

Abstract
Hepatocellular carcinoma (HCC) is frequently characterized by metabolic and immune remodeling in the tumor microenvironment. We previously discovered that liver-specific deletion of fructose-1, 6-bisphosphatase 1 (FBP1), a gluconeogenic enzyme ubiquitously suppressed in HCC tissues, promotes liver tumorigenesis and induces metabolic and immune perturbations closely resembling human HCC. However, the underlying mechanisms remain incompletely understood. Here, we reported that FBP1-deficient livers exhibit diminished amounts of natural killer (NK) cells and accelerated tumorigenesis. Using the diethylnitrosamine-induced HCC mouse model, we analyzed potential changes in the immune cell populations purified from control and FBP1-depleted livers and found that NK cells were strongly suppressed. Mechanistically, FBP1 attenuation in hepatocytes derepresses an zeste homolog 2 (EZH2)-dependent transcriptional program to inhibit PKLR expression. This leads to reduced levels of PKLR cargo proteins sorted into hepatocyte-derived extracellular vesicles (EVs), dampened activity of EV-targeted NK cells, and accelerated liver tumorigenesis. Our study demonstrated that hepatic FBP1 depletion promotes HCC-associated immune remodeling, partly through the transfer of hepatocyte-secreted, PKLR-attenuated EVs to NK cells.
Keywords: hepatocellular carcinoma, FBP1, natural killer cells, extracellular vehicles, PKLR
Graphical abstract
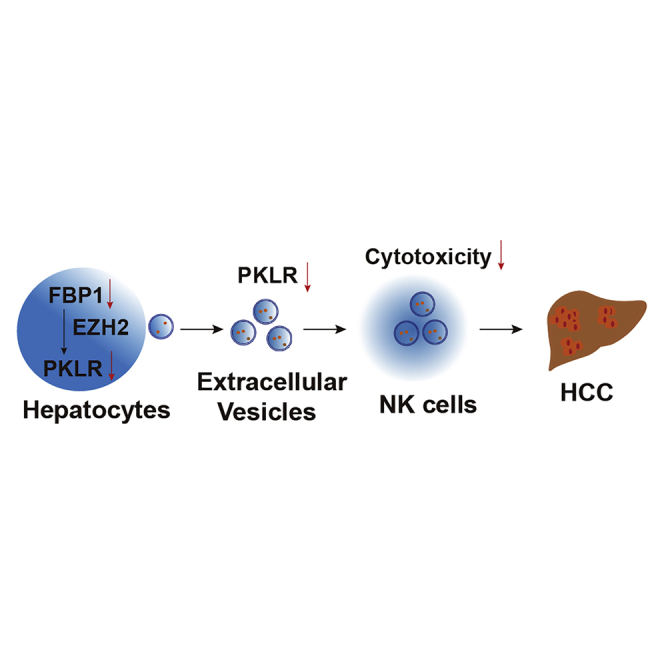
Li and colleagues observed that natural killer cells, as specialized anti-tumor immune cells, are attenuated upon tumorigenesis in the liver. This is accompanied by specific changes in the protein components of liver cell-secreted extracellular vehicles, which are intercellular communication vehicles targeting natural killer cells and regulating tumorigenesis.
Introduction
Liver cancer is among the leading malignancies worldwide and ranks third in cancer-related death in Asian countries.1,2 Hepatocellular carcinoma (HCC) is the predominant subtype of liver cancer, frequently developed from liver regions with metabolic steatosis and severe fibrosis.3 Various infiltrated immune cells form a unique and complex immune microenvironment in the neoplastic region and are generally associated with the initiation and progression of HCC pathology.4,5 For example, several studies exhibited that increased infiltrations of natural killer (NK), T, and NK T cells are positive prognostic factors in HCC, whereas the enhanced infiltration of regulatory T cells is a negative factor.6 Nonetheless, the underlying mechanisms orchestrating the immune landscape of HCC remain largely unknown.
Recently, we discovered that fructose-1, 6-bisphosphatase 1 (FBP1), a gluconeogenic enzyme harboring non-canonical functions in the cell nucleus, is uniformly suppressed in HCC tumors relative to normal adjacent tissues.7 We further demonstrated that hepatic Fbp1 deletion closely resembled the hepatic abnormalities predisposing HCC, with increased glycolysis, lipid accumulation, and enhanced fibrosis in mouse livers. Hepatic Fbp1 loss strongly promotes HCC initiation and progression, largely by inducing inflammatory responses in the peritumoral region and senescence in hepatic satellite cells, in a diethylnitrosamine (DEN)-induced HCC mouse model.8 These data implied an underappreciated link between metabolic and immune remodeling that frequently co-occurred in HCC and provided a perspective for understanding the disease biology of this common malignancy. However, little is known about the exact role of FBP1 in causing the protumorigenic immune perturbations in HCC.
NK and CD8 T cells play pivotal roles in antitumor immunity.9, 10, 11 It has been well established that NK cells perform tumor surveillance and function as cytotoxic lymphocytes in opposing multiple tumor types, including lung cancer,9 prostate cancer,12 breast cancer,13 liver cancer,7,8 etc. In the current study, we identified liver-infiltrating NK cells as the most diminished immune population in FBP1-depleted livers upon tumor initiation, suggesting that hepatic FBP1 attenuation may result in the suppression of NK-mediated tumor surveillance. We further discovered that extracellular vesicles (EVs) secreted from FBP1-depleted hepatocytes target infiltrated NK cells and induce NK dysfunction, thereby promoting tumor initiation and progression in HCC.
Results
Hepatocyte-specific FBP1 loss restrains NK cells in DEN-induced liver tumorigenesis
To decipher the role of FBP1 in modulating the immune microenvironment in HCC, we exploited a well-established HCC mouse model using DEN as the carcinogen, with hepatic FBP1 conditionally depleted as previously reported (Figure 1A). The complete abolishment of FBP1 expression was confirmed in hepatic tissues of Cre mice (Figures 1B and 1C). As expected, hepatic FBP1 loss significantly advanced tumorigenesis, demonstrated by increased numbers of surface tumors (Figures 1D and 1E), more microscopic lesions with steatosis (Figures 1F and 1G), and enhanced cell proliferation rates (Figure 1H) as early as 24 weeks in the DEN-treated Cre cohort compared to the control GFP cohort. We then examined the population changes of infiltrating leukocytes in harvested tumor tissues (Figure S1). Interestingly, we found that the NK cell population declined significantly in the FBP1-deficient livers relative to the control, whereas other leukocyte cell populations exhibited negligible variations between these two groups (Figure 1I). We further discovered that the NK cell effector function was similarly suppressed in the tumor microenvironment, as validated by IHC staining of the NK activation marker NKp46 in tumor tissues (Figure 1J). In addition, the difference in NK cell population with or without hepatic FBP1 depletion at week 36 is less than the difference at week 24 (Figure S2). These results are consistent with the notion that NK cells compose the early immune surveillance for tumorigenesis and are attenuated during later stages of tumor development.14 In conclusion, these data suggest that inhibition of NK cells may cause immune remodeling in the tumor microenvironment mediating the hepatic tumorigenesis fostered by FBP1 depletion.
Figure 1.

Hepatocyte-specific FBP1 loss restrains NK cells in DEN-induced liver tumorigenesis
(A) Flow chart of generating DEN-induced liver cancer mouse model with or without hepatic Fbp1 deletion. (B and C) The mRNA (B) and protein (C) expression levels of FBP1 in indicated mouse livers. (D−H) Representative liver images (scale bar, 1 cm) (D), the number of liver surface tumor nodules (E), representative H&E staining (scale bar, 100 μm) (F), numbers of microscopic tumors (G), and Ki67+ cells per field (H) of liver tissues from DEN-treated GFP and Cre mice at 24 weeks; n = 5. (I) Flow cytometry analysis of activated NK cells (CD3−NKP46+), B cells (B1a cells: CD19+ IgM+CD43+CD5+, B1b cells: CD19+ IgM+CD43+CD5−, B2 cells: CD19+t IgM+tCD43−CD5−, Regulatory B (Breg) cells: CD19+tCD5+tCD1d+t), T cells (CD4 cells: CD3+tCD4+t, CD8 cells: CD3+tCD8+t, γδT cell receptor (TCR) cells: CD3+tγδTCR+t), and macrophages (CD11b cells: F4/80+tCD11b+t, CD206 cells: F4/80+tCD206+t) in aforementioned liver tissues, n = 3. (J) Representative NKp46 immunohistochemical staining images (let) and quantifications (right) in 24-week DEN/GFP and DEN/Cre livers. n = 3. Scale bar, 100 μm. Data are presented as mean ± SD. Unpaired Student’s t test, ∗∗∗p < 0.001 and ∗∗∗∗p < 0.0001. See also Figures S1 and S2.
EVs secreted by FBP1-deficient hepatocytes impair NK cell function
To explore the mechanisms of NK cell inhibition upon hepatic FBP1 depletion, we first investigated cell-to-cell communication events between NK cells and hepatocytes. Different cells communicate with each other using various mediators, such as cell-cell contact, growth factors, cytokines, EVs, etc. EVs represent an important type of communication carriers in the tumor microenvironment, transferring various biomolecules between tumor and immune cells.15,16 To study this, we employed an in vitro co-culture system. Briefly, EVs released from hepatocytes were separated from cell culture supernatants containing cytokines and growth factors, which were respectively subjected to co-incubation with NK cells before examining the NK effector activities (Figure 2A). We found that silencing FBP1 expression with short hairpin (sh)RNAs did not affect EV production (Figure 2B). However, the effector function of NK cells was strongly restrained after co-incubation with EVs released by FBP1-deficient hepatocytes, based on attenuated expression of CD107a, granzyme B, and perforin (Figure 2C). In contrast, EV-free supernatants did not influence the NK effector function regardless of FBP1 status (Figure 2D). Similar results were obtained by CRISPR-mediated FBP1 knockout (KO) using 2 distinct single guide RNAs (sgRNAs) (Figures 2E and 2F). Moreover, when NK cells and hepatocytes were separated by Transwell membranes, NK cells consistently exhibited attenuated effector activities (Figures S3A−S3C), which suggests that cell-cell contact is not the key communication mediator for hepatocytes and NK cells. However, the repression of NK cell function was abolished when the co-cultured hepatocytes were pre-treated with GW4869 to block the EV release (Figure S3D). Moreover, the anti-tumor cytotoxicity of NK cells was also reduced post-co-incubation with EVs derived from FBP1-deficient hepatocytes, as demonstrated by flow cytometry analysis of K562 target cell viabilities (Figure 2G). To make the results more context relevant, we also tested Hep3B, a liver cancer cell line, as NK-targeting cells to compare the cytotoxicity of NK cells treated with EVs secreted from HHL5 cells with or without FBP1 (Figures S3E and S3H). Hep3B as NK-targeting cells exhibited similar results as K562, although overall, less cytotoxicity was observed. Interestingly, peripheral blood mononuclear cells (PBMCs) harbor comparable cytotoxic effects as purified NK cells, which can be similarly regulated by EVs derived from hepatocytes with or without FBP1 (Figures S3F and S3H). More importantly, such cytotoxic effects were blunted by NK cell depletion using a NK-specific antibody (Figure S3G). Taken together, these data suggested that EVs secreted by FBP1-deficient hepatocytes specifically impair the cytotoxic function of NK cells.
Figure 2.

EVs secreted by FBP1-deficient hepatocytes impair NK cell function
(A) Flowcharts illustrating the experimental design to examine the communication events between hepatocyte-derived EVs and NK cells. (B) Western blots confirming the knockdown efficiencies of FBP1 shRNAs. (C) Representative histograms (left) and quantifications (right) of indicated markers in NK cells incubated with equal amounts of EVs secreted from HHL5 cells with control PLKO.1 or FBP1 shRNAs. The quantification graphs display average relative frequencies of perforin+, CD107a+, or granzyme B+ NK cells. (D) Representative histograms (left) and quantifications (right) of indicated markers in NK cells incubated with equal amounts of vesicle-free supernatants from HHL5 cells with control PLKO.1 or FBP1 shRNAs. The quantification graphs display average relative frequencies of perforin+, CD107a+, or granzyme B+ NK cells. (E) Representative histograms (left) and quantifications (right) of indicated markers in NK cells incubated with equal amounts of EVs secreted from HHL5 cells with or without FBP1-targeting sgRNAs. (F) Western blots of TSG101 and CD63 expression in EVs secreted from FBP1-depleted HHL5 cells. (G) Cytotoxicity assay of primary NK cells incubated with EVs derived from hepatocytes with or without FBP1 against K562 target cells. The representative flow cytometry (FCM) profiles and quantifications of carboxyfluorescein succinimidyl amino ester+ 7-Aminoactinomycin D+ (CFSE+7AAD+) K562 cells were shown. Data are the mean ± SD. ns, no significant difference; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001 by unpaired Student’s t test. See also Figure S3.
NK cells act as major immune cell recipients of hepatocyte-derived EVs
We next investigated whether hepatocyte-derived EVs are capable of fusing with NK cells through internalization. Purified EVs under electron microscopy appeared as clusters of vesicles with 40−300 nm in diameter (Figure 3A), which peaked around 150 nm based on particle analysis (Figure 3B). To examine potential fusion events, EVs were stained with the Dir dye, purified, and co-incubated with NK cells (Figure 3C). We observed that NK cells were rapidly labeled by the Dir-stained EVs yet not by mock EV preps generated from the Dir-containing, EV-free medium (Figure 3D), indicating that the fluorescent labeling of NK cells is not due to Dir carryover but to the specific internalization of EVs. To examine whether EV activity is affected by the Dir dye, we performed flow analysis of recipient NK cell activity in the presence or absence of EV staining. As a result, there was no significant difference observed in NK cells treated with naked EVs or Dir-labeled EVs (Figure S4), suggesting that Dir staining did not affect EV activity in this context. More importantly, NK cells, but few of other immune cells in the liver, were primarily targeted by hepatocyte-derived EVs delivered through the tail vein (Figures 3E−3G), suggesting that NK cells may act as predominant immune cell recipients of hepatocyte-derived EVs, at least in normal hepatic tissues.
Figure 3.

NK cells act as major immune cell recipients of hepatocyte-derived EVs
(A) Representative image of HHL5 cell-derived EVs by transmission electron microscopy (TEM). Scale bar, 100 nm. (B) Size distribution of HHL5 cell-derived EVs using nanoparticle tracking analysis (NTA). (C) Experimental scheme illustrating EV uptake process by NK cells. (D) Flow cytometry analysis of NK cells incubated with Dir-labeled or -unlabeled, HHL5 cell-derived EVs. Dir-alone samples represent mock EV preps generated from Dir-labeled, EV-free medium. (E) Experimental scheme of EV uptake assays in various immune cell populations (NK, T, and B cells, for instance) in liver. (F and G) Representative histograms (F) and quantifications (G) of Dir fluorescence intensities in hepatic CD4 T cells, CD8 T cells, B cells, and NK cells using different concentrations of EVs. See also Figure S4.
PKLR delivered by hepatocyte-derived EVs regulates NK cell function
To elucidate the molecular mechanism by which hepatocyte-derived EVs modulate NK cell function, we performed the proteomic profiling of cargo proteins in EVs collected from hepatocytes. Interestingly, a group of central carbon metabolism-related proteins were specifically enriched in isolated EVs, especially those involved in canonical glycolysis (Figures 4A and 4B). It is noted that glucose metabolism produces most of the cellular energy to fuel immune cell growth and activation, which may help explain the functional difference between control and FBP1-depleted, EV-treated NK cells. Furthermore, we analyzed the correlation between these canonical glycolytic cargo proteins and FBP1 and found that PKLR exhibits the strongest correlation with FBP1 (Figures 4C and 4D). A previous report shows that increased expression of FBP1 in NK cells impairs NK cell function, at least in the mouse model of Kras-driven lung cancer.9 In contrast, we confirmed that EVs secreted by hepatocytes do not contain FBP1 (Figure 4E). Moreover, previous studies demonstrated that transforming growth factor β (TGF-β) is frequently upregulated in tumors and may affect tumor progression.17 To further examine the possibility of TGF-β inducing the changes of FBP1 levels in NK cells, we analyzed TGF-β expression in HCC tumor and normal tissues by immunohistochemistry (IHC) staining. We found that TGF-β expression is significantly increased in the late stage but not early stage of HCC relative to normal tissues (Figure S5A), implying that TGF-β expression may not play a role in promoting liver cancer initiation. This is consistent with previous findings that TGF-β confers protumorigenic functions only in advanced HCC.17 In addition, we also confirmed that EVs secreted by hepatocytes with or without FBP1 depletion did not affect the endogenous expression of FBP1 in NK cells (Figure S5B). Conversely, PKLR but not FBP1 was readily detected in the EVs derived from hepatocytes (Figure 4E), suggesting that PKLR, instead of FBP1, is delivered by EVs. Then we asked whether cellular FBP1 depletion may regulate PKLR protein levels in secreted EVs. Indeed, expression levels of PKLR and FBP1 are positively correlated in HCC tumor tissues, as analyzed by the Cancer Genome Atlas (TCGA) (Figure 4D), and PKLR protein levels are markedly reduced in whole-cell lysates and EVs derived from FBP1-deficient hepatocytes (Figure 4F).
Figure 4.

PKLR delivered by hepatocyte-derived EVs regulates NK cell function
(A) Pathway clustering analysis of identified proteins in hepatocyte-derived EVs based on the Dtabase for Annotation, Visualization and Integrated Discovery (DAVID) functional annotation. (B) Pathway clustering analysis of metabolism-related proteins identified in (A) based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) annotation. (C) Correlation analysis between canonical glycolytic proteins identified in (B) and FBP1 in normal liver tissues collected by TCGA and the Genotype-Tissue Expression (GTEx) project. (D) A scatterplot showing the positive correlation between PKLR and FBP1 levels in normal liver tissues collected by TCGA and the GTEx project. Transcripts per kilobase of exon model per million mapped reads (TPM) represents the normalized sequencing depth of RNA sequencing (RNA-seq). (E) Western blot analysis of whole-cell lysates (WCLs) and EVs, confirming that PKLR but not FBP1 protein is present in hepatocyte-derived EVs. (F) Western blot analysis showing FBP1 knockout (KO) in HHL5 cells results in decreased protein expression of PKLR in both WCLs and EVs. (G) Real-time qPCR analysis of PKLR mRNA levels in NK cells treated with EVs collected from hepatocytes, with or without FBP1, relative to no EV-treated control, using primers targeting multiple PKLR 3′ UTR or coding sites. (H) Representative histograms (upper) and quantifications (lower) of indicated makers in NK cells incubated with EVs secreted from HHL5 cells with PLKO.1 or PKLR shRNAs. Western blots confirming the efficiencies of PKLR knockdown were shown in (J). (I) Representative histograms (upper) and quantification (lower) of indicated makers in NK cells incubated with EVs secreted from HHL5 cells using FBP1 WT or FBP1 KO clones in the presence or absence of PKLR overexpression. Western blots confirming the efficiencies of FBP1 KO and PKLR re-expression were shown in (J). (J) Western blots confirming the efficiencies of PKLR knockdown as shown in (H) and the efficiencies of FBP1 KO and PKLR re-expression as shown in (I). (K) Analysis of glucose uptake and lactic acid production in NK cells incubated with EVs secreted from HHL5 cells with or without PKLR shRNAs or with EVs secreted from HHL5 cells using FBP1 WT or FBP1 KO clones in the presence or absence of PKLR overexpression. Data are the mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001 by unpaired Student’s t test. See also Figure S5.
Notably, PKLR mRNA levels remain unchanged in NK cells incubated with EVs derived from hepatocytes regardless of the FBP1 status, suggesting that FBP1 may signal through the delivered PKLR protein to influence NK cell function (Figure 4G). Consistently, the effector function of NK cells is suppressed by co-incubation with EVs released from PKLR-inhibited hepatocytes, as confirmed by the flow analysis of CD107a and perforin levels in examined NK cells (Figures 4H and 4J). Whereas FBP1 attenuation in hepatocytes results in secreted EVs that curb NK function, forced expression of PKLR in FBP1-deficient hepatocytes largely rescues such phenotype (Figures 4I and 4J). To elucidate how reduced PKLR in EVs inhibits NK cell function, we measured glycolytic changes in NK cells since PKLR catalyzes the conversion of phosphoenolpyruvate to pyruvate, a rate-limiting step of glycolysis.18, 19, 20 We observed that PKLR delivered by EVs upregulates the rate of glycolysis in NK cells, which could account for the inhibitory effect of PKLR-depleted EVs on NK activities. Conversely, forced PKLR expression in FBP1-deficient hepatocytes restores glycolysis in NK cells (Figure 4K). Together, these data demonstrated that PKLR, which is regulated by FBP1 in hepatocytes, is a key protein cargo delivered to NK cells via EVs, thereby regulating the glycolysis and function of target NK cells.
EZH2 suppresses PKLR expression upon FBP1 loss in hepatocytes
Next, we investigated how PKLR is exactly regulated in FBP1-deficient hepatocytes. We previously reported that a nuclear pool of FBP1 directly binds and inhibits the polycomb group protein EZH2, which is a methyltransferase inducing the trimethylation of histone H3 lysine 27 (H3K27me3) and inhibiting target gene transcription.21 To examine the possibility that FBP1 may regulate PKLR expression through EZH2, we analyzed the chromatin immunoprecipitation sequencing (ChIP-seq) data of EZH2 and H3K27me3 retrieved from the ENCODE project and found that both EZH2 and H3K27me3 signals are co-enriched at the beginning of the reading-frame region of the PKLR gene (Figure 5A). We validated this result in hepatocytes by performing ChIP-PCR to detect site-specific binding signals at the PKLR locus using a EZH2 or H3K27me3 antibody, in the presence or absence of EZH2 (Figure 5B). The reciprocal interaction between FBP1 and EZH2 further supports this mechanism (Figure 5C). Moreover, FBP1 KO reduces the mRNA levels of PKLR, as well as other EZH2 downstream genes, which can be largely restored by EZH2 knockdown (Figures 5D and 5E). Either knockdown or pharmacological inhibition of EZH2 rescues the protein expression of PKLR in FBP1-depleted hepatocytes (Figures 5F and 5G). Consistently, forced FBP1 expression abrogates the inhibitory effects of EZH2 overexpression on PKLR levels (Figure 5H). To exclude the role of other methyl transferases in this process, we treated FBP1-depleted hepatocytes with the DNA methyltransferas (DNMT) inhibitor decitabine and found that decitabine failed to rescue the protein expression of PKLR in FBP1-depleted hepatocytes (Figure S6). Together, these data demonstrated that PKLR expression is inhibited in FBP1-deficient hepatocytes through the FBP1-EZH2 axis.
Figure 5.

EZH2 suppresses PKLR expression upon FBP1 loss in hepatocytes
(A) EZH2 and H3K27me3 ChIP-seq results at the PKLR loci in HepG2 cells retrieved from the Encyclopedia of DNA Elements (ENCODE) project. At the bottom of the graph, from left to right, are exons 10 to 1 of PKLR. The red box shows that both EZH2 and H3K27me3 signals are enriched at the beginning of the PKLR genomic region. (B) EZH2 (left) and H3K27me3 (middle) ChIP qPCR results at the PKLR loci in HHL5 cells transfected with or without EZH2 shRNAs. Western blots are shown on the right. (C) Immunoprecipitation analysis by co-transfecting HHL5 cells with FBP1 and EZH2-avi tag (left) or EZH2 and FBP1-avi (right). (D) Heatmap showing FBP1 KO inhibits EZH2 target gene expression in HHL5 cells. EZH2 target genes were selected based on the ENCODE ChIP-seq data. (E) Real-time qPCR analysis of indicated genes in HHL5 FBP1 WT and KO clones followed by EZH2 knockdown. (F and G) Western blot analysis of FBP1 WT and KO HHL5 clones together with EZH2 shRNAs (F) or the EZH2 inhibitor GSK126 with indicated doses (G). (H) Western blot analysis of HHL5 cells with EZH2 and/or FBP1 overexpression. See also Figure S6.
EVs with low PKLR levels secreted by FBP1-depleted hepatocytes accelerate liver tumorigenesis in vivo
To examine the effects of EVs derived from FBP1-deficient hepatocytes on liver tumorigenesis in vivo, we utilized the DEN-induced HCC mouse model and injected EVs purified from control (FBP1 wild-type [WT]) or FBP1-depleted (FBP1 KO) hepatocytes through the tail vein (Figures 6A and 6B). Previous studies suggested that tail vein-injected EVs efficiently target hepatic tissues through circulation. As a result, DEN-treated mice in the FBP1 KO group exhibited higher tumor burden and accelerated tumorigenesis than in the WT group, based on enlarged tumor volumes, increased surface tumor numbers, elevated Ki67 expression, and reduced TUNEL staining (Figures 6C−6E). Moreover, a decreased population of NK cells but not T cells was observed in the liver and periphery blood, consistent with our previous finding that NK cells are the primary immune cell population targeted by hepatocyte-derived EVs (Figure 6F). Furthermore, activated (tumor necrosis factor [TNF]+) NK cells in the liver were specifically attenuated in the KO group (Figure 6G). Previous studies revealed that endogenous and exogenous reactive oxygen specie (ROS) induces apoptosis of NK cells.22 We next sought to ask whether the decreased population of NK cells in the FBP1 KO group results from the change of intracellular ROS levels in NK cells. To test whether ROS induces the apoptosis of NK cells in our model, we examined the effect of EVs secreted by hepatocytes on ROS production in NK cells. We injected EVs secreted by hepatocytes with or without FBP1 into C57BL/6 mice through the tail vein and found that ROS levels of hepatic NK cells isolated from these mice remained comparable (Figures S7A and S7B), indicating that ROS in NK cells may not play a role in this context.
Figure 6.

EVs with low PKLR levels secreted by FBP1-depleted hepatocytes accelerate liver tumorigenesis in vivo
(A) Experimental scheme examining the effect of hepatocyte-derived EVs in a DEN-induced HCC model using C57BL/6 mice. (B) Western blot analysis of PKLR expression in EVs secreted by indicated clones. (C) Representative image of whole livers (left) and the number of surface tumor nodules (right) in DEN-induced HCC mice after injecting EVs secreted by indicated clones, n = 6. (D and E) Representative images (left) and quantifications (right) of Ki67 (D; scale bar, 100 μm) and TUNEL staining (E; scale bar, 100 μm) in DEN-induced HCC mouse livers treated with EVs secreted by indicated clones, n = 6. (F) Quantifications of NK and T cells in mouse livers, spleens, and periphery blood harvested from DEN-treated mice after injecting EVs secreted by indicated clones, n = 6. (G) Representative histograms (top) and quantifications (bottom) of activated NK cells (TNF-α positive) in aforementioned livers and spleens, n = 6. (H) Representative IHC staining (top; scale bar, 100 μm) and correlations (bottom) among FBP1, PKLR, and infiltrated NK cells in early-stage human HCC tissues. (I) Kaplan-Meier overall survival analysis of two HCC patient groups based on PKLR and FBP1 expression levels in their tumor tissues as sequenced by TCGA. Data are presented as mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001 by unpaired Student’s t test. See also Figures S7–S9.
Recent studies showed that FBP1 can downregulate PD-L1 in cancer cells,12 which may affect the activity of tumor-infiltrating T cells. In particular, we examined whether EVs secreted by hepatocytes, with or without FBP1, affect the population of exhausted CD8 T cells in vivo. C57BL/6 mice were first subcutaneously inoculated with syngeneic Hepa1-6 cells. EVs secreted from Hepa1-6 cells, with or without FBP1, were then applied through intratumoral injection twice per week until the endpoint. We found that the amount of exhausted CD8 T cells remained unchanged regardless of the FBP1 status in EV-producing hepatocytes (Figure S7C). In addition, nontransformed hepatocytes expressed undetectable levels of programmed death ligand 1 (PD-L1) compared to HCC cells (Figure S7D). These data suggested that FBP1-mediated PD-L1 regulation may not occur during the initiation stage of liver cancer.
Consistent with the DEN-induced HCC model, the similar effect of EVs derived from FBP1-depleted cells on tumor growth was observed in a xenograft model, suggesting that these EVs also exert function in established tumors (Figures S8A−S8E). Furthermore, considering that CD8 T cells play pivotal roles in antitumor immunity, we examined the potential role of T cells in liver tumorigenesis accelerated by FBP1-depleted hepatocytes using the CB17-severe combined immunodeficiency (SCID) immunocompromised mice model. The CB17-SCID mouse strain carries dysfunctional T cells yet retains most NK cells, dendritic cells (DCs), and macrophages. We treated these mice with DEN weekly for 10 weeks and harvested their hepatic tissues 14 weeks after the first DEN treatment. EVs secreted from hepatocytes, with or without FBP1, were administrated throughout this course (Figure S8F). We observed that the CB17-SCID mouse strain showed moderate resistance to DEN and developed smaller tumors in the liver (Figure S8G). Nevertheless, consistent with the C57BL/6 model (Figures 6C and 6F), the CB17-SCID mouse livers treated with EVs secreted from FBP1-depleted hepatocytes exhibited significant increases in surface tumor numbers as accompanied by a reduced functional NK pool (Figures S8G, S8H, and S9). These in vivo data further demonstrated that EVs with low PKLR levels derived from FBP1-deficient hepatocytes promote liver tumorigenesis mainly through targeting NK cells. Ultimately, the protein levels of FBP1 and PKLR, as well as the extent of NK cell infiltration, are all positively correlated with each other in early-stage human HCC tissues (Figure 6H), and low PKLR and FBP1 levels are both associated with poor overall survival of HCC patients (Figure 6I), confirming the critical role of the FBP1-PKLR axis in regulating liver tumorigenesis.
Discussion
Recently, we and others focused on FBP, a rate-limiting gluconeogenic enzyme, and identified its multi-faceted roles in breast cancer,13 colon cancer,23 renal cell carcinoma,7 liver cancer,8 lung cancer,9 etc. FBP1 is the major isoform of FBP, in which its expression and activity are under intensive regulation at different physiopathological circumstances, and significantly contributes to the development of liver and kidney cancer.7 FBP1 is universally depleted in multiple tumor tissues, due in part to the fact that FBP1 strongly inhibits glycolysis in cancer cells through its metabolic activity.7,13 In addition, FBP1 harbors non-enzymatic functions in the cell nucleus, binding, and regulating a variety of transcription factors such as hypoxia-inducible factors,7 Myc,24 and Wnt/β-catenin.25 The pleiotropic roles of FBP1 in cancer highlight the regulatory mechanism of this enzyme in normal and transformed entities. Previous data revealed limited hypermethylation in the FBP1 promoter in both hepatic and renal tumors, suggesting the existence of additional regulatory events to inhibit FBP1 expression.23,26 We further identified EZH2, a core component of polycomb-repressive complex 2, as a critical player in suppressing FBP1 in tumorigenic livers and kidneys.21 EZH2 can be upregulated by oncogenic genotoxins that induce EZH2 acetylation and stability, which subsequently repress FBP1 to potentiate tumor initiation and progression.
In the past decade, EVs have emerged as an interesting form of cell-cell communication modules, which are lipid bilayer-enclosed vesicles containing a variety of functional biomolecules such as DNA, RNA, and protein.27,28 Through intercellular transport of biomolecules from source to target cells, another dimension of the regulatory network can be achieved in complex tissues. EVs were demonstrated to participate in the systemic homeostasis of the human body, as well as the progression of multiple diseases such as cancers,29 cardiovascular syndromes,30 neurodegenerative disorders,31 etc. By far, most EV-centered roles were attributed to the intercellular transfer of nucleotides,32 whereas the functional contribution of EV-encapsulated proteins was underappreciated. In the current study, we identified the hepatocyte-specific metabolic enzyme PKLR as a critical protein cargo sorted into EVs, transferred to nearby NK cells, and thereby composing a regulatory event toward NK cell metabolism and activity.
An array of cytotoxic lymphocytes is combined to form the first-line surveillance force against cancer. Whereas T cells and macrophages predominately regulate the immune microenvironment of pre-formed tumors, the NK cell population is the key component of the innate immune system to confront tumor initiation.9, 10, 11 Metabolic reprogramming is one of the earliest pathological events during tumor onset and is therefore coincident with the attenuation of patrolled NK cells upon tumorigenesis.8 Based on these observations, there are a growing body of evidences linking tumor-associated metabolic abnormalities to NK dysfunction. For instance, accumulated lactate in the neoplastic microenvironment reduces the cytotoxic activity of resident NK cells by inhibiting the production of interferon (IFN)-γ,33 a potent NK-stimulating cytokine. Moreover, transformed cells uptake and consume a large quantity of glucose, which is equally essential to support NK cell proliferation and activation.34 This type of metabolic competition severely limits the functionality of infiltrating NK cells. Interestingly, our study pointed to an additional route to lower the NK cytotoxic activity in terms of metabolic management, that is, through EV-mediated transport of metabolic enzymes that regulate the glycolytic activity of NK cells. We eventually identified PKLR as a key component to exert such function. Notably, the deciphered mechanisms involve no detectable transfer of FBP1 protein itself via EVs, which differs from a recent study where direct FBP1 upregulation in NK cells was observed to antagonize the glycolysis and activity of these innate immune effectors during lung cancer progression.9
Given the pivotal roles of EVs in different physiological and pathological conditions, numerous efforts have been devoted to exploiting the therapeutic application of EVs in clinics.35 The most-studied EVs from this perspective are those derived from mesenchymal stromal cells (MSCs), as multiple studies reported that the biological activities of MSCs can be mimicked by MSC-produced EVs, which are under intensive tests to treat human diseases including pulmonary hypertension, acute kidney injury, liver fibrosis, etc.36 Our data demonstrate that hepatic FBP1 depletion leads to reduced PKLR levels in hepatocyte-derived EVs, which inactivate liver-resident NK cells and promote hepatic tumorigenesis. Therefore, EVs isolated from nontransformed hepatocytes with normal PKLR levels may have the clinical potential to inhibit tumor initiation and progression by recovering NK activity in the tumor microenvironment. Nevertheless, caution is warranted to employ these purified EVs, as certain hepatic progenitor cell lines instead of primary hepatocytes are required to generate the prerequisite amount of EVs for clinic application,37 which may lead to an unpredicted immune response and rapid EV clearance in recipient patients.38 An alternative approach is to use biocompatible nanoparticles as EV mimics, given that a majority of infused EVs or nanoparticles are intercepted by the liver.37 Therefore, FBP1-incapsulated EV mimics can be applied through infusion to enhance the FBP1 level in recipient hepatocytes and upregulate PKLR expression in hepatocyte-secreted EVs, ultimately leading to the same therapeutic consequence as purified EVs.
In summary, our findings reveal that an unexpected crosstalk from the metabolic disorder to malfunctioned immunity co-existed in the HCC microenvironment and highlight the importance of gluconeogenic suppression during the tumorigenic course of this malignancy. From the therapeutic perspective, our study underscores the clinical potential to treat liver cancer with EZH2 antagonists, hepatocyte-derived EVs, or FBP1-incapsulated EV mimics.
Materials and methods
Animal model
All animal procedures were approved by the Sun Yat-sen University Institutional Animal Care and Use Committee and performed in accordance with the Declaration of Helsinki. The mouse model of HCC was constructed as previously described.8 Fbp1floxed/floxed mice were generated on the C57BL/6 background mice (Beijing Vital River Lab Animal Technology). For establishing the model of HCC in Fbp1floxed/floxed mice, the GFP and Cre mice were subjected to 23 or 35 injections of 6 mg/kg DEN (Sigma-Aldrich, St. Louis, MO, USA) into 15-day-old male mice intraperitoneally once per week (average of n = 6 mice).
For the DEN-induced HCC model of C57BL/6 mice, 50 mg/kg DEN was intraperitoneally injected into 15-day-old male mice once per week for 8 weeks. Meanwhile, tail-vein injections of EVs (25 μg in 100 μL PBS) were performed every week (average of n = 6 mice). For the DEN-induced HCC model of CB-17-SCID mice, 50 mg/kg DEN was intraperitoneally injected into 21-day-old male mice once per week for 10 weeks. Meanwhile, tail-vein injections of EVs (25 μg in 100 μL PBS) were performed every week (average of n = 5 mice).
For the xenograft model of C57BL/6 mice, Hep1-6 HCC cells (5 × 105 cells) were inoculated to subcutaneous grafts in 3- to 4-week-old male mice, which were treated with EVs secreted from Hep1-6 cells with or without FBP1 through intratumoral injections (average of n = 6 mice).
Cell culture
HHL5, HL7702, Hep3B, Hepa1-6, and K562 cell lines were obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA) and cultured in the Roswell Park Memorial Institute (RPMI) medium (Gibco, Gaithersburg, MD, USA) containing 10% fetal bovine serum (FBS; Corning Life Sciences, Corning, NY, USA), penicillin (100 U/mL; Gibco, Gaithersburg, MD, USA), and streptomycin (0.1 mg/mL) at 37°C with 5% CO2.
EV isolation and quantification
To purify EVs, cultured cells were grown to 70% confluency, washed with PBS 3 times, and cultured with serum-free RPMI for 48 h. Next, the collected medium was centrifuged at 300 × g for 10 min, followed by 2,000 × g for 10 min to deplete cell debris. The supernatants were filtered with a 0.22-μm filter membrane (SLGP033RB; Millipore, Burlington, MA, USA) and then centrifuged at 100,000 × g for 2 h at 4°C using an ultracentrifuge (Optima XE-100; Beckman Coulter, Brea, CA, USA) equipped with a SW 32 Ti rotor (369694; Beckman Coulter, Brea, CA, USA). Pelleted EVs were washed with PBS, resuspended, and quantified by a Bicinchoninic Acid (BCA) Protein Assay Kit (P0011; Beyotime, Shanghai, China).
EV labeling and uptake assay
For the EV-labeling assay, the collected EVs were resuspended with PBS and incubated with 5 μg/mL Dir (D12731; Thermo Fisher Scientific, Waltham, MA, USA), at 37°C for 1 h. The stained EVs were washed with PBS twice, accompanied by ultracentrifugation to remove the unincorporated dyes. As a control, PBS alone was mock incubated with Dir, according to the above procedures, to demonstrate that the unincorporated dyes can be completely washed off with PBS by ultracentrifugation. For the EV uptake assay, Dir-labeled or naked EVs were incubated with NK cells at 37°C for 4 h, and flow cytometry analysis was performed using a CytoFLEX S flow cytometer (Beckman Coulter, Brea, CA, USA).
Plasmids
Expression plasmids of FBP1 and PKLR were constructed by inserting corresponding cDNAs into the PCDH vector. PLKO.1-shPKLR and shFBP1 vectors were constructed by inserting shRNA sequences into polyclonal sites of the PLKO.1 vector (Addgene; 10878). FBP1 sgRNAs were cloned into the lentiCRISPR version 2 (v.2) vector (Addgene; 52961). The recognition sequences of shRNAs and sgRNAs were listed in Table S1.
Quantitative real-time PCR
Total RNA from cultured cells and EVs was extracted with Trizol (15596026; Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer’s protocol. cDNA was obtained using a Reverse Transcription Kit (RR036A; Takara, Dalian, China). The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and ribosomal 18S were used as internal reference controls. The sequences of all used primers were listed in Table S2.
Isolation and characterization of NK cells
Peripheral blood of healthy donors was from the Guangzhou Blood Center. The study protocol was approved by the Institute Research Medical Ethics Committee at Sun Yat-Sen University. Primary NK cells were isolated from periphery blood referring to standard procedures.39 Briefly, PBMCs were isolated using Ficoll through density gradient centrifugation. The middle layer was collected and washed with pre-cold PBS, then centrifuged at 450 × g for 10 min. Human NK cells were purified (or depleted) using a MACS NK Cell Isolation Kit (130-050-401; Miltenyi Biotec, Auburn, CA, USA) and incubated with HHL5 cell-derived EVs together with interleukin (IL)-2 (200 IU/mL, 200-02; Proteintech, Chicago, IL, USA). After stimulation with the Leukocyte Activation Cocktail (550583; BD Biosciences, Franklin Lakes, NJ, USA) for 5 h, NK cells were stained with various florescent antibodies and analyzed by flow cytometry as described below.
NK cell cytotoxicity assay
K562 cells (2 × 106 cells/mL) were treated with 5 μM CFSE (65-0850-84; Thermo Fisher Scientific, Waltham, MA, USA) for 6 min in the dark after washing with PBS. The purified NK cells were incubated with EVs secreted from hepatocytes with or without FBP1 for 3 days and then co-cultured with CFSE-labeled K562 cells at the ratio of 5:1 supplemented with IL-2 (200 IU/mL) for 6 h. Cells were collected and treated with 7AAD (559925; BD Biosciences, Franklin Lakes, NJ, USA), and cytotoxic target cells (CFSE+7AAD+) were analyzed by flow cytometry. As the control, NK cells or CFSE-labeled cells cultured alone were also treated with 7AAD and analyzed. NK cell cytotoxicity assays against Hep3B cells were measured using the CyQUANT LDH Cytotoxicity Assay (C20301; Thermo Fisher Scientific, Waltham, MA, USA).
Flow cytometry
Tissues harvested from mice were cut into small pieces, digested with collagenase I for 1 h, and then filtered through a 100-μm strainer.40 Leukocytes were isolated with Ficoll via gradient centrifugation. For flow cytometry analysis, cell surface markers were directly stained with fluorescent antibodies, whereas intracellular markers were stained after cells were fixed and permeabilized by a fixation/permeabilization kit (88-8824; BD Biosciences, Franklin Lakes, NJ, USA). Flow cytometry analysis was performed using CytoFLEX S (Beckman Coulter, Brea, CA, USA). Applied antibodies were listed in Table S3.
Transmission electron microscopy and nanoparticle tracking analysis (NTA)
Purified EVs were processed and observed under electron microscope according to previous reports.41 The EV size distribution was measured by NanoSight NS300, according to the instructions of the manufacturer.
Immunohistochemistry and immunofluorescence staining
Assays were performed according to previous procedures.21 For immunohistochemistry, the slides were stained using antibodies including anti-Ki67 (ab16667; Abcam, Cambridge, UK), anti-PKLR (22456-1-AP; Proteintech, Chicago, IL, USA), anti-FBP1 (HPA005857; Sigma-Aldrich, St. Louis, MO, USA), and anti-CD56 (MA1-06801; Thermo Fisher Scientific, Waltham, MA, USA), followed by a secondary antibody (K4003; Dako, Glostrup, Denmark) and 3,3’-diaminobenzidine (DAB) (K3467; Dako, Glostrup, Denmark) treatments. For TUNEL staining, a TUNEL Apoptosis Detection Kit (40306ES20; Yeason, Shanghai, China) was used following standard instructions.
Immunoprecipitation and western blot
Briefly, whole-cell lysates and EVs were lysed with RIPA lysis buffer supplemented with 1× Protease Inhibitor Cocktail (HY-K0010; MedChemExpress [MCE], Princeton, NJ, USA) on ice for 30 min, centrifuged at 12,000 × g for 30 min at 4°C, and then quantified by the BCA assay. The immunoprecipitation and western blot assays were carried out according to previous procedures.21 Anti-histone H3K27me3 (9733; Cell Signaling Technology [CST], Danvers, MA, USA), anti-TSG101 (ab125011; Abcam, Cambridge, UK), anti-PKLR (22456-1-AP; Proteintech, Chicago, IL, USA), anti-FBP1 (ab109020; Abcam, Cambridge, UK), anti-EZH2 (5246; CST, Danvers, MA, USA), anti-CD9 (ab92726; Abcam, Cambridge, UK), and anti-CD81 (ab79559; Abcam, Cambridge, UK) were used as primary antibodies. Proteins were quantified using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Proteomic analysis
Isolated EV cargo proteins were identified using proteomic analysis, according to procedures described in our previous study.8 Our raw proteomic data have been deposited in the Proteomics Identifications (PRIDE) database, and the accession ID is RRIDE: PXD026822.
ChIP
The ChIP assay was performed following standard protocols.21 Protein-DNA complexes were immunoprecipitated by antibodies recognizing H3K27me3 (9733; CST, Danvers, MA, USA), EZH2 (5246; CST, Danvers, MA, USA), or immunoglobulin G (IgG; 2729; CST, Danvers, MA, USA) and then incubated with protein A/G beads (HY-K0202; MCE, Princeton, NJ, USA). The enriched DNA fragments were detected by qRT-PCR using the following primers: 5′-CCTAGTGCACAAGTGGAAAGA-3′ and 5′-AGGCCTGAGGTCACTGTATGGGC-3′.
Statistical analysis
All statistics were performed using GraphPad Prism (GraphPad, La Jolla, CA, USA). All results were presented as mean ± SD unless otherwise indicated. Statistical analysis was performed using two-tailed unpaired Student’s t test. A survival curve was plotted with the Kaplan-Meier method and analyzed using the log-rank test.
Acknowledgments
This study was supported by the National Key Research and Development Program of China (2016YFA0502600 to B.L.), National Natural Science Foundation of China (82073074 to B.L. and 82003194 to C.P.), National Program for Support of Top-Notch Young Professionals (to J.-X.B.), Chang Jiang Scholars Program (to J.-X.B.), Special Support Program of Guangdong (to J.-X.B.), Sun Yat-sen University Young Teacher Key Cultivate Project (17ykjc24), and Guangdong Innovative and Entrepreneurial Research Team Program (2016ZT06S638).
Author contributions
Conceived the study and analyzed the data, C.P., F.L., and B.L.; designed and performed the experiments, Z.L., Y.Y., Q.C., G.L., W.P., Q.Y., J.D., Y.W., and J.-X.B.; wrote the manuscript, C.P. and B.L.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.07.015.
Contributor Information
Chaoyun Pan, Email: panchy27@mail.sysu.edu.cn.
Fuming Li, Email: fumingli@pennmedicine.upenn.edu.
Bo Li, Email: libo47@mail.sysu.edu.cn.
Supplemental information
References
- 1.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2020. CA Cancer J. Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 2.Chen W., Zheng R., Baade P.D., Zhang S., Zeng H., Bray F., Jemal A., Yu X.Q., He J. Cancer statistics in China, 2015. CA Cancer J. Clin. 2016;66:115–132. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 3.Finn R.S., Zhu A.X., Farah W., Almasri J., Zaiem F., Prokop L.J., Murad M.H., Mohammed K. Therapies for advanced stage hepatocellular carcinoma with macrovascular invasion or metastatic disease: A systematic review and meta-analysis. Hepatology. 2018;67:422–435. doi: 10.1002/hep.29486. [DOI] [PubMed] [Google Scholar]
- 4.L’Hermitte A., Pham S., Cadoux M., Couchy G., Caruso S., Anson M., Crain-Denoyelle A.-M., Celton-Morizur S., Yamagoe S., Zucman-Rossi J., et al. Lect2 Controls Inflammatory Monocytes to Constrain the Growth and Progression of Hepatocellular Carcinoma. Hepatology. 2019;69:160–178. doi: 10.1002/hep.30140. [DOI] [PubMed] [Google Scholar]
- 5.Liu L.-Z., Zhang Z., Zheng B.-H., Shi Y., Duan M., Ma L.-J., Wang Z.-C., Dong L.-Q., Dong P.-P., Shi J.-Y., et al. CCL15 Recruits Suppressive Monocytes to Facilitate Immune Escape and Disease Progression in Hepatocellular Carcinoma. Hepatology. 2019;69:143–159. doi: 10.1002/hep.30134. [DOI] [PubMed] [Google Scholar]
- 6.Kurebayashi Y., Ojima H., Tsujikawa H., Kubota N., Maehara J., Abe Y., Kitago M., Shinoda M., Kitagawa Y., Sakamoto M. Landscape of immune microenvironment in hepatocellular carcinoma and its additional impact on histological and molecular classification. Hepatology. 2018;68:1025–1041. doi: 10.1002/hep.29904. [DOI] [PubMed] [Google Scholar]
- 7.Li B., Qiu B., Lee D.S.M., Walton Z.E., Ochocki J.D., Mathew L.K., Mancuso A., Gade T.P.F., Keith B., Nissim I., Simon M.C. Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature. 2014;513:251–255. doi: 10.1038/nature13557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li F., Huangyang P., Burrows M., Guo K., Riscal R., Godfrey J., Lee K.E., Lin N., Lee P., Blair I.A., et al. FBP1 loss disrupts liver metabolism and promotes tumorigenesis through a hepatic stellate cell senescence secretome. Nat. Cell Biol. 2020;22:728–739. doi: 10.1038/s41556-020-0511-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cong J., Wang X., Zheng X., Wang D., Fu B., Sun R., Tian Z., Wei H. Dysfunction of Natural Killer Cells by FBP1-Induced Inhibition of Glycolysis during Lung Cancer Progression. Cell Metab. 2018;28:243–255.e5. doi: 10.1016/j.cmet.2018.06.021. [DOI] [PubMed] [Google Scholar]
- 10.Dyck L., Lynch L. New Job for NK Cells: Architects of the Tumor Microenvironment. Immunity. 2018;48:9–11. doi: 10.1016/j.immuni.2018.01.001. [DOI] [PubMed] [Google Scholar]
- 11.Pardoll D.M. Distinct mechanisms of tumor resistance to NK killing: of mice and men. Immunity. 2015;42:605–606. doi: 10.1016/j.immuni.2015.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang B., Zhou Y., Zhang J., Jin X., Wu H., Huang H. Fructose-1,6-bisphosphatase loss modulates STAT3-dependent expression of PD-L1 and cancer immunity. Theranostics. 2020;10:1033–1045. doi: 10.7150/thno.38137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dong C., Yuan T., Wu Y., Wang Y., Fan T.W.M., Miriyala S., Lin Y., Yao J., Shi J., Kang T., et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell. 2013;23:316–331. doi: 10.1016/j.ccr.2013.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhardwaj N. Harnessing the immune system to treat cancer. J. Clin. Invest. 2007;117:1130–1136. doi: 10.1172/JCI32136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Umezu T., Tadokoro H., Azuma K., Yoshizawa S., Ohyashiki K., Ohyashiki J.H. Exosomal miR-135b shed from hypoxic multiple myeloma cells enhances angiogenesis by targeting factor-inhibiting HIF-1. Blood. 2014;124:3748–3757. doi: 10.1182/blood-2014-05-576116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Record M., Subra C., Silvente-Poirot S., Poirot M. Exosomes as intercellular signalosomes and pharmacological effectors. Biochem. Pharmacol. 2011;81:1171–1182. doi: 10.1016/j.bcp.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 17.Chen J., Gingold J.A., Su X. Immunomodulatory TGF-β Signaling in Hepatocellular Carcinoma. Trends Mol. Med. 2019;25:1010–1023. doi: 10.1016/j.molmed.2019.06.007. [DOI] [PubMed] [Google Scholar]
- 18.Nguyen A., Loo J.M., Mital R., Weinberg E.M., Man F.Y., Zeng Z., Paty P.B., Saltz L., Janjigian Y.Y., de Stanchina E., Tavazoie S.F. PKLR promotes colorectal cancer liver colonization through induction of glutathione synthesis. J. Clin. Invest. 2016;126:681–694. doi: 10.1172/JCI83587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bidkhori G., Benfeitas R., Klevstig M., Zhang C., Nielsen J., Uhlen M., Boren J., Mardinoglu A. Metabolic network-based stratification of hepatocellular carcinoma reveals three distinct tumor subtypes. Proc. Natl. Acad. Sci. USA. 2018;115:E11874–E11883. doi: 10.1073/pnas.1807305115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Z., Zhang C., Lee S., Kim W., Klevstig M., Harzandi A.M., Sikanic N., Arif M., Ståhlman M., Nielsen J., et al. Pyruvate kinase L/R is a regulator of lipid metabolism and mitochondrial function. Metab. Eng. 2019;52:263–272. doi: 10.1016/j.ymben.2019.01.001. [DOI] [PubMed] [Google Scholar]
- 21.Liao K., Deng S., Xu L., Pan W., Yang S., Zheng F., Wu X., Hu H., Liu Z., Luo J., et al. A Feedback Circuitry between Polycomb Signaling and Fructose-1, 6-Bisphosphatase Enables Hepatic and Renal Tumorigenesis. Cancer Res. 2020;80:675–688. doi: 10.1158/0008-5472.CAN-19-2060. [DOI] [PubMed] [Google Scholar]
- 22.Brenner C., Galluzzi L., Kepp O., Kroemer G. Decoding cell death signals in liver inflammation. J. Hepatol. 2013;59:583–594. doi: 10.1016/j.jhep.2013.03.033. [DOI] [PubMed] [Google Scholar]
- 23.Chen M., Zhang J., Li N., Qian Z., Zhu M., Li Q., Zheng J., Wang X., Shi G. Promoter hypermethylation mediated downregulation of FBP1 in human hepatocellular carcinoma and colon cancer. PLoS ONE. 2011;6:e25564. doi: 10.1371/journal.pone.0025564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang B., Fan P., Zhao J., Wu H., Jin X., Wu H. FBP1 loss contributes to BET inhibitors resistance by undermining c-Myc expression in pancreatic ductal adenocarcinoma. J. Exp. Clin. Cancer Res. 2018;37:224. doi: 10.1186/s13046-018-0888-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao W., Yang S., Chen J., Zhao J., Dong J. Forced overexpression of FBP1 inhibits proliferation and metastasis in cholangiocarcinoma cells via Wnt/β-catenin pathway. Life Sci. 2018;210:224–234. doi: 10.1016/j.lfs.2018.09.009. [DOI] [PubMed] [Google Scholar]
- 26.Zhang J., Wang J., Xing H., Li Q., Zhao Q., Li J. Down-regulation of FBP1 by ZEB1-mediated repression confers to growth and invasion in lung cancer cells. Mol. Cell. Biochem. 2016;411:331–340. doi: 10.1007/s11010-015-2595-8. [DOI] [PubMed] [Google Scholar]
- 27.Cocucci E., Meldolesi J. Ectosomes and exosomes: shedding the confusion between extracellular vesicles. Trends Cell Biol. 2015;25:364–372. doi: 10.1016/j.tcb.2015.01.004. [DOI] [PubMed] [Google Scholar]
- 28.Li J., Liu K., Liu Y., Xu Y., Zhang F., Yang H., Liu J., Pan T., Chen J., Wu M., et al. Exosomes mediate the cell-to-cell transmission of IFN-α-induced antiviral activity. Nat. Immunol. 2013;14:793–803. doi: 10.1038/ni.2647. [DOI] [PubMed] [Google Scholar]
- 29.Kalluri R. The biology and function of exosomes in cancer. J. Clin. Invest. 2016;126:1208–1215. doi: 10.1172/JCI81135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martínez M.C., Andriantsitohaina R. Extracellular Vesicles in Metabolic Syndrome. Circ. Res. 2017;120:1674–1686. doi: 10.1161/CIRCRESAHA.117.309419. [DOI] [PubMed] [Google Scholar]
- 31.Riazifar M., Mohammadi M.R., Pone E.J., Yeri A., Lässer C., Segaliny A.I., McIntyre L.L., Shelke G.V., Hutchins E., Hamamoto A., et al. Stem Cell-Derived Exosomes as Nanotherapeutics for Autoimmune and Neurodegenerative Disorders. ACS Nano. 2019;13:6670–6688. doi: 10.1021/acsnano.9b01004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iqbal M.A., Arora S., Prakasam G., Calin G.A., Syed M.A. MicroRNA in lung cancer: role, mechanisms, pathways and therapeutic relevance. Mol. Aspects Med. 2019;70:3–20. doi: 10.1016/j.mam.2018.07.003. [DOI] [PubMed] [Google Scholar]
- 33.Brand A., Singer K., Koehl G.E., Kolitzus M., Schoenhammer G., Thiel A., Matos C., Bruss C., Klobuch S., Peter K., et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016;24:657–671. doi: 10.1016/j.cmet.2016.08.011. [DOI] [PubMed] [Google Scholar]
- 34.Theurich S., Tsaousidou E., Hanssen R., Lempradl A.M., Mauer J., Timper K., Schilbach K., Folz-Donahue K., Heilinger C., Sexl V., et al. IL-6/Stat3-Dependent Induction of a Distinct, Obesity-Associated NK Cell Subpopulation Deteriorates Energy and Glucose Homeostasis. Cell Metab. 2017;26:171–184.e6. doi: 10.1016/j.cmet.2017.05.018. [DOI] [PubMed] [Google Scholar]
- 35.EL Andaloussi S., Mäger I., Breakefield X.O., Wood M.J.A. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 2013;12:347–357. doi: 10.1038/nrd3978. [DOI] [PubMed] [Google Scholar]
- 36.Wiklander O.P.B., Brennan M.Á., Lötvall J., Breakefield X.O., El Andaloussi S. Advances in therapeutic applications of extracellular vesicles. Sci. Transl. Med. 2019;11:eaav8521. doi: 10.1126/scitranslmed.aav8521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sacks D., Baxter B., Campbell B.C.V., Carpenter J.S., Cognard C., Dippel D., Eesa M., Fischer U., Hausegger K., Hirsch J.A., et al. From the American Association of Neurological Surgeons (AANS), American Society of Neuroradiology (ASNR), Cardiovascular and Interventional Radiology Society of Europe (CIRSE), Canadian Interventional Radiology Association (CIRA), Congress of Neurological Surgeons (CNS), European Society of Minimally Invasive Neurological Therapy (ESMINT), European Society of Neuroradiology (ESNR), European Stroke Organization (ESO), Society for Cardiovascular Angiography and Interventions (SCAI), Society of Interventional Radiology (SIR), Society of NeuroInterventional Surgery (SNIS), and World Stroke Organization (WSO) Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int. J. Stroke. 2018;13:612–632. doi: 10.1016/j.jvir.2017.11.026. [DOI] [PubMed] [Google Scholar]
- 38.Möller A., Lobb R.J. The evolving translational potential of small extracellular vesicles in cancer. Nat. Rev. Cancer. 2020;20:697–709. doi: 10.1038/s41568-020-00299-w. [DOI] [PubMed] [Google Scholar]
- 39.Wu Y., Kuang D.-M., Pan W.-D., Wan Y.-L., Lao X.-M., Wang D., Li X.-F., Zheng L. Monocyte/macrophage-elicited natural killer cell dysfunction in hepatocellular carcinoma is mediated by CD48/2B4 interactions. Hepatology. 2013;57:1107–1116. doi: 10.1002/hep.26192. [DOI] [PubMed] [Google Scholar]
- 40.Liu R.-X., Wei Y., Zeng Q.-H., Chan K.-W., Xiao X., Zhao X.-Y., Chen M.-M., Ouyang F.-Z., Chen D.-P., Zheng L., et al. Chemokine (C-X-C motif) receptor 3-positive B cells link interleukin-17 inflammation to protumorigenic macrophage polarization in human hepatocellular carcinoma. Hepatology. 2015;62:1779–1790. doi: 10.1002/hep.28020. [DOI] [PubMed] [Google Scholar]
- 41.Chen G., Huang A.C., Zhang W., Zhang G., Wu M., Xu W., Yu Z., Yang J., Wang B., Sun H., et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature. 2018;560:382–386. doi: 10.1038/s41586-018-0392-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.