

Abstract
We generated dual-antigen receptor (DR) T cells from induced pluripotent stem cells (iPSCs) to mitigate tumor antigen escape. These cells were engineered to express a chimeric antigen receptor (CAR) for the antigen cell surface latent membrane protein 1 (LMP1; LMP1-CAR) and a T cell receptor directed to cell surface latent membrane protein 2 (LMP2), in association with human leucocyte antigen A24, to treat therapy-refractory Epstein-Barr virus-associated lymphomas. We introduced LMP1-CAR into iPSCs derived from LMP2-specific cytotoxic T lymphocytes (CTLs) to generate rejuvenated CTLs (rejTs) active against LMP1 and LMP2, or DRrejTs. All DRrejT-treated mice survived >100 days. Furthermore, DRrejTs rejected follow-up inocula of lymphoma cells, demonstrating that DRrejTs persisted long-term. We also demonstrated that DRrejTs targeting CD19 and LMP2 antigens exhibited a robust tumor suppressive effect and conferred a clear survival advantage. Co-operative antitumor effect and in vivo persistence, with unlimited availability of DRrejT therapy, will provide powerful and sustainable T cell immunotherapy.
Keywords: CD19-CAR, dual-antigen targeted CART therapy, EBV-associated lymphoma, iPSC-derived CTL, LMP1, LMP2, rejuvenated CTL, dual CAR, iPSC-CART, “off-the-shelf” T cell therapy
Graphical abstract
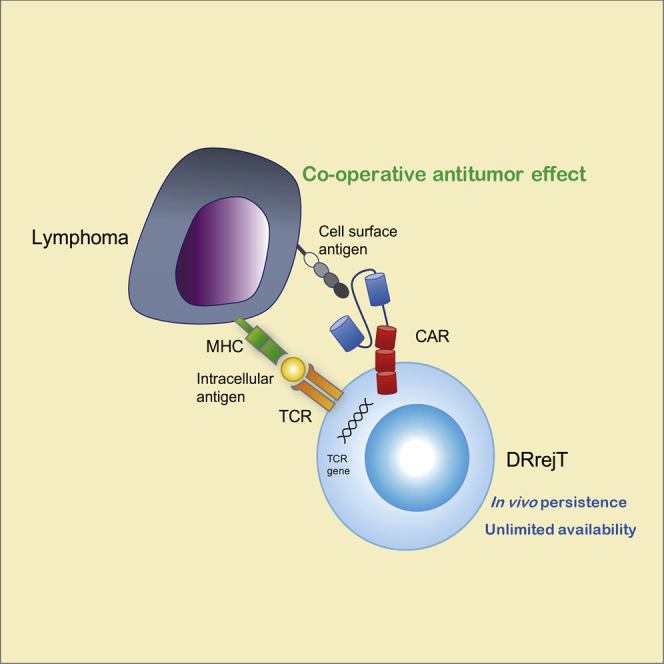
In this article, Harada and colleagues show that dual-receptor rejuvenated T cells can be generated from iPSCs to mitigate tumor antigen escape. Co-operative antitumor effect and in vivo persistence, with unlimited availability of such cells, will provide powerful and sustainable T cell immunotherapy.
Introduction
Chimeric antigen receptors (CARs) enhance effector T cell function in the tumor microenvironment. These genetically modified receptors consist of a single-chain variable fragment (scFv) as an antigen recognition module, linked to a costimulatory domain and a cytoplasmic activation domain such as the T cell receptor (TCR) CD3-ζ chain.1, 2, 3, 4, 5 Relapsed and refractory acute B lymphoblastic leukemia treated using T cells engineered to express CD19-directed CARs shows ∼90% remission rates in clinical trials.6, 7, 8, 9 Nonetheless, relapses occur in 30%–50% of patients because with continued T cell exposure to tumor, T cell exhaustion develops, with tumor antigen escape. This limits clinical utility.10, 11, 12, 13 Furthermore, CAR T cell (CART) therapy requires T cell collection, gene modification, and expansion—too long a process for some patients.14,15
Fully rejuvenated cytotoxic T lymphocytes (CTLs) as unlimited sources of therapeutic T cells may permit “off-the-shelf” therapy. Antigen-specific CTLs can be functionally rejuvenated, yielding rejuvenated CTLs (rejTs) by induced pluripotent stem cell (iPSC) technology,16, 17, 18, 19 and rejTs can survive long-term in vivo as young memory T cells.20 In contrast, peripheral blood-derived original CTLs do not prolong survival of tumor-bearing mice.20,21
We hypothesized that CARs and rejTs in combination (dual receptor rejTs [DRrejTs]) could overcome the drawbacks of conventional CTLs as well as those of CART therapy with regard to prevention of antigen escape, persistence in vivo, and an unlimited cell source. DRrejTs that show 100% CAR transgene expression and 100% antigen specificity can target dual antigens, permitting the expectation that with DRrejT incidence of tumor antigen, escape will be minimal. Latent membrane protein 2 (LMP2) antigen-specific rejTs (LMP2-rejTs) exert a strong anti-tumor effect against an aggressive and treatment-refractory lymphoma, that is, Epstein-Barr virus (EBV)-infected extranodal natural killer (NK)/T cell lymphoma (ENKL), nasal type.20 LMP2-rejTs survive >7 months in treated mice, preventing recurrence of ENKL.20 Remarkable long-term in vivo persistence of LMP2-rejTs also has been proven.20 Moreover, CAR-transduced T-iPSCs can efficiently produce unlimited numbers of DRrejTs.18,22
In this study, we demonstrate that DRrejTs can be constructed to recognize latent membrane protein 1 (LMP1) antigen via CARs and LMP2 antigen via native TCRs. In therapy, these DRrejTs are superior to peripheral blood-derived LMP1-CARTs as well as to with LMP2-rejTs: DRrejTs showed 100% survival and robustly eliminated ENKL not only once but twice (initial tumor; subsequently inoculated tumor). We also generated DRrejTs that recognize CD19 antigen via CARs and LMP2 antigen via native TCRs to confirm the reproducibility of our results. Both DRrejTs act powerfully and sustainedly against EBV-driven tumors.
Results
Lentiviral LMP1-CAR vector construction and peripheral blood LMP1-CART generation
To target LMP1+ EBV-associated lymphomas, we first examined LMP1 expression of EBV-associated lymphoma cell lines. EBV-encoded small RNA (EBER) in situ hybridization was also conducted to detect EBV sequences. The HLA-A∗2402+ EBV-infected ENKL cell line NK-YS and HLA-A∗2402+ and A∗2402− EBV-infected lymphoblastoid cell lines (LCLs) expressed LMP1 and marked for EBER. The EBV-free osteosarcoma cell line HOS did not express LMP1 (Figure 1A). We next generated a lentiviral vector encoding a third-generation CAR directed against LMP1 antigen. The construct comprises an anti-LMP1 scFv linked to sequences encoding CD28, OX40, and CD3ζ domains (Figure 1B).23 The suicide gene inducible caspase-9 (iC9) is linked to LMP1-CAR via 2A-derived nucleotides to increase the safety of CART therapy.23, 24, 25 To confirm lentiviral LMP1-CAR vector effectiveness, we transduced peripheral blood T cells with LMP1-CAR vector. Peripheral blood mononuclear cells (PBMCs) from a healthy donor were stimulated by anti-CD3 and anti-CD28 antibodies and transduced with the lentiviral LMP1-CAR vector. After 3 days, the LMP1-CAR-transduced cells were stained with biotinylated protein L, which binds to scFv of CAR, followed by streptavidin-allophycocyanin (APC) staining, and measured APC positivity by flow cytometry.26 The CAR transgene was expressed by 25.4% of LMP1-CARTs, and the percentages of CD4+ and CD8+ LMP1-CARTs were, respectively, 30.3% and 68.0% (Figure 1C). To verify antitumor activity of PBMC-derived LMP1-CARTs against LMP1-expressing tumor cells, we performed coculture experiments. PBMC-derived LMP1-CARTs were cocultured with EBV-infected LCLs that express LMP1 antigen on their surfaces27 at an effector-to-target ratio (E:T ratio) of 1:1. The percentages of LMP1-CARTs and LCLs were determined 0, 5, and 7 days after coculture began by flow cytometry to detect CD3+CD19− cells and CD3−CD19+ cells, respectively. On day 7 of coculture, activated LMP1-CARTs were abundant and proportions of LCLs were low (LMP1-CARTs/LCLs = 98.0%:0.36%), whereas LCLs were clearly abundant when control effector T cells (activated T cells) were cocultured with LCLs (activated T cells/LCLs = 17.3%:71.6%) (Figure 1D, left). At the same time, absolute cell numbers were measured using counting beads on flow cytometry. LCLs cocultured with LMP1-CARTs decreased from 95,289 ± 10,650 (day 0) to 47,533 ± 29,816 (day 7). However, LCLs cocultured with activated T cells rapidly increased from 132,298 ± 17,458 (day 0) to 978,716 ± 132,325 (day 7) (Figure 1D, right).
Figure 1.

Generation and validation of LMP1-CARTs
(A) LMP1 expression and EBV-encoded small RNA (EBER) in situ hybridization of EBV+ cell lines and EBV− cell line as negative control. Scale bars, 20 μm. (B) Schematic of the lentiviral iC9-CAR vector containing Ubc promoter. CAR contains an scFv derived from heavy (VH) and light chain (VL) variable regions of an anti-LMP1 antibody. TM, transmembrane (CD28). The vector contains the suicide gene iC9, cleavable 2A-like sequence. (C) Flow cytometric analysis of expression of LMP1-CARs in PMBC-derived LMP1-CARTs detected by protein L, which binds to the scFv of CAR, followed by streptavidin-APC staining and determination of APC signal. The data represent at least five independent experiments using four different donors. (D) PBMC-derived LMP1-CARTs and lymphoblastoid cell lines (LCLs) were cocultured (ratio 1:1). Cultures were collected, stained with anti-CD3 and anti-CD19 antibodies, and analyzed by flow cytometry on days 0, 5, and 7. The plots represent three independent experiments. Cells were counted using counting beads on flow cytometry. Graph summarizes the results of total cell number of LCLs. Error bars represent ± SEM. ∗∗∗∗p < 0.0001 by two-way ANOVA. (E) Schematic illustrations of generation of LMP2-rejTs and DRrejTs. T-iPSCs established from LMP2-specific CTL clones were differentiated into LMP2-rejTs. T-iPSCs established from LMP2-specific CTL clones were transduced with lentiviral LMP1-CAR vector and LMP1-CAR-T-iPSCs were differentiated into DRrejTs. (F) Flow cytometric analysis of LMP1-CAR transgene expression (left), CD4 and CD8 expression (center), and LMP2 antigen specificity detected by PE-conjugated HLA-A∗2402/LMP2131–139 tetramer (right) in DRrejTs. (G) Programmed cell death 1 (PD-1) expression in LMP1-CARTs and DRrejTs detected by flow cytometry. Error bars represent ± SD. ∗∗p < 0.01 by unpaired Student’s t test. The plots represent four independent experiments.
These results indicated that the lentiviral LMP1-CARs recognized LMP1 antigens on LCLs and that activated CARTs suppressed the proliferation of LCLs.
iPSC-derived DRrejTs with 100% LMP1-CAR expression and LMP2-specific native TCRs were successfully generated
We next generated iPSC-derived DRrejTs (Figure 1E). LMP2 antigen-specific CTLs (PYLFWLAAI) were generated from a healthy donor expressing human leucocyte antigen (HLA) A∗2402. The generated LMP2-specific CTLs were cloned and reprogrammed into T-iPSCs with the Sendai virus vectors encoding the Yamanaka 4 factors (OCT3/4, SOX2, KLF4, and c-MYC) and SV40 large T antigen.16, 17, 18,20 After T-iPSC establishment, the T-iPSCs were transduced with lentiviral LMP1-CAR vector.17 To select for cells without LMP1-CAR silencing, and thus with stable CAR transgene expression, LMP1-CAR-transduced T-iPSCs were repeatedly sorted for biotinylated protein L expression. After three sortings, LMP1-CAR-T-iPSCs were differentiated into LMP1-CAR-expressing LMP2-rejTs or DRrejTs (Figure 1E). To evaluate CAR transgene expression and LMP2 antigen specificity in DRrejTs, biotinylated protein L staining and LMP2 tetramer analysis were conducted. CAR-specific biotinylated protein L staining was 100%, which means that CAR transgene expression in DRrejTs was 100% (Figure 1F), a proportion much higher than that in PBMC-derived LMP1-CARTs (25.4%) (Figure 1C), suggesting that the cytotoxicity of DRrejTs against LMP1-expressing tumors could be expected to be stronger than that of PBMC-derived LMP1-CARTs. The specificity of LMP2 antigen in DRrejTs was also shown to be 99.1% by tetramer staining (Figure 1F). Furthermore, expression of programmed cell death protein 1 (PD-1), a marker of T cell exhaustion, was compared by flow cytometry with that of DRrejTs and PBMC-derived LMP1-CARTs. The average PD-1 expression was 11.51% ± 4.92% of PBMC-derived LMP1-CARTs and 0.85% ± 0.79% of DRrejTs (p = 0.0052 by an unpaired Student’s t test) (Figure 1G).
In sum, we successfully generated iPSC-derived DRrejTs that exhibited 100% expression of LMP1-CAR transgene and 99% expression of native TCRs recognizing LMP2 antigen, with almost complete absence of PD-1 expression.
DRrejTs exert greater cytotoxicity than do LMP1-CARTs against ENKL
To compare the cytotoxicity of DRrejTs with that of PBMC-derived LMP1-CARTs against NK-YS cells, we performed chromium-51 (51Cr) release assays. We identified stronger cytotoxicity against NK-YS cells with DRrejTs than with LMP1-CARTs, with specific lysis of 86.1% ± 5.9% versus 48.2% ± 13.6% (p = 0.0114, unpaired Student’s t test) and 68.3% ± 3.1% versus 31.7% ± 2.1% (p < 0.0001, unpaired Student’s t test) at E:T ratios of 5:1 and 2.5:1, respectively (Figure 2A). To compare further the in vivo cytotoxicity of peripheral blood-derived LMP1-CARTs against ENKL cells with that of DRrejTs, NK-YS cells transduced with retroviral vector encoding a green fluorescent protein (GFP)-firefly luciferase (FFLuc) fusion protein (GFP/FFLuc) were intraperitoneally engrafted into non-obese diabetic (NOD)/Shi-severe combined immunodeficiency (SCID), interleukin (IL)-2Rγ knockout (IL-2RγKO) Jic (NOG) mice (1 × 105 cells/mouse). Bioluminescence, a signal that indicates tumor growth, was monitored. Four days after tumor injection, mice were divided into three groups: a no treatment group (n = 5), a peripheral blood-derived LMP1-CART group (n = 7), and a DRrejT group (n = 8). In the two treatment groups, LMP1-CARTs or DRrejTs were intraperitoneally injected on day 0, day 7, and day 14 (5 × 106 per dose, three doses). DRrejTs more rapidly yielded a continuous anti-ENKL effect. While LMP1-CARTs suppressed ENKL proliferation to a certain degree in the short term, tumor enlargement rapidly occurred in mice treated with LMP1-CARTs, beginning 23 days after the first treatment; that is, the tumor suppressive effect of LMP1-CART did not last long (Figure 2B). Accordingly, only DRrejTs showed a significant survival advantage over both the no treatment group (p = 0.0001) and the peripheral blood-derived LMP1-CART group (p < 0.0001 by log-rank test) (Figure 2C).
Figure 2.

DRrejTs exhibit cytotoxicity against EBV-associated lymphoma superior to LMP1-CARTs and LMP2-rejTs
(A) In vitro51Cr release assay of iPSC-derived DRrejTs and PBMC-derived LMP1-CARTs (effectors) against HLA-A∗2402+ ENKL and HLA-A∗2402− LCLs (targets). Error bars represent ± SD. Data represent three independent triplicate experiments. (B) NOG mice were intraperitoneally inoculated with HLA-matched ENKL cells expressing firefly luciferase and treated either with PBMC-derived LMP1-CARTs (n = 7) or iPSC-derived DRrejTs (n = 8). No treatment indicates that mice were injected with ENKL cells but not treated with T cells (n = 5). LMP1-CARTs or DRrejTs were injected intraperitoneally once a week (3 doses). Images of all mice from each group of two independent experiments are shown. (C) Kaplan-Meier curve representing percentage survival of experimental groups: no treatment, LMP1-CARTs, or DRrejTs. ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 by log-rank test. (D) In vitro51Cr release assay of iPSC-derived DRrejTs and LMP1-CAR-transduced HPV-rejTs (LMP1-CAR/HPV-rejTs) (effectors) against HLA-A∗2402+ LCLs and LMP1− tumors (targets). Error bars represent ± SD. Data represent three independent triplicate experiments. (E) iPSC-derived DRrejTs and control rejTs were cocultured with HLA-A∗2402+ GFP/FFluc ENKL (ratio 5:1). Cultures were collected, stained with anti-CD3 antibody, and analyzed by flow cytometry on days 0, 5, and 7. The plots represent three independent experiments. Cells were counted using counting beads on flow cytometry. Graph summarizes the results of total number of NK-YS cells. Error bars represent ± SEM. ∗∗∗∗p < 0.0001 by two-way ANOVA. (F) In vitro51Cr release assay of iPSC-derived DRrejTs and LMP2-rejTs (effectors) against HLA-A∗2402+ LCL and HLA-A∗2402− LCL (targets). Error bars represent ± SD. Data represent three independent triplicate experiments. (G) iPSC-derived DRrejTs and control rejTs were cocultured with HLA-A∗2402− LCL (ratio 5:1). Cultures were collected, stained with anti-CD3 and anti-CD19 antibodies, and analyzed by flow cytometry on days 0, 5, and 7. The plots represent three independent experiments. Cells were counted using counting beads on flow cytometry. Graph summarizes the results of total cell number of LCLs. Error bars represent ± SEM. ∗∗∗∗p < 0.0001 by two-way ANOVA. E:T ratio, effector-to-target ratio. (H) In vitro51Cr release assay of DRrejT (effector) against HLA-A∗2402− ENKL and EBV− tumor (targets). Error bars represent ± SD. (I) In vitro51Cr release assay of DRrejTs (effectors) against HLA-A∗2402+ ENKL primary cells and EBV− tumors (targets). Error bars represent ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 by unpaired Student’s t test.
To compare the cytotoxicity against NK-YS cells of DRrejTs with that of non-LMP2-specific LMP1-CAR rejTs, we performed 51Cr release assays. We also confirmed stronger cytotoxicity against NK-YS cells with DRrejTs than with LMP1-CAR/HPV-rejTs, with specific lysis of 83.1% ± 2.7% versus 40.6 ± 4.5% (p = 0.0002, unpaired Student’s t test) and 70.9% ± 7.0% versus 34.1% ± 4.2% (p = 0.0014, unpaired Student’s t test) at E:T ratios of 10:1 and 5:1, respectively (Figure 2D).
For serial killing experiments, DRrejTs (CD3+ and GFP-) were cocultured with GFP/FFLuc NK-YS cells (CD3− and GFP+), at an E: T ratio of 5:1. On day 5 of coculture, DRrejTs had eliminated substantial numbers of NK-YS cells, which dropped from 8.5% (day 0) to 0.9% (day 5). In contrast, control rejTs that were not specific to EBV antigen could not eliminate the target cells and allowed the proliferation of NK-YS cells, which rose from 9.0% (day 0) to 14.7% (day 5) and 38.4% (day 7) (Figure 2E, left). The absolute cell number of GFP/FFLuc NK-YS cells cocultured with DRrejTs actually decreased from 94,861 ± 2,223 (day 0) to 5,914 ± 133 (day 7), whereas NK-YS cells cocultured with control rejTs increased from 83,896 ± 4,987 (day 0) to 114,119 ± 15,004 (day 7) (Figure 2E, right).
These data indicated that DRrejTs showed a tumor-suppressive effect against ENKL stronger than that of peripheral blood-derived LMP1-CARTs both in vitro and in vivo.
DRrejTs exhibited additive killing via LMP1-CARs
To learn whether DRrejTs exhibit a killing effect beyond that of LMP2-rejTs, the cytotoxicities of DRrejTs and LMP2-rejTs were compared by a 51Cr release assay. At an E:T ratio of 10:1, DRrejTs killed HLA-A∗2402+ LCLs (autologous LCLs) with 82.0% ± 20.1% specific lysis, whereas LMP2-rejTs showed lower levels of killing, with 62.0% ± 4.5% specific lysis (p = 0.1679, unpaired Student’s t test). LMP2-rejTs, which require HLA-restricted antigen recognition, could not kill HLA-A∗2402− LCLs (0.5% ± 1.6%, E:T ratio of 10:1; DRrejTs, p = 0.0022; LMP2-rejTs, p < 0.0001; unpaired Student’s t test) (Figure 2F).
To examine the LMP1-CAR cytotoxicity of DRrejTs, HLA-A∗2402− LCLs were cocultured with DRrejTs. On day 7 of coculture at an E:T ratio of 5:1, DRrejTs could eliminate HLA-A∗2402− LCLs through CAR recognition (CD19+ and CD3−, 11.5% day 0 to 1.4% day 7), while control rejTs not specifically responsive to EBV antigen could not eliminate HLA-A∗2402- LCLs, which thereupon proliferated (CD19+ and CD3−, 11.0% day 0 to 50.7% day 7) (Figure 2G, left). The absolute cell number of HLA-A∗2402− LCLs cocultured with DRrejTs decreased from 86,565 ± 7,941 (day 0) to 36,148 ± 4,282 (day 7), whereas that of HLA-A∗2402− LCLs cocultured with control rejTs rapidly increased from 92,450 ± 8,091 (day 0) to 370,255 ± 14,009 (day 7) (Figure 2G, right). Furthermore, we examined the LMP1-CAR cytotoxicity of DRrejTs using the HLA-A∗2402− ENKL cell line SNK-6. At an E:T ratio of 10:1, DRrejTs killed SNK-6 cells with 53.3% ± 18.0% specific lysis, whereas DRrejTs did not kill EBV− tumor cells (control target), with 8.6% ± 4.8% specific lysis (p = 0.0107, unpaired Student’s t test) (Figure 2H). Killing of LMP2-rejTs via TCRs against HLA-A∗2402+ ENKL was also strong, although additive killing via LMP1-CARs was shown, its extent was not statistically significant (Figure 2F). However, using HLA-mismatched LCLs and ENKL cell lines permitted a clear demonstration of reliable cytotoxicity of DRrejTs via LMP1-CARs (Figures 2G and 2H).
Furthermore, to prove the anti-tumor effect of DRrejTs using primary ENKL cells, we assayed the cytotoxicity of DRrejTs against HLA-A∗2402+ patient ENKL cells (42% of tumor-infiltrated bone marrow) by 51Cr release. Strong killing of DRrejTs was successfully demonstrated against HLA-A∗2402+ primary ENKL cells. At an E:T ratio of 10:1, DRrejTs killed ENKL primary cells with 60.7% ± 8.6% specific lysis, but did not kill the control target (p = 0.0008, unpaired Student’s t test) (Figure 2I).
These data suggested that DRrejTs exhibited an additional killing effect, owing to CARs, beyond that of LMP2-rejTs and also possessed cytotoxicity not only against HLA-mismatched LMP1+ tumor cells but even against primary ENKL cells.
All DRrejT-treated mice survived for more than 100 days
To assess the additive killing effect of DRrejTs over LMP2-rejTs against LMP1+ EBV-associated lymphoma cells in vivo, we also intraperitoneally engrafted NOG mice with 1 × 105 GFP/FFLuc-NK-YS cells. Four days after tumor injection, mice were divided into a control group and two treatment groups. No treatment was given in the control group (n = 5). The treatment groups (n = 5 each) were treated with LMP2-rejTs on days 0, 7, and 14 (5 × 106 cells/dose, three doses) and DRrejTs on days 0, 7, and day 14 (5 × 106 cells/dose, three doses). Tumor signals in both DRrejT- and LMP2-rejT-treated mice were suppressed, but the tumor-suppressive effect was clearly stronger in the DRrejT treatment group than in the LMP2-rejT treatment group (Figure 3A). Furthermore, DRrejTs and LMP2-rejTs conferred a significant survival advantage over the no treatment group (p = 0.0033, p = 0.03, respectively; log-rank test), and all mice treated with DRrejTs survived more than 110 days after the first rejT dose (Figure 3B). Mouse peripheral blood was stained with anti-human CD3 antibodies and HLA-A∗2402/LMP2131–139 tetramer, with analysis by flow cytometry, 101 days after the first rejT dose. Human CD3+ lymphocytes were detected in peripheral blood of both DRrejT-treated and LMP2-rejT-treated mice, but were proportionally more abundant in DRrejT-treated mice (35.8%) than in LMP2-rejT-treated mice (3.05%). In addition, tetramer+/CD3+ lymphocytes were detected in peripheral blood of DRrejT-treated mice, as well as, in smaller numbers, in that of LMP2-rejT-treated mice (Figure 3C).
Figure 3.

DRrejTs display additive anti-ENKL activity over LMP2-rejTs in vivo
(A) Bioluminescence imaging of mice treated with LMP2-rejTs or DRrejTs. ENKL-bearing mice were divided into three groups that received no treatment (n = 5), LMP2-rejTs (n = 5), or DRrejTs (n = 5). No treatment indicates that mice were injected with ENKL cells but not treated with T cells. LMP2-rejTs or DRrejTs (5 × 106 cells) were injected intraperitoneally once a week (three doses). Data represent at least two independent experiments. (B) Kaplan-Meier curve representing percentage survival of the experimental groups: no treatment, LMP2-rejTs, or DRrejTs. ∗p < 0.05, ∗∗p < 0.01 by log-rank test. ns, not significant. (C) Flow cytometric analysis of tetramer+/CD3+ population from peripheral blood of LMP2-rejT- or DRrejT-treated mice. Plots represent two independent experiments.
In conclusion, DRrejTs strongly suppressed ENKL, markedly prolonged the survival of treated mice, and persisted long-term in mouse peripheral blood.
DRrejTs could eliminate re-inoculated tumor cells
To evaluate whether DRrejTs were actually present in vivo and were still effective, we inoculated a second dose of 1 × 105 ENKL cells into mice surviving the previous experiment more than 110 days after the first rejT dose; three mice received a second dose of ENKL in the DRrejT group and two received a second dose of ENKL in the LMP2-rejT group. Both DRrejTs and LMP2-rejTs gradually eliminated re-inoculated ENKL cells over 60 days without any additional treatment (Figure 4A). Flow cytometry was also used to detect DRrejTs or LMP2-rejTs in peripheral blood of long-term surviving mice before and after re-inoculation of ENKL cells (Figure 4B). Although LMP2 tetramer+/CD3+ rejTs were not detected in peripheral blood of LMP2-rejT-treated mice before ENKL re-inoculation, they reappeared 4 days after ENKL re-inoculation (Figure 4B, upper). In contrast, LMP2 tetramer+/CD3+ DRrejTs were clearly detectable in peripheral blood of treated mice even before re-inoculation, and their population increased after re-inoculation (Figure 4B, lower). In addition, to examine whether rejTs were existing as memory T cells in vivo, CD45RA and CD62L expression in mouse peripheral blood was analyzed. The major population was shifted from an effector memory phenotype (CD45RA−CD62L−) toward a central memory phenotype (CD45RA−CD62L+) in peripheral blood of mice treated with either LMP2-rejTs or DRrejTs (Figure 4C).
Figure 4.

DRrejTs eliminate re-inoculated ENKL
(A) LMP2-rejT-treated or DRrejT-treated ENKL-inoculated mice were re-inoculated with ENKL cells. Bioluminescence imaging of re-inoculated mice: LMP2-rejT-treated mice (n = 2) or DRrejT-treated mice (n = 3). ENKL (1 × 105 cells) were injected intraperitoneally into survivors of a previous experiment, without additional rejTs. (B) Flow cytometric analysis of tetramer+/CD3+ population from peripheral blood of LMP2-rejT- or DRrejT-treated ENKL re-inoculated mice. The plots represent two independent experiments. (C) Flow cytometric analysis of memory phenotype (CD45RA and CD62L population) from peripheral blood of LMP2-rejT- or DRrejT-treated ENKL re-inoculated mice. The plots represent two independent experiments. (D) LMP2-rejTs (no iC9) and DRrejTs (with iC9) were treated with CID (80 nM) and apoptosis was measured 24 h later by flow cytometry for annexin V/7-AAD positivity. Data represent three independent triplicate experiments. Cells were counted using counting beads on flow cytometry. Graph summarizes the results of total apoptotic cell number. Error bars represent ± SD. ∗∗∗∗p < 0.0001 by two-way ANOVA. ns, not significant.
Collectively, these data demonstrated that DRrejTs persisted in the long term as young memory T cells in vivo and successfully eliminated re-inoculated ENKL cells.
iC9 safeguard system induced apoptosis in DRrejTs
To confirm effective function of the iC9 safeguard system in this therapy model, DRrejTs into which iC9 had been introduced and LMP2-rejTs were plated and treated with a chemical inducer of dimerization (CID; AP1903 or its functionally identical analog AP20187) 24 h later. In the presence of CID, dimerized iC9 directly activates caspase-3, triggering apoptosis in transduced cells.17,24,25,28, 29, 30 Annexin V-phycoerythrin (annexin V) binding and 7-aminoactinomycin D (7-AAD) uptake were examined 48 h after CID addition. CID induced apoptosis in iC9-introduced DRrejTs (with CID, 91.19%; without CID, 4.0%). In contrast, CID administration did not significantly increase apoptosis in LMP2-rejTs (with CID, 16.8%; without CID, 13.7%). The absolute number of apoptotic DRrejTs increased to 598,907 ± 63,833 with CID from 19,780 ± 2,997 without CID (p < 0.0001, two-way ANOVA), whereas it did not change in LMP2-rejTs with CID 72,271 ± 6,152 and without CID 60,776 ± 2,689 (p = 0.9064, two-way ANOVA) (Figure 4D).
Thus, the iC9 system can induce apoptosis in iC9-introduced DRrejTs and provides an effective safeguard for this therapy in the case of cytokine-release syndrome, acute graft-versus-host disease (GVHD), or other unexpected adverse effects.
DRrejTs targeting CD19 and LMP2 antigens robustly suppressed the proliferation of LCLs
To examine whether DRrejTs with another combination of target antigens also efficiently suppress tumor proliferation, we generated DRrejTs directed against CD19 antigen via CAR and against LMP2 antigen via native TCRs. LMP2-rejTs was directly transduced with retroviral CD19-CARs in this experiment. We also generated retroviral CD19-CAR-transduced human papilloma virus (HPV)-specific rejTs (CD19-CAR/HPV-rejTs) as a control targeted against a single antigen. CAR transgene expression in both CD19-CAR/HPVrejTs and DRrejTs was ∼70%–80%. After GFP/FFLuc-labeled HLA-A∗2402+ LCLs (CD19+ and LMP2+) were intraperitoneally engrafted into NOG mice on day −3, LMP2-rejTs, CD19-CARTs, or DRrejTs were injected into the mice in the treatment groups on days 0, 7, and 14. The tumor signals in mice without treatment aggressively increased with time (Figure 5A). Alternatively, LMP2-rejTs clearly suppressed tumor proliferation. Compared with mice without treatment, LMP2-rejTs and CD19-CAR/HPV-rejTs produced a significant reduction in tumor burden on day 18 (p = 0.003 and 0.0101; one-way ANOVA) (Figure 5B). DRrejTs further suppressed proliferation of EBV+ LCLs (Figure 5A) and robustly reduced tumor signal compared with untreated mice (p < 0.0001; one-way ANOVA), even with CD19-CAR/HPV-rejT-treated mice (p = 0.0349; one-way ANOVA) on day 18 (Figure 5B).
Figure 5.

DRrejTs robustly suppressed LCLs
(A) Bioluminescence imaging of mice treated with LMP2-rejTs, CD19-CAR-transduced HPV-rejTs (CD19-CAR/HPV-rejTs), or DRrejTs. EBV+ LCL-bearing mice were divided into four groups that received no treatment (n = 5), LMP2-rejTs (n = 4), CD19-CAR/HPV-rejTs (n = 4), or DRrejTs (n = 6). No treatment indicates mice that were injected with LCL cells but not treated with T cells. LMP2-rejTs, CD19-CAR/HPV-rejTs, or DRrejTs (4 × 106 cells) were injected intraperitoneally once a week (three doses). Images of all mice from each group of two independent experiments are shown. (B) Quantification of tumor burden on day 18 is represented. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001 by one-way ANOVA. (C) Kaplan-Meier curve representing percentage survival of the experimental groups: no treatment, LMP2-rejTs, CD19-CAR/HPV-rejTs, or DRrejTs. ∗p < 0.05, ∗∗p < 0.01 by log-rank test. (D) Flow cytometric analyses of peripheral blood of LMP2-rejT (day 42)-, CD19-CAR/HPV-rejT (day 23)-, or DRrejT (day 42)-treated LCL-inoculated mice. The plots represent three independent experiments. (E) CD19-CARs persisted in peripheral blood of mice treated with CD19-CAR/HPV-rejTs on day 23 or DRrejTs on day 48. Copy number of CD19-CAR-transduced T cells, positive control. Error bars represent ± SD. ∗∗∗∗p < 0.0001 by one-way ANOVA. Data represent three independent triplicate experiments.
Furthermore, LMP2-rejTs, CD19-CAR/HPV-rejTs, and DRrejTs clearly prolonged survival of mice compared to mice without treatment (p = 0.0047, p = 0.0253, and p = 0.0016 by log-rank test, respectively). DRrejTs conferred upon mice a survival advantage superior to that of mice treated with LMP2-rejTs (p = 0.0154; log-rank test). The tumor suppressive effect of DRrejTs was better, with longer survival, than that found in mice treated with CD19-CAR/HPV-rejTs (p = 0.0031; log-rank test) (Figure 5C). To confirm whether DRrejTs and LMP2-rejTs could efficiently proliferate in vivo, LMP2 tetramer+ and human CD3+ positivity in mouse peripheral blood was examined. A small LMP2 tetramer+/CD3+ population was detected in peripheral blood of a LMP2-rejT-treated mouse (0.41%) and CD19-CAR/HPV-rejT treated mice (0.89%), with a larger one in peripheral blood of a DRrejT-treated mouse (2.42%) (Figure 5D). On day 48, CD19-CAR copy number per microgram of genomic DNA in peripheral blood was 1,066,462.4 ± 37,349.9 in a mouse treated with DRrejTs, showing that DRrejTs efficiently expanded and persisted in vivo. Alternatively, CD19-CAR copy number was 123,135.1 ± 17,245.5 in a mouse treated with CD19-CAR/HPV-rejTs and significantly lower than that of DRrejT-treated mice (p < 0.0001 by one-way ANOVA). CD19-CAR-transduced T cells with 76% CAR transgene expression 4 days after CD19-CAR transduction were used as the positive control (992,987.7 ± 24,039.7) (Figure 5E).
DRrejTs targeting CD19 and LMP2 antigens thus also robustly suppressed tumor growth of LCLs, conferred a survival advantage, and efficiently expanded in vivo with high CD19-CAR transgene expression, permitting similar conclusions in two in vivo models.
Discussion
Because tumors exhibit a number of escape mechanisms, early and multi-faceted intervention is required for efficient anti-tumor therapy. EBV-associated lymphoma cells express LMP1 and LMP2 in many cases,20,31,32 good targets for CARs on the cell surface and for TCRs in association with HLA class I antigens. To target both LMP1 and LMP2, we generated iPSC-derived dual-antigen receptor bearing CTLs, namely DRrejTs.
In this study, CAR transgene expression in peripheral blood-derived LMP1-CARTs was ∼25% (Figure 1C), whereas that of iPSC-derived DRrejTs was 100% (Figure 1F). Because iPSCs are an excellent platform for gene transduction,17,18,22 transgene expression of lentiviral LMP1-CARs carrying Ubc promoter into T-iPSCs was ∼90% after the first transduction (data not shown). As the Ubc promoter provides stable expression of the transgene,17 CAR transgene expression was still high after differentiation into DRrejTs (Figure 1F). The robust cytotoxicity shown against EBV-associated lymphoma was consistent with the high transgene expression in DRrejTs (Figure 2A); DRrejTs exhibit sustained tumor suppression in vivo (Figure 2B). A clear survival advantage over peripheral blood LMP1-CARTs was also shown (Figure 2C). Furthermore, DRrejTs targeting CD19 and LMP2 antigens conferred survival in tumor-inoculated mice longer than that conferred by single antigen-targeted T cells, CD19-CAR-transduced HPV-rejTs, and LMP2-rejTs. To obtain a better result, DRrejTs might be generated from lentiviral CD19-CAR-iPSCs, resulting in stable CAR transgene expression of 100%.
Furthermore, as DRrejTs are the descendants of LMP1-CAR-transduced T-iPSCs established from LMP2-CTL clones, LMP2 antigen specificity of DRrejTs was also nearly 100% (Figure 1F). Based on LMP2-specific cytotoxicity, killing of LMP1-CARs contributed to the additive cytotoxicity of DRrejTs against EBV-associated lymphomas in vitro (Figure 2F) and in vivo (Figure 3A). As most adults are infected with EBV and are seropositive, LMP2-rejTs are theoretically stimulated effectively by engagement of native TCRs and LMP2 antigen-presenting tumor cells.33 Therefore, we think that to use LMP2-rejTs as a vehicle for CARs is reasonable in DRrejT therapy.
We further demonstrated that DRrejTs gradually eliminated even re-inoculated ENKL without additional treatment (Figure 4A). Effector and central memory T cells were detected in peripheral blood of treated mice. The population shifted from an effector to central memory phenotype after tumor re-inoculation (Figure 4C), suggesting that rejuvenated central memory phenotype DRrejTs persist strongly and prevent tumor relapse in vivo. Exclusive sampling of peripheral blood in the xenograft experiments does not consider homing of DRrejTs to secondary lymphoid organs; however, DRrejTs may home to spleen or lymph nodes and persist for the long term, causing elimination of ENKL.20
Tandem CARTs engage two antigens on glioblastomas simultaneously by inducing heterodimers, which promotes additive T cell activation when both antigens are encountered concurrently.34 However, many tumors do not express two suitable antigens on the cell surface. Although antigen recognition by DRrejTs proceeds differently from that by tandem CARTs, the powerful anti-tumor effect of DRrejTs might be attributable to its dual-antigen recognition ability, with not only LMP1 or CD19 recognition by CARs but also LMP2 recognition by native TCRs. When targeting both tumor antigens simultaneously, accelerated tumor elimination becomes possible and the emergence of resistance to therapy can be avoided.35 Using several cell lines permitted confirmation of the efficient function of DRrejTs. In addition, results of killing assays in an ENKL patient bone-marrow sample infiltrated by tumor further emphasized the likely clinical applicability of this work.
The important advantage of iPSC-derived DRrejT therapy is that once T-iPSCs are established from CTL clones and CARs are transduced into established T-iPSCs, one can unlimitedly generate therapeutic DRrejTs from T-iPSCs. Repeated administration of large numbers of DRrejTs to patients, with rapid and powerful killing by DRrejTs, would prevent antigen escape relapse. If we generate from healthy donors several iPSC lines originating from different HLA-restricted LMP2-CTLs and edit HLA class I to avoid rejection by host immune cells, DRrejT therapy will be suitable for a great number of patients and true “off-the-shelf” therapy will be feasible.
When rejTs are generated from patient peripheral blood T cells, GVHD is infrequent. However, triggering severe toxicity by CART cell expansion, modifying T cell trafficking, or effector activity remains a concern.23 To make adoptive DRrejT therapy safer, we equipped DRrejTs with an iC9 safety system. Indeed, administration of the dimerizer CID efficiently induced apoptosis in DRrejTs that included this system (Figure 4D). In clinical translation, one must pay attention to the oncogenic risk of iPSCs caused by lentiviral transduction. CAR-iPSCs can be cloned, analyzed for copy number, and selected for a minimum number of oncogenic mutations. Should an undesired event happen in spite of these validation processes, the iC9 safety system will be useful: its introduction into iPSC-derived DRrejT therapy mitigates possible toxicity and tumorigenic risk of CART therapy.17 Our work thus serves as a proof of concept that novel DRrejT therapy can overcome the drawbacks of conventional CTL and CART therapy.
Materials and methods
Cell lines
Dr. Junjiroh Tsuchiyama (Okayama University Medical School, Okayama, Japan) kindly provided the HLA-A∗2402+ ENKL cell line NK-YS.20,36 NK-YS cells were grown in Iscove’s modified Dulbecco’s medium (IMDM) (Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco), 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mM l-glutamine (Gibco), and 100 IU/mL IL-2 (Miltenyi Biotec, Bergisch Gladbach, Germany).37 Dr. Norio Shimizu (Tokyo Medical and Dental University, Tokyo, Japan) kindly provided the HLA-A∗2402− ENKL cell line SNK-6.20,38 SNK-6 cells were grown in NS-A2 medium (Nissui, Tokyo, Japan) supplemented with 10% FBS (Gibco) and 100 IU/mL IL-2 (Miltenyi Biotec). EBV+ LCLs were derived from PBMCs in our laboratory using EBV isolated from B95-8 cell lines in the presence of cyclosporin A. Donor B lymphocytes were infected with EBV and transformed into LCLs.39, 40, 41, 42 EBV+ LCLs were grown in Roswell Park Memorial Institute 1640 medium (Gibco) supplemented with 10% FBS (Gibco), 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 mM l-glutamine (Gibco). Cells from the EBV− osteosarcoma cell line HOS (ATCC, Manassas, VA, USA) were grown in IMDM (Gibco) supplemented with 10% FBS (Gibco), 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 mM l-glutamine (Gibco).
Clinical sample
Bone marrow infiltrated by ENKL primary cells from a patient diagnosed with ENKL (2014) at Juntendo University School of Medicine, Department of Hematology, were used for 51Cr release assays. ENKL was diagnosed on histopathologic study of a skin biopsy specimen (READ, Koutoubiken, Tokyo, Japan) according to World Health Organization classification guidelines. Bone marrow infiltration (42%) was also diagnosed. Bone marrow mononuclear cells, including ENKL cells, were separated and stored frozen. Cell viability in a thawed aliquot was >80%. Mononuclear cells, not further sorted or selected, were used for 51Cr release assays. Use of material and of clinical information was approved by the Research Ethics Committee for the Faculty of Medicine, Juntendo University, and was in accordance with the Declaration of Helsinki.
Immunohistochemical staining
Immunoperoxidase studies were performed on formalin-fixed paraffin sections with a horseradish peroxidase-conjugated polymer method. Anti-LMP1 mouse monoclonal antibody (CS.1-4; 1:100 dilution; M0897, Dako, Glostrup, Denmark) was used for immunostaining.43 Anti-LMP1 antibody was applied after antigen retrieval following microwave oven heating treatment.
In situ hybridization
For in situ hybridization to detect EBV RNA, the tissue was hybridized with oligo-probe EBER-1 (AGACACCGTCCTCACCACCCGGGACTTGTA) and sense probe (negative control), labeled with digoxigenin.44 After dewaxing, dehydration, and protein K digestion, sections were hybridized overnight in a solution of 50% formamide containing 5 ng of digoxigenin-labeled probes. After washing, detection was accomplished using an avidin-alkaline phosphatase conjugate, with diaminobenzidine as the chromogen.
Vector construction and virus production
DNA sequences of the variable regions of light chain (VL) and heavy chain (VH) of human anti-LMP1 are reported.45 Synthesized 2A-VL-linker-VH-hinge (GenScript, Piscataway, NJ, USA) was amplified by polymerase chain reaction (PCR) and fused to the PsiI-PflmI site of the template SFG-iC9-2A-14G2a (GD2)-CD28-OX40z by replacing GD2-CD28-OX40z with LMP1-CD28-OX40z. To generate a lentiviral CAR vector construct, the leader-LMP1-CD28-OX40z was amplified by PCR and then fused into the XbaI-XhoI site of the lentiviral Cs-Ubc-iC9-F2A-mCherry construct by replacing mCherry with LMP1-CD28-OX40. After Cs-Ubc-iC9-F2A-LMP1-CD28-OX40z was generated, lentiviral vectors pseudotyped with the vesicular stomatitis virus G glycoprotein were produced in cultured 293T cells as described.17,46 Virus supernatants were collected 48–72 h later. The pan-caspase inhibitor qVD-Oph (R&D Systems, Minneapolis, MN, USA) (20 μM) was added to the medium to protect 293T cells during lentivirus production.17 CD19-4-1BBz amplified by PCR was fused into the EcoRI-XhoI site of the MSCV-IRES-GFP-construct (Addgene, Watertown, MA, USA). After MSCV-CD19-4-1BBz-IRES-GFP was generated, 293T cells were transfected with the retroviral construct Peg-Pam-e, encoding gag-pol, and RDF, encoding the RD114 envelope, using GeneJuice transfection reagent (Sigma-Aldrich, St. Louis, MO, USA). Supernatants were collected 48–72 h later as described.28 The vector encoding GFP/FFLuc is described.47
Peripheral blood-derived CART generation
PBMCs of healthy donors were activated by anti-CD3 antibody and anti-CD28 antibody (both BioLegend) in the presence of IL-2 (100 U/mL). Three days later, activated T cells were seeded onto 24-well plates coated with RetroNectin (Takara Bio, Shiga, Japan) and LMP1-CAR lentiviral supernatants or CD19-CAR retroviral supernatants were added to each well. CARTs were cultured in NS-A2 (Nissui) with 10 ng/mL each of IL-7 and IL-15 (Miltenyi Biotec).
EBV-specific CTL generation
PBMCs were isolated from an HLA-A∗2402-positive healthy donor (the Institutional Regulation Boards for Human Ethics at Juntendo University School of Medicine and at the Institute of Medical Science, University of Tokyo, approved the experimental protocol). They were cocultured with autologous dendritic cells (DCs) presenting the LMP2-derived peptide PYLFWLAAI (Mimotopes, Victoria, Australia) or TYGPVFMSL (MBL, Nagoya, Japan) in the presence of IL-4 (400 IU/mL) and IL-7 (10 ng/mL; both Miltenyi Biotec).48,49 On day 9 of culture, T cells were restimulated with peptide-pulsed DCs. On day 16, CTLs were stained with HLA-A∗2402/LMP2131–139 tetramer or A∗2402/LMP2419–427 tetramer (MBL), and tetramer-reactive CTLs were cloned by a limiting dilution to obtain LMP2-specific CTL clones.16,17,20
T-iPSC establishment from EBV-specific CTL clones
An EBV antigen-directed LMP2-CTL clone (PYLFWLAAI) was transduced with Sendai virus vectors encoding the Yamanaka 4 factors OCT3/4, SOX2, KLF4, and c-MYC and SV40 large T antigen. Another LMP2-CTL clone (TYGPVFMSL) was transduced with Sendai virus vector encoding the Yamanaka 4 factors, NANOG, and LIN28.21 Transduced cells were transferred onto plates coated with iMatrix-511 (Nippi, Tokyo, Japan) and cultured in NS-A2. Growth medium was gradually replaced with StemFit AK03N (Ajinomoto Healthy Supply, Tokyo, Japan). After ∼14 days, iPSC colonies were picked up into new culture wells and passaged in CTS Essential 8 medium (Thermo Fisher Scientific, Waltham, MA, USA) several times until cell numbers sufficed for studies.20
LMP1-CAR transduction into T-iPSCs
T-iPSCs established from the LMP2-CTL clone (PYLFWLAAI) (1 × 105 cells) were transduced with LMP1-CAR lentiviral vector on vitronectin (Thermo Fisher Scientific)-coated plates (multiplicity of infection of 20) and cultured in CTS Essential 8 medium (Thermo Fisher Scientific). On day 9, transduced iPSCs were stained with biotinylated protein L (Thermo Fisher Scientific) followed by APC-streptavidin (BD Biosciences, San Jose, CA, USA). LMP1-CAR+ iPSCs were sorted several times by flow cytometry.
Redifferentiation of T-iPSCs into DRrejTs
Small clumps of LMP1-CAR-T-iPSCs were plated on C3H10T1/2 feeder cells in IMDM supplemented with gamma-irradiated 15% FBS (HyClone, GE Healthcare UK, Little Chalfont, UK) and a cocktail of 10 mg/mL insulin, 5.5 mg/mL transferrin, 5 ng/mL sodium selenite, 2 mM l-glutamine (Thermo Fisher Scientific), 0.45 mM α-monothioglycerol (Sigma-Aldrich), and 50 mg/mL ascorbic acid (Takeda Pharmaceutical, Tokyo, Japan) in the presence of 20 ng/mL vascular endothelial growth factor (Miltenyi Biotec). On day 14 of coculture, sac-like structures that contained hematopoietic progenitor cells were extracted and transferred onto Delta-like ligands (DLs) 1- and 4-expressing C3H10T1/2 feeder cells in minimum essential medium (MEM) α (Thermo Fisher Scientific) with FBS (HyClone) in the presence of 20 ng/mL human stem cell factor, 10 ng/mL human Fms-related tyrosine kinase 3 ligand (both Miltenyi Biotec), and 10 ng/mL IL-7. On day 28 of coculture, T-lineage cells were harvested, stimulated by irradiated PBMCs in NS-A2 in the presence of 5 mg/mL phytohemagglutinin (Sigma-Aldrich) and of IL-7 and IL-15 (Miltenyi Biotec) (10 ng/mL each). C3H10T1/2, DL1/4-expressing C3H10T1/2 feeder cell lines, and gamma-irradiated FBS (HyClone) have already been validated and are compatible with the good manufacturing practice standard for clinical use. Master cell bank stocks were made.
LMP2 specificity and LMP1-CAR transgene expression were confirmed by staining with HLA-A∗2402/LMP2131–139 tetramer and biotinylated protein L, respectively (GenScript) followed by streptavidin-APC (BD Biosciences).
Generation of DRrejTs targeting CD19 and LMP2 antigens
T-iPSCs established from LMP2-CTL clone (TYGPVFMSL) were differentiated into LMP2-rejTs as described. LMP2 specificity was confirmed by staining with HLA-A∗2402/LMP2419–427 tetramer. Next, LMP2-rejTs were transduced with MSCV-CD19-4-1BBz-IRES-GFP and GFP+ cells were sorted by flow cytometry. As a single antigen-targeted control, HPV16 E6-specific rejTs21 were transduced with MSCV-CD19-4-1BBz-IRES-GFP; GFP+ cells among CD19-CAR/HPV-rejTs were also sorted by flow cytometry. CD19-CAR transgene expression of DRrejTs and CD19-CAR/HPV-rejTs was confirmed by GFP positivity on flow cytometric analysis.
Flow cytometric analyses
Samples were processed using FACSAria II, LSRFortessa, and FACSCalibur cytometers (BD Biosciences). The results were analyzed using FlowJo software 10.5.3 (Tree Star, Ashland, OR, USA). After cells were incubated with the appropriate concentration of fluorescence-conjugated monoclonal antibody cocktail for 30 min at 4°C, they were washed with phosphate-buffered saline. Propidium iodide was added to exclude dead cells. Chromophore-conjugated monoclonal antibodies directed against surface antigens were used; they included APC mouse anti-human CD3 antibody, APC mouse anti-human CD4 antibody, APC mouse anti-human CD19 antibody, APC mouse anti-human CD279 (PD-1), APC/cyanine7 mouse anti-human CD3 antibody, fluorescein isothiocyanate (FITC) mouse anti-human CD19 antibody, phycoerythrin (PE) mouse anti-human CD4 antibody, PE mouse anti-human CD56 antibody, PE mouse anti-human CD62L antibody, Pacific Blue mouse anti-human CD8, Pacific Blue mouse anti-human CD45RA antibody (all BioLegend), and FITC mouse anti-human CD3 antibody (BD Biosciences). To detect CAR expression, CAR-transduced cells were stained with biotinylated recombinant protein L (GenScript), followed by APC-conjugated streptavidin (BD Biosciences). PE-conjugated HLA-A∗2402/LMP2131–139 tetramer, A∗2402/LMP2419–427 tetramer, or A∗2402/HPV16 E6419–427 tetramer (MBL) was used to detect antigen specific TCRs.
Coculture experiments
For serial killing experiments with PBMC-derived LMP1-CARTs, expanded LMP1-CARTs and non-transduced activated T cells (used as effector control cells) were cocultured with LCLs at the E:T ratio of 1:1 (1 × 105/1 × 105 cells) in NS-A2 with IL-7 and IL-15 (10 ng/mL each). Cultures were collected at days 0, 5, and 7 of coculture and stained with anti-CD3 and anti-CD19 antibodies. Percentages of CD3+CD19− T cells and CD19+CD3− tumor cells were evaluated by flow cytometry. For serial killing experiments with iPSC-derived DRrejTs, DRrejTs and control rejTs not specific to EBV antigen also were cocultured with HLA-A∗2402+ GFP/FFluc ENKL or HLA-A∗2402− (mismatched) LCL at the E:T ratio of 5:1 (5 × 105/1 × 105 cells) in NS-A2 with IL-7 and IL-15 (10 ng/mL each). Although CD3 is useful when separating tumor cells from rejTs, to separate these populations using CD3 and GFP/CD19 expression in coculture assays gave clearer results. Cultures were collected at days 0, 5, and 7 of coculture and stained with anti-CD3 (GFP/FFluc NK-YS) or anti-CD3 and anti-CD19 antibodies (LCLs). Percentages of CD3+ T cells and CD3− tumor cells were evaluated by flow cytometric analysis. To count absolute cell numbers by flow cytometry, CountBright absolute counting beads (Thermo Fisher Scientific) were added (20 μL).50 Absolute numbers of tumor cells were calculated according to the manufacturer’s protocol.
51Cr release assay
Cytotoxic specificities of DRrejTs, LMP1-CARTs, LMP1-CAR/HPV-rejTs, and LMP2-rejTs were analyzed in a standard 51Cr release assay at different E:T ratios.51 Target cells were labeled with 51Cr and cocultured with T cells for 20 h. After incubation, 50 μL of supernatant was transferred from each well to LumaPlate-96 wells (PerkinElmer, Waltham, MA, USA) and dried overnight. Released Cr was quantitated using a TopCount microplate scintillation counter (PerkinElmer), with % specific lysis = ([experimental Cr release] – [spontaneous Cr release]/[maximal Cr release] – [spontaneous Cr release]) × 100.
In vivo experiments
All in vivo studies were approved by the Animal Research Committees of The Institute of Medical Science, The University of Tokyo, and of Juntendo University School of Medicine.
NK-YS and EBV-infected LCLs transduced with γ-retroviral vector encoding GFP/FFluc were sorted for GFP expression by flow cytometry. Six-week-old female NOG mice (In-Vivo Science, Tokyo, Japan) were intraperitoneally engrafted with GFP/FFluc NK-YS cells (1 × 105 cells). Mice were divided into three groups, with two treatment groups and one no treatment group. Four days after tumor inoculation, mice in treatment groups were intraperitoneally injected with DRrejTs or peripheral blood-derived LMP1-CARTs to compare the anti-tumor effects of these therapeutic T cells (Figure 2B) and with DRrejTs or LMP2-rejTs to compare the anti-tumor effects of these therapeutic rejTs (Figure 3A) (5 × 106 cells, once a week, three doses) followed by intraperitoneal injection of recombinant IL-2 (1,000 U/mouse) once a week.
Six-week-old female NOG mice (In-Vivo Science) were intraperitoneally engrafted with GFP/FFluc LCLs (0.8 × 105 cells). Mice were divided into four groups, with three treatment groups and one no treatment group. Three days after tumor inoculation, mice in the three treatment groups were intraperitoneally injected with LMP2-rejTs, CD19-CAR-transduced HPV-rejTs, or DRrejTs to compare the anti-tumor effects of these therapeutic T cells (Figure 5A) (4 × 106 cells, once a week, three doses) followed by intraperitoneal injection of recombinant IL-2 (1,000 U/mouse) five times a week. Tumor-bearing mice were randomized to the different treatment groups. No blinding method was used. We repeated in vivo experiments twice to confirm the reproducibility of the results. Tumor burden was monitored by the Xenogen IVIS imaging system (Xenogen, Alameda, CA, USA) and Caliper IVIS imaging system (Caliper Life Sciences, Mountain View, CA, USA). Firefly d-luciferin substrate (OZ Biosciences, Marseille, France) was intraperitoneally injected into mice 15 min before imaging. Living Image software version 2.50 (Xenogen) and Living Image software version 4.7.2 (PerkinElmer) were used for luminescence analyses. The intensity of signal was measured as total photons (p)/s/cm2/sr as described.17,20
Quantitative real-time PCR
Genomic DNA was isolated from samples of mouse whole blood and cultured CD19-CARTs using a QIAamp DNA blood mini kit (QIAGEN, Hilden, Germany). CD19-CAR vector copy number was quantified using the StepOnePlus real-time PCR system (Applied Biosystems, Carlsbad, CA, USA). Primer and probes for the CD19-CAR transgene and GAPDH were custom ordered (Applied Biosystems) as described.52 Individual PCR reactions were normalized against GAPDH levels. Copies of transgene/microgram of DNA were calculated according to the formula: copies calculated from CD19 standard curve/input DNA (ng) × correction factor (ng detected/ng input) × 1,000 ng, as described.53
Measurement of apoptosis
On the day after 2 × 105 DRrejTs or LMP2-rejTs were plated into 24-well plates, 80 nmol/L CID/AP20187 (Clontech, Mountain View, CA, USA) was added to the wells. The cells were stained 24 h later with annexin V and 7-AAD for 15 min according to the manufacturer’s instructions (BD Biosciences). Annexin V/7-AAD+ cells were quantitated by FACSAria II flow cytometry using FlowJo software. To count absolute numbers of apoptotic cells by flow cytometry, CountBright absolute counting beads were added. Acquisition was halted at 2,000 beads.50
Statistical analysis
All data are presented as mean ± SD or SEM as stated in the figure legends. Results were analyzed by ANOVA or an unpaired Student’s t test as stated in the text. Comparison of survival curves was done using Kaplan-Meier analysis with log-rank testing. All statistical analyses were performed using Excel (Microsoft, Redmond, WA, USA) and Prism (GraphPad, San Diego, CA, USA) programs. Values of p < 0.05 were considered significant.
Acknowledgments
We thank A.S. Knisely for critical reading of the manuscript; Risa Matsuoka for technical help with cell culture; Nozomi Asano for technical help in making the LMP1-CAR vector; and Azusa Fujita, Yumiko Ishii, and Tamami Sakanishi for FACS operation. Malcolm K. Brenner provided retroviral iC9-GD2-CAR plasmid. Gianpietro Dotti and Nobuhiro Nishio provided retroviral FFluc-GFP plasmid. We also thank Motoo Watanabe and Hajime Yasuda for helpful discussions. We thank the members of the Laboratory of Radioisotope Research, Research Support Center, Juntendo University Graduate School of Medicine for technical assistance. This work was carried out in part at the Intractable Disease Research Center, Juntendo University Graduate School of Medicine. The Institutional Regulation Boards for Human Ethics at Juntendo University School of Medicine and at the Institute of Medical Science, University of Tokyo, approved the experimental protocol. This work was supported by JSPS KAKENHI (grants 16K09842 and 19K07781) and a Grant for Cross-Disciplinary Collaboration, Juntendo University (30-52).
Author contributions
M.A. planned and directed the study and wrote the manuscript. S.H. performed the experiments and wrote the manuscript. J.A. performed 51Cr assays and provided scientific discussions. M.I., S.Y., and K.O. helped in animal experiments. T.T. performed 51Cr assays and helped in animal experiments. C.I. provided retroviral CD19-CAR plasmid. T.Y., Y.N., C.S., and C.I. helped in making CAR constructs. M.O. and M.N. provided SeV vectors. Y.O., K.N., and K.O. helped in immunohistochemical staining. H.N. wrote the manuscript and provided scientific discussions. N.K. provided scientific discussions.
Declaration of interests
H.N. is a co-founder of and an advisor to Century Therapeutics. The remaining authors declare no competing interests.
Contributor Information
Miki Ando, Email: m-ando@juntendo.ac.jp.
Hiromitsu Nakauchi, Email: nakauchi@stanford.edu.
References
- 1.June C.H., Sadelain M. Chimeric antigen receptor therapy. N. Engl. J. Med. 2018;379:64–73. doi: 10.1056/NEJMra1706169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lim W.A., June C.H. The principles of engineering immune cells to treat cancer. Cell. 2017;168:724–740. doi: 10.1016/j.cell.2017.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rafiq S., Hackett C.S., Brentjens R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020;17:147–167. doi: 10.1038/s41571-019-0297-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jackson H.J., Rafiq S., Brentjens R.J. Driving CAR T-cells forward. Nat. Rev. Clin. Oncol. 2016;13:370–383. doi: 10.1038/nrclinonc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brudno J.N., Kochenderfer J.N. Chimeric antigen receptor T-cell therapies for lymphoma. Nat. Rev. Clin. Oncol. 2018;15:31–46. doi: 10.1038/nrclinonc.2017.128. [DOI] [PubMed] [Google Scholar]
- 6.Grupp S.A., Kalos M., Barrett D., Aplenc R., Porter D.L., Rheingold S.R., Teachey D.T., Chew A., Hauck B., Wright J.F., et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee D.W., Kochenderfer J.N., Stetler-Stevenson M., Cui Y.K., Delbrook C., Feldman S.A., Fry T.J., Orentas R., Sabatino M., Shah N.N., et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet. 2015;385:517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park J.H., Rivière I., Gonen M., Wang X., Sénéchal B., Curran K.J., Sauter C., Wang Y., Santomasso B., Mead E., et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N. Engl. J. Med. 2018;378:449–459. doi: 10.1056/NEJMoa1709919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turtle C.J., Hanafi L.A., Berger C., Gooley T.A., Cherian S., Hudecek M., Sommermeyer D., Melville K., Pender B., Budiarto T.M., et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Invest. 2016;126:2123–2138. doi: 10.1172/JCI85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shah N.N., Fry T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019;16:372–385. doi: 10.1038/s41571-019-0184-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Orlando E.J., Han X., Tribouley C., Wood P.A., Leary R.J., Riester M., Levine J.E., Qayed M., Grupp S.A., Boyer M., et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat. Med. 2018;24:1504–1506. doi: 10.1038/s41591-018-0146-z. [DOI] [PubMed] [Google Scholar]
- 12.Majzner R.G., Mackall C.L. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 2018;8:1219–1226. doi: 10.1158/2159-8290.CD-18-0442. [DOI] [PubMed] [Google Scholar]
- 13.Wherry E.J. T cell exhaustion. Nat. Immunol. 2011;12:492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 14.Maude S.L., Laetsch T.W., Buechner J., Rives S., Boyer M., Bittencourt H., Bader P., Verneris M.R., Stefanski H.E., Myers G.D., et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 2018;378:439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schuster S.J., Svoboda J., Chong E.A., Nasta S.D., Mato A.R., Anak Ö., Brogdon J.L., Pruteanu-Malinici I., Bhoj V., Landsburg D., et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N. Engl. J. Med. 2017;377:2545–2554. doi: 10.1056/NEJMoa1708566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nishimura T., Kaneko S., Kawana-Tachikawa A., Tajima Y., Goto H., Zhu D., Nakayama-Hosoya K., Iriguchi S., Uemura Y., Shimizu T., et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell. 2013;12:114–126. doi: 10.1016/j.stem.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 17.Ando M., Nishimura T., Yamazaki S., Yamaguchi T., Kawana-Tachikawa A., Hayama T., Nakauchi Y., Ando J., Ota Y., Takahashi S., et al. A safeguard system for induced pluripotent stem cell-derived rejuvenated T cell therapy. Stem Cell Reports. 2015;5:597–608. doi: 10.1016/j.stemcr.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ando M., Nakauchi H. “Off-the-shelf” immunotherapy with iPSC-derived rejuvenated cytotoxic T lymphocytes. Exp. Hematol. 2017;47:2–12. doi: 10.1016/j.exphem.2016.10.009. [DOI] [PubMed] [Google Scholar]
- 19.Vizcardo R., Masuda K., Yamada D., Ikawa T., Shimizu K., Fujii S., Koseki H., Kawamoto H. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8+ T cells. Cell Stem Cell. 2013;12:31–36. doi: 10.1016/j.stem.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 20.Ando M., Ando J., Yamazaki S., Ishii M., Sakiyama Y., Harada S., Honda T., Yamaguchi T., Nojima M., Ohshima K., et al. Long-term eradication of extranodal natural killer/T-cell lymphoma, nasal type, by induced pluripotent stem cell-derived Epstein-Barr virus-specific rejuvenated T cells in vivo. Haematologica. 2020;105:796–807. doi: 10.3324/haematol.2019.223511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Honda T., Ando M., Ando J., Ishii M., Sakiyama Y., Ohara K., Toyota T., Ohtaka M., Masuda A., Terao Y., et al. Sustainable tumor-suppressive effect of iPSC-derived rejuvenated T cells targeting cervical cancers. Mol. Ther. 2020;28:2394–2405. doi: 10.1016/j.ymthe.2020.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Themeli M., Rivière I., Sadelain M. New cell sources for T cell engineering and adoptive immunotherapy. Cell Stem Cell. 2015;16:357–366. doi: 10.1016/j.stem.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heczey A., Louis C.U., Savoldo B., Dakhova O., Durett A., Grilley B., Liu H., Wu M.F., Mei Z., Gee A., et al. CAR T cells administered in combination with lymphodepletion and PD-1 inhibition to patients with neuroblastoma. Mol. Ther. 2017;25:2214–2224. doi: 10.1016/j.ymthe.2017.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Straathof K.C., Pulè M.A., Yotnda P., Dotti G., Vanin E.F., Brenner M.K., Heslop H.E., Spencer D.M., Rooney C.M. An inducible caspase 9 safety switch for T-cell therapy. Blood. 2005;105:4247–4254. doi: 10.1182/blood-2004-11-4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Di Stasi A., Tey S.K., Dotti G., Fujita Y., Kennedy-Nasser A., Martinez C., Straathof K., Liu E., Durett A.G., Grilley B., et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011;365:1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zheng Z., Chinnasamy N., Morgan R.A. Protein L: A novel reagent for the detection of chimeric antigen receptor (CAR) expression by flow cytometry. J. Transl. Med. 2012;10:29. doi: 10.1186/1479-5876-10-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heslop H.E. Biology and treatment of Epstein-Barr virus-associated non-Hodgkin lymphomas. Hematology Am. Soc. Hematol. Educ. Program. 2005;2005:260–266. doi: 10.1182/asheducation-2005.1.260. [DOI] [PubMed] [Google Scholar]
- 28.Ando M., Hoyos V., Yagyu S., Tao W., Ramos C.A., Dotti G., Brenner M.K., Bouchier-Hayes L. Bortezomib sensitizes non-small cell lung cancer to mesenchymal stromal cell-delivered inducible caspase-9-mediated cytotoxicity. Cancer Gene Ther. 2014;21:472–482. doi: 10.1038/cgt.2014.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Itakura G., Kawabata S., Ando M., Nishiyama Y., Sugai K., Ozaki M., Iida T., Ookubo T., Kojima K., Kashiwagi R., et al. Fail-safe system against potential tumorigenicity after transplantation of iPSC derivatives. Stem Cell Reports. 2017;8:673–684. doi: 10.1016/j.stemcr.2017.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoyos V., Del Bufalo F., Yagyu S., Ando M., Dotti G., Suzuki M., Bouchier-Hayes L., Alemany R., Brenner M.K. Mesenchymal stromal cells for linked delivery of oncolytic and apoptotic adenoviruses to non-small-cell lung cancers. Mol. Ther. 2015;23:1497–1506. doi: 10.1038/mt.2015.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiang A.K., Tao Q., Srivastava G., Ho F.C. Nasal NK- and T-cell lymphomas share the same type of Epstein-Barr virus latency as nasopharyngeal carcinoma and Hodgkin’s disease. Int. J. Cancer. 1996;68:285–290. doi: 10.1002/(SICI)1097-0215(19961104)68:3<285::AID-IJC3>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 32.Fox C.P., Haigh T.A., Taylor G.S., Long H.M., Lee S.P., Shannon-Lowe C., O’Connor S., Bollard C.M., Iqbal J., Chan W.C., et al. A novel latent membrane 2 transcript expressed in Epstein-Barr virus-positive NK- and T-cell lymphoproliferative disease encodes a target for cellular immunotherapy. Blood. 2010;116:3695–3704. doi: 10.1182/blood-2010-06-292268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pulè M.A., Savoldo B., Myers G.D., Rossig C., Russell H.V., Dotti G., Huls M.H., Liu E., Gee A.P., Mei Z., et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 2008;14:1264–1270. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hegde M., Mukherjee M., Grada Z., Pignata A., Landi D., Navai S.A., Wakefield A., Fousek K., Bielamowicz K., Chow K.K., et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J. Clin. Invest. 2016;126:3036–3052. doi: 10.1172/JCI83416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fry T.J., Shah N.N., Orentas R.J., Stetler-Stevenson M., Yuan C.M., Ramakrishna S., Wolters P., Martin S., Delbrook C., Yates B., et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018;24:20–28. doi: 10.1038/nm.4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsuchiyama J., Yoshino T., Mori M., Kondoh E., Oka T., Akagi T., Hiraki A., Nakayama H., Shibuya A., Ma Y., et al. Characterization of a novel human natural killer-cell line (NK-YS) established from natural killer cell lymphoma/leukemia associated with Epstein-Barr virus infection. Blood. 1998;92:1374–1383. [PubMed] [Google Scholar]
- 37.Ando M., Sugimoto K., Kitoh T., Sasaki M., Mukai K., Ando J., Egashira M., Schuster S.M., Oshimi K. Selective apoptosis of natural killer-cell tumours by l-asparaginase. Br. J. Haematol. 2005;130:860–868. doi: 10.1111/j.1365-2141.2005.05694.x. [DOI] [PubMed] [Google Scholar]
- 38.Nagata H., Konno A., Kimura N., Zhang Y., Kimura M., Demachi A., Sekine T., Yamamoto K., Shimizu N. Characterization of novel natural killer (NK)-cell and γδ T-cell lines established from primary lesions of nasal T/NK-cell lymphomas associated with the Epstein-Barr virus. Blood. 2001;97:708–713. doi: 10.1182/blood.v97.3.708. [DOI] [PubMed] [Google Scholar]
- 39.Anderson M.A., Gusella J.F. Use of cyclosporin A in establishing Epstein-Barr virus-transformed human lymphoblastoid cell lines. In Vitro. 1984;20:856–858. doi: 10.1007/BF02619631. [DOI] [PubMed] [Google Scholar]
- 40.Miller G., Lipman M. Comparison of the yield of infectious virus from clones of human and simian lymphoblastoid lines transformed by Epstein-Barr virus. J. Exp. Med. 1973;138:1398–1412. doi: 10.1084/jem.138.6.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith C.A., Ng C.Y., Heslop H.E., Holladay M.S., Richardson S., Turner E.V., Loftin S.K., Li C., Brenner M.K., Rooney C.M. Production of genetically modified Epstein-Barr virus-specific cytotoxic T cells for adoptive transfer to patients at high risk of EBV-associated lymphoproliferative disease. J. Hematother. 1995;4:73–79. doi: 10.1089/scd.1.1995.4.73. [DOI] [PubMed] [Google Scholar]
- 42.Ando J., Ngo M.C., Ando M., Leen A., Rooney C.M. Identification of protective T-cell antigens for smallpox vaccines. Cytotherapy. 2020;22:642–652. doi: 10.1016/j.jcyt.2020.04.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oyama T., Ichimura K., Suzuki R., Suzumiya J., Ohshima K., Yatabe Y., Yokoi T., Kojima M., Kamiya Y., Taji H., et al. Senile EBV+ B-cell lymphoproliferative disorders: A clinicopathologic study of 22 patients. Am. J. Surg. Pathol. 2003;27:16–26. doi: 10.1097/00000478-200301000-00003. [DOI] [PubMed] [Google Scholar]
- 44.Ohshima K., Suzumiya J., Kanda M., Haraoka S., Kawasaki C., Shimazaki K., Kikuchi M. Genotypic and phenotypic alterations in Epstein-Barr virus-associated lymphoma. Histopathology. 1999;35:539–550. doi: 10.1046/j.1365-2559.1999.00784.x. [DOI] [PubMed] [Google Scholar]
- 45.Chen R., Zhang D., Mao Y., Zhu J., Ming H., Wen J., Ma J., Cao Q., Lin H., Tang Q., et al. A human Fab-based immunoconjugate specific for the LMP1 extracellular domain inhibits nasopharyngeal carcinoma growth in vitro and in vivo. Mol. Cancer Ther. 2012;11:594–603. doi: 10.1158/1535-7163.MCT-11-0725. [DOI] [PubMed] [Google Scholar]
- 46.Yamaguchi T., Hamanaka S., Kamiya A., Okabe M., Kawarai M., Wakiyama Y., Umino A., Hayama T., Sato H., Lee Y.S., et al. Development of an all-in-one inducible lentiviral vector for gene specific analysis of reprogramming. PLoS ONE. 2012;7:e41007. doi: 10.1371/journal.pone.0041007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Quintarelli C., Vera J.F., Savoldo B., Giordano Attianese G.M., Pulè M., Foster A.E., Heslop H.E., Rooney C.M., Brenner M.K., Dotti G. Co-expression of cytokine and suicide genes to enhance the activity and safety of tumor-specific cytotoxic T lymphocytes. Blood. 2007;110:2793–2802. doi: 10.1182/blood-2007-02-072843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gerdemann U., Keirnan J.M., Katari U.L., Yanagisawa R., Christin A.S., Huye L.E., Perna S.K., Ennamuri S., Gottschalk S., Brenner M.K., et al. Rapidly generated multivirus-specific cytotoxic T lymphocytes for the prophylaxis and treatment of viral infections. Mol. Ther. 2012;20:1622–1632. doi: 10.1038/mt.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rooney C.M., Leen A.M., Vera J.F., Heslop H.E. T lymphocytes targeting native receptors. Immunol. Rev. 2014;257:39–55. doi: 10.1111/imr.12133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Torres Chavez A., McKenna M.K., Canestrari E., Dann C.T., Ramos C.A., Lulla P., Leen A.M., Vera J.F., Watanabe N. Expanding CAR T cells in human platelet lysate renders T cells with in vivo longevity. J. Immunother. Cancer. 2019;7:330. doi: 10.1186/s40425-019-0804-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rooney C.M., Smith C.A., Ng C.Y., Loftin S.K., Sixbey J.W., Gan Y., Srivastava D.K., Bowman L.C., Krance R.A., Brenner M.K., Heslop H.E. Infusion of cytotoxic T cells for the prevention and treatment of Epstein-Barr virus-induced lymphoma in allogeneic transplant recipients. Blood. 1998;92:1549–1555. [PubMed] [Google Scholar]
- 52.Milone M.C., Fish J.D., Carpenito C., Carroll R.G., Binder G.K., Teachey D., Samanta M., Lakhal M., Gloss B., Danet-Desnoyers G., et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther. 2009;17:1453–1464. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kalos M., Levine B.L., Porter D.L., Katz S., Grupp S.A., Bagg A., June C.H. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]