

Abstract
Triple-negative breast cancer (TNBC) has a high propensity for organ-specific metastasis. However, the underlying mechanisms are not well understood. Here we show that the primary TNBC tumor-derived C-X-C motif chemokines 1/2/8 (CXCL1/2/8) stimulate lung-resident fibroblasts to produce the C-C motif chemokines 2/7 (CCL2/7), which, in turn, activate cholesterol synthesis in lung-colonizing TNBC cells and induce angiogenesis at lung metastatic sites. Inhibiting cholesterol synthesis in lung-colonizing breast tumor cells by pulmonary administration of simvastatin-carrying HER3-targeting nanoparticles reduces angiogenesis and growth of lung metastases in a syngeneic TNBC mouse model. Our findings reveal a novel, chemokine-regulated mechanism for the cholesterol synthesis pathway and a critical role of metastatic site-specific cholesterol synthesis in the pulmonary tropism of TNBC metastasis. The study has implications for the unresolved epidemiological observation that use of cholesterol-lowering drugs has no effect on breast cancer incidence but can unexpectedly reduce breast cancer mortality, suggesting interventions of cholesterol synthesis in lung metastases as an effective treatment to improve survival in individuals with TNBC.
Keywords: chemokine, cholesterol, FOXC1, lung fibroblasts, lung metastasis, triple-negative breast cancer
Graphical abstract
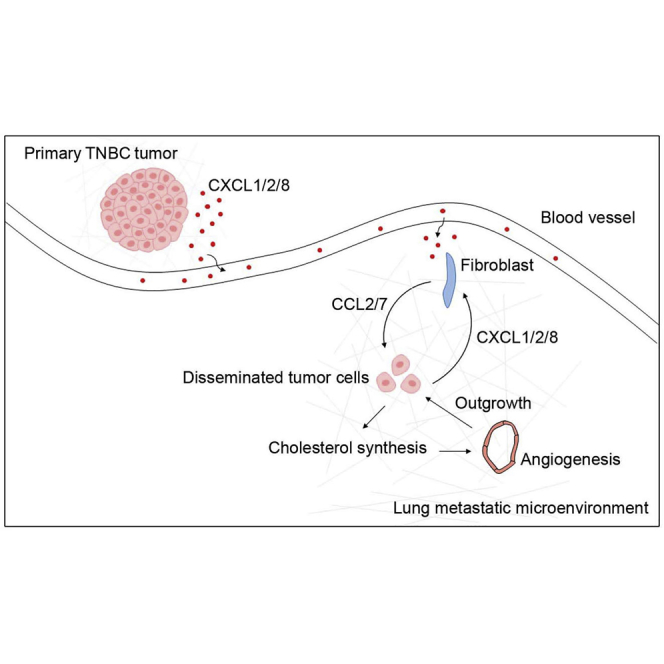
Han et al. discovered local cholesterol synthesis activation in lung-colonizing triple-negative breast cancer (TNBC) cells. They provide evidence that blockade of cholesterol synthesis in TNBC cells at metastatic sites is a potential therapeutic approach for treatment or prevention of distant recurrence in TNBC.
Introduction
Breast cancer is a highly heterogeneous disease characterized by distinct molecular and clinical features. Triple-negative breast cancer (TNBC) comprises a subgroup of tumors that lack expression of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2), as determined by immunohistochemistry.1 It is commonly associated with the basal-like feature, which is defined by a gene expression pattern resembling that of basal or myoepithelial cells of the normal breast.2,3 Clinically, basal cytokeratin markers are used to identify basal-like breast cancers (BLBCs) in TNBC.1,4 TNBC is associated with a poor prognosis and high mortality rate largely because it is prone to develop distant recurrence.4 Distinct subtypes of breast cancer have different metastatic patterns. TNBC spreads preferentially to the lungs and brain.5,6 Elucidating the biological basis of this TNBC metastatic propensity will provide clinically applicable tools for developing therapeutic interventions against latent and active metastatic disease.
Previous work suggests that the crosstalk between primary tumor cells and the microenvironmental components of distant organs, such as fibroblasts and resident and recruited immune cells, is a key factor in dictating organ-specific metastasis.7, 8, 9 This tumor cell-microenvironment interaction facilitates formation of a supportive microenvironment in a distant organ, which promotes survival and proliferation of invading primary tumor cells.9 In particular, several inflammatory factors that mediate this metastatic process have been identified, including the C-X-C motif chemokine ligand 12 (CXCL12)/C-X-C chemokine receptor type 4 (CXCR4),10 nuclear factor κB ligand,11 serum amyloid A3,12 and C-C motif chemokine ligand 2 (CCL2).13,14 Tumor-derived extracellular vesicles, such as exosomes15 and microvesicles,16 have also been shown to be involved in breast cancer organ-specific metastasis via transported microRNAs. Although much knowledge has been gained regarding breast cancer development and progression, the underlying mechanism of the organ tropism of TNBC metastasis is still not well understood.
In the present study, we attempted to systemically identify key secreted factors from breast tumor cells that interact with the lung microenvironment and govern lung metastasis formation in TNBC. We found a small set of TNBC tumor cell-derived chemokines, CXCL1/2/8, that elicit lung-resident fibroblasts to activate cholesterol synthesis in lung-colonizing TNBC cells and, therefore, lung metastasis formation. These findings may have significant clinical implications. Previous studies have shown that cholesterol-lowering drugs, such as statins, had no significant effects on the incidence of colorectal cancer,17,18 lung cancer,19,20 prostate cancer,21,22 and breast cancer.23, 24, 25 However, a protective effect of lipophilic statins (such as simvastatin) in reducing recurrence has been observed in breast cancer.26, 27, 28 More importantly, use of lipophilic statins dramatically reduced mortality in individuals with breast cancer. For example, in a meta-analysis, there was a 43% mortality reduction in individuals with breast cancer taking lipophilic statins.29 Nationwide cohort studies in Finland showed ∼50% mortality reduction.30 Dramatic reductions have also been seen in nationwide cohorts in the UK31 and Scotland.32 In line with these clinical observations, studies performed in mouse models show that a high-cholesterol diet promotes mammary gland tumor progression and lung metastasis.33 The molecular and cellular mechanisms underlying these epidemiological and animal observations remain unclear. The discovery of lung metastatic site-specific cholesterol synthesis activation and its role in metastatic growth provide important implications for the perplexing epidemiological observation that statins reduce the mortality of individuals with breast cancer but not incidence.26, 27, 28
Results
A unique set of chemokines is upregulated in TNBC cells
To identify pivotal secreted factors from TNBC cells that help engage the lung microenvironment to fuel metastatic colonization, we started with established cell models. Given that the FOXC1 transcription factor is a critical marker for TNBC/BLBC and a prognosticator for TNBC lung metastasis,34, 35, 36 we set out to compare gene expression profiles in control (FOXC1-low) and FOXC1-overexpressing TNBC cells using microarray assays (GEO: GSE73234).37 A group of chemokines was found to be among the top upregulated genes induced by FOXC1, including CXCL1/2/8/10 and CCL20 (Figure 1A). Upregulation of these chemokines was confirmed by real-time RT-PCR assays and enzyme-linked immunosorbent assays (ELISAs) (except for CXCL10; Figures S1A and S1B). mRNA upregulation of CXCL1/2/8 and CCL20 was also detected in primary TNBC human breast tumor cultures transfected with FOXC1 (Figure S1C). Analysis of The Cancer Genome Atlas (TCGA) dataset showed significant positive correlations between the levels of FOXC1 and the four chemokines in human breast tumors (Figure 1B). These positive associations were also observed in 14 other breast cancer microarray datasets (Table S1) and a microarray dataset of 51 human breast cancer cell lines38 (Figure S1D). Furthermore, CXCL1/2/8 and CCL20 showed higher expression levels in TNBC/BLBC versus the other subtypes in a microarray dataset in which medullary breast cancer was studied39 (Figure 1C). These results indicate that the chemokines CXCL1/2/8 and CCL20 are upregulated in TNBC.
Figure 1.

The chemokines CXCL1/2/8 and CCL20 are upregulated in TNBC
(A) Heatmap of CXCL1/2/8/10 and CCL20 levels from the cDNA microarrays of control and FOXC1-overexpressing MDA-MB-231 cells. (B) Pearson correlation analysis of CXCL1/2/8 and CCL20 mRNA levels versus FOXC1 in the TCGA dataset (n = 526, around 15% samples have a higher FOXC1 expression).2 Linear regression analysis was performed. (C) Differential expression of CXCL1/2/8 and CCL20 in different breast cancer subtypes in a microarray dataset.39 The molecular subtypes were determined using the single sample predictor classifier,40 and the data were normalized using distance weighted discrimination.41 (D) Real-time RT-PCR analysis of CXCL1/2/8/10 and CCL20 in control, FOXC1-overexpressing, and FOXC1-overexpressing p65 KO MDA-MB-231 cells. (E) Diagram of predicted FOXC1-binding sites in CXCL1/2/8 promoters. (F) CXCL1/2/8 promoter luciferase reporter assays. CXCL1/2/8 promoter fragments containing the FOXC1-binding sites were cloned into the pGL4 luciferase reporter vector. The resulting constructs were transfected into MDA-MB-231 cells together with pCMV6-entry (control) or pCMV6-entry-FOXC1. (G) ChIP assays using anti-FOXC1 antibody. The pull-down of the CXCL1/2/8 promoter fragments was tested by real-time RT-PCR using primers flanking the FOXC1-binding sites. The bar graph indicates mean ± SD (n = 3). ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
We then explored the mechanism by which these chemokines are upregulated in TNBC cells. Given that nuclear factor κB (NF-κB) signaling has been shown to upregulate chemokine expression in different cell types42, 43, 44, 45, 46 and that FOXC1 enhances NF-κB activity in TNBC cells,47 we reasoned that NF-κB may mediate FOXC1-induced upregulation of the four chemokines. To address this, we used CRISPR-Cas9 to perform p65 knockout (KO) in FOXC1-overexpressing MDA-MB-231 cells (Figure S1E). As shown in Figure 1D, mRNA expression of CXCL1/2/8 and CCL20 was reduced in p65 KO cells, suggesting that NF-κB is involved in upregulation of CXCL1/2/8 and CCL20 by FOXC1 in TNBC cells. However, knockdown of p65 only partially attenuated FOXC1-induced upregulation of CXCL1/2/8, whereas CCL20 induction was completely reversed by p65 knockdown. These results suggest that, in addition to involvement of p65, some other potential mechanism might also play a role in upregulation of CXCL1/2/8. Interestingly, the consensus FOXC1 DNA-binding sequence GTAAAT(/C)AA(/T/C)A(/G/T)48 is located in the CXCL1/2/8 promoters (Figure 1E), so we speculated that FOXC1 may directly bind to the promoters of the CXCL1/2/8 genes in TNBC cells. We thus cloned the CXCL1/2/8 promoter fragments containing the potential FOXC1-binding sites into the luciferase reporter vector pGL4 and performed luciferase assays. As presented in Figure 1F, luciferase activity was highly induced by FOXC1 overexpression, suggesting that FOXC1 can activate the CXCL1/2/8 promoters. Chromatin immunoprecipitation (ChIP) assays further indicated binding of FOXC1 to the CXCL1/2/8 promoter regions (Figure 1G). It is well established that p65 can upregulate CXCL1/2/8 expression by directly binding to their promoters.42,49 Our findings suggest that CXCL1/2/8 expression induction by FOXC1 in TNBC cells is partially mediated by NF-κB and may also involve direct activation of their promoters by FOXC1.
CXCL1/2/8 enhance lung metastasis in TNBC
Chemokines are known to play essential roles in cancer development and metastasis.50 To test whether CXCL1/2/8 and CCL20 expression is associated with TNBC lung metastasis, we combined and analyzed 572 human breast cancer samples from three microarray datasets51, 52, 53 and found that elevated levels of CXCL1/2/8 and CCL20 were correlated significantly with worse lung metastasis-free survival (Figure 2A, top panel). In contrast, these correlations were not observed in the bone metastasis-free survival analysis (Figure 2A, bottom panel). To examine the potential involvement of these chemokines in TNBC, we simultaneously knocked out expression of CXCL1/2/8 and CCL20 using the CRISPR-Cas9 lentiviral system in FOXC1-overexpressing MDA-MB-231 cells (Figure S2A). In orthotopic xenograft mouse models, blockade of the expression of these chemokines dramatically reduced the size of lung metastatic foci (Figure 2B, left panel) and the number of metastatic nodules (Figure 2B, right panel). These findings suggest that the chemokines CXCL1/2/8 and CCL20 collectively play an essential role in TNBC lung metastasis formation.
Figure 2.

CXCL1/2/8 regulates lung metastasis of TNBC cells
(A) Kaplan-Meier analysis of the lung and bone metastasis-free survival for CXCL1/2/8, CCL20, and FOXC1 in combined microarray datasets of human breast cancer (n = 572; GEO: GSE12276, GSE2603, and GSE2034). To remove the systematic biases, the expression values were converted to Z scores for all genes prior to combining the three datasets. (B) orthotopic xenograft mammary tumor models to evaluate the formation of lung metastasis. 1 × 106 control, FOXC1-overexpressing, or FOXC1-overexpressing chemokine-KO MDA-MB-231 cells were injected into the mammary fat pads. Mouse lungs were collected 5 weeks after injection (n = 5). Metastatic nodules in the lungs were counted (right panel), and lung sections were stained by hematoxylin and eosin (left panel). (C) ELISA analysis of CXCL1/2/8 and CCL20 levels in mouse serum. Blood samples were collected when the mice in (B) were sacrificed. The values of the concentration were calculated by the standard curves (negative values indicate extremely low levels). (D) Syngeneic mouse models to evaluate lung metastasis formation capacity. 3 × 106 EO771 cells were injected into wild-type or cxcr2+/− C57BL/6 mice through the tail vein (n = 5). Mouse lungs were collected 6 weeks after injection. Metastatic nodules were counted (right panel), and lung sections were stained by hematoxylin and eosin (left panel). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Next we explored the potential effects of these chemokines in driving metastasis. Simultaneous KO of CXCL1/2/8 and CCL20 had no effects on migration and invasion of FOXC1-overexpressing TNBC cells in vitro, as revealed by wound healing (Figure S2B) and Boyden chamber assays (Figures S2C and S2D). Cell proliferation assays (Figure S2E) and orthotopic xenograft mouse models (Figure S2F) showed that the growth of TNBC cells in vitro or in vivo was not affected by synchronous chemokine KO. To determine whether these chemokines play a role in tumor cell extravasation in the lung tissue, we injected tumor cells labeled with the green dye CMFDA (5-chloromethylfluorescein diacetate) through the tail vein. As presented in Figure S2G, inhibiting expression of the chemokines did not change the tumor cell extravasation ability. However, when tumor cell outgrowth in the lung was examined as early as 5 days after tail vein injection, the size of the metastatic foci in the chemokine KO group was reduced significantly (Figure S2H), suggesting that these chemokines may regulate outgrowth of the tumor cells disseminated in the lung.
We then tested whether elevated expression of the chemokines in the primary tumors leads to increased circulating chemokine levels in the mice used in Figure 2B. ELISAs showed that mice with chemokine KO tumors had lower circulating levels of CXCL1/2/8 compared with FOXC1-overexpressing mice (Figure 2C). Surprisingly, there was no significant elevation of circulating CCL20 in the FOXC1-overexpressing group (Figure 2C), suggesting that FOXC1-high breast tumors may not give rise to an increase in secreted or circulating CCL20 levels in vivo. Therefore, we focused on CXCL1/2/8 in the following studies.
It is known that CXCL1/2/8 share a common receptor, CXCR2.54 To confirm the role of CXCL1/2/8 in TNBC lung metastasis, we injected EO771 cells, a mouse TNBC cell line derived from tumors of C57BL/6 mice, into wild-type and cxcr2-heterozygous (cxcr2+/−) C57BL/6 mice55 through the tail vein. Lung metastasis was abolished even by cxcr2 heterozygous deficiency (Figure 2D), indicating that the CXCR2-CXCL1/2/8 signaling cascade is necessary for outgrowth of lung-colonizing TNBC cells. These data suggest an important role of primary tumor cell-derived CXCL1/2/8 in TNBC lung metastasis.
CXCL1/2/8 promote angiogenesis through lung fibroblasts
We speculated that CXCL1/2/8 might act on the tissue microenvironment to facilitate metastatic outgrowth of TNBC cells that have extravasated into the lungs. Because it has been well recognized that chemokines can regulate angiogenesis,50,56 we examined whether CXCL1/2/8 regulate angiogenesis in lung metastases samples from the mice used in Figure 2B. There was a higher density of blood vessels, as detected by staining CD31, an endothelial cell marker, in FOXC1-overexpressing MDA-MB-231 cell xenograft tumors compared with control MDA-MB-231 cell tumors (Figure 3A). This increase in vasculature was abolished in CXCL1/2/8 KO tumors (Figure 3A). These results suggest that high levels of CXCL1/2/8 in primary TNBC tumors are essential for angiogenesis in lung metastases.
Figure 3.

CXCL1/2/8 promote angiogenesis through lung fibroblasts in TNBC lung metastases
(A) Representative images of blood vessels stained with CD31 (red) in mouse lung metastases of Figure 2B. The blood vessel density was quantified by ImageJ software (n = 10 view fields; two view fields per mouse, 5 mice were examined). Magnification, 400×. (B) migration assays of isolated mouse lung endothelial cells. 2 × 105 freshly isolated mouse lung endothelial cells were suspended in Vasculife medium and placed into ECM-coated chambers. Chemokines and/or other different cells were added to the bottom chamber. The migrated cells were stained after 16 h of incubation, and the dye was extracted and measured at OD560 using a luminometer. Recombinant human CXCL1 (8 nM)/2 (2 nM)/8 (20 nM) proteins or different cell types were added to the bottom chamber of the assay system. (C) Tube formation assays of isolated mouse lung endothelial cells. Recombinant human CXCL1 (8 nM)/2 (2 nM)/8 (20 nM) proteins or the supernatant of single or co-culture of different cell types were added to the medium of endothelial cells cultured on the surface of Matrigel. (D) Migration assays of isolated mouse lung endothelial cells. Recombinant human CXCL1 (8 nM)/2 (2 nM)/8 (20 nM) proteins, antibodies, or different cell types were added to the bottom chamber. (E) Tube formation assays of the isolated mouse lung endothelial cells. Recombinant human CXCL1 (8 nM)/2 (2 nM)/8 (20 nM) proteins, neutralizing antibodies, or the supernatant from single- or co-culture of different cell types were added to the medium of endothelial cells cultured on the surface of Matrigel. The bar graph indicates mean ± SD. ∗p < 0.05, ∗∗p < 0.01.
To address whether specific host lung resident cells are involved in induction of angiogenesis by CXCL1/2/8, we isolated fibroblasts, macrophages, neutrophils, and endothelial cells from normal lung tissue of 4-week-old mice and performed lung endothelial cell migration assays. Lung fibroblasts, macrophages, and neutrophils have been shown to express Cxcr2.57, 58, 59 As illustrated in Figure 3B, when fibroblasts, but not macrophages or neutrophils, were co-cultured with FOXC1-overexpressing MDA-MB-231 cells, it led to enhanced migration of lung endothelial cells. Interestingly, FOXC1-overexpressing MDA-MB-231 cells were essential for this enhanced migration because CXCL1/2/8 proteins by themselves could not significantly increase endothelial cell migration (Figure 3B). Similar results were found in tube formation assays when conditioned media of co-cultured fibroblasts, FOXC1-overexpressing MDA-MB-231 cells, and CXCL1/2/8 proteins were added to endothelial cells seeded on top of Matrigel. The resulting tube formation capacity was increased significantly compared with the other groups (Figure 3C). In line with these results, chemokine neutralization using anti-CXCL1/2/8 antibodies inhibited lung endothelial cell migration (Figure 3D) and tube formation (Figure 3E). Similar results were obtained when cxcr2+/− lung fibroblasts replaced the cxcr2-wild-type fibroblasts (Figures 3D and 3E). These data suggest that CXCL1/2/8 mediate the cumulative effect of TNBC cells and lung fibroblasts on angiogenesis in TNBC lung metastasis.
CXCL1/2/8 stimulate CCL2/7 expression in lung fibroblasts
Given our previous results, it is reasonable to speculate that CXCL1/2/8 proteins stimulate lung fibroblasts to secrete certain factors into the in vitro culture system to induce migration of lung endothelial cells. To uncover those potential factors, we treated isolated mouse lung fibroblasts with recombinant human CXCL1/2/8 proteins and performed RNA sequencing (RNA-seq) (GEO: GSE131306). Review of the RNA-seq data using Ingenuity Pathway Analysis (IPA) identified a number of factors that were filtered by the “extracellular space” cellular localization feature (Figure 4A). To identify the potential secreted factors that mediate lung fibroblast-regulated angiogenesis, we knocked down five top upregulated genes individually using small interfering RNAs (siRNAs) (Figure S3A). In vitro angiogenesis assays showed that knockdown of chemokine Ccl2 or Ccl7 impaired the migration (Figure 4B) and tube formation (Figure 4C) capacities of mouse lung endothelial cells, suggesting that these two chemokines are important players mediating lung fibroblast-induced angiogenesis in TNBC metastasis. IPA functional analysis also revealed some hormone-related signaling pathways activated by CXCL1/2/8 in mouse lung fibroblasts, such as glucocorticoid receptor signaling (Figure S3B). These hormone-related pathways may be involved in induction of CCL2/7 in lung fibroblasts.60
Figure 4.

Lung fibroblasts increase CCL2/7 expression in response to CXCL1/2/8
(A) heatmap showing the top upregulated proteins secreted into the extracellular space. RNA-seq experiments were performed using control and human CXCL1 (8 nM)/2 (2 nM)/8 (20 nM)-treated (6 h) mouse lung fibroblasts. (B) migration assays of isolated mouse lung endothelial cells. FOXC1-overexpressing MDA-MB-231 cells and mouse lung fibroblasts transfected with different siRNAs were added to the bottom of the assay system. (C) Tube formation assays of isolated mouse lung endothelial cells. The supernatant of FOXC1-overexpressing MDA-MB-231 cells and lung fibroblasts transfected with different siRNAs were added to the medium of endothelial cells cultured on the surface of Matrigel. (D) RNAscope in situ hybridization assays of CCL2/7 mRNAs in normal human lungs and lungs with TNBC metastases. Arrow, positive red RNA signal in fibroblasts adjacent to metastatic tumor cells. (E) Real-time RT-PCR assays of Ccl2/7 mRNA expression in cancer-associated fibroblasts (CAFs) of lung metastases. (F) Real-time RT-PCR of CCL2/7 mRNAs in control cells or the human CXCL1 (8 nM)/2 (2 nM)/8 (20 nM)-treated normal human lung fibroblast cell line IMR-90. (G) Real-time RT-PCR assays of Ccl2/7 mRNA expression in fibroblasts isolated from mouse bone marrow or liver. The bar graph indicates mean ± SD (n = 3). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To further corroborate induction of CCL2/7 in lung fibroblasts in TNBC lung metastases, we performed RNAscope in situ hybridization assays in normal human lung tissue and lung tissue with TNBC metastases. As shown in Figure 4D, CCL2/7 mRNAs were upregulated dramatically in the fibroblast component of the lung tissue with TNBC metastases compared with normal lung tissues. The corresponding localization of the fibroblasts in those lung tissues was supported by vimentin immunohistochemistry (IHC) staining (Figure S3C). CCR2, the common receptor for CCL2/7, was also expressed in tumor cells in human TNBC lung metastases, as shown by IHC (Figure S3D). In addition, we isolated cancer-associated fibroblasts (CAFs) from lung metastasis samples and treated those cells with CXCL1/2/8. Ccl2/7 mRNA induction was observed in mouse lung CAFs (Figure 4E). CCL2/7 mRNA induction by CXCL1/2/8 was also detected in the normal human lung fibroblast cell line IMR-90 (Figure 4F). To assess whether the CXCL1/2/8 effect on CCL2/7 is lung fibroblast specific, we isolated fibroblasts from the mouse liver and bone marrow, two other common sites for breast cancer metastasis.6 Real-time RT-PCR analysis showed that Ccl2/7 expression was not induced significantly by CXCL1/2/8 in those fibroblasts (Figure 4G). Moreover, Ccl2/7 mRNA induction by CXCL1/2/8 was only observed in lung fibroblasts but not in macrophages, neutrophils, endothelial cells, immortalized lung epithelial cells, or TNBC tumor cells (Figure S3E). These results suggest that the CXCL1/2/8-CCL2/7 regulation axis reflects the interaction between TNBC cells and lung fibroblasts in lung metastasis-associated angiogenesis.
CCL2/7 induce cholesterol synthesis in lung-colonizing TNBC cells
Because TNBC cells and lung fibroblasts cooperate to elicit in vitro angiogenesis by lung endothelial cells, we wanted to find out whether lung fibroblast-derived CCL2/7 might interact with lung-colonizing tumor cells to promote angiogenesis. To test this, we treated FOXC1-overexpressing MDA-MB-231 cells, which express the CCL2/7 receptor CCR2 (Figure S4A), with recombinant mouse Ccl2/7 proteins for 6 h and performed RNA-seq (GEO: GSE131383). Strikingly, IPA of the gene expression profiles in control and Ccl2/7-treated cells revealed that the predominant cellular pathways activated by Ccl2/7 were related to cholesterol synthesis (Figure 5A). This result was validated by real-time RT-PCR of cholesterol synthesis-associated genes (Figure 5B). ELISA results also showed that the cholesterol concentration in TNBC cell culture supernatant was elevated after Ccl2/7 treatment (Figure S4B). Using IHC, we examined the expression of 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR) and another key cholesterol synthesis enzyme, 3-hydroxy-3-methylglutaryl-coenzyme A synthase 1 (HMGCS1), in paired samples of human primary TNBC tumors and lung metastases. As presented in Figure 5C, compared with the primary tumors, the matched lung metastases displayed markedly elevated levels of HMGCR and HMGCS1. Similar results were found in the 4T1 syngeneic TNBC mouse breast tumors and corresponding lung metastases (Figure S4C). These data suggest that lung fibroblast-derived CCL2/7 potentiate cholesterol synthesis in TNBC cells of lung metastases.
Figure 5.

CCL2/7 induce cholesterol synthesis in lung-colonizing TNBC cells
(A) Gene Ontology (GO) analysis of the upregulated genes. RNA-seq experiments were performed in control and mouse Ccl2 (10 nM)/7 (50 nM)-treated (6 h) FOXC1-overexpressing MDA-MB-231 cells. (B) Real-time RT-PCR confirming upregulation of cholesterol synthesis-associated genes identified by RNA-seq. (C) IHC of HMGCR and HMGCS1 in human primary TNBC tumors and matched lung metastases (n = 4 pairs). (D) Western blot of the nuclear accumulation of the mature SREBP2 protein in mouse Ccl2 (10 nM)/7 (50 nM)-treated FOXC1-overexpressing MDA-MB-231 cells at different time points. Lamin A/C was used as a control. (E) IHC of SREBP2 in human TNBC lung metastases. (F) Heatmap analysis of the upregulated secreted factors in the RNA-seq analysis performed in (A). The bar graph indicates mean ± SD (n = 3). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To understand the molecular mechanism underlying CCL2/7-induced cholesterol synthesis, we examined the levels of the mature form of sterol regulatory element-binding protein 2 (SREBP2), the pivotal transcription factor governing expression of cholesterol synthesis enzymes.61 Immunoblotting showed that Ccl2/7 stimulation enhanced accumulation of the mature SREBP2 protein in the nuclei of TNBC cells (Figure 5D). IHC of human TNBC lung metastases demonstrated nuclear staining of SREBP2 (Figure 5E). It was also noted that CXCL1/2/8 were among the top upregulated genes induced by Ccl2/7 in FOXC1-overexpressing MDA-MB-231 cells (Figure 5F). This finding may reflect a positive feedback regulatory loop of chemokines mediating the interaction between lung fibroblasts and neighboring tumor cells in TNBC lung metastases.
Cholesterol produced in lung-colonizing tumor cells is necessary for TNBC lung metastasis
To explore whether the cholesterol synthesized in lung-colonizing TNBC cells mediates CXCL1/2/8-induced angiogenesis in lung metastases, we first examined the effect of increased cholesterol levels on lung endothelial cell migration and tube formation in vitro. Addition of recombinant Ccl2/7 or cholesterol in FOXC1-overexpressing MDA-MB-231 cell culture dramatically increased the lung endothelial cell migration and tube formation capacities, whereas treatment with simvastatin, an HMGCR inhibitor, attenuated the Ccl2/7 effect (Figures 6A and 6B). Inhibition of cholesterol synthesis by simvastatin was confirmed by ELISAs (Figure S5A). Interestingly, addition of cholesterol, simvastatin, or Ccl2/7 alone had no effect on endothelial cell migration and tube formation in vitro (Figures 6A and 6B). It is worth mentioning that cholesterol addition had no effects on endothelial cell proliferation in in vitro assays (data not shown).
Figure 6.

Targeting cholesterol synthesis in TNBC cells in the lung to inhibit metastasis formation
(A and B) Migration (A) and tube formation (B) assays of isolated mouse lung endothelial cells. The HMGCR inhibitor simvastatin or cholesterol was added to the culture medium. (C and D) Migration (C) and tube formation (D) assays of isolated mouse lung endothelial cells. The geranylgeranyltransferase inhibitor GGTI-2133, farnesyltransferase inhibitor FTI-277, or squalene synthase inhibitor YM-53601 was added to the culture medium. (E) Syngeneic mouse models to evaluate lung metastasis formation. 0.5 × 106 4T1 cells were injected into BALB/c wild-type mice through the tail vein (n = 5). PBS or HPK-simvastatin (10 μg/mouse/daily) nanoparticles were administered intranasally beginning 48 h after tumor cell injection. Mouse lungs were collected 2 weeks after injection. Metastatic nodules were counted (right panel), and lung sections were stained by hematoxylin and eosin (left panel). (F) Representative images of CD31 staining of blood vessels in mouse lung metastases. The blood vessel density was quantified by ImageJ software (n = 15 view fields). Magnification, 400×. (G) Schematic illustrating a proposed model of the cross-talk among primary tumor cells, lung fibroblasts, and disseminated tumor cells during TNBC lung metastasis formation. ∗p < 0.05, ∗∗∗p < 0.001.
It has been recognized that, in addition to reducing cholesterol production, simvastatin also blocks synthesis of the isoprenoids geranylgeranyl pyrophosphate (GGPP) and farnesyl pyrophosphate (FPP),23,62 two critical substrates for protein prenylation that have crucial functions in biological regulation.63 To further confirm that synthesis of cholesterol, but not FPP or GGPP, is involved in endothelial cell migration and tube formation in in vitro assays, we added the geranylgeranyltransferase inhibitor GGTI-2133 or farnesyltransferase inhibitor FTI-277 to the assays. Treatment with the inhibitors had no effects on migration and tube formation of endothelial cells in vitro (Figures 6C and 6D). In contrast, addition of YM-53601, an inhibitor of squalene synthase (FDFT1) in the early steps of cholesterol synthesis, significantly blocked endothelial cell migration and tube formation (Figures 6C and 6D).
We then tested the effect of elevated cholesterol synthesis on lung metastasis formation in mouse models. To do so, 4T1 mouse TNBC cells were injected into mice via the tail vein. The mice were then treated intranasally with simvastatin, which was delivered specifically into tumor cells in the lungs by the HER3-targeted nanoparticle HerPBK10 (HPK), a recombinant multidomain polypeptide.64 The delivery efficiency of the HPK-simvastatin to lung tissue was confirmed by Alexa Fluor 680-conjugated HPK-oligo (Figure S5B). As illustrated in Figure 6C, compared with the control group, the number of lung metastatic nodules and the size of the metastatic foci were reduced in the HPK-simvastatin treatment group. CD31 staining showed that HPK-simvastatin treatment decreased blood vessel density in lung metastases (Figure 6D). We also knocked out Hmgcr expression in 4T1 cells using CRISPR-dCas9 and injected those cells into mice through the tail vein. As expected, knocking out Hmgcr impaired metastasis formation in lung tissues (Figures S5C and S5D). These results suggest that cholesterol synthesis in lung-colonizing TNBC cells is necessary for angiogenesis and, consequently, lung metastasis formation.
Discussion
In this study, we found that cholesterol synthesis is activated in lung metastatic TNBC tumor cells during metastasis formation. This activation is mediated by the interaction between lung-colonizing TNBC tumor cells and lung-resident fibroblasts. Mechanically, the TNBC tumor cell-derived chemokines CXCL1/2/8 activate lung fibroblasts to secrete additional chemokines, CCL2/7, which, in turn, induce lung-colonizing TNBC cells to increase cholesterol synthesis, promoting angiogenesis and outgrowth of lung metastases. Our findings of chemokine loop-induced cholesterol synthesis in lung-colonizing TNBC cells may help explain the epidemiological observation that statin use can decrease breast cancer distant recurrence and mortality. Our results provide a link between chemokines, cholesterol synthesis, and angiogenesis that plays an essential role in establishing the metastatic microenvironment in TNBC (Figure 6G), suggesting a potential strategy to treat lung metastasis through targeted blockade of metastatic site-specific cholesterol synthesis.
Previous work has demonstrated that elevated systemic cholesterol promotes primary breast tumor initiation and progression in mouse models.33,65, 66, 67 The effect of cholesterol in breast cancer is further supported by recent reports showing increased cholesterol synthesis in TNBC.68, 69, 70 Interestingly, epidemiological studies show that using statins can reduce breast cancer recurrence and mortality. Surprisingly, it does not affect breast cancer incidence.26, 27, 28 The biological basis for this observation remains unclear. Our results of cholesterol synthesis induction and its critical role in TNBC lung metastasis provide an explanation for the effect of statins on breast cancer distant recurrence and mortality. Indeed, blocking cholesterol synthesis specifically in lung metastatic sites using simvastatin-carrying nanoparticles in TNBC mouse models inhibited lung metastases. Consistent with our observations, recent studies show that systemic statin drug treatment inhibited breast cancer lung metastasis in xenograft mouse models.71,72 One desirable benefit of metastatic site-targeted delivery of statins is potential minimization of the drug side effect in treatment.
Here we also demonstrate a unique set of CXCL1/2/8 involved in TNBC’s interaction with the lung microenvironment. Chemokine involvement in breast cancer lung metastasis has been studied previously.50 For example, CXCL1 is a critical component of the gene signature that mediates dissemination of primary breast tumor cells to the lungs.53 An elevated circulating level of CXCL1 in individuals with breast cancer is associated with a higher propensity for circulating tumor cells to seed in lung tissue.73 Moreover, CXCL1/2-expressing breast tumor cells can recruit myeloid cells in a paracrine manner to promote tumor cell survival at primary and metastatic sites, resulting in chemoresistance and metastasis.74 Here, our findings demonstrate that those chemokines have no discernible effect on in vitro proliferation, migration, invasion, or extravasation of primary TNBC cells but instead exert an indirect effect by affecting lung fibroblasts, which, in turn, regulate TNBC cholesterol synthesis.
The indispensable roles of CAFs have been recognized during metastasis, including their participation in extracellular matrix (ECM) remodeling, tumor cell immunity, metabolism, etc.75 However, those functions are largely related to primary tumor-associated fibroblasts. To date, whether resident fibroblasts at metastatic sites are involved in metastasis formation remains poorly understood. Our results show that TNBC-derived CXCL1/2/8 can activate lung-resident fibroblasts and CAFs to secrete other chemokines, including CCL2/7. CCL2 synthesis by metastatic tumor cells and the lung stroma can recruit inflammatory monocytes to facilitate breast cancer lung metastasis.13 Interestingly, akin to the FOXC1-CXCL1/2/8-CCL2/7 signaling cascade uncovered in this study, a CXCL8-FOXC1-CCL2 signaling mechanism has been identified in hepatocellular carcinoma (HCC) cells during lung metastasis.76 Our data demonstrate a previously unidentified function of lung fibroblasts in TNBC metastasis, highlighting the key role of tissue-resident fibroblasts in organ-specific metastasis.
We further show that lung fibroblast-derived CCL2/7 act directly on lung-colonizing metastatic TNBC cells to potentiate cholesterol synthesis mediated by nuclear accumulation of mature SREBP2, the master transcription factor governing cholesterol synthesis-associated genes.61 Increased cholesterol in lung-colonizing TNBC cells leads to angiogenesis in lung metastases. Consistent with our findings, it has been reported that a high-cholesterol diet in mouse models promotes mammary tumor development and lung metastasis by inducing angiogenesis.33 Moreover, systemic 27-hydroxycholesterol, the primary metabolite of cholesterol, has also been shown to be involved in angiogenesis and metastasis in ER+ breast cancer.72,77 How cholesterol promotes angiogenesis is not well understood. We noticed that, along with cholesterol synthesis, tissue factor signaling is also highly activated by CCL2/7 in TNBC cells (Figure 5A). Given the fact that this is a well-established pathway involved in angiogenesis78 and can be activated by cholesterol,79,80 it is compelling to speculate that tissue factor signaling may mediate the effect of locally induced cholesterol production on angiogenesis in the metastatic tumor microenvironment. It is also possible that the synthesized cholesterol might be exported into the surrounding microenvironment to facilitate angiogenesis, which is supported by upregulation of the ABCA1 gene by Ccl2/7 in MDA-MB-231-FOXC1 cells in vitro (Figure S5E). Further studies are warranted to corroborate this hypothesis. Another interesting finding in our study is that CCL2/7 induce CXCL1/2/8 secretion in TNBC cells. This regulation creates a positive chemokine feedback loop between lung fibroblasts and neighboring TNBC cells at metastatic sites, resulting in potent induction of cholesterol synthesis. Thus, the chemokine loop and elevated local cholesterol production may reform the microenvironment, facilitating metastatic tumor growth.
Our study uncovers a novel mechanism for cholesterol synthesis activation for TNBC lung metastasis formation. These results suggest that disruption of the chemokine-cholesterol signaling axis or blockade of cholesterol synthesis in tumor cells at metastatic sites may be promising therapeutic strategies to treat lung metastasis or prevent recurrence in the lungs.
Materials and methods
Cell culture
The human breast cancer cell lines MDA-MB-231 and BT474 and mouse breast cancer cell lines 4T1 and EO771 were purchased from the American Type Culture Collection (ATCC) and maintained according to ATCC instructions. The normal human lung fibroblast line IMR-90 was a gift from Dr. Hisashi Tanaka (Cedars-Sinai Medical Center). The immortalized mouse lung epithelial cell line MLE15 was a gift from Dr. Barry Stripp (Cedars-Sinai Medical Center). Control and FOXC1-overexpressing MDA-MB-231 cells were established as described previously.37 KO of p65 or the chemokines CXCL1/2/8 and CCL20 in FOXC1-overexpressing MDA-MB-231 cells and KO of Hmgcr in 4T1 cells were performed using LentiCRISPRv2 (52961, Addgene).81 The cells were selected using puromycin, and the drug-resistant heterogeneous cell pool was used in the study. The guide RNA (gRNA) sequences are listed in Table S2. Primary human TNBC cells,82 mouse lung fibroblasts, and CAFs in lung metastases of xenograft tumors,83 mouse lung macrophages,84 and mouse lung endothelial cells85 were isolated as described previously. The method for isolating mouse lung fibroblasts was also used to isolate mouse liver fibroblasts. Mouse lung neutrophils were isolated using the following method. Briefly, a single-cell suspension of mouse lung tissue was prepared with collagenase/dispase (10269638001, Sigma-Aldrich). Cells were then incubated with CD16/CD32 (1:100)/1 mL PBS for 15 min, followed by incubation with CD45 phycoerythrin (PE)-Cy7 (1:200), CD11b PerCP-Cy5.5 (1:200), and Ly6G PE (1:200)/1 mL PBS for 45 min. Cells were washed once using PBS (1% fetal bovine serum [FBS]) and sorted using Becton Dickinson FACSAria II (BD Biosciences). To isolate mouse bone marrow-derived fibroblasts, bone marrow was harvested from femora and tibiae of 7-week-old mice and filtered with 40-μm-pore nylon cell strainers (BD Biosciences). Cells were cultured in DMEM/F12 supplemented with 5% fetal bovine serum, 5% Nu serum, 5 μg/mL insulin, and penicillin/streptomycin. Fresh medium was replaced every 2–3 days.
Chemicals
Recombinant human CXCL1 (275-GR-010, R&D Systems), recombinant human CXCL2 (276-GB-010, R&D Systems), recombinant human CXCL8 (208-IL-010, R&D Systems), recombinant mouse Ccl2 (479-JE-010, R&D Systems), recombinant mouse Ccl7 (456-MC-010, R&D Systems), GGTI-2133 (G5294, Sigma-Aldrich), FTI-277 (F9803, Sigma-Aldrich), YM-53601 (18113, Cayman Chemical), simvastatin (S1792, Selleckchem), and human cholesterol (C8667, Sigma-Aldrich).
Real-time RT-PCR
Total RNA was extracted using the RNeasy Mini Kit (QIAGEN) and reverse transcribed into single-stranded cDNAs using the QuantiTect Reverse Transcription Kit (QIAGEN). Real-time RT-PCR was performed using the CFX96 Real-Time System (Bio-Rad). Primers are listed in Table S3.
Western blotting
Proteins were extracted using radioimmunoprecipitation assay (RIPA) lysis buffer (Thermo Fisher Scientific), and protein concentration was determined by bicinchoninic acid (BCA) protein assays (Thermo Fisher Scientific). Nuclear proteins were extracted using NE-PER™ nuclear and cytoplasmic extraction reagents (Thermo Fisher Scientific). Proteins (50 μg) were separated on 4%–20% gradient gels and transferred onto polyvinylidene fluoride (PVDF) membranes using the Trans-Blot Turbo transfer pack (Bio-Rad) and Trans-Blot Turbo transfer system (Bio-Rad). Membranes were blocked in Odyssey blocking buffer (LI-COR Biosciences) and incubated with primary antibodies overnight at 4°C. The primary antibodies were SREBP2 (1:1,000, MAB7119, R&D Systems), Lamin A/C (1:500, SC-7292, Santa Cruz Biotechnology), HMGCR (1:1,000, MBS9406409, Mybiosource), and actin (1:1,000, SC-1616, Santa Cruz). Membranes were then incubated with IRDye 680CW or IRDye 800CW secondary antibodies (LI-COR Biosciences) for 1 h at room temperature. The membranes were scanned using the Odyssey infrared imaging system (LI-COR Biosciences).
ELISA
The concentrations of the specific chemokines and cholesterol in the supernatant of cell culture or mouse serum were measured using ELISAs according to the manufacturer’s instructions. The ELISA kits were human CXCL1 (DGR00, R&D Systems), human CXCL2 (ab184862, Abcam), human CXCL8 (D8000C, R&D Systems), human CCL20 (DM3A00, R&D Systems), and human cholesterol (MBS729689, Mybiosource).
Luciferase assay
CXCL1 (149 bp), CXCL2 (150 bp), and CXCL8 (143 bp) promoter fragments containing FOXC1-binding sites were synthesized (Thermo Fisher Scientific) and subcloned into the luciferase vector pGL4 (Promega). Cells seeded in 12-well plates were co-transfected with 500 ng luciferase plasmid and 500 ng pCMV6-entry (OriGene) or pCMV6-entry-FOXC1 (OriGene) expression plasmids using Lipofectamine 2000 (Invitrogen). Two hundred nanograms of β-galactosidase expression plasmid (Promega) was used as an internal control. After 48 h, cells were harvested, and extracts were analyzed using the Luciferase Reporter Assay System (Promega) and β-Galactosidase Enzyme Assay System (Promega) according to the manufacturer’s instructions. Luciferase activity was measured using a luminometer (GloMax-Multi Detection System, Promega).
Immunofluorescence
FOXC1-overexpressing MDA-MB-231 cells were placed into chamber slides (Thermo Fisher Scientific) at 70%–80% confluence. Cells were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100, blocked with 5% BSA, and incubated with primary anti-CCR2 antibody (1:100, NBP2-35334, Novus Biologicals) overnight at 4°C, followed by secondary antibody incubation (1:100, A21202, Life Technologies) for 1 h at room temperature. For CD31 staining of blood vessels, immunofluorescence was performed using formalin-fixed, paraffin-embedded mouse lung metastasis samples. The primary antibody used was anti-CD31 antibody (1:100, CM303A, Biocare Medical). The secondary antibody was Alexa Fluor 546 (1:200, A11081, Life Technologies). The nuclei were stained with DAPI (Life Technologies). Images were acquired using the EVOS FL Auto Imaging System (Thermo Fisher Scientific). The density of the blood vessels was analyzed using ImageJ software.
ChIP
5 × 106 cells were collected, and ChIP assays were performed using the EZ-ChIP Chromatin Immunoprecipitation Kit (EMD Millipore) according to the manufacturer’s instructions. Briefly, control or FOXC1-overexpressing MDA-MB-231 cells were fixed with 1% formaldehyde, followed by lysis and sonication of the cells. The 200- to 1,000-bp chromatin fragments were then immunoprecipitated using the anti-FOXC1 antibody (sc-21394, Santa Cruz). The protein-DNA crosslinks were then reversed, and DNA fragments were purified. The yield of the immunoprecipitated DNA fragments was quantified using a real-time RT-PCR assay. Data were analyzed using the 2-ΔΔCT method. The primers were as follows: CXCL1 forward, 5′-CTGGCTTCCACTGACGCTAA-3′; CXCL1 reverse, 5′-TCTGCTTGTTCCTTGTGTTGC-3′; CXCL2 forward, 5′-TCTAGAGGGAACCAATCCCAGG-3′; CXCL2 reverse, 5′-AGGCCATAGACACCACCAGA-3′; CXCL8 forward, 5′-GGTGCTAGTCTCTGCTCATCA-3′; CXCL8 reverse, 5′-ACTAAAGTGCCACGCTACACA-3′.
RNA-seq assay
The RNA-seq assay was performed using mouse lung fibroblasts and FOXC1-overexpressing MDA-MB-231 cells. Freshly isolated mouse lung fibroblasts were treated with PBS or recombinant human CXCL1 (8 nM), CXCL2 (2 nM), and CXCL8 (20 nM) proteins for 6 h. FOXC1-overexpressing MDA-MB-231 cells were treated with PBS or recombinant mouse Ccl2 (10 nM) and Ccl7 (50 nM) proteins for 6 h. Total RNAs were extracted from those cells using the RNeasy Mini Kit (QIAGEN). Quality control of RNAs, library preparation, and the subsequent sequencing were performed by the Genomics Core at Cedars-Sinai Medical Center. The NextSeq 500 platform (Illumina) was used. RNA-seq data were submitted to the NCBI GEO database (GEO: GSE131306 and GSE131383).
IHC
Formalin-fixed, paraffin-embedded (FFPE) primary TNBC tumors and matched lung metastasis samples from four individuals with breast cancer were selected. The institutional review board (IRB) of Cedars-Sinai Medical Center and the First Hospital of China Medical University (Shenyang, China) granted approval. The normal human lung tissue samples were purchased from US Biomax (T041a). Primary mouse breast tumors and lung metastasis samples (see Mouse models and study approval) were also used in the IHC assay. IHC staining was performed using the Vectastain ABC Kit (Vector Laboratories) and ImmPACT DAB Kit (Vector Labs). The primary antibodies were HMGCR (for human samples, 1:100, PA5-52547, Thermo Fisher Scientific), Hmgcr (for mouse samples, 1:70, MBS9406409, Mybiosource), HMGCS1 (for human and mouse samples, 1:100, GTX112346, GeneTex), CCR2 (1:100, NBP2-35334, Novus Biologicals), SREBP2 (1:100, MAB7119, R&D Systems), and Vimentin (1:100, sc-7557, Santa Cruz).
RNAscope in situ hybridization
The RNAscope 2.5 High Definition (HD)-Red in situ hybridization assay was performed according to the manufacturer’s instructions (Advanced Cell Diagnostics). Briefly, FFPE slides were baked, deparaffinized, treated with RNAscope protease, and hybridized with RNA probes. Human CCL2 and CCL7 RNA probes were designed and synthesized by Advanced Cell Diagnostics. Normal human lung tissue samples were purchased from US Biomax (T041a). Human TNBC lung metastasis samples were the same as those used in the IHC experiments.
Lung endothelial cell migration and tube formation assays
The endothelial cell migration assay was performed using the QCM 3 μm Endothelial Cell Migration Assay Fibronectin Kit (ECM200, EMD Millipore) according to the manufacturer’s instructions. Briefly, 2 × 105 freshly isolated mouse lung endothelial cells were suspended in Vasculife medium (LL-0004, Lifeline Cell Technology) and placed into ECM-coated chambers. Chemokines and/or other different cells were added to the bottom chamber. The migrated cells were stained after 16 h of incubation, and the dye was extracted and measured at optical density 560 (OD560) using a luminometer (GloMax-Multi Detection System, Promega). The endothelial cell tube formation assay was performed using the In Vitro Angiogenesis Assay Kit (ECM625, EMD Millipore) according to the manufacturer’s instructions. Briefly, 1.25 × 104 freshly isolated mouse lung endothelial cells were suspended in Vasculife medium (LL-0004, Lifeline Cell Technology) and placed on the top of ECMatrix. Tube formation was measured after 12 h of incubation.
Wound healing assay
Cells were seeded into a 6-well plate and allowed to grow to 90% confluence. The surface of each well was scratched using a 1-mL pipette tip. Cells were washed twice with culture medium to remove the detached cells. Pictures were taken at different time points, and the gap distance was measured using ImageJ software.
Tumor cell migration and invasion assay
1 × 105 cells were resuspended in 500 μL serum-free medium and seeded into the upper compartment of a Transwell chamber (Corning) or Matrigel invasion chamber (BD Biosciences) for the migration or invasion assay, respectively. The lower compartment of the chamber was filled with 750 μL complete medium (DMEM supplemented with 10% FBS). After 6 h (migration) or 16 h (invasion) of incubation, cells remaining in the upper compartment were removed with a cotton swab, and the migrated or invaded cells were stained using the HEMA3 staining kit (Thermo Fisher Scientific). Pictures were taken (EVOS FL Auto Imaging System, Thermo Fisher Scientific), and the cells were counted using ImageJ software.
Cell proliferation assay
Cells were seeded into white opaque 96-well plates at 2,000 cells per well. The cell proliferation rate was assessed using the CellTiter-Glo Luminescent Cell Viability Assay Kit (Promega) according to the manufacturer’s instructions.
Extravasation assay
The experiment was performed in accordance with the approval of the Cedars-Sinai Medical Center Institutional Animal Care and Use Committee. Cells were labeled with 5 μM CellTracker Green CMFDA (C2925, Thermo Fisher Scientific) for 45 min. 1 × 105 cells/100 μL PBS were injected into nude mice (Charles River Laboratories) through the tail vein. Forty-eight hours after injection, mice were injected with 50 μg rhodamine-lectin (RL-1102, Vector Labs)/100 μL PBS through the tail vein and euthanized 30 min later. Mouse lung tissues were collected, frozen in liquid nitrogen, and embedded in OCT compound (Tissue-Tek). Frozen sections were cut and examined using a fluorescence microscope (EVOS FL Auto Imaging System, Thermo Fisher Scientific).
HPK-simvastatin assembly
The HER3-targeting recombinant protein HPK was produced as described previously.64 Binding kinetics were assessed a priori by isothermal calorimetry (ITC) using a Malvern PEAQ-ITC. For particle assembly, simvastatin was serially diluted from DMSO into PBS until fully resuspended. The final concentration of DMSO was less than 5%. Simvastatin and HPK were incubated for 1 h at room temperature at a 1:1 mass ratio. To assess complex formation, the size of the resulting particles was assessed by dynamic light scattering (DLS) using a Malvern Zetasizer Nano ZS.
Mouse models and study approval
All animal experiments were performed in accordance with approval of the Cedars-Sinai Medical Center Institutional Animal Care and Use Committee (IACUC). In the experiment evaluating the effects of chemokines on TNBC lung metastasis (Figure 2B), 1 × 106 control, FOXC1-overexpressing, or FOXC1-overexpressing chemokine-KO MDA-MB-231 cells were suspended in serum-free medium mixed with Matrigel (BD Biosciences) at a 1:1 ratio and injected into the fourth mammary fat pads of nude mice (The Jackson Laboratory). Mice were euthanized 5 weeks after injection, and lungs were collected. For the lung metastasis formation capacity assay (Figure 2D), 3 × 106 EO771 cells in 100 μL PBS were injected into wild-type or cxcr2+/− C57BL/6 mice (stock number 006848, The Jackson Laboratory) through the tail vein. Mice were euthanized 6 weeks after injection, and lungs were collected. To assess the outgrowth capacity of tumor cells in the lungs (Figure S2H), 0.5 × 106 tumor cells of different groups in 100 μL PBS were injected into nude mice through the tail vein. Mice were euthanized 5 days after injection, and lungs were collected. To examine the expression levels of Hmgcr and Hmgcs1 proteins (Figure S4C), 0.5 × 106 4T1 cells were suspended in serum-free medium mixed with Matrigel (Sigma-Aldrich) at a 1:1 ratio and injected into the fourth mammary fat pads of BALB/c wild-type mice. Mice were euthanized 5 weeks after injection, and lungs were collected. In the experiment evaluating the effects of simvastatin on lung metastasis formation (Figure 6C), 0.5 × 106 4T1 cells in 100 μL PBS were injected into BALB/c wild-type mice through the tail vein. The mice were treated with vehicle or HPK-simvastatin (10 μg/mouse/daily; the safety of the concentration was tested in our in vitro cell model) intranasally beginning 48 h after tumor cell injection. Mice were euthanized 2 weeks after injection, and lungs were collected. To evaluate the effect of Hmgcr KO on lung metastasis formation (Figure S5D), 0.5 × 106 control or Hmgcr-KO 4T1 cells in 100 μL PBS were injected into BALB/c wild-type mice through the tail vein. Mice were euthanized 2 weeks after injection, and lungs were collected.
Statistics
Statistical analyses were performed using GraphPad Prism 6 software. Statistical comparisons were determined via independent Student’s t test. Linear regression analyses and Pearson correlation coefficients were conducted to calculate correlations. Values are represented as mean ± SD of at least three independent experiments. p < 0.05 was considered statistically significant. Kaplan-Meier survival curves were generated using GraphPad Prism 6 software.
Acknowledgments
We thank Dianhua Jiang (Cedars-Sinai Medical Center) for providing the Vimentin antibody. We thank Jing Zhang for assistance with performing the RNAscope in situ hybridization experiments. This work was supported by the National Institutes of Health (CA151610, CA129822, and CA140995), the DOD (W81XWH-15-1-0604), the Avon Foundation for Women (02-2014-063, 02-2015-060), Samuel Oschin Cancer Institute Research Development Fund (to X.C.) and the Fashion Footwear Charitable Foundation of New York, Inc., the Entertainment Industry Foundation, the Margie and Robert E. Petersen Foundation, and the Linda and Jim Lippman Research Fund (to A.G.).
Author contributions
B.H. and X.C. conceived and designed the experiments. B.H. performed experiments unless specified otherwise. T.-Y.L., B.G., and L.J. assisted with the experiments. N.D. and F.A.-V. performed biostatistics and bioinformatics analyses. Z.W., Y.Z., Y.X., and X.Z. collected primary breast tumor and matched lung metastasis samples from affected individuals and performed IHC. S.B. and N.B. isolated mouse bone marrow fibroblasts. S.S. isolated neutrophils from mouse lungs. F.A.-V. and L.M.-K. performed HPK-simvastatin assembly. B.H. and X.C. wrote, revised, and edited the manuscript. A.G. participated in experimental design and manuscript preparation.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.07.003.
Contributor Information
Bingchen Han, Email: bingchen.han@cshs.org.
Xiaojiang Cui, Email: xiaojiang.cui@cshs.org.
Supplemental information
References
- 1.Foulkes W.D., Smith I.E., Reis-Filho J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010;363:1938–1948. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 2.Koboldt D.C., Fulton R.S., McLellan M.D., Schmidt H., Kalicki-Veizer J., McMichael J.F., et al. Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perou C.M., Sørlie T., Eisen M.B., van de Rijn M., Jeffrey S.S., Rees C.A., Pollack J.R., Ross D.T., Johnsen H., Akslen L.A., et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 4.Rakha E.A., Ellis I.O. Triple-negative/basal-like breast cancer: review. Pathology. 2009;41:40–47. doi: 10.1080/00313020802563510. [DOI] [PubMed] [Google Scholar]
- 5.Harrell J.C., Prat A., Parker J.S., Fan C., He X., Carey L., Anders C., Ewend M., Perou C.M. Genomic analysis identifies unique signatures predictive of brain, lung, and liver relapse. Breast Cancer Res. Treat. 2012;132:523–535. doi: 10.1007/s10549-011-1619-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smid M., Wang Y., Zhang Y., Sieuwerts A.M., Yu J., Klijn J.G., Foekens J.A., Martens J.W. Subtypes of breast cancer show preferential site of relapse. Cancer Res. 2008;68:3108–3114. doi: 10.1158/0008-5472.CAN-07-5644. [DOI] [PubMed] [Google Scholar]
- 7.Nguyen D.X., Bos P.D., Massagué J. Metastasis: from dissemination to organ-specific colonization. Nat. Rev. Cancer. 2009;9:274–284. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- 8.Obenauf A.C., Massagué J. Surviving at a Distance: Organ-Specific Metastasis. Trends Cancer. 2015;1:76–91. doi: 10.1016/j.trecan.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peinado H., Zhang H., Matei I.R., Costa-Silva B., Hoshino A., Rodrigues G., Psaila B., Kaplan R.N., Bromberg J.F., Kang Y., et al. Pre-metastatic niches: organ-specific homes for metastases. Nat. Rev. Cancer. 2017;17:302–317. doi: 10.1038/nrc.2017.6. [DOI] [PubMed] [Google Scholar]
- 10.Zhang X.H., Wang Q., Gerald W., Hudis C.A., Norton L., Smid M., Foekens J.A., Massagué J. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell. 2009;16:67–78. doi: 10.1016/j.ccr.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lynch C.C., Hikosaka A., Acuff H.B., Martin M.D., Kawai N., Singh R.K., Vargo-Gogola T.C., Begtrup J.L., Peterson T.E., Fingleton B., et al. MMP-7 promotes prostate cancer-induced osteolysis via the solubilization of RANKL. Cancer Cell. 2005;7:485–496. doi: 10.1016/j.ccr.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 12.Hiratsuka S., Watanabe A., Sakurai Y., Akashi-Takamura S., Ishibashi S., Miyake K., Shibuya M., Akira S., Aburatani H., Maru Y. The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat. Cell Biol. 2008;10:1349–1355. doi: 10.1038/ncb1794. [DOI] [PubMed] [Google Scholar]
- 13.Qian B.Z., Li J., Zhang H., Kitamura T., Zhang J., Campion L.R., Kaiser E.A., Snyder L.A., Pollard J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475:222–225. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu X., Kang Y. Chemokine (C-C motif) ligand 2 engages CCR2+ stromal cells of monocytic origin to promote breast cancer metastasis to lung and bone. J. Biol. Chem. 2009;284:29087–29096. doi: 10.1074/jbc.M109.035899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang L., Zhang S., Yao J., Lowery F.J., Zhang Q., Huang W.C., Li P., Li M., Wang X., Zhang C., et al. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature. 2015;527:100–104. doi: 10.1038/nature15376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fong M.Y., Zhou W., Liu L., Alontaga A.Y., Chandra M., Ashby J., Chow A., O’Connor S.T., Li S., Chin A.R., et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat. Cell Biol. 2015;17:183–194. doi: 10.1038/ncb3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Setoguchi S., Glynn R.J., Avorn J., Mogun H., Schneeweiss S. Statins and the risk of lung, breast, and colorectal cancer in the elderly. Circulation. 2007;115:27–33. doi: 10.1161/CIRCULATIONAHA.106.650176. [DOI] [PubMed] [Google Scholar]
- 18.Singh H., Mahmud S.M., Turner D., Xue L., Demers A.A., Bernstein C.N. Long-term use of statins and risk of colorectal cancer: a population-based study. Am. J. Gastroenterol. 2009;104:3015–3023. doi: 10.1038/ajg.2009.574. [DOI] [PubMed] [Google Scholar]
- 19.Wang J., Li C., Tao H., Cheng Y., Han L., Li X., Hu Y. Statin use and risk of lung cancer: a meta-analysis of observational studies and randomized controlled trials. PLoS ONE. 2013;8:e77950. doi: 10.1371/journal.pone.0077950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tan M., Song X., Zhang G., Peng A., Li X., Li M., Liu Y., Wang C. Statins and the risk of lung cancer: a meta-analysis. PLoS ONE. 2013;8:e57349. doi: 10.1371/journal.pone.0057349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Babcook M.A., Joshi A., Montellano J.A., Shankar E., Gupta S. Statin Use in Prostate Cancer: An Update. Nutr. Metab. Insights. 2016;9:43–50. doi: 10.4137/NMI.S38362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murtola T.J., Tammela T.L., Lahtela J., Auvinen A. Cholesterol-lowering drugs and prostate cancer risk: a population-based case-control study. Cancer Epidemiol. Biomarkers Prev. 2007;16:2226–2232. doi: 10.1158/1055-9965.EPI-07-0599. [DOI] [PubMed] [Google Scholar]
- 23.Ahern T.P., Lash T.L., Damkier P., Christiansen P.M., Cronin-Fenton D.P. Statins and breast cancer prognosis: evidence and opportunities. Lancet Oncol. 2014;15:e461–e468. doi: 10.1016/S1470-2045(14)70119-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Undela K., Srikanth V., Bansal D. Statin use and risk of breast cancer: a meta-analysis of observational studies. Breast Cancer Res. Treat. 2012;135:261–269. doi: 10.1007/s10549-012-2154-x. [DOI] [PubMed] [Google Scholar]
- 25.Bonovas S., Filioussi K., Tsavaris N., Sitaras N.M. Statins and cancer risk: a literature-based meta-analysis and meta-regression analysis of 35 randomized controlled trials. J. Clin. Oncol. 2006;24:4808–4817. doi: 10.1200/JCO.2006.06.3560. [DOI] [PubMed] [Google Scholar]
- 26.Sakellakis M., Akinosoglou K., Kostaki A., Spyropoulou D., Koutras A. Statins and risk of breast cancer recurrence. Breast Cancer (Dove Med. Press) 2016;8:199–205. doi: 10.2147/BCTT.S116694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Manthravadi S., Shrestha A., Madhusudhana S. Impact of statin use on cancer recurrence and mortality in breast cancer: A systematic review and meta-analysis. Int. J. Cancer. 2016;139:1281–1288. doi: 10.1002/ijc.30185. [DOI] [PubMed] [Google Scholar]
- 28.Ahern T.P., Pedersen L., Tarp M., Cronin-Fenton D.P., Garne J.P., Silliman R.A., Sørensen H.T., Lash T.L. Statin prescriptions and breast cancer recurrence risk: a Danish nationwide prospective cohort study. J. Natl. Cancer Inst. 2011;103:1461–1468. doi: 10.1093/jnci/djr291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu B., Yi Z., Guan X., Zeng Y.X., Ma F. The relationship between statins and breast cancer prognosis varies by statin type and exposure time: a meta-analysis. Breast Cancer Res. Treat. 2017;164:1–11. doi: 10.1007/s10549-017-4246-0. [DOI] [PubMed] [Google Scholar]
- 30.Murtola T.J., Visvanathan K., Artama M., Vainio H., Pukkala E. Statin use and breast cancer survival: a nationwide cohort study from Finland. PLoS ONE. 2014;9:e110231. doi: 10.1371/journal.pone.0110231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cardwell C.R., Hicks B.M., Hughes C., Murray L.J. Statin use after diagnosis of breast cancer and survival: a population-based cohort study. Epidemiology. 2015;26:68–78. doi: 10.1097/EDE.0000000000000189. [DOI] [PubMed] [Google Scholar]
- 32.Mc Menamin U.C., Murray L.J., Hughes C.M., Cardwell C.R. Statin use and breast cancer survival: a nationwide cohort study in Scotland. BMC Cancer. 2016;16:600. doi: 10.1186/s12885-016-2651-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Llaverias G., Danilo C., Mercier I., Daumer K., Capozza F., Williams T.M., Sotgia F., Lisanti M.P., Frank P.G. Role of cholesterol in the development and progression of breast cancer. Am. J. Pathol. 2011;178:402–412. doi: 10.1016/j.ajpath.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jensen T.W., Ray T., Wang J., Li X., Naritoku W.Y., Han B., Bellafiore F., Bagaria S.P., Qu A., Cui X., et al. Diagnosis of Basal-Like Breast Cancer Using a FOXC1-Based Assay. J. Natl. Cancer Inst. 2015;107:djv148. doi: 10.1093/jnci/djv148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Han B., Bhowmick N., Qu Y., Chung S., Giuliano A.E., Cui X. FOXC1: an emerging marker and therapeutic target for cancer. Oncogene. 2017;36:3957–3963. doi: 10.1038/onc.2017.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taube J.H., Herschkowitz J.I., Komurov K., Zhou A.Y., Gupta S., Yang J., Hartwell K., Onder T.T., Gupta P.B., Evans K.W., et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc. Natl. Acad. Sci. USA. 2010;107:15449–15454. doi: 10.1073/pnas.1004900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han B., Qu Y., Jin Y., Yu Y., Deng N., Wawrowsky K., Zhang X., Li N., Bose S., Wang Q., et al. FOXC1 Activates Smoothened-Independent Hedgehog Signaling in Basal-like Breast Cancer. Cell Rep. 2015;13:1046–1058. doi: 10.1016/j.celrep.2015.09.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoeflich K.P., O’Brien C., Boyd Z., Cavet G., Guerrero S., Jung K., Januario T., Savage H., Punnoose E., Truong T., et al. In vivo antitumor activity of MEK and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models. Clin. Cancer Res. 2009;15:4649–4664. doi: 10.1158/1078-0432.CCR-09-0317. [DOI] [PubMed] [Google Scholar]
- 39.Sabatier R., Finetti P., Cervera N., Lambaudie E., Esterni B., Mamessier E., Tallet A., Chabannon C., Extra J.M., Jacquemier J., et al. A gene expression signature identifies two prognostic subgroups of basal breast cancer. Breast Cancer Res. Treat. 2011;126:407–420. doi: 10.1007/s10549-010-0897-9. [DOI] [PubMed] [Google Scholar]
- 40.Hu Z., Fan C., Oh D.S., Marron J.S., He X., Qaqish B.F., Livasy C., Carey L.A., Reynolds E., Dressler L., et al. The molecular portraits of breast tumors are conserved across microarray platforms. BMC Genomics. 2006;7:96. doi: 10.1186/1471-2164-7-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Benito M., Parker J., Du Q., Wu J., Xiang D., Perou C.M., Marron J.S. Adjustment of systematic microarray data biases. Bioinformatics. 2004;20:105–114. doi: 10.1093/bioinformatics/btg385. [DOI] [PubMed] [Google Scholar]
- 42.Freund A., Jolivel V., Durand S., Kersual N., Chalbos D., Chavey C., Vignon F., Lazennec G. Mechanisms underlying differential expression of interleukin-8 in breast cancer cells. Oncogene. 2004;23:6105–6114. doi: 10.1038/sj.onc.1207815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bièche I., Chavey C., Andrieu C., Busson M., Vacher S., Le Corre L., Guinebretière J.M., Burlinchon S., Lidereau R., Lazennec G. CXC chemokines located in the 4q21 region are up-regulated in breast cancer. Endocr. Relat. Cancer. 2007;14:1039–1052. doi: 10.1677/erc.1.01301. [DOI] [PubMed] [Google Scholar]
- 44.Bachmeier B.E., Mohrenz I.V., Mirisola V., Schleicher E., Romeo F., Höhneke C., Jochum M., Nerlich A.G., Pfeffer U. Curcumin downregulates the inflammatory cytokines CXCL1 and -2 in breast cancer cells via NFkappaB. Carcinogenesis. 2008;29:779–789. doi: 10.1093/carcin/bgm248. [DOI] [PubMed] [Google Scholar]
- 45.Punj V., Matta H., Schamus S., Yang T., Chang Y., Chaudhary P.M. Induction of CCL20 production by Kaposi sarcoma-associated herpesvirus: role of viral FLICE inhibitory protein K13-induced NF-kappaB activation. Blood. 2009;113:5660–5668. doi: 10.1182/blood-2008-10-186403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao L., Xia J., Wang X., Xu F. Transcriptional regulation of CCL20 expression. Microbes Infect. 2014;16:864–870. doi: 10.1016/j.micinf.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 47.Wang J., Ray P.S., Sim M.S., Zhou X.Z., Lu K.P., Lee A.V., Lin X., Bagaria S.P., Giuliano A.E., Cui X. FOXC1 regulates the functions of human basal-like breast cancer cells by activating NF-κB signaling. Oncogene. 2012;31:4798–4802. doi: 10.1038/onc.2011.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pierrou S., Hellqvist M., Samuelsson L., Enerbäck S., Carlsson P. Cloning and characterization of seven human forkhead proteins: binding site specificity and DNA bending. EMBO J. 1994;13:5002–5012. doi: 10.1002/j.1460-2075.1994.tb06827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burke S.J., Lu D., Sparer T.E., Masi T., Goff M.R., Karlstad M.D., Collier J.J. NF-κB and STAT1 control CXCL1 and CXCL2 gene transcription. Am. J. Physiol. Endocrinol. Metab. 2014;306:E131–E149. doi: 10.1152/ajpendo.00347.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roussos E.T., Condeelis J.S., Patsialou A. Chemotaxis in cancer. Nat. Rev. Cancer. 2011;11:573–587. doi: 10.1038/nrc3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bos P.D., Zhang X.H., Nadal C., Shu W., Gomis R.R., Nguyen D.X., Minn A.J., van de Vijver M.J., Gerald W.L., Foekens J.A., Massagué J. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459:1005–1009. doi: 10.1038/nature08021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang Y., Klijn J.G., Zhang Y., Sieuwerts A.M., Look M.P., Yang F., Talantov D., Timmermans M., Meijer-van Gelder M.E., Yu J., et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365:671–679. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- 53.Minn A.J., Gupta G.P., Siegel P.M., Bos P.D., Shu W., Giri D.D., Viale A., Olshen A.B., Gerald W.L., Massagué J. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lazennec G., Richmond A. Chemokines and chemokine receptors: new insights into cancer-related inflammation. Trends Mol. Med. 2010;16:133–144. doi: 10.1016/j.molmed.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Keane M.P., Belperio J.A., Xue Y.Y., Burdick M.D., Strieter R.M. Depletion of CXCR2 inhibits tumor growth and angiogenesis in a murine model of lung cancer. J. Immunol. 2004;172:2853–2860. doi: 10.4049/jimmunol.172.5.2853. [DOI] [PubMed] [Google Scholar]
- 56.Mehrad B., Keane M.P., Strieter R.M. Chemokines as mediators of angiogenesis. Thromb. Haemost. 2007;97:755–762. [PMC free article] [PubMed] [Google Scholar]
- 57.Gong L., Cumpian A.M., Caetano M.S., Ochoa C.E., De la Garza M.M., Lapid D.J., Mirabolfathinejad S.G., Dickey B.F., Zhou Q., Moghaddam S.J. Promoting effect of neutrophils on lung tumorigenesis is mediated by CXCR2 and neutrophil elastase. Mol. Cancer. 2013;12:154. doi: 10.1186/1476-4598-12-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Waugh D.J., Wilson C. The interleukin-8 pathway in cancer. Clin. Cancer Res. 2008;14:6735–6741. doi: 10.1158/1078-0432.CCR-07-4843. [DOI] [PubMed] [Google Scholar]
- 59.Bastian S., Paquet J.L., Robert C., Cremers B., Loillier B., Larrivée J.F., Bachvarov D.R., Marceau F., Pruneau D. Interleukin 8 (IL-8) induces the expression of kinin B1 receptor in human lung fibroblasts. Biochem. Biophys. Res. Commun. 1998;253:750–755. doi: 10.1006/bbrc.1998.9848. [DOI] [PubMed] [Google Scholar]
- 60.Newton R., Shah S., Altonsy M.O., Gerber A.N. Glucocorticoid and cytokine crosstalk: Feedback, feedforward, and co-regulatory interactions determine repression or resistance. J. Biol. Chem. 2017;292:7163–7172. doi: 10.1074/jbc.R117.777318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Espenshade P.J. SREBPs: sterol-regulated transcription factors. J. Cell Sci. 2006;119:973–976. doi: 10.1242/jcs.02866. [DOI] [PubMed] [Google Scholar]
- 62.Liao J.K. Isoprenoids as mediators of the biological effects of statins. J. Clin. Invest. 2002;110:285–288. doi: 10.1172/JCI16421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang M., Casey P.J. Protein prenylation: unique fats make their mark on biology. Nat. Rev. Mol. Cell Biol. 2016;17:110–122. doi: 10.1038/nrm.2015.11. [DOI] [PubMed] [Google Scholar]
- 64.Sims J.D., Taguiam J.M., Alonso-Valenteen F., Markman J., Agadjanian H., Chu D., Lubow J., Abrol R., Srinivas D., Jain A., et al. Resistance to receptor-blocking therapies primes tumors as targets for HER3-homing nanobiologics. J. Control. Release. 2018;271:127–138. doi: 10.1016/j.jconrel.2017.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nelson E.R. The significance of cholesterol and its metabolite, 27-hydroxycholesterol in breast cancer. Mol. Cell. Endocrinol. 2018;466:73–80. doi: 10.1016/j.mce.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kuzu O.F., Noory M.A., Robertson G.P. The Role of Cholesterol in Cancer. Cancer Res. 2016;76:2063–2070. doi: 10.1158/0008-5472.CAN-15-2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nelson E.R., Chang C.Y., McDonnell D.P. Cholesterol and breast cancer pathophysiology. Trends Endocrinol. Metab. 2014;25:649–655. doi: 10.1016/j.tem.2014.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen M., Zhao Y., Yang X., Zhao Y., Liu Q., Liu Y., Hou Y., Sun H., Jin W. NSDHL promotes triple-negative breast cancer metastasis through the TGFβ signaling pathway and cholesterol biosynthesis. Breast Cancer Res. Treat. 2021;187:349–362. doi: 10.1007/s10549-021-06213-8. [DOI] [PubMed] [Google Scholar]
- 69.Ehmsen S., Pedersen M.H., Wang G., Terp M.G., Arslanagic A., Hood B.L., Conrads T.P., Leth-Larsen R., Ditzel H.J. Increased Cholesterol Biosynthesis Is a Key Characteristic of Breast Cancer Stem Cells Influencing Patient Outcome. Cell Rep. 2019;27:3927–3938.e6. doi: 10.1016/j.celrep.2019.05.104. [DOI] [PubMed] [Google Scholar]
- 70.Cai D., Wang J., Gao B., Li J., Wu F., Zou J.X., Xu J., Jiang Y., Zou H., Huang Z., et al. RORγ is a targetable master regulator of cholesterol biosynthesis in a cancer subtype. Nat. Commun. 2019;10:4621. doi: 10.1038/s41467-019-12529-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Beckwitt C.H., Clark A.M., Ma B., Whaley D., Oltvai Z.N., Wells A. Statins attenuate outgrowth of breast cancer metastases. Br. J. Cancer. 2018;119:1094–1105. doi: 10.1038/s41416-018-0267-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Baek A.E., Yu Y.A., He S., Wardell S.E., Chang C.Y., Kwon S., Pillai R.V., McDowell H.B., Thompson J.W., Dubois L.G., et al. The cholesterol metabolite 27 hydroxycholesterol facilitates breast cancer metastasis through its actions on immune cells. Nat. Commun. 2017;8:864. doi: 10.1038/s41467-017-00910-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Divella R., Daniele A., Savino E., Palma F., Bellizzi A., Giotta F., Simone G., Lioce M., Quaranta M., Paradiso A., Mazzocca A. Circulating levels of transforming growth factor-βeta (TGF-β) and chemokine (C-X-C motif) ligand-1 (CXCL1) as predictors of distant seeding of circulating tumor cells in patients with metastatic breast cancer. Anticancer Res. 2013;33:1491–1497. [PubMed] [Google Scholar]
- 74.Acharyya S., Oskarsson T., Vanharanta S., Malladi S., Kim J., Morris P.G., Manova-Todorova K., Leversha M., Hogg N., Seshan V.E., et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell. 2012;150:165–178. doi: 10.1016/j.cell.2012.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kalluri R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer. 2016;16:582–598. doi: 10.1038/nrc.2016.73. [DOI] [PubMed] [Google Scholar]
- 76.Huang W., Chen Z., Zhang L., Tian D., Wang D., Fan D., Wu K., Xia L. Interleukin-8 Induces Expression of FOXC1 to Promote Transactivation of CXCR1 and CCL2 in Hepatocellular Carcinoma Cell Lines and Formation of Metastases in Mice. Gastroenterology. 2015;149 doi: 10.1053/j.gastro.2015.05.058. 1053–67.e14. [DOI] [PubMed] [Google Scholar]
- 77.Nelson E.R., Wardell S.E., Jasper J.S., Park S., Suchindran S., Howe M.K., Carver N.J., Pillai R.V., Sullivan P.M., Sondhi V., et al. 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science. 2013;342:1094–1098. doi: 10.1126/science.1241908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bluff J.E., Brown N.J., Reed M.W., Staton C.A. Tissue factor, angiogenesis and tumour progression. Breast Cancer Res. 2008;10:204. doi: 10.1186/bcr1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mandal S.K., Iakhiaev A., Pendurthi U.R., Rao L.V. Acute cholesterol depletion impairs functional expression of tissue factor in fibroblasts: modulation of tissue factor activity by membrane cholesterol. Blood. 2005;105:153–160. doi: 10.1182/blood-2004-03-0990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lesnik P., Rouis M., Skarlatos S., Kruth H.S., Chapman M.J. Uptake of exogenous free cholesterol induces upregulation of tissue factor expression in human monocyte-derived macrophages. Proc. Natl. Acad. Sci. USA. 1992;89:10370–10374. doi: 10.1073/pnas.89.21.10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sanjana N.E., Shalem O., Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jin L., Qu Y., Gomez L.J., Chung S., Han B., Gao B., Yue Y., Gong Y., Liu X., Amersi F., et al. Characterization of primary human mammary epithelial cells isolated and propagated by conditional reprogrammed cell culture. Oncotarget. 2017;9:11503–11514. doi: 10.18632/oncotarget.23817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Seluanov A., Vaidya A., Gorbunova V. Establishing primary adult fibroblast cultures from rodents. J. Vis. Exp. 2010;44:2033. doi: 10.3791/2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chavez-Santoscoy A.V., Huntimer L.M., Ramer-Tait A.E., Wannemuehler M., Narasimhan B. Harvesting murine alveolar macrophages and evaluating cellular activation induced by polyanhydride nanoparticles. J. Vis. Exp. 2012:e3883. doi: 10.3791/3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sobczak M., Dargatz J., Chrzanowska-Wodnicka M. Isolation and culture of pulmonary endothelial cells from neonatal mice. J. Vis. Exp. 2010;46:2316. doi: 10.3791/2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.