

Abstract
Mutations in the BIN1 (Bridging Interactor 1) gene, encoding the membrane remodeling protein amphiphysin 2, cause centronuclear myopathy (CNM) associated with severe muscle weakness and myofiber disorganization and hypotrophy. There is no available therapy, and the validation of therapeutic proof of concept is impaired by the lack of a faithful and easy-to-handle mammalian model. Here, we generated and characterized the Bin1mck−/− mouse through Bin1 knockout in skeletal muscle. Bin1mck−/− mice were viable, unlike the constitutive Bin1 knockout, and displayed decreased muscle force and most histological hallmarks of CNM, including myofiber hypotrophy and intracellular disorganization. Notably, Bin1mck−/− myofibers presented strong defects in mitochondria and T-tubule networks associated with deficient calcium homeostasis and excitation-contraction coupling at the triads, potentially representing the main pathomechanisms. Systemic injection of antisense oligonucleotides (ASOs) targeting Dnm2 (Dynamin 2), which codes for dynamin 2, a BIN1 binding partner regulating membrane fission and mutated in other forms of CNM, improved muscle force and normalized the histological Bin1mck−/− phenotypes within 5 weeks. Overall, we generated a faithful mammalian model for CNM linked to BIN1 defects and validated Dnm2 ASOs as a first translatable approach to efficiently treat BIN1-CNM.
Keywords: myopathy, antisense oligonucleotides, membrane curvature, dynamin, amphiphysin, therapy, MTM1, myotubular myopathy, myotubularin, t-tubule
Graphical abstract
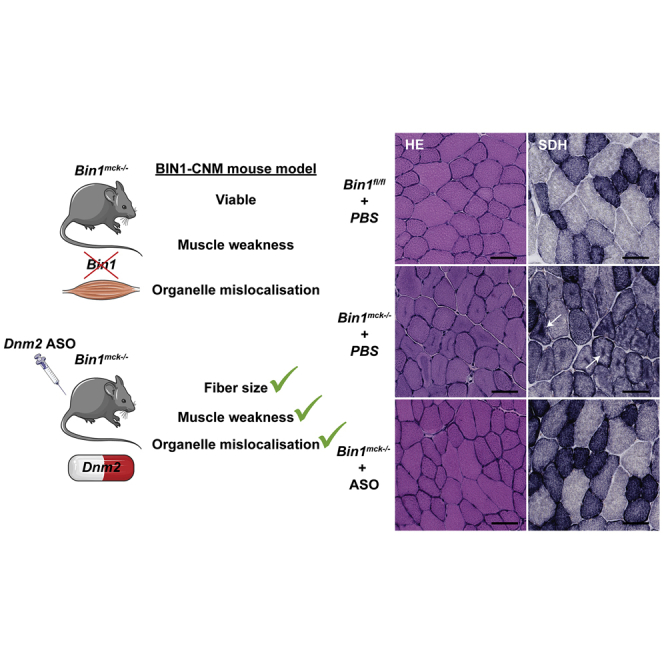
A Bin1mck−/− mouse model is generated and recapitulates most of the motor and histology defects observed in patients with autosomal recessive centronuclear myopathy caused by BIN1 mutations. Dnm2 downregulation in this model improves most phenotypes, offering promising perspectives for BIN1-CNM patient treatment.
Introduction
A plethora of muscle diseases impair different steps of muscle formation or maintenance. Centronuclear myopathies (CNMs) are rare genetic diseases associated with a severe generalized muscle weakness associated with myofiber hypotrophy and premature death.1,2 The name reflects the typical intracellular disorganization of myofibers with organelle mis-positioning, including the centralization of nuclei that are normally present at the periphery of muscle fibers.3,4 In addition, myofibers are smaller, rounder, or heterogeneous in size. Despite the significant impact on morbidity and mortality, to date there is no specific therapy available for CNM patients.
CNMs can be due to mutations in MTM1, DNM2, BIN1, or SPEG genes, while mutations in additional genes, such as RYR1, TTN, CACNA1S, ZAK, and PYROXD1, combine CNM features with other histological defects.5, 6, 7 Of note, several of these gene products directly regulate excitation-contraction coupling at the skeletal muscle triad (RYR1, CACNA1S/Cav1.1) or membrane remodeling (BIN1, DNM2), leading to the hypothesis that defects in triad structure and function form a common disease cause.8
BIN1 encodes amphiphysin 2, a protein sensing and controlling membrane curvature through its N-BAR (N-amphipathic Bin/Amphiphysin/Rvs) domain and recruiting through its SH3 (Src Homology) domain effectors like dynamins (DNM1 and DNM2), which tubulate and potentially fission membranes.9,10 While BIN1 is not the most frequent gene mutated in CNM, it is mutated in both dominant and recessive forms.11,12 Moreover, a founder mutation leads to a potentially high prevalence in the Roma population (also known as Gypsies), which is the most numerous ethnic minority in Europe, with an estimated population of 11 million.13 BIN1 mutations either modify the N-BAR domain, leading to decreased membrane tubulation, or truncate or extend the SH3 domain, altering the binding to DNM2.12,14
Regarding the understanding and treatment of BIN1-CNM, the two main bottlenecks are the absence of a faithful and easy-to-handle mammalian model and the lack of therapeutic proof of concept necessary to trigger clinical development.
A Drosophila mutant for Amph, the BIN1 ortholog, is flightless, with severe structural defects of the triads, the structural basis of excitation-contraction coupling formed by a T-tubule contacting 2 terminal cisternae of sarcoplasmic reticulum (SR).15 Downregulation of bin1 with antisense morpholino in zebrafish led to myofiber disorganization with centralized nuclei and structural and functional defects of the triads.16 A spontaneous dog model was found with a splice mutation of the muscle-specific exon 11 of BIN1 and muscle atrophy, myofibers with centralized nuclei, and radial organization of the SR.17 In mice, constitutive Bin1 knockout presented embryonic or perinatal lethality, probably due to feeding defects.18,19 Similarly, a lethal phenotype was reproduced in a skeletal-muscle-specific knockout using the Cre recombinase under the control of the human skeletal actin (HSA) promoter.18,20 Conversely, knockout of the muscle-specific exon 11 led to viable mice that did not display CNM phenotypes.20 Acute reduction of Bin1 with short hairpin RNA (shRNA) in adult mice, however, showed disruption of the triad structure linked to alterations of intracellular Ca2+ release.21
Some potential therapies for CNMs have been proposed.22 Moreover, downregulation of Dnm2 with antisense oligonucleotides (ASOs) was shown to efficiently ameliorate the skeletal muscle phenotype in MTM1- and DNM2-CNM mouse models,23,24 an approach that is currently in clinical trials (NCT04033159). However, it was never tested for BIN1-CNM due to lack of a faithful viable mouse model, albeit genetic crosses recently suggested that decreasing Dnm2 prevents the neonatal death of Bin1 knockout mice.18
Here, we generated and characterized a viable Bin1 knockout mouse faithfully reproducing most motor, histological, and structural hallmarks of CNMs and validated Dnm2 ASOs as a first translatable approach to efficiently cure this model.
Results
Generation of a viable muscle-specific Bin1 knockout mouse with decreased muscle force
To generate a viable mouse model without Bin1 expression in muscle, we crossed mice floxed for Bin1 exon 20 (Bin1fl/fl) with Bin1 heterozygous mice (Bin1mck+/−) expressing the Cre recombinase under the control of the muscle creatine kinase promoter (MCK-Cre), which expresses from 17 days post-coitum (dpc) (Figure 1A). In comparison, the HSA promoter used to obtain perinatal lethal mice expresses from 9 dpc.18,20 Bin1−/−MCK-Cre (Bin1mck−/−) mice were generated with normal Mendelian ratio and were viable to at least 12 months of age (Figures 1B and 1C), the maximum age analyzed in this study. Quantitative RT-PCR (qRT-PCR) confirmed the absence of Bin1 mRNA levels in Bin1mck−/− tibialis anterior (TA) muscle, and western blot revealed the absence of BIN1 from Bin1mck−/− animals at 8 weeks of age (Figures 1D and 1E).
Figure 1.

Muscle-specific Bin1mck−/− mice are viable with impaired TA muscle force
(A) Homozygous Bin1mck−/− mice were generated by crossing Bin1fl/fl with heterozygous floxed Bin1 mice expressing the Cre recombinase under the control of the muscle creatine kinase promoter (MCK-Cre). The represented situation refers to skeletal muscle. (B) Genotype proportion obtained 10 days after birth; expected ratio is 50/50. (C) Bin1fl/fl and Bin1mck−/− mice at 8 weeks of age. (D) Bin1 mRNA pan-isoform relative levels in TA from Bin1fl/fl and Bin1mck−/− mice at 8 weeks of age (n = 4; Student’s t test with Welch’s correction). (E) Western blot with TA muscle protein extracts probed with anti-BIN1 pan-isoform antibody. BIN1 is not detected in Bin1mck−/− muscle. (F) Body weight evolution of Bin1fl/fl and Bin1mck−/− mice (n = 8–9; two-way ANOVA and Bonferroni post hoc test). (G) Specific muscle force produced by TA of Bin1fl/fl and Bin1mck−/− at 8 weeks of age (n = 8–9; Student’s t test). Bin1mck−/− mice produce 41% less specific force than Bin1fl/fl controls. (H) Specific force produced by Bin1fl/fl and Bin1mck−/− TA when stimulated at different frequencies (n = 8–9; two-way ANOVA with Bonferroni’s post hoc test). Graphs represent mean ± SEM. ∗∗∗∗p < 0.0001; ∗∗∗p < 0.001; ∗∗p < 0.01.
Bin1mck−/− mice did not present differences in body or muscle weight at 8 weeks, while body weight was reduced at 4 months and 12 months, and TA weight ratio was reduced at 4 months only (Figures 1F and S1). Hanging capability until 8 weeks of age, endurance and fatigue in treadmill at 4 months, and performances in the string test and rotarod test at 6–7 weeks of age were comparable between Bin1fl/f and Bin1mck−/− animals, suggesting normal locomotor coordination (Figures S2A–S2E). For phenotypical and histological studies, we used TA, as this muscle is commonly affected in other CNM mouse models.23, 25, 26 We quantified the TA muscle force in situ following sciatic nerve stimulation. At 8 weeks of age and at 4 months, Bin1mck−/− developed reduced maximal and specific force compared to controls. While Bin1fl/fl mice at 8 weeks of age reached an average specific force of 18 mN/mg, Bin1mck−/− mice only produced a specific force of 10.6 mN/mg, a 41% reduction (Figures 1G and S2F). Analysis at different stimulation frequencies revealed a clear reduction at several frequencies tested in 8-week- and 4-month-old animals (Figures 1H and S2G–S2J). Therefore, the absence of BIN1 did not have a significant impact on the overall locomotor abilities, while it clearly affected the muscle force at 8 weeks and 4 months of age.
Bin1mck−/− mice develop most of the histological hallmarks of CNM
We further assessed the presence of CNM features at the histopathological level. Staining of Bin1mck−/− TA sections revealed the presence of darker hematoxylin and eosin (H&E) staining in the center of the fibers that were enriched in mitochondrial oxidative activity, as indicated in succinate dehydrogenase (SDH) and nicotinamide adenine dinucleotide dehydrogenase (NADH) staining, while Gomori staining remained normal at 8 weeks (Figures 2A and S3A). Similar defects in H&E and SDH staining were observed in 4 month and 12 month TA from Bin1mck−/− mice (Figures S3B–S3G). The gastrocnemius presented with similar histological defects as observed in the TA, while heart histology did not reveal any obvious defect at 8 weeks of age (Figure S4A). Heterozygous knockout animals (Bin1mck+/−) did not present any histological defect in TA, showing total reduction of Bin1 is required to generate the histological phenotype and confirming this is a model for recessive BIN1-CNM (Figure S4B). Bin1mck−/− TA presented smaller fibers and a strong reduction in the number of large fibers when compared to Bin1fl/f controls at 8 weeks, 4 months, and 12 months of age (Figures 2B, 2C, S3D, and S3E). Bin1mck−/− mice also presented a slight but significant increase in the percentage of fibers with high circularity at 8 weeks of age (Figure 2D). The decrease in large fibers at 8 weeks of age was in line with the reduction of fast-twitch IIb fibers and the slight but not significant increase in type I fibers (Figure S4C). Bin1mck−/− did not present increased nuclear centralization at 8 weeks of age, but the rate of TA fibers with internalized nuclei increased in 4-month- and particularly in 12-month-old Bin1mck−/− mice (Figures 2E, S3B, and S3F). Of note, the Dnm2RW/+ mouse mimicking the most common R465W mutation found in DNM2-CNM also displays CNM hallmarks but has normal nuclei positioning at 8 weeks.27 Altogether, Bin1mck−/− mice display muscle weakness and most of the histological hallmarks of CNM, highlighting the relevance of Bin1mck−/− mice as a model for BIN1-CNM.
Figure 2.

Bin1mck−/− mice develop most of the histopathological hallmarks of CNM in TA
(A) Representative H&E and SDH images of TA from Bin1fl/fl and Bin1mck−/− mice at 8 weeks of age. Bin1mck−/− muscle sections present SDH accumulations in the center of fibers (arrows; n = 3). Scale bar, 50 μm. (B) MinFeret perimeter distribution of TA fibers (n = 4–5). (C) The percentage of large fibers with MinFeret diameter higher than 40 μm is reduced in Bin1mck−/− compared to Bin1fl/fl controls (n = 4–5; Student’s t test). (D) Myofiber circularity distribution in TA; a value of 0 corresponds to a line and a value of 1 to a circle (n = 4–5). Bin1mck−/− fibers are rounder. (E) Percentage of fibers with internalized nuclei reveals no difference between Bin1mck−/− and Bin1fl/fl muscles (n = 4–5; Student’s t test). Graphs represent mean ± SEM. ∗∗p < 0.01.
Bin1mck−/− myofibers display mitochondria disorganization
To better characterize the pathomechanism linked to the defects in muscle force and histology in Bin1mck−/− mice, we performed electron microscopy on TA muscles and found a general intracellular disorganization with altered sarcomere width and spacing (Figure 3A). Nuclei position and shape were normal. Local accumulations of mitochondria were observed, together with increased levels of prohibitin protein, an inner mitochondrial membrane protein that also presented a central and sub-sarcolemma accumulation pattern in Bin1mck−/− mice (Figures 3B, 3C, and S5A). To determine the cause of mitochondrial accumulation, mitochondrial biogenesis and removal were assessed. mRNA levels from the mitochondrial biogenesis master regulator Ppargc1a were comparable between Bin1mck−/− and Bin1fl/fl mice (Figure 3D). In Bin1mck−/− muscles, electron microscopy revealed autophagosomes in regions where mitochondria accumulated, and LC3 immunofluorescence showed increased accumulation of LC3-positive dots (Figure 3E). In line with this, the protein expression level of P62 and lipidated LC3 were slightly, albeit not significantly, increased in Bin1mck−/− mice, while their gene expression in Bin1mck−/− TA muscle was mostly comparable to Bin1fl/fl littermates (Figures 3F, 3G, S5B, and S5C), suggesting mild defects in mitochondrial positioning consistent with mitochondria accumulation. Therefore, the muscle weakness observed in Bin1mck−/− muscles may arise in part from sarcomere and mitochondria disorganization.
Figure 3.

Bin1mck−/− mice present myofibril disorganization and mitochondria accumulation in TA
(A) Electron microscopy images from Bin1fl/fl and Bin1mck−/− at 8 weeks of age reveal overall myofibril disorganization with higher inter-myofibril spaces (left), while nuclear shape and position remain normal in Bin1mck−/− mice (right) (n = 2). Scale bar, 1 μm. (B) Mitochondria accumulations in Bin1mck−/− muscle observed by electron microscopy (left) and revealed with prohibitin staining (right) (n = 2). Scale bar, 1 μm for electron microscopy and 10 μm for confocal microscopy. (C) Quantification of prohibitin protein expression (n = 3; Student’s t test). (D) Ppargc1a mRNA relative levels (n = 6; Student’s t test). (E) Regions with mitochondria accumulation (arrow) in Bin1mck−/− muscle present autophagosomes (arrowhead; left), and immunofluorescence labeling reveals increased number of autophagic LC3-positive clusters (right) (n = 2). (F and G) P62 and lipidated LC3 (II) protein levels (n = 4; Student’s t test for P62 and Student’s t test with Welch’s correction for lipidated LC3). Graphs represent mean ± SEM. ∗∗p < 0.01.
Bin1mck−/− myofibers present abnormal T-tubules and deficient excitation-contraction coupling
BIN1 is a key player in the biogenesis of T-tubule in skeletal muscle.10,15 To better understand the decreased muscle force observed in Bin1mck−/− mice, T-tubules were imaged and excitation-contraction coupling assessed in isolated muscle fibers from interosseus and flexor digitorum brevis (FDB), muscles from which intact isolated fibers can be easily obtained. While Bin1mck−/− FDB histology is similar to Bin1fl/fl littermates at 8 weeks of age (Figures S6A and S6B), di-8-anneps staining of plasma membrane invaginations revealed an altered network and a decreased density of T-tubules in isolated Bin1mck−/− fibers (Figures 4A and 4B). The Ca2+ current through the T-tubule CaV1.1 Ca2+ channel/voltage sensor and the release of SR Ca2+ through RYR1 channels were simultaneously detected in isolated fibers under voltage-clamp conditions (Figures 4C–4H). The CaV1.1 current density was smaller in the Bin1mck−/− fiber, accompanied by an increased residual leak component visible in response to the lowest-amplitude pulses (Figure 4C). Mean values for the voltage dependence of the peak current in the two groups are reported in Figure 4D. In each fiber, the voltage dependence was fitted by the standard equation plus a linear outward component. Mean values from the fits revealed that the maximum conductance of the CaV1.1 channel population in the Bin1mck−/− fibers was reduced by ∼30% compared to the mean value in Bin1fl/fl fibers (Figure 4E), while corresponding mean values for the apparent reversal potential (Figure 4E), and for the half-activation voltage and steepness factor, did not statistically differ between the two groups (Figure S6C). The effect on the conductance was paralleled by a slight, though not significant, increase in the residual outward leak current (slope) in the Bin1mck−/− fibers (Figure 4E), indicating that besides affecting CaV1.1 channels, another conductance component of the plasma membrane may be affected. Rhod-2 Ca2+ transients from Bin1mck−/− fibers exhibited a reduced peak amplitude compared to Bin1fl/fl fibers (Figure 4F). The rate of SR Ca2+ release calculated from the rhod-2 transients indicated that the mean value for maximum rate was reduced by almost 60% (Figures 4G and 4H) in the Bin1mck−/− fibers, while values for the mid-activation voltage and steepness factor were unaffected (Figure S6D). Overall, myofibers lacking BIN1 have an abnormal T-tubule network and a strong defect in excitation-contraction coupling, correlating with decreased muscle force.
Figure 4.

Deficient voltage-dependent Ca2+ signaling in muscle fibers isolated from Bin1mck−/− mouse FDB
(A) Representative confocal images of the T-tubule network in muscle fibers from Bin1fl/fl and Bin1mck−/− mice at 8 weeks of age. Scale bar, 10 μm. (B) Mean values for the T-tubule network density index in the two groups (n = 3 mice per genotype, 9–13 fibers analyzed per mouse; Student’s t test). (C) Examples of CaV1.1 Ca2+ current records from muscle fibers from a Bin1fl/fl mouse (left) and from a Bin1mck−/− mouse (right) in response to 0.5-s-long depolarizing pulses to the range of indicated values (voltage increment between −40 and +50 is 10 mV). (D) Mean values for the peak Ca2+ current versus voltage in the two groups. The voltage dependence of the peak Ca2+ current in each fiber was fitted as described in Materials and methods (n = 3 mice per genotype, 5–6 fibers analyzed per mouse; Student’s t test). (E) Mean values, from fitting the voltage dependence of the peak Ca2+ current, for the maximum conductance (Gmax), reversal voltage (Vrev), and additional linear conductance (slope). Mean values for Vrev and for the two other fitted parameters (half-activation voltage and steepness factor) did not statistically differ between the two groups (n = 3 mice per genotype, 5–6 fibers analyzed per mouse; Student’s t test). (F) Representative line-averaged rhod-2 Ca2+ transients from a Bin1fl/fl and from a Bin1mck−/− muscle fiber, in response to the indicated voltage pulse protocol (same as in D). From these records, the rate of SR Ca2+ release was calculated (dCaTot/dt, see Materials and methods). (G) Mean values for the peak rate of SR Ca2+ release versus voltage in the two groups (n = 3 mice per genotype, 5–6 fibers analyzed per mouse; Student’s t test). (H) Mean values for the maximum rate of SR Ca2+ release obtained by fitting the voltage dependence of peak rate in each fiber with a Boltzmann function. Mean values for the two other fitted parameters (mid-activation voltage and steepness factor) did not statistically differ between the two groups. Data are from the same groups of fibers as in (E) (n = 3 mice per genotype, 5–6 fibers analyzed per mouse; Student’s t test). Graphs represent mean ± SEM. ∗∗p < 0.01; ∗p < 0.05.
Dnm2 downregulation improves the force production of Bin1mck−/− mice
Our previous studies suggested BIN1 is a negative regulator of DNM2 in muscle, as decreasing Dnm2 through genetic cross prevented the neonatal death of Bin1−/− mice.18 We took advantage of the Bin1mck−/− mice as a viable and faithful BIN1-CNM model to test a therapeutic approach by injecting ASOs targeting Dnm2. We selected a dose of 25 mg/kg that was previously validated as the safest and most efficient dose in other CNM mouse models.24,23,26 Bin1mck−/− muscles presented a slight increase in DNM2 protein and RNA levels (Figures 5A, 5B, and S7A). Dnm2 ASO treatment clearly reduced Dnm2 mRNA and protein levels in both ASO-treated Bin1fl/fl and Bin1mck−/− mice (Figures 5B, 5C, and S7B). While the muscle force of PBS-treated Bin1mck−/− mice was significantly different from that of PBS-treated Bin1fl/fl mice, Dnm2 downregulation slightly increased the muscle force of ASO-treated Bin1mck−/− mice to a level not significantly different from the ASO-treated Bin1fl/fl controls (Figures 5D and S8A). At low stimulation frequencies (1, 25, and 50 Hz), the force produced by ASO-treated Bin1mck−/− mice was comparable to the ASO-treated Bin1fl/fl controls and significantly increased compared to PBS-treated Bin1mck−/− mice (Figure 5E). Overall, Dnm2 downregulation increases the force production of ASO-treated Bin1mck−/− mice.
Figure 5.

Dnm2 downregulation ameliorates the TA muscle force of Bin1mck−/− mice
(A) DNM2 protein expression in TA at 8 weeks of age (n = 4; Student’s t test with Welch’s correction). (B) Dnm2 mRNA levels in TA at 8 weeks of age by qRT-PCR for pan-isoforms (n = 4; two-way ANOVA with Bonferroni’s post hoc test). (C) DNM2 protein expression in TA at 8 weeks (n = 4–7; two-way ANOVA with Bonferroni’s post hoc test). (D) Maximal specific force produced by Bin1fl/fl and Bin1mck−/− TA. Only muscle force produced by PBS-treated Bin1mck−/− muscles shows a significant decrease compared to Bin1fl/fl controls (n = 4–6; Kruskal-Wallis with Dunn’s post hoc test). (E) Specific force produced when stimulated at increasing frequencies (n = 4–6; one-way ANOVA with Tukey’s post hoc test for 1, 25, and 50 Hz frequencies and Kruskal-Wallis with Dunn’s post hoc test for 100 and 125 Hz frequencies). ∗Between Bin1fl/fl PBS and Bin1mck−/− PBS. βBetween Bin1mck−/− PBS and Bin1mck−/− ASO. Graphs in A-C show mean ± SEM, and graphs in D-E shows median ± interquartile range. ∗∗p < 0.01; ∗p < 0.05; ββp < 0.01. We found no statistical difference between Bin1mck−/− ASO and Binfl/fl ASO.
Dnm2 downregulation improves the CNM histological hallmarks of Bin1mck−/− mice
As Dnm2 downregulation increased muscle force production of Bin1mck−/− muscle, we then assessed rescue at the histopathological level. The Dnm2 ASO treatment fully normalized the central accumulations of mitochondrial oxidative activity observed in H&E, SDH, and NADH staining of Bin1mck−/− muscle sections, further confirmed by mitochondrial TOMM20 immunofluorescence (Figures 6A, 6B, and S8B–S8D) Moreover, desmin, an essential regulator of myofibril integrity, with aberrant localization in MTM1-CNM patients and mice,28,29 is mislocalized in the center of PBS-treated Bin1mck−/− fibers, and its localization is fully restored in ASO-treated Bin1mck−/− mice (Figures S8C and S8D). While myofibers from the PBS-treated Bin1mck−/− mice were smaller compared to Bin1fl/fl controls, Dnm2 ASO treatment normalized fiber size distribution and the percentage of large fibers (Figures 6C–6E). Correlating with the effect on large fibers, a slight but not significant decrease in Myh7 mRNA levels (specific for type I fiber) and increase of Myh4 mRNA levels (type IIb fiber) were noted in ASO-treated Bin1mck−/− muscles compared to PBS-treated Bin1mck−/− muscles (Figure S8E). Altogether, Dnm2 downregulation improved both motor function and the CNM histological hallmarks of Bin1mck−/− mice, highlighting Dnm2 ASO treatment as an efficient injectable therapeutic approach to treat recessive BIN1-CNM.
Figure 6.

Dnm2 downregulation rescues the TA CNM histopathological hallmarks of Bin1mck−/− mice
(A and B) Representative H&E and SDH images of TA from PBS and ASO-treated Bin1fl/fl and Bin1mck−/− mice at 8 weeks of age (n = 4–6). Scale bar, 50 μm. ASO-treated Bin1mck−/− mice do not present the central SDH accumulations seen in PBS-treated Bin1mck−/− mice (arrows). (C) MinFeret perimeter distribution of TA fibers from PBS- and ASO-treated Bin1fl/fl and Bin1mck−/− mice (n = 4–5). (D) Myofiber circularity from ASO-treated Bin1fl/fl− and Bin1mck−/− TA; a value of 0 correspond to a line and a value of 1 to a circle (n = 4–5). (E) Percentage of fibers with MinFeret diameter higher than 40 μm are comparable in PBS- and ASO-treated Bin1fl/fl and Bin1mck−/− mice (n = 4–7; one-way ANOVA with Tukey’s post hoc test). Graphs represent mean ± SEM. ∗∗p < 0.01; ∗p < 0.05.
Discussion
In this study, we generated and characterized the Bin1mck−/− mouse as a first viable mammalian model reproducing most signs of BIN1-CNM. It provided a unique opportunity to test a potential treatment. Dynamin 2 is a binding partner of BIN1 and also mutated in CNMs. Injection of ASOs targeting Dnm2 led to about 50% reduction in dynamin 2 compared to PBS-treated mice of the same genotype and to the improvement of motor and histopathological phenotypes of the Bin1mck−/− mice. In these experiments, dynamin 2 levels in both ASO-treated genotypes are below the baseline of PBS-treated Bin1fl/fl mice. These results provide novel insights in the role of BIN1 in skeletal muscle and validate a therapeutic proof of concept.
Bin1mck−/− mice represent a faithful model for autosomal recessive BIN1-CNM, with a clear decrease in muscle force, and most histological hallmarks, such as smaller and rounder myofibers with typical central accumulation of oxidative activity and a general intracellular disorganization. Alteration of the T-tubule network and the strong defect in excitation-contraction coupling most probably explain the decreased muscle force. However, such defects appear compatible with a conserved locomotor activity and survival into adulthood. In contrast to the neonatal lethality observed when Bin1 was deleted using Cre recombinase under the control of CMV (cytomegalovirus) or HSA promoters,18,19,20 Bin1mck−/− mice are viable and fertile and display a disease onset similar to patients, making this model suitable for the establishment of treatment protocols.
BIN1-CNM patients present high rates of fibers with centralized nuclei, while Bin1mck−/− mice only display increased nuclear internalization at 12 months of age and not at 8 weeks,12 suggesting progressive deterioration of muscle morphology in Bin1mck−/− mice. Similarly, in the mouse models for DNM2-CNM,26,27 the abnormal nuclei centralization seen in patients is not reproduced. Indeed, centronucleation might be an easier-to-see manifestation of the same problem that causes mislocalization of other organelles in these disorders. Furthermore, the percentage of centralized nuclei in CNM patients is highly variable and does not correlate with disease severity.30 A few studies linked centronucleation to muscle pathology.31 However, centronucleation during muscle regeneration does not lead to CNM, and muscle weakness is not often associated with central nuclei (e.g., nemaline myopathy). Therefore, the strong excitation-contraction coupling defects observed in Bin1mck−/− muscles is sufficient to cause the myopathy. Yet, several lines of evidence support the direct role of BIN1 in nuclei positioning. In particular, BIN1 controls nuclei and triad position with N-WASP in myofibers differentiated in vitro.32 Moreover, BIN1 links the nuclear envelope protein nesprin to both actin and microtubule cytoskeleton to locate nuclei in C. elegans seam cells.33 Altogether it suggests that BIN1 and DNM2 may be less important for nuclei position in mice than in humans. Conversely, BIN1 loss strongly alters mitochondria positioning in muscle, leading to accumulation of mitochondria in the center of myofibers, as typically seen in patients.3
T-tubules start forming at around 14 dpc in the mouse embryo and reach their final orientation soon after birth.34 Deleting Bin1 with the HSA promoter expressing from 9 dpc led to perinatal death,18 while using the MCK promoter expressing from 17 dpc is compatible with survival into adulthood. In addition, both the intramuscular injection of shRNA or U7 small nuclear RNA targeting Bin1 expression or splicing, respectively, in adult mice led to structural T-tubule defects and alteration of intracellular Ca2+ release.21,35 Altogether, it suggests BIN1 is a major protein for T-tubule biogenesis, as proposed in differentiating muscle cells and in cardiac muscle.10,36,37 BIN1 also has an important role in T-tubule maintenance in adults. Of note, decreasing DNM2 by about 50% in Bin1−/− mice through a genetic cross led to normal T-tubule and intracellular myofiber structure.18 Similarly, as shown here, decreasing DNM2 by about 50% in Bin1mck−/− mice with ASOs led to normal oxidative staining, fiber size, and morphology and increased muscle force. Hence, it is still unknown how T-tubules can form and/or maintain without BIN1. Either the BIN1-DNM2 balance is the major regulator, or DNM2 alone or with another BAR protein could control T-tubule biogenesis and maintenance.
Here, amelioration in all of the phenotypes tested correlates with the decrease of DNM2 protein levels. Decreasing DNM2 in a time window from 3 to 8 weeks partially improved muscle force defects while fully rescuing organelle mispositioning of Bin1mck−/− muscle fibers. This treatment period corresponds to the childhood-to-early-adulthood transition, when most BIN1-CNM patients are affected. It supports that DNM2 may be overactive in muscle lacking BIN1 and more generally that BAR-domain proteins could be negative regulators of dynamins, as shown before for dynamin 1 and endophilin.38 BIN1 and DNM2 form a complex, and the absence of BIN1 in Bin1mck−/− mice could result in an increase in DNM2 protein levels.12 In this line, DNM2 reduction via ASOs may represent a first translational approach to treat CNM related to BIN1 mutations. The existence of at least one founder BIN1 mutation in the Roma/Gypsy ethnic minority should foster further pre-clinical development. Of note, as we previously showed that DNM2 downregulation rescues mouse models for myotubular myopathy (MTM1-CNM) and dominant DNM2-CNM, it becomes clear that DNM2 ASOs could represent a common therapy to treat these different CNM forms.22 A 50% decrease in DNM2 in mice, either from embryogenesis in the Dnm2+/− knockout or upon postnatal ASO treatment,18,24,23,26 does not lead to detectable toxic effects. DNM2 downregulation is on clinical trial for MTM1 and DNM2-CNM patients older than 16 years of age (NCT04033159) by Dynacure, and our present data on this novel BIN1-CNM model support the inclusion of BIN1-CNM patients in such a trial.
BIN1 is also strongly associated with other diseases:39 BIN1 mis-splicing was proposed to be a main cause of the skeletal muscle phenotypes of myotonic dystrophy,35 decreased BIN1 plasma levels correlate with arrhythmia and with heart failure,40 BIN1 was found downregulated in several cancers including breast and colon cancers,41 and BIN1 is the second most important risk factor after APOE for late-onset Alzheimer disease.42 The therapeutic proof of concept validated here for CNMs may trigger additional avenues of research for these and other diseases.
Materials and methods
Animals
Animal care and experimentation was in accordance with French and European legislation and approved by institutional ethics committee (project numbers 2016031110589922 and 2016052510176016). Mice were placed in ventilated cages with free access to food and water in temperature-controlled rooms with 12 h daylight/dark cycles. The Bin1mck−/− mouse line was established by crossing mice floxed for exon 20 (Bin1fl/fl)18 with Bin1fl/+ mice expressing the Cre recombinase under the control of MCK-Cre (The Jackson Laboratory, Bar Harbor, ME, USA).43 Both lines were on a pure C57BL/6J background. Tail biopsies were used for genotyping testing for the floxed allele with 5′-AGTGACCTAGGACTGTACCCAGAG-3′ and 5′-ACAGGTAGGTGAAAGAGACTTGG-3′ and the Cre recombinase gene with 5′-GAACCTGATGGACATGTTCAGG-3′ and 5′-AGTGCGTTCGAACGCTAGAGCCTGT-3′. Male littermates were analyzed in this study.
ASO treatment
ASOs used in this study (Ionis Pharmaceuticals, Carlsbad, CA, USA) have phosphorothioate backbone and constrained ethyl (cET) modifications on the wings with a 3-10-3 gapmer design. They were designed to target all the murine isoforms of Dnm2 and were previously validated.23,24 Bin1fl/fl and Bin1mck−/− male mice were treated weekly from 3 weeks to 8 weeks of age with intraperitoneal injections of 25 mg/kg body weight of ASOs diluted in sterile PBS or sterile PBS only. Mice were randomly allocated in the ASO and PBS groups at weaning (3 weeks old). Mice were killed 3 days after the last injection for analysis.
RNA extraction and qRT-PCR
RNA was extracted from TA of 8-week-old mice using TRI Reagent (Molecular Research Center, Cincinnati, OH, USA) and cDNA synthesis performed with Superscript IV Transcriptase (Thermo Fisher Scientific, Waltham, MA, USA). For qPCR, cDNAs were amplified with SYBR Green Master Mix I (Roche Diagnostics, Basel, Switzerland) using 0.1 μM of forward and reverse primers in a Lightcycler 480 (Roche Diagnostics, Basel, Switzerland). Primer specificity was validated by melting curve and sequencing of amplicons. Several primers were used in previous studies and are summarized in Table S1.27,44, 45, 46 qRT-PCR was performed using technical triplicates, and relative mRNA level was calculated with Rpl27 as a housekeeping gene. The data displayed in the figures are the average of the triplicates and the result of a single experiment.
Protein expression studies
TA muscles from 8-week-old mice were lysed in radioimmunoprecipitation assay (RIPA) buffer supplemented with 1 mM PMSF and complete mini-EDTA-free protease inhibitor cocktail (Roche Diagnostics, Basel, Switzerland). Protein concentrations were determined using DC Protein Assay Kit (BioRad, Hercules, CA, USA). Samples were denaturated for 5 min at 95°C with 5× Lane Reducing Buffer (Thermo Fisher Scientific, Waltham, MA, USA) and loaded on 10% SDS-PAGE gel. Proteins were transferred to a nitrocellulose or polyvinylidene fluoride (PVDF) membrane using Transblot Turbo RTA Transfer Kit (BioRad, Hercules, CA, USA). Loading was controlled by Ponceau S staining before membrane blocking. Membranes were blocked for 1 h with 5% non-fat dry milk in 0.1% Tween 20 prior to incubations with primary and secondary antibodies. The primary and secondary antibodies used were: pan-BIN1 (rabbit, 1:1,000, R3623; homemade anti-BIN1 SH3 domain), pan-DNM2 (rabbit, 1:700, R2865; homemade anti-DNM2 PRD),47 prohibitin (rabbit, 1:500, ab28172), P62 (mouse, 1:1,000, H00008878-M01), LC3 (rabbit, 1:1,000, NB100-2220), peroxidase-coupled goat anti-rabbit (goat, 1/10,000, 112-036-04), and peroxidase-coupled goat anti-mouse (goat, 1/10,000, 115-036-068). The data displayed in the figures are the result of a single experiment.
Mouse phenotyping
Hanging test was performed weekly from 4 weeks to 8 weeks of age. Bin1fl/fl and Bin1mck−/− male mice were suspended on a cage lid for up to 60 s, and the time to fall was recorded. The test was replicated 3 times with a time interval of 5 min to rest, and the average hanging time was calculated. At 6 weeks of age, the mice performed a rotarod test with increasing speed (4–40 rpm) for a maximum of 5 min on a rotarod apparatus (Bioseb, Vitrolles, France). The mice were trained on day 1 and tested on the following 4 days, when the latency time to fail was recorded 3 times with 5 min resting intervals; the average latency was calculated for each experimental day. At 7 weeks of age, the mice performed the string test. Mice were suspended on a wire by their forelimbs and the time to reach the wire with their hindlimbs was recorded with a maximum time of 20 s; any fall was recorded as 20 s. The experiment was replicated 3 times with a resting time of 5 min. At 4 months of age, exhaustion test was performed using a treadmill apparatus (Bioseb). The experiment was initialized with 5 degrees of inclination and 25 cm/s of speed that increased up to 15 degrees of inclination and 41 cm/s over 150 min. Stimulation to keep the mouse on track was performed with an individual grip per lane delivering mild electric shocks (less than 0.2 mA). The test was stopped when mice receive more than 100 shocks/5 min or they stayed 20 consecutive seconds next to the electrodes, and maximal distance covered and latency time were recorded. Habituation for this test was done the day before the experiment for 10 min, with an inclination of 5 degrees and speed of 25 cm/s.
In situ muscle force
The force production of the TA was assessed with the Complete1300A Mouse Test System (Aurora Scientific, Aurora, ON, Canada). 8-week-, 4-month-, and 12-month-old Bin1fl/fl and Bin1mck−/− male mice were anesthetized through a triple shot cocktail by intraperitoneal injection of (1) domitor/fentanyl mix (2/0.28 mg/kg), (2) diazepam (8 mg/kg), and (3) domitor (0.28 mg/kg). The distal tendon was excised and attached to the muscle transducer while knees and feet were fixed. The sciatic nerve was stimulated by electrical pulses with increasing frequency from 1 to 125 Hz in order to measure maximal force. The specific force was determined by dividing the maximal force with the muscle weight (mg).
Histology, immunofluorescence, and electron microscopy
TA, gastrocnemius, FDB, and heart were dissected and frozen in liquid-nitrogen-cooled isopentane, and 8 μm cryosections were stained with H&E, SDH, NADH, or modified Gomori.
Fiber size distribution, circularity, and nuclei counting were determined on 8 μm cryosections from 8 week TA cryosections stained with Hoechst (Sigma-Aldrich, St. Louis, MO, USA) and Wheat Germ Agglutinin Alexa Fluor 555 conjugate (Thermo Fisher Scientific, Waltham, MA, USA) to stain nuclei and plasma membrane, respectively. Images were recorded using the Nanozoomer 2HT slide scanner (Hamamatsu, Japan), and the fiber size and nuclei counting analysis were performed using a homemade ImageJ plugin as described.48 For FDB from 8-week-old mice and TA from 4-month- and 12-month-old mice, fiber segmentation was performed using H&E images and Cellpose software for segmentation algorithm.49 Fiber MinFere diameter and nuclei counting were calculated or counted using ImageJ. For fiber circularity distribution, the fibers were classified with a value from 0 to 1, where 0 corresponds to a line and 1 to a circle, and one transversal section of the whole muscle was quantified. For immunofluorescence, 8 μm TA cryosections were fixed with paraformaldehyde 4%, blocked with fetal calf serum, and sequentially incubated with anti-prohibitin (rabbit, 1:500, ab28172), anti-LC3 (rabbit, 1:1,000, NB100-2220), anti-desmin (rabbit, 1:200, ab15200), or anti-TOMM20 (rabbit, 1:100, ab78547), and Alexa Fluor 594-coupled goat anti-rabbit (goat, 1/250, A11012) and/or Alexa Fluor 488-coupled goat anti-rabbit (goat, 1/250, A11008) and Hoechst (Sigma-Aldrich, St. Louis, MO, USA). Confocal microscopy images were recorded on an SP8 confocal microscope (Leica, Wetzlar, Germany), and image analysis and counting were performed with ImageJ.
For electron microscopy, small pieces of TA were fixed in 2.5% glutaraldehyde, 2.5% paraformaldehyde, and 50 mM Ca2+ in cacodylate buffer (0.1 M, pH 7.4). For inclusion, samples were washed in cacodylate buffer for 30 min and postfixed in 1% osmium tetroxide in 0.1 M cacodylate for 1 h at 4°C. Samples were dehydrated through graded alcohol (50%, 70%, 90%, and 100%) and propylene oxide for 30 min each and embedded in Epon 812. 70 nm ultrathin sections were cut on a Leica Ultracut microtome (Leica, Wetzlar, Germany), contrasted with uranyl acetate and lead citrate, and examined at 70 kv with a Morgagni 268D electron microscope (FEI, Electron Optics, Eindhoven, the Netherlands). Images were captured digitally by a Mega View III Camera (Soft Imaging System, Münster, Germany).
Electrophysiology, intracellular Ca2+ measurements
Single fibers were isolated from the FDB and interosseus muscles of 3 Bin1fl/fl and 3 Bin1mck−/− mice using previously described procedures.50 In brief, 8-week-old mice were anaesthetized by isoflurane inhalation and killed by cervical dislocation. Muscles were removed and incubated for 45 min at 37°C in Tyrode solution supplemented with type 1 collagenase (Sigma-Aldrich, St. Louis, MO, USA). Single fibers were then separated by gentle mechanical trituration within a culture μ-dish (Ibidi, Planegg/Martinsried, Germany).
For electrophysiology measurements, fibers from the FDB were used (5–6 fibers per mouse). The bottom of a μ-dish was first covered with a thin layer of silicone grease, and muscle trituration was performed in culture medium containing 10% fetal bovine serum (MI199; Eurobio, France). Isolated fibers were painted with silicone so that only a short portion of one fiber end remained out of the silicone, in contact with the bathing solution. The culture medium was then replaced by the experimental extracellular solution containing 140 mM TEA-methane-sulfonate, 2.5 mM CaCl2, 2 mM MgCl2, 1 mM 4-aminopyridine, 10 mM HEPES, and 0.002 mM tetrodotoxin (pH 7.2). Voltage-clamp was achieved with a glass micropipette connected to an RK-400 patch-clamp amplifier (Bio-Logic, Claix, France) in whole-cell configuration, in combination with an analog-digital converter (Digidata 1440A, Axon Instruments, Foster City, CA, USA) controlled by pClamp 9 software (Axon Instruments). The micropipette was filled with a solution containing 120 mM K-glutamate, 5 mM Na2-ATP, 5 mM Na2-phosphocreatine, 5.5 mM MgCl2, 5 mM glucose, 15 mM EGTA, 6 mM CaCl2, 0.1 mM rhod-2, and 5 mM HEPES (pH 7.2). The pipette tip was inserted into the silicone-insulated part of the fiber. It was gently crushed against the bottom of the μ-dish to ease dialysis and reduce series resistance. The fiber cytosol was dialyzed for 30 min to allow for intracellular equilibration of the solution before starting measurements.
CaV1.1 Ca2+ current and rhod-2 (Thermo Fisher Scientific, Illkirch, France) intracellular Ca2+ transients were simultaneously measured in response to 0.5-s-long depolarizing steps from a holding voltage of −80 mV. For CaV1.1 current analysis, the linear leak component of the current during the test pulses was removed by subtracting the adequately scaled current value measured during a 20 mV hyperpolarizing step. A residual leak component (referred to as slope) was found to be larger in fibers from Bin1fl/fl mice than in fibers from Bin1mck−/− mice. Thus, for each fiber, the voltage dependence of peak Ca2+ current values (normalized to fiber capacitance) was fitted with the following equation: I(V) = Gmax(V − Vrev)/(1 + [exp(V0.5 − V)/k]) + slope(V + 80), with I(V) the peak current density at the command voltage V, Gmax the maximum conductance, Vrev the apparent reversal potential, V0.5 the half-activation potential, k the steepness factor, and slope the conductance of the residual linear component.
Fluorescence imaging was conducted with a Zeiss LSM-800 system equipped with a 63× oil immersion objective (numerical aperture 1.4). For detection of rhod-2 fluorescence, excitation was from a 561 nm diode laser, and fluorescence was collected above 570 nm. Fluorescence changes were imaged using the line-scan mode (x,t) of the system with a scanning frequency of 1.02 ms per line and expressed as F/F0, where F0 is the baseline fluorescence. Quantification of the Ca2+ release flux underlying the rhod-2 Ca2+ transients was performed as previously described.50,51 In each fiber, the voltage dependence of the peak rate of Ca2+ release was fitted with a Boltzmann function.
For imaging the T-tubule network, interosseus muscle fibers were incubated for 30 min in the presence of 10 μM di-8-anepps (Thermo Fisher Scientific, Illkirch, France). Di-8-anepps was excited with a 488 nm diode laser, and fluorescence was collected above 510 nm. The T-tubule density index (9–13 fibers per mouse) was estimated as described previously.52
Statistical analysis
All experiments were performed and analyzed in a blinded manner, and the investigators were unaware of the genotype of the mice. The normal distribution of the data was assessed using the Shapiro-Wilk test and presented as mean ± standard error of the mean (SEM), with the exception of data compared with Kruskal-Wallis test, where the data are presented as median ± interquartile range. For normally distributed data, the significance of changes was examined by two-tailed Student’s t test (with or without Welch’s correction if variance was different or not) for comparison of 2 groups or by one-way ANOVA followed by Tukey’s post hoc test for comparison of more than 2 groups. In the case of not-normally distributed data, the Mann-Whitney test was used to compare 2 groups and Kruskal-Wallis followed by Dunn’s multiple comparison test to compare more than 2 groups. The significant difference of birth ratio was assessed by chi-square test. Significant differences are illustrated as ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 and ∗∗∗∗p < 0.0001.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its supplemental material.
Acknowledgments
We thank IGBMC and ICS platforms for animal care, histology, photonic and electron imaging; Shuling Guo and Brett Monia for providing the Dnm2 antisense oligonucleotide; Quentin Giraud for his help with mice; and Doulaye Dembélé for statistical analysis assistance. The creation of the Bin1fl/fl mouse was done with Phenomin-ICS, Illkirch, France. This study was supported by institute funding from Institut National de la Santé et de la Recherche Médicale, Centre National de la Recherche Scientifique, Université de Strasbourg and by grants from the Agence Nationale de la Recherche ANR-10-LABX-0030-INRT, a French State fund managed by the Agence Nationale de la Recherche under the frame program Investissements d’Avenir ANR-10-IDEX-0002-02, Réseau National des Génopoles (RNG), the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement (675392), Muscular Dystrophy Association (576154), Fondation pour la Recherche Médicale (201903007992), and AFM-Téléthon (21267, 22734, MyoNeurALP # 5.3.4.4). R.G.O. had a CIFRE fellowship obtained with Dynacure, and R.S.R. a fellowship from Fondation pour la Recherche Médicale (PLP20170939073). All experiments were done at the IGBMC.
Author contributions
J.L. designed the study; B.S.C., V.J., and J.L. supervised research; V.N., R.S.R., F.J.-F., A.M., R.G.O., N.M., V.M.L., D.R., and C.K. performed research; V.N., R.S.R., F.J-F., R.G.O., M.G., N.M., D.R., and V.J. analyzed data; R.S.R. and J.L. wrote the manuscript.
Declaration of interests
J.L. and B.S.C. are co-founders of Dynacure, B.S.C. is employed by Dynacure, and R.G.O. has a CIFRE fellowship with Dynacure. All other authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.08.006.
Supplemental information
References
- 1.Jungbluth H., Wallgren-Pettersson C., Laporte J. Centronuclear (myotubular) myopathy. Orphanet J. Rare Dis. 2008;3:26. doi: 10.1186/1750-1172-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gonorazky H.D., Bönnemann C.G., Dowling J.J. The genetics of congenital myopathies. Handb. Clin. Neurol. 2018;148:549–564. doi: 10.1016/B978-0-444-64076-5.00036-3. [DOI] [PubMed] [Google Scholar]
- 3.Romero N.B. Centronuclear myopathies: a widening concept. Neuromuscul. Disord. 2010;20:223–228. doi: 10.1016/j.nmd.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 4.Lawlor M.W., Beggs A.H., Buj-Bello A., Childers M.K., Dowling J.J., James E.S., Meng H., Moore S.A., Prasad S., Schoser B., Sewry C.A. Skeletal Muscle Pathology in X-Linked Myotubular Myopathy: Review With Cross-Species Comparisons. J. Neuropathol. Exp. Neurol. 2016;75:102–110. doi: 10.1093/jnen/nlv020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schartner V., Laporte J., Böhm J. Abnormal Excitation-Contraction Coupling and Calcium Homeostasis in Myopathies and Cardiomyopathies. J. Neuromuscul. Dis. 2019;6:289–305. doi: 10.3233/JND-180314. [DOI] [PubMed] [Google Scholar]
- 6.Lornage X., Schartner V., Balbueno I., Biancalana V., Willis T., Echaniz-Laguna A., Scheidecker S., Quinlivan R., Fardeau M., Malfatti E., et al. Clinical, histological, and genetic characterization of PYROXD1-related myopathy. Acta Neuropathol. Commun. 2019;7:138. doi: 10.1186/s40478-019-0781-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pelin K., Wallgren-Pettersson C. Update on the Genetics of Congenital Myopathies. Semin. Pediatr. Neurol. 2019;29:12–22. doi: 10.1016/j.spen.2019.01.005. [DOI] [PubMed] [Google Scholar]
- 8.Toussaint A., Cowling B.S., Hnia K., Mohr M., Oldfors A., Schwab Y., Yis U., Maisonobe T., Stojkovic T., Wallgren-Pettersson C., et al. Defects in amphiphysin 2 (BIN1) and triads in several forms of centronuclear myopathies. Acta Neuropathol. 2011;121:253–266. doi: 10.1007/s00401-010-0754-2. [DOI] [PubMed] [Google Scholar]
- 9.Hohendahl A., Roux A., Galli V. Structural insights into the centronuclear myopathy-associated functions of BIN1 and dynamin 2. J. Struct. Biol. 2016;196:37–47. doi: 10.1016/j.jsb.2016.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee E., Marcucci M., Daniell L., Pypaert M., Weisz O.A., Ochoa G.C., Farsad K., Wenk M.R., De Camilli P. Amphiphysin 2 (Bin1) and T-tubule biogenesis in muscle. Science. 2002;297:1193–1196. doi: 10.1126/science.1071362. [DOI] [PubMed] [Google Scholar]
- 11.Böhm J., Biancalana V., Malfatti E., Dondaine N., Koch C., Vasli N., Kress W., Strittmatter M., Taratuto A.L., Gonorazky H., et al. Adult-onset autosomal dominant centronuclear myopathy due to BIN1 mutations. Brain. 2014;137:3160–3170. doi: 10.1093/brain/awu272. [DOI] [PubMed] [Google Scholar]
- 12.Nicot A.S., Toussaint A., Tosch V., Kretz C., Wallgren-Pettersson C., Iwarsson E., Kingston H., Garnier J.M., Biancalana V., Oldfors A., et al. Mutations in amphiphysin 2 (BIN1) disrupt interaction with dynamin 2 and cause autosomal recessive centronuclear myopathy. Nat. Genet. 2007;39:1134–1139. doi: 10.1038/ng2086. [DOI] [PubMed] [Google Scholar]
- 13.Cabrera-Serrano M., Mavillard F., Biancalana V., Rivas E., Morar B., Hernández-Laín A., Olive M., Muelas N., Khan E., Carvajal A., et al. A Roma founder BIN1 mutation causes a novel phenotype of centronuclear myopathy with rigid spine. Neurology. 2018;91:e339–e348. doi: 10.1212/WNL.0000000000005862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gowrisankaran S., Wang Z., Morgan D.G., Milosevic I., Mim C. Cells Control BIN1-Mediated Membrane Tubulation by Altering the Membrane Charge. J. Mol. Biol. 2020;432:1235–1250. doi: 10.1016/j.jmb.2019.12.001. [DOI] [PubMed] [Google Scholar]
- 15.Razzaq A., Robinson I.M., McMahon H.T., Skepper J.N., Su Y., Zelhof A.C., Jackson A.P., Gay N.J., O’Kane C.J. Amphiphysin is necessary for organization of the excitation-contraction coupling machinery of muscles, but not for synaptic vesicle endocytosis in Drosophila. Genes Dev. 2001;15:2967–2979. doi: 10.1101/gad.207801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith L.L., Gupta V.A., Beggs A.H. Bridging integrator 1 (Bin1) deficiency in zebrafish results in centronuclear myopathy. Hum. Mol. Genet. 2014;23:3566–3578. doi: 10.1093/hmg/ddu067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Böhm J., Vasli N., Maurer M., Cowling B.S., Shelton G.D., Kress W., Toussaint A., Prokic I., Schara U., Anderson T.J., et al. Altered splicing of the BIN1 muscle-specific exon in humans and dogs with highly progressive centronuclear myopathy. PLoS Genet. 2013;9:e1003430. doi: 10.1371/journal.pgen.1003430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cowling B.S., Prokic I., Tasfaout H., Rabai A., Humbert F., Rinaldi B., Nicot A.S., Kretz C., Friant S., Roux A., Laporte J. Amphiphysin (BIN1) negatively regulates dynamin 2 for normal muscle maturation. J. Clin. Invest. 2017;127:4477–4487. doi: 10.1172/JCI90542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muller A.J., Baker J.F., DuHadaway J.B., Ge K., Farmer G., Donover P.S., Meade R., Reid C., Grzanna R., Roach A.H., et al. Targeted disruption of the murine Bin1/Amphiphysin II gene does not disable endocytosis but results in embryonic cardiomyopathy with aberrant myofibril formation. Mol. Cell. Biol. 2003;23:4295–4306. doi: 10.1128/MCB.23.12.4295-4306.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prokic I., Cowling B.S., Kutchukian C., Kretz C., Tasfaout H., Gache V., Hergueux J., Wendling O., Ferry A., Toussaint A., et al. Differential physiological roles for BIN1 isoforms in skeletal muscle development, function and regeneration. Dis. Model. Mech. 2020;13:dmm044354. doi: 10.1242/dmm.044354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tjondrokoesoemo A., Park K.H., Ferrante C., Komazaki S., Lesniak S., Brotto M., Ko J.K., Zhou J., Weisleder N., Ma J. Disrupted membrane structure and intracellular Ca2+ signaling in adult skeletal muscle with acute knockdown of Bin1. PLoS ONE. 2011;6:e25740. doi: 10.1371/journal.pone.0025740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tasfaout H., Cowling B.S., Laporte J. Centronuclear myopathies under attack: A plethora of therapeutic targets. J. Neuromuscul. Dis. 2018;5:387–406. doi: 10.3233/JND-180309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buono S., Ross J.A., Tasfaout H., Levy Y., Kretz C., Tayefeh L., Matson J., Guo S., Kessler P., Monia B.P., et al. Reducing dynamin 2 (DNM2) rescues DNM2-related dominant centronuclear myopathy. Proc. Natl. Acad. Sci. U. S. A. 2018;115:11055–11071. doi: 10.1073/pnas.1808170115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tasfaout H., Buono S., Guo S., Kretz C., Messaddeq N., Booten S., Greenlee S., Monia B.P., Cowling B.S., Laporte J. Antisense oligonucleotide-mediated Dnm2 knockdown prevents and reverts myotubular myopathy in mice. Nat. Commun. 2017;8:15661. doi: 10.1038/ncomms15661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cowling B.S., Chevremont T., Prokic I., Kretz C., Ferry A., Coirault C., Koutsopoulos O., Laugel V., Romero N.B., Laporte J. Reducing dynamin 2 expression rescues X-linked centronuclear myopathy. J. Clin. Invest. 2014;124:1350–1363. doi: 10.1172/JCI71206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Massana Muñoz X., Kretz C., Silva-Rojas R., Ochala J., Menuet A., Romero N.B., Cowling B.S., Laporte J. Physiological impact and disease reversion for the severe form of centronuclear myopathy linked to dynamin. JCI Insight. 2020;5:e137899. doi: 10.1172/jci.insight.137899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Durieux A.C., Vignaud A., Prudhon B., Viou M.T., Beuvin M., Vassilopoulos S., Fraysse B., Ferry A., Lainé J., Romero N.B., et al. A centronuclear myopathy-dynamin 2 mutation impairs skeletal muscle structure and function in mice. Hum. Mol. Genet. 2010;19:4820–4836. doi: 10.1093/hmg/ddq413. [DOI] [PubMed] [Google Scholar]
- 28.Hnia K., Tronchère H., Tomczak K.K., Amoasii L., Schultz P., Beggs A.H., Payrastre B., Mandel J.L., Laporte J. Myotubularin controls desmin intermediate filament architecture and mitochondrial dynamics in human and mouse skeletal muscle. J. Clin. Invest. 2011;121:70–85. doi: 10.1172/JCI44021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tasfaout H., Lionello V.M., Kretz C., Koebel P., Messaddeq N., Bitz D., Laporte J., Cowling B.S. Single Intramuscular Injection of AAV-shRNA Reduces DNM2 and Prevents Myotubular Myopathy in Mice. Mol. Ther. 2018;26:1082–1092. doi: 10.1016/j.ymthe.2018.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pierson C.R., Agrawal P.B., Blasko J., Beggs A.H. Myofiber size correlates with MTM1 mutation type and outcome in X-linked myotubular myopathy. Neuromuscul. Disord. 2007;17:562–568. doi: 10.1016/j.nmd.2007.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roman W., Martins J.P., Carvalho F.A., Voituriez R., Abella J.V.G., Santos N.C., Cadot B., Way M., Gomes E.R. Myofibril contraction and crosslinking drive nuclear movement to the periphery of skeletal muscle. Nat. Cell Biol. 2017;19:1189–1201. doi: 10.1038/ncb3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Falcone S., Roman W., Hnia K., Gache V., Didier N., Lainé J., Auradé F., Marty I., Nishino I., Charlet-Berguerand N., et al. N-WASP is required for Amphiphysin-2/BIN1-dependent nuclear positioning and triad organization in skeletal muscle and is involved in the pathophysiology of centronuclear myopathy. EMBO Mol. Med. 2014;6:1455–1475. doi: 10.15252/emmm.201404436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.D’Alessandro M., Hnia K., Gache V., Koch C., Gavriilidis C., Rodriguez D., Nicot A.S., Romero N.B., Schwab Y., Gomes E., et al. Amphiphysin 2 Orchestrates Nucleus Positioning and Shape by Linking the Nuclear Envelope to the Actin and Microtubule Cytoskeleton. Dev. Cell. 2015;35:186–198. doi: 10.1016/j.devcel.2015.09.018. [DOI] [PubMed] [Google Scholar]
- 34.Takekura H., Flucher B.E., Franzini-Armstrong C. Sequential docking, molecular differentiation, and positioning of T-Tubule/SR junctions in developing mouse skeletal muscle. Dev. Biol. 2001;239:204–214. doi: 10.1006/dbio.2001.0437. [DOI] [PubMed] [Google Scholar]
- 35.Fugier C., Klein A.F., Hammer C., Vassilopoulos S., Ivarsson Y., Toussaint A., Tosch V., Vignaud A., Ferry A., Messaddeq N., et al. Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat. Med. 2011;17:720–725. doi: 10.1038/nm.2374. [DOI] [PubMed] [Google Scholar]
- 36.Hong T., Yang H., Zhang S.S., Cho H.C., Kalashnikova M., Sun B., Zhang H., Bhargava A., Grabe M., Olgin J., et al. Cardiac BIN1 folds T-tubule membrane, controlling ion flux and limiting arrhythmia. Nat. Med. 2014;20:624–632. doi: 10.1038/nm.3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caldwell J.L., Smith C.E., Taylor R.F., Kitmitto A., Eisner D.A., Dibb K.M., Trafford A.W. Dependence of cardiac transverse tubules on the BAR domain protein amphiphysin II (BIN-1) Circ. Res. 2014;115:986–996. doi: 10.1161/CIRCRESAHA.116.303448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hohendahl A., Talledge N., Galli V., Shen P.S., Humbert F., De Camilli P., Frost A., Roux A. Structural inhibition of dynamin-mediated membrane fission by endophilin. eLife. 2017;6:e26856. doi: 10.7554/eLife.26856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prokic I., Cowling B.S., Laporte J. Amphiphysin 2 (BIN1) in physiology and diseases. J. Mol. Med. (Berl.) 2014;92:453–463. doi: 10.1007/s00109-014-1138-1. [DOI] [PubMed] [Google Scholar]
- 40.Hong T.T., Cogswell R., James C.A., Kang G., Pullinger C.R., Malloy M.J., Kane J.P., Wojciak J., Calkins H., Scheinman M.M., et al. Plasma BIN1 correlates with heart failure and predicts arrhythmia in patients with arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm. 2012;9:961–967. doi: 10.1016/j.hrthm.2012.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prendergast G.C., Muller A.J., Ramalingam A., Chang M.Y. BAR the door: cancer suppression by amphiphysin-like genes. Biochim. Biophys. Acta. 2009;1795:25–36. doi: 10.1016/j.bbcan.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seshadri S., Fitzpatrick A.L., Ikram M.A., DeStefano A.L., Gudnason V., Boada M., Bis J.C., Smith A.V., Carassquillo M.M., Lambert J.C., et al. CHARGE Consortium. GERAD1 Consortium. EADI1 Consortium Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA. 2010;303:1832–1840. doi: 10.1001/jama.2010.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brüning J.C., Michael M.D., Winnay J.N., Hayashi T., Hörsch D., Accili D., Goodyear L.J., Kahn C.R. A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol. Cell. 1998;2:559–569. doi: 10.1016/s1097-2765(00)80155-0. [DOI] [PubMed] [Google Scholar]
- 44.Honda M., Hidaka K., Fukada S.I., Sugawa R., Shirai M., Ikawa M., Morisaki T. Vestigial-like 2 contributes to normal muscle fiber type distribution in mice. Sci. Rep. 2017;7:7168. doi: 10.1038/s41598-017-07149-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuno A., Hosoda R., Sebori R., Hayashi T., Sakuragi H., Tanabe M., Horio Y. Resveratrol Ameliorates Mitophagy Disturbance and Improves Cardiac Pathophysiology of Dystrophin-deficient mdx Mice. Sci. Rep. 2018;8:15555. doi: 10.1038/s41598-018-33930-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thomas K.C., Zheng X.F., Garces Suarez F., Raftery J.M., Quinlan K.G., Yang N., North K.N., Houweling P.J. Evidence based selection of commonly used RT-qPCR reference genes for the analysis of mouse skeletal muscle. PLoS ONE. 2014;9:e88653. doi: 10.1371/journal.pone.0088653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cowling B.S., Toussaint A., Amoasii L., Koebel P., Ferry A., Davignon L., Nishino I., Mandel J.L., Laporte J. Increased expression of wild-type or a centronuclear myopathy mutant of dynamin 2 in skeletal muscle of adult mice leads to structural defects and muscle weakness. Am. J. Pathol. 2011;178:2224–2235. doi: 10.1016/j.ajpath.2011.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Silva-Rojas R., Treves S., Jacobs H., Kessler P., Messaddeq N., Laporte J., Böhm J. STIM1 over-activation generates a multi-systemic phenotype affecting the skeletal muscle, spleen, eye, skin, bones and immune system in mice. Hum. Mol. Genet. 2019;28:1579–1593. doi: 10.1093/hmg/ddy446. [DOI] [PubMed] [Google Scholar]
- 49.Stringer C., Wang T., Michaelos M., Pachitariu M. Cellpose: a generalist algorithm for cellular segmentation. Nat. Methods. 2021;18:100–106. doi: 10.1038/s41592-020-01018-x. [DOI] [PubMed] [Google Scholar]
- 50.Lefebvre R., Pouvreau S., Collet C., Allard B., Jacquemond V. Whole-cell voltage clamp on skeletal muscle fibers with the silicone-clamp technique. Methods Mol. Biol. 2014;1183:159–170. doi: 10.1007/978-1-4939-1096-0_9. [DOI] [PubMed] [Google Scholar]
- 51.Kutchukian C., Lo Scrudato M., Tourneur Y., Poulard K., Vignaud A., Berthier C., Allard B., Lawlor M.W., Buj-Bello A., Jacquemond V. Phosphatidylinositol 3-kinase inhibition restores Ca2+ release defects and prolongs survival in myotubularin-deficient mice. Proc. Natl. Acad. Sci. USA. 2016;113:14432–14437. doi: 10.1073/pnas.1604099113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kutchukian C., Szentesi P., Allard B., Trochet D., Beuvin M., Berthier C., Tourneur Y., Guicheney P., Csernoch L., Bitoun M., Jacquemond V. Impaired excitation-contraction coupling in muscle fibres from the dynamin2R465W mouse model of centronuclear myopathy. J. Physiol. 2017;595:7369–7382. doi: 10.1113/JP274990. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplemental material.