lines.410
8. Biological applications in other lipids
The applications of click chemistry in the major classes of protein lipid modifications have been discussed above. Other types of lipidation do exist albeit in a smaller set of proteins. Although protein fatty acylation occurs mainly through S-palmitoylation and N-myristoylation, shorter, longer, and unsaturated fatty acylation adducts have also been observed. Shorter saturated fatty acids such as octanoic (C8) or butyric (C4) acids, as well as longer saturated fatty acids such as stearic (C18) acid can be covalently attached to proteins via enzymatic or non-enzymatic mechanisms. Unsaturated fatty acids (UFA), on the other hand, can compete for palmitoylation sites, as well as generate oxidation products capable of forming adducts with proteins. Moreover, cholesterylation and GPI modification take place in the lumen of secretory organelles as mentioned previously. While cholesterylation proceeds through esterification of the C-terminus of a few known substrates, its precursors in the biosynthetic pathway generate intermediates that form protein adducts similar to UFA. The click chemistry toolbox has also been used to investigate the occurrence and biology of these relatively rarer lipid modifications.
8.1. Longer chain fatty acylation
The widespread S-acylation by fatty acids on proteins is often attributed to S-palmitoylation. However, other lipid-modified cysteines with longer fatty acyl chains such as stearate were observed in bovine heart and liver tissues.440 The ability to incorporate different lengths of fatty acyl chains into protein substrates varies across the known zDHHCs.185,292 In particular, zDHHC7 efficiently catalyzes stearoylation of SNAP25, indicating that zDHHCs facilitate lipid modifications beyond S-palmitoylation.185 Only a handful of human and viral S-stearoylated proteins are known.441–444 The S-stearoylation on viral spike proteins is essential for host-membrane fusion but is not mechanistically well-understood.445 The functional role of protein stearoylation in humans is not clearly defined as well. Here we discuss potentially stearoylated mammalian substrates and the discovery of a bacterial enzyme capable of removing this longer fatty acid modification. Modifications with very long-chain fatty acids (VLCFA) is described as well.
8.1.1. Stearoylation in mammals
Previously thought to be an S-palmitoylation substrate, stearoylation of the human transferrin receptor 1 (TFR1) was the first reported evidence for a regulatory function of stearoylation in humans.446,447 Treatment with an azide analogue of stearic acid (az-C17, Fig. 16) in HeLa cells for enrichment and proteomic analysis revealed TFR1 as the most abundant protein identified. In cultured cells, TFR1 stearoylation promotes mitochondrial fusion and function, while the loss of TFR1 stearoylation leads to mitochondrial fragmentation. CCML with az-C17 and streptavidin blotting identified zDHHC6 as a candidate enzyme that catalyzes TFR1 stearoylation. Furthermore, mitochondrial dysfunction in Drosophila lacking Parkinson disease genes Pink or Parkin can be rescued upon exogenous supplementation of stearic acid.447 In a follow up study, stearic acid was shown to regulate mitochondrial function in vivo, resulting in mitigation of fat accumulation and therefore reduces the risk for cancer and cardiovascular diseases.448
Figure 16. Chemical structures of fatty acylation probes with chain lengths longer than palmitic acid.
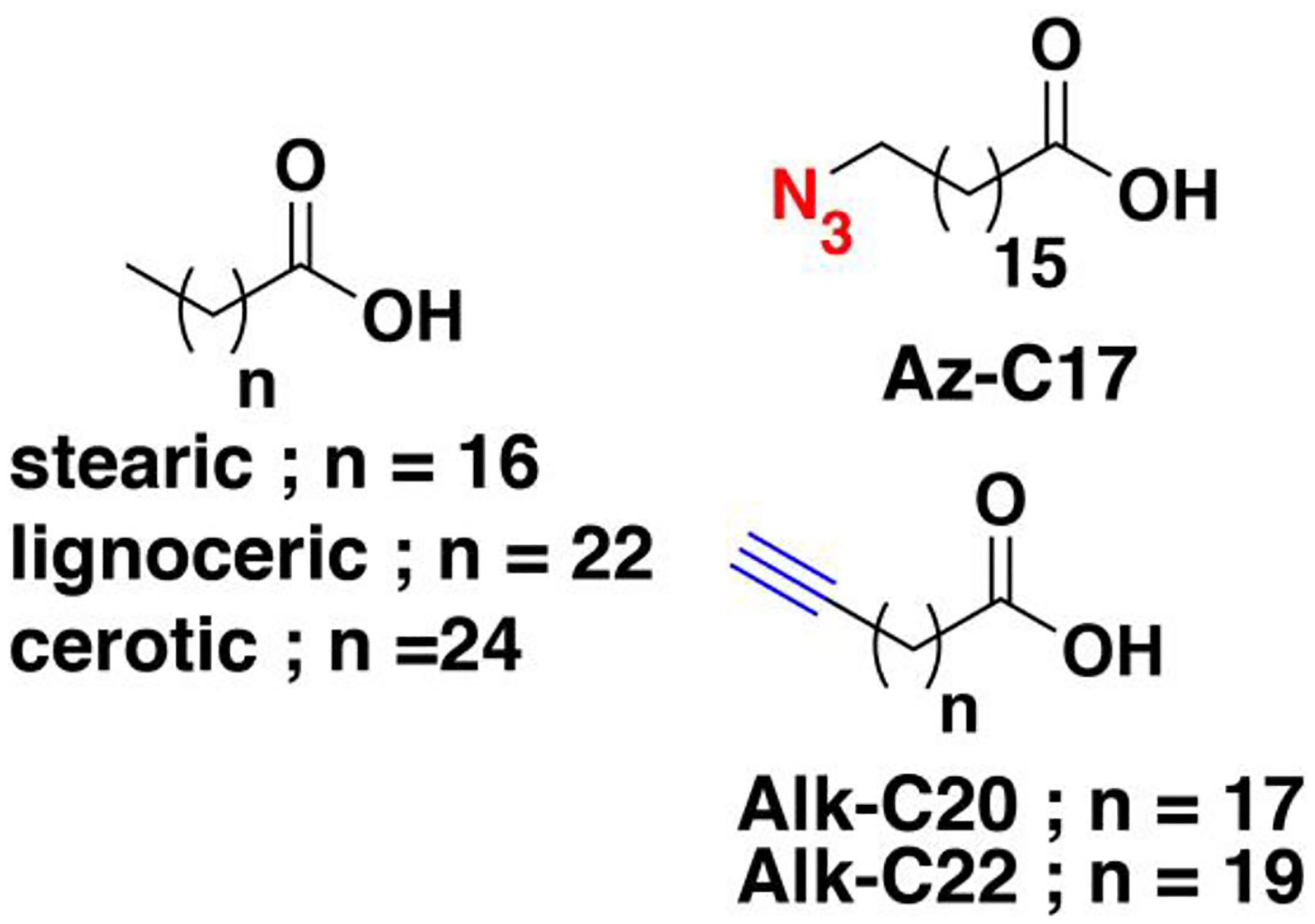
Saturated fatty acids stearic (C18:0), and very long-chain fatty acids lignoceric (C22:0) and cerotic (C24:0) are functionalized with either azide or alkyne moieties.
Another protein found to potentially incorporate stearic acid is the regulatory factor X 3 (RFX3), a transcription factor associated with ciliopathy.449 Fatty acylation on this protein appears to be a zDHHC-independent process since CCML with RFX3 co-expressed with each of the 23 known zDHHCs does not result in enhanced labeling. Furthermore, incubation of recombinant RFX3 with clickable fatty acyl-CoA analogues in an APE assay resulted in fatty acyl modification, with Alk-C18 and Alk-C18:1 exhibited a slightly more intense signal compared with Alk-C16. Therefore, RFX3 prefers the 18-carbon stearoyl and oleyl (C18:1) modifications that potentially proceeds through an autoacylation mechanism. Loss of this modification abolishes RFX3 transcriptional activity and suppresses ciliogenesis and Hh signaling, emphasizing that stearoylation is essential for RFX3 function. The dearth of studies investigating protein stearoylation in humans has hampered understanding of its functional and regulatory role, which stems from the ambiguous and interchangeable use of Alk-C16 and Alk-C18 in S-palmitoylation-centric studies. Regardless, the studies described above provide insights into the biological role and clinical relevance of protein stearoylation and suggest that this will be a fruitful area for future study.
8.1.2. Bacteria-mediated destearoylation
As previously discussed, bacterial effector proteins can alter lipid modifications on host proteins exemplified by the demyristoylation activity of S. flexneri IpaJ.368 In addition to this mechanism, S. flexneri was also discovered to release a T3SS effector protein IcsB that N-stearoylates lysine residues on host proteins (Fig. 13C).450 Stearoylated proteome profiling using Alk-C18 in infected cells identified 60 host proteins modified by SfIcsB which includes families of S-prenylated proteins such as Ras, Rho, Rab, and Rap proteins. Rho GTPases are inactivated by stearoylation at their C-termini, resulting in the disruption of the actin cytoskeleton in mammalian cells by increasing their hydrophobicity or interfering with their protein-protein interactions. S-prenylation of these Rho proteins appears to be required for recognition by SfIcsB, at least in the case of RhoA. Moreover, N-stearoylation of the host autophagy protein CHMP5 inactivates its function and hampers autophagy of the infected host. This mechanism is apparently unique to S. flexneri since CHMP5-deficient host cells infected with other bacteria such as S. typhimurium and Y. pseudotuberculosis were capable of inducing cell death. Therefore, CHMP5 in this context specifically functions in directing S. flexneri to antibacterial autophagy and this pathogen circumvents this mechanism by disarming CHMP5 function through fatty acylation by SfIcsB and escape autophagy. Concurrently, a MARTX effector protein Rho GTPase inactivation domain (RID) in Vibrio cholerae was shown to possess N-stearoylation activity.451 RID shares the same mechanism with IcsB in that it also modifies the polybasic C-terminal region of Rho GTPases but has a strong substrate preference for Rac1. Immunoprecipitation and streptavidin blot detection of Rac1 from RID-expressing mammalian cells treated with Alk-C18 validated its stearoylation. Both the core domain and the prenylated C-terminal end of Rac1 are recognized by RID. This aberrant modification of Rac1 disrupts its cytosol-membrane cycling and inhibits its function. These studies highlight a novel mechanism for how bacterial pathogens alter host cellular processes and may not only be limited to S. flexneri and V. cholerae. More efforts should be conducted in exploring other pathogens that may share similar molecular mechanisms for impairing cellular functions inside their infected hosts.
8.1.3. Very long-chain fatty acids (VLCFA)
In lipid biosynthesis, palmitic acid is the major de novo product over myristic and stearic acids, which can be further extended by elongases to generate VLCFA. Accumulation of saturated VLCFAs such as lignoceric (C24:0) and cerotic (C26:0) acids (Fig. 16) was observed during necroptosis in cells, a form of programmed cell death associated with various human diseases.452 Knockdown of the elongase ELOVL7 reduces VLCFA accumulation and necroptosis, while exogenous addition of VLCFA promotes cell death.453 Metabolic labeling with Alk-C20 and Alk-C22 with subsequent in-gel fluorescence scanning revealed a wide range of VLCFA-modified proteins. Pre-treatment with a pan inhibitor of zDHHCs resulted in only a modest increase in the cell viability of necroptotic cells. This suggests that the metabolism of VLCFA may involve other mechanisms or fatty acylation routes other than zDHHC-catalyzed processes during necroptosis.453
8.2. Shorter chain fatty acylation
Fatty acyl modification with chains longer than acetyl and propionyl but shorter than myristoyl and palmitoyl functional groups have been reported in the literature, although they occur at rarer occasions with smaller substrate scope. Octanoylation has only been described for one substrate and butyrylation may potentially occur in many bacterial and mammalian substrates through enzymatic and non-enzymatic mechanisms.
8.2.1. Butyrylation
Short-chain fatty acids (SCFAs) such as butyric acid (Fig. 17) can modulate signaling pathways and have been found to mitigate infections caused by enteric pathogens.454 Although its beneficial effect on suppressing the transcription of bacterial virulence factors is known, the molecular mechanisms by which this occurs are unclear. In order to explore the potential of butyrylation of proteins, the alkyne analogue Alk-C5 (Fig. 17) was recently synthesized and used to profile SCFA-acylated proteins in Salmonella enterica serovar Typhimurium.455 Alk-C5 labeling indicated that many proteins are acylated by this probe. Proteomic analysis identified an array of SPI-1 virulence proteins are potentially butyrylated with the transcription regulator HilA displaying the most significant enrichment. Unnatural amino acid substitution of several lysine residues indicates that site-specific acylation of HilA affects bacterial transcriptional activity and invasion in vitro, and acylation on the particular site Lys90 disarms virulence in vivo. However, gene deletion of known bacterial acyltransferase reveal that none of these enzymes catalyze butyrylation of HilA and this modification may proceed non-enzymatically. Further investigations should be carried out to either support the chemical acylation of these butyrylated proteins or characterize the acyltransferases that may catalyze this process. Regardless, this novel discovery of protein butyrylation on specific sites of a bacterial protein should prompt optimization of existing covalent inhibitors that are effective in treating infections caused by SCFA-sensitive pathogens.455
Figure 17. Chemical structures of fatty acylation probes with chain lengths shorter than myristic acid.
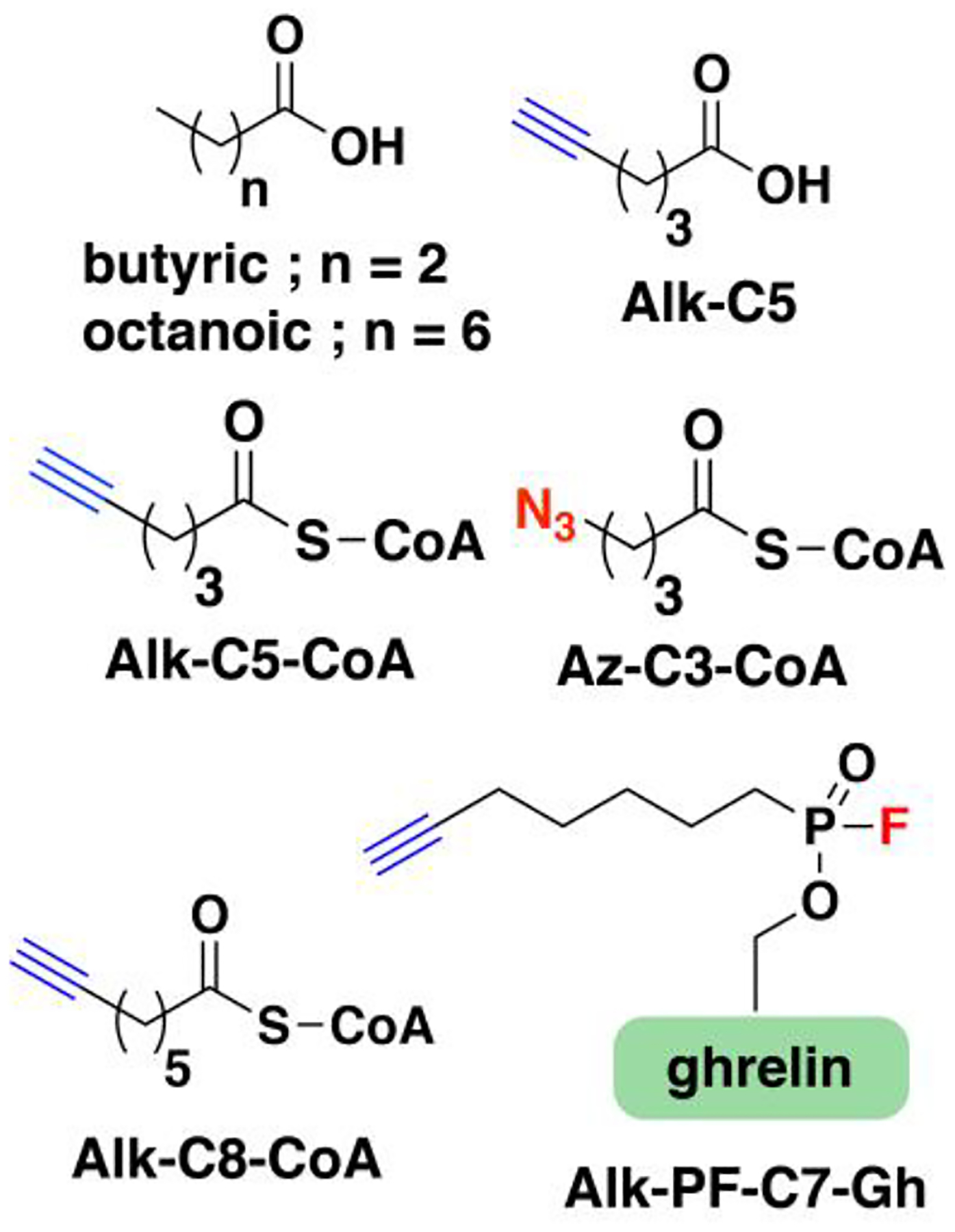
Saturated fatty acids butyric(C4:0) and octanoic (C8:0) acids are functionalized with either azide or alkyne moieties. A ghrelin probe containing an octanoyl modification, alkyne functionality, and phosphonofluoridate war head can be used to identify enzymes with octanoyl esterase activity. (Ref. 466)
In mammals, lysine acetyltransferases (KATs) such as p300 and GCN5 were found to catalyze not only acetylation but also propionylation and butyrylation of histone proteins as part of epigenetic regulation of transcription.456 Dysregulation in the activity and expression of these enzymes has been implicated in diseases in which potentially compromised pathways is not well-understood. In order to identify these protein substrates potentially acylated on lysine by these enzymes, alkyne and azide analogues of short fatty acyl-CoAs were synthesized.457 In particular, Alk-C5-CoA and Az-C3-CoA (Fig. 17) were used to profile these protein substrates in vitro.458,459 Incubation of Hela nuclear extracts with Alk-C5-CoA in the presence of p300 identified 23 known and novel substrates of this enzyme.458 In HEK293 whole lysates, treatment with Az-C3-CoA resulted in protein labeling in the presence of p300 or the mutant GCN5-T612G as shown through in-gel fluorescence imaging. Chemical proteomic analysis of lysates in the presence of the probe and either p300 or the GCN5 mutant identified 379 substrates common to both enzymes with approximately 90 protein substrates unique to each. As expected, histone proteins were highly enriched and many known acetylated proteins were identified in the analysis, The previous study performed in the nuclear extracts of HeLa identified significantly less protein substrates for p300 compared to the HEK293 whole lysate, indicating that this enzyme may function in modifying proteins outside of the nucleus. Functional annotation and pathway analysis of the p300 and GCN5 substrates reveal that these proteins are associated with diverse biological processes including gene expression, cell cycle, and cellular metabolism. While these studies have identified hundreds of potentially lysine acylated substrates with short fatty acids, they have been conducted in vitro and may not quite recapitulate the actual acylation substrates in a cellular context or in vivo. Furthermore, the probes used may ambiguously modify substrates of propionylation and butyrylation. Therefore, these data should be interpreted with caution and future studies should validate the type of modification (e. g. propionylation vs butyrylation) catalyzed by these KATs on specific proteins of interest.
8.2.2. Octanoylation
Another fatty acyl modification shorter than myristoyl and palmitoyl moieties is the octanoylation of ghrelin, an appetite-stimulating stomach hormone involved in glucose metabolism.460 Its precursor form undergoes processing steps allowing it to mature into a 28-residue peptide that is esterified with an octanoyl group on Ser3 by ghrelin O-acyltransferase (GOAT).461 To date, ghrelin is the only known and predicted octanoylated peptide substrate of GOAT. Owing to its regulatory role in metabolic processes and implications in diseases such as diabetes and obesity, effective GOAT inhibitors are constantly being sought after.462 A catalytic enzyme-linked click chemistry assay (cat-ELCCA) was developed to probe the fatty acyltransferase activity of GOAT and is amenable to inhibitor screening (Fig. 9E).463,464 In this strategy, biotinylated ghrelin substrates are immobilized on streptavidin-coated plates and incubated with GOAT and Alk-C8-CoA. Subsequent click reaction with azide-modified HRP confers signal amplification through an HRP-sensitive fluorogenic substrate. Using this method, non-peptidic small molecule antagonists of GOAT were discovered that could serve as promising drugs to treat obesity and diabetes.464
While the octanoylation of ghrelin is essential for its binding to its cognate receptor GHSR1a, the deacylated form of ghrelin also participates in metabolic processes, thereby rendering both fatty and defatty-acylation as regulators of ghrelin function.465 Several hydrolases in serum were found to display deacylase activity on octanoylated ghrelin.462 In order to identify enzymes with esterolytic activity against octanoylated ghrelin in rat serum, an activity-based ghrelin probe octanoyl containing a phosphonofluoridate warhead and alkyne handle was designed (Alk-PF-C7-Gh, Fig. 17) similar to Alk-HDFP.466 In this study, two prominent bands were observed in an in-gel fluorescence scan and identified through gel-based proteomics. One of the proteins was α2 macroglobulin (α2M), a large heterotetrameric protein with no previously known esterase activity. An HPLC-based assay validated its ability to deacylate lipidated ghrelin and inhibitor titration experiments revealed four active sites in the heterotetrameric protein. Further analysis of α2M levels in the serum suggests that it is present in sufficient amounts to be an important deacylase for ghrelin.466
In summary, ghrelin octanoylation plays an essential role in metabolic diseases and obesity. Being the only known substrate of GOAT, inhibiting GOAT activity is an attractive strategy to target diseases associated with ghrelin signaling and methods have been developed to streamline that process as discussed above. While this is true, there have been no reports on using clickable analogues of octanoic acid or those with comparable chain lengths in profiling their potential targets using any CCML strategy. Employing such methods may allow for the discovery of potential new substrates of GOAT or other proteins modified by mid-sized fatty acids through enzymatic or non-enzymatic mechanisms.
8.3. Unsaturated fatty acylation
Covalent modification of proteins with mono- (MUFAs) or polyunsaturated fatty acids (PUFAs) has also been reported for a few mammalian proteins including Src kinases, G-proteins, and Wnts.467,468 While saturated fatty acids (SFA) promote insertion of lipidated proteins into lipid rafts, MUFAs and PUFAs diminish their localization in those sites, which can partially be attributed to the distortions conferred by cis double bonds.4 This difference in saturated versus unsaturated modification of proteins represents one of the modulatory mechanisms for protein localization through fatty acylation. Furthermore, excess dietary SFAs often present risks for certain diseases while replacing with UFAs provide beneficial effects.469 Therefore, understanding the role of protein modification with SFAs vs UFAs is essential and may provide insights into these observed phenomena.
8.3.1. Monounsaturated fatty acids (MUFAs)
Wnts are a family of secreted signaling proteins that mediate communication between cells, thereby controlling cell fate and influencing developmental processes and the maintenance of adult homeostasis.470 These proteins are O-palmitoyleated (C16:1) on a serine residue by Porcupine (PORCN), a member of the membrane-bound O-acyltransferases (MBOAT) family of enzymes.471 Metabolic labeling using various alkyne-modified SFAs in PORCN- and Wnt3a-expressing cells revealed that PORCN efficiently transfers a range of fatty acid chain lengths onto Wnt3a, particularly Alk-C16.184,472 However, the potent ability of palmitoleic acid to compete with the Alk-C16 (palmitoyl probe) labeling indicates that PORCN prefers this MUFA. A recent study reported that the PORCN active site topology enforces cis-C16:1 fatty acylation on Wnts.473 Pulse labeling with either cis-Alk-C16:1 or trans-Alk-C16:1 (Fig. 18) showed that the trans isomer resulted in detrimental effects to Wnt cellular release, possibly indicating that the trans-fatty acylated Wnt is trapped in a complex with PORCN. The kink in cis-C16:1 may facilitate the release of Wnts from PORCN by lowering the energy required for sequestration by the Wnt-carrier protein. This may also explain the protracted turnover of Wnt3a in metabolic labeling studies using the saturated analogue Alk-C16.184 Therefore, in a biological milieu, Wnts are lipidated with cis-palmitoleic acid, and excess sources of SFAs and trans fats from diet may dysregulate Wnt signaling by impeding their release to the secretory pathway.473
Figure 18. Chemical structures of unsaturated fatty acylation probes.
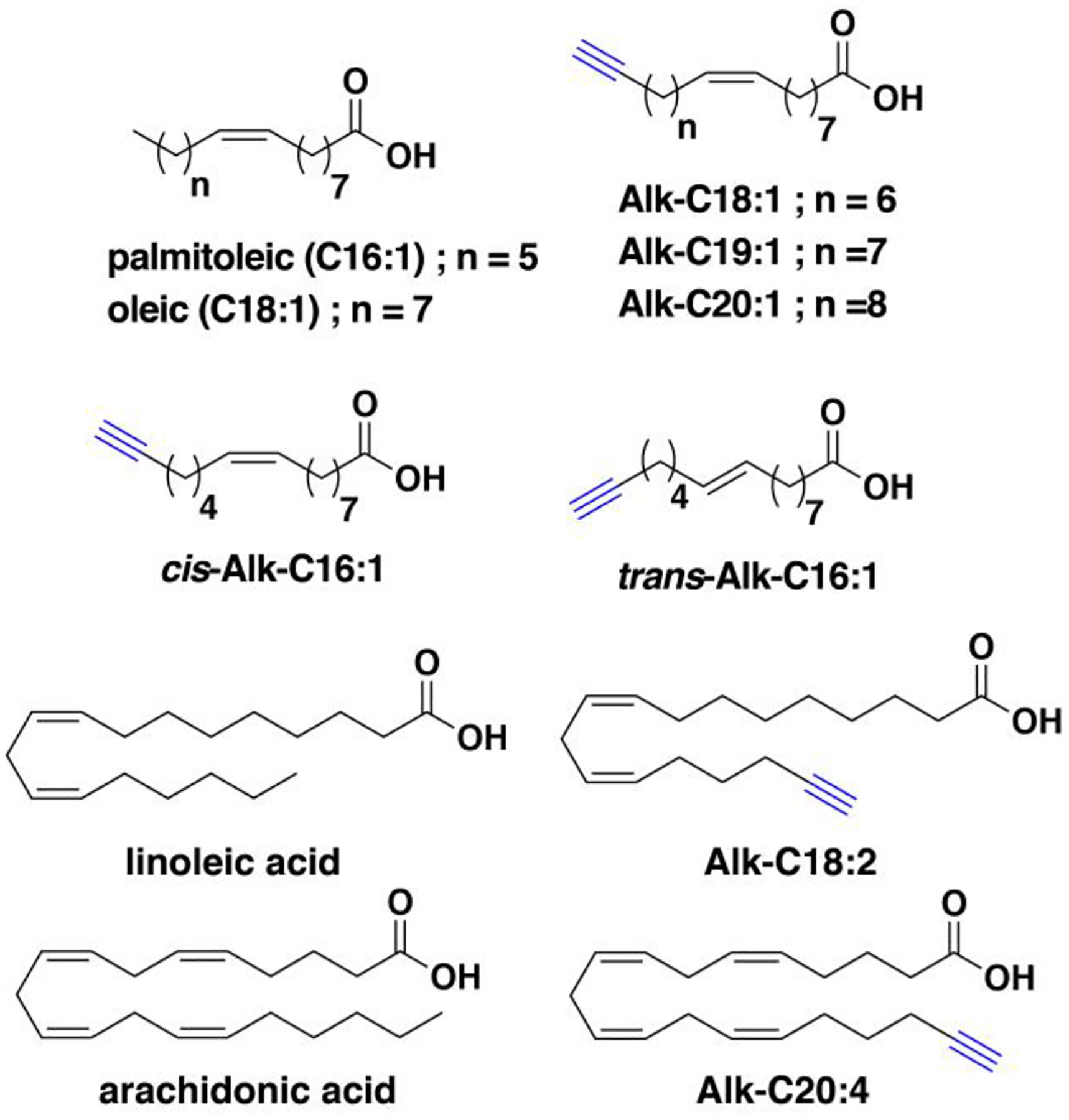
Monounsaturated fatty acids (MUFAs) palmitoleic (C16:1), oleic (C18:1), and the longer polyunsaturated (PUFAs) linoleic and arachidonic acids are modified with alkyne functionalities. The multiple double bonds in PUFAs facilitate the generation of radicals under oxidative stress that can produce electrophiles and form adducts with proteins
Some proteins were found to be modified by MUFAs longer than palmitoleic acid such as oleic acid (Fig. 18).474,475 The alkyne-modified analogue of oleic acid (Alk-C18:1) was employed to trace fatty acid metabolism, as well as to develop click chemistry-based platforms for in vitro enzymatic assays.186,187,476 With the interest of identifying proteins that can be covalently modified with oleic acid, Hang and coworkers synthesized Alk-C18:1 along with the longer MUFA probes Alk-C19:1 and Alk-C20:1 (Fig. 18).477 Metabolic labeling and fluorescence detection using these probes indicated that many protein targets are modified by these MUFAs with varying chain lengths. HRAS and IFITM3 were validated to be substrates for C18:1 modification. Furthermore, comparative proteomic analysis revealed that Alk-C18:1 and Alk-C18 labeling share a large number of modified proteins, with only 13 proteins preferentially labeled by Alk-C18. These results point to the possibility of a prevalent protein modification with MUFAs. Since the levels of palmitic and oleic acids are comparable in mammalian cells,185 this suggests that the investigation of palmitic vs oleic acid modification in a physiological context is important.
8.3.2. Polyunsaturated fatty acids (PUFAs)
Fatty acylation on proteins with PUFAs such as arachidonate (C20:4) and eicosopentaenoate (C20:5) has been described earlier with human platelets.478,479 It is not currently established whether zDHHCs catalyze these PUFA modifications but arachidonic acid was shown to effectively block zDHHC17-catalyzed Az-C16 labeling.185 While MUFAs are generally resistant to peroxidation,480 PUFAs can generate electrophiles and yield protein adducts.481 Alkyne-modified versions of linoleic acid (Alk-C18:2, Fig. 18) and arachidonic acid (Alk-C20:4, Fig. 18) were shown to undergo autooxidation processes in vitro analogous to their native counterparts.482 Quantitative proteomic analysis using these probes in a macrophage cell line identified membrane and mitochondrial proteins, which included those associated with inflammatory signaling oxidant defense.483 Interestingly, the extent of adduction correlated with the expression levels of proteins. Since 12/15-lipoxygenase (12/15-LOX) catalyzes the peroxidation of these PUFAs, the extent of protein adduction by Alk-C20:4 was investigated in peritoneal macrophages known to express high levels of 12/15-LOX.484 It is important to note that the use of Alk-C20:4 as a surrogate for C20:4 is not suitable for investigating all metabolic processes that involves this lipid.485 For example, elongation of Alk-C20:4 to Alk-C22:4 by elongases is less efficient in Jurkat cells and platelets are less able to generate LOX-dependent products of Alk-C20:4 metabolism than C20:4. Fatty acid tracing of Alk-C20:4 in mouse peritoneal macrophages in this study showed it is metabolized by LOX enzymes similarly to the native C20:4, making it a suitable C20:4 analogue for investigating the 12/15-LOX-dependent metabolism in this cell type. Proteomic analysis revealed over 200 proteins labeled by Alk-C20:4. ACADL and GAPDH were validated, which are involved in mitochondrial fatty acid β-oxidation and glycolysis, respectively. Indeed, 12/15-LOX-deficient mouse peritoneal macrophages displayed diminished glycolytic rate and mitochondrial respiration. These results aided by Alk-C20:4 labeling highlight the role of PUFAs in energy metabolism mediated by 12/15-LOX.
Under oxidative stress, PUFAs are also prone to generating shorter lipid-derived electrophiles (LDEs) such as 4-hydroxy-2-nonenal (4-HNE) and 4-oxo-2-nonenal (4-ONE) that may directly modify DNA and proteins through non-enzymatic mechanisms.486 These lipids contain an α,β-unsaturated aldehyde functionality that acts as a Michael acceptor and reacts with nucleophiles particularly thiols. Several studies have used clickable analogues of 4-HNE and 4-ONE in order to profile their cellular targets,487–489 as well as to validate and monitor their biological consequences in protein function.490,491 Moreover, these types of modification may be reversed by the lysine fatty deacylase SIRT2,492 although evidence of this phenomenon under unperturbed and physiologically relevant conditions will be necessary to ascertain its biological significance. This growing topic, however, is outside the scope of this review and will not be discussed in detail. The reader is therefore directed to other excellent reviews that cover the use of click chemistry in studying this type of non-enzymatic protein modification.493,494
8.4. Cholesterylation
The lipidation processes discussed thus far are catalyzed by transferases that are localized in the cytoplasm or cytoplasmic face of the plasma membrane. In contrast, protein cholesterylation occurs in the lumen of secretory vessels on members of the Hedgehog (Hh) family of proteins in a self-processing manner.495 These secreted morphogens play major physiological roles in embryonic development, tissue repair, and regeneration; dysregulation in the Hh pathway leads to severe abnormalities in embryonic and tissue development.496,497 Hyperactivation of Hh signaling has also been linked to certain types of cancer.498 Hh proteins are synthesized as precursors that are truncated, and subsequently N-palmitoylated on their N-terminus and auto-O-cholesterylated on their C-terminus forming an ester bond.499 The cholesterol modification on Hh is crucial for membrane tethering, receptor binding, secretion, and transport.5
8.4.1. Hh proteins and Smoothened
Given its essential role in pathways with disease relevance, clickable analogues of cholesterol were synthesized in order to characterize cholesterylated proteins. An azide-modified cholesterol (Az-Chol, Fig. 19A) was first reported by Tate and coworkers to successfully label Shh overexpressed in cultured cells.500 However, this analogue presented several drawbacks including low incorporation efficiency, non-specific labeling, and cytotoxicity. A series of alkyne-modified cholesterol analogues were designed in an effort to address the issues encountered with Az-Chol (Fig. 19A).501 The optimized form of the alkyne probes, Alk-Chol-2, successfully labeled Shh with intense signals and low cellular toxicity. The localization of cholesterylated Hh proteins were visualized in cultured cells and in developing zebrafish, the first successful imaging of lipidated proteins in vivo. Quantitative proteomic analysis on Alk-Chol-2-treated cellular models revealed subtle variations in the levels of cholesterylated Shh across multiple pancreatic cancer cell lines. Since Hh signaling is an important factor in clinical pancreatic ductal adenocarcinoma, these observed differences warrant further investigations into the potential role of cholesterylation in pancreatic cancer.501
Figure 19. Probes for O-cholesterylated proteins.
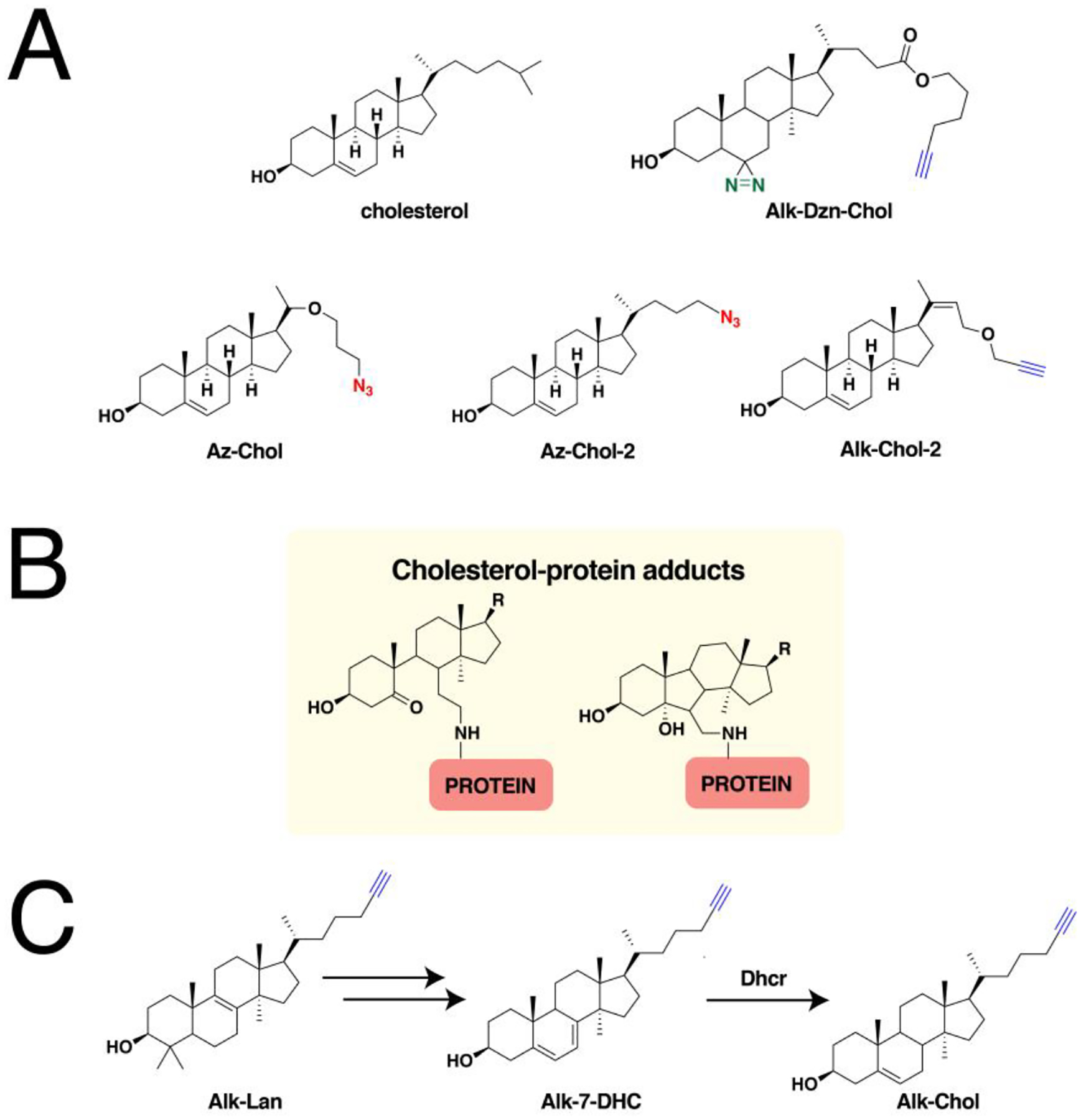
(A) The bio-orthogonal analogues of cholesterol contain azide and alkyne functionalities and may vary in length of their linkers. (B) The electrophilic species of cholesterol form adducts with proteins through their amine side chains. (C) The alkyne-modified versions of the cholesterol precursors can be efficiently metabolized to form cholesterol inside cells.
For a long time, covalent cholesteryl modification was found only in Hh proteins, although its occurrence on proteins beyond this family was proposed.502 Recent studies have shown that the Hh signal transducer and oncoprotein Smoothened (SMO) binds cholesterol within its cysteine-rich domain (CRD).503 Cholesterol immobilized on agarose resin through click chemistry efficiently isolated SMO in binding assays. Shortly thereafter, compelling evidence for the covalent cholesterol esterification of SMO emerged through metabolic labeling with Az-Chol-2.504 Proteomic analysis on Az-Chol-treated cells yielded 20 protein candidates, which were then each validated through Az-Chol labeling and immunoprecipitation. Among these proteins, SMO retained Az-Chol modification under denaturing conditions indicating covalent modification. A robust mass spectrometric analysis showed Asp95 as the site of cholesterylation, a residue situated in the vicinity of the CRD. Since protein cholesterylation is not known to be catalyzed by any cognate enzymes, it was hypothesized that cholesterol binds first to the CRD followed by auto-esterification at Asp95. Furthermore, this novel SMO cholesterylation was found to be regulated by SMO-interacting proteins Patched-1 (suppressor) and Shh (enhancer). Such lipidation is required for SMO-driven gene transcription and modulation, and loss of this modification resulted in severe developmental defects in vivo. These results open new doors for possible therapeutic interventions on treating Hh-pathway-related cancers by targeting SMO cholesterylation.504
8.4.2. Non-covalent interactors
Beyond covalent modification by cholesterol, a wide range of membrane proteins are also regulated through non-covalent interactions with this lipid. Probing these cholesterol-interacting proteins can be accomplished using cholesterol probes with photoaffinity crosslinking capabilities.505 In order to globally profile these proteins, the Cravatt group employed a diazirine- and alkyne-containing cholesterol analogue (Alk-Dzn-Chol, Fig. 19A), which confer both photoaffinity and clickable handles, respectively.506 Probe-treated cells were UV-irradiated and cell lysates were biotinylated with biotin-azide, enriched, and subjected to a SILAC-based quantitative proteomic analysis. Out of >800 proteins enriched, more than 250 cholesterol-interacting proteins displayed high selectivity and sensitivity to cholesterol—the majority of which were integral membrane proteins as expected. These results highlight the key cellular pathways that may depend on cholesterol concentration to modulate protein localization and modification. Interestingly, known sterol-interacting proteins and covalently cholesterylated Hh were not enriched in this approach; the authors noted that the introduction of these unnatural modifications may impair sterol-protein interactions and the inevitable hydrolysis of esters may impede the enrichment of covalently modified proteins.506 Indeed, this study exemplifies the use of a combination of both affinity and photocrosslinking tags in a single probe can identify interacting proteins mediated by lipid PTMs in a single experiment. Such a strategy could be useful for studying many other types of protein-lipid modifications as well.
8.4.3. Adducts of oxidized cholesterol intermediates
The biosynthesis of cholesterol also yields intermediates that can be oxidized and generate reactive electrophiles that react with amines forming protein adducts (Fig. 19B).507 High levels of 7-dehydrocholesterol (7-DHC), the immediate precursor of cholesterol, is a hallmark of the genetic neurodegenerative and developmental disease Smith-Lemli-Opitz Syndrome (SLOS).507 This is attributed to the impairment of 7-DHC reductase (DHCR7), the enzyme responsible for converting 7-DHC to cholesterol. Metabolic labeling experiments using an alkyne-modified 7-DHC (Alk-7-DHC, Fig. 19C) exhibited high levels of protein adduction in an SLOS cellular model deficient in DHCR7.508 In a follow-up study, the protein adducts of lanosterol, the precursor of sterols from squalene, were also profiled using its alkyne-modified analogue (Alk-Lan, Fig. 19C).509 Alk-Lan efficiently undergoes multistep conversion to Alk-7-DHC and Alk-Chol within neuronal cells, indicating that the alkyne moiety remains intact during Alk-Lan metabolism and that the biosynthetic enzymes can tolerate such modification of the sterol precursor. The identified Alk-Lan-modified proteins were a subset of those labeled with Alk-7-DHC in a neuronal cell line. This provides evidence that both Lan and 7-DHC form protein adducts through the same mechanism and that 7-DHC is the major source of reactive sterol electrophiles. Further efforts into characterizing the members of the sterol adductome should shed more light into the implications of this protein modification during oxidative stress and in diseases such as SLOS.
8.5. Glycosylphosphatidylinositol anchor
Glycosylphosphatidylinositol-anchored proteins (GPI-APs) are involved in secretory and endocytic pathways and are tethered to lipid membranes through a GPI glycolipid.510 The GPIs are assembled in the ER and transferred by GPI transamidases to the exposed C-termini of cleaved proprotein substrates. The structure of GPI anchors consists of a core phosphatidylinositol modified with fatty acyl groups and a conserved glycan linked to the protein through phosphoethanolamine (Fig. 20A).511 An estimate of more than 250 GPI-APs being present in eukaryotes is derived from annotations and prediction tools, making up less than 2% of the eukaryotic proteome.512,513 GPI-APs display a myriad of biological functions such as signal transduction, cell recognition and adhesion, and cell surface enzymatic reactions.511 They are also implicated in parasitic and bacterial infections, as well as in genetic blood diseases and prion pathogenesis.514
Figure 20. Chemical probes for labeling GPI-APs and structure of the GPI scaffold.
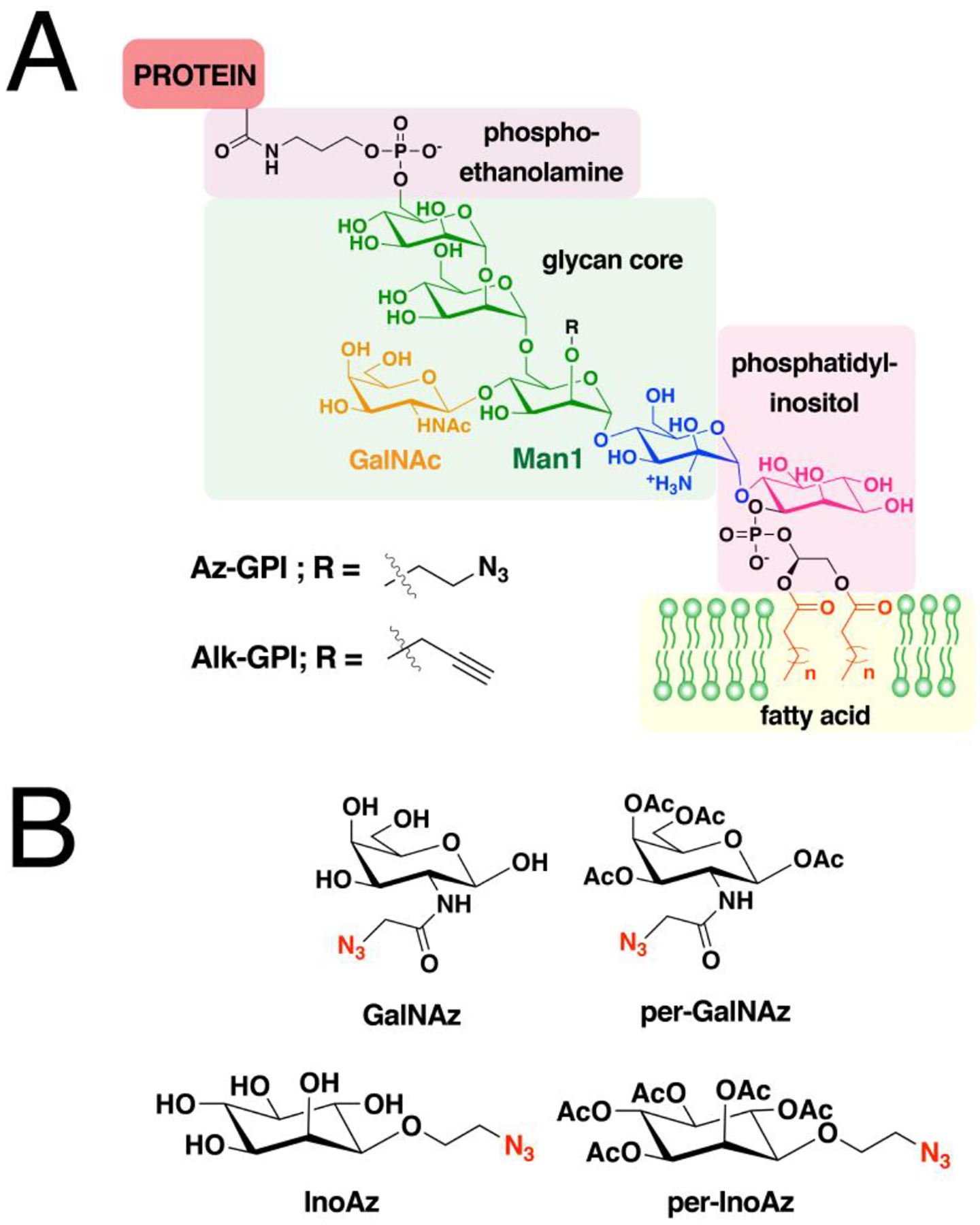
(A) Structure of the GPI assembly comprised of a glycan core linked to fatty acids through a phosphatidyl-inositol, which is also attached to the C-terminal of a protein through a phospho-ethanolamine that forms an amide bond. The lipid portion of the GPI anchors the protein to membranes. Whole GPI probes were synthesized with bio-orthogonal modifications in the glycan scaffold. (B) Structures of the azide-modified monosaccharides that can be metabolically incorporated to the glycan core of the GPI anchor.
CCML-based methods to study the biology of GPI-APs have been limited owing to the complicated structure of GPIs. There are three strategies that have been reported to generate clickable GPI probes: i.) the use of clickable fatty acid probes to tag the fatty acyl portion of GPI anchors; ii.) azide-functionalized sugar analogues to label the glycan core which is suitable live-cell copper-free click reaction; and iii.) clickable versions of the complete GPI assembly. In the first approach, the fatty acyl portion of GPIs can incorporate medium-chain saturated fatty acids in bacteria and protozoans.515 Chemical proteomic analysis of P. falciparum using Alk-C14 identified both N-myristoylated NMT substrates and GPI-anchored proteins.177 Since the myristoyl groups in GPIs are linked through an ester bond, GPI-APs can be distinguished from the N-myristoylated proteins through hydroxylamine cleavage prior to protein pulldown. This strategy yielded a list of potentially novel and previously known GPI-APs in the malaria parasite. Employing a similar strategy in T. gondii identified 52 known and predicted GPI-APs.354 Alk-C18 for S-palmitoylated proteome labeling in a macrophage cell line also enriched both S-palmitoylated proteins and GPI-APs.199 Seven predicted and annotated GPI-APs were identified in this profiling experiment and one of these proteins, CD14, was validated through CCML with Alk-C18 combined with immunoblotting. Removal of the C-terminal site of GPI attachment results in loss of Alk-C18 labeling, indicating that the lipid modification is located in the GPI-anchor. This membrane-anchored protein is critical in initiating signaling cascades that trigger immune response. Interfering with its GPI modification may therefore offer therapeutic benefits in the onset of infection.
The conserved glycan scaffold in GPIs in mammalian GPI-APs is often modified with N-acetylgalactosamine (GalNAc) as a side chain of the first mannose proximal to the phosphatidylinositol core (Man1).516 Thus, the second strategy for probe construction involves incorporation of sugar analogues into this site such as an azide analogue of GalNAc (GalNAz, Fig. 20B). Metabolic incorporation of GalNAz in GPI-APs in mammalian cells and subsequent Staudinger ligation with phosphine-biotin enabled the detection of both endogenous and recombinantly expressed GPI-APs.517 Another strategy of labeling the sugar moieties in GPI-APs is through the introduction of bio-orthogonal functional groups into the inositol portion of the glycan scaffold. An array of azide-modified inositols were also synthesized where the azide moiety was placed in various positions in the sugar molecule (e.g. InoAz, Fig. 20B), along with their peracylated analogues, i.e. all the hydroxyl groups are acylated (per-InoAz, Fig. 20B).518 Through conjugation with alkyne-biotin and subsequent streptavidin-fluorophore tagging, cell surface-localized GPI-APs on fixed cells were successfully visualized and quantified using confocal microscopy and flow cytometry, respectively. It is noteworthy that the peracylated forms of the probes (Fig. 20B) are more efficiently metabolized to label GPI-APs and exhibit lower cytotoxicity. Moreover, GPI-APs are often exposed to the exterior surface of the plasma membrane and are amenable to click reactions outside the cell. This enables the use of SPAAC chemistry with bulky reagents and overcomes the need for Cu(I) known to be toxic to cells. Indeed, super-resolution imaging was made possible on live cells that were treated with peracylated GalNAz and tagged with a DBCO-modified fluorophore in a copper-free approach.519
While a high number of GPI-APs have been identified through predictions, a more limited set of these proteins has been experimentally validated. The lower abundance of GPI-APs relative to other membrane proteins and contamination with non-GPI-APs suppress their detectability in LC-MS/MS proteomic analysis, even after membrane fractionation and selective release into the solution using phospholipases.520 In an attempt to provide an improved strategy, GalNAz was used to enrich and identify GPI-APs in mammalian cell lines.521 The intact, GalNAz-labeled cells were treated with phosphatidylinositol-specific phospholipase C (PI-PLC) to release GPI-APs and enriched using alkyne-functionalized agarose beads. A complementary method to pulldown GPI-APs using immobilized lectins was also conducted in cells that were not treated with GalNAz. Out of more than 250 predicted GPI-APs, only 33 were identified and almost 50% were shared by both methods. Polarized epithelial cells also displayed differential populations of GPI-APs in the apical and basolateral regions of the cell, with more proteins localized at the apical surface. Although fewer proteins were profiled than expected, this method employed on intact cells preserves the cell integrity and hence opens up opportunities for spatiotemporal analysis of GPI-AP expression and dynamics in cells.521
Clickable probes that are considerably more specific for GPI-APs are those that encompass the complete GPI structure similar to that synthesized in vivo. However, the reductive nature of standard hydroxyl protection strategies for the sugar molecules are not compatible with azides and alkynes during the course of their synthesis. Through the use of para-methoxybenzyl (PMB) protection, complete GPI-anchor probes functionalized with azide (Az-GPI) or alkyne (Alk-GPI) on Man1 in the glycan core were successfully generated (Fig. 20A).522 The same strategy was used to append an alkyne to the phosphate moiety in a GP anchor specific to L. donovani.523 Although efficient conjugation could be achieved with their partner click reagents, there have been no studies in the literature that used these probes in labeling GPI-APs in cells. Perhaps the arduous nature of glycan chemical synthesis and challenges in the scale up of these probes limit their accessibility. Moreover, their relatively large structures may result in poor cellular uptake. Hence, employing these clickable GPIs in biological studies of GPI-APs has yet to be reported.
9. Proteins of interest site-specifically functionalized via lipid modifying enzymes and click chemistry for biotechnological applications
Previous sections described the use of click chemistry for biological studies of proteins modified via lipidating enzymes. In the following sections, we focus on prior art that has exploited those enzymes for the modification of proteins of interests (POI) with various functionalities together with click chemistry. Protein modification via enzymatic methods provide unique advantages. The site-specificity allows homogenous product formation with controlled stoichiometry, as opposed to conventional chemical methods such as cysteine-maleimide or lysine-N-hydroxylsuccinimide ester chemistry in which conjugation of a specific site or number of amino acid residues is difficult to achieve. Precise control over the site of modification tends to cause minimal perturbation of native protein activity. For instance, modification via S-prenylation occurs at the C-terminus and N-myristoylation at the N-terminus, which are often distanced from the active sites of target proteins. Enzymatic labeling is bio-orthogonal as it occurs only on proteins that contain the specific consensus sequence, Ca1a2X for S-prenylation and MGXXXS/T/C for N-myristoylation. This allows selective labeling of the POI in mixtures such as crude lysates, which eliminates purification steps which is important in biotechnology where downstream processing of protein products is responsible for over half of the total production cost.524 Finally, enzymatic reactions enable fast modifications with high yields under mild conditions, in contrast to other site-specific methods such as unnatural amino acid incorporation which is often limited by low protein yields and technical difficulties.525
While a number of different lipid modifying enzymes exist, as noted above, the products of palmitoylation yield a hydrolysis-prone thioester linkage that is potentially susceptible to loss of the conjugated payload in various biological environments. Similarly, the ester linkages from cholesterylation and GPI-modification have related instability features, that has prompted the development of biotechnological applications that utilize lipid modifying enzymes to be focused on S-prenylation and N-myristoylation. In future research, it will be interesting to see advances that allow the expansion of the range of exploitable lipid modifying enzymes. For instance, it may be become possible to utilize palmitoylation in situations that require reversible attachment and detachment of a payload, or by enhancing the linker stability through formation of an amide bond instead of the thioester through an intramolecular S-to-N shift.495
9.1. C-terminal protein functionalization through S-prenylation
Protein S-prenylation allows site-specific protein modification of almost any protein by simple appending of a short Ca1a2X recognition motif onto the C-terminus of the POI. The use of S-prenylation for biotechnological applications has mostly evolved around FTase for a number of reasons. The synthesis of analogues of farnesyl diphosphate is much easier than that of geranylgeranyl diphosphate due to their simpler structure. FTase is known for its promiscuity that enables it to accept a wide variety of substrates including ones that incorporate a bulky aryl group or even a biotin functionality.391,526 The plasticity of FTase can be even further improved by engineering the active site via mutagenesis.391,527 While GGTase-1 accepts various analogues as well, it still exhibits more limited lipid substrate specificity compared to FTase.528 In the case of RabGGTase, an additional REP component is required for the reaction to proceed which adds additional complexity to the overall process. In this section, the development and assessment of various isoprenoid analogues (Fig. 21) and their combination with click chemistry for a variety of applications are described (Fig. 22).
Figure 21. Additional isoprenoid analogues used in enzymatic C-terminal modification of proteins.
Isoprenoids are modified with an azide, alkyne, aldehyde, alkene, ketone and fluorophore functionalities for selective bioconjugation.
Figure 22. Applications of site-specific enzymatic protein labeling through C-terminal cysteine prenylation with FTase and N-terminal acylation with NMTs.
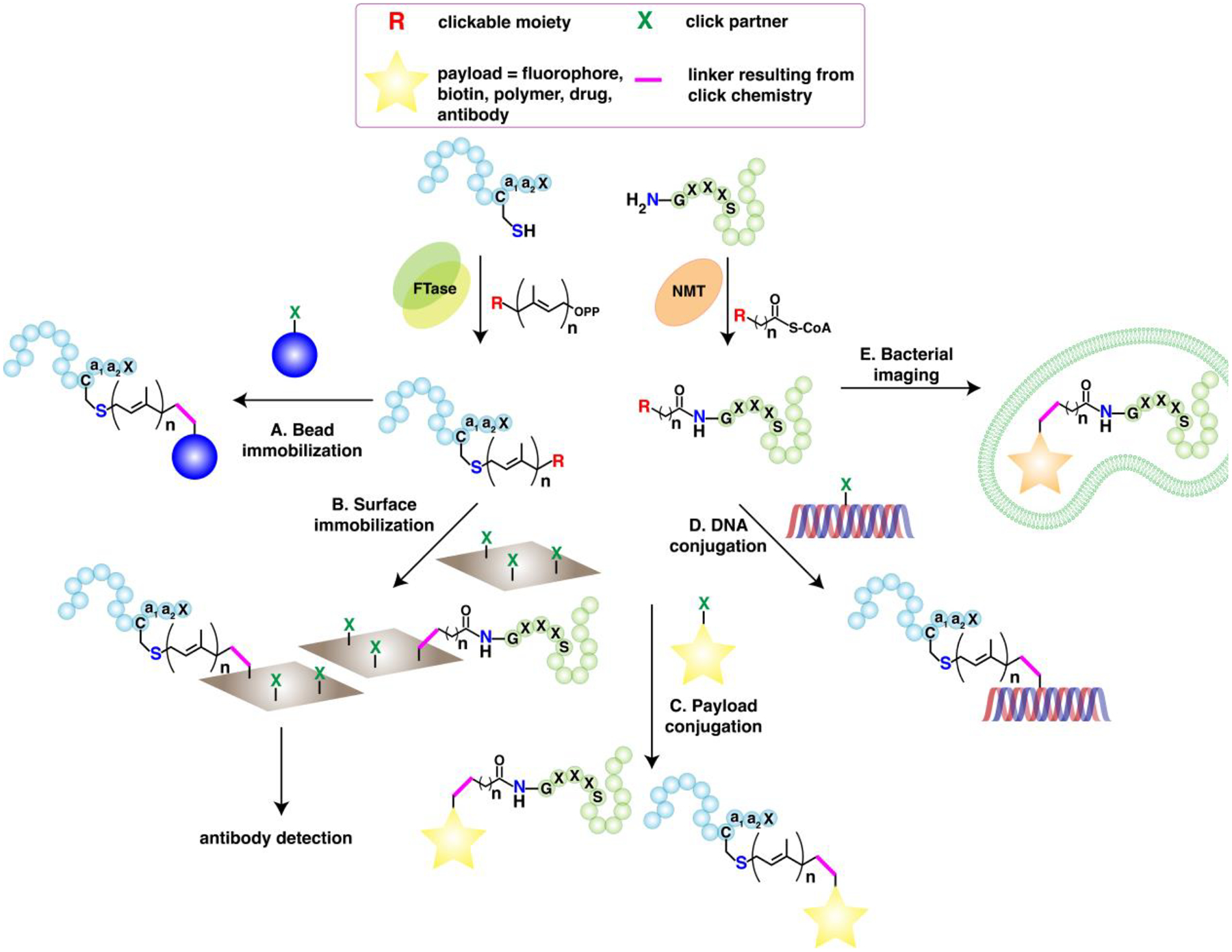
(A) Immobilization of proteins enzymatically prenylated with clickable isoprenoids on beads functionalized with clickable moieties. (B) Immobilization of tagged proteins on glass or gold surfaces coated with clickable functionalities. (C) Direct protein modification of lipidated proteins with various payloads such as fluorophores, affinity handles, polymers, drugs, or antibodies. (D) Generation of DNA-protein crosslinks through reaction between functionalized DNA and proteins prenylated with clickable isoprenoid analogues. A nucleotide base is modified with a biorthogonal moiety prior to DNA synthesis. (E) Incubation of myristoyl probes in bacteria enables visualization of N-myristoylation of heterologously expressed proteins. Bio-orthogonal fluorophores are reacted with the tagged proteins inside live bacteria.
A large number of FPP analogues which contain biorthogonal moieties for click chemistry that can be recognized as substrates by FTase have been developed by Distefano and coworkers. In their initial report, a geranylazide diphosphate (C10-Az-OPP, Fig. 21) was synthesized from geraniol.395 The product was a mixture of isomers in an approximately 1:1 ratio which was inseparable due to the facile sigmatropic interconversion of the allylic azide substrates and/or products. Nonetheless, C10-Az-OPP was confirmed as a substrate for FTase when evaluated with a peptide substrate N-dansyl-GCVIA (Dn-GCVIA). Although the incorporation of the analogue was slower than with FPP, the reaction was complete within a practical time frame (2 h at 30 °C). The azide-functionalized peptide was then reacted with a triphenylphosphine-based reagent that contains an electrophilic ester moiety. Interestingly, while the Staudinger reaction showed high product conversion, mass spectrometry analysis of the product indicated the presence of an O-alkyl imidate-linkage instead of the conventional amide-linkage (Fig. 1). Based on reports suggesting that alkyl azides yield the former while aryl azides yield the latter,529 it was assumed that the β-branched allyl azide substructure of GPP exhibits a reactivity more similar to that of aryl azides.
In a subsequent report, Rose et al. reported on C15-Az-OPP (Fig. 21).402 The new analogue showed similar properties to C10-Az-OPP, in that it was obtained as an isomeric mixture, showed similar reaction rates against Dn-GCVIA, and the product of Staudinger ligation again yielded an O-alkyl imidate-linked product instead of the amide-product as confirmed via mass spectrometry analysis.
In a more intensive study on the product formed from Staudinger ligation, Xu et al. prepared a new azide analogue 6,7-dihydrogeranylazide, which lacks the alkene present at C-6 (C10-Dh-Az-OPP, Fig. 21).530 S-prenylation of Dn-GCVIA with that analogue indicated that the absence of the third isoprene unit brings about structural perturbation when bound to the FTase active site leading to a rate of reaction that was much slower than FPP. Nonetheless, C10-Dh-Az-OPP showed similar rates to the previous C10-Az-OPP analogue which was sufficient to be completely converted to product with longer reaction times. Interestingly, mass spectrometric analysis of the reaction product between the prenylated peptide and a phosphine-based reagent indicated the formation of only alkoxyimidate products instead of the predicted amide despite the non-allylic nature of the azide. To gain a better understanding, model reactions with simpler compounds were run between the dihydroprenylazide substrate precursor (the unphosphorylated alcohol) and three model phosphine reagents. While ESI-MS analysis indicated the formation of both imidate and amide products, LCMS and NMR analysis both revealed that in fact the amide is the dominant product where in some cases the imidate form was completely undetectable. The results suggested that product analysis using mass spectrometry could give contradicting results due to potential variations in ionization efficiencies of the products. Nonetheless, the report showed evidence of alkoxyimidate products from Staudinger ligation that can form when particular reagent types were used. These observations were significant since they highlighted the potential problem of product mixtures that can sometimes occur with the Staudinger ligation.
Duckworth et al. constructed a model protein by fusing a CVIA sequence to the C-terminus of GFP, which was then selectively immobilized onto agarose beads by installation of an azide moiety via enzymatic S-prenylation followed by capture via CuAAC (Fig. 22A).531 Based on the study above, the new azide analogue (C15-Dh-Az-OPP) was composed of three isoprenoid units but lacked the alkene present in the terminal isoprene unit. Investigation of the rate of S-prenylation with Dn-GCVIA showed that 10,11-dihydrofarnesylazide diphosphate (C15-Dh-Az-OPP, Fig. 21) at saturating concentrations (10 μM) was only 2.7-fold slower than with FPP, while the previously reported C10-Dh-Az-OPP showed a significantly reduced catalytic efficiency, which indicated that the presence of a third isoprene unit and the absence of an alkene in that unit makes the azide-containing analogue a much better substrate for FTase. After enzymatic labeling with C15-Dh-Az-OPP of GFP-CVIA, CuAAC with alkyne-functionalized agarose beads was carried out for 15 h at 25 °C. Analysis of the fluorescence intensity of the beads indicated a 96% immobilization efficiency. Interestingly, 8% of GFP-CVIA was shown to be immobilized under the same click conditions even in the absence of the clickable azide, which was presumably due to nonspecific interactions caused by Cu(I)-induced activation of the alkyne-agarose.532 Selective capture could be obtained from protein mixtures containing as little as 1% GFP-N3 on account of the highly efficient click reaction.
Poulter and coworkers prepared GFP- and GST-(glutathione S-transferase) immobilized glass surfaces exploiting Staudinger ligation or CuAAC (Fig. 22B) using similar chemistry.533 Target proteins were enzymatically labeled with an azide- or alkyne-containing farnesyl diphosphate analogue (C10-Et-Az-OPP or C10-Et-Alk-OPP, Fig. 21) followed by click reaction with phosphine- or azide-derivatized glass slides that resulted in regio- and chemoselective protein immobilization. Although nonspecific binding of proteins lacking the clickable moiety was also observed, proteins covalently immobilized via click chemistry were found to be more robust; click-immobilized proteins were found remaining attached to the slides after treatment with an acidic saline solution for 2 h at 80 °C, which was sufficiently harsh to strip antibodies bound to their respective antigenic proteins, and remained bound to slides after 4 days in buffer while nonspecifically bound proteins had fully dissociated.
In a follow-up report, Poulter and coworkers described eleven additional farnesyl diphosphate analogues each bearing azide or alkyne functionality, where five were shown to be substrates for yFTase (the yeast enzyme).396 Comparison of the steady-state kinetic parameters of the analogues with Dn-GCVIA indicated that the presence of a bulky or branched w-isoprenoid unit, charged amino groups, or rigid chains may hinder their compatibility with the active site of yFTase. Azide-farnesylated Dn-GCVIA products showed two closely eluting species with identical mass determined via LC-MS, which indicated isomeric products resulting from either incorporation of both regio-isomers of the azide analogue or incorporation of a single isomer which subsequently undergoes rearrangement.
By exploiting CuAAC with a model protein GFP-CVIA site-specifically labeled with an alkyne-functionality, Duckworth et al. performed surface-immobilization and fluorophore labeling of proteins (Fig. 22A and 22C).534 A continuous fluorescence-based enzyme assay was employed to evaluate the C10-Alk-OPP as a substrate for yeast FTase (yFTase). Although farnesylation of Dn-GCVIA with the isoprenoid analogue was approximately 12-fold slower than the natural substrate FPP, GFP-CVIA was fully converted to the alkyne-product within a timeframe (3 h at 30 °C) suitable for downstream applications. The alkyne-labeled GFP was then immobilized onto azide-functionalized agarose beads under CuAAC conditions. Interestingly, while immobilization of GFP-alkyne on azide-agarose and GFP-azide on alkyne-agarose both showed highly efficient immobilization in 1 h (93% for both), conjugation to the alkyne-resin was shown to result in a higher degree of nonspecific binding. These results indicated that even for the same CuAAC reaction, immobilization characteristics could vary upon the choice of functionality carried by each clickable reagent. For instance, in proteomic studies, using an azide-resin to prevent false-capturing of undesired proteins would be especially important.532 Fluorophore labeling of GFP-alkyne was achieved with Texas Red bearing an azide, demonstrating the wide applicability of the reported site-specific labeling method. Förster resonance energy transfer (FRET) calculations gave a Förster distance of 40 Å, which was consistent with the R-distance of 37 Å predicted by a model of the GFP-Texas Red conjugate.
Distefano and coworkers synthesized protein-DNA conjugates which self-assembled into tetrahedral nanostructures (Fig. 22D).535 The model protein GFP-CVIA was farnesylated with azide functionality, which was exploited to conjugate with an alkyne-containing oligonucleotide 33 bases in length. The GFP-DNA was initially shown to form a complex with a Texas Red carrying a complementary 33-mer DNA strand evidenced by FRET. The hybridization of complementary DNA strands was further utilized in constructing a tetrahedral structured GFP-DNA nanoassembly by mixing four types of GFP-DNA conjugates and briefly annealing the mixture at 54 °C. The number of GFP molecules per tetrahedron, observed by single molecule spectroscopy, could be controlled by changing the composition of the oligonucleopeptide building blocks. Overall, the advantage of having a compact recognition tag for site-specific protein conjugation was demonstrated through its application towards GFP-DNA nanostructure assembly that requires precisely spaced and oriented proteins.
Waldmann and coworkers studied the effects of the isoprenoid analogue structure on S-prenylation and click chemistry.536 Four isoprenoid analogues were synthesized, each containing a diene (HOM-GPP, Fig. 21), either an amide-linked (AAA-GPP, Fig. 21) or aliphatic azide (APO-GPP, Fig. 21), or biotin (BGPP, Fig. 14). Affinity assays indicated all four analogues bind to the active site of FTase despite differences in length compared to the native FPP. However, while the azide and diene analogues showed product formation with Dn-GCVLS and the TKCVIM-tagged model proteins cyan fluorescent protein (CFP), RFP and small GTPase Ypt7, the biotin analogue did not show conversion. Reaction with GGTase-I failed to process the biotin analogue as well, which indicated that the complexity of analogue design involves factors beyond simply length. The azide-functionalized proteins obtained via FTase were shown to maintain solubility and functionality post-labeling and underwent Staudinger ligation with a model phosphine. The azide analogue containing the amide-linkage was shown to react much faster than the aliphatic analogue due to its stronger electrophilicity. Functionalization of the diene-labeled protein via Diels-Alder cycloaddition was shown, which required a higher excess of dienophile and low pH conditions.
Wollack et al. studied methods to improve the solubility of proteins functionalized via farnesylation.537 Following the farnesylation with a truncated alkyne-prenyl analogue (C5-Alk-OPP, Fig. 21) composed of a single isoprenoid unit, the last three amino acids of the “Ca1a2X” box were enzymatically removed, which resulted in site-specifically alkyne-labeled peptides or proteins with a net gain of only a single cysteine and nine additional non-hydrogen atoms. Although kinetic assays with OrG-RTRCVIA-OH (OrG: Orange Green) showed that the C5-alkyne analogue, C5-Alk-OPP, was a poor substrate for yFTase compared with FPP, near-complete (>85%) labeling was achieved upon extended incubation. Carboxypeptidase Y (PY) could be utilized to cleave off the -a1a2X residues without yielding undesired proteolytic degradation of the peptide. The minimalistic labeling methodology was applied for immobilization of alkyne-mCherry on azide-functionalized agarose, in which the absence of the “VIA” sequence did not affect immobilization efficiency, demonstrating the innocuousness of the method on protein stability and click reactivity. Comparison of C5-Alk-OPP and the dihydro analogue C5-Dh-Az-OPP for Hcp1 labeling demonstrated that minimizing the hydrophobicity of the lipid substrate could lead to enhanced product-solubility.
The range of exploitable click chemistries that could be employed via FTase-catalyzed labeling was expanded by Rashidian et al. who developed an aldehyde-containing analogue of FPP (C15-Ald-OPP, Fig. 21).538 The catalytic efficiency of the aldehyde analogue was shown to be comparable to previously developed azide or alkyne analogues. Dn-GCVIA was successfully farnesylated with the analogue by FTase which could subsequently undergo fluorophore addition. While the C15-Ald-OPP-labeled Dn-GCVIA rapidly formed an oxime-ligated product with aminooxy-AlexaFluor-488 in the presence of aniline, reactions with Texas-red hydrazine were shown to be unsuccessful, suggesting that low μM reaction conditions are not suitable for hydrazone formation due to its lower equilibrium constant. Farnesylation of GFP-CVIA with the aldehyde analogue manifested complete product formation within 2 h at 30 °C in the presence of FTase. Oxime ligation-induced surface labeling of aldehyde-functionalized GFP with aminooxy-agarose beads in the presence of 100 mM aniline was achieved with ~30% efficiency, while labeling with aminooxy AlexaFluor-488 and 100 mM aniline showed 60% efficiency. Overall, oxime ligation was shown an efficient bio-orthogonal functionality for labeling chemistry, which could be a potential alternative to CuAAC where copper-free conditions are necessary such as in cellular environments.
Tate and coworkers utilized site-specific labeling with GGPP analogues using RabGGTase and REP-1 in tandem with click chemistry to functionalize a series of target proteins.404 C20-Az-OPP (Fig. 14) was synthesized, which despite the interconversion between 14-azido and cis/trans 16-azido regio-isomers, was successfully transferred to a range of recombinant Rab proteins containing both single (Rab18) and dual (Rab6, GST-tagged Rab11, Rab27a, Rab38, Rab1a) S-prenylation motifs. Comparison of CuAAC, phosphine-mediated ligation, and strain-promoted cycloaddition, using the requisite biotin derivatives showed that CuAAC is the most efficient and selective click reaction. That method was then used to detect geranylgeranylated proteins in cells which is described above in Section 7.1.1.
Proteins farnesylated with the natural substrate FPP were shown to undergo oriented immobilization via thiol-ene click reactions.539 H-, N-, and K-Ras were farnesylated with FPP using FTase, followed by immobilization on thiol-poly(amidoamine) (PAMAM) functionalized SiOx/Si slides (Fig. 22B) by exposure to UV light through a photomask. Immunodetection of surface-bound Ras with an anti-Ras-Cy3 antibody revealed fluorescent micropatterns indicating successful immobilization. Interestingly, the labeling efficiency of farnesylated K-Ras was ~20 fold higher than the others, which could be attributed to the interaction between the C-terminal polylysine domain of K-Ras and the carboxylic acid groups present on the glass-bound PAMAM. Based on that observation, mCherry and Rab6A each containing 4 or 6 lysines at their C-termini were generated, respectively. Farnesylated products from both proteins underwent efficient immobilization even directly from crude lysate. The method was extendable to a co-expression system, in which the engineered mCherry was farnesylated with endogenous FPP by FTase in E. coli. The resulting modified proteins in crude lysate could be immobilized using a longer thiol linker and with extended UV exposure time. Utilizing proteins prenylated with the natural substrate FPP for biotechnological applications was an interesting approach, obviating the development and assessment of analogues that contain a click-reactive functionality. Similar work was reported recently where Distefano and coworkers exploited the site-specifically labeled lipid groups for binding of multivalent protein scaffolds to cells via hydrophobic interaction.540 While the work was unique in that it utilized protein S-prenylation for protein-conjugation without the involvement of covalent interactions, it should be noted that the work could be expanded to incorporate click chemistry for a stronger and more selective binding-interaction between multiple target substrates.
Taton and coworkers synthesized protein-oligodeoxynucleotide (protein-ODN) conjugates by combining farnesylation and copper-free click chemistry.541 Conjugation of ODN was carried out by farnesylation with either C10-Az-OPP or C15-Az-OPP of GFP or mCherry, followed by SPAAC with a DIBO-modified ODN. SPAAC proceeded over a range of pH values (3.5–12.3) and temperature (room temperature and 4 °C), and the product yield was comparable to CuAAC, demonstrating its wide applicability, simplicity, and high efficiency. ODN-conjugated proteins were utilized for supramolecular assembly via hybridization of the complementary DNA sequences. To this end, GFP and mCherry were each conjugated with complementary DBCO-ODNs and dimerized either at room temperature or 55 °C. Interestingly, while CuAAC-generated conjugates formed heterodimers under both annealing conditions, SPAAC-generated conjugates only dimerized at elevated temperatures presumably due to the more hydrophobic strained-alkyne moiety. Surface immobilization of protein-ODN conjugates on ODN-functionalized agarose beads was shown, which demonstrated the usefulness of SPAAC as an alternative to CuAAC in functionalizing proteins at low concentrations.
Rashidian et al. utilized oxime and hydrazone formation to reversibly immobilize or label proteins in crude cell lysate.62 In addition to the previously reported C15-Ald-OPP alternative substrate containing an alkyl aldehyde,538 a geranylaldehyde analogue containing an aryl aldehyde (C10-BA-OPP) was developed. Although the catalytic efficiency of the aryl analogue was 4-fold lower than the alkyl-substituted compound, higher synthetic yields gave it the advantage of being much easier to produce. Reactions of aldehyde-functionalized Dn-GCVIA and GFP-CVIA with Texas red hydrazide or AlexaFlour-488 aminooxy indicated that while the reaction rate is much faster for hydrazone formation, oxime ligation shows higher conversion due to higher association constants. Fluorophore labeling of GFP could be achieved using either hydrazone or oxime formation, in which the products displayed FRET. Reversible immobilization was achieved by utilizing the two click reactions sequentially. GFP-aldehyde was initially captured on hydrazide-functionalized agarose beads (Fig. 22A), followed by release with high concentrations of hydroxylamine, where aniline was employed as a catalyst in both reactions. The method was highly reversible with a 95% immobilization efficiency and 80% release efficiency, and could be applied to selective functionalization of proteins from crude lysate without purification. As an example, a therapeutically relevant glucose-dependent insulinotropic polypeptide (GIP-CVIM) was PEGylated from crude extract using this chemistry.
While the high chemoselectivity and reversibility of imine-based reactions make them a useful click reaction, they suffer from slow reaction kinetics especially for ketones and therefore often require catalysts. Rashidian et al. screened various catalysts for oxime and hydrazone formation and found that m-phenylenediamine (mPDA) is up to 15 times more efficient than aniline.60 At identical concentrations, mPDA was about 2-fold more efficient than aniline, but its higher water solubility allowed reactions to proceed at much higher concentrations which resulted in much greater acceleration since the rate of reaction is first order in catalyst. At pH 7, oxime ligation between an aldehyde-functionalized GFP and a dansylated aminoooxy reagent was completed in 90 s with 750 mM mPDA while 100 mM aniline only showed minimal product conversion (<7%). It was noted that both catalysts were less effective for protein aldehydes than simple model aldehydes, suggesting that the species conjugated to the aldehyde can modulate its reactivity. Protein labeling via reversible immobilization using mPDA resulted in faster and higher yields compared to aniline. It was found that Schiff base formation starts to compete with oxime ligation at high mPDA/aminooxy ratios, and therefore ratios below 250:1 were recommended. Fluorophore labeling of a therapeutically relevant protein ciliary neurotrophic factor (CNTF) via oxime ligation with aminooxy AlexaFluor-488 was achieved using 80 mM mPDA. The mPDA catalyst was shown to be effective for labeling ketone-containing proteins as well. PEGylation of an aldehyde-containing DHFR with 500 mM mPDA manifested a 2.5-fold increase in rate while 100 mM aniline had negligible enhancement. A number of other groups have reported other efficient aniline-based catalysts for this reaction as well.24
The orthogonality between CuAAC and oxime ligation to achieve simultaneous dual protein labeling was exploited by Rashidian et al.542 A tri-orthogonal molecule incorporating an aldehyde and alkyne functionality on an allylic diphosphate (C10-BA-Alk-OPP) was synthesized and confirmed as an alternative substrate for FTase by farnesylation of Dn-GCVIA and GFP-CVIA. Simultaneous oxime ligation and CuAAC with aminooxy-PEG and azido-TAMRA (Fig. 23A) was shown with GFP and CNTF that had been labeled with the tri-orthogonal analogue. Under the conditions employed, oxime ligation was found to be more efficient than CuAAC, but nonetheless both reactions reached near completion within 12 h. The method was applied towards constructing chemically self-assembled nanoring structures (CSANs) by mixing a dual-functionalized GFP labeled with aminooxy-TAMRA and azido-bis-MTX with dimeric DHFR proteins functionalized with anti-CD3 antibodies. The resulting protein nanostructure was shown to undergo selective membrane binding and energy-dependent endocytosis when treated with CD3+ T-leukemia cells, where the GFP and TAMRA components could be detected by both fluorescence and FRET measurements. In a subsequent methods article, Mahmoodi et al. provided a detailed description of the procedures for protein immobilization and release exploiting the reversible imine-base reaction and simultaneous protein labeling utilizing CuAAC and oxime click chemistry.543 In comparing m-PDA versus p-PDA in oxime and hydrazone ligation, the authors noted that at higher catalyst concentrations, m-PDA favors Schiff base formation over the desired product, while p-PDA does not exhibit such limitations and therefore allows faster reactions. However, p-PDA was shown to be prone to oxidation in air, especially in the presence of copper catalysts, which suggested that m-PDA would be more suitable for reactions that require prolonged incubations or simultaneous click conditions.
Figure 23. Simultaneous protein labeling through prenylation.
(A) Simultaneous labeling of a protein using a tri-orthogonal isoprenoid analogue containing both alkyne and aldehyde for CuAAC and oxime ligation, respectively. (Ref. 542) (B) One-pot dual labeling using orthogonal isoprenoid analogues for tail-to-tail protein bioconjugation. FTase and GGTase-I have substrate preferences for the length of isoprenoid used. (Ref. 385)
Maynard and coworkers utilized oxime and CuAAC click chemistry for chemoselective protein immobilization on gold surfaces (Fig. 22B).544 For covalent immobilization, two sets of proteins and gold surfaces were functionalized with two different types of complementary click components. Horse heart myoglobin (HHMb) was modified at its N-terminus with an aldehyde via a PLP reaction with pyridoxal 5’-phosphate, and mCherry-CVIA was farnesylated with C15-Alk-OPP. Complementary gold surfaces were functionalized with an aminooxy-alkanethiol or azide-alkanethiol by microcontact printing with a PDMS stamp. Immobilized proteins were visualized via fluorescence microscopy which demonstrated the potential of the method in applications that target uniformly oriented proteins.
Poulter and coworkers constructed uniformly oriented antibody arrays on site-specifically immobilized antibody-binding proteins (Fig. 22C).545 Truncated recombinant forms of antibody-binding proteins A, G, and L were constructed to include five antibody-binding domains from wild-type S. aureus protein, an N-terminal His6-tag, and a C-terminal CVIA motif. The chimeric proteins were farnesylated with C10-Et-Alk-OPP and immobilized onto azide-functionalized glass slides. The high selectivity and binding affinities of the immobilized antigens were shown through a series of assays involving various concentrations and mixtures of protein A, G, and L. The surface-bound antigens were utilized to fabricate an array of antibodies by a “sandwich” procedure, where glass-bottomed slides (with silicone isolators) were coated with recombinant antibody-binding protein L followed by incubation with monoclonal antibodies directed to either GFP or GST. A mixture of GFP and GST was added to the wells and the captured proteins were visualized using fluorescent antibodies, which indicated selective antibody-dependent protein detection without cross-reactivity. The antigen-bound slides could be reused by treatment with acidic buffer to strip away the antibody layer while leaving the covalently immobilized antigens attached for use in another round of protein-detection. In a follow-up paper, they showed that their “sandwich-type” antibody arrays that exhibit regioselectively immobilized antigens show superior properties compared to randomly oriented antigens immobilized on commercially epoxy-modified slides.546 As described in their previous work, a sandwich structure for detecting GFP was constructed by immobilizing an alkyne-functionalized recombinant protein L onto azide-glass slides, followed by incubation with a mouse anti-GFP IgG antibody. Captured GFP was detected with D7Light 680-labeled mouse anti-GFP IgG with high sensitivity and low cross-reactivity against the components of the sandwich array. For comparison, a sandwich structure with random orientation was constructed by immobilizing protein L on commercial epoxy-modified slides in a non-specific fashion. Regioselectively immobilized slides showed superior fluorescent outputs demonstrating the advantages that result from site-specific immobilization. Slides showed unaltered detection sensitivities after up to 6 cycles of regeneration. Poulter and coworkers subsequently expanded the scope of their immobilization methodology from glass to gold surfaces.547 Thiol or dithiocarbamate (DTC) self-assembled monolayers (SAMs) were prepared on gold slides for subsequent azide surface-functionalization. Alkyne functionalized GFP or GST, prepared by S-prenylation with an isoprenoid analogue was immobilized onto the gold surface via CuAAC and visualized with complementary fluorescent antibodies. A concentration dependent increase in immobilization was observed only under click conditions, indicating a low degree of nonspecific binding. While the immobilized proteins could be released with β-mercaptoethanol (BME) from both types of gold surfaces, proteins on DTC SAMs were shown to be much more resistant to displacement, indicating a more robust sulfur-gold linkage in the case of DTC over thiols.
Trans-cyclooctene (TCO) functionality was site-specifically incorporated by protein farnesylation for subsequent functionalization via tetrazine ligation.548 Successful labeling of an Oregon Green-CVIA peptide and GFP-CVIA showed that the developed TCO geranyl diphosphate analogue (C10-TCO-OPP) was a substrate for yFTase. The relatively compact volume of cyclooctene and hydrophobic characteristics of the analogue were thought to be the key factors of substrate-compatibility of the analogue in the FTase active site. The TCO-labeled peptide and protein both rapidly reacted with dipyridyl-tetrazine known to exhibit cell-penetrating properties, which combined with the copper-free click features of this chemistry has important implications for experiments that require protein lipid modification in live cells.
Dozier et al. examined the kinetic behavior of mutated FTases to study how the enzyme’s active site could be expanded to improve the catalytic efficiencies for bulkier FPP analogues.527 Docking experiments suggested that positions W102β, Y154β, and Y205β were important for controlling the volume of substrates that could be accommodated. Thus, those positions were altered to alanine via site directed mutagenesis which yielded mutants W102Aβ, Y154Aβ, and Y205Aβ. When tested using a Dn-GCVLS substrate, some FTase mutants were found to exhibit substantial improvements. For example, mutant W102Aβ showed a 43-fold higher catalytic efficiency for processing a benzaldehyde-containing analogue over FPP, and mutant Y205Aβ showed improved activities for a cyclooctene analogue and a dramatic 300-fold increased catalytic efficiency for a coumarin-containing analogue (C10-Cou-OPP). Some mutations however gave only minimal improvements such as the mutant Y154Aβ which displayed improved binding affinities (lower Km) for the benzaldehyde analogue which, however, was offset by a decreased kcat. Overall, it was shown that mutagenesis of FTase could be exploited to expand the variety of susceptible FPP analogues, that could potentially broaden the types of click chemistry applications associated with protein S-prenylation.
In a related work, Zhang et al. employed simultaneous enzymatic labeling of two different target proteins each selectively with different functionalities by exploiting the distinct specificities of rat GGTase-I (rGGTase-I) and rat FTase (rFTase).385 Based on a previous report which showed that rGGTase-1 has a larger binding pocket compared to that of rFTase, various isoprenoid analogues that contain a clickable functionality were evaluated as substrates for the two enzymes in search for candidates that exhibit enzyme specificity. When tested with GFP-CVIA, C10-Alk-OPP was shown to be a substrate for rFTase and an aryl ketone containing C10 isoprenoid (C10-BK-OPP) for rGGTase-I, and no cross-reactivity between the enzymes and substrates was observed. rFTase was shown to be able to prenylate CVIA substrates with C10-Alk-OPP but not related CVLL substrates while C10-BK-OPP and azide-containing C20-Az-OPP could be transferred to CVLL substrates only with rGGTase-I. Exploiting the substrate selectivity and noncomplementary Ca1a2X sequences, an all-in-one reaction was shown to successfully label GFP-CVLL and RFP-CVIA simultaneously with C10-BK-OPP and C10-Alk-OPP, respectively, in the presence of both rFTase and rGGTase-I (Fig. 23B). Utilizing the methodology further, a one pot site-specific dimerization of proteins was carried out by selectively labeling GFP-CVLL with C15-Az-OPP and GFP-CVIA with C10-Alk-OPP, followed by addition of CuAAC reagents which produced GFP-GFP homodimers in high yield. A similar reaction was carried out to synthesize GFP-RFP heterodimers demonstrating the generality of the developed method to form site-specific tail-to-tail protein dimers.
DNA-protein cross-links (DPCs) are important DNA lesions that interfere with DNA processing which leads to various mutations and cytotoxicity. However, studies on DPCs have been limited due to difficulties in obtaining structurally defined site-specific DPCs. Tretyakova and coworkers investigated the effect of DPCs on DNA replication by exploiting S-prenylation and click chemistry for the preparation of site-specific DNA-protein or -peptide crosslinks (Fig. 22D).549 SDS-PAGE and tandem mass spectrometry confirmed the successful site-specific crosslinking between prenylation-induced GFP-C15-Dh-Az-N3 and an alkyne-labeled 23-mer oligodeoxynucleotide via CuAAC. DNA-peptide crosslinks were generated using synthetic azide-containing 10- or 23-mer peptides, which were both produced in high yields. The DPCs were subjected to primer extension with translesion synthesis (TLS) polymerases hpol k, n and t either through a standing start or running start assay with an 18-mer template. In both assays, TLS polymerases showed successful extension of the primer for control templates, while complete obstruction for all three types of TLS polymerases was observed for both GFP and 23-mer conjugated DPCs. For the DPC containing the 10-mer, polymerases were shown to bypass the lesion and extend the primer with different efficiencies. Overall, these assays demonstrated how different peptide/protein sizes have an effect on primer extension near the site of DNA adduction and suggest that large DPCs would require a proteolytic mechanism for them to be processable by TLS polymerases.
Combination of S-prenylation and click chemistry was utilized for the synthesis of homogenous protein-drug conjugates. Kim and coworkers site-specifically linked a toxic monomethyl auristatin F (MMAF) warhead to repebodies, which are protein scaffolds with a high affinity for epidermal growth factor receptor (EGFR).550 Repebodies were engineered with a C-terminal CVIM, which was exploited for S-prenylation with a geranyl ketone diphosphate (GKPP). Subsequent oxime ligation with a β-glucuronide-linked and aminooxylated MMAF without the employment of a catalyst under acidic conditions produced a homogenous repebody-MMAF conjugate (RDC). The site-specific RDCs showed high serum stability in human plasma due to the hydrolysis-resistant oxime linkage which is advantageous in targeted drug delivery. Towards EGFR-positive cell lines, RDCs showed higher efficacy compared to cell-impermeable MMAF, where the RDC with the highest binding affinity showed the highest cytotoxicity. RDCs had negligible effect on EGFR-negative MCF7 cells, indicating receptor-specific cytotoxicity. Mouse studies revealed that RDCs exhibit antitumor activities, although complete tumor suppression could not be achieved presumably due to the short half-life of RDCs in blood due to the small size of the protein.
Shin and coworkers developed an LC-MS/MS based method to assess the linker stability of ADCs (antibody-drug conjugate).551 The ADC used in their study was a HER2-targeting antibody that was site-specifically conjugated with an MMAF via methods similar to those reported by Kim and coworkers, using site-specific GKPP labeling with FTase followed by payload-conjugation via oxime chemistry.550 Evaluation of the pharmacokinetic profile of the ADC was accomplished by quantifying the drug component of the ADC remaining in plasma. For this, the plasma MMAF concentrations were determined from integration of LC-MS/MS analysis previously standardized using a range of concentrations (19 – 960 ng/ml) fit to a quadratic equation. That method was used to assess the linker stability of ADCs in rat plasma, where 85 % of MMAF was found to remain bound to the ADC after incubation in rat plasma for 7 days at 37 °C. In vivo rat studies showed a similar half-life for the ADC to that of trastuzumab, demonstrating the stability of the oxime linkage.
Distefano and coworkers functionalized Designed Ankyrin Repeat Proteins (DARPins), a type of a small protein scaffold, with a fluorophore using S-prenylation and click chemistry.552 The DARPin used in the study was engineered to bind to epithelial cell adhesion molecules (EpCAM) which is a well-known marker overexpressed in certain cancer cells, and to contain a C-terminal CVIA motif to be recognized by the FTase. Site-specific labeling with C10-Az-OPP followed by SPAAC with a TAMRA-DBCO yielded functionalized DARPins with high yields (>85%). The EpCAM-binding affinities of the fluorophore functionalized DARPins were assessed by flow cytometry. A high degree of binding activity was observed in EpCAM-overexpressing MCF-7 cells which was absent in EpCAM-negative U87 cells, and the high binding affinity to MCF-7 could be turned off when cell surface EpCAMs were blocked with incubation with an excess of unlabeled DARPins. Overall, site-specific labeling of a fluorophore to DARPins allowed high retention of the protein function and did not introduce non-specific interactions.
9.2. N-terminal protein functionalization through N-myristoylation
N-myristoylation of various myristic acid analogues allows the site-specific N-terminal modification of various POIs. The acid analogue requires conversion to a myristoyl-CoA thioester by an endogenous E. coli enzyme acyl-CoA synthetase in order to be recognized as a substrate by the enzyme N-myristoyl transferase (NMT). Therefore, protein conjugation via NMT-mediated labeling and click chemistry has mostly been performed at the cellular level, either in (live) cells or crude lysates prior to POI purification. Examples of such applications are imaging of specific protein functions in (live) bacteria in which the small tag allows minimal adverse effect on the native protein function,553,554 or surface immobilization of POIs from crude lysate in which purification of the excess myristic acid analogues is unnecessary as opposed to conjugation of a molecular payload.555,556 While most of the prior work has been focused on click reactions that involve an azide, it would be interesting to see future development of new myristic acid analogues that could expand the range of utilizable click chemistries.
Tate and coworkers were the first to report site-specific labeling exploiting a combination of N-myristoylation and click chemistry.322 Az-C12-CoA (Fig. 11) was incubated with C. albicans NMT (CaNMT) and a model peptide that possesses the canonical GXXXS N-terminal N-myristoylation target motif. Incubation of equimolar substrates with a catalytic quantity of CaNMT (0.25%) led to a complete enzymatic reaction with rates comparable to that of the native substrate myristoyl-CoA. P. falciparum ADP ribosylation factor 1 (PfARF1), a natural substrate of NMT, was functionalized with an azide exploiting N-myristoylation which was subsequently captured by a phosphine-labeled biotin via Staudinger ligation with high efficiency (>99%). The method was utilized for a one-pot biotin labeling by feeding an azide-containing myristic acid to an engineered E. coli co-expressing CaNMT and PfARF1, followed by addition of biotin-phosphine to the resulting lysate. Pull-down with avidin-beads indicated successful in cellulo functionalization of PfARF1 with >99% efficiency, which indicated successful uptake of the azido-myristic acid into the cells followed by transformation to the active CoA analogue via endogenous E. coli enzyme acyl-CoA synthetase. In a subsequent report, Tate and coworkers expanded the scope of click reactions utilizing a new alkyne-analogue.557 The azide- or alkyne-containing myristoyl-CoA analogues Az-C11-CoA (Fig. 24) and Alk-C14-CoA (Fig. 11), which were designed so that the chain length and flexibility closely adhere to the native substrate, were both successfully transferred to a model peptide that contains the N-terminal region of PfARF1.
Figure 24. Additional myristoyl analogues used in enzymatic N-terminal modification of proteins.
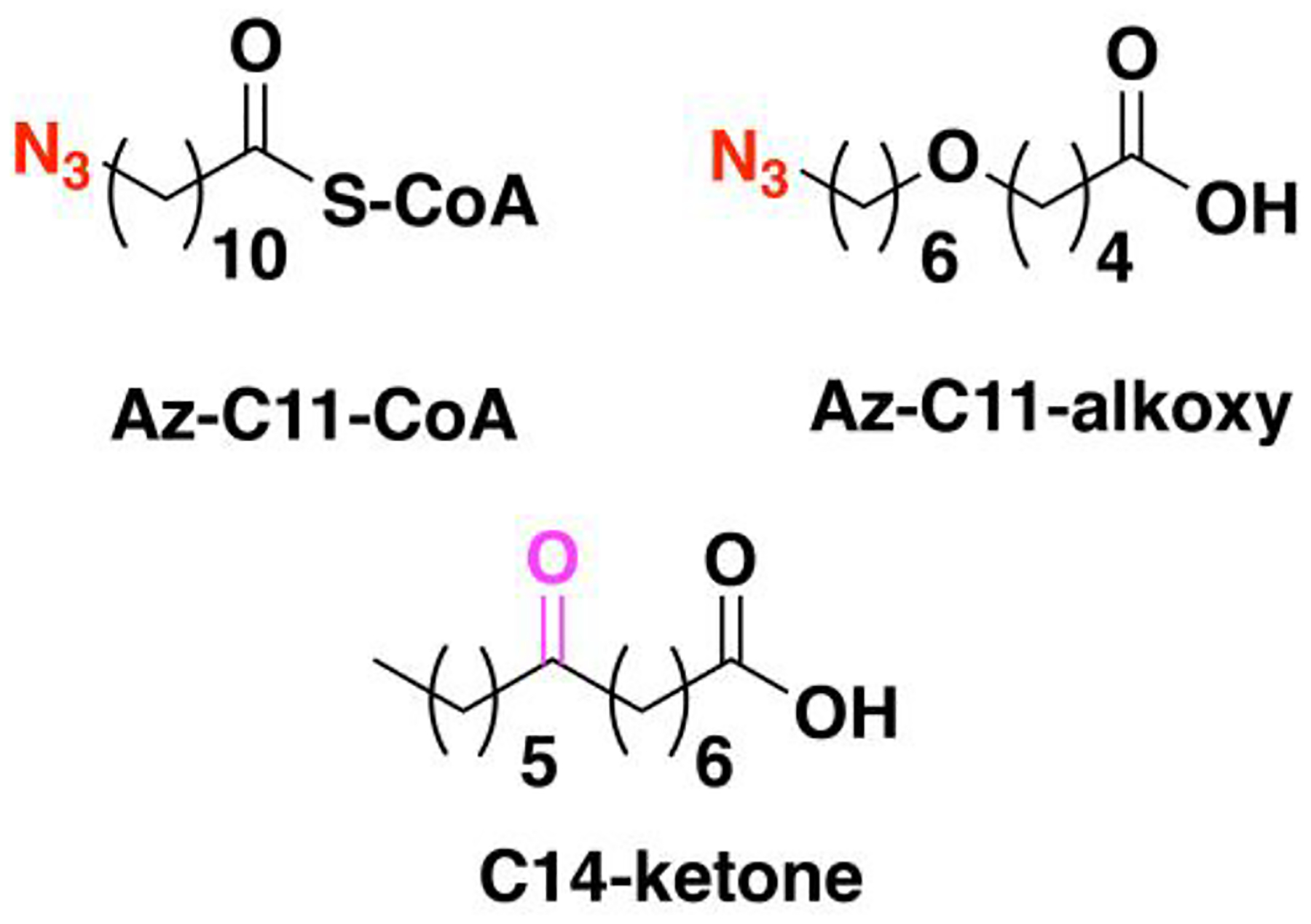
These analogues are functionalized with azides or ketones and vary in length compared to Az-C12 and Alk-C14.
While both reactions were rapid and comparable to the native substrate, the alkyne analogue was found to be two-fold faster. Both analogues were subjected to a one-pot labeling experiment in an E. coli co-expression system similar to the one described in their previous report, where the functionalized PfARF1 was subsequently reacted with biotin-capturing reagents containing either an alkyne, azide, or triarylphosphine. While capturing via both CuAAC and Staudinger ligation were shown to be successful, CuAAC was found to be more efficient similar to prior reports.23 Within CuAAC approaches, conjugating alkyne-payloads to the azide-labeled protein was shown more effective than the converse,532 presumably due to the benefits of having a large excess of activated alkyne when used as the capturing reagent. The labeling methods mentioned above were described in detail in a subsequent protocol paper, in which a multifunctional alkyne-reagent carrying a biotin and TAMRA was labelled to an azide-containing PfARF1 by CuAAC.558
Tirrell and coworkers exploited the bio-orthogonality of NMT-catalyzed protein labeling and click chemistry for fluorophore labeling and immobilization of a target protein from crude cell lysate.559 Two types of GFPs were engineered with different NMT recognition sequences, yARF-GFP which contained a motif from yeast ADP-ribosylation factor, and Fyn-GFP which featured a motif from the Src family tyrosine-protein kinase Fyn. Co-expression of each GFP with human NMT in E. coli with Az-C12, followed by lysis of the cells and CuAAC with the resulting lysate with an alkyne-TAMRA yielded selectively and site-specifically conjugated GFP-TAMRA. Incubation of the azide-labeled GFP-containing cell lysate on strained cyclooctyne-coated glass slides resulted in successful surface immobilization of GFP. While strained alkynes could undergo various side reactions, including processes involving free thiols present in GFP, SPAAC was found to be faster than those non-specific reactions, allowing control over the orientation of immobilized proteins by site-specific conjugation through the N-terminus.
NMT-mediated protein labeling was applied to calmodulin (CaM), a biologically relevant Ca2+-binding protein.555 The solvent-accessible N-terminus of CaM was engineered to display an NMT recognition sequence, derived from either human calcineurin B (CaN) or yeast ADP ribosylation factor. In addition, two types of linkers were employed to connect the NMT recognition sequence to the protein which led to a total of five engineered CaM constructs. All five were successfully labeled with an azide moiety when co-expressed in E. coli with human NMT1 in the presence of Az-C12 and were shown to maintain Ca2+-binding and CaN-activating abilities comparable to that of wild type (WT) CaM. As the NMT-motif derived from CaN showed superior activity, it was selected for one-pot fluorophore labeling by subjecting the cell lysate containing the azide-labeled construct to CuAAC with alkyne-TAMRA. A single fluorescent band was observed by SDS-PAGE analysis which indicated selective labeling of the target protein as a result of the bio-orthogonality of NMT-mediated labeling and CuAAC. Various CaM-immobilized resins were prepared and tested for their capability to bind to and purify CaN. CaM-resins prepared via SPAAC with chemoenzymatically generated azide-CaM and azadibenzocyclooctyne-functionalized resins (ADIBO-resins) showed CaN yields 10-fold higher than resins prepared by immobilizing WT CaM on NHS-ester or cyanide activated resins, demonstrating the advantage of protein immobilization in a site-specific fashion.
Tirrell and coworkers also utilized the NMT labeling system to visualize proteins in bacteria.553 The target proteins were chemotaxis proteins Tar and CheA, and cell division proteins FtsZ and FtsA, which were each co-expressed in an E. coli system with NMT and methionyl aminopeptidase. The addition of Az-C12 or Az-C11-alkoxy simultaneously during the induction stage of protein expression, followed by the addition of a bicyclononyne-containing BODIPY reagent to the subsequently produced lysate led to complete modification of all four target proteins. In situ imaging of fixed and permeabilized cells was conducted on cells expressing each of the target proteins equipped with C-terminal myc tag fusions for immunofluorescence confirmation using an anti-myc AF647-conjugated antibody. Polar localization of Tar and CheA and septal localization for FtsZ and FtsA could be visualized in both BODIPY and AlexaFluor-647 channels; the former allowed imaging of the myristoylated proteins while the latter was used to detect myc. This demonstrated the utility of NMT-catalyzed fluorophore labeling for imaging bacterial proteins in cells, which could even be applied to live cells. The details of the sample preparation and labeling protocols for live cell imaging of bacterial proteins were further described in a follow-up methods paper.554
In a related work, Zhang and coworkers assessed whether the labeling of myristic acid analogues instead of the natural substrate alters protein function using a model protein ADP-ribosylation factor (Arf1).311 Arf1 was myristoylated with five myristic acid analogues resulting in modified proteins that showed membrane-associating and GTP-hydrolyzing activities similar to endogenous Arf1 while non-myristoylated Arf1 lacked either properties. Additionally, an Arf1 labeled with Az-C14-ketone (Fig. 24) could be conjugated with a fluorescent hydrazine reagent. Importantly, in these studies, the authors described a methodology that could be used to determine the suitability of lipid-modification methods to monitor protein function in cells.
Calmodulin (CaM) conjugated Sepharose resin was prepared by a combination of N-myristoylation and click chemistry, which led to improved purification efficiencies of CaM-binding proteins.556 Azide-functionalized CaM was prepared by N-myristoylation with Az-C12, which was subsequently immobilized onto DBCO- or alkyne-functionalized Sepharose resin via SPAAC or CuAAC. DBCO-resin was shown to immobilize larger amounts of CaM compared to simple alkyne-resin and was shown to outperform commercial resin in purifying a CaM-binding protein calcineurin (CaN). CaM-resin was prepared directly from azide-CaM present in clarified lysate, which showed higher CaN purification efficiencies compared to resin prepared from purified azide-CaM. CaM-resin was tested for purification of calcium/calmodulin dependent kinase II alpha (CaMKII) which has traditionally been isolated using IMAC purification. Exploiting the CaM-resin yielded a product with higher purity and less degradation compared to purification using Ni-NTA resin. In their following methods paper, Kinzer-Ursem and coworkers described the details of CuAAC-mediated functionalization of CAM myristoylated with Az-C12.560 In addition to fluorophore labelling with an alkyne-functionalized AlexaFluor-647, protein-immobilization on gold nanoparticles (AuNP) was shown where it was noted that cyclooctyne-functionalized AuNP resulted in aggregation leading to inferior conjugation with SPAAC compared to CuAAC.
Tirrell and coworkers expanded their work from 2016 by imaging bacterial proteins in live cells with nanometer resolution.561 To exploit direct stochastic optical reconstruction microscopy (dSTORM), two cell-permeable photo-switchable rhodamine spirolactam dyes were developed. These dyes exhibited photon budgets comparable to conventional STORM dyes in the absence of additives such as oxygen-scavenging buffers or reducing agents that are usually required for facilitating the photo-switching of conventional dyes. Following the co-expression of target proteins with NMT in E. coli in the presence of Az-C12, bicyclononyne-functionalized dyes were added, which yielded live cells containing the fluorophore-labeled proteins that were then subjected to imaging without fixation or permeabilization. Chemotaxis proteins Tar and CheA and cell division proteins FtsZ and FtsA were observed with a mean localization precision in the nanometer range. While modification of the N-terminus presumably caused minimal perturbation of protein function, indicated by the successful imaging of live cells, the effect of the peptide tag and fluorophore were shown to vary for each protein of interest.
10. Conclusion
Click chemistry has played a critical role in illuminating the process of protein lipidation, as well as in discerning their roles in controlling protein function, localization, and protein-protein interactions. The ability to use metabolic labeling to incorporate lipid analogues containing functionality that can be subsequently modified for various analyses has proved to be a versatile strategy. Click reactions with affinity handles have allowed proteins to be pulled down and identified by proteomic methods. Incorporation of quantitative mass spectrometric methods into such workflows have made it possible to study the regulation of protein lipid modification and the impact of specific inhibitors. Click reactions with fluorophores have enabled the localization of lipidated proteins to be studied and their levels quantified. Overall, much information has been gained concerning the biology of protein S-prenylation using this approach. However, as mentioned, most of the currently available tools are only applicable to fixed cells and therefore efforts into applying click chemistry in molecular imaging inside live cells or tissues needs to be pursued moving forward.
In a similar way, the use of click chemistry in conjunction with protein lipidation has proved to be a powerful approach for site selective protein modification. Site-specific enzymatic incorporation, either in vitro or in cellulo, of lipid substrates bearing appropriate functional groups has enabled a diverse range of modifications to be performed on either the N-termini (via N-myristoylation) or C-termini (via S-prenylation) of target proteins. Applications include immobilization, fluorescent labeling and drug conjugation; this approach has also proved useful for the construction of more complex architectures including protein nanostructures and protein-DNA conjugates. On the other hand, enzymatic S- and N-acylation has been applied in only developing platforms for screening of fatty acylation inhibitors. There are no reports so far in using such types of lipid modification for bioconjugation strategies, which can be attributed to their lack of sequence specificity (e. g. unclear recognition motifs) and the susceptibility of S-acylated substrates to hydrolysis.
Throughout the above studies, click chemistry has played a central role. A summary of the features of different click chemistries that have been used for studying and exploiting protein lipid modification is shown Fig. 25. This graphic emphasizes that individual click reactions have different attributes that make them more or less suitable for different applications and illustrate that there is no one single reaction that is best for all applications. For example, while the alkene and tetrazine reactants are somewhat large, making it difficult to incorporate them into substrates without attenuating their enzymatic reactivity, the inverse electron demand Diels Alder (iEDDA) reaction is the fastest click reaction discovered to date. The superior kinetics of iEDDA that does not require toxic catalysts allows click reactions to be applicable for live cell studies, which could even be expanded to in vivo environments. Among click reactions that involve azides and alkynes, the copper-catalyzed version (CuAAC) is not compatible with live-cell experiments due to toxicity issues while the strain-promoted (SPAAC) reaction is considerably slower and the presence of a large hydrophobic moiety appended to a protein can create solubility/stability problems. Nonetheless, both reactions remain to be a powerful tool especially for in vitro studies due to the robustness of the reaction, and since the small size of azide and alkyne groups that can be readily introduced into the protein substrates without abrogating their reactivity. Oxime and hydrazone forming reactions are particularly attractive due to the structural innocuity of small aldehyde and ketone groups but that is partially offset by slower reaction rates, incomplete orthogonality (due to the presence of some aldehydes and ketones in cells) and hydrolytic instability; at the same time, oxime and hydrazone products manifest the highly useful feature that their formation is reversible that has been utilized for one-pot labelling and purification of protein-conjugates. Alternative ligation reactions with aldehydes that forms a stable C-C bond instead of an imine have been reported as well,67,562 which could be exploited to expand the scope of click reactions involving aldehydes.
Figure 25. Considerations in the applicability of click chemistry in protein or cellular environments.
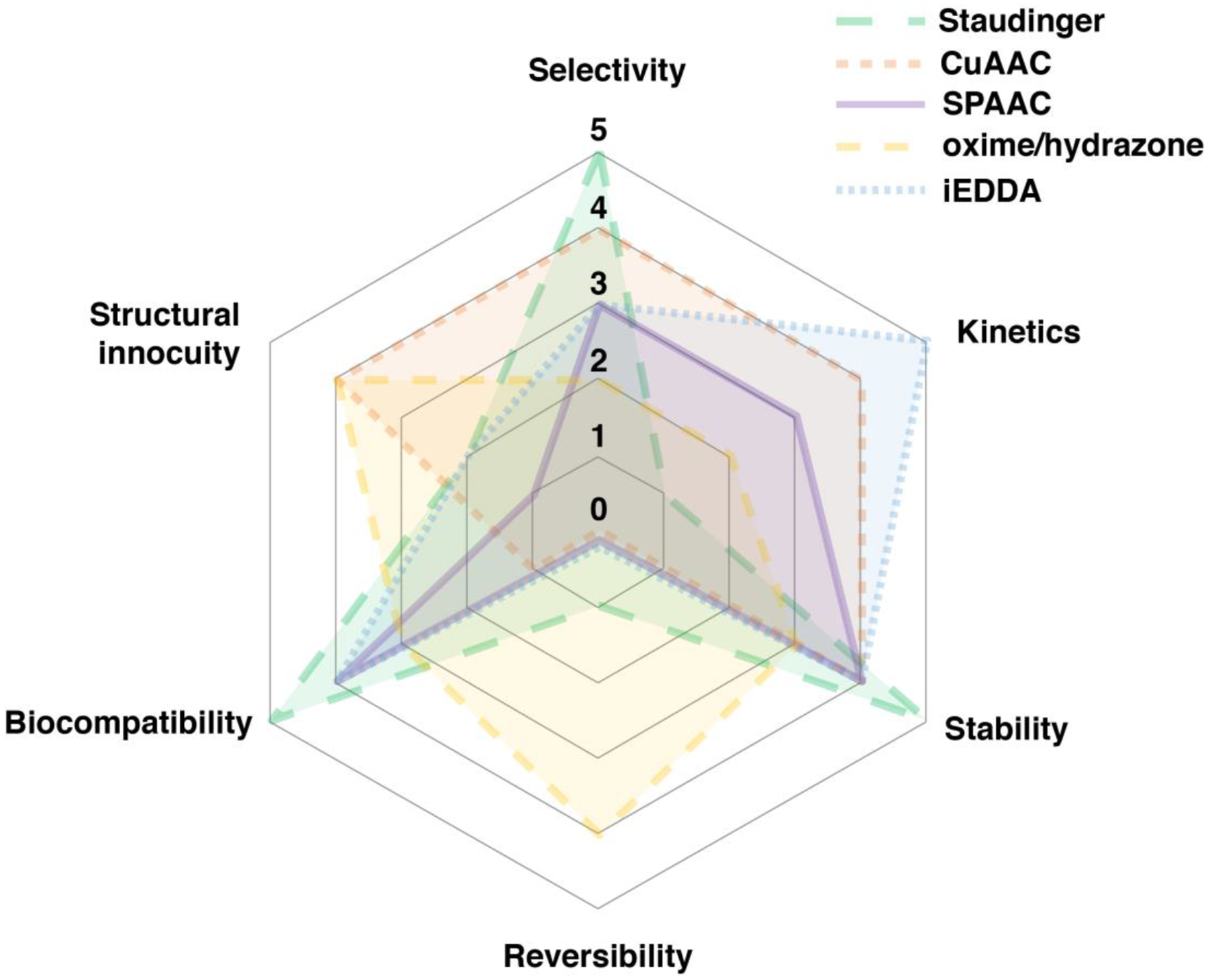
Individual reactions were given a rating from 5 (highest) to 0 (lowest) for each category based on how much they satisfy each of the following criteria. Kinetics: High rate constant; Selectivity: Selective reaction between click functionalities without side reactions with biologically endogenous functionalities; Stability: High stability of the resulting linkage; Structural Innocuity: Minimal adverse effect of the coupling partners and/or resulting linkage on cellular or protein activity; Biocompatibility: Applicability of the reaction conditions for live cell or in vivo experiments; Reversibility: Reversibility of the click chemistry product.
For studies of protein lipid modification to advance further, both the understanding of enzyme-substrate interaction and click chemistry will need to evolve. Developing enzymes that are more tolerant towards a wider variety of analogue substrates would broaden the scope of applicable click reactions, that is currently heavily focused on alkyne and azide functionalities due to their small size. In terms of potential improvements in click chemistry, new functionalities will need to be developed that allow minimalist additions to enzyme substrates that have limited impact on enzyme recognition, while exhibiting increased click reactivity to allow the detection of modified proteins present at very low concentrations in cells. The further development of click chemistries that permit experiments with live cells and that have minimal effects on the structure, function, stability and solubility of proteins will significantly broaden the types of questions that can be addressed using in cellulo metabolic labeling and in vitro modification. For instance, click reactions with rates comparable to iEDDA that do not require bulky ring systems would be particularly useful. Click reactions that proceed at equimolar concentrations compared to the target protein, even in the absence of catalysts, would open up in vivo applications where localized high concentrations are difficult to achieve. Applying two-photon chemistry to thiol-ene click chemistry could be utilized for experiments that require spatiotemporal control in in cellulo or in vivo applications due to the low toxicity and tissue penetrating capabilities of that method. While these future directions are raised here in the context of protein lipidation, they are obviously all germane to many other types of protein modifications.
Table 2.
Summary of click chemistry conditions and yields that involve site-specific protein labeling via lipid modifying enzymes
| Entry | Application | Analogue & Click Conditions | Yield | Ref |
|---|---|---|---|---|
| Farnesyl transferase (* indicates geranylgeranyl transferase type 1 or Rab geranylgeranyl transferase) | ||||
| Staudinger ligation | ||||
| 1 | Surface immobilization | C10-Et-Az-OPP 1–20 μM GFP- or GST-Azide diphenylphosphine slides 50:1 DMF/water 1.5 h |
NA | 533 |
| 2 | Proof-of-concept | C10-Az-OPP 0.9 mM Dn-GCVIA-N3 45 mM phosphine reagent 48 h |
NA | 395 |
| 3 | Proof-of-concept | C15-Az-OPP 0.9 mM Dn-GCVIA-N3 45 mM phosphine reagent 48 h |
NA | 402 |
| 4 | Proof-of-concept | AAA-GPP, APO-GPP 0.5 nmol GPP-N3 2.5 nmol phosphine |
Completion in 1.5 h | 536 |
| 25 mM HEPES pH 7.2, 40 mM NaCl, 2 mM MgCl2 8 h rt |
Completion in 4 h | |||
| CuAAC | ||||
| 5 | Surface immobilization | C15-Dh-Az-OPP 0.1 mM GFP-N3, Alkyne-beads 1 mM CuSO4, 1 mM TCEP, 0.1 mM TBTA 43 mM NaH2PO4 1 h 25 °C |
96% immobilization (>80% covalent) | 531 |
| 6 | Surface immobilization | C10-Et-Az-OPP, C10-Et-Alk-OPP 1–20 μM GFP- or GST-alkyne N3-slides 1:3 glycerol/water 1.7 mM CuSO4/TBTA/TCEP 2–7 h 4 °C |
NA | 533 |
| 7 | Surface immobilization | C10-Alk-OPP, C15-Dh-Az-OPP 50 μM GFP-alkyne and -N3 Corresponding agarose (-N3 or -alkyne) 1 mM CuSO4, 1 mM TCEP, 0.1 mM TBTA overnight 25°C 25 μM GFP-alkyne Texas red-N3 1 mM CuSO4, 1 mM TCEP, 0.1 mM TBTA 1 h 25 °C |
93% | 534 |
| 81% | ||||
| 8 | Fluorophore labeling GFP-DNA nanostructure assembly | C15-Dh-Az-OPP 20 μM GFP-N3 22 μM DNA-alkyne 50 mM NaH2PO4, pH 7.3 1 mM CuSO4, 1 mM TCEP, 0.1 mM TBTA 1 h rt |
72% | 535 |
| 9 | Surface immobilization | C5-Alk-OPP 80 μM mCherry-alkyne Agarose-N3 50 mM NaH2PO4 pH 7.3 1 mM CuSO4, 1 mM TCEP, 0.1 mM TBTA 4 h rt |
NA (presence or absence of VIA does not affect click efficiency) |
537 |
| 10 | GFP-ODN oligoconjugation | C10-Dh-Az-OPP, C15-Dh-Az-OPP 14 μM Protein-N3 17 μM DNA-alkyne 1 mM CuSO4, 1 mM TCEP, 0.2 mM TBTA 24 h rt |
>90% (GFP) >81% (mCherry) |
541 |
| 11 | Surface immobilization | C15-Alk-OPP 0.1 mM mCherry-alkyne N3-gold 1 mM CuSO4, 1 mM TCEP, 0.1 M TBTA 43 mM NaH2PO4 pH 7.3 2 h |
NA | 544 |
| 12 | Antibody immobilization | C10-Et-Alk-OPP 100 μM antibody-alkyne Glass-N3 |
NA | 545 |
| 13 | Sandwich antibody arrays | 0.7 mM CuSO4, 0.7 mM TCEP, 0.35 mM TBTA PBS pH 7.4 2 or 4 h rt |
546 | |
| 14 | Antibody arrays | C10-Et-Alk-OPP 5 or 80 μM Protein-alkyne Gold-N3 0.5 mM CuSO4, 0.5 mM TCEP, 50 μM TBTA PBS pH 7.4 4 h rt |
NA | 547 |
| 15 | Protein/peptide-DNA conjugation | C15-Dh-Az-OPP 10 μM DNA-alkyne 60 μM GFP-N3 1 mM CuSO4, 1 mM TCEP, 0.1 mM TBTA 50 mM PB pH 7.5 1.5 – 2 h rt |
70–80% | 549 |
| 16* | Protein dimerization (FTase & GGTase-1) | C10-Alk-OPP, C20-Az-OPP 50 μM GFP-N3 50 μM RFP(or GFP)-alkyne 1 mM CuSO4, 1 mM TCEP, 0.1 mM TBTA |
23% (simultaneous prenylation) 29% (separate prenylation) |
385 |
| 17* | Biotin labeling (GGTase-1 Rab GGTase) | C20-Az-OPP 1 μM REP-1-N3 50 μM Biotin-alkyne 1 mM CuSO4, 10 mM sodium ascorbate, 0.1 mM TBTA 40 min 37 °C |
NA | 404 |
| CuAAC & oxime ligation | ||||
| 18 | Simultaneous fluorophore labeling and PEGylation | C10-BA-Alk-OPP GFP-aldehyde/alkyne PEG3k-aminooxy TAMRA-N3 1 : 3.85 : 15 ratio 40 mM mPDA 1 mM CuSO4, 1 mM TCEP, 0.1 mM TBTA 15 h |
>95% for both | 542 |
| Oxime ligation | ||||
| 19 | Surface immobilization | C15-Ald-OPP 20 μM GFP-aldehyde Alkyne-agarose 100 mM aniline 0.1 M PB pH 7.0 15 min rt |
30% | 538 |
| Fluorophore labeling | 25 μM GFP-aldehyde 125 μM AF488 100 mM analine 0.1 M PB pH 7.0 30 min rt |
60% | ||
| 20 | Fluorophore labeling & PEGylation from crude lysate (model reaction conditions) |
C10-BA-OPP 54 μM GFP-aldehyde Hydrazide-agarose 100 mM aniline 0.1 M PB pH 7.0 45 min rt |
95% (Hydrazone) | 62 |
| 200 mM Hydroxylamine 100 mM aniline 0.2 M PB pH 7.0 3 h rt |
80% (Oxime) | |||
| 21 | Assessment of mPDA as catalyst (model rxn conditions) |
C10-BA-OPP 10 μM GFP-aldehyde 50 μM Dn-aminooxy 750 mM mPDA 100 mM PB pH 7.0 90 s rt |
Completion | 62 |
| 22 | Antibody-drug conjugation | GKPP 12 μM repebody-aldehyde 120 μM beta-glucuronide MMAF 200 mM Na-acetate buffer pH 4.5 10% DMSO 24 h 30 °C |
97.4% | 550 |
| SPAAC | ||||
| 23 | GFP-ODN conjugation | C10-Dh-Az-OPP, C15-Dh-Az-OPP 14 μM GFP-N3 4.6 μM ODN-DIBO H2O or 0.1 M NaCl PB pH 3.5–12.3 Overnight, rt or 4 °C |
95% (GFP) krt=6 M−1s−1 k4 °C=0.8 M−1s−1 27% (mCherry) |
541 |
| 24 | Fluorophore labeling | C10-Az-OPP DARPin-N3 TAMRA-DBCO 1 : 5 molar ratio PBS (400 mM NaCl) 3 h rt |
Near completion | 552 |
| Tetrazine ligation | ||||
| 25 | Proof of concept | C10-TCO-OPP 33 μM GFP-TCO 530 μM benzylamino-tetrazine 4 h rt + overnight 4 °C |
NA | 548 |
| 20 μM OrG-TCO 50 μM dipyridyl-tetrazine 15 min |
Completion | |||
| Thiol-ene | ||||
| 26 | Surface immobilization | FPP 0.1–100 μM Farnesylated protein Thiol-functionalized slides 2.5 mM DTE, 20% glycerol UV 365 nm 10 min 6 Jcm−2 (H, -N-, and K-Ras & Rab6A) or 20 min 12 J cm−2 (mCherry) |
NA | 539 |
| Diels-alder cycloaddition | ||||
| 27 | Proof of concept | HOM-GPP 50 μM CFP-alkene 40 eq 6-maleimidohexanoic acid 50 mM Na2HPO4 pH 6, 40 mM NaCl, 2 mM MgCl2 15 h rt |
30% | 536 |
| N-Myristoyl transferase | ||||
| Staudinger ligation | ||||
| 28 | Biotin labeling | Az-C11-CoA 3 nmol PfARF1-N3 200 μM Biotin-phosphine 2 h 37 °C |
>99% | 557 |
| CuAAC | Az-C12 (for entries 29–32) | |||
| 29 | Biotin and fluorophore labeling | 100 μg PfARF1-N3 50 μM Alkyne-biotin/TAMRA 0.5 mM CuSO4, 0.5 mM TCEP, 50 μM TBTA 1 h rt |
NA | 558 |
| 30 | Fluorophore labeling | GFP-N3 (in cell lysate) 5.2 μM TAMRA-alkyne 3.5 mM CuSO4 (Modified Invitrogen™ Click-iT Kit instructions) 50 Tris-HCl pH 8.0 1% SDS 25 min rt |
NA | 559 |
| 31 | Fluorophore labeling | CaM-N3 (in cell lysate) 5.2 μM TAMRA-alkyne 3.5 mM CuSO4 (Modified Invitrogen™ Click-iT Kit instructions) 20 min rt |
NA | 555 |
| 32 | Surface immobilization | 1, 2 or 5 mg ml−1 CAM-N3 Alkyne-resin 2 mM CuSO4, 10 mM THPTA, 10 mM sodium ascorbate |
NA | 556 |
| SPAAC | Az-C12 (for entries 33–38), Az-C11-alkoxy (for entry 38) | |||
| 33 | Surface immobilization | GFP-N3 (in 0.25–1 mg ml−1 cell lysate) Glass-DIBO or -ADIBO 40 min |
90% specificity | 559 |
| 34 | Surface immobilization | 0.5 mg ml−1 CaM-N3 (estimation in 3.5 mg ml−1 cell lysate) ADIBO-resin Overnight 4 °C |
NA | 555 |
| 35 | Surface immobilization | 1, 2 or 5 mg ml−1 CAM-N3 DBCO-resin 20 mM HEPES pH 7.5 Overnight 4 °C |
NA | 556 |
| 36 | Fluorophore labeling and live-cell imaging | Cells expressing recombinant protein-N3 20 μM BODIPY-BCN PBS 0.5–1 h, rt or 37 °C |
554 | |
| 37 | Fluorophore labeling and live-cell imaging | Cells expressing recombinant protein-N3 200 nM Rhodamine-BCN PBS 1 h 37 °C |
NA | 561 |
| 38 | Fluorophore labeling and live-cell imaging | Protein-N3 (in 0.4 mg ml−1 cell lysate) 2 μM BODIPY-BCN PBS 0.5 h, rt or 37 °C |
NA | 553 |
| Hydrazone ligation | ||||
| 39 | Fluorophore labeling | C14-ketone Arf1-ketone 1 mM Fluorescein-hydrazide PBS 16 h 4 °C |
NA | 311 |
Acknowledgements
This work was supported in part by the National Institute of Health grants RF1AG056976, R01GM084152, and R01NS107442. KFS was supported by a Doctoral Dissertation Fellowship from the University of Minnesota.
Abbreviations
- Biotin-HPDP
N-[6-(biotinamido)hexyl]-3’-(2’-pyridyldithio)propionamide
- DPBS
Dulbecco’s phosphate-buffered saline
- DTT
dithiothreitol
- ESI-MS
electrospray ionization mass spectrometry
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- LC-MS
liquid chromatography mass spectrometry
- LC-MS/MS
liquid chromatography tandem mass spectrometry
- MES
2-(N-morpholino)ethanesulfonic acid
- NMR
nuclear magnetic resonance
- PBS
phosphate-buffered saline
- RBC
Red blood cells
- RIPA
Radioimmunoprecipitation assay
- SDS
sodium dodecylsulfate
- TBTA
Tris((1-benzyl-4-triazolyl)methyl)amine
- TCEP
tris(2-carboxyethyl)phosphine
- TEA
triethylamine
- THPTA
Tris(3-hydroxypropyltriazolylmethyl)amine
- Tris-HCl
Tris(hydroxymethyl)aminomethane hydrochloride
Biographies
Kiall F. Suazo received his B.S. and M.S. in Chemistry from the University of the Philippines Diliman in 2009 and 2014, respectively, where also served as a college Chemistry instructor. He is currently a Ph.D. candidate in the Chemistry Department at the University of Minnesota supervised by Prof. Mark D. Distefano. In the Distefano group, his research involves the use of click chemistry-based probes of isoprenoids to study prenylated proteins for chemical proteomic analysis and validation of prenylation status. In collaboration with many biologists, he uses these click chemistry-based approaches to delineate key prenylated proteins associated with various diseases and characterize PTMs on candidate proteins.
Keun-Young “Kevin” Park was born in Seoul, South Korea, and grew up in Seoul and Ithaca, New York. He received his B.S (2010) and M.S (2012) degrees in Chemistry from Sungkyunkwan University, South Korea, with Professor Young-Uk Kwon working on mesoporous materials for energy applications. Then he served in the Republic of Korea Air Force as a lieutenant in charge of disaster management. He is currently back at the bench, pursuing a Ph.D. degree in Chemistry with Professor Mark. D. Distefano at the University of Minnesota. His research interests include utilizing protein modifying enzymes for the development of protein-based therapeutics.
Mark Distefano was born in Baton Rouge, LA, and grew up in California and Paris, France. He received his B.A. degree in Chemistry and Biochemistry from the University of California at Berkeley in 1985 and his Ph.D. degree from Massachusetts Institute of Technology in 1989, where he worked with Professor Christopher T. Walsh. He was a postdoctoral fellow in the laboratory of Peter B. Dervan at California Institute of Technology. He is currently Distinguished McKnight Professor of Chemistry and Medicinal Chemistry at the University of Minnesota. His current research is focused on understanding protein lipid modification including protein prenylation.
References
- (1).Hentschel A; Zahedi RP; Ahrends R Protein Lipid Modifications—More than Just a Greasy Ballast. Proteomics 2016, 16, 759–782. [DOI] [PubMed] [Google Scholar]
- (2).Wang M; Casey PJ Protein Prenylation: Unique Fats Make Their Mark on Biology. Nat. Rev. Mol. Cell Biol 2016, 17, 110–122. [DOI] [PubMed] [Google Scholar]
- (3).Jiang H; Zhang X; Chen X; Aramsangtienchai P; Tong Z; Lin H Protein Lipidation: Occurrence, Mechanisms, Biological Functions, and Enabling Technologies. Chem. Rev 2018, 118, 919–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Resh MD Fatty Acylation of Proteins: The Long and the Short of It. Prog. Lipid Res 2016, 63, 120–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ciepla P; Magee AI; Tate EW Cholesterylation: A Tail of Hedgehog. Biochem. Soc. Trans 2015, 43, 262–267. [DOI] [PubMed] [Google Scholar]
- (6).Running M The Role of Lipid Post–Translational Modification in Plant Developmental Processes. Front. Plant Sci 2014, 5, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Resh MD Covalent Lipid Modifications of Proteins. Curr. Biol 2013, 23, R431–R435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Chen B; Sun Y; Niu J; Jarugumilli GK; Wu X Protein Lipidation in Cell Signaling and Diseases: Function, Regulation, and Therapeutic Opportunities. Cell Chem. Biol 2018, 25, 817–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ganesan L; Levental I Pharmacological Inhibition of Protein Lipidation. J. Membr. Biol 2015, 248, 929–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Fraser NJ; Howie J; Wypijewski KJ; Fuller W Therapeutic Targeting of Protein S-Acylation for the Treatment of Disease. Biochem. Soc. Trans 2019, 48, 281–290. [DOI] [PubMed] [Google Scholar]
- (11).Palsuledesai CC; Distefano MD Protein Prenylation: Enzymes, Therapeutics, and Biotechnology Applications. ACS Chem. Biol 2015, 10, 51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Rashidian M; Dozier JK; Distefano MD Enzymatic Labeling of Proteins: Techniques and Approaches. Bioconjug. Chem 2013, 24, 1277–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Zhang Y; Park K-Y; Suazo KF; Distefano MD Recent Progress in Enzymatic Protein Labelling Techniques and Their Applications. Chem. Soc. Rev 2018, 47, 9106–9136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Tate EW; Kalesh KA; Lanyon-Hogg T; Storck EM; Thinon E Global Profiling of Protein Lipidation Using Chemical Proteomic Technologies. Curr. Opin. Chem. Biol 2015, 24, 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Gao X; Hannoush RN A Decade of Click Chemistry in Protein Palmitoylation: Impact on Discovery and New Biology. Cell Chem. Biol 2018, 25, 236–246. [DOI] [PubMed] [Google Scholar]
- (16).Wang Y-C; Distefano MD Synthetic Isoprenoid Analogues for the Study of Prenylated Proteins: Fluorescent Imaging and Proteomic Applications. Bioorg. Chem 2016, 64, 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Peng T; Thinon E; Hang HC Proteomic Analysis of Fatty-Acylated Proteins. Curr. Opin. Chem. Biol 2016, 30, 77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Suazo KF; Schey G; Schaber C; Odom John AR; Distefano MD Proteomic Analysis of Protein–Lipid Modifications: Significance and Application. Mass Spectrometry‐Based Chemical Proteomics. August 13, 2019, pp 317–347. [Google Scholar]
- (19).Kolb HC; Finn MG; Sharpless KB Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chemie Int. Ed 2001, 40, 2004–2021. [DOI] [PubMed] [Google Scholar]
- (20).Sletten EM; Bertozzi CR Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality. Angew. Chemie Int. Ed 2009, 48, 6974–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Sletten EM; Bertozzi CR From Mechanism to Mouse: A Tale of Two Bioorthogonal Reactions. Acc. Chem. Res 2011, 44, 666–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).McKay CS; Finn MG Click Chemistry in Complex Mixtures: Bioorthogonal Bioconjugation. Chem. Biol 2014, 21, 1075–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Oliveira BL; Guo Z; Bernardes GJL Inverse Electron Demand Diels–Alder Reactions in Chemical Biology. Chem. Soc. Rev 2017, 46, 4895–4950. [DOI] [PubMed] [Google Scholar]
- (24).Kölmel DK; Kool ET Oximes and Hydrazones in Bioconjugation: Mechanism and Catalysis. Chem. Rev 2017, 117, 10358–10376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kim E; Koo H Biomedical Applications of Copper-Free Click Chemistry: In Vitro, in Vivo, and Ex Vivo. Chem. Sci 2019, 10, 7835–7851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Saxon E; Bertozzi CR Cell Surface Engineering by a Modified Staudinger Reaction. Science 2000, 287, 2007–2010. [DOI] [PubMed] [Google Scholar]
- (27).Bednarek C; Wehl I; Jung N; Schepers U; Bräse S The Staudinger Ligation. Chem. Rev 2020, 120, 4301–4354. [DOI] [PubMed] [Google Scholar]
- (28).Lin FL; Hoyt HM; van Halbeek H; Bergman RG; Bertozzi CR Mechanistic Investigation of the Staudinger Ligation. J. Am. Chem. Soc 2005, 127, 2686–2695. [DOI] [PubMed] [Google Scholar]
- (29).Tornøe CW; Christensen C; Meldal M Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. J. Org. Chem 2002, 67, 3057–3064. [DOI] [PubMed] [Google Scholar]
- (30).Rostovtsev VV; Green LG; Fokin VV; Sharpless KB A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chemie Int. Ed 2002, 41, 2596–2599. [DOI] [PubMed] [Google Scholar]
- (31).Meldal M; Tornøe CW Cu-Catalyzed Azide−Alkyne Cycloaddition. Chem. Rev 2008, 108, 2952–3015. [DOI] [PubMed] [Google Scholar]
- (32).Neumann S; Biewend M; Rana S; Binder WH The CuAAC: Principles, Homogeneous and Heterogeneous Catalysts, and Novel Developments and Applications. Macromol. Rapid Commun 2020, 41, 1900359. [DOI] [PubMed] [Google Scholar]
- (33).Chan TR; Hilgraf R; Sharpless KB; Fokin VV Polytriazoles as Copper(I)-Stabilizing Ligands in Catalysis. Org. Lett 2004, 6, 2853–2855. [DOI] [PubMed] [Google Scholar]
- (34).Hong V; Presolski SI; Ma C; Finn MG Analysis and Optimization of Copper-Catalyzed Azide–Alkyne Cycloaddition for Bioconjugation. Angew. Chemie Int. Ed 2009, 48, 9879–9883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Presolski SI; Hong V; Cho S-H; Finn MG Tailored Ligand Acceleration of the Cu-Catalyzed Azide−Alkyne Cycloaddition Reaction: Practical and Mechanistic Implications. J. Am. Chem. Soc 2010, 132, 14570–14576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Soriano del Amo D; Wang W; Jiang H; Besanceney C; Yan AC; Levy M; Liu Y; Marlow FL; Wu P Biocompatible Copper(I) Catalysts for in Vivo Imaging of Glycans. J. Am. Chem. Soc 2010, 132, 16893–16899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Das D; Tnimov Z; Nguyen UTT; Thimmaiah G; Lo H; Abankwa D; Wu Y; Goody RS; Waldmann H; Kirill A Flexible and General Synthesis of Functionalized Phosphoisoprenoids for the Study of Prenylation in Vivo and in Vitro. ChemBioChem 2012, 13, 674–683. [DOI] [PubMed] [Google Scholar]
- (38).Uttamapinant C; Tangpeerachaikul A; Grecian S; Clarke S; Singh U; Slade P; Gee KR; Ting AY Fast, Cell-Compatible Click Chemistry with Copper-Chelating Azides for Biomolecular Labeling. Angew. Chemie Int. Ed 2012, 51, 5852–5856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Jiang H; Zheng T; Lopez-Aguilar A; Feng L; Kopp F; Marlow FL; Wu P Monitoring Dynamic Glycosylation in Vivo Using Supersensitive Click Chemistry. Bioconjug. Chem 2014, 25, 698–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Bevilacqua V; King M; Chaumontet M; Nothisen M; Gabillet S; Buisson D; Puente C; Wagner A; Taran F Copper-Chelating Azides for Efficient Click Conjugation Reactions in Complex Media. Angew. Chemie Int. Ed 2014, 53, 5872–5876. [DOI] [PubMed] [Google Scholar]
- (41).Kennedy DC; McKay CS; Legault MCB; Danielson DC; Blake JA; Pegoraro AF; Stolow A; Mester Z; Pezacki JP Cellular Consequences of Copper Complexes Used To Catalyze Bioorthogonal Click Reactions. J. Am. Chem. Soc 2011, 133, 17993–18001. [DOI] [PubMed] [Google Scholar]
- (42).Agard NJ; Prescher JA; Bertozzi CR A Strain-Promoted [3 + 2] Azide−Alkyne Cycloaddition for Covalent Modification of Biomolecules in Living Systems. J. Am. Chem. Soc 2004, 126, 15046–15047. [DOI] [PubMed] [Google Scholar]
- (43).Dommerholt J; Rutjes FPJT; van Delft FL Strain-Promoted 1,3-Dipolar Cycloaddition of Cycloalkynes and Organic Azides. Top. Curr. Chem 2016, 374, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Agard NJ; Baskin JM; Prescher JA; Lo A; Bertozzi CR A Comparative Study of Bioorthogonal Reactions with Azides. ACS Chem. Biol 2006, 1, 644–648. [DOI] [PubMed] [Google Scholar]
- (45).Mbua NE; Guo J; Wolfert MA; Steet R; Boons G-J Strain-Promoted Alkyne–Azide Cycloadditions (SPAAC) Reveal New Features of Glycoconjugate Biosynthesis. ChemBioChem 2011, 12, 1912–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Sletten EM; Bertozzi CR A Hydrophilic Azacyclooctyne for Cu-Free Click Chemistry. Org. Lett 2008, 10, 3097–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Ning X; Guo J; Wolfert MA; Boons G-J Visualizing Metabolically Labeled Glycoconjugates of Living Cells by Copper-Free and Fast Huisgen Cycloadditions. Angew. Chemie Int. Ed 2008, 47, 2253–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Chang PV; Prescher JA; Sletten EM; Baskin JM; Miller IA; Agard NJ; Lo A; Bertozzi CR Copper-Free Click Chemistry in Living Animals. Proc. Natl. Acad. Sci 2010, 107, 1821 LP–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).van Geel R; Pruijn GJM; van Delft FL; Boelens WC Preventing Thiol-Yne Addition Improves the Specificity of Strain-Promoted Azide–Alkyne Cycloaddition. Bioconjug. Chem 2012, 23, 392–398. [DOI] [PubMed] [Google Scholar]
- (50).Karver MR; Weissleder R; Hilderbrand SA Bioorthogonal Reaction Pairs Enable Simultaneous, Selective, Multi-Target Imaging. Angew. Chemie Int. Ed 2012, 51, 920–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Nikić I; Plass T; Schraidt O; Szymański J; Briggs JAG; Schultz C; Lemke EA Minimal Tags for Rapid Dual-Color Live-Cell Labeling and Super-Resolution Microscopy. Angew. Chemie Int. Ed 2014, 53, 2245–2249. [DOI] [PubMed] [Google Scholar]
- (52).Rashidian M; Keliher EJ; Bilate AM; Duarte JN; Wojtkiewicz GR; Jacobsen JT; Cragnolini J; Swee LK; Victora GD; Weissleder R; et al. Noninvasive Imaging of Immune Responses. Proc. Natl. Acad. Sci 2015, 112, 6146–6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Yang J; Šečkutė J; Cole CM; Devaraj NK Live-Cell Imaging of Cyclopropene Tags with Fluorogenic Tetrazine Cycloadditions. Angew. Chemie Int. Ed 2012, 51, 7476–7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Yang J; Liang Y; Šečkutė J; Houk KN; Devaraj NK Synthesis and Reactivity Comparisons of 1-Methyl-3-Substituted Cyclopropene Mini-Tags for Tetrazine Bioorthogonal Reactions. Chem. – A Eur. J 2014, 20, 3365–3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Engelsma SB; Willems LI; van Paaschen CE; van Kasteren SI; van der Marel GA; Overkleeft HS; Filippov DV Acylazetine as a Dienophile in Bioorthogonal Inverse Electron-Demand Diels–Alder Ligation. Org. Lett 2014, 16, 2744–2747. [DOI] [PubMed] [Google Scholar]
- (56).Cohen JD; Zou P; Ting AY Site-Specific Protein Modification Using Lipoic Acid Ligase and Bis-Aryl Hydrazone Formation. Chembiochem 2012, 13, 888–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Dirksen A; Dirksen S; Hackeng TM; Dawson PE Nucleophilic Catalysis of Hydrazone Formation and Transimination: Implications for Dynamic Covalent Chemistry. J. Am. Chem. Soc 2006, 128, 15602–15603. [DOI] [PubMed] [Google Scholar]
- (58).Dirksen A; Hackeng TM; Dawson PE Nucleophilic Catalysis of Oxime Ligation. Angew. Chemie Int. Ed 2006, 45, 7581–7584. [DOI] [PubMed] [Google Scholar]
- (59).Dirksen A; Dawson PE Rapid Oxime and Hydrazone Ligations with Aromatic Aldehydes for Biomolecular Labeling. Bioconjug. Chem 2008, 19, 2543–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Rashidian M; Mahmoodi MM; Shah R; Dozier JK; Wagner CR; Distefano MD A Highly Efficient Catalyst for Oxime Ligation and Hydrazone–Oxime Exchange Suitable for Bioconjugation. Bioconjug. Chem 2013, 24, 333–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Kalia J; Raines RT Hydrolytic Stability of Hydrazones and Oximes. Angew. Chem. Int. Ed. Engl 2008, 47, 7523–7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Rashidian M; Song JM; Pricer RE; Distefano MD Chemoenzymatic Reversible Immobilization and Labeling of Proteins without Prior Purification. J. Am. Chem. Soc 2012, 134, 8455–8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Bandyopadhyay A; Gao J Iminoboronate Formation Leads to Fast and Reversible Conjugation Chemistry of α-Nucleophiles at Neutral PH. Chem. – A Eur. J 2015, 21, 14748–14752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Nisal R; Jose GP; Shanbhag C; Kalia J Rapid and Reversible Hydrazone Bioconjugation in Cells without the Use of Extraneous Catalysts. Org. Biomol. Chem 2018, 16, 4304–4310. [DOI] [PubMed] [Google Scholar]
- (65).Agarwal P; van der Weijden J; Sletten EM; Rabuka D; Bertozzi CR A Pictet-Spengler Ligation for Protein Chemical Modification. Proc. Natl. Acad. Sci 2013, 110, 46 LP–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Drake PM; Albers AE; Baker J; Banas S; Barfield RM; Bhat AS; de Hart GW; Garofalo AW; Holder P; Jones LC; et al. Aldehyde Tag Coupled with HIPS Chemistry Enables the Production of ADCs Conjugated Site-Specifically to Different Antibody Regions with Distinct in Vivo Efficacy and PK Outcomes. Bioconjug. Chem 2014, 25, 1331–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Kudirka R; Barfield RM; McFarland J; Albers AE; de Hart GW; Drake PM; Holder PG; Banas S; Jones LC; Garofalo AW; et al. Generating Site-Specifically Modified Proteins via a Versatile and Stable Nucleophilic Carbon Ligation. Chem. Biol 2015, 22, 293–298. [DOI] [PubMed] [Google Scholar]
- (68).Zeng Y; Ramya TNC; Dirksen A; Dawson PE; Paulson JC High-Efficiency Labeling of Sialylated Glycoproteins on Living Cells. Nat. Methods 2009, 6, 207–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Spears RJ; Fascione MA Site-Selective Incorporation and Ligation of Protein Aldehydes. Org. Biomol. Chem 2016, 14, 7622–7638. [DOI] [PubMed] [Google Scholar]
- (70).Hoyle CE; Bowman CN Thiol–Ene Click Chemistry. Angew. Chemie Int. Ed 2010, 49, 1540–1573. [DOI] [PubMed] [Google Scholar]
- (71).Lowe AB Thiol–Ene “Click” Reactions and Recent Applications in Polymer and Materials Synthesis: A First Update. Polym. Chem 2014, 5, 4820–4870. [Google Scholar]
- (72).Koo SPS; Stamenović MM; Prasath RA; Inglis AJ; Du Prez FE; Barner-Kowollik C; Van Camp W; Junkers T Limitations of Radical Thiol-Ene Reactions for Polymer–Polymer Conjugation. J. Polym. Sci. Part A Polym. Chem 2010, 48, 1699–1713. [Google Scholar]
- (73).Derboven P; D’hooge DR; Stamenovic MM; Espeel P; Marin GB; Du Prez FE; Reyniers M-F Kinetic Modeling of Radical Thiol–Ene Chemistry for Macromolecular Design: Importance of Side Reactions and Diffusional Limitations. Macromolecules 2013, 46, 1732–1742. [Google Scholar]
- (74).Teders M; Henkel C; Anhäuser L; Strieth-Kalthoff F; Gómez-Suárez A; Kleinmans R; Kahnt A; Rentmeister A; Guldi D; Glorius F The Energy-Transfer-Enabled Biocompatible Disulfide–Ene Reaction. Nat. Chem 2018, 10, 981–988. [DOI] [PubMed] [Google Scholar]
- (75).Nolan MD; Scanlan EM Applications of Thiol-Ene Chemistry for Peptide Science. Frontiers in Chemistry. 2020, p 1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Grim JC; Marozas IA; Anseth KS Thiol-Ene and Photo-Cleavage Chemistry for Controlled Presentation of Biomolecules in Hydrogels. J. Control. Release 2015, 219, 95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Spicer CD; Davis BG Selective Chemical Protein Modification. Nat. Commun 2014, 5, 4740. [DOI] [PubMed] [Google Scholar]
- (78).Minguez P; Letunic I; Parca L; Bork P PTMcode: A Database of Known and Predicted Functional Associations between Post-Translational Modifications in Proteins. Nucleic Acids Res 2012, 41, D306–D311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Linder ME; Deschenes RJ Palmitoylation: Policing Protein Stability and Traffic. Nat Rev Mol Cell Biol 2007, 8, 74–84. [DOI] [PubMed] [Google Scholar]
- (80).Schmidt MFG; Schlesinger MJ Fatty Acid Binding to Vesicular Stomatitis Virus Glycoprotein: A New Type of Post-Translational Modification of the Viral Glycoprotein. Cell 1979, 17, 813–819. [DOI] [PubMed] [Google Scholar]
- (81).O’Brien PJ; Zatz M Acylation of Bovine Rhodopsin by [3H]Palmitic Acid. J. Biol. Chem 1984, 259, 5054–5057. [PubMed] [Google Scholar]
- (82).Chen ZQ; Ulsh LS; DuBois G; Shih TY Posttranslational Processing of P21 Ras Proteins Involves Palmitylation of the C-Terminal Tetrapeptide Containing Cysteine-186. J. Virol 1985, 56, 607–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Jung V; Chen L; Hofmann SL; Wigler M; Powers S Mutations in the SHR5 Gene of Saccharomyces Cerevisiae Suppress Ras Function and Block Membrane Attachment and Palmitoylation of Ras Proteins. Mol. Cell. Biol 1995, 15, 1333–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Lobo S; Greentree WK; Linder ME; Deschenes RJ Identification of a Ras Palmitoyltransferase in Saccharomyces Cerevisiae. J. Biol. Chem 2002, 277, 41268–41273. [DOI] [PubMed] [Google Scholar]
- (85).Uemura T; Mori H; Mishina M Isolation and Characterization of Golgi Apparatus-Specific GODZ with the DHHC Zinc Finger Domain. Biochem. Biophys. Res. Commun 2002, 296, 492–496. [DOI] [PubMed] [Google Scholar]
- (86).Ohno Y; Kihara A; Sano T; Igarashi Y Intracellular Localization and Tissue-Specific Distribution of Human and Yeast DHHC Cysteine-Rich Domain-Containing Proteins. Biochim. Biophys. Acta - Mol. Cell Biol. Lipids 2006, 1761, 474–483. [DOI] [PubMed] [Google Scholar]
- (87).Tabaczar S; Czogalla A; Podkalicka J; Biernatowska A; Sikorski AF Protein Palmitoylation: Palmitoyltransferases and Their Specificity. Exp. Biol. Med. (Maywood) 2017, 242, 1150–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Fukata Y; Iwanaga T; Fukata M Systematic Screening for Palmitoyl Transferase Activity of the DHHC Protein Family in Mammalian Cells. Methods 2006, 40, 177–182. [DOI] [PubMed] [Google Scholar]
- (89).Roth AF; Feng Y; Chen L; Davis NG The Yeast DHHC Cysteine-Rich Domain Protein Akr1p Is a Palmitoyl Transferase. J. Cell Biol 2002, 159, 23–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Huang K; Sanders SS; Kang R; Carroll JB; Sutton L; Wan J; Singaraja R; Young FB; Liu L; El-Husseini A; et al. Wild-Type HTT Modulates the Enzymatic Activity of the Neuronal Palmitoyl Transferase HIP14. Hum. Mol. Genet 2011, 20, 3356–3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Mitchell DA; Vasudevan A; Linder ME; Deschenes RJ Thematic Review Series: Lipid Posttranslational Modifications. Protein Palmitoylation by a Family of DHHC Protein S-Acyltransferases. J. Lipid Res 2006, 47, 1118–1127. [DOI] [PubMed] [Google Scholar]
- (92).Gottlieb CD; Zhang S; Linder ME The Cysteine-Rich Domain of the DHHC3 Palmitoyltransferase Is Palmitoylated and Contains Tightly Bound Zinc. J. Biol. Chem 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Rana MS; Kumar P; Lee C-J; Verardi R; Rajashankar KR; Banerjee A Fatty Acyl Recognition and Transfer by an Integral Membrane S-Acyltransferase. Science 2018, 359, eaao6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Rana MS; Lee C-J; Banerjee A The Molecular Mechanism of DHHC Protein Acyltransferases. Biochem. Soc. Trans 2018, 47, 157–167. [DOI] [PubMed] [Google Scholar]
- (95).Dietrich LEP; Ungermann C On the Mechanism of Protein Palmitoylation. EMBO Rep 2004, 5, 1053–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Lemonidis K; Gorleku OA; Sanchez-Perez MC; Grefen C; Chamberlain LH The Golgi S-Acylation Machinery Comprises ZDHHC Enzymes with Major Differences in Substrate Affinity and S-Acylation Activity. Mol. Biol. Cell 2014, 25, 3870–3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).González Montoro A; Chumpen Ramirez S; Valdez Taubas J The Canonical DHHC Motif Is Not Absolutely Required for the Activity of the Yeast S-Acyltransferases Swf1 and Pfa4. J. Biol. Chem 2015, 290, 22448–22459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Thomas GM; Hayashi T; Chiu S-L; Chen C-M; Huganir RL Palmitoylation by DHHC5/8 Targets GRIP1 to Dendritic Endosomes to Regulate AMPA-R Trafficking. Neuron 2012, 73, 482–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Howie J; Reilly L; Fraser NJ; Vlachaki Walker JM; Wypijewski KJ; Ashford MLJ; Calaghan SC; McClafferty H; Tian L; Shipston MJ; et al. Substrate Recognition by the Cell Surface Palmitoyl Transferase DHHC5. Proc. Natl. Acad. Sci 2014, 111, 17534 LP–17539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Zhou F; Xue Y; Yao X; Xu Y CSS-Palm: Palmitoylation Site Prediction with a Clustering and Scoring Strategy (CSS). Bioinformatics 2006, 22, 894–896. [DOI] [PubMed] [Google Scholar]
- (101).Won SJ; Cheung See Kit M; Martin BR Protein Depalmitoylases. Crit. Rev. Biochem. Mol. Biol 2018, 53, 83–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Lin DTS; Conibear E ABHD17 Proteins Are Novel Protein Depalmitoylases That Regulate N-Ras Palmitate Turnover and Subcellular Localization. Elife 2015, 4, e11306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Rocks O; Peyker A; Kahms M; Verveer PJ; Koerner C; Lumbierres M; Kuhlmann J; Waldmann H; Wittinghofer A; Bastiaens PIH An Acylation Cycle Regulates Localization and Activity of Palmitoylated Ras Isoforms. Science 2005, 307, 1746 LP–1752. [DOI] [PubMed] [Google Scholar]
- (104).Won SJ; Martin BR Temporal Profiling Establishes a Dynamic S-Palmitoylation Cycle. ACS Chem. Biol 2018, 13, 1560–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Smotrys JE; Linder ME Palmitoylation of Intracellular Signaling Proteins: Regulation and Function. Annu. Rev. Biochem 2004, 73, 559–587. [DOI] [PubMed] [Google Scholar]
- (106).Matt L; Kim K; Chowdhury D; Hell JW Role of Palmitoylation of Postsynaptic Proteins in Promoting Synaptic Plasticity. Front. Mol. Neurosci 2019, 12, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Sobocińska J; Roszczenko-Jasińska P; Ciesielska A; Kwiatkowska K Protein Palmitoylation and Its Role in Bacterial and Viral Infections. Front. Immunol 2018, 8, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Cho E; Park M Palmitoylation in Alzheimer’s Disease and Other Neurodegenerative Diseases. Pharmacol. Res 2016, 111, 133–151. [DOI] [PubMed] [Google Scholar]
- (109).Ko P-J; Dixon SJ Protein Palmitoylation and Cancer. EMBO Rep 2018, 19, e46666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Farazi TA; Waksman G; Gordon JI The Biology and Enzymology of ProteinN-Myristoylation. J. Biol. Chem 2001, 276, 39501–39504. [DOI] [PubMed] [Google Scholar]
- (111).Wright MH; Heal WP; Mann DJ; Tate EW Protein Myristoylation in Health and Disease. J. Chem. Biol 2010, 3, 19–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Martin DDO; Beauchamp E; Berthiaume LG Post-Translational Myristoylation: Fat Matters in Cellular Life and Death. Biochimie 2011, 93, 18–31. [DOI] [PubMed] [Google Scholar]
- (113).Castrec B; Dian C; Ciccone S; Ebert CL; Bienvenut WV; Le Caer J-P; Steyaert J-M; Giglione C; Meinnel T Structural and Genomic Decoding of Human and Plant Myristoylomes Reveals a Definitive Recognition Pattern. Nat. Chem. Biol 2018, 14, 671–679. [DOI] [PubMed] [Google Scholar]
- (114).Perinpanayagam MA; Beauchamp E; Martin DDO; Sim JYW; Yap MC; Berthiaume LG Regulation of Co- and Post-Translational Myristoylation of Proteins during Apoptosis: Interplay of N-Myristoyltransferases and Caspases. FASEB J 2012, 27, 811–821. [DOI] [PubMed] [Google Scholar]
- (115).Rudnick DA; McWherter CA; Rocque WJ; Lennon PJ; Getman DP; Gordon JI Kinetic and Structural Evidence for a Sequential Ordered Bi Bi Mechanism of Catalysis by Saccharomyces Cerevisiae Myristoyl-CoA:Protein N-Myristoyltransferase. J. Biol. Chem 1991, 266, 9732–9739. [PubMed] [Google Scholar]
- (116).Dian C; Pérez-Dorado I; Rivière F; Asensio T; Legrand P; Ritzefeld M; Shen M; Cota E; Meinnel T; Tate EW; et al. High-Resolution Snapshots of Human N-Myristoyltransferase in Action Illuminate a Mechanism Promoting N-Terminal Lys and Gly Myristoylation. Nat. Commun 2020, 11, 1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Towler D; Glaser L Protein Fatty Acid Acylation: Enzymatic Synthesis of an N-Myristoylglycyl Peptide. Proc. Natl. Acad. Sci 1986, 83, 2812 LP–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Farazi TA; Waksman G; Gordon JI Structures of Saccharomyces Cerevisiae N-Myristoyltransferase with Bound MyristoylCoA and Peptide Provide Insights about Substrate Recognition and Catalysis. Biochemistry 2001, 40, 6335–6343. [DOI] [PubMed] [Google Scholar]
- (119).Maurer-Stroh S; Eisenhaber F Myristoylation of Viral and Bacterial Proteins. Trends Microbiol 2004, 12, 178–185. [DOI] [PubMed] [Google Scholar]
- (120).Giang DK; Cravatt BF A Second Mammalian N-Myristoyltransferase. J. Biol. Chem 1998, 273, 6595–6598. [DOI] [PubMed] [Google Scholar]
- (121).Yang SH; Shrivastav A; Kosinski C; Sharma RK; Chen M-H; Berthiaume LG; Peters LL; Chuang P-T; Young SG; Bergo MO N-Myristoyltransferase 1 Is Essential in Early Mouse Development. J. Biol. Chem 2005, 280, 18990–18995. [DOI] [PubMed] [Google Scholar]
- (122).Stevenson FT; Bursten SL; Locksley RM; Lovett DH Myristyl Acylation of the Tumor Necrosis Factor Alpha Precursor on Specific Lysine Residues. J. Exp. Med 1992, 176, 1053 LP–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Jing H; Zhang X; Wisner SA; Chen X; Spiegelman NA; Linder ME; Lin H SIRT2 and Lysine Fatty Acylation Regulate the Transforming Activity of K-Ras4a. Elife 2017, 6, e32436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Zhang X; Spiegelman NA; Nelson OD; Jing H; Lin H SIRT6 Regulates Ras-Related Protein R-Ras2 by Lysine Defatty-Acylation. Elife 2017, 6, e25158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Spiegelman NA; Zhang X; Jing H; Cao J; Kotliar IB; Aramsangtienchai P; Wang M; Tong Z; Rosch KM; Lin H SIRT2 and Lysine Fatty Acylation Regulate the Activity of RalB and Cell Migration. ACS Chem. Biol 2019, 14, 2014–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Kosciuk T; Price IR; Zhang X; Zhu C; Johnson KN; Zhang S; Halaby SL; Komaniecki GP; Yang M; DeHart CJ; et al. NMT1 and NMT2 Are Lysine Myristoyltransferases Regulating the ARF6 GTPase Cycle. Nat. Commun 2020, 11, 1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Feldman JL; Baeza J; Denu JM Activation of the Protein Deacetylase SIRT6 by Long-Chain Fatty Acids and Widespread Deacylation by Mammalian Sirtuins. J. Biol. Chem 2013, 288, 31350–31356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Nakagawa T; Guarente L Sirtuins at a Glance. J. Cell Sci 2011, 124, 833 LP–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Park S-Y; Kim J-S A Short Guide to Histone Deacetylases Including Recent Progress on Class II Enzymes. Exp. Mol. Med 2020, 52, 204–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Cao J; Sun L; Aramsangtienchai P; Spiegelman NA; Zhang X; Huang W; Seto E; Lin H HDAC11 Regulates Type I Interferon Signaling through Defatty-Acylation of SHMT2. Proc. Natl. Acad. Sci 2019, 116, 5487 LP–5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Udenwobele DI; Su R-C; Good SV; Ball TB; Varma Shrivastav S; Shrivastav A Myristoylation: An Important Protein Modification in the Immune Response. Front. Immunol 2017, 8, 751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Martin DDO; Hayden MR Post-Translational Myristoylation at the Cross Roads of Cell Death, Autophagy and Neurodegeneration. Biochem. Soc. Trans 2015, 43, 229–234. [DOI] [PubMed] [Google Scholar]
- (133).Yuan M; Song Z; Ying M; Zhu H; He Q; Yang B; Cao J N-Myristoylation: From Cell Biology to Translational Medicine. Acta Pharmacol. Sin 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Kamiya Y; Sakurai A; Tamura S; Takahashi N; Abe K; Tsuchiya E; Fukui S; Kitada C; Fujino M Structure of Rhodotorucine A, a Novel Lipopeptide, Inducing Mating Tube Formation in Rhodosporidiumtoruloides. Biochem. Biophys. Res. Commun 1978, 83, 1077–1083. [DOI] [PubMed] [Google Scholar]
- (135).Wolda SL; Glomset JA Evidence for Modification of Lamin B by a Product of Mevalonic Acid. J. Biol. Chem 1988, 263, 5997–6000. [PubMed] [Google Scholar]
- (136).Zhang FL; Casey PJ Protein Prenylation: Molecular Mechanisms and Functional Consequences. Annu. Rev. Biochem 1996, 65, 241–269. [DOI] [PubMed] [Google Scholar]
- (137).Liu A; Du W; Liu J-P; Jessell TM; Prendergast GC RhoB Alteration Is Necessary for Apoptotic and Antineoplastic Responses to Farnesyltransferase Inhibitors. Mol. Cell. Biol 2000, 20, 6105 LP–6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Hougland JL; Lamphear CL; Scott SA; Gibbs RA; Fierke CA Context-Dependent Substrate Recognition by Protein Farnesyltransferase. Biochemistry 2009, 48, 1691–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Hougland JL; Hicks KA; Hartman HL; Kelly RA; Watt TJ; Fierke CA Identification of Novel Peptide Substrates for Protein Farnesyltransferase Reveals Two Substrate Classes with Distinct Sequence Selectivities. J. Mol. Biol 2010, 395, 176–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Maurer-Stroh S; Eisenhaber F Refinement and Prediction of Protein Prenylation Motifs. Genome Biol 2005, 6, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (141).Hicks KA; Hartman HL; Fierke CA Upstream Polybasic Region in Peptides Enhances Dual Specificity for Prenylation by Both Farnesyltransferase and Geranylgeranyltransferase Type I. Biochemistry 2005, 44, 15325–15333. [DOI] [PubMed] [Google Scholar]
- (142).Blanden MJ; Suazo KF; Hildebrandt ER; Hardgrove DS; Patel M; Saunders WP; Distefano MD; Schmidt WK; Hougland JL Efficient Farnesylation of an Extended C-Terminal C(x)3X Sequence Motif Expands the Scope of the Prenylated Proteome. J. Biol. Chem 2017, 293, 2770–2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (143).Ashok S; Hildebrandt ER; Ruiz CS; Hardgrove DS; Coreno DW; Schmidt WK; Hougland JL Protein Farnesyltransferase Catalyzes Unanticipated Farnesylation and Geranylgeranylation of Shortened Target Sequences. Biochemistry 2020, 59, 1149–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Boyartchuk VL; Ashby MN; Rine J Modulation of Ras and A-Factor Function by Carboxyl-Terminal Proteolysis. Science 1997, 275, 1796–1800. [DOI] [PubMed] [Google Scholar]
- (145).Dai Q; Choy E; Chiu V; Romano J; Slivka SR; Steitz SA; Michaelis S; Philips MR Mammalian Prenylcysteine Carboxyl Methyltransferase Is in the Endoplasmic Reticulum. J. Biol. Chem 1998, 273, 15030–15034. [DOI] [PubMed] [Google Scholar]
- (146).Hancock JF; Paterson H; Marshall CJ A Polybasic Domain or Palmitoylation Is Required in Addition to the CAAX Motif to Localize P21ras to the Plasma Membrane. Cell 1990, 63, 133–139. [DOI] [PubMed] [Google Scholar]
- (147).Hildebrandt ER; Cheng M; Zhao P; Kim JH; Wells L; Schmidt WK A Shunt Pathway Limits the CaaX Processing of Hsp40 Ydj1p and Regulates Ydj1p-Dependent Phenotypes. Elife 2016, 5, e15899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Rak A; Pylypenko O; Niculae A; Pyatkov K; Goody RS; Alexandrov K Structure of the Rab7:REP-1 Complex: Insights into the Mechanism of Rab Prenylation and Choroideremia Disease. Cell 2004, 117, 749–760. [DOI] [PubMed] [Google Scholar]
- (149).Leung KF; Baron R; Ali BR; Magee AI; Seabra MC Rab GTPases Containing a CAAX Motif Are Processed Post-Geranylgeranylation by Proteolysis and Methylation. J. Biol. Chem 2007, 282, 1487–1497. [DOI] [PubMed] [Google Scholar]
- (150).Calero M; Chen CZ; Zhu W; Winand N; Havas KA; Gilbert PM; Burd CG; Collins RN Dual Prenylation Is Required for Rab Protein Localization and Function. Mol. Biol. Cell 2003, 14, 1852–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Long SB; Casey PJ; Beese LS Reaction Path of Protein Farnesyltransferase at Atomic Resolution. Nature 2002, 419, 645–650. [DOI] [PubMed] [Google Scholar]
- (152).Taylor JS; Reid TS; Terry KL; Casey PJ; Beese LS Structure of Mammalian Protein Geranylgeranyltransferase Type-I. EMBO J 2003, 22, 5963–5974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (153).Pompliano DL; Schaber MD; Mosser SD; Omer CA; Shafer JA; Gibbs JB Isoprenoid Diphosphate Utilization by Recombinant Human Farnesyl:Protein Transferase: Interactive Binding between Substrates and a Preferred Kinetic Pathway. Biochemistry 1993, 32, 8341–8347. [DOI] [PubMed] [Google Scholar]
- (154).Dolence JM; Cassidy PB; Mathis JR; Poulter CD Yeast Protein Farnesyltransferase: Steady-State Kinetic Studies of Substrate Binding. Biochemistry 1995, 34, 16687–16694. [DOI] [PubMed] [Google Scholar]
- (155).Stirtan WG; Poulter CD Yeast Protein Geranylgeranyltransferase Type-I: Steady-State Kinetics and Substrate Binding. Biochemistry 1997, 36, 4552–4557. [DOI] [PubMed] [Google Scholar]
- (156).Terry KL; Casey PJ; Beese LS Conversion of Protein Farnesyltransferase to a Geranylgeranyltransferase. Biochemistry 2006, 45, 9746–9755. [DOI] [PubMed] [Google Scholar]
- (157).Thomä NH; Niculae A; Goody RS; Alexandrov K Double Prenylation by RabGGTase Can Proceed without Dissociation of the Mono-Prenylated Intermediate. J. Biol. Chem 2001, 276, 48631–48636. [DOI] [PubMed] [Google Scholar]
- (158).Köhnke M; Delon C; Hastie ML; Nguyen UTT; Wu Y-W; Waldmann H; Goody RS; Gorman JJ; Alexandrov K Rab GTPase Prenylation Hierarchy and Its Potential Role in Choroideremia Disease. PLoS One 2013, 8, e81758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Ochocki JD; Distefano MD Prenyltransferase Inhibitors: Treating Human Ailments from Cancer to Parasitic Infections. Medchemcomm 2013, 4, 476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (160).Berndt N; Hamilton AD; Sebti SM Targeting Protein Prenylation for Cancer Therapy. Nat Rev Cancer 2011, 11, 775–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Young SG; Yang SH; Davies BSJ; Jung H-J; Fong LG Targeting Protein Prenylation in Progeria. Sci. Transl. Med 2013, 5, 171ps3–171ps3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (162).Marakasova ES; Eisenhaber B; Maurer-Stroh S; Eisenhaber F; Baranova A Prenylation of Viral Proteins by Enzymes of the Host: Virus-Driven Rationale for Therapy with Statins and FT/GGT1 Inhibitors. BioEssays 2017, 39, 1700014. [DOI] [PubMed] [Google Scholar]
- (163).Jeong A; Suazo KF; Wood WG; Distefano MD; Li L Isoprenoids and Protein Prenylation: Implications in the Pathogenesis and Therapeutic Intervention of Alzheimer’s Disease. Crit. Rev. Biochem. Mol. Biol 2018, 53, 279–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).FDA Approves First Treatment for Hutchinson-Gilford Progeria Syndrome and Some Progeroid Laminopathies. https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-hutchinson-gilford-progeria-syndrome-and-some-progeroid-laminopathies.
- (165).Gordon LB; Kleinman ME; Miller DT; Neuberg DS; Giobbie-Hurder A; Gerhard-Herman M; Smoot LB; Gordon CM; Cleveland R; Snyder BD; et al. Clinical Trial of a Farnesyltransferase Inhibitor in Children with Hutchinson–Gilford Progeria Syndrome. Proc. Natl. Acad. Sci 2012, 109, 16666–16671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Burai L; Fabian I; Kiraly R; Szilagyi E; Brucher E Equilibrium and Kinetic Studies on the Formation of the Lanthanide(III) Complexes, [Ce(Dota)]– and [Yb(Dota)]– (H4dota = 1,4,7,10-Tetraazacyclododecane-1,4,7,10-Tetraacetic Acid). J. Chem. Soc 1998, No. 2, 243–248. [Google Scholar]
- (167).Kuchay S; Wang H; Marzio A; Jain K; Homer H; Fehrenbacher N; Philips MR; Zheng N; Pagano M GGTase3 Is a Newly Identified Geranylgeranyltransferase Targeting a Ubiquitin Ligase. Nat. Struct. Mol. Biol 2019, 26, 628–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (168).Veit M; Söllner TH; Rothman JE Multiple Palmitoylation of Synaptotagmin and the T-SNARE SNAP-25. FEBS Lett 1996, 385, 119–123. [DOI] [PubMed] [Google Scholar]
- (169).Alland L; Peseckis SM; Atherton RE; Berthiaume L; Resh MD Dual Myristylation and Palmitylation of Src Family Member P59fyn Affects Subcellular Localization. J. Biol. Chem 1994, 269, 16701–16705. [PubMed] [Google Scholar]
- (170).Maurer-Stroh S; Gouda M; Novatchkova M; Schleiffer A; Schneider G; Sirota FL; Wildpaner M; Hayashi N; Eisenhaber F MYRbase: Analysis of Genome-Wide Glycine Myristoylation Enlarges the Functional Spectrum of Eukaryotic Myristoylated Proteins. Genome Biol 2004, 5, R21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (171).Tate EW Recent Advances in Chemical Proteomics: Exploring the Post-Translational Proteome. J. Chem. Biol 2008, 1, 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (172).Hannoush RN; Sun J The Chemical Toolbox for Monitoring Protein Fatty Acylation and Prenylation. Nat. Chem. Biol 2010, 6, 498–506. [DOI] [PubMed] [Google Scholar]
- (173).Thinon E; Hang HC Chemical Reporters for Exploring Protein Acylation. Biochem. Soc. Trans 2015, 43, 253 LP–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (174).Ong S-E; Blagoev B; Kratchmarova I; Kristensen DB; Steen H; Pandey A; Mann M Stable Isotope Labeling by Amino Acids in Cell Culture, SILAC, as a Simple and Accurate Approach to Expression Proteomics. Mol. & Cell. Proteomics 2002, 1, 376 LP–386. [DOI] [PubMed] [Google Scholar]
- (175).Thompson A; Schäfer J; Kuhn K; Kienle S; Schwarz J; Schmidt G; Neumann T; Hamon C Tandem Mass Tags: A Novel Quantification Strategy for Comparative Analysis of Complex Protein Mixtures by MS/MS. Anal. Chem 2003, 75, 1895–1904. [DOI] [PubMed] [Google Scholar]
- (176).Broncel M; Serwa RA; Ciepla P; Krause E; Dallman MJ; Magee AI; Tate EW Multifunctional Reagents for Quantitative Proteome-Wide Analysis of Protein Modification in Human Cells and Dynamic Profiling of Protein Lipidation During Vertebrate Development. Angew. Chemie Int. Ed 2015, 54, 5948–5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (177).Wright MH; Clough B; Rackham MD; Rangachari K; Brannigan JA; Grainger M; Moss DK; Bottrill AR; Heal WP; Broncel M; et al. Validation of N-Myristoyltransferase as an Antimalarial Drug Target Using an Integrated Chemical Biology Approach. Nat Chem 2014, 6, 112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (178).Hang HC; Geutjes E-J; Grotenbreg G; Pollington AM; Bijlmakers MJ; Ploegh HL Chemical Probes for the Rapid Detection of Fatty-Acylated Proteins in Mammalian Cells. J. Am. Chem. Soc 2007, 129, 2744–2745. [DOI] [PubMed] [Google Scholar]
- (179).Charron G; Zhang MM; Yount JS; Wilson J; Raghavan AS; Shamir E; Hang HC Robust Fluorescent Detection of Protein Fatty-Acylation with Chemical Reporters. J. Am. Chem. Soc 2009, 131, 4967–4975. [DOI] [PubMed] [Google Scholar]
- (180).Hannoush RN; Arenas-Ramirez N Imaging the Lipidome: ω-Alkynyl Fatty Acids for Detection and Cellular Visualization of Lipid-Modified Proteins. ACS Chem. Biol 2009, 4, 581–587. [DOI] [PubMed] [Google Scholar]
- (181).Ourailidou ME; Zwinderman MRH; Dekker FJ Bioorthogonal Metabolic Labelling with Acyl-CoA Reporters: Targeting Protein Acylation. Medchemcomm 2016, 7, 399–408. [Google Scholar]
- (182).Yount JS; Moltedo B; Yang Y-Y; Charron G; Moran TM; López CB; Hang HC Palmitoylome Profiling Reveals S-Palmitoylation–Dependent Antiviral Activity of IFITM3. Nat Chem Biol 2010, 6, 610–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (183).Yount JS; Charron G; Hang HC Bioorthogonal Proteomics of 15-Hexadecynyloxyacetic Acid Chemical Reporter Reveals Preferential Targeting of Fatty Acid Modified Proteins and Biosynthetic Enzymes. Bioorg. Med. Chem 2012, 20, 650–654. [DOI] [PubMed] [Google Scholar]
- (184).Gao X; Hannoush RN Single-Cell Imaging of Wnt Palmitoylation by the Acyltransferase Porcupine. Nat. Chem. Biol 2014, 10, 61–68. [DOI] [PubMed] [Google Scholar]
- (185).Greaves J; Munro KR; Davidson SC; Riviere M; Wojno J; Smith TK; Tomkinson NCO; Chamberlain LH Molecular Basis of Fatty Acid Selectivity in the ZDHHC Family of S-Acyltransferases Revealed by Click Chemistry. Proc. Natl. Acad. Sci 2017, 114, E1365 LP–E1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (186).Thiele C; Papan C; Hoelper D; Kusserow K; Gaebler A; Schoene M; Piotrowitz K; Lohmann D; Spandl J; Stevanovic A; et al. Tracing Fatty Acid Metabolism by Click Chemistry. ACS Chem. Biol 2012, 7, 2004–2011. [DOI] [PubMed] [Google Scholar]
- (187).Thiele C; Wunderling K; Leyendecker P Multiplexed and Single Cell Tracing of Lipid Metabolism. Nat. Methods 2019, 16, 1123–1130. [DOI] [PubMed] [Google Scholar]
- (188).Lanyon-Hogg T; Faronato M; Serwa RA; Tate EW Dynamic Protein Acylation: New Substrates, Mechanisms, and Drug Targets. Trends Biochem. Sci 2017, 42, 566–581. [DOI] [PubMed] [Google Scholar]
- (189).Haberkant P; Raijmakers R; Wildwater M; Sachsenheimer T; Brügger B; Maeda K; Houweling M; Gavin A-C; Schultz C; van Meer G; et al. In Vivo Profiling and Visualization of Cellular Protein–Lipid Interactions Using Bifunctional Fatty Acids. Angew. Chemie Int. Ed 2013, 52, 4033–4038. [DOI] [PubMed] [Google Scholar]
- (190).Peng T; Hang HC Bifunctional Fatty Acid Chemical Reporter for Analyzing S-Palmitoylated Membrane Protein–Protein Interactions in Mammalian Cells. J. Am. Chem. Soc 2015, 137, 556–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (191).Zhu XG; Nicholson Puthenveedu S; Shen Y; La K; Ozlu C; Wang T; Klompstra D; Gultekin Y; Chi J; Fidelin J; et al. CHP1 Regulates Compartmentalized Glycerolipid Synthesis by Activating GPAT4. Mol. Cell 2019, 74, 45–58.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (192).Drisdel RC; Green WN Labeling and Quantifying Sites of Protein Palmitoylation. Biotechniques 2004, 36, 276–285. [DOI] [PubMed] [Google Scholar]
- (193).Forrester MT; Thompson JW; Foster MW; Nogueira L; Moseley MA; Stamler JS Proteomic Analysis of S-Nitrosylation and Denitrosylation by Resin-Assisted Capture. Nat. Biotechnol 2009, 27, 557–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (194).Blanc M; David F; Abrami L; Migliozzi D; Armand F; Bürgi J; van der Goot FG SwissPalm: Protein Palmitoylation Database. F1000Research 2015, 4, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (195).Kostiuk MA; Corvi MM; Keller BO; Plummer G; Prescher JA; Hangauer MJ; Bertozzi CR; Rajaiah G; Falck JR; Berthiaume LG Identification of Palmitoylated Mitochondrial Proteins Using a Bio-Orthogonal Azido-Palmitate Analogue. FASEB J 2008, 22, 721–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (196).Martin BR; Cravatt BF Large-Scale Profiling of Protein Palmitoylation in Mammalian Cells. Nat Meth 2009, 6, 135–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (197).Wilson JP; Raghavan AS; Yang Y-Y; Charron G; Hang HC Proteomic Analysis of Fatty-Acylated Proteins in Mammalian Cells with Chemical Reporters Reveals S-Acylation of Histone H3 Variants. Mol. Cell. Proteomics 2011, 10, M110.001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (198).Chesarino NM; Hach JC; Chen JL; Zaro BW; Rajaram MVS; Turner J; Schlesinger LS; Pratt MR; Hang HC; Yount JS Chemoproteomics Reveals Toll-like Receptor Fatty Acylation. BMC Biol 2014, 12, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (199).Sobocińska J; Roszczenko-Jasińska P; Zaręba-Kozioł M; Hromada-Judycka A; Matveichuk OV; Traczyk G; Łukasiuk K; Kwiatkowska K Lipopolysaccharide Upregulates Palmitoylated Enzymes of the Phosphatidylinositol Cycle: An Insight from Proteomic Studies. Mol. & Cell. Proteomics 2018, 17, 233 LP–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (200).Cui L; Liu M; Lai S; Hou H; Diao T; Zhang D; Wang M; Zhang Y; Wang J Androgen Upregulates the Palmitoylation of EIF3L in Human Prostate LNCaP Cells. Onco. Targets. Ther 2019, 12, 4451–4459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (201).Buddelmeijer N The Molecular Mechanism of Bacterial Lipoprotein Modification—How, When and Why? FEMS Microbiol. Rev 2015, 39, 246–261. [DOI] [PubMed] [Google Scholar]
- (202).Rangan KJ; Yang Y-Y; Charron G; Hang HC Rapid Visualization and Large-Scale Profiling of Bacterial Lipoproteins with Chemical Reporters. J. Am. Chem. Soc 2010, 132, 10628–10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (203).Jones ML; Collins MO; Goulding D; Choudhary JS; Rayner JC Analysis of Protein Palmitoylation Reveals a Pervasive Role in Plasmodium Development and Pathogenesis. Cell Host Microbe 2012, 12, 246–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (204).Foe IT; Child MA; Majmudar JD; Krishnamurthy S; van der Linden WA; Ward GE; Martin BR; Bogyo M Global Analysis of Palmitoylated Proteins in Toxoplasma Gondii. Cell Host Microbe 2015, 18, 501–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (205).Santiago-Tirado FH; Peng T; Yang M; Hang HC; Doering TL A Single Protein S-Acyl Transferase Acts through Diverse Substrates to Determine Cryptococcal Morphology, Stress Tolerance, and Pathogenic Outcome. PLoS Pathog 2015, 11, e1004908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (206).Serwa RA; Abaitua F; Krause E; Tate EW; O’Hare P Systems Analysis of Protein Fatty Acylation in Herpes Simplex Virus-Infected Cells Using Chemical Proteomics. Chem. Biol 2015, 22, 1008–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (207).Colquhoun DR; Lyashkov AE; Mohien CU; Aquino VN; Bullock BT; Dinglasan RR; Agnew BJ; Graham DRM Bioorthogonal Mimetics of Palmitoyl-CoA and Myristoyl-CoA and Their Subsequent Isolation by Click Chemistry and Characterization by Mass Spectrometry Reveal Novel Acylated Host-Proteins Modified by HIV-1 Infection. Proteomics 2015, 15, 2066–2077. [DOI] [PubMed] [Google Scholar]
- (208).Martin BR; Wang C; Adibekian A; Tully SE; Cravatt BF Global Profiling of Dynamic Protein Palmitoylation. Nat Meth 2012, 9, 84–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (209).Liu Y; Patricelli MP; Cravatt BF Activity-Based Protein Profiling: The Serine Hydrolases. Proc. Natl. Acad. Sci. U. S. A 1999, 96, 14694–14699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (210).Medina-Cleghorn D; Nomura DK Exploring Metabolic Pathways and Regulation through Functional Chemoproteomic and Metabolomic Platforms. Chem. Biol 2014, 21, 1171–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (211).Wei X; Adak S; Zayed M; Yin L; Feng C; Speck SL; Kathayat RS; Zhang Q; Dickinson BC; Semenkovich CF Endothelial Palmitoylation Cycling Coordinates Vessel Remodeling in Peripheral Artery Disease. Circ. Res 2020, 0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (212).Wright MH; Paape D; Price HP; Smith DF; Tate EW Global Profiling and Inhibition of Protein Lipidation in Vector and Host Stages of the Sleeping Sickness Parasite Trypanosoma Brucei. ACS Infect. Dis 2016, 2, 427–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (213).Emmer BT; Nakayasu ES; Souther C; Choi H; Sobreira TJP; Epting CL; Nesvizhskii AI; Almeida IC; Engman DM Global Analysis of Protein Palmitoylation in African Trypanosomes. Eukaryot. Cell 2011, 10, 455 LP–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (214).Hernandez JL; Davda D; Majmudar JD; Won SJ; Prakash A; Choi AI; Martin BR Correlated S-Palmitoylation Profiling of Snail-Induced Epithelial to Mesenchymal Transition. Mol. Biosyst 2016, 12, 1799–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (215).Miura GI; Buglino J; Alvarado D; Lemmon MA; Resh MD; Treisman JE Palmitoylation of the EGFR Ligand Spitz by Rasp Increases Spitz Activity by Restricting Its Diffusion. Dev. Cell 2006, 10, 167–176. [DOI] [PubMed] [Google Scholar]
- (216).Thinon E; Fernandez JP; Molina H; Hang HC Selective Enrichment and Direct Analysis of Protein S-Palmitoylation Sites. J. Proteome Res 2018, 17, 1907–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (217).Yount JS; Zhang MM; Hang HC Visualization and Identification of Fatty Acylated Proteins Using Chemical Reporters. Curr. Protoc. Chem. Biol 2011, 3, 65–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (218).Hannoush RN Profiling Cellular Myristoylation and Palmitoylation Using ω-Alkynyl Fatty Acids BT - Chemical Genomics and Proteomics: Reviews and Protocols; Zanders ED, Ed.; Humana Press: Totowa, NJ, 2012; pp 85–94. [DOI] [PubMed] [Google Scholar]
- (219).Martin BR Nonradioactive Analysis of Dynamic Protein Palmitoylation. Curr. Protoc. Protein Sci 2013, 73, 14.15.1–14.15.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (220).Ji Y; Bachschmid MM; Costello CE; Lin C S- to N-Palmitoyl Transfer During Proteomic Sample Preparation. J. Am. Soc. Mass Spectrom 2016, 27, 677–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (221).Percher A; Ramakrishnan S; Thinon E; Yuan X; Yount JS; Hang HC Mass-Tag Labeling Reveals Site-Specific and Endogenous Levels of Protein S-Fatty Acylation. Proc. Natl. Acad. Sci 2016, 113, 4302 LP–4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (222).Yount JS; Karssemeijer RA; Hang HC S-Palmitoylation and Ubiquitination Differentially Regulate Interferon-Induced Transmembrane Protein 3 (IFITM3)-Mediated Resistance to Influenza Virus. J. Biol. Chem 2012, 287, 19631–19641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (223).McMichael TM; Zhang L; Chemudupati M; Hach JC; Kenney AD; Hang HC; Yount JS The Palmitoyltransferase ZDHHC20 Enhances Interferon-Induced Transmembrane Protein 3 (IFITM3) Palmitoylation and Antiviral Activity. J. Biol. Chem 2017, 292, 21517–21526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (224).Spence JS; He R; Hoffmann H-H; Das T; Thinon E; Rice CM; Peng T; Chandran K; Hang HC IFITM3 Directly Engages and Shuttles Incoming Virus Particles to Lysosomes. Nat. Chem. Biol 2019, 15, 259–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (225).Benfield CTO; MacKenzie F; Ritzefeld M; Mazzon M; Weston S; Tate EW; Teo BH; Smith SE; Kellam P; Holmes EC; et al. Bat IFITM3 Restriction Depends on S-Palmitoylation and a Polymorphic Site within the CD225 Domain. Life Sci. Alliance 2020, 3, e201900542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (226).Chesarino NM; Compton AA; McMichael TM; Kenney AD; Zhang L; Soewarna V; Davis M; Schwartz O; Yount JS IFITM3 Requires an Amphipathic Helix for Antiviral Activity. EMBO Rep 2017, 18, 1740–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (227).Tsukamoto T; Li X; Morita H; Minowa T; Aizawa T; Hanagata N; Demura M Role of S-Palmitoylation on IFITM5 for the Interaction with FKBP11 in Osteoblast Cells. PLoS One 2013, 8, e75831–e75831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (228).Hach JC; McMichael T; Chesarino NM; Yount JS Palmitoylation on Conserved and Nonconserved Cysteines of Murine IFITM1 Regulates Its Stability and Anti-Influenza A Virus Activity. J. Virol 2013, 87, 9923–9927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (229).Hicks SW; Charron G; Hang HC; Galán JE Subcellular Targeting of Salmonella Virulence Proteins by Host-Mediated S-Palmitoylation. Cell Host Microbe 2011, 10, 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (230).Melvin WJ; McMichael TM; Chesarino NM; Hach JC; Yount JS IFITMs from Mycobacteria Confer Resistance to Influenza Virus When Expressed in Human Cells. Viruses 2015, 7, 3035–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (231).Zhang MM; Wu P-YJ; Kelly FD; Nurse P; Hang HC Quantitative Control of Protein S-Palmitoylation Regulates Meiotic Entry in Fission Yeast. PLOS Biol 2013, 11, e1001597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (232).Roth AF; Papanayotou I; Davis NG The Yeast Kinase Yck2 Has a Tripartite Palmitoylation Signal. Mol. Biol. Cell 2011, 22, 2702–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (233).Bhattacharyya R; Barren C; Kovacs DM Palmitoylation of Amyloid Precursor Protein Regulates Amyloidogenic Processing in Lipid Rafts. J. Neurosci 2013, 33, 11169–11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (234).Aramsangtienchai P; Spiegelman NA; Cao J; Lin H S-Palmitoylation of Junctional Adhesion Molecule C Regulates Its Tight Junction Localization and Cell Migration. J. Biol. Chem 2017, 292, 5325–5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (235).Konitsiotis AD; Chang S-C; Jovanović B; Ciepla P; Masumoto N; Palmer CP; Tate EW; Couchman JR; Magee AI Attenuation of Hedgehog Acyltransferase-Catalyzed Sonic Hedgehog Palmitoylation Causes Reduced Signaling, Proliferation and Invasiveness of Human Carcinoma Cells. PLoS One 2014, 9, e89899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (236).Ampah KK; Greaves J; Shun-Shion AS; Asnawi AW; Lidster JA; Chamberlain LH; Collins MO; Peden AA S-Acylation Regulates the Trafficking and Stability of the Unconventional Q-SNARE STX19. J. Cell Sci 2018, 131, jcs212498. [DOI] [PubMed] [Google Scholar]
- (237).Zhang MM; Tsou LK; Charron G; Raghavan AS; Hang HC Tandem Fluorescence Imaging of Dynamic S-Acylation and Protein Turnover. Proc. Natl. Acad. Sci 2010, 107, 8627 LP–8632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (238).Liu K; Yang P-Y; Na Z; Yao SQ Dynamic Monitoring of Newly Synthesized Proteomes: Up-Regulation of Myristoylated Protein Kinase A During Butyric Acid Induced Apoptosis. Angew. Chemie Int. Ed 2011, 50, 6776–6781. [DOI] [PubMed] [Google Scholar]
- (239).Akimzhanov AM; Boehning D Rapid and Transient Palmitoylation of the Tyrosine Kinase Lck Mediates Fas Signaling. Proc. Natl. Acad. Sci 2015, 112, 11876 LP–11880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (240).Holland SM; Collura KM; Ketschek A; Noma K; Ferguson TA; Jin Y; Gallo G; Thomas GM Palmitoylation Controls DLK Localization, Interactions and Activity to Ensure Effective Axonal Injury Signaling. Proc. Natl. Acad. Sci 2016, 113, 763 LP–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (241).Kim Y-C; Lee SE; Kim SK; Jang H-D; Hwang I; Jin S; Hong E-B; Jang K-S; Kim H-S Toll-like Receptor Mediated Inflammation Requires FASN-Dependent MYD88 Palmitoylation. Nat. Chem. Biol 2019, 15, 907–916. [DOI] [PubMed] [Google Scholar]
- (242).Segal-Salto M; Sapir T; Reiner O Reversible Cysteine Acylation Regulates the Activity of Human Palmitoyl-Protein Thioesterase 1 (PPT1). PLoS One 2016, 11, e0146466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (243).Segal-Salto M; Hansson K; Sapir T; Kaplan A; Levy T; Schweizer M; Frotscher M; James P; Reiner O Proteomics Insights into Infantile Neuronal Ceroid Lipofuscinosis (CLN1) Point to the Involvement of Cilia Pathology in the Disease. Hum. Mol. Genet 2017, 26, 1678. [DOI] [PubMed] [Google Scholar]
- (244).Pei Z; Xiao Y; Meng J; Hudmon A; Cummins TR Cardiac Sodium Channel Palmitoylation Regulates Channel Availability and Myocyte Excitability with Implications for Arrhythmia Generation. Nat. Commun 2016, 7, 12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (245).Zoltewicz SJ; Lee S; Chittoor VG; Freeland SM; Rangaraju S; Zacharias DA; Notterpek L The Palmitoylation State of PMP22 Modulates Epithelial Cell Morphology and Migration. ASN Neuro 2012, 4, 409–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (246).Ramsey J; Renzi EC; Arnold RJ; Trinidad JC; Mukhopadhyay S Palmitoylation of Sindbis Virus TF Protein Regulates Its Plasma Membrane Localization and Subsequent Incorporation into Virions. J. Virol 2017, 91, e02000–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (247).Lynes EM; Raturi A; Shenkman M; Sandoval CO; Yap MC; Wu J; Janowicz A; Myhill N; Benson MD; Campbell RE; et al. Palmitoylation Is the Switch That Assigns Calnexin to Quality Control or ER Ca2+ Signaling. J. Cell Sci 2013, 126, 3893 LP–3903. [DOI] [PubMed] [Google Scholar]
- (248).Chan P; Han X; Zheng B; DeRan M; Yu J; Jarugumilli GK; Deng H; Pan D; Luo X; Wu X Autopalmitoylation of TEAD Proteins Regulates Transcriptional Output of the Hippo Pathway. Nat. Chem. Biol 2016, 12, 282–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (249).Noland CL; Gierke S; Schnier PD; Murray J; Sandoval WN; Sagolla M; Dey A; Hannoush RN; Fairbrother WJ; Cunningham CN Palmitoylation of TEAD Transcription Factors Is Required for Their Stability and Function in Hippo Pathway Signaling. Structure 2016, 24, 179–186. [DOI] [PubMed] [Google Scholar]
- (250).Roberts BJ; Johnson KE; McGuinn KP; Saowapa J; Svoboda RA; Mahoney MG; Johnson KR; Wahl JK Palmitoylation of Plakophilin Is Required for Desmosome Assembly. J. Cell Sci 2014, 127, 3782 LP–3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (251).Antinone SE; Ghadge GD; Lam TT; Wang L; Roos RP; Green WN Palmitoylation of Superoxide Dismutase 1 (SOD1) Is Increased for Familial Amyotrophic Lateral Sclerosis-Linked SOD1 Mutants. J. Biol. Chem 2013, 288, 21606–21617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (252).Jiang M; Hu J; White FKH; Williamson J; Klymchenko AS; Murthy A; Workman SW; Tseng G-N S-Palmitoylation of Junctophilin-2 Is Critical for Its Role in Tethering the Sarcoplasmic Reticulum to the Plasma Membrane. J. Biol. Chem 2019, 294, 13487–13501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (253).Chen B; Zheng B; DeRan M; Jarugumilli GK; Fu J; Brooks YS; Wu X ZDHHC7-Mediated S-Palmitoylation of Scribble Regulates Cell Polarity. Nat. Chem. Biol 2016, 12, 686–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (254).Hernandez JL; Davda D; Cheung See Kit M; Majmudar JD; Won SJ; Gang M; Pasupuleti SC; Choi AI; Bartkowiak CM; Martin BR APT2 Inhibition Restores Scribble Localization and S-Palmitoylation in Snail-Transformed Cells. Cell Chem. Biol 2017, 24, 87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (255).Schroeder GN; Aurass P; Oates C V; Tate, E. W.; Hartland, E. L.; Flieger, A.; Frankel, G. Legionella Pneumophila Effector LpdA Is a Palmitoylated Phospholipase D Virulence Factor. Infect. Immun 2015, 83, 3989 LP–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (256).Ren W; Jhala US; Du K Proteomic Analysis of Protein Palmitoylation in Adipocytes. Adipocyte 2013, 2, 17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (257).Ticho AL; Malhotra P; Manzella CR; Dudeja PK; Saksena S; Gill RK; Alrefai WA S-Acylation Modulates the Function of the Apical Sodium-Dependent Bile Acid Transporter in Human Cells. J. Biol. Chem 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (258).Kim SW; Kim DH; Park KS; Kim MK; Park YM; Muallem S; So I; Kim HJ Palmitoylation Controls Trafficking of the Intracellular Ca2+ Channel MCOLN3/TRPML3 to Regulate Autophagy. Autophagy 2019, 15, 327–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (259).Sada R; Kimura H; Fukata Y; Fukata M; Yamamoto H; Kikuchi A Dynamic Palmitoylation Controls the Microdomain Localization of the DKK1 Receptors CKAP4 and LRP6. Sci. Signal 2019, 12, eaat9519. [DOI] [PubMed] [Google Scholar]
- (260).Yao H; Lan J; Li C; Shi H; Brosseau J-P; Wang H; Lu H; Fang C; Zhang Y; Liang L; et al. Inhibiting PD-L1 Palmitoylation Enhances T-Cell Immune Responses against Tumours. Nat. Biomed. Eng 2019, 3, 306–317. [DOI] [PubMed] [Google Scholar]
- (261).Yao H; Li C; He F; Song T; Brosseau J-P; Wang H; Lu H; Fang C; Shi H; Lan J; et al. A Peptidic Inhibitor for PD-1 Palmitoylation Targets Its Expression and Functions. RSC Chem. Biol 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (262).Ebersole B; Petko J; Woll M; Murakami S; Sokolina K; Wong V; Stagljar I; Lüscher B; Levenson R Effect of C-Terminal S-Palmitoylation on D2 Dopamine Receptor Trafficking and Stability. PLoS One 2015, 10, e0140661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (263).Coleman DT; Gray AL; Kridel SJ; Cardelli JA Palmitoylation Regulates the Intracellular Trafficking and Stability of C-Met. Oncotarget 2016, 7, 32664–32677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (264).Runkle KB; Kharbanda A; Stypulkowski E; Cao X-J; Wang W; Garcia BA; Witze ES Inhibition of DHHC20-Mediated EGFR Palmitoylation Creates a Dependence on EGFR Signaling. Mol. Cell 2016, 62, 385–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (265).Wegleiter T; Buthey K; Gonzalez-Bohorquez D; Hruzova M; bin Imtiaz MK; Abegg A; Mebert I; Molteni A; Kollegger D; Pelczar P; et al. Palmitoylation of BMPR1a Regulates Neural Stem Cell Fate. Proc. Natl. Acad. Sci 2019, 116, 25688 LP–25696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (266).Zingler P; Särchen V; Glatter T; Caning L; Saggau C; Kathayat RS; Dickinson BC; Adam D; Schneider-Brachert W; Schütze S; et al. Palmitoylation Is Required for TNF-R1 Signaling. Cell Commun. Signal 2019, 17, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (267).Poggi M; Kara I; Brunel J-M; Landrier J-F; Govers R; Bonardo B; Fluhrer R; Haass C; Alessi M-C; Peiretti F Palmitoylation of TNF Alpha Is Involved in the Regulation of TNF Receptor 1 Signalling. Biochim. Biophys. Acta - Mol. Cell Res 2013, 1833, 602–612. [DOI] [PubMed] [Google Scholar]
- (268).Wang W; Runkle KB; Terkowski SM; Ekaireb RI; Witze ES Protein Depalmitoylation Is Induced by Wnt5a and Promotes Polarized Cell Behavior. J. Biol. Chem 2015, 290, 15707–15716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (269).Stypulkowski E; Asangani IA; Witze ES The Depalmitoylase APT1 Directs the Asymmetric Partitioning of Notch and Wnt Signaling during Cell Division. Sci. Signal 2018, 11, eaam8705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (270).Zhang M; Zhou L; Xu Y; Yang M; Xu Y; Komaniecki GP; Kosciuk T; Chen X; Lu X; Zou X; et al. A STAT3 Palmitoylation Cycle Promotes TH17 Differentiation and Colitis. Nature 2020, 586, 434–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (271).Cao Y; Qiu T; Kathayat RS; Azizi S-A; Thorne AK; Ahn D; Fukata Y; Fukata M; Rice PA; Dickinson BC ABHD10 Is an S-Depalmitoylase Affecting Redox Homeostasis through Peroxiredoxin-5. Nat. Chem. Biol 2019, 15, 1232–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (272).Cui Y; Ma L Sequential Use of Milk and Bovine Serum Albumin for Streptavidin-Probed Western Blot. Biotechniques 2018, 65, 125–126. [DOI] [PubMed] [Google Scholar]
- (273).Fokin VV Click Imaging of Biochemical Processes in Living Systems. ACS Chem. Biol 2007, 2, 775–778. [DOI] [PubMed] [Google Scholar]
- (274).Ayana R; Yadav P; Kumari R; Ramu D; Garg S; Pati S; Singh S Identification and Characterization of a Novel Palmitoyl Acyltransferase as a Druggable Rheostat of Dynamic Palmitoylome in L. Donovani. Front. Cell. Infect. Microbiol 2018, 8, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (275).Siddiqui MA; Singh S; Malhotra P; Chitnis CE Protein S-Palmitoylation Is Responsive to External Signals and Plays a Regulatory Role in Microneme Secretion in Plasmodium Falciparum Merozoites. ACS Infect. Dis 2020, 6, 379–392. [DOI] [PubMed] [Google Scholar]
- (276).Kilian N; Zhang Y; LaMonica L; Hooker G; Toomre D; Ben Mamoun C; Ernst AM Palmitoylated Proteins in Plasmodium Falciparum-Infected Erythrocytes: Investigation with Click Chemistry and Metabolic Labeling. BioEssays 2020, 42, 1900145. [DOI] [PubMed] [Google Scholar]
- (277).Zaręba-Kozioł M; Bartkowiak-Kaczmarek A; Figiel I; Krzystyniak A; Wojtowicz T; Bijata M; Wlodarczyk J Stress-Induced Changes in the S-Palmitoylation and S-Nitrosylation of Synaptic Proteins. Mol. Cell. ProteomicsCellular Proteomics 2019, 18, 1916–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (278).Dumoulin A; Dagane A; Dittmar G; Rathjen FG S-Palmitoylation Is Required for the Control of Growth Cone Morphology of DRG Neurons by CNP-Induced CGMP Signaling. Front. Mol. Neurosci 2018, 11, 345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (279).Ernst AM; Syed SA; Zaki O; Bottanelli F; Zheng H; Hacke M; Xi Z; Rivera-Molina F; Graham M; Rebane AA; et al. S-Palmitoylation Sorts Membrane Cargo for Anterograde Transport in the Golgi. Dev. Cell 2018, 47, 479–493.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (280).Gao X; Arenas-Ramirez N; Scales SJ; Hannoush RN Membrane Targeting of Palmitoylated Wnt and Hedgehog Revealed by Chemical Probes. FEBS Lett 2011, 585, 2501–2506. [DOI] [PubMed] [Google Scholar]
- (281).Gao X; Hannoush RN Method for Cellular Imaging of Palmitoylated Proteins with Clickable Probes and Proximity Ligation Applied to Hedgehog, Tubulin, and Ras. J. Am. Chem. Soc 2014, 136, 4544–4550. [DOI] [PubMed] [Google Scholar]
- (282).Gao X; Hannoush RN Single-Cell in Situ Imaging of Palmitoylation in Fatty-Acylated Proteins. Nat. Protoc 2014, 9, 2607–2623. [DOI] [PubMed] [Google Scholar]
- (283).Yang Y; Dong W; Guo Y; Rioux RM Cu(i)-Catalyzed Aerobic Cross-Dehydrogenative Coupling of Terminal Alkynes with Thiols for the Construction of Alkynyl Sulfides. Green Chem 2013, 15, 3170–3175. [Google Scholar]
- (284).Witten AJ; Ejendal KFK; Gengelbach LM; Traore MA; Wang X; Umulis DM; Calve S; Kinzer-Ursem TL Fluorescent Imaging of Protein Myristoylation during Cellular Differentiation and Development. J. Lipid Res 2017, 58, 2061–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (285).Coleman DT; Soung YH; Surh Y-J; Cardelli JA; Chung J Curcumin Prevents Palmitoylation of Integrin B4 in Breast Cancer Cells. PLoS One 2015, 10, e0125399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (286).Rodgers UR; Lanyon-Hogg T; Masumoto N; Ritzefeld M; Burke R; Blagg J; Magee AI; Tate EW Characterization of Hedgehog Acyltransferase Inhibitors Identifies a Small Molecule Probe for Hedgehog Signaling by Cancer Cells. ACS Chem. Biol 2016, 11, 3256–3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (287).Dodge ME; Moon J; Tuladhar R; Lu J; Jacob LS; Zhang L; Shi H; Wang X; Moro E; Mongera A; et al. Diverse Chemical Scaffolds Support Direct Inhibition of the Membrane Bound O-Acyltransferase Porcupine. J. Biol. Chem 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (288).Proffitt KD; Madan B; Ke Z; Pendharkar V; Ding L; Lee MA; Hannoush RN; Virshup DM Pharmacological Inhibition of the Wnt Acyltransferase PORCN Prevents Growth of WNT-Driven Mammary Cancer. Cancer Res 2013, 73, 502 LP–507. [DOI] [PubMed] [Google Scholar]
- (289).Madan B; Ke Z; Harmston N; Ho SY; Frois AO; Alam J; Jeyaraj DA; Pendharkar V; Ghosh K; Virshup IH; et al. Wnt Addiction of Genetically Defined Cancers Reversed by PORCN Inhibition. Oncogene 2016, 35, 2197–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (290).Zhan T; Rindtorff N; Boutros M Wnt Signaling in Cancer. Oncogene 2017, 36, 1461–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (291).Yadav P; Ayana R; Garg S; Jain R; Sah R; Joshi N; Pati S; Singh S Plasmodium Palmitoylation Machinery Engineered in E. Coli for High-Throughput Screening of Palmitoyl Acyl-Transferase Inhibitors. FEBS Open Bio 2019, 9, 248–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (292).Fukata M; Fukata Y; Adesnik H; Nicoll RA; Bredt DS Identification of PSD-95 Palmitoylating Enzymes. Neuron 2004, 44, 987–996. [DOI] [PubMed] [Google Scholar]
- (293).Zheng B; DeRan M; Li X; Liao X; Fukata M; Wu X 2-Bromopalmitate Analogues as Activity-Based Probes To Explore Palmitoyl Acyltransferases. J. Am. Chem. Soc 2013, 135, 7082–7085. [DOI] [PubMed] [Google Scholar]
- (294).Davda D; El Azzouny MA; Tom CTMB; Hernandez JL; Majmudar JD; Kennedy RT; Martin BR Profiling Targets of the Irreversible Palmitoylation Inhibitor 2-Bromopalmitate. ACS Chem. Biol 2013, 8, 1912–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (295).Zheng B; Zhu S; Wu X Clickable Analogue of Cerulenin as Chemical Probe to Explore Protein Palmitoylation. ACS Chem. Biol 2015, 10, 115–121. [DOI] [PubMed] [Google Scholar]
- (296).Ganesan L; Shieh P; Bertozzi CR; Levental I Click-Chemistry Based High Throughput Screening Platform for Modulators of Ras Palmitoylation. Sci. Rep 2017, 7, 41147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (297).Konitsiotis AD; Jovanović B; Ciepla P; Spitaler M; Lanyon-Hogg T; Tate EW; Magee AI Topological Analysis of Hedgehog Acyltransferase, a Multipalmitoylated Transmembrane Protein. J. Biol. Chem 2015, 290, 3293–3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (298).Lanyon-Hogg T; Masumoto N; Bodakh G; Konitsiotis AD; Thinon E; Rodgers UR; Owens RJ; Magee AI; Tate EW Click Chemistry Armed Enzyme-Linked Immunosorbent Assay to Measure Palmitoylation by Hedgehog Acyltransferase. Anal. Biochem 2015, 490, 66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (299).Lanyon-Hogg T; Patel NV; Ritzefeld M; Boxall KJ; Burke R; Blagg J; Magee AI; Tate EW Microfluidic Mobility Shift Assay for Real-Time Analysis of Peptide N-Palmitoylation. SLAS Discov. Adv. Sci. Drug Discov 2017, 22, 418–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (300).Lanyon-Hogg T; Ritzefeld M; Sefer L; Bickel JK; Rudolf AF; Panyain N; Bineva-Todd G; Ocasio CA; O’Reilly N; Siebold C; et al. Acylation-Coupled Lipophilic Induction of Polarisation (Acyl-CLIP): A Universal Assay for Lipid Transferase and Hydrolase Enzymes. Chem. Sci 2019, 10, 8995–9000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (301).Morrison E; Wegner T; Zucchetti AE; Álvaro-Benito M; Zheng A; Kliche S; Krause E; Brügger B; Hivroz C; Freund C Dynamic Palmitoylation Events Following T-Cell Receptor Signaling. Commun. Biol 2020, 3, 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (302).Salaun C; Greaves J; Tomkinson NCO; Chamberlain LH The Linker Domain of the SNARE Protein SNAP25 Acts as a Flexible Molecular Spacer That Ensures Efficient S-Acylation. J. Biol. Chem 2020, 295, 7501–7515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (303).Martinez A; Traverso JA; Valot B; Ferro M; Espagne C; Ephritikhine G; Zivy M; Giglione C; Meinnel T Extent of N-Terminal Modifications in Cytosolic Proteins from Eukaryotes. Proteomics 2008, 8, 2809–2831. [DOI] [PubMed] [Google Scholar]
- (304).Martin DDO; Vilas GL; Prescher JA; Rajaiah G; Falck JR; Bertozzi CR; Berthiaume LG Rapid Detection, Discovery, and Identification of Post-Translationally Myristoylated Proteins during Apoptosis Using a Bio-Orthogonal Azidomyristate Analog. FASEB J 2008, 22, 797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (305).Yap MC; Kostiuk MA; Martin DDO; Perinpanayagam MA; Hak PG; Siddam A; Majjigapu JR; Rajaiah G; Keller BO; Prescher JA; et al. Rapid and Selective Detection of Fatty Acylated Proteins Using ω-Alkynyl-Fatty Acids and Click Chemistry. J. Lipid Res 2010, 51, 1566–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (306).Storck EM; Serwa RA; Tate EW Chemical Proteomics: A Powerful Tool for Exploring Protein Lipidation. Biochem. Soc. Trans 2013, 41, 56 LP–61. [DOI] [PubMed] [Google Scholar]
- (307).Martin DDO; Ahpin CY; Heit RJ; Perinpanayagam MA; Yap MC; Veldhoen RA; Goping IS; Berthiaume LG Tandem Reporter Assay for Myristoylated Proteins Post-Translationally (TRAMPP) Identifies Novel Substrates for Post-Translational Myristoylation: PKC∊, a Case Study. FASEB J 2012, 26, 13–28. [DOI] [PubMed] [Google Scholar]
- (308).Boyle PC; Schwizer S; Hind SR; Kraus CM; De la Torre Diaz S; He B; Martin GB Detecting N-Myristoylation and S-Acylation of Host and Pathogen Proteins in Plants Using Click Chemistry. Plant Methods 2016, 12, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (309).Takamitsu E; Fukunaga K; Iio Y; Moriya K; Utsumi T Cell-Free Identification of Novel N-Myristoylated Proteins from Complementary DNA Resources Using Bioorthogonal Myristic Acid Analogues. Anal. Biochem 2014, 464, 83–93. [DOI] [PubMed] [Google Scholar]
- (310).Rampoldi F; Sandhoff R; Owen RW; Gröne H-J; Porubsky S A New, Robust, and Nonradioactive Approach for Exploring N-Myristoylation. J. Lipid Res 2012, 53, 2459–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (311).Gao H; Sun W; Song Z; Yu Y; Wang L; Chen X; Zhang Q A Method to Generate and Analyze Modified Myristoylated Proteins. Chembiochem 2017, 18, 324–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (312).Goya Grocin A; Serwa RA; Morales Sanfrutos J; Ritzefeld M; Tate EW Whole Proteome Profiling of N-Myristoyltransferase Activity and Inhibition Using Sortase A. Mol. & Cell. Proteomics 2019, 18, 115 LP–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (313).Kinoshita-Kikuta E; Tanikawa A; Hosokawa T; Kiwado A; Moriya K; Kinoshita E; Koike T; Utsumi T A Strategy to Identify Protein-N-Myristoylation-Dependent Phosphorylation Reactions of Cellular Proteins by Using Phos-Tag SDS-PAGE. PLoS One 2019, 14, e0225510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (314).Kosciuk T; Lin H N-Myristoyltransferase as a Glycine and Lysine Myristoyltransferase in Cancer, Immunity, and Infections. ACS Chem. Biol 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (315).Thinon E; Serwa RA; Broncel M; Brannigan JA; Brassat U; Wright MH; Heal WP; Wilkinson AJ; Mann DJ; Tate EW Global Profiling of Co- and Post-Translationally N-Myristoylated Proteomes in Human Cells. Nat Commun 2014, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (316).Thinon E; Morales-Sanfrutos J; Mann DJ; Tate EW N-Myristoyltransferase Inhibition Induces ER-Stress, Cell Cycle Arrest, and Apoptosis in Cancer Cells. ACS Chem. Biol 2016, 11, 2165–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (317).Broncel M; Serwa RA; Ciepla P; Krause E; Dallman MJ; Magee AI; Tate EW Myristoylation Profiling in Human Cells and Zebrafish. Data Br 2015, 4, 379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (318).Zhang X; Khan S; Jiang H; Antonyak MA; Chen X; Spiegelman NA; Shrimp JH; Cerione RA; Lin H Identifying the Functional Contribution of the Defatty-Acylase Activity of SIRT6. Nat. Chem. Biol 2016, 12, 614–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (319).Jiang H; Khan S; Wang Y; Charron G; He B; Sebastian C; Du J; Kim R; Ge E; Mostoslavsky R; et al. SIRT6 Regulates TNF-[Agr] Secretion through Hydrolysis of Long-Chain Fatty Acyl Lysine. Nature 2013, 496, 110–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (320).Aramsangtienchai P; Spiegelman NA; He B; Miller SP; Dai L; Zhao Y; Lin H HDAC8 Catalyzes the Hydrolysis of Long Chain Fatty Acyl Lysine. ACS Chem. Biol 2016, 11, 2685–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (321).Spiegelman NA; Hong JY; Hu J; Jing H; Wang M; Price IR; Cao J; Yang M; Zhang X; Lin H A Small-Molecule SIRT2 Inhibitor That Promotes K-Ras4a Lysine Fatty-Acylation. ChemMedChem 2019, 14, 744–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (322).Heal WP; Wickramasinghe SR; Bowyer PW; Holder AA; Smith DF; Leatherbarrow RJ; Tate EW Site-Specific N-Terminal Labelling of Proteinsin Vitro and in Vivo Using N-Myristoyl Transferase and Bioorthogonal Ligation Chemistry. Chem. Commun 2008, No. 4, 480–482. [DOI] [PubMed] [Google Scholar]
- (323).Meyer-Schaller N; Chou Y-C; Sumara I; Martin DDO; Kurz T; Katheder N; Hofmann K; Berthiaume LG; Sicheri F; Peter M The Human Dcn1-like Protein DCNL3 Promotes Cul3 Neddylation at Membranes. Proc. Natl. Acad. Sci 2009, 106, 12365 LP–12370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (324).Govatati S; Pichavaram P; Janjanam J; Guo L; Virmani R; Rao GN Myristoylation of LMCD1 Leads to Its Species-Specific Derepression of E2F1 and NFATc1 in the Modulation of CDC6 and IL-33 Expression During Development of Vascular Lesions. Arterioscler. Thromb. Vasc. Biol 2020, 40, 1256–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (325).Kumar S; Parameswaran S; Sharma RK Novel Myristoylation of the Sperm-Specific Hexokinase 1 Isoform Regulates Its Atypical Localization. Biol. Open 2015, 4, 1679 LP–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (326).Rampoldi F; Bonrouhi M; Boehm ME; Lehmann WD; Popovic ZV; Kaden S; Federico G; Brunk F; Gröne H-J; Porubsky S Immunosuppression and Aberrant T Cell Development in the Absence of N-Myristoylation. J. Immunol 2015, 195, 4228 LP–4243. [DOI] [PubMed] [Google Scholar]
- (327).Rampoldi F; Brunk F; Bonrouhi M; Federico G; Krunic D; Porubsky S; Gröne H-J; Popovic ZV Deficiency of N-Myristoylation Reveals Calcineurin Activity as Regulator of IFN-γ-Producing Γδ T Cells. J. Leukoc. Biol 2017, 101, 1005–1014. [DOI] [PubMed] [Google Scholar]
- (328).Stackpole EE; Akins MR; Fallon JR N-Myristoylation Regulates the Axonal Distribution of the Fragile X-Related Protein FXR2P. Mol. Cell. Neurosci 2014, 62, 42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (329).Demetriadou A; Morales-Sanfrutos J; Nearchou M; Baba O; Kyriacou K; Tate EW; Drousiotou A; Petrou PP Mouse Stbd1 Is N-Myristoylated and Affects ER–Mitochondria Association and Mitochondrial Morphology. J. Cell Sci 2017, 130, 903 LP–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (330).Liang J; Xu Z-X; Ding Z; Lu Y; Yu Q; Werle KD; Zhou G; Park Y-Y; Peng G; Gambello MJ; et al. Myristoylation Confers Noncanonical AMPK Functions in Autophagy Selectivity and Mitochondrial Surveillance. Nat. Commun 2015, 6, 7926. [DOI] [PubMed] [Google Scholar]
- (331).Kallemeijn WW; Lueg GA; Faronato M; Hadavizadeh K; Goya Grocin A; Song O-R; Howell M; Calado DP; Tate EW Validation and Invalidation of Chemical Probes for the Human N-Myristoyltransferases. Cell Chem. Biol 2019, 26, 892–900.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (332).Zhu F; Xie N; Jiang Z; Li G; Ma L; Tong T The Cellular Senescence-Inhibited Gene Is Essential for PPM1A Myristoylation To Modulate Transforming Growth Factor β Signaling. Mol. Cell. Biol 2018, 38, e00414–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (333).Zha J; Weiler S; Oh KJ; Wei MC; Korsmeyer SJ Posttranslational N-Myristoylation of BID as a Molecular Switch for Targeting Mitochondria and Apoptosis. Science 2000, 290, 1761 LP–1765. [DOI] [PubMed] [Google Scholar]
- (334).Martin DDO; Heit RJ; Yap MC; Davidson MW; Hayden MR; Berthiaume LG Identification of a Post-Translationally Myristoylated Autophagy-Inducing Domain Released by Caspase Cleavage of Huntingtin. Hum. Mol. Genet 2014, 23, 3166–3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (335).Martin DDO; Kay C; Collins JA; Nguyen YT; Slama RA; Hayden MR A Human Huntingtin SNP Alters Post-Translational Modification and Pathogenic Proteolysis of the Protein Causing Huntington Disease. Sci. Rep 2018, 8, 8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (336).Das U; Kumar S; Sharma JRD and R. K Inhibition of Protein N-Myristoylation: A Therapeutic Protocol in Developing Anticancer Agents. Current Cancer Drug Targets. 2012, pp 667–692. [DOI] [PubMed] [Google Scholar]
- (337).Kim S; Yang X; Li Q; Wu M; Costyn L; Beharry Z; Bartlett MG; Cai H Myristoylation of Src Kinase Mediates Src-Induced and High-Fat Diet–Accelerated Prostate Tumor Progression in Mice. J. Biol. Chem 2017, 292, 18422–18433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (338).Kim S; Alsaidan OA; Goodwin O; Li Q; Sulejmani E; Han Z; Bai A; Albers T; Beharry Z; Zheng YGG; et al. Blocking Myristoylation of Src Inhibits Its Kinase Activity and Suppresses Prostate Cancer Progression. Cancer Res 2017, canres.0981.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (339).Beauchamp E; Yap MC; Iyer A; Perinpanayagam MA; Gamma JM; Vincent KM; Lakshmanan M; Raju A; Tergaonkar V; Tan SY; et al. Targeting N-Myristoylation for Therapy of B-Cell Lymphomas. Nat. Commun 2020, 11, 5348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (340).Li Q; Alsaidan OA; Ma Y; Kim S; Liu J; Albers T; Liu K; Beharry Z; Zhao S; Wang F; et al. Pharmacologically Targeting the Myristoylation of the Scaffold Protein FRS2α Inhibits FGF/FGFR-Mediated Oncogenic Signaling and Tumor Progression. J. Biol. Chem 2018, 293, 6434–6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (341).Bowyer PW; Tate EW; Leatherbarrow RJ; Holder AA; Smith DF; Brown KA N-Myristoyltransferase: A Prospective Drug Target for Protozoan Parasites. ChemMedChem 2008, 3, 402–408. [DOI] [PubMed] [Google Scholar]
- (342).Tate EW; Bell AS; Rackham MD; Wright MH N-Myristoyltransferase as a Potential Drug Target in Malaria and Leishmaniasis. Parasitology 2014, 141, 37–49. [DOI] [PubMed] [Google Scholar]
- (343).Ritzefeld M; Wright MH; Tate EW New Developments in Probing and Targeting Protein Acylation in Malaria, Leishmaniasis and African Sleeping Sickness. Parasitology 2018, 145, 157–174. [DOI] [PubMed] [Google Scholar]
- (344).Poulin B; Patzewitz E-M; Brady D; Silvie O; Wright MH; Ferguson DJP; Wall RJ; Whipple S; Guttery DS; Tate EW; et al. Unique Apicomplexan IMC Sub-Compartment Proteins Are Early Markers for Apical Polarity in the Malaria Parasite. Biol. Open 2013, 2, 1160 LP–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (345).Schlott AC; Mayclin S; Reers AR; Coburn-Flynn O; Bell AS; Green J; Knuepfer E; Charter D; Bonnert R; Campo B; et al. Structure-Guided Identification of Resistance Breaking Antimalarial N-Myristoyltransferase Inhibitors. Cell Chem. Biol 2019, 26, 991–1000.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (346).Price HP; Hodgkinson MR; Wright MH; Tate EW; Smith BA; Carrington M; Stark M; Smith DF A Role for the Vesicle-Associated Tubulin Binding Protein ARL6 (BBS3) in Flagellum Extension in Trypanosoma Brucei. Biochim. Biophys. Acta - Mol. Cell Res 2012, 1823, 1178–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (347).Roberts AJ; Torrie LS; Wyllie S; Fairlamb AH Biochemical and Genetic Characterization of Trypanosoma Cruzi N-Myristoyltransferase. Biochem. J 2014, 459, 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (348).Herrera LJ; Brand S; Santos A; Nohara LL; Harrison J; Norcross NR; Thompson S; Smith V; Lema C; Varela-Ramirez A; et al. Validation of N-Myristoyltransferase as Potential Chemotherapeutic Target in Mammal-Dwelling Stages of Trypanosoma Cruzi. PLoS Negl. Trop. Dis 2016, 10, e0004540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (349).Roberts AJ; Fairlamb AH The N-Myristoylome of Trypanosoma Cruzi. Sci. Rep 2016, 6, 31078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (350).Wright MH; Paape D; Storck EM; Serwa RA; Smith DF; Tate EW Global Analysis of Protein N-Myristoylation and Exploration of N-Myristoyltransferase as a Drug Target in the Neglected Human Pathogen Leishmania Donovani. Chem. Biol 2015, 22, 342–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (351).Rackham MD; Yu Z; Brannigan JA; Heal WP; Paape D; Barker KV; Wilkinson AJ; Smith DF; Leatherbarrow RJ; Tate EW Discovery of High Affinity Inhibitors of Leishmania Donovani N-Myristoyltransferase. Medchemcomm 2015, 6, 1761–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (352).Corpas-Lopez V; Moniz S; Thomas M; Wall RJ; Torrie LS; Zander-Dinse D; Tinti M; Brand S; Stojanovski L; Manthri S; et al. Pharmacological Validation of N-Myristoyltransferase as a Drug Target in Leishmania Donovani. ACS Infect. Dis 2019, 5, 111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (353).Bell AS; Yu Z; Hutton JA; Wright MH; Brannigan JA; Paape D; Roberts SM; Sutherell CL; Ritzefeld M; Wilkinson AJ; et al. Novel Thienopyrimidine Inhibitors of Leishmania N-Myristoyltransferase with On-Target Activity in Intracellular Amastigotes. J. Med. Chem 2020, 63, 7740–7765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (354).Broncel M; Dominicus C; Vigetti L; Nofal SD; Bartlett EJ; Touquet B; Hunt A; Wallbank BA; Federico S; Matthews S; et al. Profiling of Myristoylation in Toxoplasma Gondii Reveals an N-Myristoylated Protein Important for Host Cell Penetration. Elife 2020, 9, e57861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (355).Foe IT; Onguka O; Amberg-Johnson K; Garner RM; Amara N; Beatty W; Yeh E; Bogyo M The Toxoplasma Gondii Active Serine Hydrolase 4 Regulates Parasite Division and Intravacuolar Parasite Architecture. mSphere 2018, 3, e00393–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (356).Kemp LE; Rusch M; Adibekian A; Bullen HE; Graindorge A; Freymond C; Rottmann M; Braun-Breton C; Baumeister S; Porfetye AT; et al. Characterization of a Serine Hydrolase Targeted by Acyl-Protein Thioesterase Inhibitors in Toxoplasma Gondii. J. Biol. Chem 2013, 288, 27002–27018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (357).Lodge JK; Jackson-Machelski E; Toffaletti DL; Perfect JR; Gordon JI Targeted Gene Replacement Demonstrates That Myristoyl-CoA: Protein N-Myristoyltransferase Is Essential for Viability of Cryptococcus Neoformans. Proc. Natl. Acad. Sci 1994, 91, 12008 LP–12012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (358).Lodge JK; Johnson RL; Weinberg RA; Gordon JI Comparison of Myristoyl-CoA:Protein N-Myristoyltransferases from Three Pathogenic Fungi: Cryptococcus Neoformans, Histoplasma Capsulatum, and Candida Albicans. J. Biol. Chem 1994, 269, 2996–3009. [PubMed] [Google Scholar]
- (359).Weinberg RA; McWherter CA; Freeman SK; Wood DC; Gordon JI; Lee SC Genetic Studies Reveal That MyristoylCoA:Protein N-Myristoyltransferase Is an Essential Enzyme in Candida Albicans. Mol. Microbiol 1995, 16, 241–250. [DOI] [PubMed] [Google Scholar]
- (360).Fang W; Robinson DA; Raimi OG; Blair DE; Harrison JR; Lockhart DEA; Torrie LS; Ruda GF; Wyatt PG; Gilbert IH; et al. N-Myristoyltransferase Is a Cell Wall Target in Aspergillus Fumigatus. ACS Chem. Biol 2015, 10, 1425–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (361).Fedoryshchak RO; Ocasio CA; Strutton B; Mattocks J; Corran AJ; Tate EW Wheat Pathogen Zymoseptoria Tritici N-Myristoyltransferase Inhibitors: On-Target Antifungal Activity and an Unusual Metabolic Defense Mechanism. RSC Chem. Biol 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (362).Zhu AY; Zhou Y; Khan S; Deitsch KW; Hao Q; Lin H Plasmodium Falciparum Sir2A Preferentially Hydrolyzes Medium and Long Chain Fatty Acyl Lysine. ACS Chem. Biol 2012, 7, 155–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (363).Charlton TM; Kovacs-Simon A; Michell SL; Fairweather NF; Tate EW Quantitative Lipoproteomics in Clostridium Difficile Reveals a Role for Lipoproteins in Sporulation. Chem. Biol 2015, 22, 1562–1573. [DOI] [PubMed] [Google Scholar]
- (364).Nimchuk Z; Marois E; Kjemtrup S; Leister RT; Katagiri F; Dangl JL Eukaryotic Fatty Acylation Drives Plasma Membrane Targeting and Enhances Function of Several Type III Effector Proteins from Pseudomonas Syringae. Cell 2000, 101, 353–363. [DOI] [PubMed] [Google Scholar]
- (365).Boyle PC; Martin GB Greasy Tactics in the Plant–Pathogen Molecular Arms Race. J. Exp. Bot 2015, 66, 1607–1616. [DOI] [PubMed] [Google Scholar]
- (366).Ribet D; Cossart P Pathogen-Mediated Posttranslational Modifications: A Re-Emerging Field. Cell 2010, 143, 694–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (367).Burnaevskiy N; Fox TG; Plymire DA; Ertelt JM; Weigele BA; Selyunin AS; Way SS; Patrie SM; Alto NM Proteolytic Elimination of N-Myristoyl Modifications by the Shigella Virulence Factor IpaJ. Nature 2013, 496, 106–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (368).Burnaevskiy N; Peng T; Reddick LE; Hang HC; Alto NM Myristoylome Profiling Reveals a Concerted Mechanism of ARF GTPase Deacylation by the Bacterial Protease IpaJ. Mol. Cell 2015, 58, 110–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (369).Kim BS; Satchell KJF MARTX Effector Cross Kingdom Activation by Golgi-Associated ADP-Ribosylation Factors. Cell. Microbiol 2016, 18, 1078–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (370).Li H; Dou J; Ding L; Spearman P Myristoylation Is Required for Human Immunodeficiency Virus Type 1 Gag-Gag Multimerization in Mammalian Cells. J. Virol 2007, 81, 12899 LP–12910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (371).P G; Le Seyec J; Rumin S; Guguen-Guillouzo C Myristylation of the Hepatitis B Virus Large Surface Protein Is Essential for Viral Infectivity. Virology 1995, 213, 292–299. [DOI] [PubMed] [Google Scholar]
- (372).MacLean CA; Clark B; McGeoch DJ Gene UL11 of Herpes Simplex Virus Type 1 Encodes a Virion Protein Which Is Myristylated. J. Gen. Virol 1989, 70, 3147–3157. [DOI] [PubMed] [Google Scholar]
- (373).Ohta H; Takamune N; Kishimoto N; Shoji S; Misumi S N-Myristoyltransferase 1 Enhances Human Immunodeficiency Virus Replication through Regulation of Viral RNA Expression Level. Biochem. Biophys. Res. Commun 2015, 463, 988–993. [DOI] [PubMed] [Google Scholar]
- (374).Marc D; Masson G; Girard M; van der Werf S Lack of Myristoylation of Poliovirus Capsid Polypeptide VP0 Prevents the Formation of Virions or Results in the Assembly of Noninfectious Virus Particles. J. Virol 1990, 64, 4099–4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (375).Corbic Ramljak I; Stanger J; Real-Hohn A; Dreier D; Wimmer L; Redlberger-Fritz M; Fischl W; Klingel K; Mihovilovic MD; Blaas D; et al. Cellular N-Myristoyltransferases Play a Crucial Picornavirus Genus-Specific Role in Viral Assembly, Virion Maturation, and Infectivity. PLoS Pathog 2018, 14, e1007203–e1007203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (376).Mousnier A; Bell AS; Swieboda DP; Morales-Sanfrutos J; Pérez-Dorado I; Brannigan JA; Newman J; Ritzefeld M; Hutton JA; Guedán A; et al. Fragment-Derived Inhibitors of Human N-Myristoyltransferase Block Capsid Assembly and Replication of the Common Cold Virus. Nat. Chem 2018, 10, 599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (377).Bell AS; Mills JE; Williams GP; Brannigan JA; Wilkinson AJ; Parkinson T; Leatherbarrow RJ; Tate EW; Holder AA; Smith DF Selective Inhibitors of Protozoan Protein N-Myristoyltransferases as Starting Points for Tropical Disease Medicinal Chemistry Programs. PLoS Negl. Trop. Dis 2012, 6, e1625–e1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (378).Casey PJ; Solski PA; Der CJ; Buss JE P21ras Is Modified by a Farnesyl Isoprenoid. Proc. Natl. Acad. Sci. U. S. A 1989, 86, 8323–8327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (379).Casey PJ; Thissen JA; Moomaw JF Enzymatic Modification of Proteins with a Geranylgeranyl Isoprenoid. Proc. Natl. Acad. Sci. U. S. A 1991, 88, 8631–8635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (380).Kinsella BT; Maltese WA Rab GTP-Binding Proteins with Three Different Carboxyl-Terminal Cysteine Motifs Are Modified in Vivo by 20-Carbon Isoprenoids. J. Biol. Chem 1992, 267, 3940–3945. [PubMed] [Google Scholar]
- (381).London N; Lamphear CL; Hougland JL; Fierke CA; Schueler-Furman O Identification of a Novel Class of Farnesylation Targets by Structure-Based Modeling of Binding Specificity. PLOS Comput. Biol 2011, 7, e1002170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (382).Wang Y-C; Dozier JK; Beese LS; Distefano MD Rapid Analysis of Protein Farnesyltransferase Substrate Specificity Using Peptide Libraries and Isoprenoid Diphosphate Analogues. ACS Chem. Biol 2014, 9, 1726–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (383).Berger BM; Kim JH; Hildebrandt ER; Davis IC; Morgan MC; Hougland JL; Schmidt WK Protein Isoprenylation in Yeast Targets COOH-Terminal Sequences Not Adhering to the CaaX Consensus. Genetics 2018, 210, 1301 LP–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (384).Krzysiak AJ; Rawat DS; Scott SA; Pais JE; Handley M; Harrison ML; Fierke CA; Gibbs RA Combinatorial Modulation of Protein Prenylation. ACS Chem. Biol 2007, 2, 385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (385).Zhang Y; Blanden MJ; Sudheer C; Gangopadhyay SA; Rashidian M; Hougland JL; Distefano MD Simultaneous Site-Specific Dual Protein Labeling Using Protein Prenyltransferases. Bioconjug. Chem 2015, 26, 2542–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (386).Hovlid ML; Edelstein RL; Henry O; Ochocki J; DeGraw A; Lenevich S; Talbot T; Young VG; Hruza AW; Lopez-Gallego F; et al. Synthesis, Properties, and Applications of Diazotrifluropropanoyl-Containing Photoactive Analogs of Farnesyl Diphosphate Containing Modified Linkages for Enhanced Stability. Chem. Biol. Drug Des 2010, 75, 51–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (387).Kyro K; Manandhar SP; Mullen D; Schmidt WK; Distefano MD Photoaffinity Labeling of Ras Converting Enzyme Using Peptide Substrates That Incorporate Benzoylphenylalanine (Bpa) Residues: Improved Labeling and Structural Implications. Bioorg. Med. Chem 2011, 19, 7559–7569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (388).Vervacke JS; Funk AL; Wang Y-C; Strom M; Hrycyna CA; Distefano MD Diazirine-Containing Photoactivatable Isoprenoid: Synthesis and Application in Studies with Isoprenylcysteine Carboxyl Methyltransferase. J. Org. Chem 2014, 79, 1971–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (389).Davies BSJ; Yang SH; Farber E; Lee R; Buck SB; Andres DA; Spielmann HP; Agnew BJ; Tamanoi F; Fong LG; et al. Increasing the Length of Progerin’s Isoprenyl Anchor Does Not Worsen Bone Disease or Survival in Mice with Hutchinson-Gilford Progeria Syndrome. J. Lipid Res 2009, 50, 126–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (390).Onono FO; Morgan MA; Spielmann HP; Andres DA; Subramanian T; Ganser A; Reuter CWM A Tagging-via-Substrate Approach to Detect the Farnesylated Proteome Using Two-Dimensional Electrophoresis Coupled with Western Blotting. Mol. Cell. Proteomics 2010, 9, 742–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (391).Nguyen UTT; Guo Z; Delon C; Wu Y; Deraeve C; Fränzel B; Bon RS; Blankenfeldt W; Goody RS; Waldmann H; et al. Analysis of the Eukaryotic Prenylome by Isoprenoid Affinity Tagging. Nat. Chem. Biol 2009, 5, 227–235. [DOI] [PubMed] [Google Scholar]
- (392).Hougland JL; Fierke CA Getting a Handle on Protein Prenylation. Nat. Chem. Biol 2009, 5, 197–198. [DOI] [PubMed] [Google Scholar]
- (393).Ali N; Jurczyluk J; Shay G; Tnimov Z; Alexandrov K; Munoz MA; Skinner OP; Pavlos NJ; Rogers MJ A Highly Sensitive Prenylation Assay Reveals in Vivo Effects of Bisphosphonate Drug on the Rab Prenylome of Macrophages Outside the Skeleton. Small GTPases 2015, 6, 202–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (394).Kho Y; Kim SC; Jiang C; Barma D; Kwon SW; Cheng J; Jaunbergs J; Weinbaum C; Tamanoi F; Falck J; et al. A Tagging-via-Substrate Technology for Detection and Proteomics of Farnesylated Proteins. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 12479–12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (395).Rose MW; Xu J; Kale TA; O’Doherty G; Barany G; Distefano MD Enzymatic Incorporation of Orthogonally Reactive Prenylazide Groups into Peptides Using Geranylazide Diphosphate via Protein Farnesyltransferase: Implications for Selective Protein Labeling. Pept. Sci 2005, 80, 164–171. [DOI] [PubMed] [Google Scholar]
- (396).Labadie GR; Viswanathan R; Poulter CD Farnesyl Diphosphate Analogues with ω-Bioorthogonal Azide and Alkyne Functional Groups for Protein Farnesyl Transferase-Catalyzed Ligation Reactions. J. Org. Chem 2007, 72, 9291–9297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (397).Hosokawa A; Wollack JW; Zhang Z; Chen L; Barany G; Distefano MD Evaluation of an Alkyne-Containing Analogue of Farnesyl Diphosphate as a Dual Substrate for Protein-Prenyltransferases. Int. J. Pept. Res. Ther 2007, 13, 345–354. [Google Scholar]
- (398).Crick DC; Andres DA; Waechter CJ Farnesol Is Utilized for Protein Isoprenylation and the Biosynthesis of Cholesterol in Mammalian Cells. Biochem. Biophys. Res. Commun 1995, 211, 590–599. [DOI] [PubMed] [Google Scholar]
- (399).Bentinger M; Grünler J; Peterson E; Swiezewska E; Dallner G Phosphorylation of Farnesol in Rat Liver Microsomes: Properties of Farnesol Kinase and Farnesyl Phosphate Kinase. Arch. Biochem. Biophys 1998, 353, 191–198. [DOI] [PubMed] [Google Scholar]
- (400).Meigs TE; Simoni RD Farnesol as a Regulator of HMG-CoA Reductase Degradation: Characterization and Role of Farnesyl Pyrophosphatase. Arch. Biochem. Biophys 1997, 345, 1–9. [DOI] [PubMed] [Google Scholar]
- (401).Rizzo WB Fatty Aldehyde and Fatty Alcohol Metabolism: Review and Importance for Epidermal Structure and Function. Biochim. Biophys. Acta 2014, 1841, 377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (402).Rose MW; Rose ND; Boggs J; Lenevich S; Xu J; Barany G; Distefano MD Evaluation of Geranylazide and Farnesylazide Diphosphate for Incorporation of Prenylazides into a CAAX Box-Containing Peptide Using Protein Farnesyltransferase. J. Pept. Res 2005, 65, 529–537. [DOI] [PubMed] [Google Scholar]
- (403).Chan LN; Hart C; Guo L; Nyberg T; Davies BSJ; Fong LG; Young SG; Agnew BJ; Tamanoi F A Novel Approach to Tag and Identify Geranylgeranylated Proteins. Electrophoresis 2009, 30, 3598–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (404).Berry AFH; Heal WP; Tarafder AK; Tolmachova T; Baron RA; Seabra MC; Tate EW Rapid Multilabel Detection of Geranylgeranylated Proteins by Using Bioorthogonal Ligation Chemistry. ChemBioChem 2010, 11, 771–773. [DOI] [PubMed] [Google Scholar]
- (405).DeGraw AJ; Palsuledesai C; Ochocki JD; Dozier JK; Lenevich S; Rashidian M; Distefano MD Evaluation of Alkyne-Modified Isoprenoids as Chemical Reporters of Protein Prenylation. Chem. Biol. Drug Des 2010, 76, 460–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (406).Placzek AT; Gibbs RA New Synthetic Methodology for the Construction of 7-Substituted Farnesyl Diphosphate Analogs. Org. Lett 2011, 13, 3576–3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (407).Jennings BC; Danowitz AM; Wang Y-C; Gibbs RA; Distefano MD; Fierke CA Analogs of Farnesyl Diphosphate Alter CaaX Substrate Specificity and Reactions Rates of Protein Farnesyltransferase. Bioorg. Med. Chem. Lett 2016, 26, 1333–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (408).Goldstein JL; Brown MS Regulation of the Mevalonate Pathway. Nature 1990, 343, 425. [DOI] [PubMed] [Google Scholar]
- (409).Ahmadi M; Suazo KF; Distefano MD Optimization of Metabolic Labeling with Alkyne-Containing Isoprenoid Probes BT - Protein Lipidation: Methods and Protocols; Linder ME, Ed.; Springer New York: New York, NY, 2019; pp 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (410).Suazo KF; Hurben AK; Liu K; Xu F; Thao P; Sudheer C; Li L; Distefano M Metabolic Labeling of Prenylated Proteins Using Alkyne- Modified Isoprenoid Analogues. Curr. Protoc. Chem. Biol 2018, In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (411).Charron G; Tsou LK; Maguire W; Yount JS; Hang HC Alkynyl-Farnesol Reporters for Detection of Protein S-Prenylation in Cells. Mol. Biosyst 2011, 7, 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (412).Charron G; Li MMH; MacDonald MR; Hang HC Prenylome Profiling Reveals S-Farnesylation Is Crucial for Membrane Targeting and Antiviral Activity of ZAP Long-Isoform. Proc. Natl. Acad. Sci 2013, 110, 11085–11090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (413).Ochocki JD; Distefano MD Prenyltransferase Inhibitors: Treating Human Ailments from Cancer to Parasitic Infections. Med. Chem. Commun 2013, 4, 476–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (414).Nallan L; Bauer KD; Bendale P; Rivas K; Yokoyama K; Hornéy CP; Pendyala PR; Floyd D; Lombardo LJ; Williams DK; et al. Protein Farnesyltransferase Inhibitors Exhibit Potent Antimalarial Activity. J. Med. Chem 2005, 48, 3704–3713. [DOI] [PubMed] [Google Scholar]
- (415).Suazo KF; Schaber C; Palsuledesai CC; Odom John AR; Distefano MD Global Proteomic Analysis of Prenylated Proteins in Plasmodium Falciparum Using an Alkyne-Modified Isoprenoid Analogue. Sci. Rep 2016, 6, 38615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (416).Mathews ES; Jezewski AJ; Odom John AR Protein Prenylation and Hsp40 in Thermotolerance of Plasmodium Falciparum Malaria Parasites. bioRxiv 2019, 842468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (417).Gisselberg JE; Zhang L; Elias JE; Yeh E The Prenylated Proteome of Plasmodium Falciparum Reveals Pathogen-Specific Prenylation Activity and Drug Mechanism-of-Action. Mol. Cell. Proteomics 2017, 16, S54–S64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (418).Storck EM; Morales-Sanfrutos J; Serwa RA; Panyain N; Lanyon-Hogg T; Tolmachova T; Ventimiglia LN; Martin-Serrano J; Seabra MC; Wojciak-Stothard B; et al. Dual Chemical Probes Enable Quantitative System-Wide Analysis of Protein Prenylation and Prenylation Dynamics. Nat. Chem 2019, 11, 552–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (419).Seabra MC; Brown MS; Goldstein JL Retinal Degeneration in Choroideremia: Deficiency of Rab Geranylgeranyl Transferase. Science 1993, 259, 377 LP–381. [DOI] [PubMed] [Google Scholar]
- (420).Suazo KF; Jeong A; Ahmadi M; Brown C; Qu W; Li L; Distefano MD Metabolic Labeling with an Alkyne Probe Reveals Similarities and Differences in the Prenylomes of Several Brain-Derived Cell Lines and Primary Cells. Sci. Rep 2021, 11, 4367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (421).Qu W; Suazo KF; Liu W; Cheng S; Jeong A; Hottman D; Yuan L-L; Distefano MD; Li L Neuronal Protein Farnesylation Regulates Hippocampal Synaptic Plasticity and Cognitive Function. Mol. Neurobiol 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (422).Palsuledesai CC; Ochocki JD; Markowski TW; Distefano MD A Combination of Metabolic Labeling and 2D-DIGE Analysis in Response to a Farnesyltransferase Inhibitor Facilitates the Discovery of New Prenylated Proteins. Mol. Biosyst 2014, 10, 1094–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (423).Berg TJ; Gastonguay AJ; Lorimer EL; Kuhnmuench JR; Li R; Fields AP; Williams CL Splice Variants of SmgGDS Control Small GTPase Prenylation and Membrane Localization. J. Biol. Chem 2010, 285, 35255–35266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (424).Schuld NJ; Vervacke JS; Lorimer EL; Simon NC; Hauser AD; Barbieri JT; Distefano MD; Williams CL The Chaperone Protein SmgGDS Interacts with Small GTPases Entering the Prenylation Pathway by Recognizing the Last Amino Acid in the CAAX Motif. J. Biol. Chem 2014, 289, 6862–6876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (425).Brandt AC; McNally L; Lorimer EL; Unger B; Koehn OJ; Suazo KF; Rein L; Szabo A; Tsaih S-W; Distefano MD; et al. Splice Switching an Oncogenic Ratio of SmgGDS Isoforms as a Strategy to Diminish Malignancy. Proc. Natl. Acad. Sci 2020, 117, 3627 LP–3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (426).ten Klooster JP; Hordijk PL Targeting and Localized Signalling by Small GTPases. Biol. Cell 2007, 99, 1–12. [DOI] [PubMed] [Google Scholar]
- (427).Akula MK; Ibrahim MX; Ivarsson EG; Khan OM; Kumar IT; Erlandsson M; Karlsson C; Xu X; Brisslert M; Brakebusch C; et al. Protein Prenylation Restrains Innate Immunity by Inhibiting Rac1 Effector Interactions. Nat. Commun 2019, 10, 3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (428).Kang R; Wan J; Arstikaitis P; Takahashi H; Huang K; Bailey AO; Thompson JX; Roth AF; Drisdel RC; Mastro R; et al. Neural Palmitoyl-Proteomics Reveals Dynamic Synaptic Palmitoylation. Nature 2008, 456, 904–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (429).Nishimura A; Linder ME Identification of a Novel Prenyl and Palmitoyl Modification at the CaaX Motif of Cdc42 That Regulates RhoGDI Binding. Mol. Cell. Biol 2013, 33, 1417 LP–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (430).Wilkins JA; Kaasik K; Chalkley RJ; Burlingame AL Characterization of Prenylated C-Terminal Peptides Using a Thiopropyl-Based Capture Technique and LC-MS/MS. Mol. Cell. Proteomics 2020, 19, 1005 LP–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (431).Palsuledesai CC; Ochocki JD; Kuhns MM; Wang Y-C; Warmka JK; Chernick DS; Wattenberg EV; Li L; Arriaga EA; Distefano MD Metabolic Labeling with an Alkyne-Modified Isoprenoid Analog Facilitates Imaging and Quantification of the Prenylome in Cells. ACS Chem. Biol 2016, 11, 2820–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (432).Diaz-Rodriguez V; Hsu E-T; Ganusova E; Werst ER; Becker JM; Hrycyna CA; Distefano MD A-Factor Analogues Containing Alkyne- and Azide-Functionalized Isoprenoids Are Efficiently Enzymatically Processed and Retain Wild-Type Bioactivity. Bioconjug. Chem 2018, 29, 316–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (433).Gaebler A; Penno A; Kuerschner L; Thiele C A Highly Sensitive Protocol for Microscopy of Alkyne Lipids and Fluorescently Tagged or Immunostained Proteins. J. Lipid Res 2016, 57, 1934–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (434).Scott-Solomon E; Kuruvilla R Prenylation of Axonally Translated Rac1 Controls NGF-Dependent Axon Growth. Dev. Cell 2020, 53, 691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (435).Shirakawa R; Goto-Ito S; Goto K; Wakayama S; Kubo H; Sakata N; Trinh DA; Yamagata A; Sato Y; Masumoto H; et al. A SNARE Geranylgeranyltransferase Essential for the Organization of the Golgi Apparatus. EMBO J. e104120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (436).Moudgil DK; Westcott N; Famulski JK; Patel K; Macdonald D; Hang H; Chan GKT A Novel Role of Farnesylation in Targeting a Mitotic Checkpoint Protein, Human Spindly, to Kinetochores. J. Cell Biol 2015, 208, 881–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (437).Silva PMA; Delgado ML; Ribeiro N; Florindo C; Tavares ÁA; Ribeiro D; Lopes C; do Amaral B; Bousbaa H; Monteiro LS Spindly and Bub3 Expression in Oral Cancer: Prognostic and Therapeutic Implications. Oral Dis 2019, 25, 1291–1301. [DOI] [PubMed] [Google Scholar]
- (438).Ivanov SS; Charron G; Hang HC; Roy CR Lipidation by the Host Prenyltransferase Machinery Facilitates Membrane Localization of Legionella Pneumophila Effector Proteins. J. Biol. Chem 2010, 285, 34686–34698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (439).Reinicke AT; Hutchinson JL; Magee AI; Mastroeni P; Trowsdale J; Kelly AP A Salmonella Typhimurium Effector Protein SifA Is Modified by Host Cell Prenylation and S-Acylation Machinery. J. Biol. Chem 2005, 280, 14620–14627. [DOI] [PubMed] [Google Scholar]
- (440).DeMar JC; Anderson RE Identification and Quantitation of the Fatty Acids Composing the CoA Ester Pool of Bovine Retina, Heart, and Liver. J. Biol. Chem 1997, 272, 31362–31368. [DOI] [PubMed] [Google Scholar]
- (441).Fujimoto T; Stroud E; Whatley RE; Prescott SM; Muszbek L; Laposata M; McEver RP P-Selectin Is Acylated with Palmitic Acid and Stearic Acid at Cysteine 766 through a Thioester Linkage. J. Biol. Chem 1993, 268, 11394–11400. [PubMed] [Google Scholar]
- (442).Zeng F-Y; Kaphalia BS; Ansari GAS; Weigel PH Fatty Acylation of the Rat Asialoglycoprotein Receptor: The Three Subunits From Active Receptors Contain Covalently Bound Palmitate and Stearate. J. Biol. Chem 1995, 270, 21382–21387. [DOI] [PubMed] [Google Scholar]
- (443).Liang X; Lu Y; Neubert TA; Resh MD Mass Spectrometric Analysis of GAP-43/Neuromodulin Reveals the Presence of a Variety of Fatty Acylated Species. J. Biol. Chem 2002, 277, 33032–33040. [DOI] [PubMed] [Google Scholar]
- (444).Kordyukova LV; Serebryakova MV; Baratova LA; Veit M Site-Specific Attachment of Palmitate or Stearate to Cytoplasmic versus Transmembrane Cysteines Is a Common Feature of Viral Spike Proteins. Virology 2010, 398, 49–56. [DOI] [PubMed] [Google Scholar]
- (445).Wang M; Ludwig K; Böttcher C; Veit M The Role of Stearate Attachment to the Hemagglutinin-Esterase-Fusion Glycoprotein HEF of Influenza C Virus. Cell. Microbiol 2016, 18, 692–704. [DOI] [PubMed] [Google Scholar]
- (446).Alvarez E; Gironès N; Davis RJ Inhibition of the Receptor-Mediated Endocytosis of Diferric Transferrin Is Associated with the Covalent Modification of the Transferrin Receptor with Palmitic Acid. J. Biol. Chem 1990, 265, 16644–16655. [PubMed] [Google Scholar]
- (447).Senyilmaz D; Virtue S; Xu X; Tan CY; Griffin JL; Miller AK; Vidal-Puig A; Teleman AA Regulation of Mitochondrial Morphology and Function by Stearoylation of TFR1. Nature 2015, 525, 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (448).Senyilmaz-Tiebe D; Pfaff DH; Virtue S; Schwarz KV; Fleming T; Altamura S; Muckenthaler MU; Okun JG; Vidal-Puig A; Nawroth P; et al. Dietary Stearic Acid Regulates Mitochondria in Vivo in Humans. Nat. Commun 2018, 9, 3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (449).Chen B; Niu J; Kreuzer J; Zheng B; Jarugumilli GK; Haas W; Wu X Auto-Fatty Acylation of Transcription Factor RFX3 Regulates Ciliogenesis. Proc. Natl. Acad. Sci 2018, 115, E8403 LP–E8412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (450).Liu W; Zhou Y; Peng T; Zhou P; Ding X; Li Z; Zhong H; Xu Y; Chen S; Hang HC; et al. Nε-Fatty Acylation of Multiple Membrane-Associated Proteins by Shigella IcsB Effector to Modulate Host Function. Nat. Microbiol 2018, 3, 996–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (451).Zhou Y; Huang C; Yin L; Wan M; Wang X; Li L; Liu Y; Wang Z; Fu P; Zhang N; et al. Nε-Fatty Acylation of Rho GTPases by a MARTX Toxin Effector. Science 2017, 358, 528 LP–531. [DOI] [PubMed] [Google Scholar]
- (452).Parisi LR; Li N; Atilla-Gokcumen GE Very Long Chain Fatty Acids Are Functionally Involved in Necroptosis. Cell Chem. Biol 2017, 24, 1445–1454.e8. [DOI] [PubMed] [Google Scholar]
- (453).Parisi LR; Sowlati-Hashjin S; Berhane IA; Galster SL; Carter KA; Lovell JF; Chemler SR; Karttunen M; Atilla-Gokcumen GE Membrane Disruption by Very Long Chain Fatty Acids during Necroptosis. ACS Chem. Biol 2019, 14, 2286–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (454).Rivera-Chávez F; Zhang LF; Faber F; Lopez CA; Byndloss MX; Olsan EE; Xu G; Velazquez EM; Lebrilla CB; Winter SE; et al. Depletion of Butyrate-Producing Clostridia from the Gut Microbiota Drives an Aerobic Luminal Expansion of Salmonella. Cell Host Microbe 2016, 19, 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (455).Zhang ZJ; Pedicord VA; Peng T; Hang HC Site-Specific Acylation of a Bacterial Virulence Regulator Attenuates Infection. Nat. Chem. Biol 2020, 16, 95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (456).Ali I; Conrad RJ; Verdin E; Ott M Lysine Acetylation Goes Global: From Epigenetics to Metabolism and Therapeutics. Chem. Rev 2018, 118, 1216–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (457).Yang C; Mi J; Feng Y; Ngo L; Gao T; Yan L; Zheng YG Labeling Lysine Acetyltransferase Substrates with Engineered Enzymes and Functionalized Cofactor Surrogates. J. Am. Chem. Soc 2013, 135, 7791–7794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (458).Yu-Ying Y; Markus G; Howard HC Identification of Lysine Acetyltransferase P300 Substrates Using 4-Pentynoyl-Coenzyme A and Bioorthogonal Proteomics. Bioorg. Med. Chem. Lett 2011, 21, 4976–4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (459).Han Z; Chou C; Yang X; Bartlett MG; Zheng YG Profiling Cellular Substrates of Lysine Acetyltransferases GCN5 and P300 with Orthogonal Labeling and Click Chemistry. ACS Chem. Biol 2017, 12, 1547–1555. [DOI] [PubMed] [Google Scholar]
- (460).Heppner KM; Tong J; Kirchner H; Nass R; Tschöp MH The Ghrelin O-Acyltransferase–Ghrelin System: A Novel Regulator of Glucose Metabolism. Curr. Opin. Endocrinol. Diabetes Obes 2011, 18. [DOI] [PubMed] [Google Scholar]
- (461).Hougland JL Ghrelin Octanoylation by Ghrelin O-Acyltransferase: Unique Protein Biochemistry Underlying Metabolic Signaling. Biochem. Soc. Trans 2019, 47, 169–178. [DOI] [PubMed] [Google Scholar]
- (462).Cleverdon ER; McGovern-Gooch KR; Hougland JL The Octanoylated Energy Regulating Hormone Ghrelin: An Expanded View of Ghrelin’s Biological Interactions and Avenues for Controlling Ghrelin Signaling. Mol. Membr. Biol 2016, 33, 111–124. [DOI] [PubMed] [Google Scholar]
- (463).Garner AL; Janda KD Cat-ELCCA: A Robust Method To Monitor the Fatty Acid Acyltransferase Activity of Ghrelin O-Acyltransferase (GOAT). Angew. Chemie Int. Ed 2010, 49, 9630–9634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (464).Garner AL; Janda KD A Small Molecule Antagonist of Ghrelin O-Acyltransferase (GOAT). Chem. Commun 2011, 47, 7512–7514. [DOI] [PubMed] [Google Scholar]
- (465).Ibrahim Abdalla MM Ghrelin - Physiological Functions and Regulation. Eur. Endocrinol 2015, 11, 90–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (466).Eubanks LM; Stowe GN; De Lamo Marin S; Mayorov AV; Hixon MS; Janda KD Identification of A2 Macroglobulin as a Major Serum Ghrelin Esterase. Angew. Chemie Int. Ed 2011, 50, 10699–10702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (467).Liang X; Nazarian A; Erdjument-Bromage H; Bornmann W; Tempst P; Resh MD Heterogeneous Fatty Acylation of Src Family Kinases with Polyunsaturated Fatty Acids Regulates Raft Localization and Signal Transduction. J. Biol. Chem 2001, 276, 30987–30994. [DOI] [PubMed] [Google Scholar]
- (468).Takada R; Satomi Y; Kurata T; Ueno N; Norioka S; Kondoh H; Takao T; Takada S Monounsaturated Fatty Acid Modification of Wnt Protein: Its Role in Wnt Secretion. Dev. Cell 2006, 11, 791–801. [DOI] [PubMed] [Google Scholar]
- (469).Flock MR; Kris-Etherton PM Diverse Physiological Effects of Long-Chain Saturated Fatty Acids: Implications for Cardiovascular Disease. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16. [DOI] [PubMed] [Google Scholar]
- (470).Mikels AJ; Nusse R Wnts as Ligands: Processing, Secretion and Reception. Oncogene 2006, 25, 7461–7468. [DOI] [PubMed] [Google Scholar]
- (471).Ching W; Hang HC; Nusse R Lipid-Independent Secretion of a Drosophila Wnt Protein. J. Biol. Chem 2008, 283, 17092–17098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (472).Miranda M; Galli LM; Enriquez M; Szabo LA; Gao X; Hannoush RN; Burrus LW Identification of the WNT1 Residues Required for Palmitoylation by Porcupine. FEBS Lett 2014, 588, 4815–4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (473).Tuladhar R; Yarravarapu N; Ma Y; Zhang C; Herbert J; Kim J; Chen C; Lum L Stereoselective Fatty Acylation Is Essential for the Release of Lipidated WNT Proteins from the Acyltransferase Porcupine (PORCN). J. Biol. Chem 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (474).Schey KL; Gutierrez DB; Wang Z; Wei J; Grey AC Novel Fatty Acid Acylation of Lens Integral Membrane Protein Aquaporin-0. Biochemistry 2010, 49, 9858–9865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (475).Montigny C; Decottignies P; Le Maréchal P; Capy P; Bublitz M; Olesen C; Møller JV; Nissen P; le Maire M S-Palmitoylation and S-Oleoylation of Rabbit and Pig Sarcolipin. J. Biol. Chem 2014, 289, 33850–33861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (476).Gaebler A; Milan R; Straub L; Hoelper D; Kuerschner L; Thiele C Alkyne Lipids as Substrates for Click Chemistry-Based in Vitro Enzymatic Assays. J. Lipid Res 2013, 54, 2282–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (477).Thinon E; Percher A; Hang HC Bioorthogonal Chemical Reporters for Monitoring Unsaturated Fatty-Acylated Proteins. ChemBioChem 2016, 17, 1800–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (478).Muszbek L; Laposata M Covalent Modification of Proteins by Arachidonate and Eicosapentaenoate in Platelets. J. Biol. Chem 1993, 268, 18243–18248. [PubMed] [Google Scholar]
- (479).Hallak H; Muszbek L; Laposata M; Belmonte E; Brass LF; Manning DR Covalent Binding of Arachidonate to G Protein Alpha Subunits of Human Platelets. J. Biol. Chem 1994, 269, 4713–4716. [PubMed] [Google Scholar]
- (480).Puca AA; Andrew P; Novelli V; Anselmi CV; Somalvico F; Cirillo NA; Chatgilialoglu C; Ferreri C Fatty Acid Profile of Erythrocyte Membranes As Possible Biomarker of Longevity. Rejuvenation Res 2007, 11, 63–72. [DOI] [PubMed] [Google Scholar]
- (481).Refsgaard HHF; Tsai L; Stadtman ER Modifications of Proteins by Polyunsaturated Fatty Acid Peroxidation Products. Proc. Natl. Acad. Sci 2000, 97, 611 LP–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (482).Beavers WN; Serwa R; Shimozu Y; Tallman KA; Vaught M; Dalvie ED; Marnett LJ; Porter NA ω-Alkynyl Lipid Surrogates for Polyunsaturated Fatty Acids: Free Radical and Enzymatic Oxidations. J. Am. Chem. Soc 2014, 136, 11529–11539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (483).Beavers WN; Rose KL; Galligan JJ; Mitchener MM; Rouzer CA; Tallman KA; Lamberson CR; Wang X; Hill S; Ivanova PT; et al. Protein Modification by Endogenously Generated Lipid Electrophiles: Mitochondria as the Source and Target. ACS Chem. Biol 2017, 12, 2062–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (484).Isobe Y; Kawashima Y; Ishihara T; Watanabe K; Ohara O; Arita M Identification of Protein Targets of 12/15-Lipoxygenase-Derived Lipid Electrophiles in Mouse Peritoneal Macrophages Using Omega-Alkynyl Fatty Acid. ACS Chem. Biol 2018, 13, 887–893. [DOI] [PubMed] [Google Scholar]
- (485).Robichaud PP; Poirier SJ; Boudreau LH; Doiron JA; Barnett DA; Boilard E; Surette ME On the Cellular Metabolism of the Click Chemistry Probe 19-Alkyne Arachidonic Acid. J. Lipid Res 2016, 57, 1821–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (486).Schopfer FJ; Cipollina C; Freeman BA Formation and Signaling Actions of Electrophilic Lipids. Chem. Rev 2011, 111, 5997–6021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (487).Vila A; Tallman KA; Jacobs AT; Liebler DC; Porter NA; Marnett LJ Identification of Protein Targets of 4-Hydroxynonenal Using Click Chemistry for Ex Vivo Biotinylation of Azido and Alkynyl Derivatives. Chem. Res. Toxicol 2008, 21, 432–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (488).Galligan JJ; Rose KL; Beavers WN; Hill S; Tallman KA; Tansey WP; Marnett LJ Stable Histone Adduction by 4-Oxo-2-Nonenal: A Potential Link between Oxidative Stress and Epigenetics. J. Am. Chem. Soc 2014, 136, 11864–11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (489).Sun R; Fu L; Liu K; Tian C; Yang Y; Tallman KA; Porter NA; Liebler DC; Yang J Chemoproteomics Reveals Chemical Diversity and Dynamics of 4-Oxo-2-Nonenal Modifications in Cells. Mol. & Cell. Proteomics 2017, 16, 1789 LP–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (490).Long MJC; Parvez S; Zhao Y; Surya SL; Wang Y; Zhang S; Aye Y Akt3 Is a Privileged First Responder in Isozyme-Specific Electrophile Response. Nat. Chem. Biol 2017, 13, 333–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (491).Long MJC; Rogg C; Aye Y An Oculus to Profile and Probe Target Engagement In Vivo: How T-REX Was Born and Its Evolution into G-REX. Acc. Chem. Res 2020. [DOI] [PubMed] [Google Scholar]
- (492).Cui Y; Li X; Lin J; Hao Q; Li XD Histone Ketoamide Adduction by 4-Oxo-2-Nonenal Is a Reversible Posttranslational Modification Regulated by Sirt2. ACS Chem. Biol 2017, 12, 47–51. [DOI] [PubMed] [Google Scholar]
- (493).Ullery JC; Marnett LJ Protein Modification by Oxidized Phospholipids and Hydrolytically Released Lipid Electrophiles: Investigating Cellular Responses. Biochim. Biophys. Acta - Biomembr 2012, 1818, 2424–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (494).Wright MH; Sieber SA Chemical Proteomics Approaches for Identifying the Cellular Targets of Natural Products. Nat. Prod. Rep 2016, 33, 681–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (495).Mann RK; Beachy PA Novel Lipid Modifications of Secreted Protein Signals. Annu. Rev. Biochem 2004, 73, 891–923. [DOI] [PubMed] [Google Scholar]
- (496).Heretsch P; Tzagkaroulaki L; Giannis A Modulators of the Hedgehog Signaling Pathway. Bioorg. Med. Chem 2010, 18, 6613–6624. [DOI] [PubMed] [Google Scholar]
- (497).Sasai N; Toriyama M; Kondo T Hedgehog Signal and Genetic Disorders. Front. Genet 2019, 10, 1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (498).Rubin LL; de Sauvage FJ Targeting the Hedgehog Pathway in Cancer. Nat. Rev. Drug Discov 2006, 5, 1026–1033. [DOI] [PubMed] [Google Scholar]
- (499).Hall TMT; Porter JA; Young KE; Koonin EV; Beachy PA; Leahy DJ Crystal Structure of a Hedgehog Autoprocessing Domain: Homology between Hedgehog and Self-Splicing Proteins. Cell 1997, 91, 85–97. [DOI] [PubMed] [Google Scholar]
- (500).Heal WP; Jovanovic B; Bessin S; Wright MH; Magee AI; Tate EW Bioorthogonal Chemical Tagging of Protein Cholesterylation in Living Cells. Chem. Commun 2011, 47, 4081–4083. [DOI] [PubMed] [Google Scholar]
- (501).Ciepla P; Konitsiotis AD; Serwa RA; Masumoto N; Leong WP; Dallman MJ; Magee AI; Tate EW New Chemical Probes Targeting Cholesterylation of Sonic Hedgehog in Human Cells and Zebrafish. Chem. Sci 2014, 5, 4249–4259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (502).Porter JA; Young KE; Beachy PA Cholesterol Modification of Hedgehog Signaling Proteins in Animal Development. Science 1996, 274, 255 LP–259. [DOI] [PubMed] [Google Scholar]
- (503).Byrne EFX; Sircar R; Miller PS; Hedger G; Luchetti G; Nachtergaele S; Tully MD; Mydock-McGrane L; Covey DF; Rambo RP; et al. Structural Basis of Smoothened Regulation by Its Extracellular Domains. Nature 2016, 535, 517–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (504).Xiao X; Tang J-J; Peng C; Wang Y; Fu L; Qiu Z-P; Xiong Y; Yang L-F; Cui H-W; He X-L; et al. Cholesterol Modification of Smoothened Is Required for Hedgehog Signaling. Mol. Cell 2017, 66, 154–162.e10. [DOI] [PubMed] [Google Scholar]
- (505).Tukachinsky H; Kuzmickas RP; Jao CY; Liu J; Salic A Dispatched and Scube Mediate the Efficient Secretion of the Cholesterol-Modified Hedgehog Ligand. Cell Rep 2012, 2, 308–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (506).Hulce JJ; Cognetta AB; Niphakis MJ; Tully SE; Cravatt BF Proteome-Wide Mapping of Cholesterol-Interacting Proteins in Mammalian Cells. Nat. Methods 2013, 10, 259–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (507).Porter NA; Xu L; Pratt DA Reactive Sterol Electrophiles: Mechanisms of Formation and Reactions with Proteins and Amino Acid Nucleophiles. Chemistry. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (508).Windsor K; Genaro-Mattos TC; Kim H-YH; Liu W; Tallman KA; Miyamoto S; Korade Z; Porter NA Probing Lipid-Protein Adduction with Alkynyl Surrogates: Application to Smith-Lemli-Opitz Syndrome. J. Lipid Res 2013, 54, 2842–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (509).Tallman KA; Kim H-YH; Korade Z; Genaro-Mattos TC; Wages PA; Liu W; Porter NA Probes for Protein Adduction in Cholesterol Biosynthesis Disorders: Alkynyl Lanosterol as a Viable Sterol Precursor. Redox Biol 2017, 12, 182–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (510).Mayor S; Riezman H Sorting GPI-Anchored Proteins. Nat. Rev. Mol. Cell Biol 2004, 5, 110–120. [DOI] [PubMed] [Google Scholar]
- (511).Paulick MG; Bertozzi CR The Glycosylphosphatidylinositol Anchor: A Complex Membrane-Anchoring Structure for Proteins. Biochemistry 2008, 47, 6991–7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (512).Poisson G; Chauve C; Chen X; Bergeron A FragAnchor: A Large-Scale Predictor of Glycosylphosphatidylinositol Anchors in Eukaryote Protein Sequences by Qualitative Scoring. Genomics Proteomics Bioinforma 2007, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (513).Pierleoni A; Martelli PL; Casadio R PredGPI: A GPI-Anchor Predictor. BMC Bioinformatics 2008, 9, 392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (514).Taylor DR; Hooper NM GPI-Anchored Proteins in Health and Disease BT - Post-Translational Modifications in Health and Disease; Vidal CJ, Ed.; Springer New York: New York, NY, 2011; pp 39–55. [Google Scholar]
- (515).Paul KS; Jiang D; Morita YS; Englund PT Fatty Acid Synthesis in African Trypanosomes: A Solution to the Myristate Mystery. Trends Parasitol 2001, 17, 381–387. [DOI] [PubMed] [Google Scholar]
- (516).Ferguson MAJ; Hart GW; Kinoshita T Glycosylphosphatidylinositol Anchors, 3rd ed.; Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY), 2015. [Google Scholar]
- (517).Vainauskas S; Cortes LK; Taron CH In Vivo Incorporation of an Azide-Labeled Sugar Analog to Detect Mammalian Glycosylphosphatidylinositol Molecules Isolated from the Cell Surface. Carbohydr. Res 2012, 362, 62–69. [DOI] [PubMed] [Google Scholar]
- (518).Lu L; Gao J; Guo Z Labeling Cell Surface GPIs and GPI-Anchored Proteins through Metabolic Engineering with Artificial Inositol Derivatives. Angew. Chemie Int. Ed 2015, 54, 9679–9682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (519).Mateos-Gil P; Letschert S; Doose S; Sauer M Super-Resolution Imaging of Plasma Membrane Proteins with Click Chemistry. Front. Cell Dev. Biol 2016, 4, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (520).Elortza F; Mohammed S; Bunkenborg J; Foster LJ; Nuhse TS; Brodbeck U; Peck SC; Jensen ON Modification-Specific Proteomics of Plasma Membrane Proteins: Identification and Characterization of Glycosylphosphatidylinositol-Anchored Proteins Released upon Phospholipase D Treatment. J Proteome Res 2006, 5. [DOI] [PubMed] [Google Scholar]
- (521).Cortes LK; Vainauskas S; Dai N; McClung CM; Shah M; Benner JS; Corrêa IR Jr.; VerBerkmoes NC; Taron CH Proteomic Identification of Mammalian Cell Surface Derived Glycosylphosphatidylinositol-Anchored Proteins through Selective Glycan Enrichment. Proteomics 2014, 14, 2471–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (522).Swarts BM; Guo Z Chemical Synthesis and Functionalization of Clickable Glycosylphosphatidylinositol Anchors. Chem. Sci 2011, 2, 2342–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (523).Ding N; Li X; Chinoy ZS; Boons G-J Synthesis of a Glycosylphosphatidylinositol Anchor Derived from Leishmania Donovani That Can Be Functionalized by Cu-Catalyzed Azide-Alkyne Cycloadditions. Org. Lett 2017, 19, 3827–3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (524).Roque ACA; Lowe CR; Taipa MÂ Antibodies and Genetically Engineered Related Molecules: Production and Purification. Biotechnol. Prog 2004, 20, 639–654. [DOI] [PubMed] [Google Scholar]
- (525).Young TS; Schultz PG Beyond the Canonical 20 Amino Acids: Expanding the Genetic Lexicon. J. Biol. Chem 2010, 285, 11039–11044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (526).Subramanian T; Pais JE; Liu S; Troutman JM; Suzuki Y; Leela Subramanian K; Fierke CA; Andres DA; Spielmann HP Farnesyl Diphosphate Analogues with Aryl Moieties Are Efficient Alternate Substrates for Protein Farnesyltransferase. Biochemistry 2012, 51, 8307–8319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (527).Dozier JK; Khatwani SL; Wollack JW; Wang Y-C; Schmidt-Dannert C; Distefano MD Engineering Protein Farnesyltransferase for Enzymatic Protein Labeling Applications. Bioconjug. Chem 2014, 25, 1203–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (528).Zahn TJ; Whitney J; Weinbaum C; Gibbs RA Synthesis and Evaluation of GGPP Geometric Isomers: Divergent Substrate Specificities of FTase and GGTase I. Bioorg. Med. Chem. Lett 2001, 11, 1605–1608. [DOI] [PubMed] [Google Scholar]
- (529).Restituyo JA; Comstock LR; Petersen SG; Stringfellow T; Rajski SR Conversion of Aryl Azides to O-Alkyl Imidates via Modified Staudinger Ligation. Org. Lett 2003, 5, 4357–4360. [DOI] [PubMed] [Google Scholar]
- (530).Xu J; DeGraw AJ; Duckworth BP; Lenevich S; Tann C-M; Jenson EC; Gruber SJ; Barany G; Distefano MD Synthesis and Reactivity of 6,7-Dihydrogeranylazides: Reagents for Primary Azide Incorporation into Peptides and Subsequent Staudinger Ligation. Chem. Biol. Drug Des 2006, 68, 85–96. [DOI] [PubMed] [Google Scholar]
- (531).Duckworth BP; Xu J; Taton TA; Guo A; Distefano MD Site-Specific, Covalent Attachment of Proteins to a Solid Surface. Bioconjug. Chem 2006, 17, 967–974. [DOI] [PubMed] [Google Scholar]
- (532).Speers AE; Cravatt BF Profiling Enzyme Activities In Vivo Using Click Chemistry Methods. Chem. Biol 2004, 11, 535–546. [DOI] [PubMed] [Google Scholar]
- (533).Gauchet C; Labadie GR; Poulter CD Regio- and Chemoselective Covalent Immobilization of Proteins through Unnatural Amino Acids. J. Am. Chem. Soc 2006, 128, 9274–9275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (534).Duckworth BP; Zhang Z; Hosokawa A; Distefano MD Selective Labeling of Proteins by Using Protein Farnesyltransferase. ChemBioChem 2007, 8, 98–105. [DOI] [PubMed] [Google Scholar]
- (535).Duckworth BP; Chen Y; Wollack JW; Sham Y; Mueller JD; Taton TA; Distefano MD A Universal Method for the Preparation of Covalent Protein–DNA Conjugates for Use in Creating Protein Nanostructures. Angew. Chemie Int. Ed 2007, 46, 8819–8822. [DOI] [PubMed] [Google Scholar]
- (536).Nguyen UTT; Cramer J; Gomis J; Reents R; Gutierrez‐Rodriguez M; Goody RS; Alexandrov K; Waldmann H Exploiting the Substrate Tolerance of Farnesyltransferase for Site‐Selective Protein Derivatization. ChemBioChem 2007, 8, 408–423. [DOI] [PubMed] [Google Scholar]
- (537).Wollack JW; Silverman JM; Petzold CJ; Mougous JD; Distefano MD A Minimalist Substrate for Enzymatic Peptide and Protein Conjugation. Chembiochem 2009, 10, 2934–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (538).Rashidian M; Dozier JK; Lenevich S; Distefano MD Selective Labeling of Polypeptides Using Protein Farnesyltransferase via Rapid Oxime Ligation. Chem. Commun 2010, 46, 8998–9000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (539).Weinrich D; Lin P; Jonkheijm P; Nguyen UTT; Schröder H; Niemeyer CM; Alexandrov K; Goody R; Waldmann H Oriented Immobilization of Farnesylated Proteins by the Thiol‐Ene Reaction. Angew. Chemie Int. Ed 2010, 49, 1252–1257. [DOI] [PubMed] [Google Scholar]
- (540).Wang Y; Kilic O; Csizmar CM; Ashok S; Hougland JL; Distefano MD; Wagner CR Engineering Reversible Cell–Cell Interactions Using Enzymatically Lipidated Chemically Self-Assembled Nanorings. Chem. Sci 2021, 12, 331–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (541).Khatwani SL; Kang JS; Mullen DG; Hast MA; Beese LS; Distefano MD; Taton TA Covalent Protein-Oligonucleotide Conjugates by Copper-Free Click Reaction. Bioorg. Med. Chem 2012, 20, 4532–4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (542).Rashidian M; Kumarapperuma SC; Gabrielse K; Fegan A; Wagner CR; Distefano MD Simultaneous Dual Protein Labeling Using a Triorthogonal Reagent. J. Am. Chem. Soc 2013, 135, 16388–16396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (543).Mahmoodi MM; Rashidian M; Zhang Y; Distefano MD Application of Meta- and Para-Phenylenediamine as Enhanced Oxime Ligation Catalysts for Protein Labeling, PEGylation, Immobilization, and Release. Curr. Protoc. Protein Sci 2015, 79, 15.4.1–15.4.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (544).Tolstyka ZP; Richardson W; Bat E; Stevens CJ; Parra DP; Dozier JK; Distefano MD; Dunn B; Maynard HD Chemoselective Immobilization of Proteins by Microcontact Printing and Bioorthogonal Click Reactions. Chembiochem 2013, 14, 2464–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (545).Seo J; Lee S; Poulter CD Regioselective Covalent Immobilization of Recombinant Antibody-Binding Proteins A, G, and L for Construction of Antibody Arrays. J. Am. Chem. Soc 2013, 135, 8973–8980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (546).Seo J; Poulter CD Sandwich Antibody Arrays Using Recombinant Antibody-Binding Protein L. Langmuir 2014, 30, 6629–6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (547).Choi S; Seo J; Bohaty RFH; Poulter CD Regio- and Chemoselective Immobilization of Proteins on Gold Surfaces. Bioconjug. Chem 2014, 25, 269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (548).Wollack JW; Monson BJ; Dozier JK; Dalluge JJ; Poss K; Hilderbrand SA; Distefano MD Site-Specific Labeling of Proteins and Peptides with Trans-Cyclooctene Containing Handles Capable of Tetrazine Ligation. Chem. Biol. Drug Des 2014, 84, 140–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (549).Yeo JE; Wickramaratne S; Khatwani S; Wang Y-C; Vervacke J; Distefano MD; Tretyakova NY Synthesis of Site-Specific DNA–Protein Conjugates and Their Effects on DNA Replication. ACS Chem. Biol 2014, 9, 1860–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (550).Lee J; Choi H; Yun M; Kang Y; Jung J; Ryu Y; Kim TY; Cha Y; Cho H; Min J; et al. Enzymatic Prenylation and Oxime Ligation for the Synthesis of Stable and Homogeneous Protein–Drug Conjugates for Targeted Therapy. Angew. Chemie Int. Ed 2015, 54, 12020–12024. [DOI] [PubMed] [Google Scholar]
- (551).Lee BI; Park M-H; Byeon J-J; Shin S-H; Choi J; Park Y; Park Y-H; Chae J; Shin YG Quantification of an Antibody-Conjugated Drug in Fat Plasma by an Affinity Capture LC-MS/MS Method for a Novel Prenyl Transferase-Mediated Site-Specific Antibody-Drug Conjugate. Molecules 2020, 25, 1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (552).Zhang Y; Auger S; Schaefer JV; Plückthun A; Distefano MD Site-Selective Enzymatic Labeling of Designed Ankyrin Repeat Proteins Using Protein Farnesyltransferase BT - Bioconjugation: Methods and Protocols; Massa S, Devoogdt N, Eds.; Springer New York: New York, NY, 2019; pp 207–219. [DOI] [PubMed] [Google Scholar]
- (553).Ho SH; Tirrell DA Chemoenzymatic Labeling of Proteins for Imaging in Bacterial Cells. J. Am. Chem. Soc 2016, 138, 15098–15101. [DOI] [PubMed] [Google Scholar]
- (554).Ho SH; Tirrell DA N-Myristoyl Transferase (NMT)-Catalyzed Labeling of Bacterial Proteins for Imaging in Fixed and Live Cells BT - Enzyme-Mediated Ligation Methods; Nuijens T, Schmidt M, Eds.; Springer New York: New York, NY, 2019; pp 315–326. [DOI] [PubMed] [Google Scholar]
- (555).Kulkarni C; Lo M; Fraseur JG; Tirrell DA; Kinzer-Ursem TL Bioorthogonal Chemoenzymatic Functionalization of Calmodulin for Bioconjugation Applications. Bioconjug. Chem 2015, 26, 2153–2160. [DOI] [PubMed] [Google Scholar]
- (556).Fraseur JG; Kinzer-Ursem TL Next Generation Calmodulin Affinity Purification: Clickable Calmodulin Facilitates Improved Protein Purification. PLoS One 2018, 13, e0197120–e0197120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (557).Heal WP; Wickramasinghe SR; Leatherbarrow RJ; Tate EW N-Myristoyl Transferase-Mediated Protein Labelling in Vivo. Org. Biomol. Chem 2008, 6, 2308–2315. [DOI] [PubMed] [Google Scholar]
- (558).Heal WP; Wright MH; Thinon E; Tate EW Multifunctional Protein Labeling via Enzymatic N-Terminal Tagging and Elaboration by Click Chemistry. Nat. Protoc 2012, 7, 105–117. [DOI] [PubMed] [Google Scholar]
- (559).Kulkarni C; Kinzer‐Ursem TL; Tirrell DA Selective Functionalization of the Protein N Terminus with N‐Myristoyl Transferase for Bioconjugation in Cell Lysate. ChemBioChem 2013, 14, 1958–1962. [DOI] [PubMed] [Google Scholar]
- (560).Ejendal KFK; Fraseur JG; Kinzer-Ursem TL Protein Labeling and Bioconjugation Using N-Myristoyltransferase BT - Bioconjugation: Methods and Protocols; Massa S, Devoogdt N, Eds.; Springer New York: New York, NY, 2019; pp 149–165. [DOI] [PubMed] [Google Scholar]
- (561).Ho SH; Tirrell DA Enzymatic Labeling of Bacterial Proteins for Super-Resolution Imaging in Live Cells. ACS Cent. Sci 2019, 5, 1911–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (562).Kudirka RA; Barfield RM; McFarland JM; Drake PM; Carlson A; Bañas S; Zmolek W; Garofalo AW; Rabuka D Site-Specific Tandem Knoevenagel Condensation–Michael Addition To Generate Antibody–Drug Conjugates. ACS Med. Chem. Lett 2016, 7, 994–998. [DOI] [PMC free article] [PubMed] [Google Scholar]