

Abstract
Preclinical studies have demonstrated a complex cross-talk between Notch and estrogen signaling in ERα-positive breast cancer. Gamma-secretase inhibitors (GSIs) are investigational agents that block the cleavage and activation of Notch receptors. In animal models of endocrine-resistant breast cancer, combinations of tamoxifen and GSIs produce additive or synergistic efficacy, while decreasing the intestinal toxicity of GSIs. However, results of a clinical trial of a GSI-endocrine therapy combination in the metastatic setting have not been published to date, nor had the safety of such combinations been investigated with longer term treatment. We conducted a phase 1b dose escalation trial (NCT01149356) of GSI RO4929097 with exemestane in patients with ERα+, metastatic breast cancer (MBC).
Study objectives:
to determine the safety, tolerability and maximum tolerated dose (MTD) or recommended phase 2 dose (RP2D) of RO4929097 when administered in combination with exemestane in patients with estrogen receptor positive metastatic breast cancer.
Results:
We enrolled 15 patients with MBC. Of 14 evaluable patients, one had a partial response, 6 had stable disease and 7 progressive disease. Twenty % of patients had stable disease for ≥ 6 months. Common toxicities included nausea (73.3%), anorexia (60%), hyperglycemia (53.3%), hypophosphatemia (46.7%), fatigue (66.7%) and cough (33.0%). Grade 3 toxicities were uncommon, and included hypophosphatemia (13%) and rash (6.3%). Rash was the only DLT observed at 140 mg/d. Results suggest a possible recommended phase 2 dose of 90 mg/d. Ten patients with evaluable archival tissue showed expression of PKCα, which correlated with expression of Notch4. Mammospheres from a PKCα-expressing, endocrine-resistant T47D cell line were inhibited by a GSI-fulvestrant combination.
Conclusions:
Our data indicate that combinations including endocrine therapy and Notch inhibitors deserve further investigation in endocrine-resistant ERα-positive breast cancer.
Keywords: Endocrine-refractory metastatic breast cancer, Notch, gamma secretase inhibitor
Micro-abstract
We conducted a phase 1b trial of Notch inhibitor GSI RO4929097 with exemestane in 15 patients with ER+, metastatic breast cancer. The combination was well-tolerated, with clinical responses in 7/14 evaluable patients. Two patients had stable disease for ≥ 6 months. Our results suggest that combinations of Notch inhibitors and endocrine therapy deserve further investigation in endocrine-refractory breast cancer.
Introduction
The Notch signaling pathway is the object of intense clinical and pre-clinical investigation as a possible therapeutic target in oncology. This is due to the importance of Notch signaling in controlling multiple cell fate decisions in tumors (proliferation, survival, metabolism), in cancer stem cells (CSC) and in endothelial cells. Theoretically, inhibition of Notch in tumors where the pathway is active may produce growth inhibition, apoptosis, while simultaneously inhibiting CSC and angiogenesis. The first class of pharmacological Notch inhibitors to reach the clinic was γ-secretase inhibitors (GSIs). GSIs include different chemical classes of molecules that inhibit γ-secretase, an intramembranous multisubunit aspartyl protease that catalyzes the cleavage of multiple (>90) membrane proteins, including Notch receptors and the β-amyloid precursor in Alzheimer’s disease. A phase 3 clinical trial of GSI Semagacestat in Alzheimer’s disease failed due to unacceptable long-term toxicity upon chronic administration. GSIs have received significant interest in oncology over the past decade 1, 2, and while several of these agents were abandoned, two of them, PF-03084014 (nirogacestat) and AL101 are now in advanced clinical development 3. GSIs have significant activity in a variety of preclinical tumor models, including several breast cancer models 2, 4–6. Their primary dose-limiting toxicity in animals and humans is goblet cell metaplasia of the intestinal mucosa, which produces diarrhea and malabsorption. This toxicity is target-mediated, since canonical Notch signaling is necessary for proper cell fate determination in intestinal epithelial stem cells 7. Intermittent administration regimens decrease this toxicity, as does the combination of GSIs with corticosteroids 8 or tamoxifen 9.
We and others have demonstrated that Notch signaling cross-talks with estrogen in breast cancer models. In ERα+ breast cancer cells, estrogen inhibits the activation of Notch1 via ERα 10. Estrogen deprivation or treatment with Selective Estrogen Receptor Modulators (SERMS) results in Notch re-activation 10. Conversely, Notch1 is capable of activating ERα-dependent transcription in the absence of estrogen, by recruiting transcriptional co-activators and protein kinase IKKα to estrogen-responsive elements in the genome 11. Interestingly, in endothelial cells estrogen activates Notch1 via ERβ, suggesting that Notch participates in the pro-angiogenic activity of estrogen 12. Consistent with these findings, expression of the cleaved, intracellular form of Notch1 (Notch1IC) in ERα+ breast cancer cells produces estrogen-independent and tamoxifen resistant growth, inducing a “lumino-basal” phenotype that resembles basal-like breast cancer cells, while retaining ERα expression 13. PKCα, a protein kinase commonly expressed in endocrine-resistant breast cancer and marker of poor prognosis in ERα positive and negative breast cancer, induces endocrine resistance at least in part by increasing expression of Notch4. Like Notch1IC, Notch4IC is also sufficient to cause estrogen-independent and tamoxifen-resistant growth in ERα-positive breast cancer cells 9. More recently, Gelsomino et al. showed that mutations in the ESR1 gene encoding ERα associated with endocrine resistance promote a CSC phenotype through activation of Notch4 14. A pilot clinical trial of short term GSI MK0752 in combination with letrozole or tamoxifen in early stage ERα+ breast cancer patients showed that this regimen was safe and produced molecular changes in tumors consistent with the inhibition of proliferation and “stemness” and the induction of apoptosis 15. Based on these findings, we designed a phase 1b study of GSI-exemestane combination in the metastatic setting.
Materials and Methods
Study objectives:
The primary objective of was to determine the safety, tolerability and maximum tolerated dose (MTD) or recommended phase 2 dose (RP2D) of RO4929097 when administered in combination with exemestane in patients with estrogen receptor positive metastatic breast cancer. Secondary objectives were:
To determine the Overall Tumor Response Rates (ORR), defined as the proportion of patients who achieved Complete Response (CR) or Partial Response (PR) confirmed by a second evaluation 6 weeks later. Response and progression were measured by Solid Tumor Response Criteria (RECIST). Patients received a dedicated contrasted CT scan of the chest, abdomen and pelvis and a bones scan at time of enrollment and 6 weeks later. If no evidence of progression was observed at 6 weeks, patients received a CT scan and bone scan every 9 weeks until progression. Response in patients with bone lesions only was categorized as not assessable unless there is progressive disease, in which case response was considered as progressive disease.
To determine the Overall Survival (number of deaths at final analysis).
To perform a pharmacokinetics evaluation of RO4929097 when administered in combination with exemestane.
To evaluate candidate biomarkers PKCα and Notch4 in available pre-treatment surgical specimens. Additionally, we conducted preclinical experiments to evaluate the in vitro activity of RO4929097 in mammosphere generation assays in a Notch4-dependent endocrine-resistant cell line model.
Study design:
The study was a Phase Ib trial in patients with ER+/HER2− MBC, who had the following characteristics: 1) disease that relapsed during treatment or within 6 months of discontinuation of an adjuvant nonsteroidal aromatase inhibitor or Tamoxifen, or 2) MBC that progressed during treatment with 1st or 2nd line endocrine therapy. Previous chemotherapy was allowed (without limit on lines). An interval of at least 4 weeks since discontinuation of chemotherapy (6 weeks for nitrosoureas or mitomycin C) or at least 2 weeks since discontinuation of radiotherapy was required prior to entering the study. At the time this study was opened, CDK4/6 inhibitors, mTOR inhibitors and PI3KCA inhibitors were investigational, hence no patient was pre-treated with these agents. Patients with visceral and non-visceral metastatic disease were eligible for enrollment, and CNS metastases were not an exclusion criterion. All premenopausal patients were to receive Goserelin (3.6 mg q28 days Sub-Q).
GSI RO4929097 was provided by the NCI CTEP program. GSI RO4929097 was administered orally daily for 3 consecutive days, followed by 4 days off on a 21 day cycle. Exemestane was used at 25 mg/d. Doses of RO4929097 were escalated on a standard 3+3 design over 5 doses (20, 30, 45, 90, and 140 mg). Patients were fully evaluated for dose limiting toxicity (DLT) for 3 weeks. Patients were treated until disease progression or occurrence of unacceptable toxicity.
Toxicity was assessed on a weekly basis, including safety visits with a study nurse, serum chemistry and hematology. Patients were followed for 4 weeks after removal from study for toxicity. Patients removed from study for unacceptable adverse events were followed until resolution or stabilization of the adverse event. Patients who had been removed from study were followed for overall survival and had their charts reviewed every six months until death. Toxicity grading was based on the CTEP Version 4.0 of the CTCAE. Any of the following events occurring during Cycle 1 were considered a dose limiting toxicity (DLT) event when possibly, probably or definitively classified as drug-related, according to NCI CTEP Adverse Event Reporting Requirements:
CTCAE Grade 4 neutropenia (ANC <0.5 10 9/L) lasting for > 5 days.
Febrile neutropenia (i.e.: CTCAE Grade 3 or 4 neutropenia (ANC <1.0 10 9/L) associated with a fever 38.5C) or documented infection associated with Grade 3–4 neutropenia
CTCAE Grade 4 thrombocytopenia < 25 × 10 9/L or CTCAE Grade 3 < 50–25 × 10 9/L thrombocytopenia with bleeding.
CTCAE Grade 3–4 non-hematologic toxicity (including grade 3 diarrhea) despite symptomatic therapy where existing
Inability to initiate Cycle 2 within 2 weeks of scheduled treatment due to slow recovery from study drug-induced toxicities and failure to meet re-treatment criteria.
Patients with asymptomatic grade 3 hypophosphatemia, hypocalcemia, hypomagnesemia were considered a DLT on this if they had not resolved within in 72 hrs of appropriate therapy or were associated with new EKG findings.
Statistical plan for phase Ib clinical trial:
Dose escalation for MTD determination followed a standard 3+3 design: Dose escalation was designed to proceed as long as 0 in 3 patients or 1 or fewer patients in 6 patients treated experienced a DLT. If 2 or more DLT’s were observed in 3 or 6 patients treated, the MTD will be exceeded. The MTD was defined as the highest dose level tested in which fewer than 2 patients in 6 treated at that dose level experienced DLT. The MTD determined in the trial will be the recommended phase 2 dose for subsequent testing. Since the declaration of the MTD requires 6 patients, the minimum sample size was 6 patients and the maximum sample size required for this phase I study was 36 patients.
Pharmacokinetics evaluation:
A full PK assessment for the combination of RO4929097 and exemestane was evaluated. This was conducted to identify any potential effects that the combination therapy may have on the pharmacokinetics of RO4929097. Blood samples for full PK assessment of RO4929097 were collected from all patients on the phase I clinical trial on Cycle 1 Days 1 and 2. The PK full sampling time points were: immediately pre-dose and at (0.5, 1, 2, 4, 6 and 24 hrs after dosing). Additional samples were collected on day 10 for a trough level at pre-dose and a 2 hr post dose. A final sample was also collected for trough level on Cycle 2 Day 1.
A validated liquid chromatography triple quadrupole mass spectrometry (LC/MS/MS) method was used to determine levels of RO4929097 in plasma. The method was validated per ICH/FDA guidelines and was conducted as follows. Plasma samples were prepared for chromatographic injections by solid-supported liquid extraction (SLE). Calibration and quality control (QC) samples were made by adding known amounts of RO4929097 to blank plasma. All calibration, QC and unknown patient samples were prepared in the following manner. 100 μl of plasma was added to a 96-well sample prep plate and diluted with 100 μl of 1% formic acid. 10 μl of 10 μg/ml RO4929097-D6, internal standard, was added to each well. Well contents were aliquoted to an Isolute 96 well SLE 200mg plate (Biotage AB, Charlotte, NC) and loaded under positive pressure. 1ml of ethyl acetate was added to each well for elution of analytes under positive pressure. Ethyl acetate was evaporated under a gentle stream of air and then wells were reconstituted with 200 μl of 0.1% acetic acid for injection. 10 μl of each sample was injected into a Thermo Accela/TSQ Quantum LC/MS/MS system (Thermo Scientific, San Jose, CA). Mobile phase consisted of water and methanol, both containing 0.1% acetic acid. Gradient pumping conditions were run at 400 μl/min for 5 minutes. Samples were maintained at 4°C in the autosampler during sequences and RO4929097 was separated using a Luna C18 (2 × 50mm, 3 μm) column (Phenomenex, Inc., Torrance, CA). Column eluate entered the mass spectrometer via electrospray ionization. Multiple reaction monitoring (MRM) was employed utilizing the following molecular transitions; 470 ➜180 and 476 ➜196 for RO4929097 and RO4929097-D6, respectively. Peaks were detected and integration was performed with Thermo LC Quan software. Calibration curves were generated for each run and unknowns back-calculated from the corresponding regression line using LC Quan. The assay is linear from 5 – 2500 ng/ml and recovery from plasma is on average greater than 70%. Inter and intra-assay variability was less than 6% with a relative mean error of less than 12%. Plasma RO4929097 concentration-time data was analyzed using Phoenix WinNonlin 6.3 (Pharsight Corp., Mountain View, CA) using non-compartmental methods.
Biomarker evaluation:
For 10 of the patients we had sufficient tissue availability to evaluate expression of PKCα and Notch4, which we have previously shown are strongly correlated with each other at the mRNA and protein level. Tissue processing and Q-RT-PCR were performed as previously described 9 using 10 5 μm sections from the surgical specimens. IHC for PKCα and Notch4 were performed as previously described using full-face sections of the original surgical specimens 9. Scoring was performed as described 16 by two independent observers and scores were averaged.
Cell Culture and mammosphere experiments :
T47D:PKCα cells were generated in the laboratory of Dr. Debra Tonetti (UIC), by transfection with the pSPKCα plasmid by electroporation 17. Individual G418-resistant clones were selected and clone F2 was determined to overexpress PKCα by Western blot. The cell line was authenticated as T47D by STR (short tandem repeat) analysis by an independent contract laboratory (Genetica DNA Laboratories – LabCorp). Cells were cultured as previously described9. Secondary mammosphere cultures were established from cell lines as previously described18 and treated with GSI, fulvestrant or combinations thereof at concentrations determined through pilot experiments. Mammospheres were counted after 7 days as previously described18, and contrast microphotographs of randomly selected fields were taken.
Statistical analysis of biomarker and preclinical studies:
The biomarker studies were designed to be hypothesis generating and were not powered to reach statistical significance. It is anticipated that promising findings will be validated in the context of future studies. Exploratory statistical analysis was performed using SPSS v19.0. Spearman correlation tests were used to evaluate mRNA expression levels and IHC scores. Statistical significance was set at p < 0.05 (two-sided).
Results
Patient Demographics:
Table 1 shows the demographic characteristics of our patients. A total of 15 patients were enrolled. Median age was 56. Average number of chemotherapies received in the adjuvant or metastatic setting was 3.3. Six/15 patients were pre-treated with chemotherapy. Eleven patients had received adjuvant endocrine therapy. One patient received this as 1st line endocrine therapy in the metastatic setting. Eleven patients received this investigational regimen as 2nd line endocrine therapy in the metastatic setting. Three patients received this investigational regimen as 3rd line endocrine therapy in the metastatic setting.
Table 1:
patient demographics
| Age | |
| Median | 55 |
| Range | 39–68 |
|
| |
| Race | |
| Caucasian | 14 (93.3%) |
| African-American | 0 (0.0%) |
| Other | 1 (26.7%) |
|
| |
| ECOG PS | |
| 0 | 11 (73.3%) |
| 1 | 4 (26.7%) |
|
| |
| Menopausal Status | |
| Premenopausal | 0 (0.0%) |
| Postmenopausal | 15 (100%) |
|
| |
| Disease Location | |
| Visceral | 10 (66.7%) |
| Non-visceral | 5 (33.3%) |
|
| |
| Prior Adjuvant Endocrine Therapy | |
| Nonsteroidal AI | 7 (46.7%) |
| Tamoxifen | 4 (26.7%) |
|
| |
| Prior Metastatic Endocrine Therapy | |
| Nonsteroidal AI | 9 (60.0%) |
| Tamoxifen | 1 (6.7%) |
| Faslodex | 4 (26.7%) |
|
| |
| Prior Metastatic Chemotherapy | 6 (40.0%) |
Toxicity:
Adverse events were defined and evaluated according to the NCI Common Terminology Criteria for Adverse Events (CTCAE version 4.0). The timelines of patient follow-up at each dose level are summarized in Figure 1. Toxicity was limited (Table 2), with the only Dose-Limiting Toxicity (DLT) experienced at the highest dose of 140 mg. This DLT was cutaneous rash. The most common toxicities observed were nausea (73.3%), anorexia (60%), hyperglycemia (53.3%), hypophosphatemia (46.7%), fatigue (66.7%) and cough (33.0%). Grade 3 toxicities included hypophosphatemia (13.0%) and rash (6.3%). Importantly, no secretory diarrhea was observed at any dose levels, consistent with our preclinical results that estrogen deprivation ameliorates the intestinal toxicity of GSIs. We were unable to determine an MTD due to limited access to study drug. Adverse events are summarized in Table 2.
Figure 1.
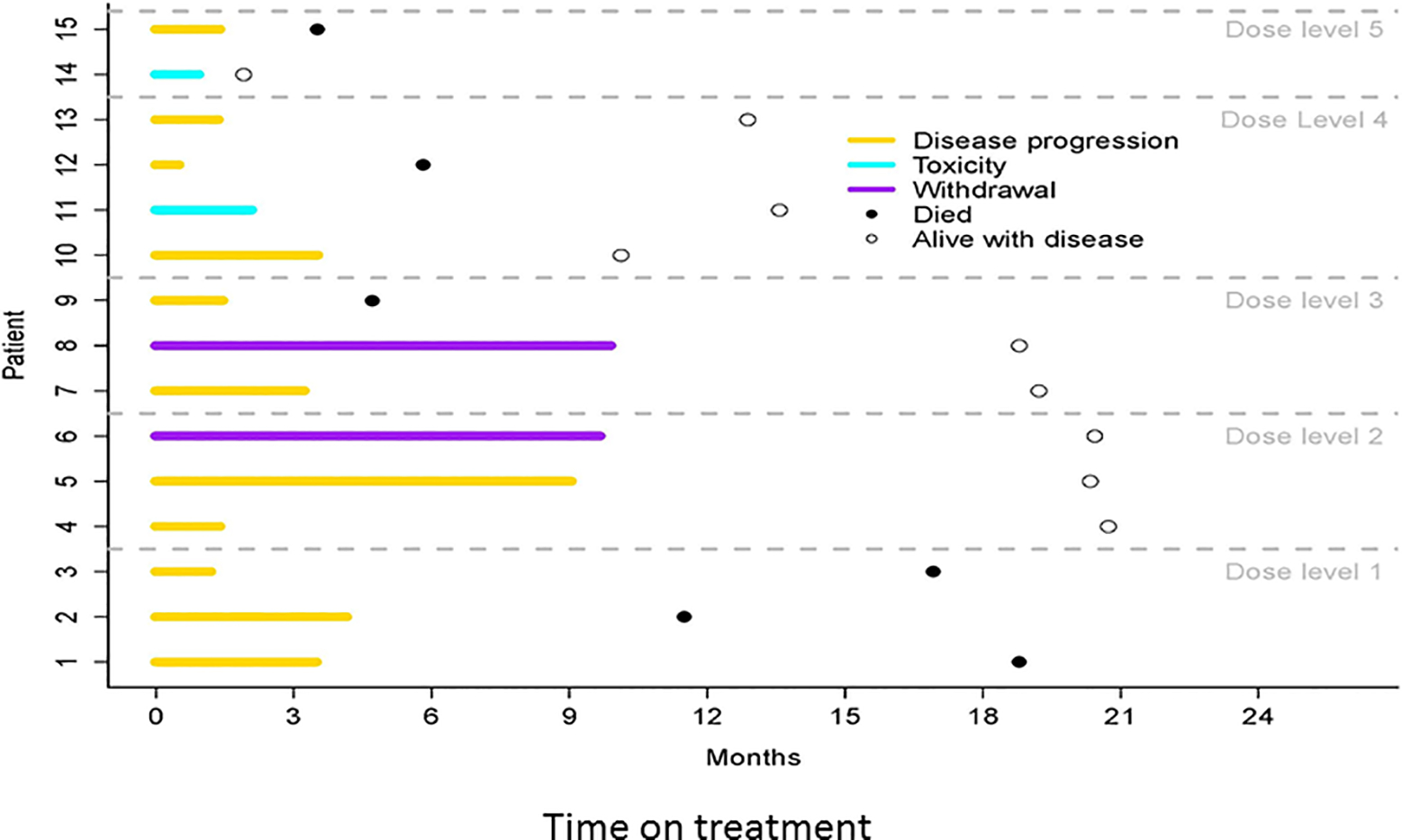
Timeline of clinical responses of individual patients on this study at each dose level.
Table 2:
Most Frequent Adverse Events
| Adverse event | Grade 3 AE N (%) |
All Grades AE N (%) |
|---|---|---|
| Nausea | 0 | 11 (73.3) |
| Anorexia | 0 | 9 (60) |
| Fatigue | 1 (6.0) | 10 (66.7) |
| Hyperglycemia | 1 (6.0) | 8 (53.3) |
| Hypophosphatemia | 2 (13.4) | 7 (46.7) |
| Cough | 0 | 4 (26.6) |
| EKC QT prolonged interval | 0 | 1 (6.0) |
| Rash | 1 (6.0)* | 1 (6.0) |
DLT
Clinical response rate:
Two patients discontinued due to toxicity, 11 due to disease progression and 2 withdrew from the study. Clinical responses were observed in 8/14 evaluable patients. These include one partial response (PR), 7 stable diseases (SD) and 6 progressions of disease. No complete responses (CR) were observed. The total clinical benefit rate (CR+PR+SD ≥ 6 months) was 20%, with a progression-free survival (PFS) of 3.2 months. Estimated overall survival (OS) at 6 months was 79% (60–100%).
Pharmacokinetics:
Pharmacokinetics analyses are shown in Figure 2. Data was compared to previously reported findings for the pharmacokinetics of RO4929097. AUC and Cmax appear to correlate with the dose administered. (Figure 2). The terminal half-life calculated is also similar to previously published reports. Patients achieved nearly unquantifiable levels of the drug six days after dosing. Trough levels on different sampling days can be seen in Figure 3. A summary of the pharmacokinetic parameters can be found in Table 3.
Figure 2.
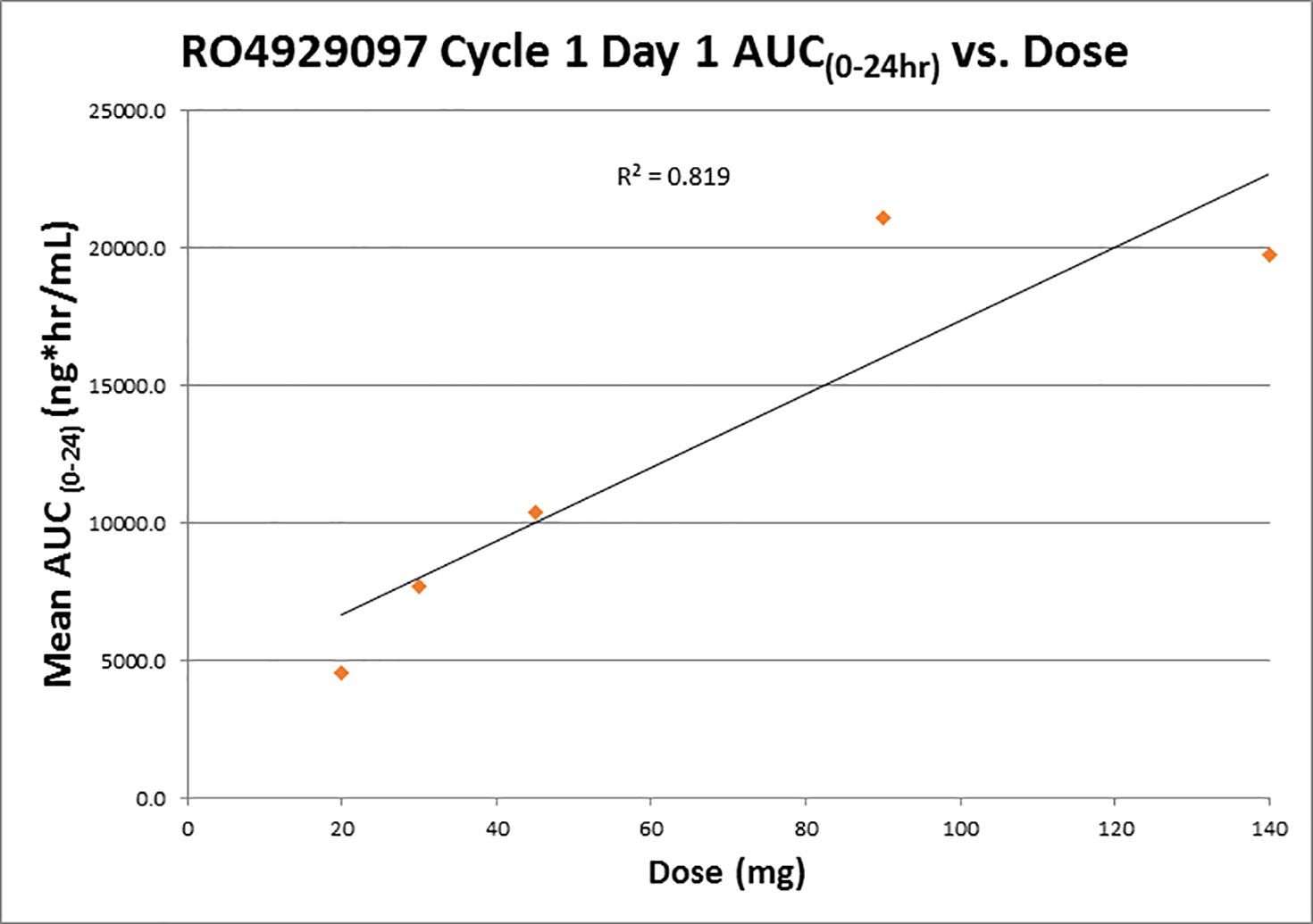
Correlation between AUC(0–24h) and dose in treatment cycle 1.
Figure 3.
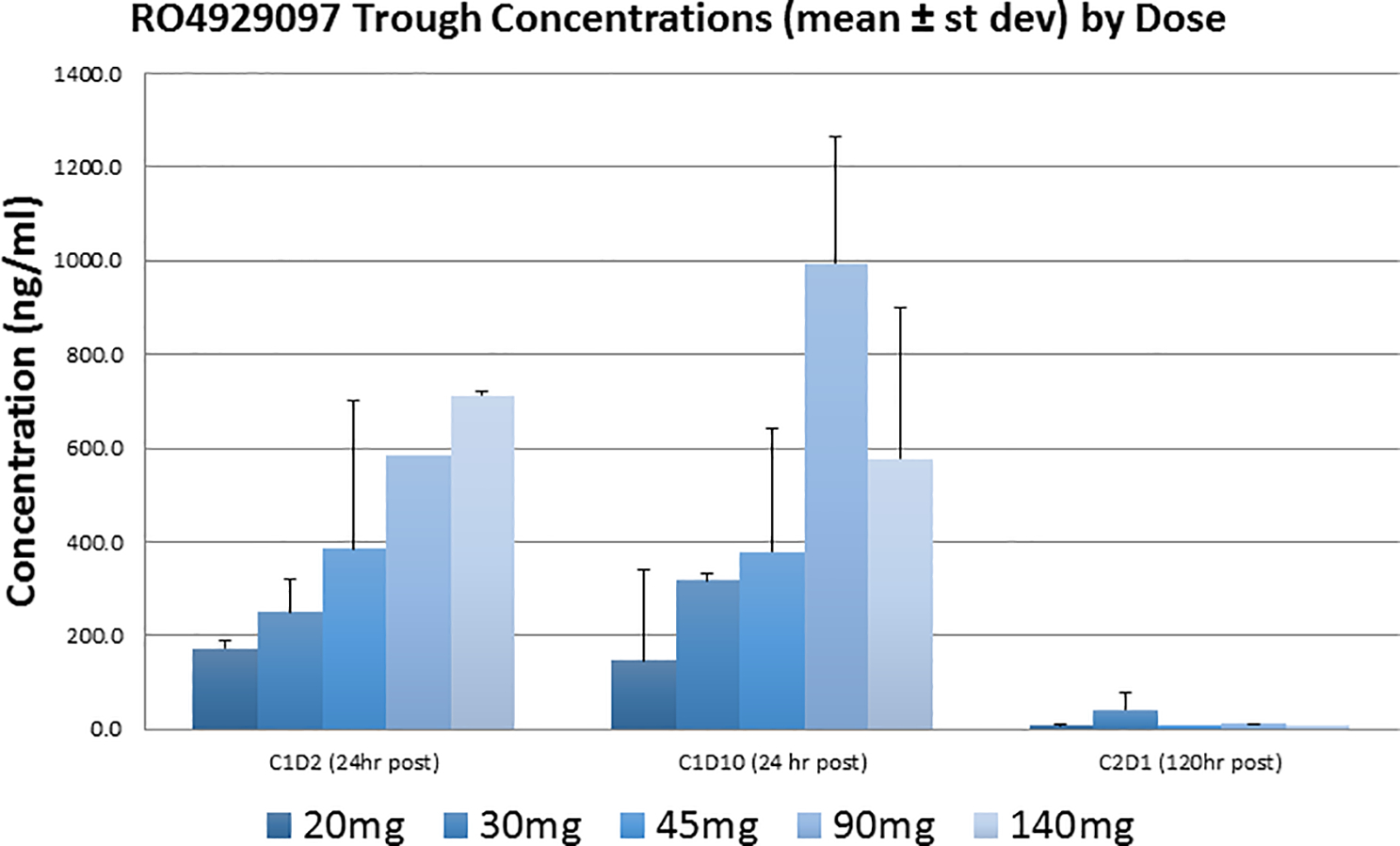
Trough concentration of RO4929097 versus dose, evaluated at cycle 1, dosing 1 (C1D1), 24 hours post dosing, cycle 1, dosing 10 (C1D10), 24 hours post dosing and C1D1, 120 hours post dosing.
Table 3:
RO4929097 Pharmacokinetics Parameters Summary
| Cohort | AUC (0–24hr) (hr*ng/mL) Mean (±st dev) |
t1/2 (hr) Mean (±st dev) |
Cmax (ng/mL) mean (±st dev) |
Tmax (hr) mean (range) |
Vol of Dist (L) mean (±st dev) |
Clearance (L/hr) mean (±st dev) |
|---|---|---|---|---|---|---|
| 20mg (n=3) | 4550.1 (±1279.5) | 26.0 (±19.7) | 265.9 (±52.3) | 3.0 (1–6) | 61.0 (±23.2) | 2.3 (±1.5) |
| 30mg (n=2) | 7710.4 (±3898.2) | 20.7 (±10.9) | 1045.7 (±707.4) | 0.5 | 64.4 (±41.8) | 2.1 (±0.3) |
| 45mg (n=2) | 10390.7 (±7775.7) | 23.1 (±0.4) | 1107.6 (±942.8) | 1.5 (1–2) | 94.0 (±74.5) | 2.8 (±2.2) |
| 90mg (n=1) | 21077.0 | 13.6 | 2081.2 | 4.3 | 54.2 | 2.8 |
| 140mg (n=2) | 19725.4 (±1046.4) | 33.4 (±2.4) | 1326.5 (±121.1) | 1.5 (1–2) | 124.7 (±0) | 2.6 (±0.2) |
Pharmacokinetics data was compared to previously reported findings for the pharmacokinetics of RO4929097 19, 20. AUC and Cmax appear to correlate with the dose administered (Figure 2). The terminal half-life calculated is also similar to previously published reports. Patients achieved nearly unquantifiable levels of the drug six days after dosing. Trough levels on different sampling days can be seen in Figure 3. A summary of the pharmacokinetic parameters can be found in Table 3. These results indicate that, at least in this dose range, the pharmacokinetics of RO4929097 is probably not appreciably affected by co-treatment with exemestane. We were unable to establish an MTD. However, based on data shown in Figure 3 and Table 3, it would appear that increasing the dose from 90 to 140 mg/d does not increase the AUC and may decrease trough concentrations upon repeated administration. This may be due to CYP3A4 auto-induction, which has been observed in other phase 1 trials 19, 21, and may argue in favor of a recommended phase 2 dose of 90 mg/d. The average trough concentrations of RO4929097 at a dose of 90 mg/d varied between 600 and 1000 ng/ml. This translates to 1.28 to 2.13 μM (mw 469.4). The reported in vitro IC50 of RO4929097 in cell-based assays for Notch1 cleavage is 5 nM 22, and we have observed IC50s in the nanomolar range as well 23. However, biological activity of RO4929097 as a single agent as determined by inhibition of mammosphere formation (a surrogate for CSC) may require near-complete inhibition of all four Notch paralogs. In our hands in triple-negative breast cancer models, this was observed at concentrations between 1 and 10 μM 23. Our mammosphere studies suggest that lower GSI concentrations may suffice in combination with endocrine therapy. These considerations emphasizes the importance of verifying biological activity by molecular markers in target tissues, ideally through research biopsies, in future trials.
RO4929097 is highly plasma protein-bound, and concomitant administration of vismodegib significantly increases drug exposure 20. However, exemestane, which is also 90% protein-bound 24 does not appear to alter the pharmacokinetics of RO4929097. Both drugs bind α−1 acid globulin (AGG) and albumin 20, 24. It is possible that the affinity of RO4929097 for these proteins is higher than that of exemestane, or that binding sites do not overlap. Whether RO4929097 affects the pharmacokinetics of exemestane remains to be determined. Future studies of highly protein-bound GSIs in combination with exemestane should evaluate free and protein-bound GSI.
Biomarker studies:
Protein kinase C α (PKCα) has been reported to be expressed in 73% of ER+ tumors that recurred on tamoxifen and to predict tamoxifen resistance 25. We have previously reported that PKCα induces expression of Notch4, and that expression of these two genes is strongly correlated in ER+ breast cancers 9. Notch4 was necessary and sufficient to cause endocrine resistance in vitro 9. Thus, we explored expression of PKCα and Notch4 by immunohistochemistry in surgical specimens from patients enrolled in this study. Out of 10 evaluable tumors, 10 (100%) stained positive for PKCα (2+ or 3+). Of these, 5 also showed intense nuclear or perinuclear staining (Figure 4A). Nuclear Notch4 expression (2+ or 3+) was observed in 7/10 specimens, while 3/10 showed cytoplasmic staining. Interestingly, we consistently observed nuclear localization of both PKCα and Notch4 at the invading edges of tumors, while inside tumor nodules both signals were predominantly cytoplasmic (Figure 4A). Nuclear PKCα scores were strongly correlated with nuclear Notch4 scores (r = 0.741, p = 0.014). This suggests that the activation status of Notch4 may depend on microenvironmental signals. Similarly, mRNA levels of PKCα and Notch4 were strongly correlated (r = 0.715, p = 0.046, Figure 5). This is consistent with our previously published data 9. These observations are consistent with the hypothesis that Notch4 contributes to the endocrine-resistant phenotype 14.
Figure 4.
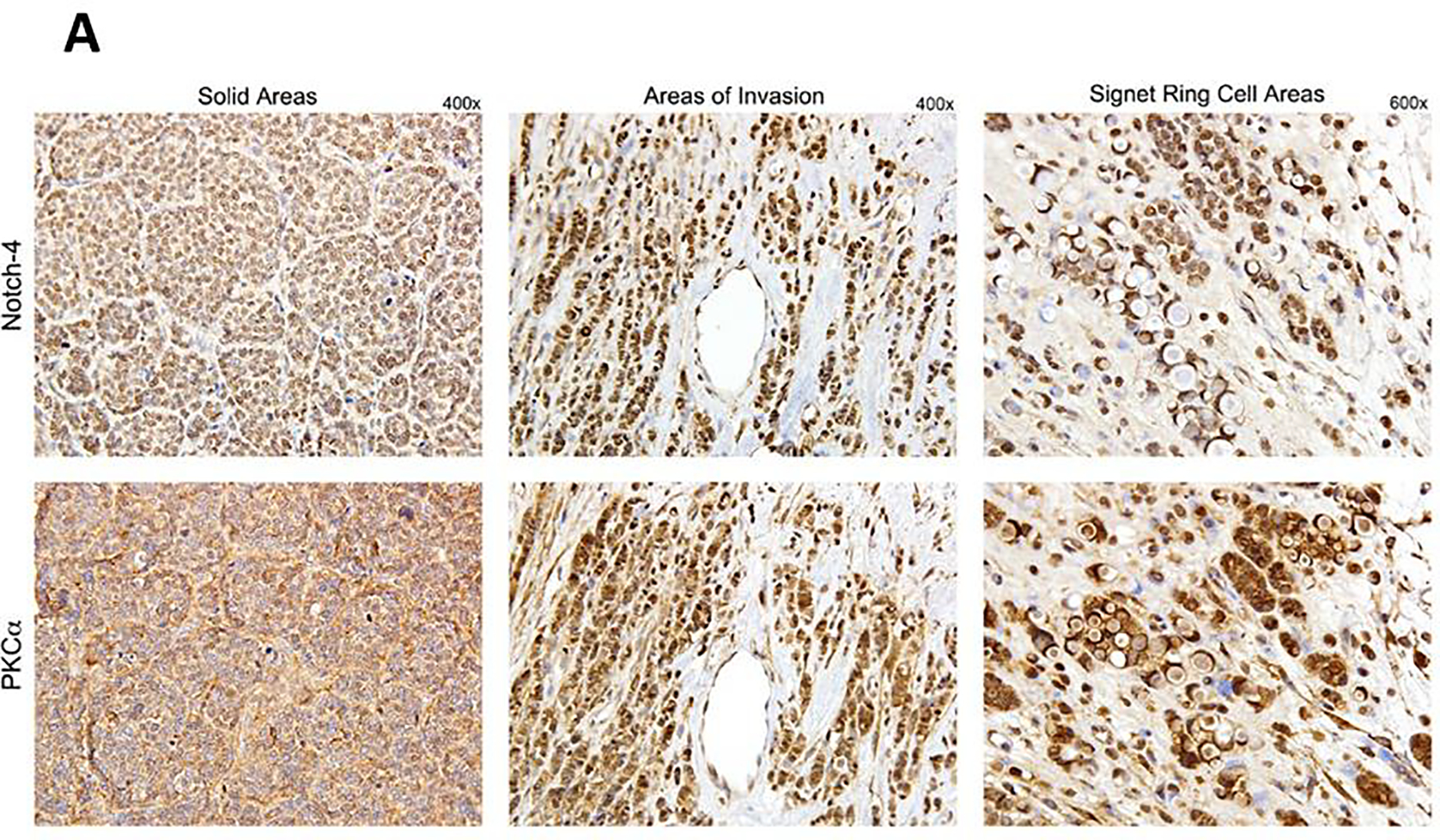
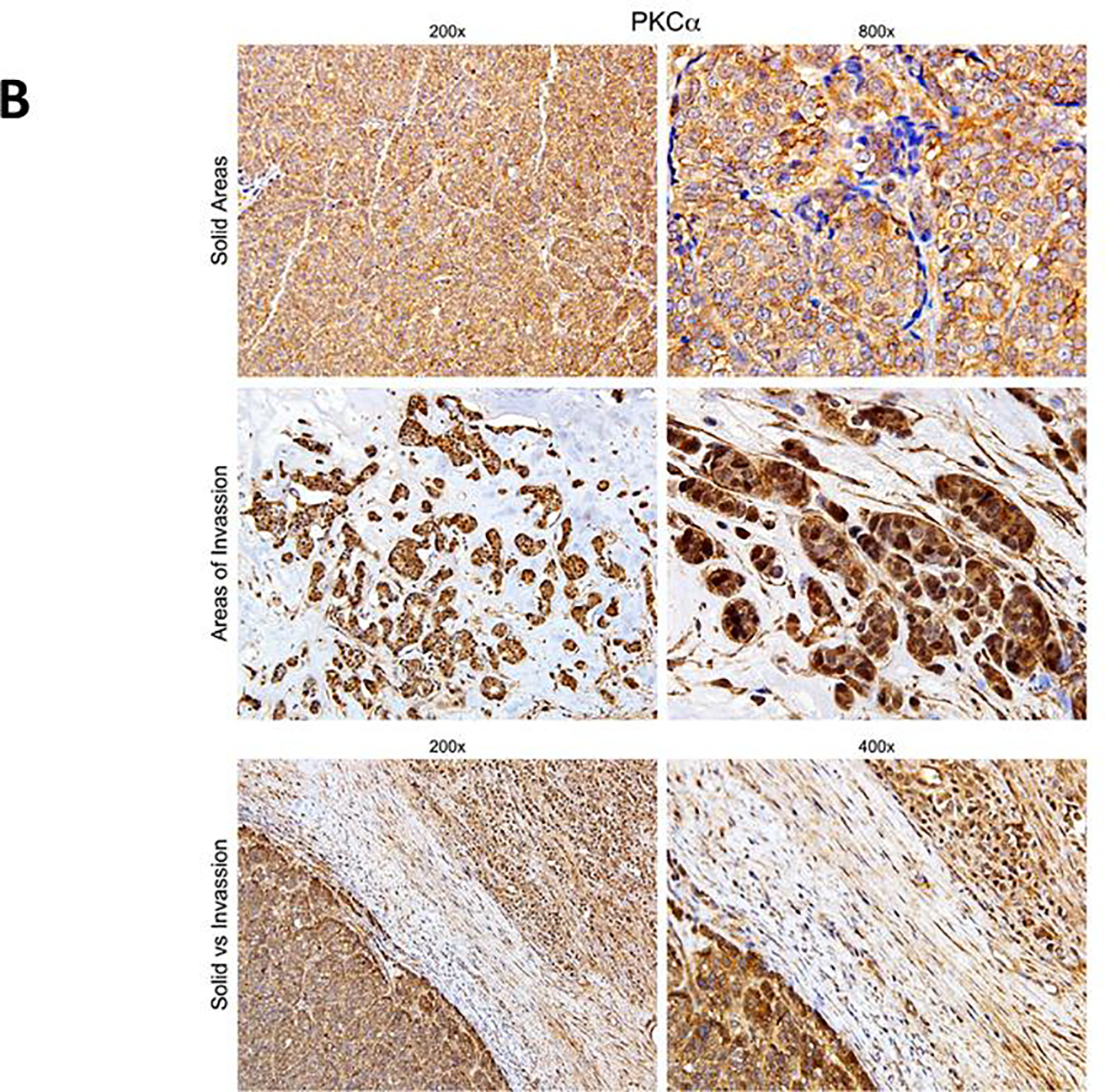
Immunohistochemistry images showing expression of Notch4 and PKCα in representative tissue samples from patients enrolled in this study. A: the cellular localization of Notch4 and PKCα coincides, and is primarily cytoplasmic in solid areas of tumors and nuclear in areas of invasion at the tumor edge. B: a different case showing striking differences in cellular localization of PKCα in different areas of the tumor. The bottom panels show a field where both invading front and a solid tumor nodule are visible, each with its distinctive PKCα localization.
Figure 5.
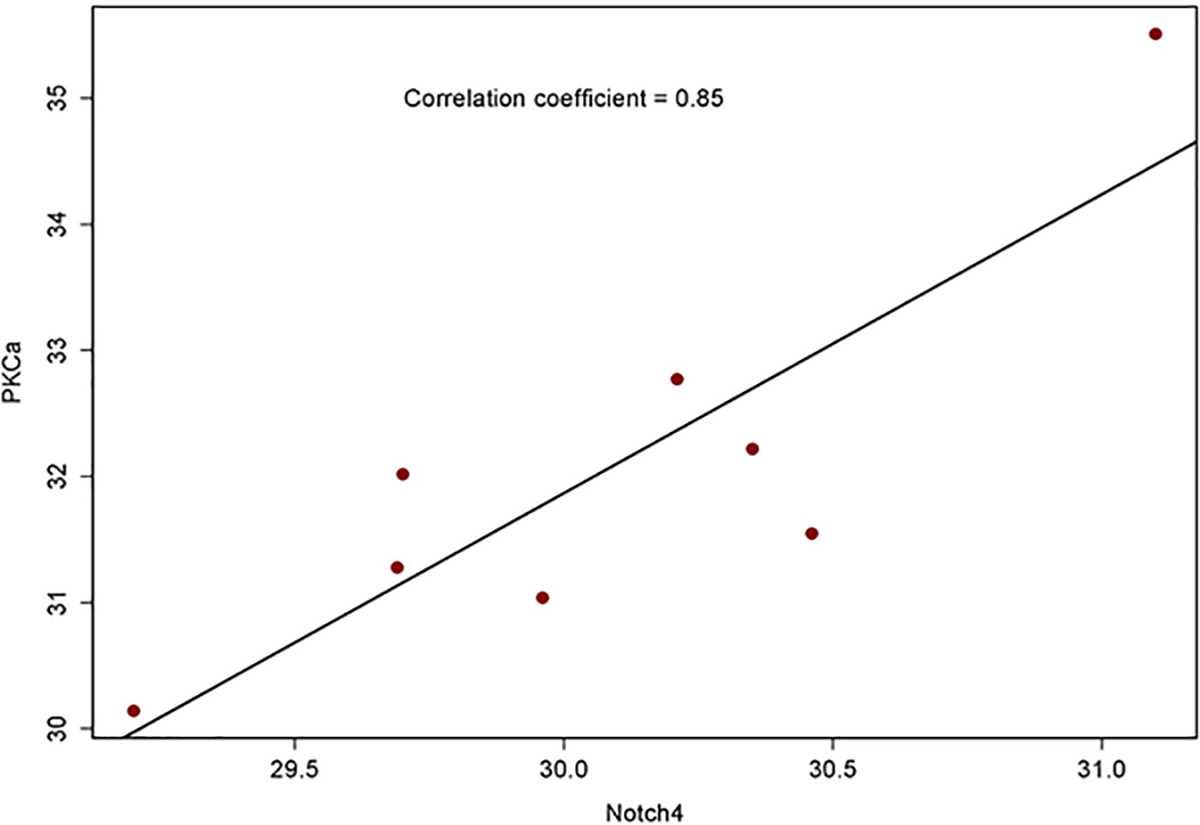
Correlation between mRNA levels of Notch4 and PKCα as determined by RT-PCR in the same samples examined by immunohistochemistry.
Notch4 has been reported to be resistant to some GSIs 26. The mechanism of Notch4 cleavage is poorly understood, and the γ-secretase cleavage site in this Notch paralog is unknown. Given the potential importance of Notch4 in the endocrine resistant phenotype, GSIs or other Notch inhibitors used in endocrine-resistant breast cancer should have Notch4 inhibitory activity. Simoes et al. 27 have shown that RO4929097 does inhibit Notch4 cleavage.
Mammosphere studies:
Cancer stem cells (CSC) in ERα+ breast cancers have been reported to be Notch-dependent (reviewed in 3, 28 29). Mammosphere formation is a surrogate for CSC activity. We sought to determine whether RO4929097 inhibits mammosphere formation in an endocrine-resistant cell line model in the presence or absence of endocrine therapy. We used fulvestrant in these in vitro experiments, rather than simple estrogen deprivation, because Notch1 can induce ERα-dependent transcription in the absence of estrogen 11. We hypothesize that future clinical trials of combinations between Notch inhibitors and endocrine therapy should explore selective estrogen receptor disruptors (SERDs) rather than aromatase inhibitors. Mammosphere experiments in an endocrine-resistant cell line engineered to express PKCα were consistent with the notion that combined inhibition of Notch and ER is superior to either agent alone (Figure 6A–B). Mammospheres generated from T47D:PKCα cells were partially sensitive to fulvestrant as a single agent. RO4929097 alone (5 μM) significantly decreased mammosphere numbers, consistent with the results of Simoes et al. 27. The combination GSI plus fulvestrant was significantly more potent than either agent alone, nearly abolishing mammosphere growth. Mammosphere size was also dramatically reduced (Figure 6B).
Figure 6.


Mammosphere inhibitory activity of RO4929097 alone (5 μM) and in combination with fulvestrant (30 nM) in T47D:PKCα cells. A: mammosphere counts in 3 independent experiments +/− standard deviations. B: representative microphotographs of phase contrast images from mammosphere cultures. Two fields per treatment were photographed.
Discussion
Recurrent, ER+ breast cancer remains a major cause of morbidity and mortality, and it is generally considered incurable, despite the introduction of CDK4/6 inhibitors, everolimus and PI3K inhibitors 30. There is strong evidence for an oncogenic role of Notch signaling, and Notch4 in particular, in this subgroup of breast cancers. Combinations of endocrine therapy and GSIs that inhibit Notch4 are safe and effective in preclinical models, in which endocrine therapy (tamoxifen or ovariectomy) decrease the intestinal toxicity of GSIs 9.
This was the first clinical study of an endocrine therapy-GSI combination in ERα-positive breast cancer in the metastatic setting. Our data suggest that this combination is safe and reasonably well tolerated, with preliminary hints of efficacy despite the pharmacokinetic liability of RO4929097. The safety profile of this combination appears to be favorable. Consistent with our data in preclinical models, secretory diarrhea was not observed in any of our patients, even after 6 months of treatment. Interestingly, low-grade hyperglycemia was a common and unexpected side effect. Systemic loss of Notch signaling and pharmacological Notch inhibition increase glucose tolerance and insulin sensitivity 31. Assuming that the hyperglycemia observed in some of our patients is an on-target effect, it may be a rebound phenomenon due to the intermittent treatment schedule. Further investigation is warranted on the metabolic effects of Notch inhibitors in a clinical setting. In preclinical models of ERα+ breast cancer, Notch1 has a dose-dependent effect on glucose utilization 32. Specifically, hyperactivation of Notch signaling triggers glycolysis via AKT activation. Severe deficiency of Notch signaling also triggers glycolysis by decreasing mitochondrial respiratory activity.
Despite considerable evidence that Notch signaling plays important pathogenic roles in a number of malignancies and the efficacy of GSIs and other Notch pathway inhibitors in preclinical models, early phase clinical trials of these agents have rarely shown preliminary evidence of single agent efficacy, even in T-LL patients with gain-of-function Notch1 mutations 1, 3. This is similar to what has been observed with other agents targeting developmental pathways. There are several, non-mutually exclusive possible reasons for these results. Given their pervasive biological importance, highly conserved developmental pathways are involved in intricate cross-talks with other signaling pathways and gene regulatory networks. Single pathway inhibition is likely to trigger feedback compensatory mechanisms and/or selection of resistant clones. In T-cell lymphoblastic leukemia (T-LL), the most studied disease where Notch1 acts as an oncogene, the Notch, PI3K and MEK pathways are intricately interconnected. In T-LL mouse models with or without Kras mutations, treatment with PI3K inhibitor GDC-0941 is effective, and its efficacy is increased by MEK inhibitor PD0325901. However, treatment is invariably followed by the emergence of resistant clones that no longer have oncogenic Notch mutations 33. This suggests that Notch1-driven clones are addicted to the PI3K pathway, and mutations that upregulate this pathway can compensate for loss of constitutively active Notch. Sub-lethal inhibition of Notch leads to compensatory PI3K activation 33. Mutant Notch1 activates the PI3K pathway through a number of mechanisms, and constitutive activation of PI3K due to PTEN loss of function leads to GSI resistance 34. Conversely, in triple negative breast cancer dual inhibition of mTORC1/mTORC2 causes resistance mediated by Notch activation 35.
Additionally, it is possible that Notch inhibition in some breast cancer may affect primarily CSC, rather than resulting in bulk tumor cytotoxicity and radiological remissions. In a preclinical model of HER2-enriched breast cancer, Pandya et al. showed that GSI MRK003 alone had virtually no effect on tumor volume, nor did it increase the anti-tumor activity of trastuzumab. However, when used in combination with trastuzumab, this GSI abrogated tumor recurrence, resulting in permanent cures 36. Such effects, most likely due to inhibition of CSC, would be difficult to detect in a short-term clinical trial using standard radiological criteria. Our mammosphere results are consistent with an anti-CSC effect, and confirm that concomitant inhibition of Notch and estrogen signaling is at least additive in blocking the growth of PKCα-positive, endocrine-resistant mammospheres.
The optimal use of Notch inhibitors in oncology will require an understanding of the cross-talk between Notch and other pathways in specific indications and mechanism-based, rational combinations. We and others have described a complex cross-talk between Notch and estrogen signaling in ERα-positive breast cancer cells. In MCF-7 and T47D cells, estrogen inhibits the activation of Notch1 via ERα 10. Estrogen deprivation or treatment with SERMS causes re-activation of Notch1, which induces Notch4 expression 10. In turn, Notch1IC is capable of activating ERα-dependent transcription in the absence of estrogen and in the presence of tamoxifen, by recruiting MAML1 and p300 to ERα-dependent promoters 11. The result is Notch-dependent, estrogen-independent and tamoxifen-resistant growth. Haughian et al. 13 confirmed these findings, determining that Notch suppression maintains the hormone-responsiveness of luminal breast cancers. Landor et al. 32 showed that increased Notch1IC enhances glycolysis and promotes aggressive growth in MCF-7 cells. Yun et al. showed that PKCα drives endocrine resistance via upregulation of Notch4 in T47D and MCF-7 cells 9. Expression of Notch4IC in T47D cells was sufficient to cause estrogen-independent, tamoxifen-resistant growth 9. Lombardo et al. 37 showed that Nicastrin (a subunit of γ-secretase) and Notch4 drive endocrine therapy resistance and epithelial-mesenchymal transition in MCF7 cells. Conversely, in endothelial cells there is some evidence that estrogen activates Notch1 signaling via ERβ 12. Notch1 activity in endothelial cells is necessary for productive angiogenesis 38. These observations provide a strong scientific rationale to evaluate combinations of endocrine therapy and Notch inhibitors, including GSIs. In mouse models, such combinations are more effective than single agents. GSI MRK003 reverses the endocrine resistance of PKCα-overexpressing, Notch4-positive ER+ cells and causes tumor regression without significant toxicity 9. That particular GSI, like RO4929097, is capable of inhibiting Notch4 9, which has been shown to be resistant to other GSIs. Interestingly, estrogen suppression greatly reduced the intestinal toxicity of GSI in T47D xenograft models 9.
Notch4 inhibition may be critical in the setting of endocrine resistant breast cancer. Harrison et al. 26 showed that Notch4 is enriched in ER+ breast CSC and drives their self-renewal. Simoes et al. 27 showed that endocrine resistance in luminal tumors is driven by Jagged1-Notch4-dependent CSC. The same study showed that RO4929097, unlike other GSIs, inhibits Notch4 cleavage in ER+ cancer cells 27. More recently, Gelsomino et al. showed that in ER+ breast cancer lines expressing ESR1 mutations Y537 N, Y537S, D538G, Notch signaling and particularly Notch4 mediate increased mammosphere-forming efficiency, self-renewal and migration in CSC 14. Our results are completely consistent with these observations.
In patients with early stage ERα-positive breast cancer treated with letrozole or tamoxifen, the addition of short-term GSI MK0752 to endocrine therapy in the neo-adjuvant setting caused no significant toxicity and significantly affected Ki67 expression in tumors, modulating numerous genes associated with cancer cell “stemness” and metastasis 15.
This study has several limitations. Sample size was small, preventing an adequate examination of patient heterogeneity and individual variability of response. Limited drug availability prevented us from firmly establishing an MTD. We did not evaluate whether RO4929097 affects the pharmacokinetics of exemestane. Follow-up time was relatively short (6 months). The anti-CSC activity of RO4929097 was evaluated in a Notch4-dependent model but not in a wide range of models. We have previously shown that a different GSI had broad anti-CSC activity in different ER+ models, including primary patient-derived CSC 18. Additional studies will be required to characterize in detail the anti-CSC activity of exemestane/GSI combinations.
The strong statistical and functional correlation between PKCα and Notch4 expression observed in different studies suggests that co-expression of these candidate biomarkers may identify a subset of patients more likely to respond to endocrine therapy plus Notch inhibitor combinations. Immunohistochemistry in evaluable cases showed a striking co-localization of Notch4 and PKCα, which appear either in the cytoplasm (in solid areas of tumors) or in the nucleus (at the invading front). We do not know the mechanism of this different localization. The biological and clinical significance of this phenomenon is unclear, but it may indicate that the tumor microenvironment modulates the activation of Notch4. Taken together, our results indicate that combinations of Notch inhibitors with endocrine therapy deserve further clinical investigation in luminal breast cancers. The choice of drugs to be tested in future trials will depend among other things on our understanding of the cross-talk between Notch and estrogen signaling, the mechanism of Notch-induced endocrine resistance and the role of Notch4 as a key target in this disease subset.
Conclusions
There are now several second- and third-generation Notch inhibitors at various stages of clinical development. Our results indicate that these agents should be explored in endocrine-refractory breast cancer in combination with endocrine therapy. Additional combinations with PI3K inhibitors, mTOR inhibitors or CDK inhibitors deserve further investigation.
Acknowledgements
We are grateful to all the patients who enrolled in this trial. We acknowledge the support of the NCI’s Cancer Therapeutics Evaluation Program, which also provided us with RO4949097. We acknowledge the participation in this study of Drs. Susan Minton, formerly at Moffitt Cancer Center and He Zhu, formerly at University of Mississippi Medical Center, who have since retired.
Footnotes
The authors declare no conflicts of interest related to this study.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Takebe N, Miele L, Harris PJ, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nature reviews. Clinical oncology. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Monticone G, Miele L. Notch Pathway: A Journey from Notching Phenotypes to Cancer Immunotherapy. Advances in experimental medicine and biology. 2021;1287:201–222. [DOI] [PubMed] [Google Scholar]
- 3.Majumder S, Crabtree JS, Golde TE, Minter LM, Osborne BA, Miele L. Targeting Notch in oncology: the path forward. Nat Rev Drug Discov. 2021;20:125–144. [DOI] [PubMed] [Google Scholar]
- 4.Espinoza I, Miele L. Deadly crosstalk: Notch signaling at the intersection of EMT and cancer stem cells. Cancer letters. 2013;341:41–45. [DOI] [PubMed] [Google Scholar]
- 5.Espinoza I, Pochampally R, Xing F, Watabe K, Miele L. Notch signaling: targeting cancer stem cells and epithelial-to-mesenchymal transition. OncoTargets and therapy. 2013;6:1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Espinoza I, Miele L. Notch inhibitors for cancer treatment. Pharmacol Ther. 2013:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Es JH, van Gijn ME, Riccio O, et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature. 2005;435:959–963. [DOI] [PubMed] [Google Scholar]
- 8.Real PJ, Ferrando AA. NOTCH inhibition and glucocorticoid therapy in T-cell acute lymphoblastic leukemia. Leukemia. 2009;23:1374–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yun J, Pannuti A, Espinoza I, et al. Crosstalk between PKCalpha and Notch-4 in endocrine-resistant breast cancer cells. Oncogenesis. 2013;2:e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rizzo P, Miao H, D’Souza G, et al. Cross-talk between notch and the estrogen receptor in breast cancer suggests novel therapeutic approaches. Cancer Res. 2008;68:5226–5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hao L, Rizzo P, Osipo C, et al. Notch-1 activates estrogen receptor-alpha-dependent transcription via IKKalpha in breast cancer cells. Oncogene. 2010;29:201–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caliceti C, Aquila G, Pannella M, et al. 17beta-estradiol enhances signalling mediated by VEGF-A-delta-like ligand 4-notch1 axis in human endothelial cells. PloS one. 2013;8:e71440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haughian JM, Pinto MP, Harrell JC, et al. Maintenance of hormone responsiveness in luminal breast cancers by suppression of Notch. Proc Natl Acad Sci U.S.A. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gelsomino L, Panza S, Giordano C, et al. Mutations in the estrogen receptor alpha hormone binding domain promote stem cell phenotype through notch activation in breast cancer cell lines. Cancer letters. 2018;428:12–20. [DOI] [PubMed] [Google Scholar]
- 15.Albain K, Covington KR, Gallagher BT, et al. Identification of a notch-driven breast cancer stem cell gene signature for anti-notch therapy in an ER+ presurgical window model. Abstracts of the 37th San Antonio Breast Cancer Symposium. 2014. [Google Scholar]
- 16.Zhu H, Bhaijee F, Ishaq N, et al. Correlation of Notch1, pAKT and nuclear NF-kappaB expression in triple negative breast cancer. American journal of cancer research. 2013;3:230–239. [PMC free article] [PubMed] [Google Scholar]
- 17.Reifel-Miller AE, Conarty DM, Valasek KM, Iversen PW, Burns DJ, Birch KA. Protein kinase C isozymes differentially regulate promoters containing PEA-3/12-O-tetradecanoylphorbol-13-acetate response element motifs. The Journal of biological chemistry. 1996;271:21666–21671. [DOI] [PubMed] [Google Scholar]
- 18.Grudzien P, Lo S, Albain KS, et al. Inhibition of Notch signaling reduces the stem-like population of breast cancer cells and prevents mammosphere formation. Anticancer Res. 2010;30:3853–3867. [PubMed] [Google Scholar]
- 19.Tolcher AW, Messersmith WA, Mikulski SM, et al. Phase I study of RO4929097, a gamma secretase inhibitor of Notch signaling, in patients with refractory metastatic or locally advanced solid tumors. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012;30:2348–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu J, Lorusso PM, Matherly LH, Li J. Implications of plasma protein binding for pharmacokinetics and pharmacodynamics of the gamma-secretase inhibitor RO4929097. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:2066–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diaz-Padilla I, Hirte H, Oza AM, et al. A phase Ib combination study of RO4929097, a gamma-secretase inhibitor, and temsirolimus in patients with advanced solid tumors. Invest New Drugs. 2013;31:1182–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luistro L, He W, Smith M, et al. Preclinical profile of a potent gamma-secretase inhibitor targeting notch signaling with in vivo efficacy and pharmacodynamic properties. Cancer Research. 2009;69:7672–7680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ran Y, Hossain F, Pannuti A, et al. gamma-Secretase inhibitors in cancer clinical trials are pharmacologically and functionally distinct. EMBO Mol Med. 2017;9:950–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.FDA. Aromasin package insert 1999.
- 25.Tonetti DA, Morrow M, Kidwai N, Gupta A, Badve S. Elevated protein kinase C alpha expression may be predictive of tamoxifen treatment failure. British Journal of Cancer. 2003;88:1400–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harrison H, Farnie G, Howell SJ, et al. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. 2010;70:709–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simoes BM, O’Brien CS, Eyre R, et al. Anti-estrogen Resistance in Human Breast Tumors Is Driven by JAG1-NOTCH4-Dependent Cancer Stem Cell Activity. Cell Rep. 2015;12:1968–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crabtree JS, Miele L. Breast Cancer Stem Cells. Biomedicines. 2018;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harrison H, Farnie G, Brennan KR, Clarke RB. Breast cancer stem cells: something out of notching? Cancer Res. 2010;70:8973–8976. [DOI] [PubMed] [Google Scholar]
- 30.Hartkopf AD, Grischke EM, Brucker SY. Endocrine-Resistant Breast Cancer: Mechanisms and Treatment. Breast Care (Basel). 2020;15:347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bi P, Shan T, Liu W, et al. Inhibition of Notch signaling promotes browning of white adipose tissue and ameliorates obesity. Nat Med. 2014;20:911–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Landor SK, Mutvei AP, Mamaeva V, et al. Hypo- and hyperactivated Notch signaling induce a glycolytic switch through distinct mechanisms. Proc Natl Acad Sci U.S.A. 2011;108:18814–18819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dail M, Wong J, Lawrence J, et al. Loss of oncogenic Notch1 with resistance to a PI3K inhibitor in T-cell leukaemia. Nature. 2014;513:512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hales EC, Taub JW, Matherly LH. New insights into Notch1 regulation of the PI3K-AKT-mTOR1 signaling axis: targeted therapy of gamma-secretase inhibitor resistant T-cell acute lymphoblastic leukemia. Cellular signalling. 2014;26:149–161. [DOI] [PubMed] [Google Scholar]
- 35.Bhola NE, Jansen VM, Koch JP, et al. Treatment of Triple-Negative Breast Cancer with TORC1/2 Inhibitors Sustains a Drug-Resistant and Notch-Dependent Cancer Stem Cell Population. Cancer Res. 2016;76:440–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pandya K, Meeke K, Clementz AG, et al. Targeting both Notch and ErbB-2 signalling pathways is required for prevention of ErbB-2-positive breast tumour recurrence. Br.J Cancer. 2011;105:796–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lombardo Y, Faronato M, Filipovic A, Vircillo V, Magnani L, Coombes RC. Nicastrin and Notch4 drive endocrine therapy resistance and epithelial to mesenchymal transition in MCF7 breast cancer cells. Breast Cancer Res. 2014;16:R62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gu JW, Rizzo P, Pannuti A, Golde T, Osborne B, Miele L. Notch signals in the endothelium and cancer “stem-like” cells: opportunities for cancer therapy. Vasc.Cell 2012;4:7. doi: 10.1186/2045-824X-4-7.:7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]