

Abstract
CAR T cell therapy has shown great promise for the treatment of B cell malignancies. However, antigen-negative escape variants often cause disease relapse, necessitating the development of multi-antigen-targeting approaches. We propose that a T cell receptor (TCR)-based strategy would increase the number of potential antigenic targets, as peptides from both intracellular and extracellular proteins can be recognized. Here, we aimed to isolate a broad range of promising TCRs targeting multiple antigens for treatment of B cell malignancies. As a first step, 28 target genes for B cell malignancies were selected based on gene expression profiles. Twenty target peptides presented in human leukocyte antigen (HLA)-A∗01:01, -A∗24:02, -B∗08:01, or -B∗35:01 were identified from the immunopeptidome of B cell malignancies and used to form peptide-HLA (pHLA)-tetramers for T cell isolation. Target-peptide-specific CD8 T cells were isolated from HLA-mismatched healthy donors and subjected to a stringent stepwise selection procedure to ensure potency and eliminate cross-reactivity. In total, five T cell clones specific for FCRL5 in HLA-A∗01:01, VPREB3 in HLA-A∗24:02, and BOB1 in HLA-B∗35:01 recognized B cell malignancies. For all three specificities, TCR gene transfer into CD8 T cells resulted in cytokine production and efficient killing of multiple B cell malignancies. In conclusion, using this systematic approach we successfully identified three promising TCRs for T cell therapy against B cell malignancies.
Keywords: TCR, T cell therapy, gene therapy, B cell malignancies, multiple myeloma, VPREB3, FCRL5, BOB1
Graphical abstract
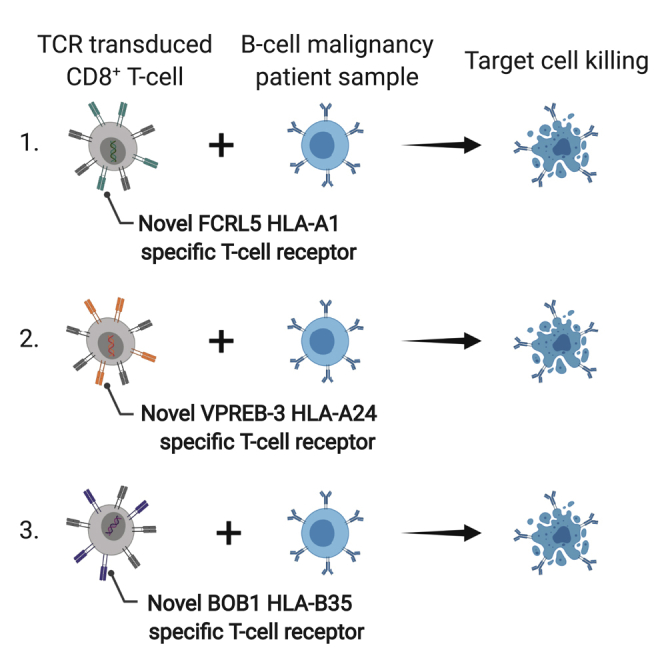
TCR gene therapy is an attractive approach to complement CAR T cell therapies and generate multi-antigen-targeting treatments for B cell malignancies. Following identification of target genes and epitopes Meeuwsen et al. identified three novel TCRs specific for FCRL5, VPREB3, and BOB1. T cells modified with these TCRs specifically lyse patient-derived B cell malignancies.
Introduction
Adoptive T cell therapy for the treatment of B cell malignancies has shown great promise over the past decade. CD19-targeting chimeric antigen receptor (CAR) T cell therapies have induced complete remission (CR) in 70%–97% of patients with relapsed or refractory acute lymphoblastic leukemia (ALL).1,2 In aggressive relapsed or refractory diffuse large B cell lymphoma (DLBCL) and transformed follicular lymphoma, CR rates after CD19 CAR therapy are approximately 50%.3,4 In responding patients, antigen-loss escape variants are frequently observed, hampering long-term relapse-free survival.5,6 Multi-antigen-targeting T cell therapy may reduce outgrowth of antigen-escape variants and enhance long-term remission rates of patients, as simultaneous loss of multiple antigens is more unlikely.7,8 However, in order to generate multi-antigen-targeting T cell therapies, additional potent and safe T cell therapies need to be developed.
Target antigens for immunotherapy of cancer should be expressed by the malignant cells, while expression in essential healthy tissues must be absent.9,10 B cells are considered a non-essential tissue; therefore, antigens expressed by the healthy B cell lineage could be safe targets for the treatment of B cell malignancies. This was confirmed upon CD19 CAR T cell therapy, which resulted in depletion of healthy B cells, which was clinically managed by immunoglobulin administration.11
CAR T cells target epitopes of proteins located on the cell surface.12 In contrast to CARs, T cell receptors (TCRs) recognize peptides derived from proteins independent of cellular localization that are presented in human leukocyte antigen (HLA) molecules on the cell surface.13 Therefore, TCR-based therapy theoretically allows targeting of all proteins, including those involved in essential intracellular pathways. So far, limited availability of safe and high-affinity TCRs has hampered clinical progress of TCR gene therapy. Furthermore, a large collection of high-affinity TCRs targeting different peptide-HLA (pHLA) complexes would be required to allow TCR gene therapy for all patients.
T cells with high-affinity TCRs for self-peptides presented in self-HLA alleles are deleted from the T cell repertoire to prevent autoimmunity. Therefore, high-avidity T cells targeting non-mutated peptides cannot be isolated from HLA-matched individuals.14,15 In contrast, high-avidity T cells targeting non-mutated peptides have successfully been isolated from the T cell repertoire of HLA-mismatched (allogeneic) healthy donors.15, 16, 17, 18, 19, 20, 21, 22, 23 Previous efforts have mainly focused on identification of TCRs specific for peptides presented in HLA-A∗02:01 and HLA-B∗07:02.15, 16, 17, 18, 19,24 Here, to allow application of TCR gene therapy for individuals with other HLA genotypes, we aimed to identify TCRs specific for peptides presented in HLA alleles A∗01:01 (A1), A∗24:02 (A24), B∗08:01 (B8), and B∗35:01 (B35). In this study, we performed a systematic approach for simultaneous identification of clinically relevant TCRs targeting multiple epitopes. We started by identifying genes with expression restricted to B cell malignancies and the healthy B cell lineage. Target peptides derived from these genes presented in target HLA alleles HLA-A1, -A24, -B8, or -B35 were selected from the immunopeptidome of B cell malignancies. To allow identification of high-avidity T cells targeting these epitopes, T cells were isolated from the allogeneic (allo) T cell repertoire using HLA-mismatched healthy donor peripheral blood mononuclear cells (PBMCs). Multiple efficacy and safety screenings resulted in the selection of clinically relevant TCRs recognizing antigens in HLA-A1, HLA-A24, and HLA-B35 presented on B cell malignancies. Finally, upon TCR gene transfer, selected TCRs induced specific lysis of different B cell malignancies, including a multiple myeloma (MM) cell line and patient-derived ALL, chronic lymphocytic leukemia (CLL), mantle cell lymphoma (MCL), and hairy cell leukemia (HCL).
Results
Target gene selection for treatment of B cell malignancies
To ensure targeting of malignant B cells while limiting the risk of toxicity, data obtained from an in-house-generated microarray database were used to identify genes expressed in B cell malignancies with expression restricted to the B cell lineage. This database was previously generated and validated for selection of target antigens valuable for immunotherapy of hematological malignancies with lineage-restricted expression.25 Gene expression was measured by probe fluorescence represented as mean fluorescence intensity (MFI), with a lower detection limit of MFI = 50. Potent T cell recognition occurs when target antigens are highly expressed in B cell malignancies, and, therefore, a threshold of gene expression MFI ≥ 250 in primary ALL, CLL, or MM was set for each probe (Figure S1). To prevent recognition of healthy tissues (excluding B cells), a threshold was set at MFI = 100 to define very low expression, and all genes expressed at MFI > 100 in healthy tissues were excluded. In total, 28 genes highly expressed in primary B cell malignancies with very low or no expression in healthy tissues were selected as target genes (Figure 1). Of these genes, 14 were highly expressed (MFI ≥ 250) in either ALL, CLL, or MM, while 13 genes were expressed in two types and the BOB1 encoding gene POU2AF1 was expressed in all three types of primary B cell malignancies.
Figure 1.

Gene expression of target genes selected for treatment of B cell malignancies
Gene expression was retrieved from an Illumina HT12.0 microarray dataset.25 Per gene, the average MFI of samples is shown for different types of patient-derived (primary) B cell malignancies, BMMCs, B cells (CD19pos), ALL cell lines, and EBV-LCLs. The final column represents the highest gene expression as measured in any healthy tissue other than B cells, PBMCs, and BMMCs as included in the microarray dataset and shown in Figure S1. Genes are clustered according to the type of B cell malignancy in which they are expressed (MFI ≥ 250). Abbreviations: MFI, mean fluorescence intensity; ALL, acute lymphoblastic leukemia; CLL, chronic lymphocytic leukemia; MM, multiple myeloma; BMMC, bone marrow mononuclear cells; EBV-LCL; Epstein-Barr virus-transformed lymphoblastoid cell lines.
Identification of candidate epitopes from the immunopeptidome of malignant B cells
To identify target peptides for TCR-based recognition of B cell malignancy antigens, the HLA-presented peptide repertoire of B cell malignancies was determined. Patient material was obtained from nine ALL patients, two CLL patients, one HCL patient, and one follicular lymphoma patient expressing HLA-A1, -A24, -B8, and/or -B35 (Table S1). Since materials from MM patients lacked sufficient cell numbers for peptide elution, the MM cell line UM9 was included instead. Cells from each malignant cell population were lysed, and peptides were eluted before subsequent separation by high-performance liquid chromatography (HPLC) and mass spectrometry analysis. Per sample, between 539 and 82,504 unique peptides with an ion score (IS) ≥ 20 were identified (Table S1). The ion score indicates the confidence of correctly matching the observed mass spectrum of a peptide to the reference database spectrum. Therefore, a cutoff of ion score ≥ 20 was used to ensure a high chance of correct peptide identification. Eluted peptides were matched to HLA alleles by combining predicted HLA binding (netMHC3.4) with the HLA typing of the material from which the peptides originated. Twenty peptides were identified to be derived from one of the target genes and presented by target HLA alleles HLA-A1, -A24, -B8, or -B35 (Table 1; Table S2). Peptide sequences of these peptides were validated by comparing mass spectra of eluted peptides to spectra of synthetically generated peptides (Figure S2). After peptide verification, target HLA binding was confirmed by stable pHLA-monomer refolding (data not shown). From these monomers, phycoerythrin (PE)-labeled pHLA-tetramers were generated and used for T cell isolation.
Table 1.
Target gene-derived peptides presented in HLA-A1, -A24, -B8, or -B35 by B cell malignancies
| Protein | HLA | Assigned peptide no. | Sequence | Affinity (nM)a | SB/WBa |
|---|---|---|---|---|---|
| RALGPS2 | A1 | p242 | LTDSEKGNSY | 24 | SB |
| FCRL5 | A1 | p243 | LTEGHSGNYY | 15 | SB |
| FCRL5 | A1 | p263 | TTENSGNYY | 9 | SB |
| MIST | A1 | p248 | ESEYADTHY | 102 | WB |
| TLR10 | A1 | p265 | YLDHNSFDY | 8 | SB |
| IGJ | A1 | p268 | YTAVVPLVY | 7 | SB |
| BLK | A24 | p246 | AYIERMNSI | 75 | WB |
| VPREB3 | A24 | p269 | YYCSVGYGF | 37 | SB |
| RALGPS2 | A24 | p258 | QYIEELQKF | 78 | WB |
| TLR10 | B8 | p247 | ELFKRTIQL | 54 | WB |
| TLR10 | B8 | p256 | LPHLKTLIL | 26 | SB |
| IGLL1 | B8 | p251 | HGLLRPTAA | 588 | <WBb |
| KLHL14 | B8 | p239 | DMNTKRAIHTL | 396 | WB |
| FCRL2 | B8 | p252 | IVKIKVQEL | 252 | WB |
| FAM129C | B8 | p255 | LPALRAQTL | 32 | SB |
| FAM129C | B8 | p271 | YLRLLDAL | 726 | <WBb |
| RALGPS2 | B8 | p261 | TLKIRAEVL | 43 | SB |
| TNFRSF13B | B35 | p259 | SADQVALVY | 11 | SB |
| BOB1 | B35 | p233 | APAPTAVVL | 196 | WB |
| BOB1 | B35 | p236 | LPHQPLATY | 6 | SB |
Affinity (nM) for the respecitive HLA molecule and predicted strong binding (SB) or weak binding (WB) according to NetMHC 3.4.
HLA-B8 most likely origin of peptides, highest predicted binding.
Isolation and selection of T cell clones with on-target functional specificity
To isolate high-avidity T cells recognizing target peptides, pHLA-tetramers were incubated with PBMCs from healthy donors negative for the target HLA alleles (Table S3). pHLA-tetramer-bound cells were enriched, and pHLA-tetramerpos CD8pos T cells were single-cell sorted and clonally expanded. In total, 12,336 (range, 192–2,640) T cells were sorted from 13 healthy donors (Table 2). On average, 59% (mean; range, 14%–83%) of the T cell clones expanded. Expanded T cell clones were initially screened for on-target functional specificity. The HLA-negative myeloid leukemia cell line K562 was transduced (Td) with target HLA alleles and mixed into two pools containing HLA-A1 and HLA-A24 or HLA-B8 and HLA-B35. All target peptides were mixed and loaded onto K562 before incubation with the T cell clones (Table 1). As measured by cytokine production, unloaded K562 recognition was frequently observed revealing off-target recognition, and these clones were discarded to prevent off-target toxicity (Table 2). Additionally, 34%–98% of expanded T cell clones were not reactive and were discarded due to lack of target peptide recognition. On-target recognition was defined when only peptide-mix-loaded K562 cells were recognized. From these 13 donors, 23 T cell clones recognized only HLA-A1/A24pos K562 cells loaded with the target peptide mix (Table 2). In addition, 23 T cell clones only recognized target-peptide-loaded HLA-B8/B35pos K562 cells (Table 2). In summary, 46 T cell clones demonstrated specific, on-target recognition and were selected for further screening.
Table 2.
Identification of target peptide-specific T cell clones from tetramerpos CD8pos sorted T cells using healthy donor PBMCs
| Donora | T cells sorted (n)b | Clones exp. (n)c | Reactivity of T cell clonesd |
Peptide specificity of T cell clonese |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Not reactive (n) | Off-target (n) | HLA-A1/A24 (n) | HLA-B8/B35 (n) | p236 BOB1 B35 | p243 FCRL5 A1 | p247 TLR10 B8 | p248 MIST A1 | p252 FCRL2 B8 | p256 TLR10 B8 | p259 TNFRSF13B B35 | p261 RALGPS2 B8 | p263 FCRL5 A1 | p265 TLR10 A1 | p269 VPREB3 A24 | |||
| 3 | 288 | 239 | 233 | 6 | 0 | 0 | |||||||||||
| 4 | 192 | 156 | 101 | 54 | 1 | 0 | 1x | ||||||||||
| 5 | 192 | 129 | 44 | 83 | 0 | 2 | 2x | ||||||||||
| 6 | 528 | 433 | 235 | 180 | 6 | 12 | 6x | 1x | 2x | 3x | 1x | 1x | 3x | 1x | |||
| 7 | 288 | 207 | 181 | 26 | 0 | 0 | |||||||||||
| 8 | 384 | 311 | 289 | 22 | 0 | 0 | |||||||||||
| 12 | 864 | 639 | 320 | 314 | 5 | 0 | 1x | 2x | 2x | ||||||||
| 13 | 1,728 | 1,002 | 891 | 108 | 2 | 1 | 1x | 2x | |||||||||
| 14 | 1,296 | 384 | 330 | 53 | 0 | 1 | 1x | ||||||||||
| 15 | 864 | 449 | 399 | 49 | 0 | 1 | 1x | ||||||||||
| 16 | 1,920 | 1,018 | 907 | 105 | 3 | 3 | 1x | 2x | 1x | 2x | |||||||
| 17 | 2,640 | 360 | 351 | 9 | 0 | 0 | |||||||||||
| 18 | 1,152 | 192 | 143 | 40 | 6 | 3 | 2x | 2x | 1x | 2x | 2x | ||||||
| Total | 12,336 | 5,519 | 4,424 | 1,049 | 23 | 23 | 6x | 2x | 1x | 7x | 5x | 3x | 5x | 3x | 2x | 5x | 7x |
Arbitrary donor numbers assigned to healthy donors from which PBMCs were obtained, in accordance with Table S3.
Number of pHLA-tetramerpos CD8pos T cells single-cell sorted from each donor.
Number of single-cell-sorted T cells that expanded and were tested for peptide specificity.
Expanded T cell clones were stimulated with a 1:1 mixture of HLA-A1 and HLA-A24 Td K562 cells or HLA-B8 and B35 Td K562 cells, unloaded or loaded with a mixture of all target peptides (100 nM). Reactivity was assessed after overnight co-culture based on IFN-γ ELISA; for T cell clones not producing IFN-γ, GM-CSF was used instead. Recognition was categorized as: not reactive, off-target recognition, HLA-A1/A24-restricted peptide-specific recognition (HLA-A1/A24), and HLA-B8/B35-restricted peptide-specific recognition (HLA-B8/B35).
The number of peptide-specific T cell clones per donor is indicated. Specificity was determined as demonstrated in Figure 2. No T cell clones were identified for peptides not included in the table.
Defining T cell specificity and confirming recognition of endogenously processed target antigen
To determine the peptide specificity of each selected T cell clone, target peptides were assorted into combinatorial mixes revealing unique recognition patterns for all specificities. Mixes were loaded onto HLA-A1/A24pos or HLA-B8/B35pos K562 cells accordingly and incubated with each T cell clone. For example, T cell clones 1F3.4 and 6B10.12 recognized two peptide mixes loaded on HLA-A1/A24pos K562 cells (Figure 2A). This recognition pattern revealed specificity for peptide 243 (p243) derived from FCRL5 presented in the context of HLA-A1 (Table 1). Following this approach, all 46 T cell clones were tested, revealing 11 different peptide specificities (Table 2). Since recognition of endogenously processed and presented antigen correlates with T cell avidity and recognition of malignant cells,26 T cell clones were also screened for recognition of K562 cells Td with target genes and target HLA alleles. T cell clones demonstrating no or weak recognition of endogenously processed and presented antigen were discarded. In Figure 2, 23 T cell clones recognizing eight different B cell-specific peptides that exhibited strong endogenous recognition are shown. These 23 T cell clones were selected for further investigation of clinical relevance.
Figure 2.

Peptide specificity and target gene recognition by selected T cell clones
T cell clones were stimulated with the appropriate HLA-expressing K562 cells in an effector:target (E:T) ratio of 1:6 loaded with combinatorial combinations of target peptides to determine peptide specificity (upper part) and K562 cells Td with target gene and HLA (bottom part) to determine recognition of endogenously processed and presented peptide. IFN-γ (in black) or GM-CSF (in gray) production by T cell clones categorized as high endogenous recognition. GM-CSF production is shown for clones that do not produce IFN-γ. T cell clones isolated from different donors were tested in separate experiments. (A) T cell clones recognizing HLA-A1 or -A24 presented target peptides. (B) T cell clones recognizing HLA-B8 or -B35 presented target peptides; peptides 233 and 236 were not part of combinatorial mixes. For clone 1C5.6 and 4H5.6, specificity for p236 was determined by pHLA-tetramer stain (data not shown). Abbreviations: n.p., not performed
Recognition of B cell malignancies identifies T cell clones that are candidates for TCR sequencing
To identify candidates for TCR sequencing, the 23 selected T cell clones were screened for recognition of B cell malignancies. T cell clones were tested for cytokine production upon stimulation with patient-derived CLL material, ALL cell lines, or MM cell lines. Per specificity, the appropriate cell types were selected based on gene expression profiles (Figure 1). Target gene expression in materials used was determined by quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR) (Figure S3). Despite recognition of endogenously processed antigen in target gene Td k562 cells, MIST HLA-A1-, FCRL2 HLA-B8-, RALGPS2 HLA-B8-, and TNFRSF13B HLA-B35-restricted T cell clones failed to produce cytokine upon co-culture with malignant B cells (data not shown). FCRL5 HLA-A1-specific T cell clones 1F3.4 and 7E8.12, recognizing p243 and p263, respectively, were co-cultured with FCRL5-expressing patient-derived CLL samples. T cell clone 6B10.12 did not expand and therefore could not be included. Recognition of HLA-A1pos CLL samples was observed for clone 1F3.4 (Figure 3A) but not for clone 7E8.12 (data not shown). Allo-HLA T cell clones were included as positive controls for stimulatory capacity and HLA expression of the target cells. Furthermore, 3 out of 6 VPREB3 HLA-A24-restricted T cell clones (6A2.18, 6E2.18, and 10G3.16) efficiently recognized all VPREB3-expressing HLA-A24pos ALL cell lines, whereas HLA-A24neg ALL cell lines were not recognized (Figure 3B). In addition, BOB1 HLA-B35-restricted T cell clone 1C5.6 demonstrated efficient recognition of all POU2AF1 (BOB1)-expressing HLA-B35pos ALL and MM cell lines tested (Figure 3C). BOB1 HLA-B35-restricted T cell clone 4H5.6 did not recognize these B cell malignancies (data not shown). Of note, malignant cell recognition by the VPREB3 HLA-A24 and BOB1 HLA-B35 T cell clones correlated with a high avidity as measured by peptide titration (Figure S4). To summarize, 5 out of 22 tested T cell clones recognized B cell malignancies and were therefore promising candidates for TCR gene therapy. These T cell clones were specific for FCRL5 in HLA-A1, VPREB3 in HLA-A24, and BOB1 in HLA-B35.
Figure 3.

Recognition of malignant B cells by selected T cell clones
IFN-γ production by T cell clones after overnight stimulation with various target cells. Values and error bars represent mean and standard deviations of technical duplicates. Target cells were positive (+), negative (−), or transduced (Td) with target HLA. Target gene expression was measured by qRT-PCR and is depicted below targets as expression relative to housekeeping genes (HKG set to 1). Allo HLA T cell clones were included as positive controls for HLA expression. (A) FCRL5 HLA-A1-specific T cell clone stimulated with patient-derived CLL samples, in an E:T ratio of 1:20 to correct for cell size and negative control K562 cells in E:T ratio 1:6. T cell clone 6B10.12 could not be tested due to lack of expansion. (B) VPREB3 HLA-A24 clones stimulated with different ALL cell lines and K562 cells in E:T ratio 1:6. (C) BOB1 HLA-B35 clone stimulated with different ALL cell lines, MM cell lines UM9 and U266, and K562 cells in E:T ratio 1:6.
Safety screenings reveal high on-target specificity of selected T cell clones
To study the specificity of the 5 selected T cell clones, safety profiles were investigated. Cross-reactivity with other HLA alleles was assessed using an Epstein-Barr virus-transformed lymphoblastoid cell line (EBV-LCL) panel expressing all HLA-I alleles with an allele frequency > 1% in the white population (Figure 4; Table S4).27 Additionally, cross-reactivity with other peptides presented in target HLA alleles was determined by stimulating with HLA-A1 and -B8 or HLA-A24 and -B35 Td cell lines of non-B cell origins (Figure 4). The respective allo HLA-A1, -A24, and -B35 T cell clones recognized the HLA Td cell lines (Figure S5), demonstrating that sufficient HLA was expressed to allow T cell recognition. T cell clones 1F3.4 (FCRL5 HLA-A1), 6A2.18 (VPREB3 HLA-A24), 10G3.16 (VPREB3 HLA-A24), and 1C5.6 (BOB1 HLA-B35) did not demonstrate cross-reactivity with any of the target cells included in the screenings (Figures 4A–4C). However, T cell clone 6E2.18 (VPREB3 HLA-A24) produced IFN-γ when stimulated with EBV-LCLs from donor URN (EBV-LCL URN) and upon stimulation with HLA-A24/B35 Td Caski cells (Figure 4B). This was a result of cross-reactivity with two possible pHLA complexes. Recognition of EBV-LCL URN resulted from cross-reactivity with a peptide presented in HLA-B∗08:01 or -B∗50:01, since these HLA alleles were expressed by EBV-LCL URN but not by other EBV-LCLs in the panel. Recognition of HLA-A24/B35 Td Caski cells was caused by cross-reactivity with a peptide in HLA-A24 or -B35, since untransduced Caski cells were not recognized (data not shown). In summary, most T cell clones targeting FCRL5 in HLA-A1, VPREB3 in HLA-A24, or BOB1 in HLA-B35 revealed promising safety profiles for clinical application in TCR gene therapy of B cell malignancies.
Figure 4.

Safety screenings for selected T cell clones
IFN-γ production by T cells measured by ELISA after overnight co-culture; technical duplicates are depicted. T cell clones stimulated with an EBV-LCL panel not expressing target HLA alleles (left panels). Target gene and HLA Td K562 cells were included as positive control for T cell function. T cells were stimulated in E:T ratio 1:6. T cell clones stimulated with NBS and VH10 fibroblast cell lines, HeLa, and Caski cervix carcinoma cell lines, EGI-1 bile duct carcinoma cell lines, HCT116 colon carcinoma cell line, MZ1851 and MZ1257 renal cell carcinoma cell lines, SJNB-8 neuroblastoma cell line, A549 and H292 lung carcinoma cell lines Td with HLA-A1 and HLA-B8 (+HLA-A1/B8) or HLA-A24 and HLA-B35 (+HLA-A24/B35) (right panels). Lack of FCRL5, VPREB3, and POU2AF1 (BOB1) expression in these cell lines was confirmed by qRT-PCR (data not shown). (A) Safety screenings of FCRL5 HLA-A1-specific T cell clones. (B) Safety screenings of VPREB3 HLA-A24 T cell clones. (C) Safety screenings of the BOB1 HLA-B35 T cell clone.
TCR gene transfer to CD8 T cells reveals promising candidates for TCR gene therapy of B cell malignancies
To study the potential for clinical application in TCR gene therapy, TCRs of the five selected T cell clones were sequenced and retrovirally transferred to healthy donor CD8 T cells. Additionally, the TCR of T cell clone 6B10.12 (FCRL5 HLA-A1), which was not screened for recognition of B cell malignancies due to lack of in vitro expansion, was included. All six identified TCRs, including the TCRs from T cell clones 6A2.18 and 6E2.18, which were isolated from the same donor, had unique TCR sequences (data not shown). Moreover, all TCRs were functional upon transfer into CD8 T cells as demonstrated by pHLA-tetramer binding. The intensity of the staining, however, was weaker in TCR Td CD8 T cells than in the parental T cell clones (Figure 5A). To determine clinical relevance of identified TCRs, cytokine production by TCR Td T cells after co-culture with primary B cell malignancies of multiple origins was assessed. HCL, MCL, ALL, and CLL patient-derived samples were included depending on availability of material expressing target HLA in the LUMC biobank. TMD8 and U266 were used as representatives for DLBCL and MM, respectively, as primary malignant cells were not available for these diseases.
Figure 5.

Target recognition by TCR Td CD8 T cells
(A) TCR Td CD8 T cells and parental T cell clones stained with pHLA-tetramer-PE, CD8, and mTCR. TCR Td T cells were enriched for mTCR by MACS and gated on CD8 and mTCR expression. Parental T cell clones were gated on CD8. MFIs of tetramer stains are depicted. Untransduced T cells were included as negative controls for tetramer staining. Left panel: FCRL5 HLA-A1-specific T cells; middle panel: VPREB3 HLA-A24-specific T cells; right panel: BOB1 HLA-B35-specific T cells. (B) IFN-γ production after overnight co-culture of CD8 T cells Td with identified TCRs, CMV (pp65-NLV-HLA-A2) TCR as negative control, and allo-HLA T cell clones as positive controls. Target cells were patient-derived B cell malignancy samples, diffuse large B cell lymphoma cell line TMD8, and MM cell lines U266, HLA Td K562 cells with (100 nM) or without target peptide. Target cells were positive (+), negative (−), or Td with target HLA. Target gene expression was measured by qRT-PCR and is depicted below targets as expression relative to housekeeping genes (HKG set to 1). To correct for cell size, different E:T ratios were used: E:T 1:12 for patient-derived B cell malignancies, E:T 1:6 for cell lines. Graphs are separated based on specificities; upper panels: FCRL5 HLA-A1 TCR T cells; middle panels: VPREB3 HLA-A24 TCR T cells; bottom panels: BOB1 HLA-B35 TCR T cells. Data are representative of two independent experiments; values and error bars represent mean and standard deviations of technical duplicates. Abbreviations: HCL, hairy cell leukemia; MCL, mantle cell lymphoma.
The results depicted in Figure 5B demonstrate that FCRL5 HLA-A1-specific TCR 6B10.12 Td T cells efficiently recognized FCRL5 expressing HLA-A1pos patient-derived HCL, MCL, and CLL samples, as well as HLA-A1 Td DLBCL cell line TMD8. Reactivity by TCR 6B10.12 Td T cells was more efficient than by TCR 1F3.4 Td T cells (Figure 5B), which corresponded with the difference observed in tetramer binding (Figure 5A).
VPREB3 HLA-A24-specific TCR 6A2.18 Td T cells consistently outperformed TCRs 6E2.18 and 10G3.16 for recognition of HLA-A24pos patient-derived ALL and CLL samples, DLBCL cell line TMD8, and MM cell line U266 (Figure 5B), although TCR 6E2.18 Td T cells showed higher tetramer binding than TCR 6A2.18 Td T cells (Figure 5A).
Finally, BOB1 HLA-B35-restricted TCR 1C5.6 Td T cells efficiently recognized patient-derived HLA-B35pos ALL, CLL, and MCL samples as well as HLA-B35 Td MM and DLBCL cell lines (Figure 5B). Based on these results, TCRs 6B10.12 (FCRL5 HLA-A1), 6A2.18 (VPREB3 HLA-A24), and 1C5.6 (BOB1 HLA-B35) were selected to be the most potent TCRs identified. Since TCR 6B10.12 was the most potent FCRL5 HLA-A1-specific TCR, but safety of the parental T cell clones had not been examined due to lack of in vitro expansion, safety screenings were performed using endogenous TCRαβ knockout (KO) CD8 T cells Td with TCR 6B10.12 (Figure S6). Here, no cross-reactivity for TCR 6B10.12 was identified; therefore, this TCR and TCRs 6A2.18 (VPREB3 HLA-A24) and 1C5.6 (BOB1 HLA-B35) are promising candidates to further investigate relevance for TCR gene therapy of B cell malignancies.
High-avidity TCRs show strong promise for therapy of B cell malignancies
To gain better insight in the value of the identified TCRs for TCR gene therapy of B cell malignancies, TCR functionality was investigated further. Peptide titration experiments demonstrated that the identified TCRs are of high avidity for the target pHLA complexes, requiring between 364 and 1,068 pg/mL peptide for a half-maximum response (Figure S7). Despite high-avidity interactions between the selected TCRs and target pHLA complexes, TCR 6B10.12 (FCRL5 HLA-A1) and 1C5.6 (BOB1 HLA-B35) are dependent on expression of the CD8 co-receptor. Transfer into CD4 T cells induced pHLA-tetramer binding, but no IFN-γ was produced upon stimulation with antigen Td K562 cells (Figure S8). In contrast, TCR 6A2.18 (VPREB3 HLA-A24) Td CD4 T cells produced cytokine upon stimulation with antigen Td K562 cells in addition to pHLA-tetramer binding (Figure S8). However, stimulation with patient-derived B cell malignancy materials demonstrated reduced functionality of TCR 6A2.18 Td CD4 T cells compared to CD8 T cells (data not shown); therefore, presence of the CD8 co-receptor was still beneficial for TCR 6A2.18 Td T cells.
To investigate the anti-tumor reactivity of the identified TCRs, in vitro cytotoxicity assays were performed with patient-derived HCL, CLL, MCL, and ALL samples, diffuse large B cell lymphoma cell line TMD8, and MM cell line U266, demonstrating that TCR 6B10.12 (FCRL5 HLA-A1) Td T cells induced specific lysis of HCL, CLL, MCL, and DLBCL (Figure 6A). Additionally, TCR 6A2.18 Td T cells (VPREB3 HLA-A24) mediated potent lysis of patient-derived ALL, CLL, DLBCL, and MM (Figure 6B), and TCR 1C5.6 Td T cells (BOB1 HLA-B35) efficiently killed ALL, CLL, MCL, DLBCL, and MM (Figure 6C). No lysis of antigen-negative target HLA Td K562 cells (Figure 6) or fibroblasts and keratinocytes endogenously expressing the HLA restriction alleles was observed (Figure S9), demonstrating that no general de novo cross-reactivities were induced by transfer of the three selected TCRs into donor-derived CD8 T cells.
Figure 6.

Antigen-specific killing of B cell malignancies by TCR Td CD8 T cells
Killing by CD8 T cells Td with selected TCRs (in pink), CMV (pp65-NLV-HLA-A2) TCR Td CD8 T cells (in black) as negative control, and allo-HLA-A1, -A24 or -B35 T cell clones (in gray) as positive controls. Target cells were patient-derived HCL, CLL, MCL, and ALL; diffuse large B cell lymphoma cell line TMD8; MM cell line U266; and antigen-negative target HLA Td K562 cells. Target cells endogenously expressed target HLA (HLApos) or were Td with target HLA alleles (+HLA). Killing was measured by 51CR release assay after 6-h co-culture in different E:T ratios. Values and error bars represent mean and standard deviations of technical triplicates. Experiments are representative of two independent experiments. (A) Killing by FCRL5 HLA-A1-specific TCR 6B10.12 Td T cells. (B) Killing by VPREB3 HLA-A24-specific TCR 6A2.18 Td T cells. (C) Killing by BOB1 HLA-B35-specific TCR 1C5.6 Td T cells.
Finally, we investigated the in vivo killing capacity of TCR 1C5.6 (BOB1 HLA-B35) Td CD8 T cells in a previously established xenograft model for treatment of established MM.16 NSG mice were inoculated with BOB1-expressing, HLA-B35 Td MM cell line U266. Upon treatment with BOB1 HLA-B35-restricted TCR 1C5.6 Td CD8 T cells, a strong anti-tumor effect was observed (Figure S10). Tumors in TCR 1C5.6-treated mice reached their minimal size 6 days after T cell infusion, when the mean tumor burden was 148-fold lower in 1C5.6 TCR-treated mice compared to control TCR-treated mice. Despite near-complete tumor eradication, U266 regrows after day 6 post T cells likely due to absence of the required human cytokine environment.
In conclusion, TCR 6B10.12 (FCRL5 HLA-A1), 6A2.18 (VPREB3 HLA-A24), and 1C5.6 (BOB1 HLA-B35) are promising TCRs for further clinical development for application in TCR gene therapy of B cell malignancies.
Discussion
In this study we aimed to extend the options for cellular immunotherapy of B cell malignancies. We performed a broad and stepwise approach to identify multiple TCRs specific for different target genes and HLA alleles. As a first step, target genes with restricted expression in B cell malignancies and the B cell lineage were selected. Twenty HLA-A1, -A24, -B8, or -B35 binding peptides derived from these genes were identified by mass-spectrometry-based immunopeptidomics. T cell clones were isolated for 11 of these 20 peptides, and multiple efficacy and safety screenings revealed the 6 most promising T cell clones for which TCRs were sequenced. Despite confirmed presentation of all selected epitopes, only the most potent FCRL5-, VPREB3-, and BOB1-specific T cell clones were of sufficient avidity to recognize antigen levels naturally expressed by B cell malignancies. Upon transfer to CD8 T cells, three TCRs directed against peptides derived from FCRL5, VPREB3, and BOB1 demonstrated potent lytic activity against multiple B cell malignancies.
The first selected TCR was specific for a peptide derived from FCRL5 presented in HLA-A1. FCRL5 is a cell surface receptor involved in regulation of B cell receptor signaling through binding to IgG.28 In this study, we aimed to identify TCRs targeting selected B cell malignancy antigens regardless of their cellular localization. However, the cell surface localization of FCRL5 would also permit CAR-mediated targeting, which could additionally be exploited as a therapeutic strategy. In healthy B cells, FCRL5 is expressed by naive and memory peripheral blood B cells as well as on plasma cells, while expression in germinal center B cells is low.29 FCRL5 is expressed in almost all cases of CLL and has previously been proposed as an immunotherapeutic target for CLL.29 Similarly, in our study, FCRL5 expression was observed in CLL and was additionally expressed in HCL, MCL, and DLBCL.
The second TCR targets a peptide derived from VPREB3 in HLA-A24. The exact function of VPREB3 is still unknown, but VPREB3 has been suggested to play a role in the intracellular assembly of the pre-B cell receptor and thus in B cell development. In healthy B cells, VPREB3 is expressed in precursor B cells as well as in a subset of germinal center B cells.30 VPREB3 was also reported to be expressed in all cases of Burkitt lymphoma and in a subset DLBCL. In DLBCL, VPREB3 expression co-occurred with c-MYC abnormalities and was therefore associated with an aggressive phenotype.30 In this study, we additionally observed VPREB3 expression and TCR targeting of ALL and CLL. Furthermore, despite low VPREB3 expression, DLBCL cell line TMD8 and MM cell line U266 were lysed by TCR Td T cells, indicating broad applicability for VPREB3-restricted TCRs.
The third TCR targets a peptide in HLA-B35 derived from POU2AF1, which encodes the BOB1 protein. BOB1 is an intracellular transcription factor regulating both B cell development amd function and is broadly expressed throughout the healthy B cell lineage.31,32 Additionally, POU2AF1 (BOB1) is expressed in all types of B cell malignancies and has been suggested to play a role in survival of malignant cells.33,34 We previously identified a TCR targeting a BOB1-derived peptide in HLA-B∗07:02, which demonstrated specific lysis of B cell malignancies of multiple origins, including MM.16 Here we similarly observed potent lysis of B cell malignancies of multiple origins and strong in vivo anti-tumor efficacy by CD8 T cells Td with the HLA-B35-restricted TCR.
All three candidate TCRs demonstrated promising safety profiles, as no cross-reactivities were identified using parental T cell clones 6A2.18 (VPREB3 HLA-A24) and 1C5.6 (BOB1 HLA-B35) or TCR 6B10.12 (FCRL5 HLA-A1) Td CD8 T cells. Cross-reactivities with peptides presented in target HLA alleles were investigated in a safety screening including tumor cell lines derived from a variety of cellular origins, thereby ensuring broad gene-expression profiles. However, as not all genes will be expressed in the included cell lines, potential cross-reactivities could have remained unidentified. To further investigate the safety profile of the identified TCRs, peptide library scanning remains to be performed in additional preclinical studies. In addition to TCR intrinsic cross-reactivities, de novo cross-reactivities could potentially occur when transferring TCRs into CD8 T cells, resulting from mixed dimer formation between chains of the introduced TCR and endogenous TCR.35,36 Here, TCR transfer into CD8 T cells confirmed a lack of general de novo cross-reactivity when stimulated with antigen-negative K562, fibroblast, and keratinocytes. As the risk for de novo cross-reactivities remains a valid concern for the safety of TCR gene therapy, future TCR gene therapy is expected to shift focus to generating T cell products with a knockout of the endogenous TCR or insertion of the introduced TCR into the TCR locus. Deletion of the endogenous TCR will additionally increase the expression of the introduced TCR by eliminating competition for CD3 and thereby lead to more potent antitumor efficacy.37
The three TCRs identified in this study were specific for peptides presented in the context of HLA-A1, -A24, or -B35. These HLA alleles have relative allele frequencies in the worldwide population of 17%, 21%, and 8%, respectively. Future clinical application of the TCRs is restricted to individuals expressing these HLA genotypes. However, since the target genes are expressed in a broad range of B cell malignancies, a large group of patients is still eligible for therapy with these TCRs.
Since tumors are often of heterogenous nature, tumor cells negative for non-essential antigens can be present. Upon single-antigen-targeting immunotherapy regimens, this can result in escape of antigen-negative variants.5 This limitation could be overcome by multi-antigen-targeting therapy.7,8 CAR T cells can be used to treat all malignancies expressing the target antigen, independent of HLA genotype of patients. However, the number of suitable CAR antigens is limited and might not suffice to prevent escape variants. Additionally, for MM the number of cell-surface antigens expressed by malignant cells but not by healthy cell types of non-B cell origin is low.
In contrast to CARs, TCRs are not limited by the cellular localization of antigens. In this study, cellular localization was not included as a selection criterion for target antigens, which resulted in identification of TCRs recognizing peptides derived from cell surface as well as intracellular proteins. Moreover, targeting intracellular proteins that are essential for cell survival, like the transcription factor BOB1 for which we here identified an HLA-B35-restricted TCR, could prevent escape through antigen loss. The benefits of CAR and TCR therapy can complement each other and should be combined in multi-antigen-targeting therapies. Generating a library of CARs and TCRs will allow selection of multiple relevant receptors based on antigen expression and HLA genotypes of patients. Our research has contributed to this goal by identification of three clinically relevant TCRs. The approach described in this study can similarly be applied to identify TCRs for treatment of solid tumors. For these cancers, tumor-associated antigens and neo-antigens are promising targets, and TCR-based therapy will likely be the most valuable option, as cell-surface expression of tumor-specific antigens is generally absent.
In summary, we applied a broad and systematic approach to identify clinically relevant TCRs for the treatment of B cell malignancies. This resulted in identification of three TCRs that induced potent lysis of B cell malignancies of different origins and are therefore promising TCRs for future application in multi-antigen-targeting T cell therapy for B cell malignancies.
Material and methods
Target gene selection, HT12 microarray
A detailed description of protocols followed for sample collection, purification, RNA isolation, and Illumina HT-12.0 microarray measurements can be found in Pont et al.25 Gene expression analysis was performed using gene expression in patient-derived ALL, CLL, and MM; ALL cell lines; EBV-LCLs; healthy B cells; bone marrow mononuclear cells (BMMCs); total PBMCs; and multiple healthy hematopoietic and non-hematopoietic cell types (Figure S1).
Peptide elution and candidate peptide identification
To identify peptides derived from candidate genes that are presented in HLA on the cell surface, the HLA peptidome of primary B cell malignancy samples of different origins was established as previously described.38 In short, after informed consent, apheresis material was obtained at time of diagnosis. HLA typing of the material was performed, and cell pellets were stored at −80°C until use. Cell pellets (0.1 × 109 to 610 × 109 cells) were lysed, and pHLA complexes were purified by immunoaffinity using the anti HLA-I W6/32 antibody. Peptides were separated from HLA molecules using acid, and the peptide-containing fraction was obtained by size filtration. Peptide-containing fractions were separated by strong cation exchange chromatography and freeze-dried. Peptide fractions were lyophilized, dissolved in 95/3/0.1 water/acetonitrile/formic acid v/v/v, and subsequently analyzed with nanoHPLC-tandem mass spectrometry (MS/MS). Peptide and protein identification from tandem mass spectra was performed by proteome discoverer version 2.1 (Thermo Fisher Scientific) using the Mascot node and the UniProt Homo sapiens database. Synthetic peptides were ordered for potential target peptides meeting the following selection criteria: (1) peptides derived from one of the candidate genes; (2) for which a cross query between patient HLA type and predicted HLA binding according to NetMHC version 3.4 indicates binding to HLA-A1, -A24, -B8, or -B35; (3) with a minimal Mascot ion score ≥ 20; (4) with an amino acid length between 8 and 11 amino acids; (5) that were rank 1 peptides. (6) Cysteine-containing peptides were excluded, and (7) the peptide sequence had to be unique for the candidate gene. For peptides meeting all selection criteria, synthetic peptides were ordered, measured by mass spectrometry, and spectra of the synthetic and eluted peptides were compared to confirm peptide identification. Peptides for which the spectra did not match were excluded from the selection.
Generation of pHLA-tetramers
Synthetic peptides were generated in-house using standard Fmoc chemistry. Recombinant HLA-A1, -A24, -B8, or -B35 heavy chains (HCs) and human beta-2 microglobulin (B2M) were produced in-house in Escherichia coli. PE-labeled pHLA-tetramers were produced as previously described with minor modifications.39
T cell isolation and culture
Buffy coats were obtained from healthy donors negative for HLA-A1, HLA-A24, HLA-B8, and HLA-B35 after informed consent (Sanquin). PBMCs were isolated using Ficoll gradient separation and incubated with pHLA-tetramers for 1 h at 4°C or 15 min at 37°C. Cells were washed, and pHLA-tetramer-bound cells were enriched by magnetic-activated cell sorting (MACS) using anti-PE beads (Miltenyi Biotec). The positive fraction was stained with CD8 Alexa Fluor 700 (Invitrogen/Catlag) and fluorescein isothiocyanate (FITC)-labeled CD4, CD14, and CD19 (BD Pharmingen). pHLA-tetramerpos, CD8pos cells were single-cell sorted using an Aria III cell sorter (BD Biosciences) in a 96-well round-bottom plate containing 5 × 104 irradiated PBMCs (35 Gy) and 5 × 103 EBV-LCL-JY cells (50 Gy) in 100 μL T cell medium (TCM) with 0.8 μg/mL phytohemagglutinin (PHA; Oxoid Microbiology Products, Thermo Fisher Scientific). TCM contains IMDM (Lonza), 1% penicillin/streptomycin (Pen/Strep; Lonza), 1.5% glutamine (Lonza), 100 IU/mL IL-2 (Proleukin; Novartis Pharma), 5% fetal bovine serum (FBS; Gibco, Life Technologies), and 5% human serum. T cell clones were restimulated every 10–15 days with irradiated feeder cells and PHA or cryopreserved until further use.
Target cell culture and generation
Cell lines were cultured in IMDM (Lonza), 1% Pen/Strep (Lonza), 1.5% glutamine (Lonza), and 10% FBS (Gibco, Life Technologies). ALL cell lines were cultured as described.40 Primary malignant samples were defrosted and rested overnight (O/N) at 37°C in medium containing 10% human serum before use in experiments. HLA and target gene Td target cells were generated by retroviral transduction with HLA alone or with target gene and HLA combined. Retroviral transduction was performed as previously described.19 Candidate genes and HLA alleles were expressed in MP71 retroviral backbone vectors with marker genes truncated nerve growth factor receptor (NGF-R), CD34, or mouse CD19 (mCD19). Transduced cells were MACS or fluorescence-activated cell sorting (FACS) enriched for marker gene and/or HLA-I expression using HLA-ABC FITC (Serotec), NGF-R PE (BD/Pharmingen), mCD19 PE (BD), or CD34 PE (BD/Pharmingen).
T cell recognition assay
Target cell recognition was determined by incubating 5,000 T cells with target cells in effector:target (E:T) ratio 1:6, unless indicated otherwise, in a 384-well flat-bottom tissue culture plate. T cells were washed before use in experiments to remove expansion-related cytokines. After overnight incubation, recognition was determined by measuring IFN-γ and/or granulocyte-macrophage colony-stimulating factor (GM-CSF) production in supernatants by ELISA (Sanquin and R&D Systems). Supernatants were tested undiluted, 1:5 diluted (Figure 2; Figure S4), or in multiple dilutions to calculate concentrations using optical density values in the linear part of the standard curve (all other experiment). Supernatants were transferred using a Hamilton Microlab STAR Liquid Handling System (Hamilton). Peptide-loaded target cells were loaded with 100 nM per peptide or decreasing peptide concentrations starting at 1 μM for peptide titration experiments. Initial screenings to determine peptide recognition of pHLA-tetramerpos, CD8pos sorted and expanded T cells were performed in a high-throughput manner. The 96-well T cell cultures were split into 4 wells of a 384-well plate using a Hamilton Microlab STAR Liquid Handling System (Hamilton). Peptide-mix-loaded or -unloaded target cells were added manually. T cell clones were defined as reactive when IFN-γ and/or GM-CSF production was >100 pg/mL upon target cell stimulation. T cell clones were considered peptide specific when no cytokine production (<30 pg/mL) was measured for unloaded target cells. T cells meeting these selection criteria were picked manually and transferred to a 24-well plate for restimulation and further expansion. T cell-mediated cytotoxicity was measured using 51Cr-release experiments. Target cells were incubated 1 h at 37°C with 100 μCi Na2 51CrO4 (PerkinElmer). Target cells were washed and co-cultured with T cells at various E:T ratios for 6 h in 96-well U-bottom culture plates. Supernatants were harvested and transferred to 96-well LumaPlates (PerkinElmer). Spontaneous and maximum 51Cr release was determined using TCM alone or TCM containing 1% Triton X-100 (Sigma-Aldrich), respectively. 51Cr-release was measured in counts per minute (cpm) using a 2450 Microbeta2 plate counter (PerkinElmer). Percentage target cell killing was calculated using
qRT-PCR
Total RNA was isolated from 0.5–5 × 106 cells using the Small Scale Kit or ReliaPrep RNA cell mini prep system according to manufacturer’s protocol (Ambion, Promega, respectively). Total RNA was converted to cDNA using Moloney murine leukemia virus reverse transcriptase and oligo (dT) primer (Invitrogen by Thermo Fisher Scientific). qRT-PCR was performed using Fast Start TaqDNA Polymerase (Roche) and EvaGreen (Biotum), and gene expression was measured on the Lightcycler 480 (Roche). Forward and reverse primers used are depicted in Table S5. Target gene expression was calculated relative to the average expression of housekeeping genes GUSB, PSMB4, and VPS29.
TCR identification
TCRα and TCRβ sequences of T cell clones were identified as previously described with minor modifications.41 mRNA was isolated from 1 × 106 cells using the Dynabeads mRNA DIRECT kit (Invitrogen by Thermo Fisher Scientific). TCR cDNA was generated using reverse primers in the TCR constant alfa and beta regions, SMARTScribe Reverse Transcriptase (Takara, Clontech), and a SA.rt template switching oligo forward primer.42 Barcoded TCR PCR product was generated in two rounds of PCR. In the first PCR, alfa and beta TCR products were generated; in a second PCR, the first PCR product was used to include a barcode sequence that allowed discrimination between TCRs of different T cell clones. PCR products of different T cell clones were pooled, after which TCR sequences were identified by HiSeq (GenomeScan). HiSeq data were analyzed using MiXCR and ImMunoGeneTics (IMGT) database to determine the Vα/Vβ family. V(D)J segments of the TCRα and TCRβ were codon optimized and cloned into the modified MP71-TCR-flex retroviral vector. To increase expression and preferential pairing of the introduced TCRαβ chain, the MP71-TCR-flex vector contains codon-optimized and cysteine-modified murine TCRαβ constant domains and P2A sequence to link TCR chains.43 Phoenix-AMPHO (ATCC) cells were transfected; after 48 and 72 h, virus supernatant was harvested and stored at −80°C.
TCR transfer to healthy donor T cells
CD8 and CD4 T cells were separately isolated from healthy donor PBMCs by MACS using anti-CD8 or anti-CD4 microbeads (Miltenyi Biotec). PBMCs were obtained after informed consent. T cells were activated with irradiated autologous PBMCs (35 Gy) and 0.8 μg/mL PHA. On day 2, retroviral supernatants were added to 24-well suspension culture plates (Greiner Bio-One) precoated with 30 mg/mL retronectin (Takara) and blocked with 2% human serum albumin (Sanquin). Plates were spun down for 20 min, 2,000 × g at 4°C. Virus supernatant was removed, and 0.3 × 106 activated T cells were transferred to each well. After overnight incubation, T cells were transferred to a 24-well culture plate (Costar). On day 7 after T cell activation, TCR Td T cells were MACS enriched using anti-mouse TCR-Cβ (mTCR) APC antibody (BD PharMingen) followed by anti-APC MicroBeads (Miltenyi Biotec) according to manufacturer’s protocol. TCR Td T cells were functionally tested between days 10 and 12 after activation. For safety screening of TCR 6B10.12, endogenous TCRαβ knockout of healthy donor CD8 T cells was performed prior to TCR Td as described by Morton et al.37 To assess TCR expression and tetramer binding, cells were stained using mTCR APC antibody and PE-labeled pHLA-tetramers. Cells were measured on the LSR II (BD Bioscience), and data were analyzed with FlowJo software.
In vivo antitumor efficacy of BOB1 HLA-B35 TCR
Female NOD scid gamma (NSG) mice (NOD.Cg-Prkdc(scid)Il2rg(tm1Wjl)/SzJ, The Jackson Laboratory) were injected intravenously (i.v.) with 2 × 106 U266 MM cells; U266 were Td with Luciferase-tdTomato Red and HLA-B35-NGFR and enriched to reach >98% purity. Tumor growth was measured 1–2 times per week after intraperitoneal (i.p.) injection of 150 μL 7.5 mM d-luciferine (Cayman Chemical) using a CCD camera (IVIS Spectrum, PerkinElmer). On day 21, mice were i.v. injected with 5 × 106 T cells, which were Td with 1C5.6 (BOB1 HLA-B35) TCR (n = 4) or irrelevant cytomegalovirus (CMV) pp65-NLV HLA-A2 specific TCR (n = 3). The TCR Td T cells were enriched for mTCR expression before infusion. This study was approved by the national Ethical Committee for Animal Research (AVD116002017891) and was performed in accordance with Dutch laws for animal experiments.
Study approval
Healthy donor and patient material from the Leiden University Medical Center Biobank for Hematological Diseases were used in this study. The study was approved by the Institutional Review Board of the Leiden University Medical Center (approval number B16.039). Materials were obtained after written informed consent in accordance with the Declaration of Helsinki.
Acknowledgments
The authors thank the operators of the LUMC Flow Cytometry Core Facility (Leiden University Medical Center, Leiden, the Netherlands) for providing expert technical assistance in flow cytometric cell sorting. The research in this study was funded by the Dutch Cancer Society (KWF kankerbestrijding, Amsterdam, the Netherlands) under grant number KWF UL-2014-6831. The graphical abstract was created using BioRender.com.
Author contributions
Conceptualization, J.H.F.F. and M.H.M.H.; Methodology, L.J., M.G.D.K., P.A.v.V., and M.H.M.H.; Validation, M.H.M., M.G.D.K., and D.M.v.d.S.; Formal analysis, L.J., M.G.D.K., D.M.v.d.S., M.G., and P.A.v.V.; Investigation, M.H.M., A.K.W., R.S.H., M.G.D.K., D.F.G.R., D.M.v.d.S., C.K., and A.H.d.R.; Resources, P.A.v.V.; Data curation, M.G.D.K., D.M.v.d.S., A.H.d.R., and M.G.; Writing – original draft, M.H.M.; Writing – review & editing, L.T.M., J.H.F.F., and M.H.M.H.; Visualization, M.H.M.; Supervision, J.H.F.F. and M.H.M.H.; Project administration, M.H.M. and M.H.M.H.; Funding acquisition, M.H.M.H.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.08.010.
Contributor Information
Miranda H. Meeuwsen, Email: m.h.meeuwsen@lumc.nl.
Mirjam H.M. Heemskerk, Email: m.h.m.heemskerk@lumc.nl.
Supplemental information
References
- 1.Lee D.W., Kochenderfer J.N., Stetler-Stevenson M., Cui Y.K., Delbrook C., Feldman S.A., Fry T.J., Orentas R., Sabatino M., Shah N.N., et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turtle C.J., Hanafi L.A., Berger C., Gooley T.A., Cherian S., Hudecek M., Sommermeyer D., Melville K., Pender B., Budiarto T.M., et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Invest. 2016;126:2123–2138. doi: 10.1172/JCI85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Locke F.L., Ghobadi A., Jacobson C.A., Miklos D.B., Lekakis L.J., Oluwole O.O., Lin Y., Braunschweig I., Hill B.T., Timmerman J.M., et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1-2 trial. Lancet Oncol. 2019;20:31–42. doi: 10.1016/S1470-2045(18)30864-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abramson J.S., Gordon L.I., Palomba M.L., Lunning M.A., Arnason J.E., Forero-Torres A., Wang M., Maloney D.G., Sehgal A., Andreadis C., et al. Updated safety and long term clinical outcomes in TRANSCEND NHL 001, pivotal trial of lisocabtagene maraleucel (JCAR017) in R/R aggressive NHL. J. Clin. Oncol. 2018;36(15, suppl):7505. [Google Scholar]
- 5.Grupp S.A., Kalos M., Barrett D., Aplenc R., Porter D.L., Rheingold S.R., Teachey D.T., Chew A., Hauck B., Wright J.F., et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park J.H., Rivière I., Gonen M., Wang X., Sénéchal B., Curran K.J., Sauter C., Wang Y., Santomasso B., Mead E., et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018;378:449–459. doi: 10.1056/NEJMoa1709919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang N., Hu X., Cao W., Li C., Xiao Y., Cao Y., Gu C., Zhang S., Chen L., Cheng J., et al. Efficacy and safety of CAR19/22 T-cell cocktail therapy in patients with refractory/relapsed B-cell malignancies. Blood. 2020;135:17–27. doi: 10.1182/blood.2019000017. [DOI] [PubMed] [Google Scholar]
- 8.Ruella M., Barrett D.M., Kenderian S.S., Shestova O., Hofmann T.J., Perazzelli J., Klichinsky M., Aikawa V., Nazimuddin F., Kozlowski M., et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J. Clin. Invest. 2016;126:3814–3826. doi: 10.1172/JCI87366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morgan R.A., Yang J.C., Kitano M., Dudley M.E., Laurencot C.M., Rosenberg S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lamers C.H., Klaver Y., Gratama J.W., Sleijfer S., Debets R. Treatment of metastatic renal cell carcinoma (mRCC) with CAIX CAR-engineered T-cells-a completed study overview. Biochem. Soc. Trans. 2016;44:951–959. doi: 10.1042/BST20160037. [DOI] [PubMed] [Google Scholar]
- 11.Kochenderfer J.N., Dudley M.E., Feldman S.A., Wilson W.H., Spaner D.E., Maric I., Stetler-Stevenson M., Phan G.Q., Hughes M.S., Sherry R.M., et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gross G., Waks T., Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA. 1989;86:10024–10028. doi: 10.1073/pnas.86.24.10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vyas J.M., Van der Veen A.G., Ploegh H.L. The known unknowns of antigen processing and presentation. Nat. Rev. Immunol. 2008;8:607–618. doi: 10.1038/nri2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roex M.C.J., Hageman L., Veld S.A.J., van Egmond E., Hoogstraten C., Stemberger C., Germeroth L., Einsele H., Falkenburg J.H.F., Jedema I. A minority of T cells recognizing tumor-associated antigens presented in self-HLA can provoke antitumor reactivity. Blood. 2020;136:455–467. doi: 10.1182/blood.2019004443. [DOI] [PubMed] [Google Scholar]
- 15.Amir A.L., van der Steen D.M., van Loenen M.M., Hagedoorn R.S., de Boer R., Kester M.D., de Ru A.H., Lugthart G.J., van Kooten C., Hiemstra P.S., et al. PRAME-specific Allo-HLA-restricted T cells with potent antitumor reactivity useful for therapeutic T-cell receptor gene transfer. Clin. Cancer Res. 2011;17:5615. doi: 10.1158/1078-0432.CCR-11-1066. 1525. [DOI] [PubMed] [Google Scholar]
- 16.Jahn L., Hombrink P., Hagedoorn R.S., Kester M.G., van der Steen D.M., Rodriguez T., Pentcheva-Hoang T., de Ru A.H., Schoonakker M.P., Meeuwsen M.H., et al. TCR-based therapy for multiple myeloma and other B-cell malignancies targeting intracellular transcription factor BOB1. Blood. 2017;129:1284–1295. doi: 10.1182/blood-2016-09-737536. [DOI] [PubMed] [Google Scholar]
- 17.Mensali N., Ying F., Sheng V.O., Yang W., Walseng E., Kumari S., Fallang L.E., Kolstad A., Uckert W., Malmberg K.J., et al. Targeting B-cell neoplasia with T-cell receptors recognizing a CD20-derived peptide on patient-specific HLA. OncoImmunology. 2016;5:e1138199. doi: 10.1080/2162402X.2016.1138199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jahn L., Hagedoorn R.S., van der Steen D.M., Hombrink P., Kester M.G., Schoonakker M.P., de Ridder D., van Veelen P.A., Falkenburg J.H., Heemskerk M.H. A CD22-reactive TCR from the T-cell allorepertoire for the treatment of acute lymphoblastic leukemia by TCR gene transfer. Oncotarget. 2016;7:71536–71547. doi: 10.18632/oncotarget.12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jahn L., Hombrink P., Hassan C., Kester M.G., van der Steen D.M., Hagedoorn R.S., Falkenburg J.H., van Veelen P.A., Heemskerk M.H. Therapeutic targeting of the BCR-associated protein CD79b in a TCR-based approach is hampered by aberrant expression of CD79b. Blood. 2015;125:949–958. doi: 10.1182/blood-2014-07-587840. [DOI] [PubMed] [Google Scholar]
- 20.Wilde S., Sommermeyer D., Frankenberger B., Schiemann M., Milosevic S., Spranger S., Pohla H., Uckert W., Busch D.H., Schendel D.J. Dendritic cells pulsed with RNA encoding allogeneic MHC and antigen induce T cells with superior antitumor activity and higher TCR functional avidity. Blood. 2009;114:2131–2139. doi: 10.1182/blood-2009-03-209387. [DOI] [PubMed] [Google Scholar]
- 21.Stronen E., Abrahamsen I.W., Gaudernack G., Wälchli S., Munthe E., Buus S., Johansen F.E., Lund-Johansen F., Olweus J. Dendritic cells engineered to express defined allo-HLA peptide complexes induce antigen-specific cytotoxic T cells efficiently killing tumour cells. Scand. J. Immunol. 2009;69:319–328. doi: 10.1111/j.1365-3083.2008.02223.x. [DOI] [PubMed] [Google Scholar]
- 22.Savage P., Gao L., Vento K., Cowburn P., Man S., Steven N., Ogg G., McMichael A., Epenetos A., Goulmy E., Stauss H.J. Use of B cell-bound HLA-A2 class I monomers to generate high-avidity, allo-restricted CTLs against the leukemia-associated protein Wilms tumor antigen. Blood. 2004;103:4613–4615. doi: 10.1182/blood-2003-11-3903. [DOI] [PubMed] [Google Scholar]
- 23.Rammensee H.G., Bevan M.J. Evidence from in vitro studies that tolerance to self antigens is MHC-restricted. Nature. 1984;308:741–744. doi: 10.1038/308741a0. [DOI] [PubMed] [Google Scholar]
- 24.Stanislawski T., Voss R.H., Lotz C., Sadovnikova E., Willemsen R.A., Kuball J., Ruppert T., Bolhuis R.L., Melief C.J., Huber C., et al. Circumventing tolerance to a human MDM2-derived tumor antigen by TCR gene transfer. Nat. Immunol. 2001;2:962–970. doi: 10.1038/ni1001-962. [DOI] [PubMed] [Google Scholar]
- 25.Pont M.J., Honders M.W., Kremer A.N., van Kooten C., Out C., Hiemstra P.S., de Boer H.C., Jager M.J., Schmelzer E., Vries R.G., et al. Microarray Gene Expression Analysis to Evaluate Cell Type Specific Expression of Targets Relevant for Immunotherapy of Hematological Malignancies. PLoS ONE. 2016;11:e0155165. doi: 10.1371/journal.pone.0155165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jahn L., van der Steen D.M., Hagedoorn R.S., Hombrink P., Kester M.G., Schoonakker M.P., de Ridder D., van Veelen P.A., Falkenburg J.H., Heemskerk M.H. Generation of CD20-specific TCRs for TCR gene therapy of CD20low B-cell malignancies insusceptible to CD20-targeting antibodies. Oncotarget. 2016;7:77021–77037. doi: 10.18632/oncotarget.12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amir A.L., D’Orsogna L.J., Roelen D.L., van Loenen M.M., Hagedoorn R.S., de Boer R., van der Hoorn M.A., Kester M.G., Doxiadis I.I., Falkenburg J.H., et al. Allo-HLA reactivity of virus-specific memory T cells is common. Blood. 2010;115:3146–3157. doi: 10.1182/blood-2009-07-234906. [DOI] [PubMed] [Google Scholar]
- 28.Franco A., Kraus Z., Li H., Seibert N., Dement-Brown J., Tolnay M. CD21 and FCRL5 form a receptor complex with robust B-cell activating capacity. Int. Immunol. 2018;30:569–578. doi: 10.1093/intimm/dxy052. [DOI] [PubMed] [Google Scholar]
- 29.Polson A.G., Zheng B., Elkins K., Chang W., Du C., Dowd P., Yen L., Tan C., Hongo J.A., Koeppen H., Ebens A. Expression pattern of the human FcRH/IRTA receptors in normal tissue and in B-chronic lymphocytic leukemia. Int. Immunol. 2006;18:1363–1373. doi: 10.1093/intimm/dxl069. [DOI] [PubMed] [Google Scholar]
- 30.Rodig S.J., Kutok J.L., Paterson J.C., Nitta H., Zhang W., Chapuy B., Tumwine L.K., Montes-Moreno S., Agostinelli C., Johnson N.A., et al. The pre-B-cell receptor associated protein VpreB3 is a useful diagnostic marker for identifying c-MYC translocated lymphomas. Haematologica. 2010;95:2056–2062. doi: 10.3324/haematol.2010.025767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hess J., Nielsen P.J., Fischer K.D., Bujard H., Wirth T. The B lymphocyte-specific coactivator BOB.1/OBF.1 is required at multiple stages of B-cell development. Mol. Cell. Biol. 2001;21:1531–1539. doi: 10.1128/MCB.21.5.1531-1539.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim U., Qin X.F., Gong S., Stevens S., Luo Y., Nussenzweig M., Roeder R.G. The B-cell-specific transcription coactivator OCA-B/OBF-1/Bob-1 is essential for normal production of immunoglobulin isotypes. Nature. 1996;383:542–547. doi: 10.1038/383542a0. [DOI] [PubMed] [Google Scholar]
- 33.Wang T., Birsoy K., Hughes N.W., Krupczak K.M., Post Y., Wei J.J., Lander E.S., Sabatini D.M. Identification and characterization of essential genes in the human genome. Science. 2015;350:1096–1101. doi: 10.1126/science.aac7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hodson D.J., Shaffer A.L., Xiao W., Wright G.W., Schmitz R., Phelan J.D., Yang Y., Webster D.E., Rui L., Kohlhammer H., et al. Regulation of normal B-cell differentiation and malignant B-cell survival by OCT2. Proc. Natl. Acad. Sci. USA. 2016;113:E2039–E2046. doi: 10.1073/pnas.1600557113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Loenen M.M., de Boer R., Amir A.L., Hagedoorn R.S., Volbeda G.L., Willemze R., van Rood J.J., Falkenburg J.H., Heemskerk M.H. Mixed T cell receptor dimers harbor potentially harmful neoreactivity. Proc. Natl. Acad. Sci. USA. 2010;107:10972–10977. doi: 10.1073/pnas.1005802107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bendle G.M., Linnemann C., Hooijkaas A.I., Bies L., de Witte M.A., Jorritsma A., Kaiser A.D.M., Pouw N., Debets R., Kieback E., et al. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat. Med. 2010;16:565–570. doi: 10.1038/nm.2128. [DOI] [PubMed] [Google Scholar]
- 37.Morton L.T., Reijmers R.M., Wouters A.K., Kweekel C., Remst D.F.G., Pothast C.R., Falkenburg J.H.F., Heemskerk M.H.M. Simultaneous Deletion of Endogenous TCRalphabeta for TCR Gene Therapy Creates an Improved and Safe Cellular Therapeutic. Mol. Ther. 2020;28:64–74. doi: 10.1016/j.ymthe.2019.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van der Lee D.I., Reijmers R.M., Honders M.W., Hagedoorn R.S., de Jong R.C., Kester M.G., van der Steen D.M., de Ru A.H., Kweekel C., Bijen H.M., et al. Mutated nucleophosmin 1 as immunotherapy target in acute myeloid leukemia. J. Clin. Invest. 2019;129:774–785. doi: 10.1172/JCI97482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burrows S.R., Kienzle N., Winterhalter A., Bharadwaj M., Altman J.D., Brooks A. Peptide-MHC class I tetrameric complexes display exquisite ligand specificity. J. Immunol. 2000;165:6229–6234. doi: 10.4049/jimmunol.165.11.6229. [DOI] [PubMed] [Google Scholar]
- 40.Nijmeijer B.A., Szuhai K., Goselink H.M., van Schie M.L., van der Burg M., de Jong D., Marijt E.W., Ottmann O.G., Willemze R., Falkenburg J.H. Long-term culture of primary human lymphoblastic leukemia cells in the absence of serum or hematopoietic growth factors. Exp. Hematol. 2009;37:376–385. doi: 10.1016/j.exphem.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 41.van Bergen C.A., van Luxemburg-Heijs S.A., de Wreede L.C., Eefting M., von dem Borne P.A., van Balen P., Heemskerk M.H., Mulder A., Claas F.H., Navarrete M.A., et al. Selective graft-versus-leukemia depends on magnitude and diversity of the alloreactive T cell response. J. Clin. Invest. 2017;127:517–529. doi: 10.1172/JCI86175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koning M.T., Kiełbasa S.M., Boersma V., Buermans H.P.J., van der Zeeuw S.A.J., van Bergen C.A.M., Cleven A.H.G., Kluin P.M., Griffioen M., Navarrete M.A., Veelken H. ARTISAN PCR: rapid identification of full-length immunoglobulin rearrangements without primer binding bias. Br. J. Haematol. 2017;178:983–986. doi: 10.1111/bjh.14180. [DOI] [PubMed] [Google Scholar]
- 43.Linnemann C., Heemskerk B., Kvistborg P., Kluin R.J., Bolotin D.A., Chen X., Bresser K., Nieuwland M., Schotte R., Michels S., et al. High-throughput identification of antigen-specific TCRs by TCR gene capture. Nat. Med. 2013;19:1534–1541. doi: 10.1038/nm.3359. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.