

Abstract
Recent studies have implicated mitochondrial dysfunction as a trigger of inflammatory bowel diseases, including Crohn’s disease (CD) and ulcerative colitis (UC). We have investigated the role of the mitochondria gate-keeper protein, the voltage-dependent-anion channel 1 (VDAC1), in gastrointestinal inflammation and tested the effects of the newly developed VDAC1-interacting molecules, VBIT-4 and VBIT-12, on UC induced by dextran sulfate sodium (DSS) or trinitrobenzene sulphonic acid (TNBS) in mice. VDAC1, which controls metabolism, lipids transport, apoptosis, and inflammasome activation, is overexpressed in the colon of CD and UC patients and DSS-treated mice. VBIT-12 treatment of cultured colon cells inhibited the DSS-induced VDAC1 overexpression, oligomerization, and apoptosis. In the DSS-treated mice, VBIT-12 suppressed weight loss, diarrhea, rectal bleeding, pro-inflammatory cytokine production, crypt and epithelial cell damage, and focal inflammation. VBIT-12 also inhibited the infiltration of inflammatory cells, apoptosis, mtDNA release, and activation of caspase-1 and NRLP3 inflammasome to reduce the inflammatory response. The levels of the ATP-gated P2X7-Ca2+/K+ channel and ER-IP3R-Ca2+ channel, and of the mitochondrial anti-viral protein (MAVS), mediating NLRP3 inflammasome assembly and activation, were highly increased in DSS-treated mice, but not when VBIT-12 treated. We conclude that UC may be promoted by VDAC1-overexpression and may therefore be amenable to treatment with novel VDAC1-interacting molecules. This VDAC1-based strategy exploits a completely new target for UC treatment and opens a new avenue for treating other inflammatory/autoimmune diseases.
Keywords: apoptosis, IBD, mitochondria, VDAC1, NLRP3 inflammasome, mtDNA, MAVS
Graphical abstract
VDAC1 is overexpressed in the colon of inflammatory bowel disease (IBD) patients and DSS-induced colitis mice. DSS-induced colitis pathophysiology includes weight loss, diarrhea, rectal bleeding, pro-inflammatory cytokines production, epithelial cell damage, apoptosis, mtDNA release, and NRLP3 inflammasome activation. All were prevented in mice treated with the VDAC1-interacting molecule, VBIT-12. These findings point to VDAC1’s association with IBD and as a potential therapeutic target.
Introduction
Inflammatory bowel disease (IBD) is a group of chronic inflammatory conditions of the gastrointestinal tract, leading to multiple symptoms such as diarrhea, bloody stools, abdominal pain, and weight loss.1 Two main forms of IBD are Crohn’s disease (CD) and ulcerative colitis (UC), which each have characteristic but diverse clinical, histopathological, endoscopic, and radiological features.1 Specifically, CD typically presents with transmural inflammation that may affect any part of the gastrointestinal tract, and disease presentation is dependent upon both the location and severity of inflammation. On the other hand, UC primarily affects the mucosa in the colon and rectum.
Although there is no cure for IBD, a variety of therapeutics (e.g., corticosteroids, immune modulators, antibiotics, amino salicylates, and biologic therapies) are employed to help manage the symptoms of disease. Nonetheless, the effects of these treatments are variable and increase the potential risk of morbidity in patients over time.1 The precise etiology of IBD is unclear, but it is thought to be triggered by a complex interaction between genetic, environmental, and immune regulatory factors, including mitochondrial DNA (mtDNA).2,3
Gut epithelial cells have high energy requirements, and mitochondrial dysfunction can disrupt the maintenance of mucosal and intestinal stem cells (ISCs)4 and lead to epithelial cell apoptosis and to intestinal diseases including IBD.5,6 Intriguingly, the intermediates of the mitochondrial Krebs cycle, succinate and citrate, act as pro-inflammatory modulators of dendritic cells and macrophages.7 Mitochondria in the epithelium of IBD patients have lower ATP levels and higher levels of mitochondrial reactive oxygen species (mtROS), as well as evidence of misfolded proteins, ultrastructural changes, inflammasome activation, and pro-inflammatory cytokine production.6
Mitochondria serve as an essential signaling hub in the regulation of both innate and adaptive immunity.8, 9, 10 mtROS are necessary for the release of neutrophil extracellular traps in a process called NETosis, by which the cell death of low-density granulocytes releases oxidized nuclear DNA, as well as mtDNA to trap bacteria.2 Release of mtDNA into the cytosol triggers inflammasome activation and pro-inflammatory responses through the cGAS-STING cytosolic DNA-sensing pathway.3
During active IBD, mtDNA is released into the serum and acts as a key pro-inflammatory factor and damage-associated molecular pattern (DAMP) for immune cell activation, with a pathogenic role in several inflammatory diseases.11 The mechanism for this release is poorly understood, but we recently demonstrated that a large pore formed by oligomerization of the voltage-dependent anion channel 1 (VDAC1) mediates mtDNA movement across the outer mitochondrial membrane (OMM).12
VDAC1 is the most abundant of the three mammalian isoforms found. The strategic location in the OMM allows the protein to mediate the transfer of nucleotides and metabolites such as pyruvate, malate, succinate, NADH/NAD, ions such as Ca2+, and ROS between the mitochondrion and the cytoplasm. VDAC1 is also involved in the transport of fatty acids, cholesterol, and hemes, and regulates the cell redox state.13, 14, 15
VDAC1 has now been estimated to interact with over 100 proteins, some of which are involved in key cellular functions including metabolism, apoptosis, signal transduction, anti-oxidation, or regulation of DNA and RNA.13 As a result, VDAC1 has been implicated in cell death13, 14, 15, 16 and intracellular communication between the ER and mitochondria, involving transfer of Ca2+ and phospholipids, as well as mitochondrial fission, autophagy, and inflammation13,17 and inflammasome activation.18
One major role of VDAC1 involves regulation of the release of mitochondrial pro-apoptotic proteins such as cytochrome c (cyto c), from the intermembrane space (IMS) to the cytosol, and apoptosis regulation via interactions with apoptosis regulatory proteins such as Bcl-2, Bcl-xL, and hexokinase (HK).13, 14, 15 We recently demonstrated that VDAC1 exists in a dynamic equilibrium, where inducers of apoptosis by upregulating VDAC1 expression shift the protein state from monomers toward oligomers. The oligomer forms a large channel in the mitochondrial membrane that enables the release of pro-apoptotic proteins, leading to cell death16,19 and the transport mtDNA.12
In order to counteract VDAC1 oligomerization and cell death,16,19 we developed new molecules, VBIT-4 and VBIT-12, that interact with VDAC1 to prevent oligomerization and subsequent apoptosis.20 We validated their efficacy in diseases where VDAC1 is overexpressed as type 2 diabetes (T2D)21 and in a mouse model of systemic lupus erythematosus (SLE).12 In the SLE model, VBIT-4 prevented VDAC1 oligomerization and reduced mtDNA release, type-Ι interferon signaling, the neutrophil extracellular traps formation, and disease severity.12
Here, we demonstrate that VDAC1 is overexpressed in the colon of IBD patients and in mice with dextran sulfate sodium (DSS)-induced colitis. DSS-induced colitis in mice22 resembles UC patients23 in several pathophysiological and morphological features, including loss of weight, diarrhea, rectal bleeding, production of pro-inflammatory cytokines, crypt and epithelial cell damage, focal inflammation, cellular infiltration, and ulceration. Mice treated with DSS also exhibit swollen mitochondria, increased generation of mtROS, and decreased ATP levels in the colonic mucosa that can be ameliorated by anti-tumor necrosis factor-α (TNF-α) treatment.24
Remarkably, mice treated with VBIT-4 and VBIT-12 did not develop DSS-induced colitis pathology in the colon tissue, and treatment inhibited the infiltration of inflammatory cells, apoptosis, mtDNA release, and NRLP3 inflammasome activation and reduced the inflammatory response.
Results
VDAC1 is overexpressed in colon tissue from patients with chronic colitis, CD of the ileocecal junction, and UC (Figure 1A). This overexpression was 6- to 8-fold higher relative to the levels in healthy donors (Figure 1B).
Figure 1.

VDAC1 is overexpressed in human colon pathologies and in DSS-treated cells, which result in VDAC1 oligomerization, apoptosis, and mtDNA release that are inhibited by VBIT-12
Immunohistochemical (IHC) staining of VDAC1 was performed on tissue microarray slides obtained from Biomax US (Cat No. CO245). The array contains colon sections from healthy (H, 4 samples), colitis (Co, 4 samples), ulcerative colitis (UC, 4 samples), and Crohn’s disease of the ileocecal junction (CD, 4 samples). (A) Representative sections of the indicated tissues were IHC stained for VDAC1 using specific antibodies. Scale bars are 20 or 50 μm, as indicated. (B) Quantitation of VDAC1 expression levels in the whole area of the provided sections. Staining intensity was quantified using a panoramic microscope and HistoQuant software. Results, mean ± SEM, ∗∗p < 0.01, ∗∗∗p < 0.001, NS not significant (n = 4 for each group). (C–E) Representative images of colon tissue sections from control and DSS-induced colitis mice, IHC, stained for VDAC1 (C; n = 6 for control and n = 7 for DSS). Scale bar is 50 μm. (D–I) CT-26 cells were incubated with the indicated DSS concentration for 48 h in the presence or absence of VBIT-12 (10 or 20 μM) (n = 3). Then samples were analyzed for VDAC1 expression level by immunoblotting with acting serving as loading control (D), oligomerization (E), apoptosis (assayed by PI staining and flow cytometry) (F), intracellular Ca2+ levels (G), mtROS (H), and mtDNA release (I); all were assayed as described in the Materials and methods section. Results, mean ± SEM, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, n = 3, NS, not significant. (J) Proposed coupling of VDAC1 overexpression induced by pathological conditions (IBD) and VDAC1 oligomerization mediating the release of apoptogenic proteins from the IMS, leading to apoptosis and the release of mtDNA, leading to inflammasome activation. These processes are inhibited by the VDAC1-interacting molecules, VBIT-4 and VBIT-12, via preventing VDAC1 oligomerization.
Similarly, VDAC1 levels, as measured by immunohistochemistry (IHC), were much higher in colon sections from DSS-treated mice than in untreated controls (Figure 1C). The increase in VDAC1 levels in IBD led us to test the effects VBIT-4 and VBIT-12 that interact with the overexpressed VDAC1 to prevent its oligomerization and subsequent apoptosis20 in DSS-induced colitis.
The direct interaction of VBIT-4 and VBIT-12 with purified VDAC1 was demonstrated by their effects on the channel conductance of bilayer-reconstituted VDAC1, with VBIT-12 reducing the channel conductance by more than VBIT-4 (Figure S1).
Preventing DSS-induced mitochondria dysfunction, VDAC1 oligomerization, apoptosis, and mtDNA release
First, we tested the effects of DSS and VBIT-12 in an in-vitro model using two cell lines in which DSS was shown to induce cytotoxicity.25 DSS reduced the viability of a murine colorectal carcinoma-derived cell line, CT-26, and of a human colon-cancer-derived intestinal epithelial cell (IEC) line Caco-2, as analyzed by the XTT (2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide) assay, but this was ameliorated by VBIT-12 (Figures S2A and S2B). DSS treatment also induced VDAC1 overexpression, VDAC1 oligomerization, and apoptotic cell death and increased mtROS production and intracellular Ca2+ levels, which were all inhibited by VBIT-12 (Figures 1D–1H; Figure S2C), as reported previously for apoptosis induction.20 VBIT-12 added to the cells in the absence of DSS had no effect on either of the tested mitochondria-related activities.
VBIT-12 inhibited the cytosolic release of mtDNA in CT-26 cells treated with either DSS- or H2O2 (Figures 1I; Figure S2D). To demonstrate the requirement of VDAC1 for mtDNA release, we measured mtDNA release in CT-26 cells in which VDAC1 expression was silenced by a VDAC1-specific siRNA significantly decreased H2O2-induced release of mtDNA (Figure S2E).
The results obtained with DSS are similar to those found with other apoptosis inducers13 and are summarized in a proposed model (Figure 1J). Stress and pathological conditions lead to VDAC1 overexpression and oligomerization. The oligomer then forms a channel that facilitates the release of pro-apoptotic proteins with subsequent apoptosis and mtDNA release leading to inflammasome activation. VBIT-12, by preventing VDAC1 oligomerization, protects against apoptosis and mtDNA release (Figure 1J).
DSS-induced colitis pathology was decreased by VBIT-4 and VBIT-12 treatment
Next, we tested VBIT-12 effects on the DSS-induced colitis mouse model. Mice were provided DSS in their drinking water for 5 days and subjected to VBIT-12 treatment (added to the water) either from day 2 to 14 (D2) or from day 5 to 14 (D5; Figure 2A). Representative pictures show colons isolated from mice untreated or treated with 2% (Figure 2B) or 2.5% (Figure S3B) DSS, and with or without VBIT-12 treatment. DSS treatment decreased the colon length by about 45%, and by 22% when VBIT-12 treatment was started at day 2, and 26% when started at day 5 (Figure 2C). Similar results were obtained with 2.5% DSS and VBIT-12 and VBIT-4 treatment (Figures S3A–S3C). Similar results were obtained in trinitrobenzene sulphonic acid (TNBS)-induced colitis in which colon length decreased by about 30% that was prevented by VBIT-12 treatment (Figures S4B and S4C).
Figure 2.

VBIT-12 protects against DSS-induced UC pathology
(A) Experimental protocol for DSS-induced colitis (see Materials and methods). Mice (8 mice/group) were provided with 2% DSS in drinking water for 5 days. VBIT-12 (20 mg/kg) was given from day 2 (D2) or day 5 (D5), after the start of DSS treatment. On day 14, mice were sacrificed and monitored for colitis. (B) Representative images of colons, (C) colon length and (D) spleen weight. (E) The disease activated index (DAI) and the inflammation score (see Materials and methods section). The p value in blue color represents the significance relative to DSS-treated mice. (F) Representative images of colon tissue sections stained with H&E with superficial inflammation labeled by a dashed line. Scale bar is 50 μm. (G) Ki-67 staining; Scale bar, 50 μm. (H) Quantitative analysis of Ki-67. Red, yellow, blue, white, and orange arrows point to crypts, lamina propria, submucosa, lumen, and the muscularis mucosae, respectively. Results show means ± SEM, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, with p value in blue color represents the significance relative to DSS-treated mice. NS, not significant.
Body weight decreased by 32% within 10 days of DSS treatment, but only by 10% in VBIT-12-treated mice (Figures S3D and S3E). Similarly, the increase in spleen weight (35%) in the DSS-treated group barely changed in the VBIT-12-treated mice (Figure 2D; Figure S3F). VBIT-12 was found to be more effective than VBIT-4 in protecting against DSS-induced colon pathology (Figure S3G). Body weight in TNBS-treated mice decreased by 21% and 10% within 5 and 7 days post-TNBS treatment, but only by 13% and 6% for VBIT-12 treated mice starting at day 1 or 2 post-TNBS treatment (Figure S4D).
The clinical signs of the disease were scored according to changes in body weight, stool consistency, stool color, and occult bleeding (Table S2) and are presented as the disease activity index (DAI) for day 10 and day 14 (Figure 2E). The results show that VBIT-12 significantly decreased DAI when given on the second day of DSS treatment and was more effective than when given from the fifth day of DSS treatment (Figure 2E). Similar results were obtained when mice were treated with 2.5% DSS and VBIT-4 or VBIT-12 (Figure S3G). DAI was highly increased in TNBS-induced colitis mice, and VBIT-12 given at day 1 was more effective than when given at day 2 post-TNBS treatment (Figure S4E).
VBIT-12 given for 7 days to control mice not exposed to DSS or TNBS had no effect on colon length, body weight, or colon morphology (data not shown).
Histological analysis of H&E-stained control colon sections demonstrates the expected structure of villi in the crypts, lumen, lamina propria, muscularis mucosae, and submucosa (Figure 2F; Figures S5A and S5B). In contrast, colon sections of the DSS-treated mice show severe morphological aberrations, including swollen villi and a high level of inflammatory cell infiltration. These abnormalities were either dramatically reduced or absent after VBIT-12 treatment (Figure 2F). The effects of VBIT-12 on inflammation are reflected by the inflammation parameters score (Table S3) of the H&E-stained colon sections (see Figure S6). VBIT-12 significantly ameliorated inflammation and supplementation from the second day was more effective than on the fifth/last day of DSS treatment (Figures 2E; Figure S3G). Additionally, IHC staining showed that the levels of proliferation marker Ki-67, which were highly increased in DSS-treated mice colons, were decreased in VBIT-12-treated mice (Figures 2G and 2H).
Inflammatory cell infiltration was visualized by H&E staining of colon sections obtained from TNBS-induced colitis and was less than in DSS-induced colitis. VBIT-12 given at day 1 or 2 post-TNBS treatment significantly reduced the inflammation parameters score (Figure S7).
The colon epithelium comprises a single layer of various subtypes of IECs, including absorptive enterocytes, mucus-producing goblet cells that secrete the mucus layer that protects the epithelium from the luminal contents. Additional colon epithelial cells are the enteroendocrine cells that secrete various gastrointestinal hormones including pancreozymin and enteroglucagon, among others.26 To assess the amount and distribution of goblet cells in the tissue and to verify the effects of VBIT-12 treatment, we stained colon sections with periodic acid-Schiff (PAS). We found that DSS elevated PAS staining in mucin-filled goblet cells and revealed a large amount of secreted adherent mucus in the lumen (Figure 3A). A similar increase was seen by immunostaining with anti-mucin-2 antibodies (MUC2; Figures 3B and 3C). VBIT-12 treatment significantly decreased both PAS and anti-mucin-2 staining (Figures 3A–3C). Notably, increased MUC2 levels have been reported during the regenerative phase (9 days post-DSS treatment) of the model.27 This indicates that specific adaptations of the mucus layer maintain the protective capacities during DSS-induced colitis.
Figure 3.

DSS-induced alterations in epithelial cells that were prevented by VBIT-12
Mice were treated with 2% DSS with and without VBIT-12, as described in Figure 2. Representative images of colon tissue sections stained for Goblet cells with periodic acid-Schiff (PAS) (A), IHC stained with anti-MUC2 antibodies (B and C), or IF stained with anti-chromogranin A (D and E). Scale bars are 20 or 50 μm, as indicated. Quantification of chromogranin A-positive cells in the different groups (E). Nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole) (blue). IHC staining of α-SMA of colon sections and their quantitative analysis (F and G). Scale bars are 10, 20, or 50 μm, as indicated. The arrows indicate the strong staining of fibroblasts in the mucosa, submucosa, and muscularis layers. Quantitative analysis (C, E, and G). IF analysis of vimentin and E-cadherin expression levels in colon sections from control and DSS-treated mice (H). Arrows point to the high expression of vimentin in cells subjacent to the basement membrane. Results, means ± SEM (n = 3), ∗p < 0.05, ∗∗p < 0.01 ∗∗∗p < 0.001. NS, not significant.
Next, we verified the effect of VBIT-12 treatment on enteroendocrine cells, as visualized with anti-chromogranin A antibodies (Figures 3D and 3E). The increase in the number of chromogranin A-expressing cells seen in the colon from DSS-treated mice was attenuated with VBIT-12 treatment, with higher efficacy seen when given at day 2 than at day 5 of DSS treatment (Figures 3D and 3E).
Furthermore, we analyzed the expression levels of alpha-smooth muscle actin (α-SMA), vimentin, and E-cadherin in colon sections from DSS-treated mice with or without VIBT-12 treatment (Figures 3F–3H; Figure S8). Specific antibodies directed to α-SMA primarily stained the muscularis mucosa region in both control and DSS-treated mice. However, DSS treatment increased the number of α-SMA-positive cells exhibiting the long, spindle-shaped morphology characteristic of treatment (Figures 3F–3H). Specific antibodies directed to α-SMA primarily stained the muscularis mucosa region in both control and DSS-treated mice. However, DSS treatment increased the number of fibroblasts (Figures 3F and 3G). This observation is supported by the strong expression of the fibroblastic marker, vimentin, in cells subjacent to the basement membrane, juxtaposed against the bottom of the epithelial cells in DSS-treated mice (Figures 3H; Figure S8). The results indicate that the levels of α-SMA were increased in the colon of DSS-treated mice but were reduced by VBIT-12 treatment (Figure 3F and 3G).
Similar results were obtained with TNF-α and vimentin staining of colon sections obtained from TNBS-induced colitis with VBIT-12 given at day 1 or day 2 post-TNBS treatment, which reduced the expression levels to that of the control (Figure S9). The increased expression of both α-SMA and vimentin was not observed in colon sections from VBIT-12-treated DSS- and TNBS-treated mice (Figures 3F–3H; Figure S7 and S9).
The levels of the cell-cell adhesion mediator, E-cadherin, were reduced in DSS-induced colons, while VBIT-12 treatment restored E-cadherin expression to that of control mice (Figures 3H; Figure S8).
Sirius red staining detected increased collagen expression in colon sections from DSS-treated mice, and VBIT-12 given from the second day of DSS treatment reduced tissue fibrosis, as reflected by lower collagen levels (Figure S10).
Collectively, these results indicate that pathological changes in the colon from DSS-treated mice were attenuated by VBIT-12 and to a lesser extent by VBIT-4. DSS administration induces clinical signs of disease as soon as 1 day post-treatment, with changes in the expression of tight junction proteins28 and increased expression of pro-inflammatory cytokines.29 Thus, when starting VBIT-12 treatment after 2 days of DSS treatment, the mice are already in the disease stage.
DSS-induced colon cell apoptosis is reduced by VBIT-12
As DSS increases gut epithelial cell apoptosis,30 we analyzed the effect of VBIT-12 on several apoptosis parameters (Figures 4A–4F; Figure S11). Specifically, DSS markedly increased DNA fragmentation in the epithelial cells, as assayed by the TUNEL (terminal deoxynucleotidyl transferase dUTP nick-end labeling) staining, and this was largely suppressed by VBIT-12 (Figure 4A). Similar results were obtained in mice treated with 2.5% DSS, which showed increased TUNEL and caspase-3 staining that was prevented by VBIT-12 (Figures S11A and S11B).
Figure 4.

Overexpression of VDAC1 and pro-apoptotic proteins in the colon of DSS-treated mice was reduced by VBIT-12
(A) Representative TUNEL staining of colonic sections obtained from control and DSS-treated mice, with and without treatment with VBIT-12. Scale bar is 50 μm. Cells stained green/yellow are apoptotic cells. (B and C) Immunoblotting of apoptosis-related proteins in colon proteins extracted from the various groups, using specific antibodies for VDAC1, Bax, t-Bid, caspase-8, caspase-3, and p53, with β-actin used as a loading control (B), and their quantitative analysis, with p value in blue color represents the significance relative to DSS-treated mice (C). (D) IF staining of p53, scale bar is 50 μm. (E and F) VDAC1 IHC staining (E) and quantitative analysis (F). (G) IP3R and VDAC1 IF-staining. (H) Colon sections were subjected to an in situ PLA using anti-VDAC1 and anti-IP3R antibodies. The ligation products appear in red; nuclei were DAPI-stained. Scale bars are 50 μm. Results, means ± SEM (n = 3–4), ∗p < 0.05, ∗∗p < 0.01; ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, with p value in blue color represents the significance relative to DSS-treated mice. NS, not significant.
Similar results were obtained with cleaved caspase-3 staining of colon sections obtained from TNBS-induced colitis with VBIT-12 was given at day 1 or 2 post-TNBS treatment that reduced both cleaved caspase-3 and VDAC1 expression (Figures S11C and S11D). DSS-induced apoptosis was further demonstrated by the increased expression of pro-apoptotic proteins such as VDAC1, caspase-1, caspase-3, caspase-8, t-Bid, Bax, and p53, and the decreased levels of the anti-apoptotic protein Bcl-2 (Figures 4B–4F; Figures S12 and S13). VBIT-12 treatment, particularly on day 2 of DSS treatment, prevented these changes in the protein expression levels.
The pro-apoptotic protein p53 is activated in epithelial and inflammatory cells during the process of colitis. p53 protein levels were increased in the crypt IECs from UC patients.31 In agreement with these findings, p53 levels were increased in DSS-treated mice by 4-fold, which was attenuated with VBIT-12 treatment (Figures 4B–4D; Figure S14). The elevated p53 induced by DSS treatment is mainly extranuclear, mostly co-localized with the mitochondrial protein ATP synthase-5a (Figure S14). The translocation of cytoplasmic p53 to mitochondria in response to a stress signal was demonstrated in several studies.32, 33, 34 These findings suggest that VBIT-12 prevented DSS-induced apoptotic cell death.
VDAC1 involves structural and functional tethering in the ER-mitochondria-associated membranes (MAM).17 Staining colon sections from DSS-treated mice with anti-VDAC1 and anti-IP3R antibodies revealed a massive increase in the levels of both protein that was co-localized (Figure 4G). VBIT-12 treatment prevented the increase in expression of both proteins. By using an in situ proximity ligation assay (PLA),35 we confirmed that DSS increased MAM in the colon (Figure 4H).
VBIT-12 prevents DSS-induced inflammation and increased serum mtDNA levels
The inflammatory response is a central pathological process in IBD,36 and a key mediator of inflammation, including that in IBD, is the nuclear factor κB (NF-κB).37 Many inflammation triggers, including released mtDNA, are known to activate NF-κB and many effectors of inflammation such as inflammatory cytokines are activated either directly or indirectly by NF-κB. Moreover, the activation of the NF-κB pathway has been implicated in the pathogenesis of IBD as induced by DSS.38 Thus, we focused on how VBIT-12 treatment affected NF-κB and other NF-κB-related pathways in IBD.
Immunostaining of colon sections with anti-NF-κB antibodies revealed a dramatic increase in NF-κB expression in epithelial cells of DSS-treated mice (Figures 5B and 5D; Figure S15). Moreover, anti-p65-NF-κB antibody staining showed that DSS-treatment led to increased levels of phosphorylated NF-κB (p65) and its localization in the nucleus of colon cells (Figures 5C and 5D). VBIT-12 administration significantly reduced the levels of both NF-κB and p-p65 in the colon of DSS-colitis-induced mice (Figures 5B–5D).
Figure 5.

VBIT-12 reduces inflammation signaling in DSS-induced UC
(A–D) Colon sections from DSS-treated mice with and without VBIT-12 treatment were subjected to IF staining for TNF-α (A), IHC staining for NF-κB (B), or IF staining for p-P65, with the inset showing nucleus-cytoplasm distribution of p-p65 (C) and quantified staining intensity for TNF-α, NF-κB, and p-P65 (D). (E and F) IF staining of iNOS (E) and its quantitative analysis (F). Scale bars are, 10, 20, or 50 μm, as indicated. (G) MPO activity in protein extracts obtained from control and DSS-treated mice with and without VBIT-12. (H) Real-time PCR analysis of DSS-induced proinflammatory cytokine production, as described in the Supplemental Information. Results, means ± SEM (n = 3) ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, with p value in blue color represents the significance relative to DSS-treated mice, with p value in blue color represents the significance relative to DSS-treated mice. NS, not significant.
Similar results were obtained with NF-κB-p-p65 staining of colon sections obtained from TNBS-induced colitis with VBIT-12 given at day 1 or day 2 post-TNBS treatment, which reduced its expression to the level of control mice (Figure S16).
We monitored the major mediators of inflammation in IBD and specifically in UC, namely, TNF-α, and inducible nitric oxide synthase (iNOS; Figures 5A, 5D, 5E, 5F; Figure S9), myeloperoxidase (MPO)39 (Figure 5G). These play key roles in the development of UC.40 TNF-α exerts a variety of beneficial functions in the intestine, playing a critical role not only in maintaining intestinal integrity but conversely also in the pathogenesis of intestinal inflammation.
Upregulation of iNOS has been closely associated with the initiation and maintenance of intestinal inflammation in IBD.41 Similarly, the increased expression of iNOS seen by immunofluorescence staining of the DSS-treated mice was decreased by VBIT-12 (Figures 5E and 5F).
In order to confirm these findings, we used real-time PCR and demonstrated that the mRNA levels of iNOS, TNF-α, interleukin-1β (IL-1β), and IL-6 were increased in DSS-treated mice colons and that administration of VBIT-12 on days 2 or 5 of DSS treatment strongly reduced the induced expression of the pro-inflammatory mediator in the colon (Figure 5H).
Increases in MPO and NO levels are markers of an aggravated inflammatory reaction, with NO levels increasing over 100-fold in UC patients41 and the level of neutrophil MPO being proportional to neutrophil infiltration in inflamed tissue.42 As DSS treatment induces an influx of neutrophils into the lamina propria and submucosa, we analyzed colonic MPO activity as a marker for neutrophil infiltration and inflammation. Intestinal MPO activity in DSS-treated mice was about 8-fold higher than in the control mice and was significantly reduced by VBIT-12 treatment (Figure 5G).
The nucleotide-binding and oligomerization domain (NOD)-like receptors (NLRs) are pattern-recognition receptors, with the family member, NLR pyrin domain, containing 3 (NLRP3) inflammasome emerging as a crucial regulator of intestinal homeostasis that is activated in colitis.43 In contrast to the low expression of NLRP3 in the colon of control mice, a very high level of expression of NLRP3 was seen in the epithelial and stromal cells of DSS-treated mice. Treatment with VBIT-12 suppressed the NLRP3 expression induced by DSS-treatment (Figures 6A and 6B).
Figure 6.

DSS-induced MAVS expression, inflammation, and serum mtDNA were all prevented by VBIT-12
Colon sections were obtained from control and DSS-treated mice with and without treatment with VBIT-12 and subjected to IF staining for NRLP3 (A and B); scale bar is 50 μm. (C) Protein extracts from frozen colon obtained from control and DSS-treated mice with and without treatment with VBIT-12 were subjected to immunoblotting with anti-MAVS or anti-caspase-1 antibodies. RU indicates relative expression. (D) IHC staining for MAVS and its quantification (E); scale bar is 50 μm. (F) IF staining for caspase-1; scale bar is 50 μm. (G and H) Caspase-1 activity (G). Immunoblotting of HIF-1α and MIF with their relative expression presented as relative units (RU, H). (I) Levels of mtDNA in serum were measured by qPCR. Results, mean ± SEM (n = 3) ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001. NS, not significant.
Importantly, the activation of the NLRP3 inflammasome requires MAVS to recruit the NLRP3 inflammasome during RNA viral infections and direct its translocation to the mitochondria.44,45 The very low expression of MAVS seen in control mice was much higher in the colons of DSS-treated mice and VBIT-12 suppressed MAVS expression (Figures 6C–6E).
NLRP3 inflammasome is composed of NLRP3, adaptor molecule ASC, and caspase-1, with its activated caspase-1 converting the proinflammatory cytokines into their active forms.46 The expression levels of caspase-1 and its activity were highly increased in the colons of DSS-treated mice and were suppressed when the mice were treated with VBIT-12 applied on day 2 of DSS treatment (Figures 6C, 6F, and 6G).
The overall results demonstrate that VBIT-12 alleviates the severity of DSS-induced colitis in mice, as reflected by suppresses the infiltration of inflammatory cells, the decreased activity of MPO, and the lower expression of pro-inflammatory mediators.
Acute inflammation of the mucosa leads to tissue hypoxia.47 Tissue and cell survival, metabolism, and other functions under low oxygenation conditions are regulated by the transcription factor, the hypoxia inducible factor (HIF).48 It has been suggested that HIF augments colitis through a macrophage migration inhibitory factor (MIF)-dependent inflammatory signaling cascade.49 Immunoblotting of HIF-1α and MIF revealed that VBIT-12 suppressed DSS-induced elevation of HIF-1α and MIF (Figure 6H).
Finally, the inflammatory effect of mtDNA was demonstrated in several studies.3,11 mtDNA levels were about ∼5.5-fold higher in DSS-treated mice serum relative to control mice and were decreased to the levels of control mice when DSS mice were treated with VBIT-12 starting 2 days after the DSS-treatment and by 40% when given on the last day of treatment (Figure 6I). Thus, VBIT-12 given to DSS-treated mice suppresses mucosal inflammation, as demonstrated by decreasing the expression of NF-κB and the inflammatory factors IL-6, IL-1β, TNF-α, iNOS, MPO activity, and the activation of NLRP3 inflammasome, as well as the release of mtDNA.
Inflammatory and autoimmune diseases have been identified as prime examples of the link between metabolism and inflammation, with immunometabolism presenting the interface between immunity and metabolism,50,51 including in IBD.47
In this context, acetyl-glucosamine (GlcNAc) formed during bacterial infection and degradation of the bacterial cell wall peptidoglycan in lysosomes leads to the detachment of the glycolytic enzyme HK from the mitochondria, which activates the NLRP3 inflammasome.51 IHC and IF staining of colon sections showed a dramatic increase in HK-I expression in DSS-treated mice, which was reduced by VBIT-12 to the levels found in control mice (Figures 7A–7C).
Figure 7.

DSS-induced UC alterations in the expression of metabolism-related enzymes and P2X7 receptor that were prevented by VBIT-12 treatment
Representative colon sections obtained from control or DSS-treated mice with and without treatment with VBIT-12 were IHC stained for HK-I (A) and quantitative analysis (B). IF staining of HK-I and cytochrome c (cyto c, C). Immunoblotting of HK-I, citrate synthase (CS), and ATPsyn5a with corresponding antibodies (D) with expression levels quantified, with p value in blue color represents the significance relative to DSS-treated mice (E). IHC staining of P2X7 (F) and quantitative analysis (G). Scale bars are 50 μm. Results, mean ± SEM (n = 3) ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, with p value in blue color represents the significance relative to DSS-treated mice. NS, not significant.
Cyto c expression was also elevated in the cytoplasm of DSS-treated mice, indicating the induction of apoptosis (Figures 7C; Figure S17). Co-staining of HK-I and Cyto c indicated a cytoplasmic localization of HK-I suggesting its detachment from the mitochondria to the cytoplasm (Figures 7C; Figure S17). The increase in the levels of cyto c was also suppressed by VBIT-12 treatment.
The levels of the glycolytic enzyme HK-I, the Kreb’s cycle enzyme, citrate synthase (CS), and ATP synthase subunit 5a were increased 2.8-, 1.6-, and 1.5-fold, respectively, in DSS-treated mice (Figures 7D and 7E). This increase was eliminated by VBIT-12 treatment given on day 2 and also, albeit to a lesser extent, when given on day 5 (Figures 7A–7E).
During intestinal inflammation, ATP is released continuously into the extracellular space, generally as a result of direct cellular injury and, specifically, through hemi-channels expressed by infiltrating inflammatory cells such as neutrophils.52 ATP acts as a classical DAMP signal and is involved in inflammasome activation by signaling through purinergic receptors such as P2X7, expressed on IECs and lymphocytes.53 Our results demonstrate that the P2X7R expression level is increased in colon sections obtained from DSS-treated mice, but not in those treated with VBIT-12 (Figures 7F and 7G).
The presented results, as discussed below, demonstrate that VBIT-12 comprehensively protects against DSS-induced disease-associated pathologies including increases in apoptosis, pro-inflammatory cytokines, and NLRP3 inflammasome activation, and therefore suggests that VBIT-12 has potential for the treatment of IBD.
Discussion
The chronic inflammatory disorders of the gastrointestinal system that comprise IBD afflict 1.6 million North Americans and 5 million people worldwide, with nearly 15% growth over the last 5 years.1 The diverse and complex interaction between genetic, environmental, and immunoregulatory factors thought to be responsible for its pathology complicates the treatment. Currently, there are a number of therapeutic strategies, including reinforcing epithelial barrier function, inhibition of proinflammatory cytokines, and blocking inflammatory cell trafficking.54 Although anti-TNF-α antibody therapy shows remarkable efficacy in treating IBD, many patients either do not respond to anti-TNF-α treatment or exhibit a reduced response over time.55 This highlights the need for new molecular targets for IBD therapy.
Previously, we reported that the mitochondrial gatekeeper, VDAC1, is overexpressed in several diseases including cancer,56 Alzheimer’s disease,57 β-cells in T2D,21 autoimmune diseases such as lupus,12 and non-alcoholic steatohepatitis (NASH).58 Moreover, upregulation of VDAC1 expression constitutes a mechanism by which apoptosis inducers can trigger apoptosis.59 VDAC1 overexpression promotes oligomerization and the formation of large channels that can facilitate mitochondrial pro-apoptotic protein release.16 In order to prevent VDAC1 oligomerization-mediated apoptosis and/or mitochondrial dysfunction, we developed two VDAC1 oligomerization inhibitors: VBIT-4 and VBIT-12.20 Their efficacy as inhibitors of VDAC1 oligomerization, apoptosis, and associated processes has been validated in T2D21 and lupus models.12
Here, we demonstrate for the first time that VDAC1 is overexpressed in the colon of IBD patients and in DSS-induced or TNBS-induced colitis mouse models and that the VDAC1-interacting molecule, VBIT-12, can ameliorate the colonic histological damage and inflammation seen in the DSS-induced colitis mouse model. Although it is not clear whether VDAC1 overexpression precedes the IBD colon pathology, VDAC1-induced apoptosis would contribute to barrier defects.
Our data indicate that VBIT-12 suppresses DSS-induced overexpressed VDAC1, mitochondrial dysfunction, apoptosis, mtDNA release, and NRLP3 inflammasome activation in the epithelial cells of the colon. These effects were achieved by a number of different cellular pathways: (1) mitochondria-mediated apoptosis, (2) expression and activation of pro-inflammatory cytokines, (3) cytosolic mtDNA- and ROS-associated inflammatory response, (4) MAVS mediating NRLP3 assembly, and (5) extracellular ATP and intracellular Ca2+-signaling.
Extracellular ATP and intracellular Ca2+-signaling
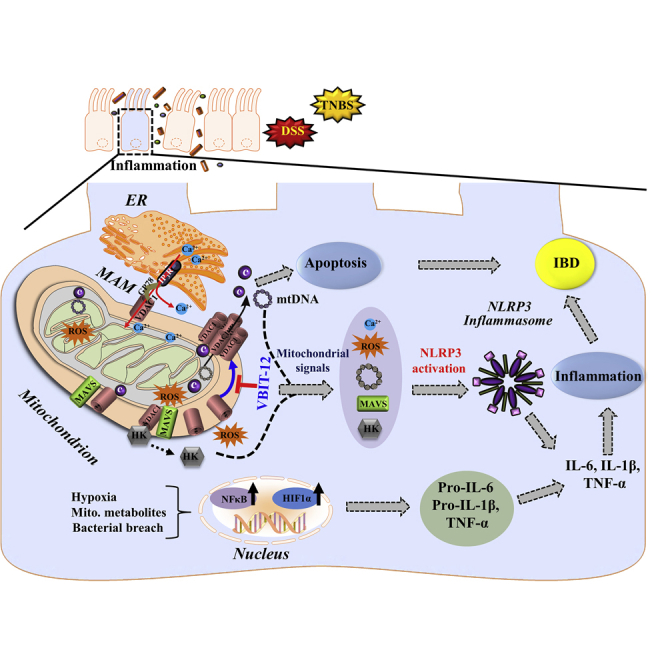
All these pathways are associated with NRLP3 inflammasome activation, inflammation, and IBD development. Activation of the NLRP3 inflammasome is thought to be one of the key steps in the development of inflammation in IBD, although the precise mechanism remains unclear.60 Recent evidence now suggests that mitochondria sense and integrate various danger signals and then relay the signals to the NLRP3 inflammasome.18 The functions of the mitochondria and VDAC1 in inflammasome activation in IBD are summarized in Figure 8 and discussed below.
-
1.
Mitochondria-mediated apoptosis: DSS-treatment induced apoptosis in the colon,30 as confirmed here by colon TUNEL staining and increased expression of caspase-3, caspase-8, t-Bid, p53, and Bax. These DSS-induced changes in apoptosis-related factors were prevented by VBIT-12. The protective effects of VBIT-12 against apoptosis and mitochondria dysfunction suggest that the overexpressed VDAC1 and its oligomerization play a central role in the DSS-induced pathology. Understanding the mechanism(s) that cause VDAC1 overexpression under pathological conditions requires further study. However, based on our findings and previous reports on IBD, we propose that VDAC1 overexpression, and the resulting mitochondrial dysfunction trigger inflammation in IBD (Figure 8).
-
2.
Expression and activation of pro-inflammatory cytokines: The inflamed intestine seen in IBD patients is heavily infiltrated by inflammatory cells that release a large amount of pro-inflammatory mediators such as cytokines and NO.39 NF-κB signaling in the gut epithelium has been recognized as a critical regulator of epithelial integrity and intestinal immune homeostasis associated with the pathogenesis of human IBD via induction of pro-inflammatory gene transcription.39 NF-κB regulates the expression of many inflammatory cytokines including TNF-α, IL-1β, IL-6, and IL-8, which can cause the gut epithelial damage seen in IBD.39
Figure 8.

Schematic model of the function of mitochondria, VDAC1, and associated proteins in the apoptosis, inflammasome activation, and inflammatory response leading to IBD, and their prevention by VBIT-12
VDAC-1 is involved in numerous mitochondria-associated functions including epithelial metabolism, inflammation, and apoptosis, all impaired in IBD. VDAC1 overexpression induced by stress conditions (DSS) triggers mitochondria dysfunction and four major signaling pathways: (A) Mitochondrial signals (mtDNA release, increased mtROS and Ca2+ levels, HK detachment, and MAVS assembly) that induce NLRP3 assembly and activation, leading to pro-inflammatory activation and IBD development. MAVS, which interacts with VDAC1, stabilizes the inflammasome complex and activates NLRP3 on the MAMs. In addition, intracellular Ca2+ signaling is controlled by the MAM tether comprising VDAC1, IP3R, and other proteins. These signaling molecules facilitate the inflammasome assembly and activation, leading to pro-caspase-1 activation and pro-inflammatory cytokine maturation. (B) The apoptosis pathway is induced by oligomerization of the overexpressed VDAC1 to form a large channel-mediating cyto c release, apoptosome formation, caspase activation, and apoptotic cell death. The same oligomeric VDAC1 channel allows the release of mtDNA. (C) The HIF-α/NF-κB/MIF-regulated gene expression pathway may be induced by mtROS/hypoxia. The activation of HIF1-α and of NF-κB by the bacterial flora of the gut increases the production of the pro-inflammatory cytokines. (D) NLRP3 activation is triggered by various stress signals including the activation of P2X7R by ATP release from cells, leading to K+ efflux and influx of Ca2+, both of which contribute to the activation of NLRP3, which activates caspase-1 and initiates an intestinal inflammatory cascade. In this model, the overexpression and oligomerization of VDAC1 acts as a common functional junction for all these activated pathways in IBD. Hence, inhibition of VDAC1 oligomerization by VBIT-12 suppresses apoptosis and inflammation to prevent the IBD pathology.
The increased expression of NF-κB and the phosphorylated p65 seen in the DSS-treated mice led to an increase in NLRP3 inflammasome levels. Our findings indicate that VBIT-12 suppresses the DSS-induced induction of NF-κB, the prime regulator of NLRP3 expression.61 In epithelia and immune cells, NF-κB primes the expression of pro-IL-1β and pro-IL-18 that are activated by cleavage by the activated NLRP-caspase-1 inflammasome. Caspase-1 has been identified as a central mediator of DSS-induced colitis.62 In the present study, we provide evidence that the levels of NLRP3 expression and of caspase-1 and its activation were increased in colons of DSS-treated mice, but not in VBIT-12-treated mice.
-
3.
Cytosolic mtDNA- and ROS-associated inflammatory response: Release of mtROS and mtDNA into the cytosol are key upstream events implicated in NLRP3 activation. HIF-1α, which is activated by hypoxia, enhances colitis through the MIF-dependent inflammatory signaling cascade49 that regulates IL-1α, IL-1β, and IL-18 release via activation of the NLRP3 inflammasome. Oxidized mtDNA when released into the cytosol binds to NLRP3 and activates the NLRP3 inflammasome.3,63
Recently, we demonstrated that in a mouse model of systemic lupus erythematosus, VDAC1 oligomerizes to form a large channel that promotes mtDNA release, stimulates type-I interferon responses, and NETosis that were all decreased with VBIT-4 treatment, including disease severity.12 Similarly, the results presented here demonstrate that VBIT-12 decreases the DSS-induced release of mtDNA in mice and in cells expressing VDAC1, suggesting that VDAC1 is required for mtDNA release.
-
4.
MAVS mediating NRLP3 assembly: Mitochondria have been suggested to act as docking sites for inflammasome assembly. The association of NLRP3 inflammasome with the mitochondria was proposed to involve MAVS, which was shown to mediate NLRP3 inflammasome assembly and activation during RNA viral infections.44,45 MAVS recruits NLRP3 and directs it to the mitochondria for inflammasome activation.44,45 MAVS has been shown to interact with VDAC1,64 and the increased MAVS expression seen in DSS mice was suppressed after VBIT-12 treatment.
-
5.
Extracellular ATP and intracellular Ca2+signaling: NLRP3 is associated with the ER membranes of resting cells although, upon activation, the complex becomes associated with the MAM,18 which is crucial for ER-mitochondrial Ca2+-signaling. Dysfunctional epithelial mitochondria and ER stress have each been associated with IBD, with MAM being important in Ca2+ homeostasis and protein folding.17 Here, we report for the first time that DSS-induced colitis resulted in a high increase in IP3R levels, which was co-localized with the overexpressed VDAC1, suggesting an increase in MAM.
A manifestation of IBD is increased ER stress in the intestinal epithelium,65 where impaired ER function leads to an accumulation of proteins. This then triggers Ca2+ release from the ER and activates NF-κB and NF-κB-dependent gene expression.66 The increase in IP3R levels and VDAC1 seen in the MAM supports the involvement of intracellular Ca2+ stores in inflammasome activation.
Finally, NLRP3 inflammasome activation by extracellular ATP is mediated through the ATP-gated P2X7R channel, which mediates Ca2+ influx67 and K+ efflux68 that activate the NLRP3 inflammasome. ATP-activated P2X7R contributes to elevated levels of pro-inflammatory cytokines such as IL-1β, TNF-α, and IL-17 in the tissue microenvironment.53
The major source of ATP is the mitochondria. VDAC1, as the ATP transporter,59 controls the cellular and extracellular ATP levels, and thereby, activation of P2X7R. The increased levels of P2X7R in DSS-induced colitis were suppressed by VBIT-12, supporting the involvement of VDAC1 in P2X7R activation of the pro-inflammatory cascade via Ca2+ influx67 and K+ efflux68 (Figure 8). In CD patients, P2X7R expression is increased,53 and pharmacologic P2-receptor antagonists inhibit inflammation including in IBD.
Remarkably, all these inflammatory pathways shown to activate the NLRP3 inflammasome were activated by DSS but suppressed by VBIT-12 treatment.
The VDAC1 requirement for inflammasome activation was supported by the findings that tunicamycin-induced inflammasome activation was impaired in cells with downregulated VDAC1, but not VDAC2.69 Also, downregulation of VDAC1 in THP1 cells results in a considerable decrease in caspase-1 activation and IL-1β secretion after stimulation with various NLRP3 inflammasome activators.18 Here, we suggest that the VDAC1 overexpressed in the colon of IBD patients and DSS-treated mice is involved in complex interactions and signaling pathways leading to activation of the NLRP3 inflammasome, which then induces a pro-inflammatory response and IBD (Figure 8).
This VDAC1-based scenario introduces a completely new potential target for IBD therapy, provides proof of concept for UC, and opens a new avenue for treating other inflammatory/autoimmune diseases that may be amenable to the use of novel VDAC1-interacting molecules.
Materials and methods
See the Supplemental Materials for Materials.
DSS-induced colitis in mice
Female, specific pathogen free (SPF) C57BL/6 (H-2b), 6- to 7-week-old mice (Envigo, Israel) were kept in SPF static cages with a 12-h light/dark cycle. Chow pellets and tap water were available ad libitum. The experimental protocols were approved by the Institutional Animal Care and Use Committee of Ben Gurion University, and all the procedures were performed in accordance with the approved guidelines. Animals were provided ad libitum a commercial rodent diet and free access to either regular or DSS-containing drinking water. DSS was prepared in drinking water (2.0% or 2.5%, w/v), and was supplied for 5 days, followed subsequently by 8 days of regular drinking water until study termination on day 14 (Figure 3A). Mice were weighed and examined for health before disease induction and thereafter, and clinical signs of colitis such as body weight change, stool consistency, and fecal blood content were scored on a daily basis, starting from study day 1 to the time of scheduled euthanasia. At the end of the study, surviving animals were euthanized by CO2 asphyxiation. Immediately after euthanasia, the entire large intestine from the ileo-cecal junction to the anus were removed and weighed, and the length of the colon was measured using a ruler. Then, a section of each colon was frozen in liquid nitrogen and the rest was fixed overnight with 4% formaldehyde in phosphate-buffered saline (PBS, pH 7.4), dehydrated with graded alcohol, placed in xylene, and embedded in paraffin and sectioned (5 μm). Blood was collected, and serum was prepared and stored at −20°C until it was processed for mtDNA measurement.
DSS-induced colitis score
Colitis severity was scored according to macroscopic and microscopic assessments, as presented in the Supplemental Information and in Tables S2 and S3.
Histological, immunohistochemistry, immunofluorescence, and immunoblotting analyses of colon tissue
Formalin-fixed, paraffin-embedded sections of colon obtained from control or DSS-treated mice with or without VBIT-4 or VBIT-12 treatment were H&E stained and probed with appropriate antibodies via IHC or immunofluorescence, using the antibodies listed in Table S1, as described in the Supplemental Information.
TUNEL assay, PLA, and Sirius red staining
Fixed colon sections in paraffin were processed for TUNEL, PLA, or Sirius red staining, as described in the Supplemental Information.
MPO activity assay
Colon samples were snap-frozen immediately after removal in liquid nitrogen and stored at −80°C. For MPO measurement, the frozen samples were homogenized by a mechanical homogenizer in 500 μL, 50 mM hexadecyltrimethylammonium bromide buffer (Sigma-Aldrich, St. Louis, MO, USA) in 50 mM potassium phosphate buffer at pH 6, and sonicated and centrifuged at 15,000 × g for 20 min. The supernatant was used for protein determination and stored at −20°C. MPO activity assay was performed from thawed samples in a 96-well plate in triplicates. Briefly, 10 μL samples were combined with 80 μL 0.75 mM H2O2 and 110 μL −3,3′,5,5′-tetramethyl-benzidine (TMB) solution (2.9 mM TMB in 14.5% DMSO and 150 mM sodium phosphate buffer at pH 5.4), and the plate was incubated at 37°C for 5 min. The reaction was stopped by adding 50 μL of 2M H2SO4, and absorption was measured at 450 nm to estimate the MPO activity. MPO activities were expressed in unit/mg of wet tissue.
Caspase-1 activity assay
Frozen colon tissues from control, DSS-treated and DSS-treated mice subjected to VBIT-12 treatment were homogenized in lysis buffer (BioVision, Milpitas, CA, USA) using a homogenizer. Subsequently, homogenated tissue was centrifuged at 10,000 × g for 10 min. Protein concentration in the supernatant was determined. Caspase-1 activity was assessed using a Caspase-1 Fluorometric Assay kit (BioVision).
Cell culture, DSS treatment, cell viability assay, mtROS production, intracellular Ca2+, and cell death
Human and mouse colon-cancer-derived Caco-2 (HTB-37) and CT-26 (CRL-2638), respectively, were from ATCC (Manassas, Virginia). Caco-2 cells were maintained using Eagle’s minimum essential medium (EMEM) supplemented with 20% inactivated fetal bovine serum (FBS) and CT-26, using 25 mM glucose Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (vol/vol) of inactivated FBS, (Biological Industries, Beit Haemek, Israel). Both culture media were supplemented with 100 U/mL penicillin and 100 μg/mL streptomycin. Cells were grown at 37°C in 5% CO2 in a humidified incubator.
DSS treatment
Cells (60% confluence) were incubated for 48 h in a serum-free growth medium with different concentrations of DSS (dissolved in culture media and filter-sterilized using a 0.45-μm filter) in triplicates. Cells were treated with different concentrations of DSS (1%–3%) in the absence or presence of VBIT-12 and analyzed for cell viability, VDAC1 expression levels, VDAC1 oligomerization, ROS production, mtDNA release, and apoptosis as described below.
Cell viability
This was assayed using the XTT according to the manufacturer’s instructions. Briefly, cells (5 × 103 cells/well) were seeded in 96-well plates and were treated with DSS as above.
mtROS production
mtROS production was monitored by using MitoSOX Red (5 μM), a mitochondrial superoxide indicator for live-cell imaging, according to the manufacturer’s protocol (Invitrogen, Grand Island, NY, USA). Fluorescence was measured using an iCyt flow cytometer and analyzed using ec800 software (SONY Biotechnology, San Jose, CA, USA).
Cellular Ca2+ analysis
Fluo-4-AM was used to monitor changes in cytosolic Ca2+ levels in CT-26 cells as described in the Supplemental Information.
Cell death
Cell death was analyzed using propidium iodide (PI) staining, performed according to the recommended protocol, and subjected to flow cytometry analysis.
VDAC1 overexpression and oligomerization assays
Cells were treated with the DSS as above, harvested, washed with PBS, pH 8.3, and then were incubated at 1 mg/mL with or without EGS (100 μM, 15 min). Lysates (20 μg protein for measuring VDAC1 expression levels and 60–80 μg protein for visualization of VDAC1 oligomerization) were subjected to SDS-PAGE and immunoblotting using anti-VDAC1 antibodies. Analyses of immuno-reactive VDAC1 monomers, dimers, and multimer bands was performed using FUSION-FX (Vilber Lourmat, France).
Cell siRNA treatment, RNA preparation, q-RT-PCR, and mtDNA release quantification
Details are presented in the Supplemental Information section and the primers used are listed in Table S4.
Statistics and data analysis
The mean ± SEM of results obtained from at least three independent experiments are presented. The significance of differences for cell-based assays was calculated by a two-tailed Student’s t test and for IBD-mouse model by using a one-way ANOVA with post hoc tests. Significance of differences is reported as ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p < 0.0001. NS, not significant.
Acknowledgments
This research was supported by a grants from the National Institute for Biotechnology in the Negev (NIBN) and Ben-Gurion University of the Negev, Israel.
Author contributions
A.V. , B.A and S.P. carried out all of the experiments and analysis, A.K.-S. contributed to the data analysis and figures preparation. E.O. contributed to the interpretation of the results and helped in writing the paper and preparing the summary model, and J.H.C. assisted in writing the final version. V.S.-B. designed the experimental strategy, interpreted the results, and wrote the paper.
Declaration of interests
The authors declare no competing financial interests. A patent application pertaining to the results presented in the paper has been filed.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.06.024.
Supplemental information
References
- 1.Colombel J.F., Mahadevan U. Inflammatory Bowel Disease 2017: Innovations and Changing Paradigms. Gastroenterology. 2017;152:309–312. doi: 10.1053/j.gastro.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 2.Lood C., Blanco L.P., Purmalek M.M., Carmona-Rivera C., De Ravin S.S., Smith C.K., Malech H.L., Ledbetter J.A., Elkon K.B., Kaplan M.J. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 2016;22:146–153. doi: 10.1038/nm.4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.West A.P., Shadel G.S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 2017;17:363–375. doi: 10.1038/nri.2017.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berger E., Rath E., Yuan D., Waldschmitt N., Khaloian S., Allgäuer M., Staszewski O., Lobner E.M., Schöttl T., Giesbertz P., et al. Mitochondrial function controls intestinal epithelial stemness and proliferation. Nat. Commun. 2016;7:13171. doi: 10.1038/ncomms13171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Novak E.A., Mollen K.P. Mitochondrial dysfunction in inflammatory bowel disease. Front. Cell Dev. Biol. 2015;3:62. doi: 10.3389/fcell.2015.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jackson D.N., Theiss A.L. Gut bacteria signaling to mitochondria in intestinal inflammation and cancer. Gut Microbes. 2019;11:1–20. doi: 10.1080/19490976.2019.1592421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martínez-Reyes I., Chandel N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020;11:102. doi: 10.1038/s41467-019-13668-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Angajala A., Lim S., Phillips J.B., Kim J.H., Yates C., You Z., Tan M. Diverse Roles of Mitochondria in Immune Responses: Novel Insights Into Immuno-Metabolism. Front. Immunol. 2018;9:1605. doi: 10.3389/fimmu.2018.01605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Breda C.N.S., Davanzo G.G., Basso P.J., Saraiva Câmara N.O., Moraes-Vieira P.M.M. Mitochondria as central hub of the immune system. Redox Biol. 2019;26:101255. doi: 10.1016/j.redox.2019.101255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mills E.L., Kelly B., O’Neill L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017;18:488–498. doi: 10.1038/ni.3704. [DOI] [PubMed] [Google Scholar]
- 11.Boyapati R.K., Dorward D.A., Tamborska A., Kalla R., Ventham N.T., Doherty M.K., Whitfield P.D., Gray M., Loane J., Rossi A.G., et al. Mitochondrial DNA Is a Pro-Inflammatory Damage-Associated Molecular Pattern Released During Active IBD. Inflamm. Bowel Dis. 2018;24:2113–2122. doi: 10.1093/ibd/izy095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim J., Gupta R., Blanco L.P., Yang S., Shteinfer-Kuzmine A., Wang K., Zhu J., Yoon H.E., Wang X., Kerkhofs M., et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science. 2019;366:1531–1536. doi: 10.1126/science.aav4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shoshan-Barmatz V., Maldonado E.N., Krelin Y. VDAC1 at the crossroads of cell metabolism, apoptosis and cell stress. Cell Stress. 2017;1:11–36. doi: 10.15698/cst2017.10.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shoshan-Barmatz V., De Pinto V., Zweckstetter M., Raviv Z., Keinan N., Arbel N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Aspects Med. 2010;31:227–285. doi: 10.1016/j.mam.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 15.Geula S., Ben-Hail D., Shoshan-Barmatz V. Structure-based analysis of VDAC1: N-terminus location, translocation, channel gating and association with anti-apoptotic proteins. Biochem. J. 2012;444:475–485. doi: 10.1042/BJ20112079. [DOI] [PubMed] [Google Scholar]
- 16.Keinan N., Tyomkin D., Shoshan-Barmatz V. Oligomerization of the mitochondrial protein voltage-dependent anion channel is coupled to the induction of apoptosis. Mol. Cell. Biol. 2010;30:5698–5709. doi: 10.1128/MCB.00165-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marchi S., Patergnani S., Pinton P. The endoplasmic reticulum-mitochondria connection: one touch, multiple functions. Biochim. Biophys. Acta. 2014;1837:461–469. doi: 10.1016/j.bbabio.2013.10.015. [DOI] [PubMed] [Google Scholar]
- 18.Zhou R., Yazdi A.S., Menu P., Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 19.Weisthal S., Keinan N., Ben-Hail D., Arif T., Shoshan-Barmatz V. Ca(2+)-mediated regulation of VDAC1 expression levels is associated with cell death induction. Biochim. Biophys. Acta. 2014;1843:2270–2281. doi: 10.1016/j.bbamcr.2014.03.021. [DOI] [PubMed] [Google Scholar]
- 20.Ben-Hail D., Begas-Shvartz R., Shalev M., Shteinfer-Kuzmine A., Gruzman A., Reina S., De Pinto V., Shoshan-Barmatz V. Novel Compounds Targeting the Mitochondrial Protein VDAC1 Inhibit Apoptosis and Protect against Mitochondrial Dysfunction. J. Biol. Chem. 2016;291:24986–25003. doi: 10.1074/jbc.M116.744284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang E., Mohammed Al-Amily I., Mohammed S., Luan C., Asplund O., Ahmed M., Ye Y., Ben-Hail D., Soni A., Vishnu N., et al. Preserving Insulin Secretion in Diabetes by Inhibiting VDAC1 Overexpression and Surface Translocation in β Cells. Cell Metab. 2019;29:64–77.e6. doi: 10.1016/j.cmet.2018.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wirtz S., Neufert C., Weigmann B., Neurath M.F. Chemically induced mouse models of intestinal inflammation. Nat. Protoc. 2007;2:541–546. doi: 10.1038/nprot.2007.41. [DOI] [PubMed] [Google Scholar]
- 23.Livingstone S., Sorsiah M., Peter M., Eilish D., Margaret H., Morris L., et al. The dextran sulphate sodium (DSS) model of colitis: an overview. Comp. Clin. Pathol. 2010;19:235–239. [Google Scholar]
- 24.Heller S., Penrose H.M., Cable C., Biswas D., Nakhoul H., Baddoo M., Flemington E., Crawford S.E., Savkovic S.D. Reduced mitochondrial activity in colonocytes facilitates AMPKα2-dependent inflammation. FASEB J. 2017;31:2013–2025. doi: 10.1096/fj.201600976R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Araki Y., Sugihara H., Hattori T. In vitro effects of dextran sulfate sodium on a Caco-2 cell line and plausible mechanisms for dextran sulfate sodium-induced colitis. Oncol. Rep. 2006;16:1357–1362. [PubMed] [Google Scholar]
- 26.Crosnier C., Stamataki D., Lewis J. Organizing cell renewal in the intestine: stem cells, signals and combinatorial control. Nat. Rev. Genet. 2006;7:349–359. doi: 10.1038/nrg1840. [DOI] [PubMed] [Google Scholar]
- 27.Renes I.B., Boshuizen J.A., Van Nispen D.J., Bulsing N.P., Büller H.A., Dekker J., Einerhand A.W. Alterations in Muc2 biosynthesis and secretion during dextran sulfate sodium-induced colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2002;282:G382–G389. doi: 10.1152/ajpgi.00229.2001. [DOI] [PubMed] [Google Scholar]
- 28.Poritz L.S., Garver K.I., Green C., Fitzpatrick L., Ruggiero F., Koltun W.A. Loss of the tight junction protein ZO-1 in dextran sulfate sodium induced colitis. J. Surg. Res. 2007;140:12–19. doi: 10.1016/j.jss.2006.07.050. [DOI] [PubMed] [Google Scholar]
- 29.Yan Y., Kolachala V., Dalmasso G., Nguyen H., Laroui H., Sitaraman S.V., Merlin D. Temporal and spatial analysis of clinical and molecular parameters in dextran sodium sulfate induced colitis. PLoS ONE. 2009;4:e6073. doi: 10.1371/journal.pone.0006073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vetuschi A., Latella G., Sferra R., Caprilli R., Gaudio E. Increased proliferation and apoptosis of colonic epithelial cells in dextran sulfate sodium-induced colitis in rats. Dig. Dis. Sci. 2002;47:1447–1457. doi: 10.1023/a:1015931128583. [DOI] [PubMed] [Google Scholar]
- 31.Sipos F., Molnár B., Zágoni T., Berczi L., Tulassay Z. Growth in epithelial cell proliferation and apoptosis correlates specifically to the inflammation activity of inflammatory bowel diseases: ulcerative colitis shows specific p53- and EGFR expression alterations. Dis. Colon Rectum. 2005;48:775–786. doi: 10.1007/s10350-004-0831-5. [DOI] [PubMed] [Google Scholar]
- 32.Marchenko N.D., Zaika A., Moll U.M. Death signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J. Biol. Chem. 2000;275:16202–16212. doi: 10.1074/jbc.275.21.16202. [DOI] [PubMed] [Google Scholar]
- 33.Mihara M., Erster S., Zaika A., Petrenko O., Chittenden T., Pancoska P., Moll U.M. p53 has a direct apoptogenic role at the mitochondria. Mol. Cell. 2003;11:577–590. doi: 10.1016/s1097-2765(03)00050-9. [DOI] [PubMed] [Google Scholar]
- 34.Zhao Y., Chaiswing L., Velez J.M., Batinic-Haberle I., Colburn N.H., Oberley T.D., St Clair D.K. p53 translocation to mitochondria precedes its nuclear translocation and targets mitochondrial oxidative defense protein-manganese superoxide dismutase. Cancer Res. 2005;65:3745–3750. doi: 10.1158/0008-5472.CAN-04-3835. [DOI] [PubMed] [Google Scholar]
- 35.Söderberg O., Gullberg M., Jarvius M., Ridderstråle K., Leuchowius K.J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L.G., Landegren U. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods. 2006;3:995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- 36.Park J.H., Peyrin-Biroulet L., Eisenhut M., Shin J.I. IBD immunopathogenesis: A comprehensive review of inflammatory molecules. Autoimmun. Rev. 2017;16:416–426. doi: 10.1016/j.autrev.2017.02.013. [DOI] [PubMed] [Google Scholar]
- 37.Zaidi D., Wine E. Regulation of Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells (NF-κβ) in Inflammatory Bowel Diseases. Front Pediatr. 2018;6:317. doi: 10.3389/fped.2018.00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feng J., Guo C., Zhu Y., Pang L., Yang Z., Zou Y., Zheng X. Baicalin down regulates the expression of TLR4 and NFkB-p65 in colon tissue in mice with colitis induced by dextran sulfate sodium. Int. J. Clin. Exp. Med. 2014;7:4063–4072. [PMC free article] [PubMed] [Google Scholar]
- 39.Abraham C., Medzhitov R. Interactions between the host innate immune system and microbes in inflammatory bowel disease. Gastroenterology. 2011;140:1729–1737. doi: 10.1053/j.gastro.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Atreya R., Neurath M.F. Involvement of IL-6 in the pathogenesis of inflammatory bowel disease and colon cancer. Clin. Rev. Allergy Immunol. 2005;28:187–196. doi: 10.1385/CRIAI:28:3:187. [DOI] [PubMed] [Google Scholar]
- 41.Kolios G., Valatas V., Ward S.G. Nitric oxide in inflammatory bowel disease: a universal messenger in an unsolved puzzle. Immunology. 2004;113:427–437. doi: 10.1111/j.1365-2567.2004.01984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dou W., Zhang J., Ren G., Ding L., Sun A., Deng C., Wu X., Wei X., Mani S., Wang Z. Mangiferin attenuates the symptoms of dextran sulfate sodium-induced colitis in mice via NF-κB and MAPK signaling inactivation. Int. Immunopharmacol. 2014;23:170–178. doi: 10.1016/j.intimp.2014.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Du X., Chen W., Wang Y., Chen C., Guo L., Ju R., Li J., Zhang D., Zhu L., Ye C. Therapeutic efficacy of carboxyamidotriazole on 2,4,6-trinitrobenzene sulfonic acid-induced colitis model is associated with the inhibition of NLRP3 inflammasome and NF-κB activation. Int. Immunopharmacol. 2017;45:16–25. doi: 10.1016/j.intimp.2017.01.015. [DOI] [PubMed] [Google Scholar]
- 44.Subramanian N., Natarajan K., Clatworthy M.R., Wang Z., Germain R.N. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell. 2013;153:348–361. doi: 10.1016/j.cell.2013.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Park S., Juliana C., Hong S., Datta P., Hwang I., Fernandes-Alnemri T., Yu J.W., Alnemri E.S. The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J. Immunol. 2013;191:4358–4366. doi: 10.4049/jimmunol.1301170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Broz P., Monack D.M. Molecular mechanisms of inflammasome activation during microbial infections. Immunol. Rev. 2011;243:174–190. doi: 10.1111/j.1600-065X.2011.01041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lanis J.M., Kao D.J., Alexeev E.E., Colgan S.P. Tissue metabolism and the inflammatory bowel diseases. J. Mol. Med. (Berl.) 2017;95:905–913. doi: 10.1007/s00109-017-1544-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Manresa M.C., Taylor C.T. Hypoxia Inducible Factor (HIF) Hydroxylases as Regulators of Intestinal Epithelial Barrier Function. Cell. Mol. Gastroenterol. Hepatol. 2017;3:303–315. doi: 10.1016/j.jcmgh.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shah Y.M., Ito S., Morimura K., Chen C., Yim S.H., Haase V.H., Gonzalez F.J. Hypoxia-inducible factor augments experimental colitis through an MIF-dependent inflammatory signaling cascade. Gastroenterology. 2008;134:2036–2048. doi: 10.1053/j.gastro.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mathis D., Shoelson S.E. Immunometabolism: an emerging frontier. Nat. Rev. Immunol. 2011;11:81. doi: 10.1038/nri2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wolf A.J., Reyes C.N., Liang W., Becker C., Shimada K., Wheeler M.L., Cho H.C., Popescu N.I., Coggeshall K.M., Arditi M., Underhill D.M. Hexokinase Is an Innate Immune Receptor for the Detection of Bacterial Peptidoglycan. Cell. 2016;166:624–636. doi: 10.1016/j.cell.2016.05.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eltzschig H.K., Eckle T., Mager A., Küper N., Karcher C., Weissmüller T., Boengler K., Schulz R., Robson S.C., Colgan S.P. ATP release from activated neutrophils occurs via connexin 43 and modulates adenosine-dependent endothelial cell function. Circ. Res. 2006;99:1100–1108. doi: 10.1161/01.RES.0000250174.31269.70. [DOI] [PubMed] [Google Scholar]
- 53.Neves A.R., Castelo-Branco M.T., Figliuolo V.R., Bernardazzi C., Buongusto F., Yoshimoto A., Nanini H.F., Coutinho C.M., Carneiro A.J., Coutinho-Silva R., de Souza H.S. Overexpression of ATP-activated P2X7 receptors in the intestinal mucosa is implicated in the pathogenesis of Crohn’s disease. Inflamm. Bowel Dis. 2014;20:444–457. doi: 10.1097/01.MIB.0000441201.10454.06. [DOI] [PubMed] [Google Scholar]
- 54.Katsanos K.H., Papadakis K.A. Inflammatory Bowel Disease: Updates on Molecular Targets for Biologics. Gut Liver. 2017;11:455–463. doi: 10.5009/gnl16308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roda G., Jharap B., Neeraj N., Colombel J.F. Loss of Response to Anti-TNFs: Definition, Epidemiology, and Management. Clin. Transl. Gastroenterol. 2016;7:e135. doi: 10.1038/ctg.2015.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shoshan-Barmatz V., Ben-Hail D., Admoni L., Krelin Y., Tripathi S.S. The mitochondrial voltage-dependent anion channel 1 in tumor cells. Biochim. Biophys. Acta. 2015;1848(10 Pt B):2547–2575. doi: 10.1016/j.bbamem.2014.10.040. [DOI] [PubMed] [Google Scholar]
- 57.Cuadrado-Tejedor M., Vilariño M., Cabodevilla F., Del Río J., Frechilla D., Pérez-Mediavilla A. Enhanced expression of the voltage-dependent anion channel 1 (VDAC1) in Alzheimer’s disease transgenic mice: an insight into the pathogenic effects of amyloid-β. J. Alzheimers Dis. 2011;23:195–206. doi: 10.3233/JAD-2010-100966. [DOI] [PubMed] [Google Scholar]
- 58.Pittala S., Krelin Y., Kuperman Y., Shoshan-Barmatz V. A Mitochondrial VDAC1-Based Peptide Greatly Suppresses Steatosis and NASH-Associated Pathologies in a Mouse Model. Mol. Ther. 2019;27:1848–1862. doi: 10.1016/j.ymthe.2019.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shoshan-Barmatz V., Krelin Y., Shteinfer-Kuzmine A., Arif T. Voltage-Dependent Anion Channel 1 As an Emerging Drug Target for Novel Anti-Cancer Therapeutics. Front. Oncol. 2017;7:154. doi: 10.3389/fonc.2017.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Swanson K.V., Deng M., Ting J.P. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019;19:477–489. doi: 10.1038/s41577-019-0165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bauernfeind F.G., Horvath G., Stutz A., Alnemri E.S., MacDonald K., Speert D., Fernandes-Alnemri T., Wu J., Monks B.G., Fitzgerald K.A., et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009;183:787–791. doi: 10.4049/jimmunol.0901363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Siegmund B., Lehr H.A., Fantuzzi G., Dinarello C.A. IL-1 beta -converting enzyme (caspase-1) in intestinal inflammation. Proc. Natl. Acad. Sci. USA. 2001;98:13249–13254. doi: 10.1073/pnas.231473998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shimada K., Crother T.R., Karlin J., Dagvadorj J., Chiba N., Chen S., Ramanujan V.K., Wolf A.J., Vergnes L., Ojcius D.M., et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36:401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guan K., Zheng Z., Song T., He X., Xu C., Zhang Y., Ma S., Wang Y., Xu Q., Cao Y., et al. MAVS regulates apoptotic cell death by decreasing K48-linked ubiquitination of voltage-dependent anion channel 1. Mol. Cell. Biol. 2013;33:3137–3149. doi: 10.1128/MCB.00030-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cao S.S., Zimmermann E.M., Chuang B.M., Song B., Nwokoye A., Wilkinson J.E., Eaton K.A., Kaufman R.J. The unfolded protein response and chemical chaperones reduce protein misfolding and colitis in mice. Gastroenterology. 2013;144:989–1000.e6. doi: 10.1053/j.gastro.2013.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pahl H.L., Baeuerle P.A. Endoplasmicreticulum-induced signal transduction and gene expression. Trends Cell Biol. 1997;7:50–55. doi: 10.1016/S0962-8924(96)10050-7. [DOI] [PubMed] [Google Scholar]
- 67.Lee G.S., Subramanian N., Kim A.I., Aksentijevich I., Goldbach-Mansky R., Sacks D.B., Germain R.N., Kastner D.L., Chae J.J. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature. 2012;492:123–127. doi: 10.1038/nature11588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pétrilli V., Papin S., Dostert C., Mayor A., Martinon F., Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007;14:1583–1589. doi: 10.1038/sj.cdd.4402195. [DOI] [PubMed] [Google Scholar]
- 69.Menu P., Mayor A., Zhou R., Tardivel A., Ichijo H., Mori K., Tschopp J. ER stress activates the NLRP3 inflammasome via an UPR-independent pathway. Cell Death Dis. 2012;3:e261. doi: 10.1038/cddis.2011.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.