

Abstract
CD19-directed chimeric antigen receptor (CAR) T cells have yielded impressive response rates in refractory/relapse B cell acute lymphoblastic leukemia (B-ALL); however, most patients ultimately relapse due to poor CAR T cell persistence or resistance of either CD19+ or CD19− B-ALL clones. CD22 is a pan-B marker whose expression is maintained in both CD19+ and CD19− relapses. CD22-CAR T cells have been clinically used in B-ALL patients, although relapse also occurs. T cells engineered with a tandem CAR (Tan-CAR) containing in a single construct both CD19 and CD22 scFvs may be advantageous in achieving higher remission rates and/or preventing antigen loss. We have generated and functionally validated using cutting-edge assays a 4-1BB-based CD22/CD19 Tan-CAR using in-house-developed novel CD19 and CD22 scFvs. Tan-CAR-expressing T cells showed similar in vitro expansion to CD19-CAR T cells with no increase in tonic signaling. CRISPR-Cas9-edited B-ALL cells confirmed the bispecificity of the Tan-CAR. Tan-CAR was as efficient as CD19-CAR in vitro and in vivo using B-ALL cell lines, patient samples, and patient-derived xenografts (PDXs). Strikingly, the robust antileukemic activity of the Tan-CAR was slightly more effective in controlling the disease in long-term follow-up PDX models. This Tan-CAR construct warrants a clinical appraisal to test whether simultaneous targeting of CD19 and CD22 enhances leukemia eradication and reduces/delays relapse rates and antigen loss.
Keywords: B-ALL, tandem CAR T cells, CD19, CD22, patient-derived xenografts, relapse
Graphical abstract
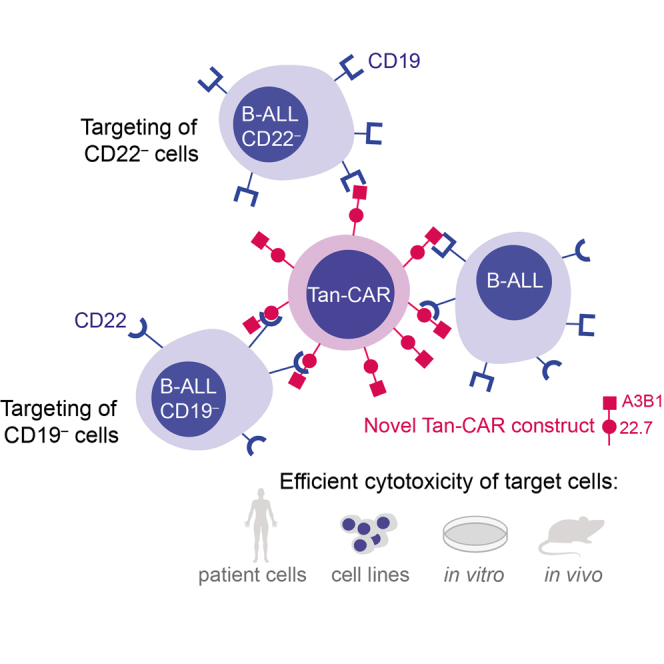
Zanetti and colleagues have developed a novel tandem chimeric antigen receptor (Tan-CAR), targeting both CD19 and CD22 antigens. They showed its specific and effective antileukemic activity against B cell acute lymphoblastic leukemia cells using cutting-edge in vitro and in vivo assays.
Introduction
B cell acute lymphoblastic leukemia (B-ALL) is an aggressive hematologic malignancy characterized by the clonal expansion of CD19+ B cell precursors.1 B-ALL is the most common malignancy in children, and although less prevalent in adults, it is associated with an unfavorable prognosis.2 Although >90% of patients achieve complete remission after first-line treatment, the prognosis of those with refractory/relapse (R/R) B-ALL is dismal, with a 5-year overall survival of <20%.2,3
Adoptive transfer of T cells engineered to express artificial chimeric antigen receptors (CARs) targeting tumor cell surface-associated antigens (Ag) represents a revolutionary approach in cancer immunotherapy.4 CD19 represents the ideal CAR T cell therapy for B-ALL, because it is homogeneously expressed on malignant cells, its off-target expression is limited to normal B cells, and CD19-CAR T cell-mediated B cell aplasia is easily manageable clinically through the administration of gamma-immunoglobulins (γ-Igs).5 CD19-CAR T cells have revolutionized the treatment of R/R B-ALL with complete response rates of ∼80%–90%; however, 40%–60% of patients treated with CD19-targeted immunotherapy still relapse after 1 year.3,6 Two major types of relapse have been distinguished7, 8, 9, 10: relapse that remains CD19+, typically linked to poor T cell function or loss of CAR T cell persistence, and relapse CD19−, in which the disease recurs with loss of CD19, representing a novel “stem cell origin-related” mechanism of tumor escape.
Another promising target for CAR T cell therapy in B-ALL is CD22. CD22 is a pan B cell surface Ag expressed in most cases of B-ALL.11 CD22-targeted immunotherapy has been developed and tested in several studies,12, 13, 14, 15 and the results from initial clinical trials in children with either CD19+ or CD19− R/R B-ALL are promising, but relapses are also common12,14 and are in a proportion of patients associated with a downregulation of CD22 expression.12 R/R B-ALL thus remains clinically challenging.
To overcome leukemia immunoediting during CAR T cell therapy for the treatment of B-ALL, compensatory strategies such as dual-Ag or multi-Ag targeting by CARs will likely be needed.10,16 One potential strategy to reduce immunological pressure over a single Ag and to offset tumor Ag-loss relapse involves generating T cells with one CAR molecule containing two different binding domains in tandem (Tan-CAR),17 which appears to enhance the potency and antitumor activity in vivo when compared with single CARs.18, 19, 20 Several clinical trials exploring combinatorial anti-CD19 and anti-CD22 strategies are under way to optimize response rates and reduce the risk of leukemic cell escape to CAR T cell therapy in B-ALL.17,21
We have developed, characterized, and functionally validated a 4-1BB-based CD22/CD19 Tan-CAR using in-house-developed novel CD19 and CD22 monoclonal antibodies (mAbs). Here, we report a specific and efficient in vitro and in vivo elimination of B-ALL cell lines, primary B-ALL cells, and B-ALL patient-derived xenografts (PDXs) cells with our Tan-CAR construct that is on par with CD19-CAR, but is slightly more effective in vivo in controlling the disease in long-term follow-up B-ALL PDXs.
Results
Generation and expression of CD22/CD19 Tan-CARs on human T cells
We sought to evaluate a strategy to reduce immunological pressure over a single Ag (CD19) in B-ALL by modifying T cells with one CAR molecule containing both CD19- and CD22-binding domains, generating a CD22/CD19 Tan-CAR. To compare the effectiveness of the CD22/CD19 Tan-CAR and CD19-CAR, we designed two second-generation Tan-CARs: a short (S) and a long (L) version (Figure 1A). We used a proprietary anti-CD22 single-chain fragment variable (scFv)15 and a clinically validated anti-CD19 scFv.22, 23, 24, 25 Constructs S and L differ only in the length of the flexible linker sequence connecting the anti-CD22 scFv and anti-CD19 scFv (Figure 1A). We maintained the original linkers between the VH and VL domains from each single CAR. The hinge, transmembrane (TM), and signaling domains were identical for CD19-CAR and Tan-CARs constructs, each encoding a CD8-derived hinge and TM domain, a 4-1BB signaling domain, and a CD3ζ-derived signaling domain. As an experimental control, we used an “empty” CD8TM-4-1BB-CD3ζ CAR construct without a scFv (Mock-CAR) (Figure 1A). To assess the transduction efficiency and to track the CAR expression, we incorporated a GFP reporter gene after a 2A ribosomal skip sequence at the C-terminal of the CAR sequence.
Figure 1.

Generation, transduction, expansion, and detection of Tan-CAR T cells
(A) Scheme of the CAR constructs used: (S) and (L) denote short -(G4S)4- and long -(G4S)7- size for the inter-scFv linker, respectively. (B) T cell activation after 48-h exposure to anti-CD3/CD28, plus IL-7 and IL-15 evaluated by CD25 and CD69 expression by FACS (left panel) and by light microscopy analysis of activated T cell clusters (right panel, magnification ×40) (n = 5). (C) Transduction efficiency (left panel) and expansion (right panel) of activated T cells transduced with the indicated CARs (n = 5). The arrow represents the time of CAR T cell harvesting for CAR detection in the surface of T cells. (D) Representative flow cytometry plots of CAR expression on human T cells detected as GFP+ (top panels), anti-scFv (second row), CD19-Fc/anti-Fc-PE (third row), and anti-HisTag-APC or CD22-HisTag/anti-HisTag-APC (bottom row). CAR-transduced T cells are shown in green. (E) Representative CD19 and CD22 mean fluorescence intensity (MFI) quantification by FACS of the GFP+ CAR T cells shown in (D). MFI values are indicated in the upper right corner. (F) Representative CAR detection on human CD4+ and CD8+ T cells. (G) VCN representing the number of integrated copies of the provirus (CAR vector) per transduced genome. Each symbol represents a different donor (n = 3). See also Figure S2.
Human T cells were activated as described in the Method details section, and successful T cell activation was determined 48 h later by measuring CD25 and CD69 expression by fluorescence-activated cell sorting (FACS) and by observing the formation of activated T cell clusters with light microscopy (Figure 1B). T cells were transduced with CAR-expressing lentivectors and expanded in the presence of interleukin-7 (IL-7) and IL-15.7,15,24,26,27 The results showed that the transduction efficiency of Tan-CARs was lower than that of the Mock-CAR or CD19-CAR (Figure 1C). Expression of Mock- and CD19-CARs on the surface of transduced T cells ranged from 30% to 50%, whereas the expression of Tan(S)-CAR ranged from 5% to 26% and Tan(L)-CAR ranged from 1% to 13%. Of note, retroviral infection substantially increased the transduction efficiencies of T cells with the Tan(S)-CAR to ∼30%–40% (Figures S2A–S2C), facilitating the clinical translation of this Tan(S)-CAR. All CAR T cells had an identical proliferative capacity and expanded 200-fold over a 15-day period, with the exception of the Tan(L)-CAR, which expanded 50% less (Figure 1C). CAR transduction efficiency in human T cells was analyzed by FACS using GFP reporter expression, and CAR expression was detected on the surface of transduced T cells using an anti-scFv antibody (Figure 1D). To verify that both scFvs were intact, we incubated CAR T cells with human recombinant CD19-Fc protein or CD22-HisTag protein. Both Tan-CAR constructs retained the ability to bind CD19 and CD22 Ags, but Tan(L)-CAR showed a slightly lower binding on the surface. As expected, CD19-CAR bound to CD19-Fc but not CD22-HisTag. Mock-CAR surface expression was detected using an anti-HisTag antibody (Figures 1D and 1E). Binding of Tan-CARs to recombinant CD22 was similar to that observed for the single CD22-CAR.15 The expression of all CARs could be easily detected in both CD4+ and CD8+ T cells (Figure 1F). These observations were reproduced in CAR T cells generated from n = 5 healthy donors (HDs). In addition, we analyzed the vector copy number (VCN) of the CAR vector (provirus) in the T cells. We found 6–7 proviral copies genome integrated in Mock-CAR and CD19-CAR, whereas for Tan(S)-CAR up to an average of 12 proviral copies were found per transduced cell, indicating a proper genome integration of the Tan(S)-CAR vector into the T cells (Figure 1G). Finally, kinetics of T cell volume of the CAR T cells together with analysis of the expression of activation (CD69 and CD25), differentiation (CCR7, CD27, and CD45RO), and inhibition (LAG3, TIM3, and PD-1) markers on the indicated CAR T cells throughout the 21-day in vitro expansion revealed no differences in tonic signaling of the different CARs as compared to activated untransduced T cells (Figure S3). In all subsequent experiments, we normalized the rates of transduction efficiency between vectors by diluting with non-transduced T cells to functionally compare CAR T cells with equal transduction rates.
CD22/CD19 Tan-CAR T cells display a highly efficient and bispecific cytotoxicity
We next evaluated and compared the antileukemic activity of Tan-CARs by measuring the cytotoxicity and secretion of pro-inflammatory cytokines against B-ALL cell lines in vitro. Both Tan(S)-CAR and Tan(L)-CAR displayed a comparable massive cytotoxicity activity against SEM and NALM6 cell lines after 48 h, at a 1:1 effector:target (E:T) ratio (Figure 2A). Of note, Tan(S)-CAR had a slightly stronger activity than Tan(L)-CAR against SEM cells at a lower E:T ratio (Figure 2B). Both Tan-CARs produced equivalent amounts of IL-2, interferon γ (IFN-γ), and tumor necrosis factor α (TNF-α) (Figure 2C). This, together with the lower expansion of Tan(L)-CAR (Figure 1C), made us choose the Tan(S)-CAR for subsequent experiments.
Figure 2.

Robust antileukemic efficacy and specificity of both Tan(S)- and Tan(L)-CAR T cells in vitro
(A and B) Absolute number (A) and percentage (B) of alive target cells (SEM or NALM6) after 48-h incubation with the indicated CAR T cells and E:T ratios. Results in (B) are normalized to Mock-CAR data. PBMCs from n = 3 independent HDs. (C) Production of the pro-inflammatory cytokines IL-2, IFN-γ, and TNF-α by CAR T cells after 48-h exposure to SEM or NALM6 target cells at 1:1 E:T ratio. PBMCs from n = 3 independent HDs. (D) Different CD22/CD19 combinatorial phenotypes of CRISPR/Cas9-edited SEM cells. (E and F) Absolute number (E) and percentage (F) of alive target cells after 48-h incubation with the indicated CAR T cells and E:T ratios. Results in (F) are normalized to Mock-CAR data. PBMCs from n = 5 independent HDs. (G) Production of IL-2, IFN-γ, and TNF-α by the indicated CAR T cells after 48-h exposure to the indicated phenotypes of SEM cells (n = 5). Data are shown as means ± SEMs. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; 1-way ANOVA with the Tukey post hoc test. See also Figures S1 and S3.
To test the bispecific functionality of Tan(S)-CAR T cells, we generated CRISPR/Cas9-edited CD19-knockout (KO), CD22-KO, and DO-KO SEM cells, and validated the expression of CD19 and CD22 in each cell line by FACS (Figure 2D). We then used the resulting transgenic SEM cells in cytotoxicity assays with CD19-CAR and Tan(S)-CAR. Results using single CD22-CAR T cells are shown in Velasco-Hernandez et al. 15 As shown in Figures 2E and 2F, CD19-CAR and Tan(S)-CAR T cells eliminated wild-type (WT) and CD22-KO SEM cells, but not DO-KO SEM cells. In addition, Tan(S)-CAR T cells also eliminated CD19-KO SEM cells, suggesting that Tan(S)-CAR T cells specifically eliminate both CD19+CD22− and CD19−CD22+ leukemic cells.
We next surveyed cytokine secretion (at a 1:1 ratio) after 48 h. Of note, the production of pro-inflammatory cytokines by Tan(S)-CAR T cells was significantly lower than that of CD19-CAR T cells (when exposed to CD19+ target cells: SEM WT and SEM CD22-KO; Figure 2G). Neither CD19-CAR nor Tan(S)-CAR T cells secreted pro-inflammatory cytokines in co-culture with SEM DO-KO cells. Also, cytokine levels were undetectable in the Mock-CAR group regardless of the phenotype of the target cells (Figure 2G). Collectively, Tan(S)-CAR T cells recognize both Ags and display bispecific in vitro cytotoxicity activity on par with that of CD19-CAR T cells.
Tan(S)-CAR T cells are as efficient as CD19-CAR T cells in vivo
We next determined the in vivo activity of Tan(S)-CAR T cells in two xenograft mouse models with different aggressiveness based on either NALM6 (Figures 3A–3E) or SEM (Figures 3F–3I) B-ALL cell lines. NALM6-Luc+ or SEM-Luc+ cells were intratibially (i.t.) injected into NSG mice (n = 6/group), followed 4 days later by CAR T cells at 15% of transduction. A schematic of the experimental design is shown in Figure 3A. Mice were followed up weekly by bioluminescence imaging (BLI). Analysis showed that Tan(S)-CAR T cells and CD19-CAR T cells had equivalent activity against NALM6 (Figures 3B and 3C) and SEM (Figures 3F and 3G) growth in NSG mice, whereas mice treated with Mock-CAR failed to control the disease. Mice were sacrificed at day 14 (“highly aggressive” NALM6) and 35 (“less aggressive” SEM), and bone marrow (BM) and peripheral blood (PB) were collected and analyzed by FACS for the detection of residual leukemic cells and the presence of T cells (Figures 3D, 3E, 3H, and 3I). Mice treated with Mock-CAR showed a massive expansion of leukemic cells, whereas mice treated with either CD19-CAR T or Tan(S)-CAR T cells showed leukemic eradication accompanied by circulating T cells. Targeting CD19 and CD22 simultaneously does not compromise in vivo the antileukemic efficacy of the Tan(S)-CAR.
Figure 3.

Tan(S)-CAR T cells are as efficient as CD19-CAR T cells in vivo using both NALM6 and SEM B-ALL cell lines
(A) Scheme of the experimental design. NSG mice (n = 6/group) were intra-BM transplanted with 1 × 105 Luc-expressing NALM6 or SEM cells. Four days later, 4 × 106 Mock-CAR, CD19-CAR, or Tan(S)-CAR T cells were i.v. injected. Leukemic burden was monitored weekly by BLI. Mice were sacrificed and FACS analyzed for leukemic burden and T cell persistence when Mock-treated mice were fully leukemic by BLI. (B) IVIS imaging of NALM6 leukemic burden monitored by BLI at the indicated time points. (C) Average radiance quantification (p/sec/cm2/sr) for NALM6 at the indicated time points. (D) NALM6 leukemic burden quantified by FACS in BM and PB at sacrifice of mice treated with Mock-CAR, CD19-CAR, and Tan(S)-CAR, respectively. Each dot represents a mouse. (E) Representative FACS plots showing NALM6 cells (blue) and T cell persistence (red) in BM at sacrifice of mice treated with Mock-CAR, CD19-CAR, and Tan(S)-CAR, respectively. (F–I) Identical analysis to (B)–(E) for SEM target cells. Data are shown as means ± SEMs. ∗p < 0.05, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; 1-way ANOVA with the Tukey post hoc test.
Tan(S)-CAR T cells and CD19-CAR T cells efficiently eliminate primary and PDX B-ALL cells in vitro
We next tested the function of Tan(S)-CAR and CD19-CAR T cells after co-culture with three human primary B-ALL samples and three PDX samples of B-ALL that exhibit distinct expression levels of target Ags (Figures 4A and 4B; Table 1). Notably, T cells transduced with either Tan(S)-CAR or CD19-CAR showed equivalent cytotoxicity activity (Figures 4C and 4D) and production of IFN-γ and TNF-α (Figures 4E and 4F). Of note, however, a significantly higher production of IL-2 was consistently observed with Tan(S)-CAR (Figures 4E and 4F) and does not correlate with a differential T cell proliferation (Figure 4G), suggesting a functional advantage of the Tan(S)-CAR over the single CD19-CAR. In a more clinically applicable setting, PB-derived T cells from a B-ALL patient (patient 4 [Pt#4]) were used as effector cells. Magnetic-activated cell sorting (MACS)-sorted CD3+ T cells were activated, transduced with either Mock-CAR or Tan(S)-CAR, and exposed for 24 h to both autologous B-ALL CD19+ blasts and SEM cells (2:1 ratio). B-ALL patient-derived T cells were efficiently transduced, with similar levels to healthy donor-derived T cells (Figure 4H), and effectively and specifically eliminated both autologous CD19+ blasts and allogenic SEM cells (Figure 4I).
Figure 4.

Tan(S)-CAR T cells eliminate primary and PDX B-ALL cells in vitro
(A and B) CD22 and CD19 FACS expression in primary B-ALL blasts (n = 3) (A) and B-ALL PDX cells (n = 3) (B). (C and D) Absolute number of live primary B-ALL blasts (C) and B-ALL PDX cells (D) after 24-h incubation with the indicated CAR T cells at 2:1 E:T ratio. (E and F) Production of the pro-inflammatory cytokines IL-2, IFN-γ, and TNF-α by the indicated CAR T cells after 24-h exposure to either primary B-ALL blasts (E) or B-ALL PDX cells (F) at 2:1 E:T ratio. PBMCs from n = 3 independent HDs. (G) Absolute number of live T cells after 24-h incubation with the indicated primary B-ALL blasts or B-ALL PDX cells at 2:1 E:T ratio. (H and I) Autologous cytotoxicity experiment. Representative FACS plots of CAR T cells produced from MACS-sorted T cells from the PB of a B-ALL patient (Pt #4) showing the transduction efficiency at day 6 (H). Absolute number of live SEM cells (left panel) or live primary B-ALL blasts from the same patient (patient 4) after 24-h incubation with the Tan(S)-CAR T cells at 2:1 E:T ratio (I). Data are shown as means ± SEMs. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; 2-way ANOVA with the Tukey post hoc test.
Table 1.
Clinical and biological features of the primary B-ALL samples used in this study
| Sample | Molecular | Disease stage | Blasts, % | Age, years | Gender | CD19 MFI | CD22 MFI |
|---|---|---|---|---|---|---|---|
| Pt#1 | ETV6/RUNX1 | diagnosis | 90 | 12 | female | 9,749 | 3,848 |
| Pt#2 | ETV6/RUNX1 | diagnosis | 96 | 5 | male | 22,826 | 7,570 |
| Pt#3 | ETV6/RUNX1 | diagnosis | 97 | 2 | male | 12,104 | 3,581 |
| Pt#4 | NUP214-ABL1 | diagnosis | 95 | 24 | male | + | + |
| Pt#5 | PAX5-MLLT3 | relapse after CD19-CAR T cell therapy and alloSCT | 92 | 31 | male | − | + |
| PDX#1 | high-hyperdiploid | relapse | 97 | 3.5 | male | 14,963 | 7,881 |
| PDX#2 | low-hypodiploid | diagnosis | 97 | 12 | male | 25,459 | 15,192 |
| PDX#3 | TCF3-PBX1 | diagnosis | 98 | 7 | female | 4,510 | 2,028 |
| PDX#4 | high-hyperdiploid | diagnosis | 92 | 2.5 | male | 15,554 | 6,167 |
alloSCT, allogenic stem cell transplantation; MFI, mean fluorescence intensity.
Simultaneous targeting of CD22 and CD19 Ags controls the disease in long-term follow-up B-ALL PDXs
To further explore the in vivo activity of Tan(S)-CAR versus CD19-CAR T cells, we used clinically relevant PDX models of B-ALL. NSG mice (n = 5–9/group) were intravenously (i.v.) transplanted with 0.5 × 106 B-ALL cells (PDX#3) or 1 × 106 B-ALL cells (PDX#4), and CAR T cells were infused when B-ALL engraftment was detectable in BM (day 17 for PDX#3 or day 31 for PDX#4). A schematic of the experimental design is shown in Figure 5A. One day before CAR T cell infusion, the leukemic (CD19+CD22+CD10+) engraftment was determined in the PB and BM, and mice were subsequently randomized based on BM leukemic burden to receive i.v. 5 × 106 Mock-CAR, CD19-CAR, or Tan(S)-CAR T cells. Leukemic burden and CAR T cell persistence was monitored in PB biweekly by FACS. BM aspirates were FACS analyzed when Mock-treated mice were sacrificed (week 4) and at the endpoint (week 13). As expected, mice treated with Mock-CAR T cells succumbed quickly to the disease, and had to be sacrificed with >40% and >80% of the leukemic graft in PB and BM, respectively (Figure 5B). By contrast, CD19-CAR and Tan(S)-CAR T cells were both capable of controlling the disease by eliminating leukemic cells in BM within 4 weeks after CAR T cell infusion (Figure 5B). For the PDX#4 model, disease recurrence was followed up biweekly from week 4 (minimal residual disease negativity [MRD−]) to week 13, when many mice had to be sacrificed because termination criteria had been reached. Six weeks after CAR T cell infusion, circulating leukemic cells began to emerge in mice treated with CD19-CAR, and both leukemic burden and the number of relapsing mice (>1% blasts in BM or >0.1% in PB) increased over time in this group of CD19-CAR-treated mice (Figure 5C). In contrast, leukemic cells in PB were observed in only one mouse treated with Tan(S)-CAR 13 weeks after CAR T cell infusion (Figure 5C). Importantly, T cell persistence in PB was observed throughout the 13 weeks for both CAR-19 and Tan(S)-CAR T cells (Figure 5C). Accordingly, the disease-free survival (DFS) for Tan(S)-CAR-treated mice at week 13 was double that of CD19-CAR-treated mice (86% versus 43%; p = 0.08; Figure 5D). Detailed BM analysis at the endpoint confirmed that 4 of 7 (57%) of the CD19-CAR-treated mice and only 1 of 7 (14%) of the Tan(S)-CAR-treated mice presented leukemic cells (>1%) (Figure 5E). Of note, FACS analysis of the BM revealed a diminished expression of CD19 in some cells in 1 of 4 mice (25%) that relapsed after CD19-CAR T cell infusion (Figure 5E). Our pre-clinical results suggest that simultaneous targeting of CD22 and CD19 may have a longer therapeutic effect in B-ALL patients.
Figure 5.

Tan(S)-CAR is very effective in controlling the disease in a long-term follow-up B-ALL PDX model
(A) Scheme showing the experimental design. NSG mice (n = 5–9/group) were i.v. transplanted with 0.5 × 106 or 1 × 106 of B-ALL cells from PDX#3 or PDX#4, respectively. Upon B-ALL engraftment detectable in BM, mice were randomized, and 5 × 106 Mock-CAR, CD19-CAR, or Tan(S)-CAR T cells were i.v. injected. Leukemic burden and CAR T cell persistence was monitored in PB biweekly by FACS. BM aspirates were FACS analyzed when Mock-treated mice were sacrificed (week 4) and at the endpoint (week 13). (B) Upper panels, leukemic burden in PB and BM at the indicated time points after CAR T cell infusion. Bottom panels, representative BM FACS analysis showing CD19 and CD22 expression for both PDXs before CAR T cell infusion (1 day before CAR T cell infusion [−0.1]) and at the time Mock-treated mice were sacrificed (week 4). The gating strategy is shown on the left. Indicated percentages are referred to the total live single cells in each sample. The complete gating strategy is shown in Figure S1B. (C) Follow-up at the indicated time points after CAR T cell infusion of leukemic progression/relapse (left panel) and persistent T cells (right panel) of CD19-CAR-treated versus Tan(S)-CAR-treated mice transplanted with PDX#4 (n = 7 mice/group). (D) DFS curves for CD19-CAR-treated versus Tan(S)-CAR-treated mice transplanted with PDX#4. The log-rank (Mantel-Cox) test was used to calculate significance. (E) Leukemic burden at sacrifice/endpoint (week 13 after CAR T cell infusion) in BM from CD19-CAR-treated versus Tan(S)-CAR-treated mice transplanted with PDX#4 (n = 7 mice/group). A mouse is considered in relapse when the percentage of blasts in BM is >1% (horizontal dotted line) or >0.1% in PB. Each dot represents a mouse. The bottom panels show the expression of CD19 and CD22 by FACS analysis of B-ALL cells for each independent CD19-CAR-treated and Tan(S)-CAR-treated mouse. Indicated percentages are referred to the total live single cells in each sample. The complete gating strategy is shown in Figure S1B. Data are shown as means ± SEMs. ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; 1-way ANOVA with the Tukey post hoc test. See also Figure S1.
Finally, to explore further the in vivo bispecificity of the Tan(S)-CAR in a clinically relevant model of CD19 or CD22 resistance, we next assessed the ability of the Tan(S)-CAR to control CD19−CD22+ and CD19+CD22− B-ALL cells in vivo. We used both SEM CD19-KO and SEM CD22-KO as well as CD19− primary B-ALL cells from a patient relapsed after CD19-CAR treatment (Pt#5). Tan(S)-CAR T cells fully controlled the in vivo growth of SEM CD22-KO and partially that of SEM CD19-KO (Figures 6A and 6B). Strikingly, we observed a fully eradication of the in vivo growth of CD19−CD22+ primary B-ALL cells (Figure 6C) in mice infused with Tan(S)-CAR and CD22-CAR T cells (Figure 6D). The experiment was terminated at day 49 due to graft-versus-host disease associated with a high number of human T cells in PB and BM (data not shown). These results demonstrate the in vivo bispecificity of the Tan(S)-CAR presented in this work, positioning it as an effective asset for clinical translation.
Figure 6.

In vivo bispecificity of Tan(S)-CAR T cells
(A) Average radiance quantification (p/sec/cm2/sr) from mice transplanted with SEM CD22-KO (n = 4–5/group) or SEM CD19-KO (n = 9–10/group) at the indicated time points after Mock- or Tan(S)-CAR T cell infusion. Data from n = 2 independent experiments. (B) Representative BLIs of SEM CD22-KO- and SEM CD19-KO-transplanted mice from week 2 to week 4 after CAR T cell infusion. (C) CD19 and CD22 expression of the CD19−CD22+ blasts from the B-ALL patient (Pt#5) used for the in vivo bispecificity experiment. (D) NSG mice (n = 6/group) were i.v. transplanted with 1 × 106 of CD19−CD22+ B-ALL cells (Pt#5). Upon detectable B-ALL engraftment in BM, mice were randomized to receive 5 × 106 Mock-CAR, CD19-CAR, CD22-CAR, or Tan(S)-CAR T cells. The leukemic burden in BM before CAR T cell infusion (day 0) and at endpoint of the experiment (day 49) is shown. Data are shown as means ± SEMs. ∗p < 0.05, ∗∗p < 0.01, 2-way ANOVA (mixed-effects model) with Šídáks multiple comparisons test.
Discussion
CD19-CAR T cells have produced unprecedented results in multiple clinical trials.6,28 However, only one CD19-CAR T cell product is currently approved in Europe and the United States for the treatment of R/R B-ALL in patients younger than 25 years old, tisagenlecleucel (Kymriah). The long-term follow-up of CD19-CAR-treated patients shows that the durability of the response is limited and relapse rates are ∼50% after 12–18 months.6,8,28 Relapse is likely not unique to therapeutic approaches targeting CD19, as it was also observed with other agents targeting CD22.12,14 Poor CAR T cell persistence and/or leukemic cell resistance resulting from Ag loss or modulation are among the most common limitations of single targeted CAR T cell therapies,9,17 and guide the search for innovative CAR T cell approaches.
Activation and exponential expansion of CAR T cells following infusion are essential for successful clinical responses.29,30 In fact, leukemia recurrence after CAR T cell therapy, especially when occurring a few months after achieving initial complete response, is often associated with limited CAR T cell persistence. Accordingly, strategies to improve CAR T cell persistence independently of the CAR T cell design and manufacturing process are being tested clinically.29,30 Early relapse with Ag+ disease and loss of CAR T cell persistence presents a potential opportunity for the reinfusion of CAR T cells. However, later relapses are frequently associated with either loss of the target Ag or biological resistance to the CD19-directed CAR. Established mechanisms of the loss of CD19 expression include alternative splicing, low Ag density, epitope masking, interruption in the transport of CD19 to the cell surface, and cell lineage switching.9,10,17 Of note, rather than a complete Ag loss, partial Ag loss due to downregulation has been reported in patients treated with CD22-CAR T cells.12
CAR T cells against single Ags are likely insufficient for effective and durable (years) long-term antileukemic responses due to CAR-mediated immune pressure and patient-specific intrinsic vulnerabilities to CAR resistance. Of note, previous administration of inotuzumab ozogamicin, a toxic-conjugated anti-CD22 mAb clinically used to induce complete remission in relapsing B-ALL patients, has been suggested to limit/impair the subsequent expansion of autologous CD19-CAR T cells.31 For this reason, CAR constructs incorporating multi-Ag targeting are being investigated to allow simultaneous co-targeting of more than a single Ag, extending the activity of CAR T cells to several phenotypic subpopulations of the disease. This can be achieved by (1) generating two or more T cell products, each one expressing different CARs and infusing them together or sequentially (co-administration); (2) using a bicistronic vector that encodes two different CARs on the same cell (bicistronic CAR T cells); (3) simultaneously engineering T cells with two different CAR constructs (co-transduction), which will generate three CAR T subsets consisting of dual and single CAR-expressing cells; or (4) encoding two Ag-recognizing scFvs on the same chimeric protein using a single vector (Tan-CAR T cells), which will mediate T cell activation in response to either one of the two target Ags.17 Importantly, there is evidence that pooled infusions of single CARs are inferior to Tan-CARs or dual CARs in preclinical models,20,32 and that Tan-CARs can induce additive cytokine secretion upon encounter of both targets simultaneously, boosting antitumor efficacy with an enhancement of the immune synapse.20
In B cell malignancies, targeting pan-B Ags beyond CD19, such as CD20 and CD22, has the advantage of low cumulative risk of on-target off-tumor toxicity. In this line, two preclinical studies with bispecific CARs for B-ALL—CD22/CD19-CAR or CD20/CD19-CAR—have reported in vitro and in vivo eradication of PDX or primary B-ALL cells.12,33 Based on this encouraging preclinical data, several clinical trials with Tan-CARs are under way for B-ALL.9,21 Preliminary data from a Phase I clinical trial with a CD22/CD19-CAR demonstrate that CD22/CD19-CAR T cell therapy is safe and mediates a robust antileukemic activity in patients with R/R B-ALL; however, relapse occurred in three of six enrolled patients: two patients as CD19+CD22+ and one patient as CD19−CD22low.34 Follow-up data on clinical trials with different Tan-CARs are still limited, but in the future, they will help inform whether dual-Ag-targeted approaches are sufficient to prevent/delay disease relapse and whether targeting more than one Ag will be an effective strategy for enhancing clinical outcomes.
Developing functional multitargeted constructs is no easy task, as acknowledged by Qin and colleagues.35 In our drive to improve CD19-CAR T cell therapy for B-ALL, we developed a Tan-CAR to simultaneously target CD19 and CD22 based on our proprietary single-CAR constructs and the clinical validation of our CD19-CAR.15,22, 23, 24, 25 We compared two versions of Tan-CARs in vitro and we observed that both had similar antileukemic activity and cytokine production. However, transduction efficiencies with the Tan-CAR carrying a longer scFv linker (Tan(L)-CAR) were lower due to size constraints, and T cells transduced with this Tan(L)-CAR displayed a lower proliferation; therefore, the Tan(S)-CAR was selected for further characterization and comparison with CD19-CAR. The different functionality between the structure of our Tan(S)-CAR and those LoopTan-CARs previously reported by Qin et al.35 may respond to a distinct VH/VL combination, scFv order, length and flexibility/rigidity of the linkers, and extracellular spacer length, which have all been suggested to affect the expression and the activity/potency of Tan-CAR constructs.33,35,36 In addition, another not minor difference may be the nature of our CD22 scFv that is unique in targeting a distal membrane epitope of CD22.15 Further configurational studies will reveal the functional differences among the distinct Tan-CARs available.
Demonstration of the ability to bind/recognize target Ags by both scFvs in our Tan(S)-CAR was achieved using human recombinant CD19 and CD22 proteins. In vitro pro-inflammatory cytokine release and cytotoxicity activity also established Tan-CAR functionality. Moreover, our results indicate that the Tan(S)-CAR performs as well as the CD19-CAR in vitro and in vivo against both B-ALL cell lines and patient leukemic cells. Although previous preclinical studies suggested that both scFvs may act synergistically,18, 19, 20 this was not observed in the present study, which concurs with the previous study by Qin and colleagues,35 suggesting that this effect may be dependent on the Ag/epitope targeted or the affinity/avidity of the scFvs. Similar to Qin and colleagues,35 our Tan(S)-CAR displayed a potency/efficacy comparable to that of the single CD19-CAR, but slightly less potent/efficient than the single CD22-CAR.15 An encouraging observation is that the current Tan(S)-CAR seems more effective in vivo than a CD19-CAR in controlling the disease in a long-term follow-up B-ALL PDX model. Whether the superior ability of the Tan(S)-CAR to produce IL-2 in the presence of B-ALL primary samples and B-ALL PDXs contributes to this observation needs further investigation. These data fit well with a study by Schneider and colleagues,36 who observed that a higher ability to produce IL-2 leads to better antileukemia CAR activity in NSG mice. These data indicate that our Tan(S)-CAR warrants a clinical appraisal to test whether simultaneous targeting of CD19 and CD22 Ags offers more durable clinical responses with reduced risk of Ag loss than the standard-of-care CD19-CAR approach. In addition, the excellent performance of the Tan(S)-CAR in vitro and in vivo also positions them as promising assets for clinical translation.
Materials and methods
Reagents, drugs, and antibodies
Advanced DMEM, IMDM, RPMI-1640, l-glutamine, penicillin/streptomycin (P/S), and insulin-transferrin-selenium (ITS) were purchased from GIBCO/Invitrogen (Waltham, MA, USA). StemSpan serum-free expansion medium (SFEM) was purchased from STEMCELL Technologies (Vancouver, Canada). Phosphate-buffered saline was purchased from Merck Life Science SL (Darmstadt, Germany). Human (h) stem cell factor (SCF), hFMS-like tyrosine kinase 3 ligand (FLT3-L), hIL-3, hIL-7, and hIL-15 were purchased from Miltenyi Biotec (Bergisch Gladbach, Germany). Fetal bovine serum (FBS) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Anti-CD3 (OKT3) and anti-CD28 (CD28.2) mAbs, 7-amino-actinomycin D (7-AAD), and fluorescein isothiocyanate (FITC)-, phycoerythrin (PE)-, peridin chlorophyll (PerCP)-, allophycocyanin (APC)-, phycoerythrin/cyanine7 (PE/Cy7)-, Brilliant Violet 421 (BV421)-, and BV510-conjugated mAbs specific for human CD3 (SK7), CD19 (HIB19), CD22 (HIB22), CD10 (HI10a), CD13 (WM15), CD45 (HI30), HLA-ABC (G46-2.6), CD25 (M-A251), CCR7 (150503), CD27 (L128), CD45RO (UCHL1), LAG3 (T47-530), TIM3 (7D3), PD-1 (MIH4), and isotype-matched negative control mAbs, were purchased from BD Biosciences (Franklin Lakes, NJ, USA), CD69 (REA824) from Miltenyi Biotec, and anti-His (J095G46) from BioLegend (San Diego, CA, USA).
Cell lines
The B-ALL cell lines SEM and NALM6 were obtained from the DSMZ cell line bank (Braunschweig, Germany). Luciferase (Luc)/GFP-expressing NALM6 cells were kindly provided by Prof. R.J. Brentjens (Memorial Sloan Kettering Cancer Center, NY, USA). Stable Luc-expressing SEM cell lines were generated with the lentiviral pUltra-Chili-Luc backbone (Addgene #48688) using a spinfection (centrifugation) protocol, as previously described.37 Briefly, 1 × 106 cells were seeded on a 6-well plate with 1.2 mL viral supernatant. Plates were then centrifuged at 900 × g for 1 h at 28°C, after which the plate was placed in an incubator at 37°C for 3 h and fresh media was added. Cells were incubated for an additional 48 h after a medium exchange. Finally, cherry-positive cells were isolated by FACS (>99% purity) and luminescence was checked. CD19-KO, CD22-KO, and double-KO (DO-KO) SEM cells were generated by CRISPR-mediated genome editing. Briefly, 200,000 cells were electroporated (Neon transfector, ThermoFisher Scientific, Waltham, MA, USA) with a Cas9 protein/tracrRNA/crRNA complex (IDT, Coralville, IA, USA). Two guides were designed for each gene: CD19-exon 2.1 CAGGCCTGGGAATCCACATG and CD19-exon 14.1 AGAACATGGATAATCCCGAT and CD22-exon 3.2 TCAATGACAGTGGTCAGCTG and CD22-exon 9 CAGGTGTAGTGGGAGACGGG. Cells were allowed to recover after electroporation, and CD19− or CD22− cells were isolated by FACS sorting (>99% purity).15
Human samples
PB mononuclear cells (PBMCs) were isolated from buffy coats of HDs by Ficoll-Hypaque gradient centrifugation (GE Healthcare, Chicago, IL, USA).38 Buffy coats were obtained from the Catalan Blood and Tissue Bank upon institutional review board approval (HCB/2018/0030). BM aspirates were obtained from five B-ALL patients (Table 1). Human samples were obtained after written informed consent in accordance with the Declaration of Helsinki. All of the experimental studies were approved by the institutional review board of the Ethics Committee on Clinical Research of the Clinic Hospital of Barcelona (HCB/2017/0781).
CD19-CAR and CD22/CD19 Tan-CAR vector, lentiviral production, T cell transduction, activation, and expansion
CAR Ag-binding domain (scFv) sequences were derived from the mouse hybridoma A3B1 for CD1922,23,25 and hCD22.7 for CD22.15 We used a clinically validated pCCL second-generation lentiviral CD19-CAR backbone, which contains the A3B1 scFv, the hinge and human CD8 TM domain, and human 4-1BB and CD3ζ endodomains.22, 23, 24,26,27 Two second-generation lentiviral bispecific, tandem CD22/CD19-CARs were designed and referred to as Tan(S)- and Tan(L)-CARs; these included the hCD22.7 scFv and the A3B1 scFv linked in sequence by a flexible (glycine4serine)4–7 interchain linker, followed by the hinge and the human CD8 TM domain and the human 4-1BB and CD3ζ endodomains. Constructs S and L differ only in the length of the flexible interchain linker sequence connecting the anti-CD22 scFv and the anti-CD19 scFv (Figure 1A). An identical lentiviral vector with the CD8 TM-4-1BB-CD3ζ domains linked to a His-Tag (Mock-CAR) was used as an experimental control.15 All of the CARs were fused to a GFP reporter gene by a 2A ribosomal skip sequence (T2A) at the C-terminal CAR sequence, for tracing the transduction efficiency and CAR expression.
CAR-expressing viral particles pseudotyped with VSV-G were generated by the transfection of HEK293T cells with pCCL, VSV-G, and psPAX2 vectors using polyethylenimine (Polysciences Inc., Warrington, PA, USA). Supernatants were collected at 48 and 72 h after transfection and concentrated by ultracentrifugation following the standard procedure. T cells were activated by plate coating with anti-CD3 and anti-CD28 mAbs (1 μg/mL) in complete RPMI medium for 2 days, and were then transduced with a CAR-expressing lentivirus at a multiplicity of infection of 10 in the presence of hIL-7 and hIL-15 (10 ng/mL).15,24,26,27 T cells from a B-ALL patient were purified using anti-hCD3 magnetic beads (Miltenyi Biotec) by AutoMACs according to the manufacturer’s instructions. T cells were expanded in complete RPMI medium plus hIL-7 and hIL-15 for up to 5–7 days. CAR transduction efficiency in T cells was analyzed by FACS. Vector copy number (VCN) was determined by quantitative PCR using Light Cycler 480 SYBRGreen I Master (Roche, Basel, Switzerland), as in Ortiz-Maldonado et al.25 Briefly, pairs of primers were designed against GATA2 (control gene, GATA2_F: 5′tggcgcacaactacatggaa3′; GATA2_R: 5′cgagtcgaggtgattgaagaaga3′) and WPRE sequence (part of the provirus WPRE_F: 5′gtcctttccatggctgctc3′; WPRE_R: 5′ccgaagggacgtagcaga3′). Absolute quantification method was used to determine the VCN/genome. VCN results were adjusted to the percentage of transduction of each CAR determined by FACS analysis. The same cassettes of CD19-CAR and Tan(S)-CAR were cloned into the SFG retroviral backbone kindly provided by Dr. Maksim Mamonkin, and retrovirus production and transduction were performed following standard procedures.39
CAR surface detection
Cell surface expression of Mock-CAR was confirmed by binding to anti-His-APC. Cell surface expression of the CD19-CAR and Tan-CARs was confirmed by binding to an AffiniPure F(ab′)2 fragment goat anti-mouse IgG (H+L)-APC and an anti-human IgG (H+L)-PE (both from Jackson ImmunoResearch, Westgrove, PA, USA) after prior incubation with human recombinant CD19-Fc (R&D Systems, Abingdon, UK). Tan-CARs were also confirmed by binding to an anti-His-APC after prior incubation with human recombinant CD22-His (ThermoFisher Scientific).
In vitro CAR T cell cytotoxicity assays and cytokine release determination
Target cells (B-ALL cell lines, primary B-ALL cells, and B-ALL PDXs cells; 1 × 105 target cells/well) were incubated with Mock-, CD19-, or Tan-CAR T cells at different E:T ratios for the indicated time periods. Cell lines were cultured in complete RPMI medium and primary cells/PDXs were cultured in StemSpan SFEM supplemented with 20% heat-inactivated FBS, P/S, ITS, hSCF (100 ng/mL), hFLT3-L (100 ng/mL), hIL-3 (10 ng/mL), and hIL-7 (10 ng/mL). At each time point, cells were collected, washed, and stained with anti-CD3, anti-CD19, anti-CD10, and anti-CD22 mAbs, and 7-AAD. CAR T cell-mediated cytotoxicity was determined by analyzing the residual living (7-AAD−CD3−GFP−CD10+) target cells at each time point and E:T ratio (Figure S1A). BD TruCount absolute count tubes (BD Biosciences) were used for absolute cell counting. The quantification of IL-2, IFN-γ, and TNF-α was measured by enzyme-linked immunosorbent assay (ELISA) using the OptEIA Human ELISA Kit (BD Biosciences) on supernatants harvested after 1–2 days of co-culture at a 1:1 E:T ratio. ELISA determinations were performed in triplicate.
In vivo CAR T cell-mediated cytotoxicity assay with B-ALL cell lines and PDX samples
Ten-week-old non-obese diabetic (NOD) Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (The Jackson Laboratory, Bar Harbor, ME, USA) were bred and housed under pathogen-free conditions. Animal experimentation procedures were approved by the local ethics committee (HRH-17-0029-P1). For B-ALL cell lines, NSG mice were i.t. injected with 1 × 105 of either NALM6-Luc+ cells or SEM-Luc+ cells24, followed 4 days later by an i.v. infusion of 4 × 106 CAR T cells. Mice were followed up weekly by BLI using an in vivo imaging system (IVIS, Lumina III; Perkin-Elmer, Waltham, MA, USA). Mice were sacrificed at day 14 (NALM6-injected mice) and day 35 (SEM-injected mice), and PB and BM samples were collected and analyzed by FACS to assess leukemic burden and CAR T cell persistence.
For PDXs, 0.5–1.0 × 106 PDX B-ALL cells were i.v. injected in sublethally irradiated (2 Gy) NSG mice, followed by i.v. infusion of 5 × 106 CAR T cells at the indicated time points. B-ALL engraftment was monitored in PB every other week, and BM aspirates were analyzed 4 weeks after CAR T cell infusion and at sacrifice by FACS. MRD− was defined as <0.1% BM blasts (identified as CD45+HLA-ABC+CD3−GFP−CD10+ by FACS) (Figure S1B). Relapse was defined as the reappearance of blasts in either PB (>0.1%) or BM (>1%) after complete remission.
Statistical analysis
Data were plotted as means ± SEMs. One-way analysis of variance with Tukey’s post hoc test was used unless stated otherwise. All of the analyses were performed with Prism software, version 8.0 (GraphPad Prism Software, San Diego, CA, USA).
Acknowledgments
We thank Dr. Meritxell Vinyoles for technical support and Dr. Maksim Mamonkin, who kindly provided the SFG retroviral backbone. We thank CERCA/Generalitat de Catalunya and Fundació Josep Carreras-Obra Social la Caixa for core support. Financial support for this work was obtained from the European Research Council (CoG-2014-646903, PoC-2018-811220), the Spanish Ministry of Economy and Competitiveness (SAF2016-80481R, PID2019-108160RB-I00), the Health Institute Carlos III (ISCIII/FEDER, PI17/01028, PI20/00822, to C.B.), the Fundación Uno entre Cienmil, the Obra Social La Caixa (LCF/PR/HR19/52160011), the Leo Messi Foundation, and the “Heroes hasta la médula” initiative to P.M. S.R.Z. and T.V.-H. were supported by Marie Skłodowska Curie Fellowships (GA795833 and GA792923, respectively). D.S.-M. and R.T.-R. were supported by postdoctoral fellowships from ISCIII (Sara Borrell) and the Spanish Association against Cancer (AECC), respectively.
Author contributions
S.R.Z. conceived the study, designed and performed the experiments, analyzed and interpreted the data, prepared the figures, and wrote the manuscript. T.V.-H. designed and performed the experiments, analyzed and interpreted the data, prepared the figures, and wrote the manuscript. F.G.-A., V.M.D., P.A.R., H.R.-H., D.S.-M., N.T., M.L.B., P.P., and R.T.-R. performed the experiments. O.M., A.B., J.L.F., P.B., M.J., and I.J. provided the human primary samples and PDXs. C.B. supported the study technically and financially. P.M. conceived the study, designed the experiments, interpreted the data, wrote the manuscript, and financially supported the study.
Declaration of interests
S.R.Z., T.V.-H., F.G.-A., D.S.-M., and P.M. are inventors of a CD22scFv pending European patent (file no. 20382175.6). P.M. is the co-founder of OneChain Immunotherapeutics (OCI). V.M.D. is employed by OCI.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.08.033.
Contributor Information
Samanta Romina Zanetti, Email: samzanetti@gmail.com.
Talia Velasco-Hernandez, Email: tvelasco@carrerasresearch.org.
Pablo Menéndez, Email: pmenendez@carrerasresearch.org.
Supplemental information
References
- 1.Katz A.J., Chia V.M., Schoonen W.M., Kelsh M.A. Acute lymphoblastic leukemia: an assessment of international incidence, survival, and disease burden. Cancer Causes Control. 2015;26:1627–1642. doi: 10.1007/s10552-015-0657-6. [DOI] [PubMed] [Google Scholar]
- 2.Gavralidis A., Brunner A.M. Novel Therapies in the Treatment of Adult Acute Lymphoblastic Leukemia. Curr. Hematol. Malig. Rep. 2020;15:294–304. doi: 10.1007/s11899-020-00591-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Capitini C.M. CAR-T immunotherapy: how will it change treatment for acute lymphoblastic leukemia and beyond? Expert Opin. Orphan Drugs. 2018;6:563–566. doi: 10.1080/21678707.2018.1529562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Waldman A.D., Fritz J.M., Lenardo M.J. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat. Rev. Immunol. 2020;20:651–668. doi: 10.1038/s41577-020-0306-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scheuermann R.H., Racila E. CD19 antigen in leukemia and lymphoma diagnosis and immunotherapy. Leuk. Lymphoma. 1995;18:385–397. doi: 10.3109/10428199509059636. [DOI] [PubMed] [Google Scholar]
- 6.Rafei H., Kantarjian H.M., Jabbour E.J. Recent advances in the treatment of acute lymphoblastic leukemia. Leuk. Lymphoma. 2019;60:2606–2621. doi: 10.1080/10428194.2019.1605071. [DOI] [PubMed] [Google Scholar]
- 7.Aldoss I., Bargou R.C., Nagorsen D., Friberg G.R., Baeuerle P.A., Forman S.J. Redirecting T cells to eradicate B-cell acute lymphoblastic leukemia: bispecific T-cell engagers and chimeric antigen receptors. Leukemia. 2017;31:777–787. doi: 10.1038/leu.2016.391. [DOI] [PubMed] [Google Scholar]
- 8.Ruella M., Maus M.V. Catch Me If You Can: Leukemia Escape after CD19-Directed T Cell Immunotherapies. Comput. Struct. Biotechnol. J. 2016;14:357–362. doi: 10.1016/j.csbj.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shah N.N., Fry T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019;16:372–385. doi: 10.1038/s41571-019-0184-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song M.K., Park B.B., Uhm J.E. Resistance Mechanisms to CAR T-Cell Therapy and Overcoming Strategy in B-Cell Hematologic Malignancies. Int. J. Mol. Sci. 2019;20:E5010. doi: 10.3390/ijms20205010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nitschke L. CD22 and Siglec-G: B-cell inhibitory receptors with distinct functions. Immunol. Rev. 2009;230:128–143. doi: 10.1111/j.1600-065X.2009.00801.x. [DOI] [PubMed] [Google Scholar]
- 12.Fry T.J., Shah N.N., Orentas R.J., Stetler-Stevenson M., Yuan C.M., Ramakrishna S., Wolters P., Martin S., Delbrook C., Yates B., et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018;24:20–28. doi: 10.1038/nm.4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haso W., Lee D.W., Shah N.N., Stetler-Stevenson M., Yuan C.M., Pastan I.H., Dimitrov D.S., Morgan R.A., FitzGerald D.J., Barrett D.M., et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121:1165–1174. doi: 10.1182/blood-2012-06-438002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pan J., Niu Q., Deng B., Liu S., Wu T., Gao Z., Liu Z., Zhang Y., Qu X., Zhang Y., et al. CD22 CAR T-cell therapy in refractory or relapsed B acute lymphoblastic leukemia. Leukemia. 2019;33:2854–2866. doi: 10.1038/s41375-019-0488-7. [DOI] [PubMed] [Google Scholar]
- 15.Velasco-Hernandez T., Zanetti S.R., Roca-Ho H., Gutierrez-Aguera F., Petazzi P., Sánchez-Martínez D., Molina O., Baroni M.L., Fuster J.L., Ballerini P., et al. Efficient elimination of primary B-ALL cells in vitro and in vivo using a novel 4-1BB-based CAR targeting a membrane-distal CD22 epitope. J. Immunother. Cancer. 2020;8:e000896. doi: 10.1136/jitc-2020-000896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Navai S.A., Ahmed N. Targeting the tumour profile using broad spectrum chimaeric antigen receptor T-cells. Biochem. Soc. Trans. 2016;44:391–396. doi: 10.1042/BST20150266. [DOI] [PubMed] [Google Scholar]
- 17.Shah N.N., Maatman T., Hari P., Johnson B. Multi Targeted CAR-T Cell Therapies for B-Cell Malignancies. Front. Oncol. 2019;9:146. doi: 10.3389/fonc.2019.00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grada Z., Hegde M., Byrd T., Shaffer D.R., Ghazi A., Brawley V.S., Corder A., Schönfeld K., Koch J., Dotti G., et al. TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol. Ther. Nucleic Acids. 2013;2:e105. doi: 10.1038/mtna.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hegde M., Corder A., Chow K.K., Mukherjee M., Ashoori A., Kew Y., Zhang Y.J., Baskin D.S., Merchant F.A., Brawley V.S., et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol. Ther. 2013;21:2087–2101. doi: 10.1038/mt.2013.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hegde M., Mukherjee M., Grada Z., Pignata A., Landi D., Navai S.A., Wakefield A., Fousek K., Bielamowicz K., Chow K.K., et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J. Clin. Invest. 2016;126:3036–3052. doi: 10.1172/JCI83416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao J., Song Y., Liu D. Clinical trials of dual-target CAR T cells, donor-derived CAR T cells, and universal CAR T cells for acute lymphoid leukemia. J. Hematol. Oncol. 2019;12:17. doi: 10.1186/s13045-019-0705-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Castella M., Boronat A., Martín-Ibáñez R., Rodríguez V., Suñé G., Caballero M., Marzal B., Pérez-Amill L., Martín-Antonio B., Castaño J., et al. Development of a Novel Anti-CD19 Chimeric Antigen Receptor: A Paradigm for an Affordable CAR T Cell Production at Academic Institutions. Mol. Ther. Methods Clin. Dev. 2018;12:134–144. doi: 10.1016/j.omtm.2018.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Castella M., Caballero-Baños M., Ortiz-Maldonado V., González-Navarro E.A., Suñé G., Antoñana-Vidósola A., Boronat A., Marzal B., Millán L., Martín-Antonio B., et al. Point-Of-Care CAR T-Cell Production (ARI-0001) Using a Closed Semi-automatic Bioreactor: Experience From an Academic Phase I Clinical Trial. Front. Immunol. 2020;11:482. doi: 10.3389/fimmu.2020.00482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zanetti S.R., Romecin P.A., Vinyoles M., Juan M., Fuster J.L., Cámos M., Querol S., Delgado M., Menendez P. Bone marrow MSC from pediatric patients with B-ALL highly immunosuppress T-cell responses but do not compromise CD19-CAR T-cell activity. J. Immunother. Cancer. 2020;8:e001419. doi: 10.1136/jitc-2020-001419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ortiz-Maldonado V., Rives S., Castella M., Alonso-Saladrigues A., Benitez-Ribas D., Caballero-Banos M., Baumann T., Cid J., Garcia-Rey E., Llanos C., et al. CART19-BE-01: A Multicenter Trial of ARI-0001 Cell Therapy in Patients with CD19(+) Relapsed/Refractory Malignancies. Mol. Ther. 2021;29:636–644. doi: 10.1016/j.ymthe.2020.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baroni M.L., Sanchez Martinez D., Gutierrez Aguera F., Roca Ho H., Castella M., Zanetti S.R., Velasco Hernandez T., Diaz de la Guardia R., Castaño J., Anguita E., et al. 41BB-based and CD28-based CD123-redirected T-cells ablate human normal hematopoiesis in vivo. J. Immunother. Cancer. 2020;8:e000845. doi: 10.1136/jitc-2020-000845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sánchez-Martínez D., Baroni M.L., Gutierrez-Agüera F., Roca-Ho H., Blanch-Lombarte O., González-García S., Torrebadell M., Junca J., Ramírez-Orellana M., Velasco-Hernández T., et al. Fratricide-resistant CD1a-specific CAR T cells for the treatment of cortical T-cell acute lymphoblastic leukemia. Blood. 2019;133:2291–2304. doi: 10.1182/blood-2018-10-882944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sermer D., Brentjens R. CAR T-cell therapy: Full speed ahead. Hematol. Oncol. 2019;37(Suppl 1):95–100. doi: 10.1002/hon.2591. [DOI] [PubMed] [Google Scholar]
- 29.Maude S.L., Frey N., Shaw P.A., Aplenc R., Barrett D.M., Bunin N.J., Chew A., Gonzalez V.E., Zheng Z., Lacey S.F., et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nie Y., Lu W., Chen D., Tu H., Guo Z., Zhou X., Li M., Tu S., Li Y. Mechanisms underlying CD19-positive ALL relapse after anti-CD19 CAR T cell therapy and associated strategies. Biomark. Res. 2020;8:18. doi: 10.1186/s40364-020-00197-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baruchel A., Krueger J., Balduzzi A., Bittencourt H., Moerloose B.D., Peters C., Bader P., Buechner J., Boissel N., Hiramatsu H., et al. 2020. Tisagenlecleucel for pediatric/young adult patients with relapsed/refractory B-cell acute lymphoblastic leukemia: preliminary report of B2001X focusing on prior exposure to blinatumomab and inotuzumab.https://eha25-eha.web.indrina.com/abstracts/page/list/5db0358bf4d01d313c664553/programelement/5eb52c8d5275843e2bdece84 [Google Scholar]
- 32.Ruella M., Barrett D.M., Kenderian S.S., Shestova O., Hofmann T.J., Perazzelli J., Klichinsky M., Aikawa V., Nazimuddin F., Kozlowski M., et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J. Clin. Invest. 2016;126:3814–3826. doi: 10.1172/JCI87366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martyniszyn A., Krahl A.C., André M.C., Hombach A.A., Abken H. CD20-CD19 Bispecific CAR T Cells for the Treatment of B-Cell Malignancies. Hum. Gene Ther. 2017;28:1147–1157. doi: 10.1089/hum.2017.126. [DOI] [PubMed] [Google Scholar]
- 34.Dai H., Wu Z., Jia H., Tong C., Guo Y., Ti D., Han X., Liu Y., Zhang W., Wang C., et al. Bispecific CAR-T cells targeting both CD19 and CD22 for therapy of adults with relapsed or refractory B cell acute lymphoblastic leukemia. J. Hematol. Oncol. 2020;13:30. doi: 10.1186/s13045-020-00856-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qin H., Ramakrishna S., Nguyen S., Fountaine T.J., Ponduri A., Stetler-Stevenson M., Yuan C.M., Haso W., Shern J.F., Shah N.N., Fry T.J. Preclinical Development of Bivalent Chimeric Antigen Receptors Targeting Both CD19 and CD22. Mol. Ther. Oncolytics. 2018;11:127–137. doi: 10.1016/j.omto.2018.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schneider D., Xiong Y., Wu D., Nӧlle V., Schmitz S., Haso W., Kaiser A., Dropulic B., Orentas R.J. A tandem CD19/CD20 CAR lentiviral vector drives on-target and off-target antigen modulation in leukemia cell lines. J. Immunother. Cancer. 2017;5:42. doi: 10.1186/s40425-017-0246-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodriguez-Perales S., Torres-Ruiz R., Suela J., Acquadro F., Martin M.C., Yebra E., Ramirez J.C., Alvarez S., Cigudosa J.C. Truncated RUNX1 protein generated by a novel t(1;21)(p32;q22) chromosomal translocation impairs the proliferation and differentiation of human hematopoietic progenitors. Oncogene. 2016;35:125–134. doi: 10.1038/onc.2015.70. [DOI] [PubMed] [Google Scholar]
- 38.Zanetti S.R., Ziblat A., Torres N.I., Zwirner N.W., Bouzat C. Expression and Functional Role of α7 Nicotinic Receptor in Human Cytokine-stimulated Natural Killer (NK) Cells. J. Biol. Chem. 2016;291:16541–16552. doi: 10.1074/jbc.M115.710574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gomes-Silva D., Srinivasan M., Sharma S., Lee C.M., Wagner D.L., Davis T.H., Rouce R.H., Bao G., Brenner M.K., Mamonkin M. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood. 2017;130:285–296. doi: 10.1182/blood-2017-01-761320. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.