

Abstract
The allosteric transition within tetrameric hemoglobin (Hb) that allows both full binding to four oxygen molecules in the lung and full release of four oxygens in hypoxic tissues would earn Hb the moniker of ‘honorary enzyme’. However, the allosteric model for oxygen binding in hemoglobin overlooked the essential role of blood flow in tissue oxygenation that is essential for life (aka autoregulation of blood flow). That is, blood flow, not oxygen content of blood, is the principal determinant of oxygen delivery under most conditions. With the discovery that hemoglobin carries a third biologic gas, nitric oxide (NO) in the form of S-nitrosothiol (SNO) at β-globin Cys93 (βCys93), and that formation and export of SNO to dilate blood vessels are linked to hemoglobin allostery through enzymatic activity, this title is honorary no more. This chapter reviews evidence that hemoglobin formation and release of SNO is a critical mediator of hypoxic autoregulation of blood flow in tissues leading to oxygen delivery, considers the physiological implications of a 3-gas respiratory cycle (O2/NO/CO2) and the pathophysiological consequences of its dysfunction. Opportunities for therapeutic intervention to optimize oxygen delivery at the level of tissue blood flow are highlighted.
Keywords: S-nitroso-hemoglobin, hypoxic vasodilation, oxygen delivery, S-nitrosothiol, erythrocyte, allostery
1. Background
1.1. Hemoglobin, blood gas transport and blood flow
The history of medical science is rich with discoveries relating to blood and hemoglobin (Hb), from the recognition of blood circulation through the body by the pumping action of the heart by Harvey (Harvey, 1628; Ribatti, 2009) and the identification of red blood cells (RBCs) by Leeuwenhoeck using one of the first crude microscopes (Leewenhoeck, 1682). But it was only with the discovery of oxygen gas and the recognition of its role as an oxidizer in the burning of carbohydrate and fat fuels that the reason for respiration and blood circulation was identified with the delivery of the oxygen for cellular metabolism (Sternbach and Varon, 2005). It is now commonly appreciated that delivery of oxygen via circulating hemoglobin is foundational to multicellular life.
Interest in blood and blood cells has continued throughout the development of the medical sciences. Because RBCs are one of the most numerous cell types in the body and are readily available for research, and the hemoglobin they contain is nearly pure already, RBCs and hemoglobin are often the test subjects for new technologies and approaches. The development of the fluid-mosaic model of membrane structure derives in large part from studies of RBCs (Singer and Nicolson, 1972) and concepts of protein structure-function, enzymatic cooperativity and state-dependent allostery derive from classic studies of oxygen binding and release from hemoglobin by Bohr, Haldane and others a century ago (Bohr et al., 1904; Douglas et al., 1912; Henderson, 1920), and from subsequent X-ray crystallography studies by Perutz that provided a molecular mechanism (Perutz, 1962, 1990).
Briefly, hemoglobin consists of a tetrameric complex of two copies each of two proteins, α-globin and some form of β-globin (Perutz, 1978). Multiple distinct β-globin-family genes are present in mammalian genomes and are expressed differentially throughout development (Bulger et al., 1999), and the β-globin isoform within a hemoglobin tetramer determines the hemoglobin type. In adult humans, the predominant Hb form is HbA, which contains β-globin itself. In early development prior to blood circulation, embryos contain HbE, which uses ε-globin, while in later development through shortly after birth, fetal RBCs contain HbG, which contains γ-globin. Adult RBCs also have lower amounts of HbD, which instead contains δ-globin, as well as a small amount of residual HbG.
Each globin protein is bound to a heme cofactor containing an iron atom center, so that each Hb contains four such iron atoms (Perutz, 1962). Hb carries 4 O2 bound to these 4 iron atoms. Whereas iron and oxygen typically combine to form rust where the iron is bound to multiple individual oxygen atoms in a very low energy state, each heme-iron interacts with a single diatomic oxygen molecule and binds loosely enough to be able to release the oxygen gas into hypoxic tissues. The amino acid residues that coordinate and orient heme are therefore among the only residues that are invariant among globin chains (Olson et al., 1988; Perutz, 1962); an adjacent cysteine residue at position 93 in the β-globin isoforms that can interact with heme (Salgado et al., 2011) is also invariant (Nagai et al., 1985) as will be discussed below.
Oxygen binding affinity for heme iron is tightly controlled through intricate molecular machinery within the Hb complex (Perutz, 1990). Functionally, in the presence of high oxygen (atmospheric levels are 20.9%), Hb is fully bound to oxygen and adopts a conformation termed the relaxed or R-state (Perutz, 1962). But when oxygen levels are reduced (as in a hypoxic tissue), Hb releases O2 gas as the complex transitions to the tense or T-state (Perutz, 1962). The sequential binding of oxygen to heme iron within the lung (that is, under high O2 gas partial pressure) is highly cooperative through inducing the allosteric shift from the deoxy R-state to the oxygenated T-state by increasing the affinity of remaining unliganded hemes for O2 (Douglas et al., 1912; Perutz, 1970). Conversely, in tissues in need of O2 (low O2 partial pressure), sequential unloading of each O2 molecule reduces the affinity of Hb for the remaining O2, allowing full unloading under hypoxic conditions (Perutz, 1978). Curiously, despite this highly cooperative full unloading of Hb-bound O2 under full hypoxia in a test tube, the oxygen saturation of venous blood returning to the right heart and lungs from tissues is ~70%. Thus under normal circumstances, Hb operates (on average) between fully liganded at the lung (~99% O2 saturation, all four hemes liganded) and just over one free, unliganded heme per tetramer after leaving target tissues. In addition to ligand-driven allostery, Hb also interacts with other molecules, most notably CO2, 2,3-diphosphoglycerate and protons (i.e., acidic pH), that also modify the allosteric transition and heme affinity for O2 (Bohr et al., 1904; Chanutin and Curnish, 1967). Overall, this Hb-centric model suggests that control of O2 delivery is driven mainly by oxygen levels in lung versus in oxygen-consuming end organs and tissues. But perhaps surprisingly, oxygen delivery is driven by another more important factor, namely blood flow, whose role has been missing from classic models.
That the smallest blood vessels are not simply passive conduits for blood passage has been appreciated for more than a century. In 1878, Severini appears to be the first to have described that small blood vessels change diameter in response to local oxygen levels (Severini, 1878). Seminal experiments by August Krogh (Krogh, 1919, 1922) demonstrated that while the capillaries within in a tissue (muscle in his case) are closely spaced to minimize the distance that gasses must diffuse, those capillaries exhibit dynamic changes between an open state with RBC flow and a closed state where RBCs are excluded or stationary. In a metabolically inactive resting muscle, few capillaries are open at any one moment. Observation over time, however, revealed that the capillaries that were open to flow soon closed and others that had been closed then open to take their place in perfusing the tissue, so that all regions of the tissue receive intermittent direct (nearby) blood flow with longer periods of more distant flow. In this way, the basal level of O2 delivery is maintained to support cell viability. Stimulating the muscle (and thus oxygen demand to support aerobic metabolism) led to profound increase in the number of open capillaries (until at very high activity, essentially all capillaries are open to flow) and thus to a great decrease in the average distance for gas diffusion, thereby increasing tissue O2 and nutrient delivery as well as CO2 removal (Krogh, 1919, 1922).
Just how blood flow through the tissue microcirculation is controlled spatially and temporally remained unknown for some time. Current understanding includes multiple distinct mechanisms that act in different vessels and at different timescales, including pre-capillary sphincters, shunt vessels and metabolic and electrical coupling of tissue and vasculature (Fung and Zweifach, 1971; Gutterman et al., 2016). For the purposes of this discussion, we are particularly concerned with how microcirculatory blood flow responds to instantaneous, local tissue oxygen demand within the Hb-centric model of the respiratory cycle. In classic physiology experiments in the 1960’s, Arthur Guyton and colleagues demonstrated that tissue blood flow increases linearly with desaturation of hemoglobin, a response that maintains sufficient oxygen delivery to match an increase in metabolic activity (Figure 1A). In their experiment, these researchers connected one lung, through a pump system maintaining constant blood pressure, to one leg of an anesthetized canine, and controlled the inspired oxygen concentration given to that lung to induce tissue hypoxia while measuring blood flow in the leg muscle (Ross et al., 1962). They observed that blood flow rose (that is, tissue resistance to blood flow fell) in direct proportion to decreases in HbO2 saturation (not PO2). Because the leg muscles were inactive during this procedure and the CO2 levels of the input blood was low, the authors argued that metabolic CO2 was unlikely to be the driver of hypoxic vasodilation to increase blood flow, and similarly that tissue release of a circulating vasodilatory substance was also unlikely, in that other tissues under normoxia did not exhibit vasodilation too. Instead, they concluded that some oxygen sensor was required to respond to oxygen lack within the hypoxic tissue itself, but the identity of this sensor remained unknown.
Figure 1.
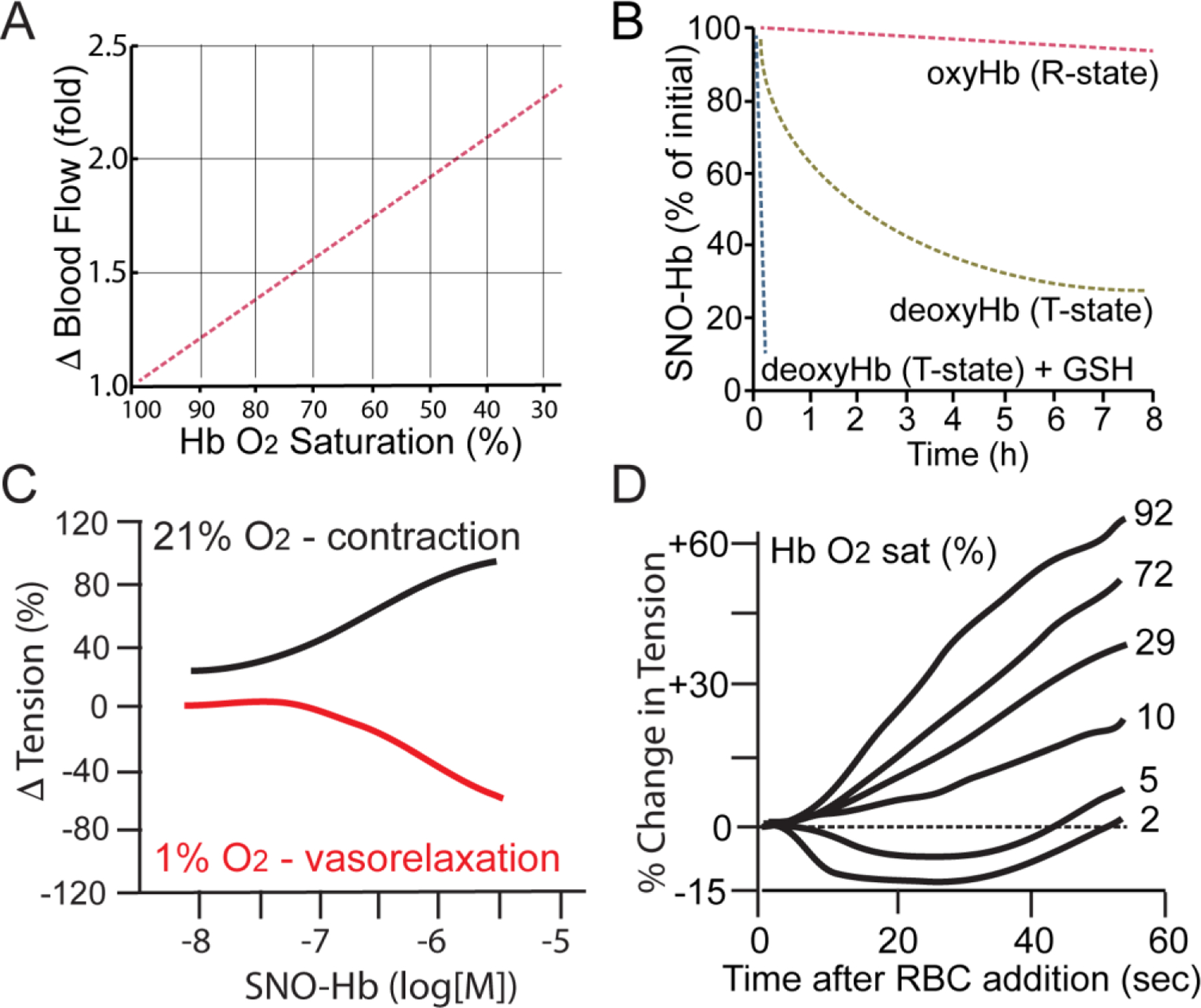
Role of allostery in hypoxic vasodilation. SNO-Hb mediates tissue blood flow increase in hypoxia in proportion to Hb deoxygenation (see also Figure 3). A. Limb blood flow increases linearly with decreasing oxygen saturation of the perfusing blood. Adapted from (Ross et al., 1962). B. Oxygenation-state allostery controls the reactivity of SNO-Hb. Isolated SNO-Hb in the oxygenated R-state is stable, but is destabilized and reactive in the in hypoxic T-state; loss of SNO from T-state Hb is potentiated by glutathione (GSH) serving as a SNO acceptor. Adapted from (Jia et al., 1996). C. SNO-Hb induces hypoxic vasodilation. Isolated SNO-Hb produces dose-dependent contraction of isolated aorta in an organ bath under room air (21% oxygen, black) but vasodilation under hypoxia (1% oxygen, red). Adapted from (McMahon et al., 2000). D. Human RBCs dilate blood vessels in proportion to Hb desaturation. Adapted from (McMahon et al., 2002).
1.2. NO and vasodilation
Experiments in the early 1980’s by Furchgott and colleagues showed that vasorelaxant drugs act on large blood vessels through a mechanism that requires endothelial cell release of a substance they named endothelium-derived relaxation factor (EDRF) (Furchgott and Zawadzki, 1980). In classic experiments, Moncada and colleagues and Ignarro and colleagues demonstrated that EDRF had properties of a gaseous free radical consistent with being nitric oxide (NO) (Ignarro et al., 1987; Palmer et al., 1987). Later, Hibbs and Moncada would show that the body can synthesize NO gas from the amino acid arginine (Hibbs et al., 1987; Moncada et al., 1989; Palmer and Moncada, 1989). Separately, Murad and colleagues had determined that the long-known effect of nitroglycerin (used in the treatment of angina) to produce vasorelaxation was likely due to generation of NO, which activated soluble guanylyl cyclase to generate cGMP (Arnold et al., 1977). Thus, vasorelaxation in the control of blood pressure is regulated through an endothelial cell– G protein-coupled receptor – endothelial nitric oxide synthase – NO – cGMP signaling cascade (Vallance and Moncada, 1994). This quickly became the established signaling mechanism for NO in the vasculature and beyond, and RBCs in this model served to scavenge NO and block vasodilation. However, in the Guyton hypoxic vasodilation experiments, (Ross et al., 1962), RBCs were clearly identified with increases in blood flow, whereas scavenging NO would decrease blood flow, so NO gas does not have the properties needed to be a mediator of hypoxic vasodilation to maintain oxygen delivery.
1.3. Classic role of heme in blocking NO bioactivity
One main way that EDRF and NO were compared during their identification was by showing that both are equivalently inhibited by hemoglobin (Ignarro et al., 1987; Palmer et al., 1987). Indeed, when NO reacts with oxygenated hemoglobin (HbFeO2) it is rapidly converted to inactive nitrate (NO3−), thus limiting arterial vasodilation (Owusu et al., 2012; Su et al., 2020). In addition, NO can bind vacant hemes in hemoglobin, forming an inactive FeNO complex.. However, if NO sequestration were the entire story, Hb/RBCs would paradoxically impair oxygen delivery. NO does in fact bind to vacant hemes, but mainly in deoxygenated venous blood returning to the lung, which does not affect blood flow. Here it participates not in NO elimination, but rather in a reaction pathway for regenerating NO-bioactivity through S-nitrosothiols (SNOs), as will be described below. Altogether, the original model for NO in which Hb serves as an NO scavenger via hemes is incomplete.
1.4. S-nitrosylation as a means to preserve NO bioactivity
Fortunately, NO bioactivity and signaling capacity is not limited to NO itself activating a single target protein (soluble guanylyl cyclase) through a single mechanism (binding to heme). In fact, the scope of NO signaling is far broader than was imagined in the traditional cGMP paradigm. In 1992, Stamler and colleagues reported the existence of endogenous S-nitrosothiols (SNOs), where NO is attached to the sulfur atom of a thiol (SH) as in cysteine residues in proteins (Stamler et al., 1992c), and demonstrated that these SNOs exhibit biological activities (Stamler et al., 1992a; Stamler et al., 1992c) that are resistant to heme inactivation. In particular, SNO-proteins or SNO-donor molecules added to blood vessels produce vasodilation through smooth muscle relaxation (Stamler et al., 1992b; Stamler et al., 1992c) and do so in blood effectively (Keaney et al., 1993; Palmer et al., 2007). Since that discovery, S-nitrosothiols have been identified on thousands of proteins (e.g., (Li et al., 2021)) and on numerous cellular thiols (e.g., glutathione, coenzyme A, cysteine), and S-nitrosylation has emerged as a bona fide cellular signaling modality emanating from signaling pathways that generate NO (Hess et al., 2005; Hess et al., 2001; Hess and Stamler, 2012).
Initial resistance to these ideas stemmed from questions surrounding chemistry of SNO formation under physiological conditions. SNO-forming reactions of the free radical NO gas (NO•) with a thiol formally entails redox chemistry to remove one electron from the NO• to form nitrosonium (NO+) or from free thiol (RSH) to form thiyl radical (RS•). Transition metal catalysts enable this chemistry (e.g., Fe3+–NO is in equilibrium with Fe2+–NO+ through charge transfer) (Stamler et al., 1992c). Notably, hemoglobin catalyzes the conversion of FeNO to SNO, representing SNO synthase activity (Angelo et al., 2006; Nagababu et al., 2006). Structural and redox requirements for this reaction are met through the allosteric transition of mixed valency hemoglobins (e.g., Hb[Fe(III)NO][Fe(II)]3), which form SNO (e.g., SNO-Hb[Fe(II)O2) upon oxygenation.
Once converted to SNO, NO bioactivity is independent of heme-based inhibition. The ability to convert NO gas into SNO thus preserves NO bioactivity, since SNO is able to activate soluble guanylyl cyclase too (Diesen et al., 2008; Ignarro and Gruetter, 1980). More importantly, formation of SNO also greatly expands the range of bioactivity of NO by introducing a completely distinct mode of action for NO-derived signals (Hess et al., 2005; Hess et al., 2001).
1.5. Enzymatic activity of an ‘honorary enzyme’.
Nitrosothiol addition to a protein is termed S-nitrosylation and acts as a post-translational modification, much like phosphorylation or ubiquitination, and can modify functions of target proteins (Hess et al., 2005). These changes can include binding to an active site cysteine to inhibit an enzymatic activity directly, altering the stability or partner binding of the target protein to alter function, to simply shifting the protein allosteric state to change function (Hess et al., 2005). Examples have been demonstrated for these regulatory effects across all classes of proteins (~25,000 SNO sites in 10,000 proteins). Protein S-nitrosylation thus forms the basis for ubiquitous signaling by NO.
One fundamental and key property of S-nitrosothiols that differs substantially from the more-familiar protein phosphorylation paradigm is that under appropriate conditions, the SNO nitroso group can transfer to another thiol without input of additional energy (Hess et al., 2001). While originally conceived of as a stochastic chemical process, it has become clear that transfer of SNO groups between proteins can be greatly facilitated by protein-protein interactions, and thus SNO-proteins that transfer their SNO to defined sites on specific target proteins can be categorized as “transnitrosylase” enzymes (Stomberski et al., 2019). Increasing numbers of protein-based SNO-transfer reactions are being identified, and are mediated by S-nitrosylated proteins include GAPDH and S100A8/S100A9 (Jia et al., 2014; Kornberg et al., 2010). Moreover, emerging evidence is suggesting that even NO donors work through enzymes to S-nitrosylate proteins.
Hemoglobin was one of the first proteins to be identified as being modified by SNO (Jia et al., 1996) and the first to show enzymatic activity in S-nitrosylation of proteins (Angelo et al., 2006; Pawloski et al., 2001; Singel and Stamler, 2005). The primary site of S-nitrosylation of the hemoglobin tetramer has been mapped to the β-globin Cys93 residue (βCys93) (Chan et al., 1998; Stamler et al., 1997a). As noted earlier, the βCys93 residue is among the very small set of absolutely conserved residues in the β-globin isoforms, along with the residues coordinating heme iron (Perutz et al., 1965). Importantly, de novo S-nitrosylation of βCys93 is catalyzed by the β–globin heme (SNO synthase activity) and SNO-hemoglobin can then transfer its NO group to proteins in the RBC membrane representing transnitrosylase activity (Pawloski et al., 2001). Thus, whereas hemoglobin was originally viewed as a model allosteric protein without catalytic activity, leading to it being termed an “honorary enzyme” (Imai, 1999; Wyman Jr and Allen, 1951), in fact hemoglobin provides undergirding for the enzymatic model of S-nitrosylation (Figure 2A–E) (Seth et al., 2018; Zhou et al., 2021), entailing multi-enzyme machinery including: 1. Heme-NO generation (e.g., nitrite reductase activity); 2 conversion of NO to SNO (SNO synthase activity) and SNO transfer among proteins (transnitrosylase activity). But whereas the concerted actions of 3 enzymes is usually required (Seth et al., 2018), Hb is self-sufficient in all three enzymatic activities (Angelo et al., 2006; Pawloski et al., 2001).
Figure 2.
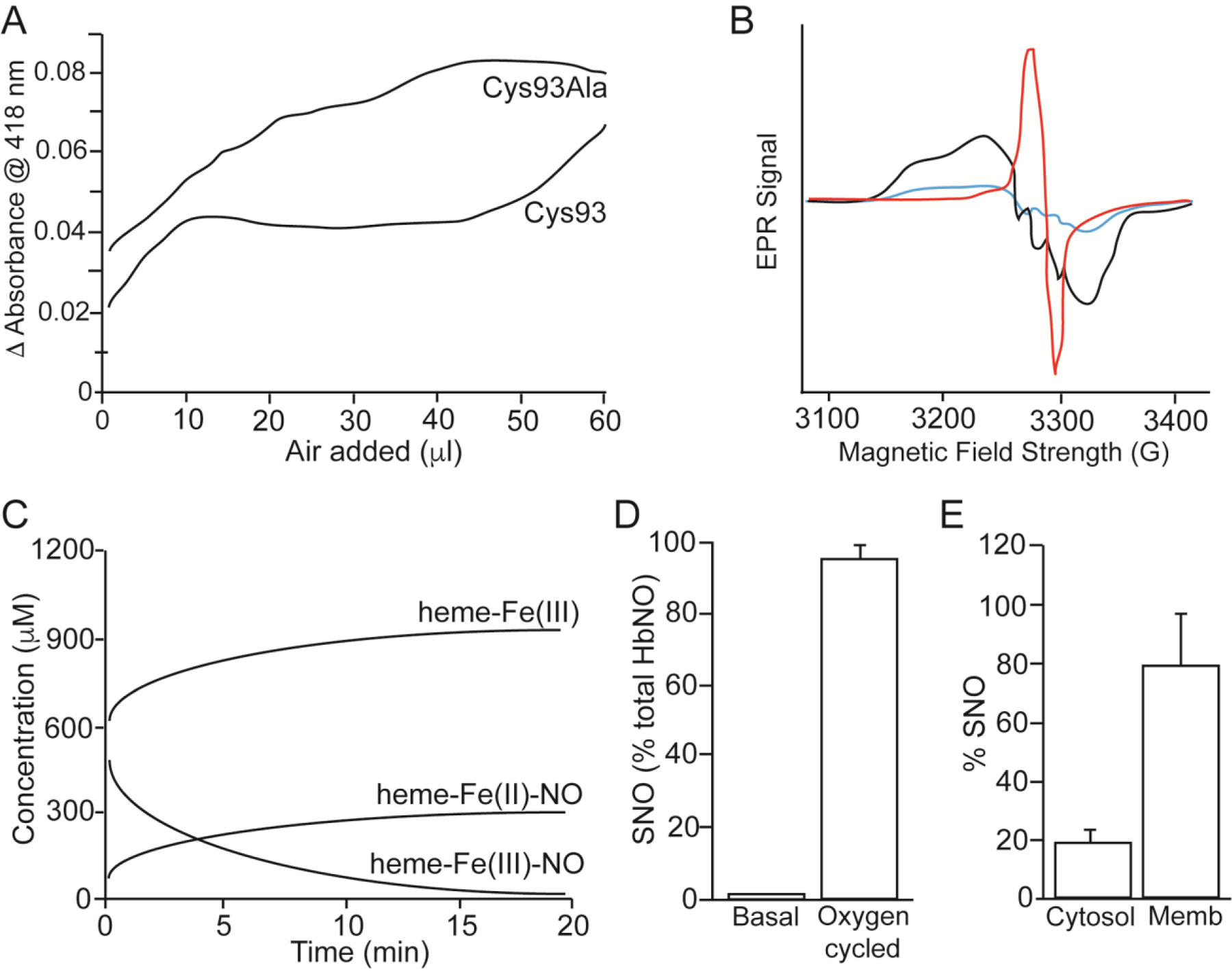
Hb as a SNO-generating enzyme. I. Converting FeNO to SNO through SNO synthase activity (A-D). A. Absorbance spectroscropy of Hb-bound FeNO as a function of oxygen (room air) added to the sample, comparing recombinant human Hb and the Cys93Ala mutant, showing that the Cys93Ala mutant lacks a ‘plateau phase’ (from ~15–45 ul) representing loss of FeNO in production of SNO, whereas the wildtype Hb demonstrates O2-dependent displacement of NO from heme. Adapted from (Gow and Stamler, 1998). B. Electron Paramagnetic Resonance (EPR) measurements of nitrite/Hb under deoxy (black)/oxy (red)/deoxy (blue) cycling. FeNO Hb (black) produced from nitrite under deoxy conditions; loss of this FeNO spectrum upon oxygenation (red), consistent with transfer of NO from heme to Cys to form SNO (note: a newly formed Cys radical is observed; red); and regeneration of FeNO under deoxygenation (blue), consistent with release of NO from SNO upon return to the deoxygenated state (blue), as described by (Pezacki et al., 2001). Adapted from (McMahon et al., 2002). C. Absorbance spectroscropy of metHb over time under deoxygenated conditions in the presence of NO, demonstrating increase in Fe(II)-nitrosyl Hb (heme-Fe(II)NO), and decrease in Fe(III) nitrosyl Hb (heme-Fe(III)NO), indicative of reductive nitrosylation via heme-Fe(III) that precedes SNO formation. Contemporaneous measurements confirm the formation of SNO-Hb. Adapted from (Luchsinger et al., 2003). D. Hb is SNO synthase. Treatment of Hb by cycling between deoxygenated state to oxygenated state in the presence of nitrite leads to near-100% formation of SNO-Hb. Adapted from (Angelo et al., 2006). II. Hb is a transnitrosylase. E. Treatment of deoxygenated RBCs with 1 μM NO (to create FeNO) and subsequent oxygenation (to promote SNO-Hb formation in cytosol) followed by deoxygnation promotes transfer of SNO to membrane-assocated proteins (including AE-1). Adapted from (Pawloski et al., 2001).
1.6. The 3-gas respiratory cycle
The ability of hemoglobin to carry SNO on the βCys93 residue and the ability of SNO to mediate NO bioactivity suggests that Hb βCys93-SNO functions as a store and dispenser of NO bioactivity, and has the properties expected of the proposed mediator of the hypoxic autoregulator. Indeed, Hb βCys93-SNO specifically functions to release vasoactive SNO in the hypoxic tissue microvasculature to increase microvessel blood flow and oxygen delivery. Evidence supporting this model includes the ability of isolated SNO-Hb and SNO-replete RBCs to increase microvascular blood flow under hypoxic conditions (Reynolds et al., 2013; Stamler et al., 1997a); the inverse proportional dependence of increased microvessel blood flow on decreased Hb O2-saturation (Reynolds et al., 2007); the proportionality of release of SNO by RBCs with Hb deoxygenation (Doctor et al., 2005); the presence of arterial-venous gradients in SNO-Hb consistent with dispensing SNO in hypoxic tissues (McMahon et al., 2002); the in vitro ability of RBCs to relax aortic strips being entirely dependent on hypoxic conditions (RBC vasocontrict in normoxia) (Jia et al., 1996; McMahon et al., 2002) and is lost by storage-mediated denitrosylation but restored by renitrosylation (Reynolds et al., 2007). Most importantly, genetic evidence has been obtained from the βCys93Ala (C93A) mouse model, where mice carry human HbA and HbF (knock-in of the α-globin, β-globin and γ-globin genes) and have a mutation in β-globin (Cys93Ala) where HbA cannot be SNO-modified at βCys93 (Premont et al., 2021; Premont and Stamler, 2020). In the C93A mice, basal tissue oxygenation is reduced to compared to control C93 mice, and tissue blood flow does not increase normally as mice are exposed to a reduced oxygen environment (Zhang et al., 2015). Profound hypoxic phenotypes in these mice will be described more completely in a later section, as will the biochemical basis for hypoxic vasodilation by SNO-Hb, which lies in ‘thermodynamic linkage’ between hemoglobin conformation and reactivity of Cys93 under deoxygenated conditions.
Interestingly, RBCs can synthesize their own NO. RBCs have been shown to express eNOS protein and to contain NO synthetic activity that is blocked by pharmacological NOS inhibitors such as L-NAME (Cortese-Krott and Kelm, 2014). Recent evidence from endothelial-specific vs. RBC-specific eNOS knockout mice demonstrates that both endothelial and RBC eNOS contribute to NO-mediated blood pressure regulation (Leo et al., 2021). The role of RBC vs. endothelial eNOS in SNO-Hb formation or function was, unfortunately, not assessed, but seems likely to be an important contributor to the overall formation of Hb βCys93-SNO within RBCs. Furthermore, nitrite anion produced by NO synthases in the body may likely serve as a source of NO in RBCs that converts to SNO-Hb (Angelo et al., 2006).
Thus, hemoglobin functions to facilitating the transport of three physiological gasses, O2, CO2 and NO, and the respiratory cycle should be conceived as a three-gas cycle (McMahon et al., 2002). While O2 is transported from air into metabolically-active tissues and metabolic CO2 from tissues to the atmosphere as waste, NO is synthesized by endothelial cells, platelets, white blood cells and within RBCs themselves, circulated as SNO-Hb, and delivered into hypoxic tissues along with O2. Importantly, transport of O2 and NO (as S-nitrosothiol) are linked allosterically, such that O2 release into hypoxic tissue also liberates SNO in a graded manner: more O2 release into more deeply hypoxic tissue leads to more SNO release.
2. Biochemistry of S-nitroso-hemoglobin
2.1. Hb allostery is linked to SNO stability vs release
The β-globin Cys93 residue (βCys93) is the most reactive cysteine thiol in the hemoglobin tetramer (Stamler et al., 1997a; Sun et al., 2019) and the major site that is modified in SNO-Hb (Chan et al., 1998; Stamler et al., 1997a), reflecting a surrounding motif for S-nitrosylation (Stamler et al., 1997b). Studies have shown that the reactivity of βCys93 varies with the conformational state of the Hb tetramer and is thus regulated through the allosteric transition: in the oxygenated R-state of Hb, βCys93 free thiol is more reactive and SNO-βCys93 is protected from solvent (i.e, reaction with other thiols), compared to Hb in the deoxygenated T-state (Jia et al., 1996; Stamler et al., 1997a). Therefore, SNO-Hb is relatively stable under high oxygen saturation in the arteries, but becomes increasingly more reactive as Hb encounters reduced oxygen in the microcirculation (Figure 1B). One consequence is that this reactivity varies with the allosteric state of the hemoglobin pool, and thus SNO release within a given RBC will be a relatively linear function of Hb O2 saturation rather than a sharp transition (as at 50% O2 saturation, for example) (Figure 1C,D). For SNO-Hb, this results in release of SNO to other thiols with the linear responsivity expected of the Guyton blood flow hypoxic-autoregulation sensor (Premont et al., 2020; Ross et al., 1962). For this reason, SNO-Hb was proposed to mediate flow autoregulation directly, through hypoxia-dependent release of vasodilatory S-nitrosothiol (Jia et al., 1996; Stamler et al., 1997a).
It is important to keep in mind that under normal physiological conditions, S-nitroso-hemoglobin (SNO-Hb) comprises only <1% of the Hb pool (Hausladen et al., 2007). That is, most Hb molecules do not have SNO attached and do not participate in SNO-mediated functions. Nevertheless, most SNO-Hb bioactivity resides within the membrane compartment associated with band 3 (Pawloski et al., 2001), where SNO-Hb represents a large fraction of the total Hb. Thus, the activity of Hb is compartmentalized.
One implication of this hypoxic SNO-release and normoxic SNO-rebinding mechanism is that arterial blood is expected to have higher SNO-Hb than venous blood, as SNO is liberated from Hb as RBCs pass through hypoxic tissues and used for vasorelaxation. Comparison of arterial and venous SNO-Hb levels have revealed a significant drop in SNO-Hb level in venous blood (Jennings, 2013; McMahon et al., 2002; Reynolds et al., 2007) when blood gases are performed under airtight conditions and samples analyzed while maintaining Hb oxygenation state (Figure 4A). Moreover, SNO levels within RBCs are directly proportional to HbO2 saturation across a wide range of O2 concentrations, reflective of arterial–venous differences in vivo (Doctor et al., 2005). It must be noted, however, that some other groups have seemingly failed to measure such arterial–venous differences (Bailey et al., 2017), but their measurements were performed in room air where such differences in O2/SNO have been shown to be eliminated (McMahon et al., 2002). It has also been shown that these failures are attributable to the SNO quantification methods used (Hausladen et al., 2007; Stamler et al., 2017).
Figure 4.
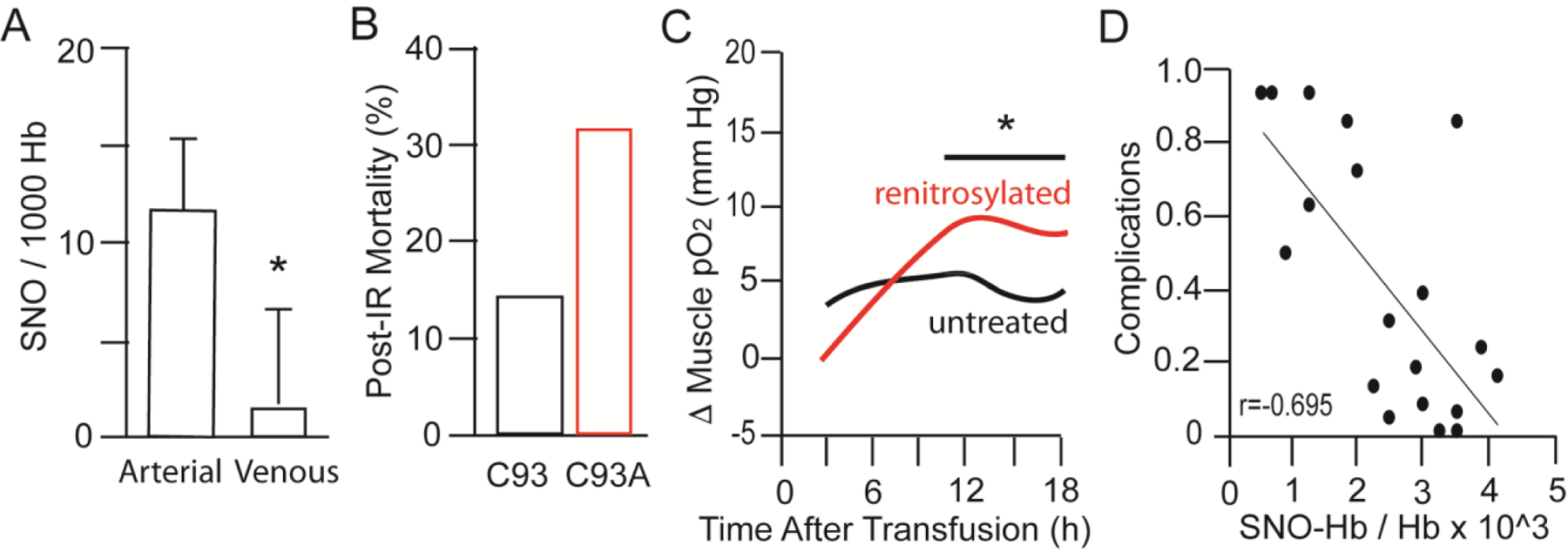
Clinical manifestations of SNO-Hb function. A. Arterial-venous difference in SNO-Hb content in normal humans breathing room air. SNO-Hb is high in oxygenated arterial blood and lower in deoxygenated venous blood (protected from air) consistent with allosteric regulation of SNO bioactivity. Adapted from (McMahon et al., 2002). B. Mice lacking βCys93 SNO-Hb exhibit elevated rate of death after myocardial infarction consequent upon ischemia-reperfusion injury. Adapted from (Zhang et al., 2016). C. Tissue oxygenation is elevated in sheep transfused with 2 units of blood renitrosylated by treatment with ethyl nitrite vs blood left untreated and deficient in SNO-Hb. Adapted from (Reynolds et al., 2013). D. Reduced complications in pediatric bypass patients after transfusion correlates with patient SNO-Hb level post-transfusion. Adapted from (Matto et al., 2018).
2.2. SNO synthase function of hemoglobin micropopulations
While NO gas can bind directly to heme iron to form iron-nitrosyl (FeNO), it cannot react directly with a cysteine thiol to form an S-nitrosothiol. Instead NO must be redox-activated through loss of one electron to form nitrosonium ion (NO+) (Hogg, 2002; Singel and Stamler, 2005; Stamler et al., 1992d). This is readily accomplished through NO binding to ferric (Fe3+) heme, as noted above. More specifically, while heme in Hb is mainly held at a redox state near Fe2+ overall, this redox level varies with the allosteric and ligation state of Hb, with the R-state being more shifted toward Fe3+ and the deoxy state toward Fe2+ (Sánchez et al., 2017). Oxygen bound in R-state Hb exists formally as superoxide O2− (Fe3+− O2−) reflecting change transfer between Fe2+ and O2 (van Zwieten et al., 2014). Similarly, Fe3+–NO is in equilibrium with Fe2+–NO+ (Nagababu et al., 2006; Salgado et al., 2011), providing a clear conceptual route to the redox activation of NO required for SNO synthesis (Singel and Stamler, 2005). Indeed, it has been shown that allosteric transition of Hb from T-state to R-state in the presence of NO gas is able to convert that NO to Hb-bound SNO (Angelo et al., 2006; Doctor et al., 2005; Gow and Stamler, 1998; Luchsinger et al., 2003; McMahon et al., 2002; Pawloski et al., 2001). That is, Hb functions as a SNO-synthase enzyme (Figure 2A–D).
Compared to canonical Hb[Fe(II)4]/Hb[Fe(II)O2]4 within the cytosol, which constitutes the majority of Hb, most SNO-Hb forms within mixed valency micropopulations (Angelo et al., 2006; Herold and Rock, 2003; Nagababu et al., 2006; Singel and Stamler, 2005). These SNO-generating micropopulations include ferric-heme that can oxidize NO, for example, Hb[Fe(II)3Fe(III)1] (Figure 2C). It had been proposed that oxidation of NO may occur in the ferrous complex of HbFe(II)NO, entailing electron transfer to a ferric heme within the same dimer (Angelo et al., 2006; Luchsinger et al., 2003; Singel and Stamler, 2005). Moreover, it has been shown that Hb β–chains preferentially support SNO-Hb formation (Angelo et al., 2006; Hausladen et al., 2007; Pezacki et al., 2001; Salgado et al., 2009; Singel and Stamler, 2005), and further that this function is enhanced upon upon oxygenation. That is, oxygenation of Hb-βFe(II)NO supports both the structural and redox requirements for S-nitrosylation (Angelo et al., 2006). Taken together, these data support the idea that oxidation of NO within β-Fe(II)NO complexes is allosterically coupled to migration of NO from hemes to thiols. In addition, distinct cytosolic vs. membrane-associated pools (De Rosa et al., 2008; Pawloski et al., 2005; Singel and Stamler, 2005) each account for 50% of SNO-Hb. Thus, ~1% of total Hb residing within the membrane (including hemichromes) accounts of 50% of SNO-Hb. Notably, Hb binds to the Band 3 anion exporter in the RBC membrane (as well as other membrane proteins), defining a subset of Hb molecules closely apposed to the RBC membrane, and it has also been shown that this membrane-associated pool of Hb is enriched in metHb (Fe3+ forms) (De Rosa et al., 2008) and that metHb reacts more efficiently with NO to form SNO-Hb compared to oxyHb or deoxy-Hb (Herold and Rock, 2003). As NO diffuses into RBCs, therefore, these membrane-associated Hbs are the first molecules that NO is likely to encounter. Furthermore, the native level of SNO-Hb in human RBCs freshly-isolated from venous blood is approximately 1 SNO per 1000 Hb tetramers (Reynolds et al., 2007), and this SNO-Hb is also enriched in Hb located near the RBC membrane (Pawloski et al., 2001). Thus, a model for Hb function as a SNO synthase involves NO gas binding to β-globin Fe3+ heme in Hb decorating the RBC membrane and being converted to SNO at βCys93 as Hb undergoes the oxygenation-dependent allosteric transition to the R-state (Angelo et al., 2006).
The SNO synthase function of Hb may also rely on Hb acting as a nitrite reductase enzyme to convert nitrite to FeNO (Doyle et al., 1981; Huang et al., 2005; Nagababu et al., 2006). This activity requires that nitrite bind to Fe(II) heme in Hb, and generates NO and Fe(III) heme in Hb (metHb) as products (Doyle et al., 1981; Huang et al., 2005; Nagababu et al., 2006). NO formed in this way will not exist free in solution but rather will be bound to heme iron and thereby contribute to formation of mixed valency micropopulations (Angelo et al., 2006) that support formation of SNO-Hb (Angelo et al., 2006; McMahon et al., 2002). Comparing oxygenated Hb and deoxygenated Hb, it was found that the Hb nitrite reductase activity was maximal at the oxygen p50 where oxy/deoxy-Hb states are in an equimolar ratio (because while the reductase activity is higher in the oxygenated state, the ability of nitrite to bind to heme to be a substrate necessitates that heme is not liganded by oxygen) (Huang et al., 2005). This redox/structural feature of Hb holds true also for O2 conversation into superoxide, which is maximal at p50, and far exceeds amounts of NO generated from nitrite. Thus, any NO generated through nitrite reductase activity would either be sequestered by heme or eliminated by superoxide.
It should be noted that such p50-dependence of nitrite reduction has been touted as critical to achieve hypoxic vasodilation by generating NO (Huang et al., 2005). However, the inability of NO to escape superoxide and hemes would seem to eliminate this possibility, and the whole notion of p50 is misguided because hypoxic vasodilation actually increases linearly from p100 to p0 (Ross et al., 1962), excluding a role for nitrite reductase activity in direct formation of free NO (McMahon et al., 2003). In fact, no experiments have demonstrated that NO produced from nitrite by Hb in native concentrations, actually escapes RBCs to produce any vasoactivity (Premont et al., 2020; Stamler et al., 2017). With that said, Fe(III)NO generated from nitrite is positioned to be converted into SNO-Hb during Hb deoxygenation/oxygenation, as noted above (Angelo et al., 2006; Luchsinger et al., 2003; Nagababu et al., 2006), and RBCs have been shown to release SNO under hypoxia (i.e., in the form of GSNO formed from glutathione added outside the RBCs) (Lipton et al., 2001). Thus, the most parsimonious explanation for NO-bioactivity is SNO-based.
2.3. SNO transnitrosylase function of hemoglobin
Once SNO is formed on Hb, it must be transferred to other target proteins in order to have a biological effect. Because the transfer of SNO from one protein to another requires precise positioning of the donor SNO and the recipient thiol group (likely requiring direct protein-protein contact), this transfer is considered to be enzymatic (Nakamura and Lipton, 2013; Seth and Stamler, 2011). That is, SNO-Hb as a donor of SNO acts as a SNO transferase enzyme. Due to the allosteric regulation of SNO βCys93 reactivity, this occurs preferentially when Hb is in its deoxygenated T-state (Lipton et al., 2001; Stamler et al., 1997a). Accordingly, SNO-Hb is a poor donor of SNO until the Hb is under hypoxic conditions. SNO at non-βCys93 sites in Hb will not be subject to this limitation, and could be transferred equivalently regardless of oxygenation level and allosteric state of the Hb. For this reason, SNO-Hb acts as a selective source of vasodilatory SNO under hypoxia, whereas the vasoconstrictive action of Hb through NO gas scavenging by heme predominates under oxygenated conditions.
SNO from Hb can be transferred to the band III anion transporter AE-1 (Pawloski et al., 2001) and to protein disulfide isomerase (Kallakunta et al., 2013) at the membrane, but likely other proteins as well (Figure 2E). In the case of AE-1 and protein disulfide isomerase (PDI), however, the transfer of SNO to intracellular sites on these proteins has been linked to the further transfer of SNO to sites on their extracellular side (Kallakunta et al., 2013; Pawloski et al., 2001), leading to a ‘bucket brigade’ coupled-equilibria model where increased reactivity of SNO-βCys93 Hb within hypoxic RBCs drives SNO to new sites on partner proteins especially at the membrane, leading to RBCs becoming coated with SNO during the moment they are in the smallest microvessels.
SNO from Hb also can be transferred to the thiols of cellular metabolites within RBCs that mediate biological activities. One prominent target is glutathione (GSH) (Figure 1B). It has been shown via direct liquid chromatography/mass spectroscopy that GSH added to blood can be converted to SNO-glutathione (GSNO) when the blood is subjected to hypoxia (Lipton et al., 2001) but not normoxia. These results are fully consistent with SNO-Hb being stable even in hypoxic conditions in the absence of GSH, but very rapidly undergoes loss of SNO when excess GSH is added (Bonaventura et al., 1999; Doctor et al., 2005; Jia et al., 1996), suggesting that Hb can only exert bioactivity through SNOs. That RBC-resident SNO-Hb is coupled with extracellular GSH/GSNO is clear from the aforementioned study, where hypoxia induces RBC-mediated production of GSNO in extracellular plasma (Doctor et al., 2005; Lipton et al., 2001). Whether GSNO is exported as such from RBCs, or formed by reaction of GSH with membrane SNOs, remains unresolved. But outside the RBC, GSNO has access to the endothelia. GSNO acts as both vasorelaxant and bronchorelaxant (Gaston et al., 1994; Gaston et al., 1993; Jansen et al., 1992; Smith et al., 1994) through its ability to transfer the SNO group to target proteins that regulate smooth muscle contraction. Another potential role for SNO comes from studies of SNO-cysteine, which appears to act in a SNO-transfer-independent manner as a signaling molecule to activate the voltage-gated Kv1.1 potassium channel via direct binding (Gaston et al., 2020) and hydrogen sulfide has been implicated in SNO export from RBCs as well (Ivanovic-Burmazovic and Filipovic, 2019). Whether GSNO has equivalent direct roles remains unexplored.
That RBC-borne SNO affects endothelial cells is unsurprising, since in the smallest capillaries the RBC membrane makes intimate contact with the vessel wall as both deform during passage. SNO groups could readily transfer from RBC membrane proteins to endothelial membrane proteins during this contact. More surprising is that in larger vessels, RBC-derived SNO dilates smooth muscle. At the minimum, SNO on endothelial cell membrane proteins must control signaling that regulates smooth muscle contraction, but this also suggests that SNO transports through the membrane out of RBCs and into/onto endothelia, and further into smooth muscle cells (and beyond) through SNO transferases in coupled equilibria.
2.4. SNO/FeNO recycling upon oxygenation/deoxygenation
Once an RBC has passed through a capillary in a hypoxic tissue, it recovers both O2 and NO. Excess O2 rebinds to Hb through countercurrent exchange in venules apposed to arterioles, so that the O2-saturation of mixed venous blood reaching the right heart is 70–75% (that is, of the 4 O2 molecules carried by Hb from the lungs, only 1 is actually delivered to tissues on average). This level of O2 binding is sufficient to return Hb to the R-state in 75% of molecules whereas 25% remain in deoxy (T)-state available to pick up NO on vacant hemes during transit to the lungs. In the lungs, the allosteric transition to R-state is completed and, SNO-Hb enters the arterial circuit where it is then exposed to oxygen gradients in smaller vessels that release NO from SNO-Hb (McMahon et al., 2002; Stamler et al., 1997a). Released NO takes two routes (McMahon et al., 2000; Pezacki et al., 2001; Singel and Stamler, 2005): i. export out of the RBC via transfer to other thiols as described above (Pawloski et al., 2001); ii. auto-capture at hemes within the β-chain to form βFe(III)NO. In this way, a majority of SNO-Hb is converted to βFe(III)NO (Pezacki et al., 2001; Salgado et al., 2009) in T-state and thereby conserved during arterial-venous transit. This recycling of SNO/FeNO is critical, as the quantity of SNO on Hb is very high compared to amounts of NO/SNO required for vasodilation (1/1000th of that carried) and compared to the body’s capacity to synthesize NO (Castillo et al., 1996). In fact, the majority of NO in RBCs is carried in the form of SNO/Fe(III)NO (Hausladen et al., 2007; Salgado et al., 2009; Singel and Stamler, 2005) and recycled repeatedly, whereas only a very small fraction of SNO is exported to induce vasodilation (McMahon et al., 2002).
3. Physiological Roles of S-nitroso-hemoglobin
3.1. The RBC/endothelial cell unit
SNO-Hb has been reported to have numerous physiological roles. These roles are based on studies with purified Hb and isolated RBCs, following addition of SNO-Hb or SNO-repleted RBCs into animals, and using mutant mice with inability to carry SNO at βCys93 (Premont et al., 2021; Premont and Stamler, 2020). The foundational physiological role of SNO-Hb is to preserve and transport NO bioactivity in the form of S-nitrosothiol, from its source of synthesis (endothelium or elsewhere) around the body to produce SNO-dependent responses. Because SNO release from βCys93 is dependent on the T-(high spin)-state of Hb, SNO delivery by SNO-Hb is primarily to hypoxic tissues. SNO from Hb in the T-state then moves to nearby thiols, and since most SNO-Hb within an RBC is concentrated at the RBC membrane (Pawloski et al., 2001), much of this liberated SNO is transferred to membrane proteins including AE1 and PDI, but also to low molecular weight thiols like glutathione. Through coupled equilibria, membrane protein SNO groups are thought to be transferred from the inside domains of the membrane proteins to their extracellular domains, leading to an RBC that is decorated by extracellular SNO (Pawloski et al., 2001), which serves as a source of vasodilatory activity. As this RBC transits a capillary in a hypoxic tissue, some of these SNO groups are proposed to be transferred to proteins on capillary endothelia, leading to capillary changes that facilitate the transit of RBCs that follow (Premont et al., 2020). At this time, the identity of endothelial proteins that accept SNO from hypoxic RBCs remain unknown, but may include endothelial cell-surface tissue transglutaminase (Lai et al., 2017), which can accept and deliver SNO.
3.2. O2 delivery to hypoxic tissues
Blood flow, not oxygen carrying by Hb, is the principal determinant of oxygen delivery under normal physiological conditions (Premont et al., 2020). In this regard, the first role of SNO released from SNO-βCys93 Hb is to increase the flow of RBCs through hypoxic tissues, where the extent of flow increase is proportional to the depth of hypoxia (Ross et al., 1962). This increased flow maximizes O2 delivery, matching oxygen delivery to metabolic oxygen demand. This has been most clearly demonstrated in mice with a mutation in Hb that is unable to carry SNO at βCys93 (Premont and Stamler, 2020; Zhang et al., 2015; Zhang et al., 2016). In mutant mice, SNO-Hb, tissue oxygenation and hypoxia-induced blood flow are reduced at baseline (Figure 3A–D), and survival through the gestation and neonatal periods is reduced (Zhang et al., 2015). The ability to maintain tissue oxygenation by increasing tissue blood flow (reactive hyperemia) during an acute episode of progressive systemic hypoxia (breathing air with reduced O2) is lost (Zhang et al., 2015). Mutant mice also succumb more quickly to exposure to a reduced (5%) oxygen environment than do control mice (Zhang et al., 2015). Cardiac biomechanical functions appear normal in mutant mice breathing room air, but exhibit increased dysfunction with exposure to progressive systemic hypoxia whereas mutant mice exhibit reduced T-wave area, a sign of reduced coronary blood flow/ischemia, even at baseline (Zhang et al., 2015). Mutant mice subjected to ischemia-reperfusion injury exhibit elevated heart injury and mortality, and survivor mice were found to have collateral coronary arteries feeding the left heart (Zhang et al., 2016). This development of collaterals is also a sign of adaptation to developmental hypoxia. Mutant mice subjected to a pressure overload heart failure model developed worse heart failure, evidenced by reduced heart function, increased hypertrophy and reduced survival (Zhang et al., 2016) (Figure 4B). Thus, these mice display defects in tissue oxygenation and hypoxia-induced blood flow that are exacerbated by hypoxic stress, and despite compensatory adaptations within RBCs to increase SNO at non-βCys93 sites.
Figure 3.
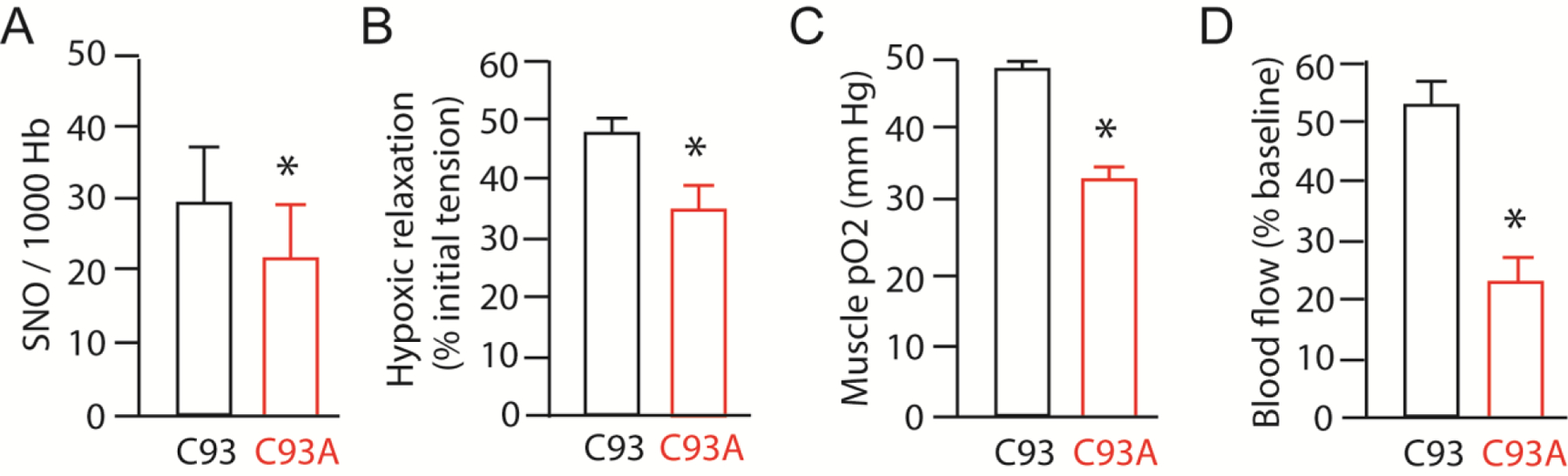
SNO(Cys93)-Hb induces hypoxic vasodilation and tissue oxygen delivery. A. RBCs derived from C93A mutant animals have diminished SNO-Hb (Alfred Hausladen,JSS unpublished data). B. RBCs derived from C93A mutant animals cannot effectively dilate blood vessels under hypoxia. C. Mice bearing RBCs with wildtype human Hb (C93) exhibit higher muscle oxygen level (pO2) compared to mice bearing mutant Hb unable to carry SNO at Cys93 (Cys93Ala). D. Mice bearing RBCs with mutant human Hb (C93) exhibit impaired autoregulation of blood flow in peripheral limbs. Panels B-D adapted from (Zhang et al., 2015).
3.3. Ventilation control
Central control of ventilation is mediated primarily by brainstem and carotid body detection of CO2 and acidic pH (Levitsky, 2018). A role for SNO-βCys93 Hb in ventilatory control was identified using βCys93 mutant mice. When mice are exposed to a hypoxic environment, they increase their rate of respiration acutely, but this increase is only transient; conversely, when such hypoxic mice are returned to normal room air, their respiratory rate paradoxically increases acutely and then subsides, a response termed short-term potentiation of breathing. βCys93 mutant mice completely lack this short-term potentiation of breathing response (Gaston et al., 2014), indicating a role for SNO-Hb in modulating ventilatory drive.
3.4. Pulmonary arterial pressure.
While hypoxia causes peripheral vasodilation, it raises blood pressure in the lung. This serves to improve ‘ventilation/perfusion matching’ in the lungs in order to oxygenate RBCs. βCys93/Ala mutant mice are subject to hypoxia (Zhang et al., 2015) and we can confirm, preliminarily, that SNO-Hb deficiency in these mice may cause pulmonary arterial hypertension.
4. Clinical Roles of S-nitroso-hemoglobin
4.1. SNO-Hb as a large pool of SNO compared to NO/SNO synthesis rate
One important aspect of NO biology to bear in mind when considering any pathophysiology associated with S-nitrosylation is that the rate of NO production by NOS enzymes in the human body is limited and rather low. Estimates suggest that total NOS activity to convert arginine to NO is approximately 1 μmol/kg/h (Castillo et al., 1996). Of the NO bioactivity in the blood, about half derives from eNOS (mainly from vascular endothelium, but also from RBCs (Leo et al., 2021)), and the remainder comes from nNOS/iNOS but also dietary and microbiome sources of nitrates and nitrites (Jia et al., 1996; Zhang et al., 2020). Thus, despite the fact that SNO-Hb under ordinary circumstances represents only a tiny fraction of total Hb (only 1–2 in 1000 Hb are SNO-modified) (Hausladen et al., 2007), the amount of Hb in the blood is so high that this amount of SNO represents a vast store of NO bioactivity compared to daily NO production. Conditions leading to depletion of SNO-Hb therefore can lead to long-term deficits due to the extended time necessary to replenish SNO-Hb stocks.
4.2. Altitude sickness and SNO-Hb
One “normal” physical condition that can lead to dramatic loss of SNO-Hb is acute hypoxia associated with high altitude. Although “thin” mountain air has the same gas composition as that at sea level (~20% oxygen), the overall air pressure and particularly the oxygen partial pressure are reduced so less oxygen is available to bind to Hb. As a practical limit, elevations much over 5000 meters are uninhabitable without supplemental oxygen, and mountain climbers must acclimate to high altitude conditions and even bring along stored oxygen in order to function at such heights and beyond. Under such conditions of low oxygen partial pressure, all tissues sense hypoxia and therefore utilize SNO-Hb in an effort to maintain normal oxygen delivery to counteract the reduced oxygen saturation of arterial blood. Rapid ascent to high altitude (or mimicking this in a hypobaric or hypoxic chamber) therefore results in depletion of SNO-Hb (Janocha et al., 2011), which will exacerbate tissue hypoxia. Measurement of SNO content of Hb during staged ascent to high altitude has demonstrated a dramatic loss of SNO-Hob content immediately after ascent, but a partial replenishment over days at high altitude (Janocha et al., 2011), likely reflecting upregulation of NO production (Ho et al., 2012).
For populations that live at very high altitude, trans-generational hypoxia has led to multiple adaptations to increase oxygen delivery to tissues despite low ambient oxygen. These adaptations range from enhanced erythropoiesis leading to elevated hematocrit, Hb content and oxygen carrying capacity, to more rapid breathing at rest and increased tissue microvascularization (Beall, 2007; Beall et al., 2012). Interestingly, distinct high-altitude populations have been shown to utilize different strategies, choosing different trade-offs in oxygen capture from air, oxygen transport in blood, and oxygen delivery in tissues to make maximal use of the low oxygen available (Beall, 2007; Beall et al., 2012). For instance, increasing RBC numbers through erythropoeisis increases oxygen carrying capacity per unit of blood, but also leads to detrimental increases in blood viscosity and subsequently elevated blood pressure (Beall et al., 2012). Of particular interest is the finding that SNO-Hb is highly elevated in high altitude Tibetans compared to US lowlanders (2400 nM at high altitude vs 10 nM at sea level), as are plasma nitrite (12 μM vs 0.5 μM) and nitrate (~200 μM vs ~25 μM), and urinary nitrate (105 mM vs 8 mM) (Erzurum et al., 2007), consistent with very high NO/SNO flux. More direct methodologies including photolysis-chemiluminescence (McMahon et al., 2002; Singel and Stamler, 2005) and phosphine-based labeling (Seneviratne et al., 2013) show ~micromolar SNOs in RBCs and plasma at sea levels, highlighting an equilibrium between SNO pools that are otherwise independent of nitrite and nitrate.
Failure to adapt to hypobaric hypoxia at high altitude can lead to acute or chronic altitude sickness (also called “mountain” sickness) (Ulloa and Cook, 2020). Acute altitude sickness arises within a day after ascent to an altitude above about 2000 meters, particularly when the ascent is very rapid and the difference between starting and destination altitudes is great. Symptoms include shortness of breath, cough, headache and nausea. In most people, symptoms subside within a day or two, but can be exacerbated by exertion and dehydration. The ailment can progress to life-threatening High Altitude Pulmonary Edema (HAPE), where fluid buildup in the lungs blocks efficient gas exchange and dramatically worsens the systemic hypoxia (Ulloa and Cook, 2020). Treatment is usually evacuation to lower altitude, or supplemental oxygen. Mechanistically, blood vessels within the lung normally respond to local hypoxia through the process of hypoxic ventilation-perfusion matching, where less well oxygenated regions of the lung (as in an infection) are serviced with reduced blood flow via pulmonary vessel vasoconstriction, so that blood passing through the lungs is sent to the most oxygenated sections (Dunham-Snary et al., 2017). Under chronic hypobaric hypoxia at altitude, however, this regulation goes awry as all sections of the lung appear hypoxic so all pulmonary vessels vasoconstrict, elevating pulmonary blood pressure. This vicious cycle reduces gas exchange in the lung, exacerbating tissue hypoxia and leading to death if untreated. While the role of SNO-Hb has not been established in hypoxia-driven pulmonary hypertension or altitude sickness directly, patients with idiopathic pulmonary hypertension and secondary hypoxia exhibit reduced levels of SNO-Hb compared to matched controls (McMahon et al., 2005), and RBCs show reduced hypoxic vasodilatory activity in the lungs, suggesting that hypoxia-driven loss of SNO-Hb may also be a crucial factor in altitude-dependent pulmonary dysfunction.
4.3. Blood transfusion and SNO-Hb
Clinical guidelines for blood transfusion due to blood loss are generally based on a threshold value of low Hb/hematocrit, although the value to be use remains controversial (Tomic Mahecic et al., 2020; Trentino et al., 2020). However, no matter what threshold is used, guidelines are to add back as little as needed, since full restoration or over-restoration leads to elevation of bad outcomes (Storch et al., 2019). Importantly, it is well-recognized that RBCs stored ex vivo for transfusion undergo detrimental changes over time in storage that collectively are referred to as “storage lesion” (Hess, 2010). These changes include RBC hemolysis, reduced RBC rheology and flexible deformability, loss of cellular factors such as ATP and 2,3-diphosphoglycerate, and more (Hess, 2010). The consequence of any of these changes with regard the role of RBCs to deliver oxygen to hypoxic tissues in not known. Ultimately, the time that RBCs can be banked is limited (under 2 months) not based off functional measure, but rather amounts of hemolysis. However, due to the practicalities of blood banking, a first-in first-out approach is generally used, so a patient needing a transfusion often gets the oldest erythrocytes in the bank, essentially guaranteeing that they have some degree of storage lesion deficits. The relative benefits of administering “fresh” stored blood versus “old” blood to various patient groups have been assessed in numerous retrospective studies, with some studies reporting benefit from fresh blood but most not. Randomized clinical trial comparisons of “fresh” RBCs – generally considered as within 5–7 days of donation, but sometimes even up to 14 days – versus older stored RBCs find little difference in morbidity or mortality endpoints for various patient groups, leading to meta-analyses and clinical recommendations asserting that there is no benefit difference (Shah et al., 2018; Zhou et al., 2019). Notably, immediate tissue oxygenation itself is rarely assessed as an outcome in these studies, however. In transfusion studies where tissue oxygenation has been measured, transfusion did not increase basal tissue oxygen saturation (StO2) (Podbregar et al., 2016; Weinberg et al., 2013), suggesting that “fresh” and “old” RBCs are both impaired, with older RBCs potentially more impaired.
Less widely appreciated is the finding that banked RBCs are also markedly deficient in SNO-Hb content (Bennett-Guerrero et al., 2007; Reynolds et al., 2007), and even “fresh” RBCs (5–7 days of storage) are SNO depleted. Blood collected for transfusion loses SNO from Hb within hours on the day of collection (Bennett-Guerrero et al., 2007; Reynolds et al., 2007) and is maximally depleted by 2 days. This extremely rapid loss of SNO-Hb negates the vast majority of studies comparing “fresh” to “old” stored RBCs with regard to the role of SNO-dependent hypoxic vasodilation and oxygen delivery, as both ages of RBCs are equally SNO-Hb deficient. Indeed, direct comparison of RBC vasorelaxation activity over time after collection demonstrated a loss of this bioactivity within 3 hours of blood collection (Bennett-Guerrero et al., 2007). Thus, by transfusion of stored RBCs (“fresh” or “old”) into a patient in desperate need of tissue oxygenation following blood loss, the patient is actually being given RBCs that may appear to carry oxygen well (high oxygen saturation) but really are deficient in hypoxic vasodilation of tissue microvessels that mediate oxygen delivery to tissues (Figure 4C,D). Furthermore, these SNO-depleted RBCs will also reduce the effectiveness of the patient’s own SNO-replete RBCs through dilution. To reiterate, the only storage lesion known to affect the primary role of RBCs in oxygen delivery is SNO-Hb loss, and transfusion studies with fresh and old RBCs (many showing increased morbidity and mortality) are, in all cases, SNO-deficient
To correct this SNO-Hb deficit, stored erythrocytes have been repleted experimentally with SNO and then tested for restoration of oxygen delivery function. One approach is to pretreat stored erythrocytes with a SNO donor to increase SNO-Hb content immediately prior to transfusion (Reynolds et al., 2013). Alternatively, a gaseous SNO donor, ethyl nitrite (ENO), can be added to inspired air to add SNO to Hb in erythrocytes as they circulate in the bloodstream through the lung (Brandler et al., 2005). For example, CO2 insufflation of the peritoneal cavity in swine, which should deplete SNO-Hb through an allosteric transition, led to reduced liver and spleen blood flow, while addition of ENO to the peritoneal cavity increased blood flow to these organs (Ali et al., 2005). This increased blood flow after ENO treatment was associated with elevated SNO-Hb (Shimazutsu et al., 2009). More generally, tissue oxygenation has been shown to be improved when SNO-repleted erythrocytes are transfused into several animal models (Reynolds et al., 2013; Reynolds et al., 2018; Yurcisin et al., 2013) or into human patients (Reynolds et al., 2018), compared to untreated (SNO-depleted) RBCs. Thus SNO-repletion of transfused RBCs is a promising avenue to increase the effectiveness of stored RBCs to deliver oxygen in patients with insufficient Hb or hematocrit.
4.4. Microvascular disease and SNO-Hb (PAH, PAD, dementia, etc)
Reduced tissue microvessel blood flow and/or poor oxygen delivery into tissues are common features of many diseases (Foster et al., 2003; Gutterman et al., 2016). While there has been a great focus on adaptive responses of cells and tissues to hypoxia through such mediators as HIFs (Chen et al., 2020), much less attention has been focused on responses that actively mediate oxygen delivery through tissue microvasculature, and no drugs are available to augment oxygen delivery directly. Typically, hypoxemia is treated with increased inspired oxygen and even hyperbaric oxygen (Choudhury, 2018), even though normal oxygen saturation in arterial blood in persons breathing room air is near 100% already and the saturation of mixed venous blood is normally ~70% (i.e., the majority of O2 recirculates without delivery). This fact demonstrates that there is great potential for drugs that increase oxygen delivery to tissues.
As noted previously, tissues utilize a variety of mechanisms, acting at multiple levels of the vasculature, to regulate blood flow through tissues. These include endothelial (and RBC) NO relaxing large blood vessels to regulate blood pressure, and endothelial and RBC release of vasoactive mediators including ATP and adenosine (which act through NO), cyclooxygenase and prostaglandin metabolites, the metabolite and electrical coupling of microvascular endothelia, and pre-capillary sphincters and shunt collaterals. Of these many means to alter tissue blood flow, only SNO-βCys93 Hb is directly regulated by tissue hypoxia (as reflected in Hb allostery). Therefore, it seems likely that SNO-Hb is dysregulated in diseases with a prominent vasoconstrictive effect resulting in hypoxia or ischemia.
Pulmonary Arterial Hypertension (PAH) is a constrictive disease of the pulmonary system that results in elevated blood pressure in the pulmonary artery, leading to dysfunction and eventually failure of the right heart (Dodson et al., 2018). While genetic causes are known, most cases are idiopathic. Studies in a newborn rat model of neonatal pulmonary hypertension have suggested a protective role for SNO in PAH (Jankov et al., 2018). RBCs from PAH patients are deficient in SNO-Hb content and are impaired in ability to induce vasorelaxation in aorta ex vivo (McMahon et al., 2005). Treatment of PAH patients with an inhaled SNO-donor gas, ethyl nitrite, was able to increase the SNO level of Hb within circulating RBCs, and these repleted RBCs demonstrated restored aorta vasorelaxation ability in an ex vivo bioassay (McMahon et al., 2005). Mice lacking βCys93 exhibit elevated pulmonary blood pressure and right heart hypertrophy with age consistent with PAH (Rongli Zhang, JSS unpublished), and that is further accentuated by hypoxia, suggesting a causative role for low SNO-Hb.
Peripheral Arterial Disease (PAD) is a vasoocclusive disease of peripheral arteries, and is frequently due to atherosclerosis associated with traditional cardiovascular risk factors such as obesity and hypercholesterolemia, but is also a complication of diseases such as type II diabetes (Zemaitis et al., 2021). NO has been suggested to be protective or therapeutic, and supplementation of patients with L-arginine, the substrate for nitric oxide synthases and precursor of NO, improves symptoms on PAD (Kashyap et al., 2017), although long-term L-arginine supplementation may lose effectiveness (Wilson et al., 2007). Notably, microvascular disease is a frequent complication of PAD and preliminary evidence indicates that PAD patients have reduced SNO-Hb compared to normal controls (Premont et al., 2020), suggesting that SNO itself (instead of just NO) may be important in PAD.
4.5. Vaso-occlusive disease and SNO-Hb
Sickle cell anemia is a vaso-occlusive disease that leads to tissue hypoxia. Mutation of β-globin Glu6Val leads to aggregation of the mutant Hb tetramer, termed HbS instead of HbA (Sedrak and Kondamudi, 2021). This aggregation then leads to deformation of the RBC into a characteristic sickle shape such that it does not pass through capillaries readily (Sedrak and Kondamudi, 2021). Aggregations of sickled RBCs can block microvascular flow, leading to painful episodes of tissue hypoxia that can lead to death (Sedrak and Kondamudi, 2021). RBCs from sickle cell disease patients exhibit a deficit in SNO-Hb content and an impairment of hypoxic vasodilation assayed in vitro (Pawloski et al., 2005) and sickle cell RBC adhesion in vivo (McMahon et al., 2019). Reduced SNO-Hb activity correlated with disease severity, and renitrosylation of HbS in vitro increased hypoxic vasodilation activity (Pawloski et al., 2005). Thus, in addition to mechanical deficits affecting RBC structure and deformability leading to indirect reductions in oxygen delivery (RBCs that fail to reach tissue capillaries cannot release O2 there), the sickle cell mutation also appears to affect SNO-mediated microvascular vasorelaxation and tissue oxygen delivery directly.
4.6. Vasodilation in sepsis and SNO-Hb
Sepsis is the result of pathogen growth within the body and particularly in the bloodstream, where infection-driven induction of chemokines results in widespread expression and activation of inducible NOS (iNOS). This excessive NO production produces a general vasodilation leading to low blood pressure and inadequate organ perfusion (Singer et al., 2016). One consequence of this elevated NO in septic shock is excessive S-nitrosylation of Hb (Liu et al., 2004) leading to hypoxia-independent release of SNO from Hb (Crawford et al., 2004), which will promote widespread and non-specific vasodilation of large vessels and reduced blood pressure. A recent study in rabbits reported that shock induced by injection of bacterial lipopolysaccharide was associated with a defect in SNO release from RBCs, a reduced arterial–venous SNO-level difference, and an increased level of microvascular blood flow heterogeneity (Yao et al., 2019), suggesting a concurrent deficit in tissue oxygen delivery.
4.7. Transplant organ support
Organs for transplant are often obtained from brain dead donors. In an effort to stabilize organ function following the trauma leading to brain death, such donors are often kept on ventilation to maintain organs alive for some time prior to organ harvest. Over time, the animal becomes hemodynamically unstable and organs fail. Experiments in swine with induced brain death have revealed that loss of brain function leads to dysregulation of NO synthesis and depletion of SNO-Hb, during time on mechanical ventilation (Yurcisin et al., 2013). Supplementation of inspired air with 20 ppm ENO gas normalized SNO-Hb and improved deficits in blood flow and tissue resistance. Importantly, ENO also improved markers of organ function, suggesting that normalizing microvascular blood flow and oxygen delivery after brain death may better preserve organ function prior to harvest for transplant (Yurcisin et al., 2013).
5. Current perspectives and future directions
Hemoglobin was originally conceived of as an irreversible sink for NO that eliminates NO bioactivity to constrict blood vessels, and this activity was foundational in the discovery of NO as a bioactive molecule. But this viewpoint opposed hemoglobin’s role in oxygen delivery. Accumulating evidence now indicates that Hb is in fact the primary source of NO bioactivity essential for mammalian life (Premont and Stamler, 2020) and it serves as a paradigm for ubiquitous NO-based signaling by S-nitrosylation (Premont et al., 2021) —including ~25,000 sites in >10,000 proteins across phylogeny—to regulate all main classes of proteins (Leo et al., 2021). This perspective is consistent with Hb’s role in oxygen delivery. Moreover, whereas S-nitrosylation of Hb was at first disputed, and subsequently argued by many to be chemically mediated and stochastic, S-nitrosylation in Hb is in fact precisely regulated through enzymatic activity that represents three classes of enzymes found in other cells (Seth et al., 2018): i. NO generating enzymes (Hb can generate FeNO though nitrite reductase activity); SNO generating enzymes (Hb is a SNO synthase that coverts NO to SNO (Angelo et al., 2006); and SNO transferirng enzymes (Hb is a transnitrosylase that binds Band 3 and exchanges NO to produce SNO-Band 3). Thus, Hb is a de facto enzyme that serves as the “spinal column” for the circulatory system to convert NO into SNO, whereby RBCs, can carry and release SNO to optimize oxygen delivery in the respiratory cycle. Absent C93, animals cannot oxygenate tissues adequately despite carrying normal amounts of oxygen (Zhang et al., 2015), consistent with the view that blood flow (not oxygen content) is the primary determinant of oxygen delivery (Premont et al., 2020). These finding explain the invariant nature of Cys93 in Hb, which serves as the primary site of S-nitrosylation; its functional linkage to the allosteric transition in Hb; and the enzymatic activity of Hb that is essential for mammalian respiratory function.
One very intriguing possibility arises from the recognition that SNO may be transported not only through the blood circulation, but into and throughout cells within organs, including liver, lungs, heart and brain. Distinct work using the C. elegans model has demonstrated with SNO derived from microbiome bacteria is able to be incorporated into C. elegans tissue proteins (Seth et al., 2019). Additionally, repletion of SNO-Hb using an inhaled SNO-donor drug in brain dead animals improved organ function throughout the body. These studies and others raise the idea that SNO derived from RBCs (perhaps along with other blood-borne SNO-proteins such as albumin) provides a means to spread SNO from sites of synthesis to multiple target organs to affect physiological functions. This potential is entirely untapped for therapeutic benefit.
Acknowledgements:
Funding:
This work was supported by the National Institutes of Health grants HL075443, HL128192, HL126900 and DK119506, and American Heart Association-Allen Brain Health Initiative grant 19PABHI34580006 to JSS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest:
JSS has patents relating to nitrosylation of hemoglobin and is a founder of SNOBio. CWRU and UHCMC have management plans in place. The other authors declare no conflicts.
References Cited:
- Ali NA, Eubanks WS, Stamler JS, Gow AJ, Lagoo-Deenadayalan SA, Villegas L, El-Moalem HE, Reynolds JD, 2005. A Method to Attenuate Pneumoperitoneum-Induced Reductions in Splanchnic Blood Flow. Annals of Surgery 241 (2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelo M, Singel DJ, Stamler JS, 2006. An S-nitrosothiol (SNO) synthase function of hemoglobin that utilizes nitrite as a substrate. Proc Natl Acad Sci U S A 103 (22), 8366–8371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold WP, Mittal CK, Katsuki S, Murad F, 1977. Nitric oxide activates guanylate cyclase and increases guanosine 3′:5′-cyclic monophosphate levels in various tissue preparations. Proceedings of the National Academy of Sciences 74 (8), 3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey DM, Rasmussen P, Overgaard M, Evans KA, Bohm AM, Seifert T, Brassard P, Zaar M, Nielsen HB, Raven PB, Secher NH, 2017. Nitrite and S-Nitrosohemoglobin Exchange Across the Human Cerebral and Femoral Circulation: Relationship to Basal and Exercise Blood Flow Responses to Hypoxia. Circulation 135 (2), 166–176. [DOI] [PubMed] [Google Scholar]
- Beall CM, 2007. Two routes to functional adaptation: Tibetan and Andean high-altitude natives. Proc Natl Acad Sci U S A 104 Suppl 1, 8655–8660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beall CM, Laskowski D, Erzurum SC, 2012. Nitric oxide in adaptation to altitude. Free Radic Biol Med 52 (7), 1123–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett-Guerrero E, Veldman TH, Doctor A, Telen MJ, Ortel TL, Reid TS, Mulherin MA, Zhu H, Buck RD, Califf RM, McMahon TJ, 2007. Evolution of adverse changes in stored RBCs. Proceedings of the National Academy of Sciences 104 (43), 17063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohr C, Hasselbalch K, Krogh A, 1904. Uber einen in biologischer Beziehung wichtigen Einfluss, den die kohlens urespannung des Blutes auf dessen Sauerstoffbindung Ubt. Skand. Arch. Physiol 16, 402–412. [Google Scholar]
- Bonaventura C, Ferruzzi G, Tesh S, Stevens RD, 1999. Effects of S-nitrosation on oxygen binding by normal and sickle cell hemoglobin. J Biol Chem 274 (35), 24742–24748. [DOI] [PubMed] [Google Scholar]
- Brandler MD, Powell SC, Craig DM, Quick G, McMahon TJ, Goldberg RN, Stamler JS, 2005. A Novel Inhaled Organic Nitrate That Affects Pulmonary Vascular Tone in a Piglet Model of Hypoxia-Induced Pulmonary Hypertension. Pediatric Research 58 (3), 531–536. [DOI] [PubMed] [Google Scholar]
- Bulger M, van Doorninck JH, Saitoh N, Telling A, Farrell C, Bender MA, Felsenfeld G, Axel R, Groudine M, 1999. Conservation of sequence and structure flanking the mouse and human beta-globin loci: the beta-globin genes are embedded within an array of odorant receptor genes. Proc Natl Acad Sci U S A 96 (9), 5129–5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo L, Beaumier L, Ajami AM, Young VR, 1996. Whole body nitric oxide synthesis in healthy men determined from [15N] arginine-to-[15N]citrulline labeling. Proceedings of the National Academy of Sciences 93 (21), 11460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan NL, Rogers PH, Arnone A, 1998. Crystal structure of the S-nitroso form of liganded human hemoglobin. Biochemistry 37 (47), 16459–16464. [DOI] [PubMed] [Google Scholar]
- Chanutin A, Curnish RR, 1967. Effect of organic and inorganic phosphates on the oxygen equilibrium of human erythrocytes. Archives of Biochemistry and Biophysics 121 (1), 96–102. [DOI] [PubMed] [Google Scholar]
- Chen P-S, Chiu W-T, Hsu P-L, Lin S-C, Peng IC, Wang C-Y, Tsai S-J, 2020. Pathophysiological implications of hypoxia in human diseases. Journal of Biomedical Science 27 (1), 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury R, 2018. Hypoxia and hyperbaric oxygen therapy: a review. Int J Gen Med 11, 431–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortese-Krott MM, Kelm M, 2014. Endothelial nitric oxide synthase in red blood cells: key to a new erythrocrine function? Redox Biol 2, 251–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford JH, Chacko BK, Pruitt HM, Piknova B, Hogg N, Patel RP, 2004. Transduction of NO-bioactivity by the red blood cell in sepsis: novel mechanisms of vasodilation during acute inflammatory disease. Blood 104 (5), 1375–1382. [DOI] [PubMed] [Google Scholar]
- De Rosa MC, Alinovi CC, Galtieri A, Russo A, Giardina B, 2008. Allosteric properties of hemoglobin and the plasma membrane of the erythrocyte: New insights in gas transport and metabolic modulation. IUBMB Life 60 (2), 87–93. [DOI] [PubMed] [Google Scholar]
- Diesen DL, Hess DT, Stamler JS, 2008. Hypoxic vasodilation by red blood cells: evidence for an s-nitrosothiol-based signal. Circ Res 103 (5), 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doctor A, Platt R, Sheram ML, Eischeid A, McMahon T, Maxey T, Doherty J, Axelrod M, Kline J, Gurka M, Gow A, Gaston B, 2005. Hemoglobin conformation couples erythrocyte S-nitrosothiol content to O2 gradients. Proc Natl Acad Sci U S A 102 (16), 5709–5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson MW, Brown LM, Elliott CG, 2018. Pulmonary Arterial Hypertension. Heart Fail Clin 14 (3), 255–269. [DOI] [PubMed] [Google Scholar]
- Douglas CG, Haldane JS, Haldane JBS, 1912. The laws of combination of hæmoglobin with carbon monoxide and oxygen. The Journal of Physiology 44 (4), 275–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle MP, Pickering RA, DeWeert TM, Hoekstra JW, Pater D, 1981. Kinetics and mechanism of the oxidation of human deoxyhemoglobin by nitrites. J Biol Chem 256 (23), 12393–12398. [PubMed] [Google Scholar]
- Dunham-Snary KJ, Wu D, Sykes EA, Thakrar A, Parlow LRG, Mewburn JD, Parlow JL, Archer SL, 2017. Hypoxic Pulmonary Vasoconstriction: From Molecular Mechanisms to Medicine. CHEST 151 (1), 181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erzurum SC, Ghosh S, Janocha AJ, Xu W, Bauer S, Bryan NS, Tejero J, Hemann C, Hille R, Stuehr DJ, Feelisch M, Beall CM, 2007. Higher blood flow and circulating NO products offset high-altitude hypoxia among Tibetans. Proc Natl Acad Sci U S A 104 (45), 17593–17598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster MW, McMahon TJ, Stamler JS, 2003. S-nitrosylation in health and disease. Trends Mol Med 9 (4), 160–168. [DOI] [PubMed] [Google Scholar]
- Fung YC, Zweifach BW, 1971. Microcirculation: Mechanics of Blood Flow in Capillaries. Annual Review of Fluid Mechanics 3 (1), 189–210. [Google Scholar]
- Furchgott RF, Zawadzki JV, 1980. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 288 (5789), 373–376. [DOI] [PubMed] [Google Scholar]
- Gaston B, Drazen JM, Jansen A, Sugarbaker DA, Loscalzo J, Richards W, Stamler JS, 1994. Relaxation of human bronchial smooth muscle by S-nitrosothiols in vitro. Journal of Pharmacology and Experimental Therapeutics 268 (2), 978. [PubMed] [Google Scholar]
- Gaston B, May WJ, Sullivan S, Yemen S, Marozkina NV, Palmer LA, Bates JN, Lewis SJ, 2014. Essential role of hemoglobin beta-93-cysteine in posthypoxia facilitation of breathing in conscious mice. J Appl Physiol (1985) 116 (10), 1290–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaston B, Reilly J, Drazen JM, Fackler J, Ramdev P, Arnelle D, Mullins ME, Sugarbaker DJ, Chee C, Singel DJ, Loscalzo J, Stamler JS, 1993. Endogenous nitrogen oxides and bronchodilator S-nitrosothiols in human airways. Proc Natl Acad Sci U S A 90 (23), 10957–10961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaston B, Smith L, Bosch J, Seckler J, Kunze D, Kiselar J, Marozkina N, Hodges CA, Wintrobe P, McGee K, Morozkina TS, Burton ST, Lewis T, Strassmaier T, Getsy P, Bates JN, Lewis SJ, 2020. Voltage-gated potassium channel proteins and stereoselective S-nitroso-l-cysteine signaling. JCI Insight 5 (18). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gow AJ, Stamler JS, 1998. Reactions between nitric oxide and haemoglobin under physiological conditions. Nature 391 (6663), 169–173. [DOI] [PubMed] [Google Scholar]
- Gutterman DD, Chabowski DS, Kadlec AO, Durand MJ, Freed JK, Ait-Aissa K, Beyer AM, 2016. The Human Microcirculation. Circulation Research 118 (1), 157–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey W, 1628. Exercitatio Anatomica de Motu Cordis et Sanguinis in Animalibus. Fitzeri, William, Frankfort. [PubMed] [Google Scholar]
- Hausladen A, Rafikov R, Angelo M, Singel DJ, Nudler E, Stamler JS, 2007. Assessment of nitric oxide signals by triiodide chemiluminescence. Proc Natl Acad Sci U S A 104 (7), 2157–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson LJ, 1920. THE EQUILIBRIUM BETWEEN OXYGEN AND CARBONIC ACID IN BLOOD. Journal of Biological Chemistry 41 (3), 401–430. [Google Scholar]
- Herold S, Rock G, 2003. Reactions of deoxy-, oxy-, and methemoglobin with nitrogen monoxide. Mechanistic studies of the S-nitrosothiol formation under different mixing conditions. J Biol Chem 278 (9), 6623–6634. [DOI] [PubMed] [Google Scholar]
- Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS, 2005. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol 6 (2), 150–166. [DOI] [PubMed] [Google Scholar]
- Hess DT, Matsumoto A, Nudelman R, Stamler JS, 2001. S-nitrosylation: spectrum and specificity. Nat Cell Biol 3 (2), E46–49. [DOI] [PubMed] [Google Scholar]
- Hess DT, Stamler JS, 2012. Regulation by S-nitrosylation of protein post-translational modification. J Biol Chem 287 (7), 4411–4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess JR, 2010. Red cell changes during storage. Transfusion and Apheresis Science 43 (1), 51–59. [DOI] [PubMed] [Google Scholar]
- Hibbs JB Jr., Taintor RR, Vavrin Z, 1987. Macrophage cytotoxicity: role for L-arginine deiminase and imino nitrogen oxidation to nitrite. Science 235 (4787), 473–476. [DOI] [PubMed] [Google Scholar]
- Ho JJ, Man HS, Marsden PA, 2012. Nitric oxide signaling in hypoxia. J Mol Med (Berl) 90 (3), 217–231. [DOI] [PubMed] [Google Scholar]
- Hogg N, 2002. The biochemistry and physiology of S-nitrosothiols. Annu Rev Pharmacol Toxicol 42, 585–600. [DOI] [PubMed] [Google Scholar]
- Huang Z, Shiva S, Kim-Shapiro DB, Patel RP, Ringwood LA, Irby CE, Huang KT, Ho C, Hogg N, Schechter AN, Gladwin MT, 2005. Enzymatic function of hemoglobin as a nitrite reductase that produces NO under allosteric control. J Clin Invest 115 (8), 2099–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G, 1987. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proceedings of the National Academy of Sciences 84 (24), 9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignarro LJ, Gruetter CA, 1980. Requirement of thiols for activation of coronary arterial guanylate cyclase by glyceryl trinitrate and sodium nitrite: possible involvement of S-nitrosothiols. Biochim Biophys Acta 631 (2), 221–231. [DOI] [PubMed] [Google Scholar]
- Imai K, 1999. The haemoglobin enzyme. Nature 401 (6752), 437, 439. [DOI] [PubMed] [Google Scholar]
- Ivanovic-Burmazovic I, Filipovic MR, 2019. Saying NO to H2S: A Story of HNO, HSNO, and SSNO–. Inorganic Chemistry 58 (7), 4039–4051. [DOI] [PubMed] [Google Scholar]
- Jankov RP, Daniel KL, Iny S, Kantores C, Ivanovska J, Ben Fadel N, Jain A, 2018. Sodium nitrite augments lung S-nitrosylation and reverses chronic hypoxic pulmonary hypertension in juvenile rats. American Journal of Physiology-Lung Cellular and Molecular Physiology 315 (5), L742–L751. [DOI] [PubMed] [Google Scholar]
- Janocha AJ, Koch CD, Tiso M, Ponchia A, Doctor A, Gibbons L, Gaston B, Beall CM, Erzurum SC, 2011. Nitric oxide during altitude acclimatization. N Engl J Med 365 (20), 1942–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen A, Drazen J, Osborne JA, Brown R, Loscalzo J, Stamler JS, 1992. The relaxant properties in guinea pig airways of S-nitrosothiols. J Pharmacol Exp Ther 261 (1), 154–160. [PubMed] [Google Scholar]
- Jennings ML, 2013. Transport of H2S and HS− across the human red blood cell membrane: rapid H2S diffusion and AE1-mediated Cl−/HS− exchange. American Journal of Physiology-Cell Physiology 305 (9), C941–C950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J, Arif A, Terenzi F, Willard B, Plow EF, Hazen SL, Fox PL, 2014. Target-selective protein S-nitrosylation by sequence motif recognition. Cell 159 (3), 623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia L, Bonaventura C, Bonaventura J, Stamler JS, 1996. S-nitrosohaemoglobin: a dynamic activity of blood involved in vascular control. Nature 380 (6571), 221–226. [DOI] [PubMed] [Google Scholar]
- Kallakunta VM, Slama-Schwok A, Mutus B, 2013. Protein disulfide isomerase may facilitate the efflux of nitrite derived S-nitrosothiols from red blood cells. Redox Biol 1, 373–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashyap VS, Lakin RO, Campos P, Allemang M, Kim A, Sarac TP, Hausladen A, Stamler JS, 2017. The LargPAD Trial: Phase IIA evaluation of l-arginine infusion in patients with peripheral arterial disease. J Vasc Surg 66 (1), 187–194. [DOI] [PubMed] [Google Scholar]
- Keaney JF Jr., Simon DI, Stamler JS, Jaraki O, Scharfstein J, Vita JA, Loscalzo J, 1993. NO forms an adduct with serum albumin that has endothelium-derived relaxing factor-like properties. J Clin Invest 91 (4), 1582–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg MD, Sen N, Hara MR, Juluri KR, Nguyen JV, Snowman AM, Law L, Hester LD, Snyder SH, 2010. GAPDH mediates nitrosylation of nuclear proteins. Nat Cell Biol 12 (11), 1094–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, 1919. The supply of oxygen to the tissues and the regulation of the capillary circulation. J Physiol 52 (6), 457–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, 1922. The Anatomy and Physiology of Capillaries. Yale University Press, New Haven. [Google Scholar]
- Lai TS, Lindberg RA, Zhou HL, Haroon ZA, Dewhirst MW, Hausladen A, Juang YL, Stamler JS, Greenberg CS, 2017. Endothelial cell-surface tissue transglutaminase inhibits neutrophil adhesion by binding and releasing nitric oxide. Sci Rep 7 (1), 16163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leewenhoeck A, 1682. Microscopical Observations (Phil Trans Royal Soc 117:378). John Nourse, London. [Google Scholar]
- Leo F, Suvorava T, Heuser SK, Li J, LoBue A, Barbarino F, Piragine E, Schneckmann R, Hutzler B, Good ME, Fernandez BO, Vornholz L, Rogers S, Doctor A, Grandoch M, Stegbauer J, Weitzberg E, Feelisch M, Lundberg JO, Isakson BE, Kelm M, Cortese-Krott MM, 2021. Red Blood Cell and Endothelial eNOS Independently Regulate Circulating Nitric Oxide Metabolites and Blood Pressure. Circulation 144 (11), 870–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitsky MG, 2018. Pulmonary Physiology, 9e ed. McGraw-Hill Education, New York. [Google Scholar]
- Li S, Yu K, Wu G, Zhang Q, Wang P, Zheng J, Liu Z-X, Wang J, Gao X, Cheng H, 2021. pCysMod: Prediction of Multiple Cysteine Modifications Based on Deep Learning Framework. Frontiers in Cell and Developmental Biology 9 (117). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton AJ, Johnson MA, Macdonald T, Lieberman MW, Gozal D, Gaston B, 2001. S-nitrosothiols signal the ventilatory response to hypoxia. Nature 413 (6852), 171–174. [DOI] [PubMed] [Google Scholar]
- Liu L, Yan Y, Zeng M, Zhang J, Hanes MA, Ahearn G, McMahon TJ, Dickfeld T, Marshall HE, Que LG, Stamler JS, 2004. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell 116 (4), 617–628. [DOI] [PubMed] [Google Scholar]
- Luchsinger BP, Rich EN, Gow AJ, Williams EM, Stamler JS, Singel DJ, 2003. Routes to S-nitroso-hemoglobin formation with heme redox and preferential reactivity in the beta subunits. Proc Natl Acad Sci U S A 100 (2), 461–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matto F, Kouretas PC, Smith R, Ostrowsky J, Cina AJ, Hess DT, Stamler JS, Reynolds JD, 2018. S-Nitrosohemoglobin Levels and Patient Outcome After Transfusion During Pediatric Bypass Surgery. Clinical and Translational Science 11 (2), 237–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon TJ, Ahearn GS, Moya MP, Gow AJ, Huang Y-CT, Luchsinger BP, Nudelman R, Yan Y, Krichman AD, Bashore TM, Califf RM, Singel DJ, Piantadosi CA, Tapson VF, Stamler JS, 2005. A nitric oxide processing defect of red blood cells created by hypoxia: Deficiency of S-nitrosohemoglobin in pulmonary hypertension. Proceedings of the National Academy of Sciences of the United States of America 102 (41), 14801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon TJ, Moon RE, Luschinger BP, Carraway MS, Stone AE, Stolp BW, Gow AJ, Pawloski JR, Watke P, Singel DJ, Piantadosi CA, Stamler JS, 2002. Nitric oxide in the human respiratory cycle. Nat Med 8 (7), 711–717. [DOI] [PubMed] [Google Scholar]
- McMahon TJ, Pawloski JR, Hess DT, Piantadosi CA, Luchsinger BP, Singel DJ, Stamler JS, 2003. S-nitrosohemoglobin is distinguished from other nitrosovasodilators by unique oxygen-dependent responses that support an allosteric mechanism of action. Blood 102 (1), 410–411; author reply 412–413. [DOI] [PubMed] [Google Scholar]
- McMahon TJ, Shan S, Riccio DA, Batchvarova M, Zhu H, Telen MJ, Zennadi R, 2019. Nitric oxide loading reduces sickle red cell adhesion and vaso-occlusion in vivo. Blood Adv 3 (17), 2586–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon TJ, Stone AE, Bonaventura J, Singel DJ, Stamler JS, 2000. Functional coupling of oxygen binding and vasoactivity in S-nitrosohemoglobin. J Biol Chem 275 (22), 16738–16745. [DOI] [PubMed] [Google Scholar]
- Moncada S, Palmer RM, Higgs EA, 1989. The biological significance of nitric oxide formation from L-arginine. Biochem Soc Trans 17 (4), 642–644. [DOI] [PubMed] [Google Scholar]
- Nagababu E, Ramasamy S, Rifkind JM, 2006. S-Nitrosohemoglobin: A mechanism for its formation in conjunction with nitrite reduction by deoxyhemoglobin. Nitric Oxide 15 (1), 20–29. [DOI] [PubMed] [Google Scholar]
- Nagai K, Perutz MF, Poyart C, 1985. Oxygen binding properties of human mutant hemoglobins synthesized in Escherichia coli. Proceedings of the National Academy of Sciences 82 (21), 7252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Lipton SA, 2013. Emerging role of protein-protein transnitrosylation in cell signaling pathways. Antioxid Redox Signal 18 (3), 239–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson JS, Mathews AJ, Rohlfs RJ, Springer BA, Egeberg KD, Sligar SG, Tame J, Renaud JP, Nagai K, 1988. The role of the distal histidine in myoglobin and haemoglobin. Nature 336 (6196), 265–266. [DOI] [PubMed] [Google Scholar]
- Owusu BY, Stapley R, Patel RP, 2012. Nitric oxide formation versus scavenging: the red blood cell balancing act. The Journal of Physiology 590 (20), 4993–5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer LA, Doctor A, Chhabra P, Sheram ML, Laubach VE, Karlinsey MZ, Forbes MS, Macdonald T, Gaston B, 2007. S-nitrosothiols signal hypoxia-mimetic vascular pathology. J Clin Invest 117 (9), 2592–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer RM, Moncada S, 1989. A novel citrulline-forming enzyme implicated in the formation of nitric oxide by vascular endothelial cells. Biochem Biophys Res Commun 158 (1), 348–352. [DOI] [PubMed] [Google Scholar]
- Palmer RMJ, Ferrige AG, Moncada S, 1987. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 327 (6122), 524–526. [DOI] [PubMed] [Google Scholar]
- Pawloski JR, Hess DT, Stamler JS, 2001. Export by red blood cells of nitric oxide bioactivity. Nature 409 (6820), 622–626. [DOI] [PubMed] [Google Scholar]
- Pawloski JR, Hess DT, Stamler JS, 2005. Impaired vasodilation by red blood cells in sickle cell disease. Proceedings of the National Academy of Sciences of the United States of America 102 (7), 2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perutz MF, 1962. Relation between structure and sequence of haemoglobin. Nature 194, 914–917. [DOI] [PubMed] [Google Scholar]
- Perutz MF, 1970. Stereochemistry of Cooperative Effects in Haemoglobin: Haem–Haem Interaction and the Problem of Allostery. Nature 228 (5273), 726–734. [DOI] [PubMed] [Google Scholar]
- Perutz MF, 1978. Hemoglobin structure and respiratory transport. Sci Am 239 (6), 92–125. [DOI] [PubMed] [Google Scholar]
- Perutz MF, 1990. Mechanisms regulating the reactions of human hemoglobin with oxygen and carbon monoxide. Annu Rev Physiol 52, 1–25. [DOI] [PubMed] [Google Scholar]
- Perutz MF, Kendrew JC, Watson HC, 1965. Structure and function of haemoglobin: II. Some relations between polypeptide chain configuration and amino acid sequence. Journal of Molecular Biology 13 (3), 669–678. [Google Scholar]
- Pezacki JP, Ship NJ, Kluger R, 2001. Release of nitric oxide from S-nitrosohemoglobin. Electron transfer as a response to deoxygenation. J Am Chem Soc 123 (19), 4615–4616. [DOI] [PubMed] [Google Scholar]
- Podbregar M, Gavric AU, Podbregar E, Mozina H, Stefanovic S, 2016. Red blood cell transfusion and skeletal muscle tissue oxygenation in anaemic haematologic outpatients. Radiology and oncology 50 (4), 449–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premont RT, Reynolds JD, Zhang R, Stamler JS, 2020. Role of Nitric Oxide Carried by Hemoglobin in Cardiovascular Physiology: Developments on a Three-Gas Respiratory Cycle. Circ Res 126 (1), 129–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premont RT, Reynolds JD, Zhang R, Stamler JS, 2021. Red Blood Cell-Mediated S-Nitrosohemoglobin-Dependent Vasodilation: Lessons Learned from a beta-Globin Cys93 Knock-in Mouse. Antioxid Redox Signal 34 (12), 936–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premont RT, Stamler JS, 2020. Essential Role of Hemoglobin betaCys93 in Cardiovascular Physiology. Physiology (Bethesda) 35 (4), 234–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds JD, Ahearn GS, Angelo M, Zhang J, Cobb F, Stamler JS, 2007. S-nitrosohemoglobin deficiency: A mechanism for loss of physiological activity in banked blood. Proceedings of the National Academy of Sciences 104 (43), 17058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds JD, Bennett KM, Cina AJ, Diesen DL, Henderson MB, Matto F, Plante A, Williamson RA, Zandinejad K, Demchenko IT, Hess DT, Piantadosi CA, Stamler JS, 2013. S-nitrosylation therapy to improve oxygen delivery of banked blood. Proceedings of the National Academy of Sciences 110 (28), 11529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds JD, Jenkins T, Matto F, Nazemian R, Farhan O, Morris N, Longphre JM, Hess DT, Moon RE, Piantadosi CA, Stamler JS, 2018. Pharmacologic Targeting of Red Blood Cells to Improve Tissue Oxygenation. Clinical Pharmacology & Therapeutics 104 (3), 553–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribatti D, 2009. William Harvey and the discovery of the circulation of the blood. Journal of Angiogenesis Research 1 (1), 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross JM, Fairchild HM, Weldy J, Guyton AC, 1962. Autoregulation of blood flow by oxygen lack. American Journal of Physiology-Legacy Content 202 (1), 21–24. [DOI] [PubMed] [Google Scholar]
- Salgado MT, Nagababu E, Rifkind JM, 2009. Quantification of intermediates formed during the reduction of nitrite by deoxyhemoglobin. J Biol Chem 284 (19), 12710–12718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgado MT, Ramasamy S, Tsuneshige A, Manoharan PT, Rifkind JM, 2011. A New Paramagnetic Intermediate Formed during the Reaction of Nitrite with Deoxyhemoglobin. Journal of the American Chemical Society 133 (33), 13010–13022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez M, Sabio L, Gálvez N, Capdevila M, Dominguez-Vera JM, 2017. Iron chemistry at the service of life. IUBMB Life 69 (6), 382–388. [DOI] [PubMed] [Google Scholar]
- Sedrak A, Kondamudi NP, 2021. Sickle Cell Disease, StatPearls. StatPearls Publishing Copyright © 2021, StatPearls Publishing LLC., Treasure Island (FL). [Google Scholar]
- Seneviratne U, Godoy LC, Wishnok JS, Wogan GN, Tannenbaum SR, 2013. Mechanism-based triarylphosphine-ester probes for capture of endogenous RSNOs. J Am Chem Soc 135 (20), 7693–7704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth D, Hess DT, Hausladen A, Wang L, Wang YJ, Stamler JS, 2018. A Multiplex Enzymatic Machinery for Cellular Protein S-nitrosylation. Mol Cell 69 (3), 451–464 e456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth D, Stamler JS, 2011. The SNO-proteome: causation and classifications. Curr Opin Chem Biol 15 (1), 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth P, Hsieh PN, Jamal S, Wang L, Gygi SP, Jain MK, Coller J, Stamler JS, 2019. Regulation of MicroRNA Machinery and Development by Interspecies S-Nitrosylation. Cell 176 (5), 1014–1025 e1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severini L, 1878. Ricerche sulla innervazione dei vasi sanguigni. Tipografia Boncompagne, Perugia. [Google Scholar]
- Shah A, Brunskill SJ, Desborough MJ, Doree C, Trivella M, Stanworth SJ, 2018. Transfusion of red blood cells stored for shorter versus longer duration for all conditions. The Cochrane database of systematic reviews 12 (12), Cd010801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazutsu K, Uemura K, Auten KM, Baldwin MF, Belknap SW, La Banca F, Jones MC, McClaine DJ, McClaine RJ, Eubanks WS, Stamler JS, Reynolds JD, 2009. Inclusion of a Nitric Oxide Congener in the Insufflation Gas Repletes S-Nitrosohemoglobin and Stabilizes Physiologic Status During Prolonged Carbon Dioxide Pneumoperitoneum. Clinical and Translational Science 2 (6), 405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singel DJ, Stamler JS, 2005. Chemical physiology of blood flow regulation by red blood cells: the role of nitric oxide and S-nitrosohemoglobin. Annu Rev Physiol 67, 99–145. [DOI] [PubMed] [Google Scholar]
- Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, Hotchkiss RS, Levy MM, Marshall JC, Martin GS, Opal SM, Rubenfeld GD, van der Poll T, Vincent JL, Angus DC, 2016. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 315 (8), 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer SJ, Nicolson GL, 1972. THE FLUID MOSAIC MODEL OF THE STRUCTURE OF CELL MEMBRANES Reprinted with permission from Science, Copyright AAA, 18 February 1972, Volume 175, pp. 720–731, in: Day SB, Good RA (Eds.), Membranes and Viruses in Immunopathology. Academic Press, pp. 7–47. [DOI] [PubMed] [Google Scholar]
- Smith MP, Humphrey SJ, Kerr SW, Mathews WR, 1994. In vitro vasorelaxant and in vivo cardiovascular effects of S-nitrosothiols: comparison to and cross tolerance with standard nitrovasodilators. Methods and findings in experimental and clinical pharmacology 16 (5), 323–335. [PubMed] [Google Scholar]
- Stamler JS, Jaraki O, Osborne J, Simon DI, Keaney J, Vita J, Singel D, Valeri CR, Loscalzo J, 1992a. Nitric oxide circulates in mammalian plasma primarily as an S-nitroso adduct of serum albumin. Proc Natl Acad Sci U S A 89 (16), 7674–7677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamler JS, Jia L, Eu JP, McMahon TJ, Demchenko IT, Bonaventura J, Gernert K, Piantadosi CA, 1997a. Blood flow regulation by S-nitrosohemoglobin in the physiological oxygen gradient. Science 276 (5321), 2034–2037. [DOI] [PubMed] [Google Scholar]
- Stamler JS, Reynolds JD, Hess DT, 2017. Letter by Stamler et al Regarding Article, “Nitrite and S-Nitrosohemoglobin Exchange Across the Human Cerebral and Femoral Circulation: Relationship to Basal and Exercise Blood Flow Responses to Hypoxia”. Circulation 135 (24), e1135–e1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamler JS, Simon DI, Jaraki O, Osborne JA, Francis S, Mullins M, Singel D, Loscalzo J, 1992b. S-nitrosylation of tissue-type plasminogen activator confers vasodilatory and antiplatelet properties on the enzyme. Proc Natl Acad Sci U S A 89 (17), 8087–8091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamler JS, Simon DI, Osborne JA, Mullins ME, Jaraki O, Michel T, Singel DJ, Loscalzo J, 1992c. S-nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proc Natl Acad Sci U S A 89 (1), 444–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamler JS, Singel DJ, Loscalzo J, 1992d. Biochemistry of nitric oxide and its redox-activated forms. Science 258 (5090), 1898–1902. [DOI] [PubMed] [Google Scholar]
- Stamler JS, Toone EJ, Lipton SA, Sucher NJ, 1997b. (S)NO signals: translocation, regulation, and a consensus motif. Neuron 18 (5), 691–696. [DOI] [PubMed] [Google Scholar]
- Sternbach GL, Varon J, 2005. The discovery and rediscovery of oxygen. The Journal of Emergency Medicine 28 (2), 221–224. [DOI] [PubMed] [Google Scholar]
- Stomberski CT, Hess DT, Stamler JS, 2019. Protein S-Nitrosylation: Determinants of Specificity and Enzymatic Regulation of S-Nitrosothiol-Based Signaling. Antioxid Redox Signal 30 (10), 1331–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storch EK, Custer BS, Jacobs MR, Menitove JE, Mintz PD, 2019. Review of current transfusion therapy and blood banking practices11This article reflects the views of the author and should not be construed to represent FDA’s views or policies. Blood Reviews 38, 100593. [DOI] [PubMed] [Google Scholar]
- Su H, Liu X, Du J, Deng X, Fan Y, 2020. The role of hemoglobin in nitric oxide transport in vascular system. Medicine in Novel Technology and Devices 5, 100034. [Google Scholar]
- Sun CW, Yang J, Kleschyov A, Zhuge Z, Carlstrom M, Pernow J, Wajih N, Isbell TS, Oh JY, Cabrales P, Tsai A, Townes T, Kim-Shapiro DB, Patel RP, Lundberg JO, 2019. Hemoglobin beta93 Cysteine is Not Required for Export of Nitric Oxide Bioactivity From the Red Blood Cell. Circulation 139 (23), 2654–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomic Mahecic T, Dünser M, Meier J, 2020. RBC Transfusion Triggers: Is There Anything New? Transfusion Medicine and Hemotherapy 47 (5), 361–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trentino KM, Farmer SL, Isbister JP, Sanfilippo FM, Leahy MF, Hofmann A, Shander A, Murray K, 2020. Restrictive Versus Liberal Transfusion Trials: Are They Asking the Right Question? Anesthesia & Analgesia 131 (6). [DOI] [PubMed] [Google Scholar]
- Ulloa NA, Cook J, 2020. Altitude Induced Pulmonary Hypertension, StatPearls. StatPearls Publishing LLC., Treasure Island (FL). [PubMed] [Google Scholar]
- Vallance P, Moncada S, 1994. Nitric oxide--from mediator to medicines. J R Coll Physicians Lond 28 (3), 209–219. [PMC free article] [PubMed] [Google Scholar]
- van Zwieten R, Verhoeven AJ, Roos D, 2014. Inborn defects in the antioxidant systems of human red blood cells. Free Radical Biology and Medicine 67, 377–386. [DOI] [PubMed] [Google Scholar]
- Weinberg JA, MacLennan PA, Vandromme-Cusick MJ, Magnotti LJ, Kerby JD, Rue LW 3rd, Angotti JM, Garrett CA, Hendrick LE, Croce MA, Fabian TC, Barnum SR, Patel RP, 2013. The deleterious effect of red blood cell storage on microvascular response to transfusion. The journal of trauma and acute care surgery 75 (5), 807–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson AM, Harada R, Nair N, Balasubramanian N, Cooke JP, 2007. L-Arginine supplementation in peripheral arterial disease: No benefit and possible harm. Circulation 116 (2), 188–195. [DOI] [PubMed] [Google Scholar]
- Wyman J Jr, Allen DW, 1951. The problem of the heme interactions in hemoglobin and the basis of the bohr effect. Journal of Polymer Science 7 (5), 499–518. [Google Scholar]
- Yao B, Liu DW, Chai WZ, Wang XT, Zhang HM, 2019. Microcirculation dysfunction in endotoxic shock rabbits is associated with impaired S-nitrosohemoglobin-mediated nitric oxide release from red blood cells: a preliminary study. Intensive Care Med Exp 7 (1), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurcisin BM, Davison TE, Bibbs SM, Collins BH, Stamler JS, Reynolds JD, 2013. Repletion of S-Nitrosohemoglobin Improves Organ Function and Physiological Status in Swine After Brain Death. Annals of Surgery 257 (5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemaitis MR, Boll JM, Dreyer MA, 2021. Peripheral Arterial Disease, StatPearls. StatPearls Publishing Copyright © 2021, StatPearls Publishing LLC., Treasure Island (FL). [PubMed] [Google Scholar]
- Zhang R, Hess DT, Qian Z, Hausladen A, Fonseca F, Chaube R, Reynolds JD, Stamler JS, 2015. Hemoglobin betaCys93 is essential for cardiovascular function and integrated response to hypoxia. Proc Natl Acad Sci U S A 112 (20), 6425–6430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Hess DT, Reynolds JD, Stamler JS, 2016. Hemoglobin S-nitrosylation plays an essential role in cardioprotection. J Clin Invest 126 (12), 4654–4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Deng Y, Yang X, Xue H, Lang Y, 2020. The Relationship Between Protein S-Nitrosylation and Human Diseases: A Review. Neurochemical Research 45 (12), 2815–2827. [DOI] [PubMed] [Google Scholar]
- Zhou H-L, Premont RT, Stamler JS, 2021. The manifold roles of protein S-nitrosylation in the life of insulin. Nature Reviews Endocrinology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Xu Z, Wang Y, Sun L, Zhou W, Liu X, 2019. Association between storage age of transfused red blood cells and clinical outcomes in critically ill adults: A meta-analysis of randomized controlled trials. Medicina Intensiva 43 (9), 528–537. [DOI] [PubMed] [Google Scholar]