

Abstract
CD19-targeting chimeric antigen receptor (CAR) T cells have become an important therapeutic option for patients with relapsed and refractory B cell malignancies. However, a significant portion of patients still do not benefit from the therapy owing to various resistance mechanisms, including high expression of multiple inhibitory immune checkpoint receptors. Here, we report a lentiviral two-in-one CAR T approach in which two checkpoint receptors are downregulated simultaneously by a dual short hairpin RNA cassette integrated into a CAR vector. Using this system, we evaluated CD19-targeting CAR T cells in the context of four different checkpoint combinations—PD-1/TIM-3, PD-1/LAG-3, PD-1/CTLA-4, and PD-1/TIGIT—and found that CAR T cells with PD-1/TIGIT downregulation uniquely exerted synergistic antitumor effects. Importantly, functional and phenotypic analyses suggested that downregulation of PD-1 enhances short-term effector function, whereas downregulation of TIGIT is primarily responsible for maintaining a less differentiated/exhausted state, providing a potential mechanism for the observed synergy. The PD-1/TIGIT-downregulated CAR T cells generated from diffuse large B cell lymphoma patient-derived T cells also showed robust antitumor activity and significantly improved persistence in vivo. The efficacy and safety of PD-1/TIGIT-downregulated CD19-targeting CAR T cells are currently being evaluated in adult patients with relapsed or refractory large B cell lymphoma (ClinicalTrials.gov: NCT04836507).
Keywords: CD19, CAR T, shRNA, immune checkpoints, PD-1, TIGIT, clinical trial
Graphical abstract
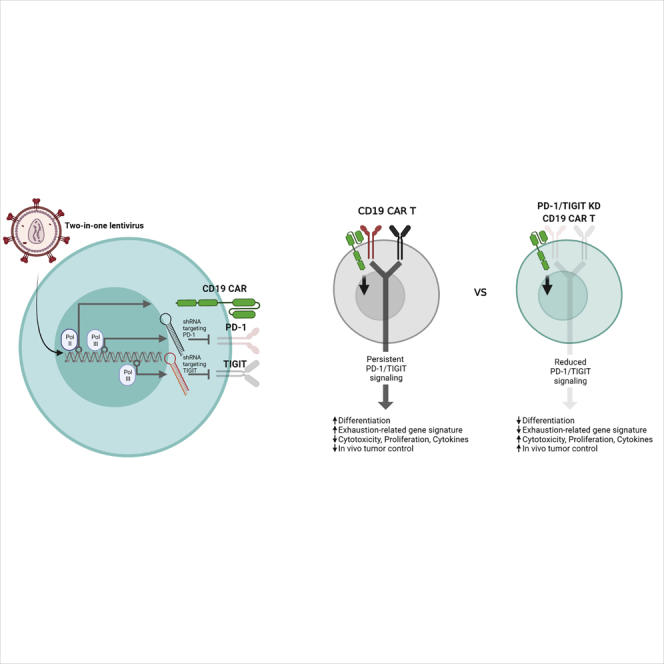
A two-in-one lentiviral anti-CD19 CAR T cell vector containing a dual PD-1/TIGIT shRNA cassette displays superior effector function and early memory phenotype persistence in mouse xenograft models through mechanisms distinctly coordinated by PD-1 and TIGIT.
Introduction
CD19-specific chimeric antigen receptor (CAR) T cell therapy has proven highly effective in the treatment of relapsed or refractory (R/R) B cell malignancies such as acute lymphoblastic leukemia (ALL) and non-Hodgkin’s lymphomas (NHLs).1, 2, 3, 4, 5, 6 However, in a recent long-term clinical follow-up, the percentages of a 3-year duration of response of CD19 CAR T therapy were 51% for all evaluable treatments.7,8 Treatment failure is often attributed to the loss of CD19 expression in cancer cells,9 but it can also involve the limited expansion, poor persistence, and dysfunctional state of infused CAR T cells.10, 11, 12
T cells express inhibitory immune checkpoint receptors (ICRs) that are essential for maintaining peripheral tolerance and regulating immune responses.13,14 However, in chronic inflammatory conditions and cancer, persistent ICR expression leads to dysfunction of antigen-specific T cells, which includes reduced cytolytic activity, cytokine secretion, survival, and proliferation.13,15, 16, 17 Consistent with this, monoclonal antibody-mediated blockade of either cytotoxic T lymphocyte-associated protein 4 (CTLA-4) or programmed cell death protein 1 (PD-1) is effective against many cancers.16,18 However, single ICR blockade therapy only yields a sustained response in a small subset of patients and often fades over time.19 Instead, recent murine and human studies have shown that simultaneous or sequential blockade of multiple ICRs can have synergistic effects on disease control.16,20, 21, 22, 23, 24, 25
In the context of CAR T therapy, previous preclinical and clinical studies have evaluated the co-administration of PD-1-blocking antibody, demonstrating enhanced antitumor activity and in vivo expansion of CAR T cells.26, 27, 28 Genetically, CAR T cells have been modified to secrete single-chain variable fragments (scFvs) against PD-129 and to express dominant-negative27,30 or chimeric-switch receptors.31, 32, 33 In addition, CRISPR-Cas9-based knockout and short hairpin RNA (shRNA)-based knockdown have been used to achieve sustained downregulation of ICRs in a cell-intrinsic manner.27,34, 35, 36, 37, 38, 39 However, most of these genetic approaches target a single ICR, and few studies have examined the therapeutic effects and cellular mechanisms of CAR T cells with multiple cell-intrinsic ICR blockade.40
Here, we describe a dual shRNA-based approach that allows for the simultaneous downregulation of two ICRs in CAR T cells. With this, we found that different combinations of ICR downregulation differentially affected the activity of CAR T cells, with some being detrimental. In particular, the downregulation of PD-1 and TIGIT (T cell immunoreceptor with immunoglobulin [Ig] and ITIM domains) was the most effective, significantly enhancing the efficacy of CAR T cells generated from a healthy donor and NHL patient T cells. Importantly, transcriptomic and cellular analysis suggested that the downregulation of PD-1 and TIGIT in CAR T cells distinctly affect their effector function and differentiation, which likely accounts for the synergistic improvement in activity compared to the downregulation of PD-1 or TIGIT alone.
Results
PD-1 downregulation using a two-in-one lentiviral vector system enhances the antitumor activity of CD19-targeting CAR T cells
Given that PD-1 is one of the major ICRs, we first designed a two-in-one lentiviral vector that expresses shRNA against PD-1 alongside a 41BB-based second-generation CD19-targeting CAR (Figure 1A). We tested three different PD-1-targeting shRNAs (shRNA #1–3; Figure S1A) and three different RNA polymerase III (RNA Pol III) promoters (mouse U6, human U6, and human H1)41 and found that the 19PBBz construct expressing shPD-1 #1 driven by mouse U6 efficiently downregulated PD-1 (Figures 1B and 1D) and had no negative effect on CAR expression (Figure S1B) or CAR T cell expansion (Figures 1C and 1E). We next cocultured 19PBBz CAR T cells with CD19-positive target cells with or without PD-L1 expression (Nalm-6, Nalm-6-PD-L1, K562-CD19, and K562-CD19-PD-L1 cells; Figure S2) and found that 19PBBz CAR T cells exhibited enhanced cytotoxic activity (Figure 1F) and proliferation (Figure 1G) compared to control CAR T cells (19GBBz and 19BBz, with and without the mU6-shGFP cassette, respectively), which depended on target PD-L1 expression. Moreover, under repeated antigen exposure, 19PBBz CAR T cells produced significantly higher amounts of interleukin (IL)-2 and tumor necrosis factor (TNF)-α, but not interferon (IFN)-γ, compared to 19GBBz CAR T cells (Figure 1H). 19PBBz cells also exhibited improved in vivo antitumor activity in mouse xenograft models of disseminated human blood cancer (Nalm-6-GL-PD-L1 and Nalm-6-GL) in a manner dependent on target PD-L1 expression (Figure 1I; Figure S3). Importantly, PD-1 expression levels in CAR T cells isolated from mice on day 43 indicated that our two-in-one vector system persistently downregulated PD-1 under in vivo conditions (Figure 1J).
Figure 1.

PD-1 downregulation enhances the antitumor function of CAR T cells
(A) Schematic representation of the lentiviral two-in-one vector carrying a CD19-CAR and shRNA expressing module. (B) PD-1 expression levels of CAR T cells with different shPD-1 candidates as determined by flow cytometry on day 2 after stimulation with gamma-irradiated Nalm-6-GL cells. Gray denotes the isotype control. Data are the pooled mean ± SD from three independent experiments, each performed in triplicate. (C) Cell counts from the homeostatic expansion of LNGFR+ CAR T cells with PD-1 downregulation candidates on days 3 and 6 after cell seeding. Data are the pooled mean ± SD from three independent experiments performed in triplicates. (D) The effects of hH1-, hU6-, and mU6-shPD1 on PD-1 expression was analyzed by flow cytometry 2 days after stimulation with gamma-irradiated K562-CD19 cells. Data are the pooled mean ± SD from two independent experiments performed in triplicates. (E) Cell counts from the homeostatic expansion of CAR T cells with each RNA Pol III promoter after LNGFR+ isolation on days 3 and 6 after cell seeding. Data are the pooled mean ± SD from two independent experiments performed in triplicates. (F) CAR T cells were incubated with GFP-expressing Nalm-6-GL or Nalm-6-GL-PD-L1 cells at a 1:1, 0.3:1, and 0.1:1 effector-to-target (E:T) ratio. GFP intensity was measured every 2 h using the IncuCyte S3 live cell imaging system. The relative percentage of total integrated GFP intensity was calculated as (GFP intensity at each time point/GFP intensity at 0 h) × 100. Representative mean ± SD from two independent experiments performed in triplicates. (G) CAR T cells were incubated with gamma-irradiated K562-CD19 or K562-CD19-PD-L1 cells at a 1:3 E:T ratio and counted on day 7. Data are the pooled mean ± SD from two independent experiments performed in triplicates. (H) 3 × 105 19GBBz or 19PBBz T cells were incubated with 9 × 106 gamma-irradiated Nalm-6-GL-PD-L1-CD80 cells every 6 days (first and second stimulation) and supernatants were collected on day 2 after each respective stimulation. Data are the pooled mean ± SD from four independent experiments. (I) NSG mice were injected intravenously with 1 × 106 Nalm-6-GL-PD-L1 leukemia cells. 5 days later, 1 × 106 CAR T cells were injected intravenously. Tumor burden was monitored based on the bioluminescence intensity from the IVIS imaging system. Data are from n = 3 mice (mock) and n = 5 mice (19BBz, 19GBBz, and 19PBBz). (J) PD-1 expression levels of CAR T cells from Nalm-6-GL-PD-L1-bearing mice at day 43. Gray denotes isotype control. Data are the mean ± SD from three mice per group. Statistical analysis was done by one-way ANOVA for (C), (E), (F), and (H) and an unpaired two-tailed t test for (G) and (J). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; ns, not significant.
After optimizing the shRNA expression cassette targeting PD-1, we wondered whether this system has differential effects depending on the costimulatory domain within the CAR. Because CD28 has been shown as the major target of PD-1,42 we also generated a 19P28z construct that replaces the 41BB costimulatory domain in 19PBBz with CD28 and confirmed similar CAR expression levels between 19P28z, 19PBBz, and their controls (19G28z and 19GBBz; Figure S4A). We then confirmed that, as with 19PBBz, 19P28z exhibited reduced PD-1 protein expression compared to its control upon stimulation with gamma-irradiated K562-CD19 (Figure 2A). When we evaluated the in vitro cytotoxicity, we found similarly improved cytotoxicity of freshly prepared 19P28z and 19PBBz CAR T cells compared to their controls against Nalm-6-GL-PD-L1 cells (Figure 2B). However, when we compared the cytotoxicity of CAR T cells that were stimulated with Nalm-6-PD-L1-CD80 for 6 days, we found that 19P28z CAR T cells showed reduced cytotoxicity compared to 19PBBz CAR T cells (Figure 2C). Additionally, 19PBBz was the only construct capable of robust proliferation upon repeated stimulation (Figure 2D). Consistent with these in vitro observations, 19PBBz CAR T cells demonstrated superior in vivo antitumor activity compared to 19P28z CAR T cells in mouse xenograft models of disseminated human blood cancer (Nalm-6-GL-PD-L1) and exhibited sustained, high-level accumulation of CAR T cells in the blood for up to 43 days (Figures 2E and 2F).
Figure 2.

CAR costimulatory domains differentially affect the in vivo persistence of PD-1-downregulated CAR T cells
(A) PD-1 expression level of CAR T cells with CD28 or 41BB costimulatory domains 2 days after stimulation with gamma-irradiated K562-CD19 cells. Numbers denote the geometric mean fluorescence intensity (gMFI) of PD-1. (B) Unstimulated and (C) stimulated CAR T cells were incubated with Nalm-6-GL-PD-L1 cells at a 1:1, 0.3:1, and 0.1:1 E:T ratio and analyzed using the IncuCyte S3 system. Stimulated CAR T cells were generated by coincubation with Nalm-6-PD-L1-CD80 cells for 6 days prior to cytotoxicity assay. Data are the representative mean ± SD from two independent experiments performed in triplicates. (D) CAR T cells were incubated with gamma-irradiated Nalm-6-GL-PD-L1-CD80 or K562-CD19-PD-L1 cells at a 1:3 E:T ratio and counted on day 6 after each stimulation. Data are the mean ± SD from two independent experiments performed in triplicates. (E) NSG mice were injected intravenously with 1 × 106 Nalm-6-GL-PD-L1 leukemia cells. 5 days later, 1 × 106 CAR T cells were injected intravenously. Tumor burden was monitored based on the bioluminescence intensity from the IVIS imaging system. Data are from n = 3 mice (mock), n = 5 mice (19G28z and 19GBBz), and n = 4 mice (19P28z and 19PBBz). (F) The number of CAR T cells in mouse blood was determined on days 20 and 43 after CAR T cell injection. Data are mean ± SD from three mice per group. Statistical analysis for (A)–(D) and (F) was done by one-way ANOVA. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; ns, not significant.
Collectively, our results show that the modification of the lentiviral vector to express an additional shRNA cassette that specifically targets PD-1 enhances the antitumor activity of CD19-targeting CAR T cells against PD-L1-positive cancer cells. In addition, this system had superior activity with a 41BB costimulatory domain compared to CD28, exhibiting a delayed exhaustion phenotype and longer persistence in vivo.
Simultaneous downregulation of PD-1 and TIGIT further enhances the in vivo functionality of CD19-targeting CAR T cells
Multiple ICRs are frequently expressed on intratumoral T cells from NHL patients,43 and PD-1/PD-L1 blockade is known to induce compensatory upregulation of alternative ICRs.20,44 We reasoned that simultaneous downregulation of additional ICRs would further enhance the antitumor activity of PD-1-downregulated CAR T cells. We chose four ICRs, that is, T cell immunoglobulin and mucin domain-containing protein 3 (TIM-3), lymphocyte-activation gene 3 (LAG-3), TIGIT, and CTLA-4, and confirmed that they are expressed on activated T cells in vitro (Figure S5) and selected shRNA sequences that individually downregulated their indicated receptors without affecting CAR expression (Figures S6–S8). We next modified our two-in-one lentiviral vector to include a second shRNA cassette (Figure 3A) and generated CD19-targeting dual-shRNA CAR vectors with four ICR combinations: PD-1/TIM-3, PD-1/LAG-3, PD-1/TIGIT, and PD-1/CTLA-4. We confirmed that PD-1 downregulation was similar across all constructs, and that the secondary shRNAs also specifically downregulated their respective ICRs without affecting CAR expression (Figure 3B; Figures S9A and S9B). To assess the effect of dual ICR downregulation on antitumor activity, we injected 1 × 106 CAR T cells into mice bearing Nalm-6-GL-PD-L1 leukemia cells that express ligands for TIGIT (CD112/155), LAG-3 (human leukocyte antigen [HLA]-DR), and TIM-3 (galectin-9), but not CTLA-4 (CD80) (Figure 2; Figures S9C–S9E). Unexpectedly, we found that three of the four combinations (PD-1/TIM-3, PD-1/LAG-3, and PD-1/CTLA-4) failed to improve the in vivo efficacy of CD19-targeting CAR T cells compared with single downregulation of PD-1; in the case of PD-1/LAG-3, dual downregulation severely compromised antitumor activity (Figures 3C and 3D; Figure S10A). However, the PD-1/TIGIT combination reduced the frequency of leukemia relapse and improved the survival of mice compared to single PD-1 downregulation (Figures 3C and 3D). Furthermore, dual PD-1/TIGIT downregulation outperformed single PD-1 downregulation in a stress test with two lower doses (0.5 × 106 and 0.25 × 106) of CAR T cells in the same mouse model (Figures 3E and 3F; Figure S10B).
Figure 3.

Simultaneous downregulation of PD-1 and TIGIT further enhances the in vivo functionality of CD19-targeting CAR T cells
(A) Schematic representation of the engineered two-in-one vector system carrying dual shRNA cassettes for two ICRs. (B) Dual downregulation efficiency of each ICR in CAR T cells stimulated for 48 h with gamma-irradiated K562-CD19 cells. FACS plots are representative data from two independent experiments performed in duplicates, and the bar graphs are the pooled mean ± SD. (C) NSG mice were injected intravenously with 1 × 106 Nalm-6-GL-PD-L1 leukemia cells. 5 days later, 1 × 106 CAR T cells with each dual downregulation (shGFP, shPD-1/shGFP, shPD-1/shTIM-3, shPD-1/shLAG-3, shPD-1/shTIGIT, shPD-1/shCTLA-4) were injected intravenously. Tumor burden was monitored based on the bioluminescence intensity from the IVIS imaging system. Data are from n = 3 mock, shPD-1/shTIM-3, and shPD-1/shLAG-3, n = 4 shGFP, shPD-1/shGFP, and shPD-1/shCTLA-4, and n = 5 shPD-1/shTIGIT. (D) Kaplan-Meier survival analysis with the log-rank (Mantel-Cox) test comparing each CAR T treated mice from (C). (E) NSG mice were injected intravenously with 1 × 106 Nalm-6-GL-PD-L1 leukemia cells. 5 days later, 0.5 × 106 or 0.25 × 106 CAR T cells with PD-1 (shPD-1/shGFP) or PD-1/TIGIT (shPD-1/shTIGIT) downregulation were injected intravenously. Tumor burden was monitored based on the bioluminescence intensity from the IVIS imaging system. Data are from n = 7 mice for the 0.5 × 106 dose groups and n = 6 mice for the 0.25 × 106 dose groups. (F) Kaplan-Meier survival analysis with log-rank (Mantel-Cox) test comparing CAR T treated mice from (E). Statistical analysis for (B) was done by one-way ANOVA. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; ns, not significant.
These results suggest that the effects of simultaneous cell-intrinsic downregulation of two ICRs vary depending on the combinations, with some being detrimental. Notably, dual downregulation of PD-1/TIGIT enhances the in vivo antitumor activity of CD19-targeting CAR T cells compared to single downregulation of PD-1.
Downregulation of PD-1 and TIGIT distinctly affects the in vitro function of CAR T cells
To investigate the mechanistic basis of the cell-intrinsic PD-1/TIGIT dual downregulation effect, we generated CD19-targeting CAR T cells with single (19PBBz and 19TBBz) or dual (19PTBBz) downregulation of each receptor in parallel (Figure S11). Under repeated antigen exposure with gamma-irradiated Nalm-6-PD-L1-CD80 cells, 19PTBBz CAR T cells exhibited the greatest expansion after secondary stimulation, followed by 19PBBz and 19TBBz CAR T cells, which showed similarly improved expansion compared with 19GBBz CAR T cells, respectively (Figure 4A). Interestingly, the proliferative advantage of the 19PTBBz cells compared to 19PBBz was abrogated in the presence of CD226 blocking antibody (Figure 4B). To further confirm the functional importance of CD226 signaling, we knocked out the CD226 gene using CRISPR-Cas9, which yielded a mixture of CD226+ and CD226− 19PTBBz CAR T cells (Figures S12A and S12B). Indeed, after the second stimulation of the CAR T mixture, intracellular immunostaining analysis revealed a reduced production of IL-2 in the CD226− population compared with the CD226+ population (Figures S12C and S12D). Notably, we found that activated CAR T cells also express high levels of CD112 and CD155 (Figure S13), the ligands for CD226 and TIGIT, which can potentially contribute to costimulatory CD226 signaling in cis or in trans between adjacent CAR T cells.45,46
Figure 4.

Downregulation of PD-1 and TIGIT distinctly affects the in vitro function of CAR T cells
(A) 1 × 106 CAR T cells with single or dual downregulation were incubated with 3 × 106 gamma-irradiated Nalm-6-GL-PD-L1-CD80 cells every 6 days (first and second stimulation) and counted on day 6 after each respective stimulation. Data are the pooled mean ± SD from two independent experiments performed in triplicates. (B) 1 × 106 first-stimulated day 6 mice (19PBBz or 19PTBBz) were incubated with 3 × 106 gamma-irradiated Nalm-6-GL-PD-L1-CD80 cells with or without 10 μg/mL CD226 blockade antibody for 6 days and counted. Data are the mean ± SD from one experiment performed in triplicates. (C) The expression of CD45RO and CCR7 was measured to distinguish the differentiation state of CD4+ and CD8+ 19GBBz, 19PBBz, 19TBBz, or 19PTBBz cells on day 16 (10 days after the second stimulation). Data are the mean ± SD from 3 donors. TCM, CD45RO+CCR7+; TEM, CD45RO+CCR7−. (D) IncuCyte-based cytotoxicity kinetics of first stimulation CAR T cells against Nalm-6-GL-PD-L1 cells on day 6 at the indicated ratios. Data are the representative mean ± SD from two independent experiments performed in triplicates. Filled black dots represent co-culture with untransduced T cells. Statistical analysis for (A)–(C) was done by one-way ANOVA and for (D) by an unpaired two-tailed t test. ∗∗p < 0.01, ∗∗∗p < 0.0001, ∗∗∗∗p < 0.0001; ns, not significant.
Next, we analyzed the composition of memory subsets of each CAR T construct based on the expression of C-C motif chemokine receptor 7 (CCR7) and CD45RO. Although most freshly prepared CAR T cells have a central memory (TCM) phenotype, both CD4+ and CD8+ 19GBBz CAR T cells dramatically differentiated into the effector memory (TEM) phenotype upon repeated in vitro stimulation (Figure S14).47,48 Interestingly, the degree of differentiation was lower in the two TIGIT-downregulated CAR T cells, 19PTBBz and 19TBBz, compared with 19GBBz and 19PBBz, resulting in a higher TCM/TEM ratio in both CD4+ and CD8+ populations (Figure 4C; Figure S15).
We next compared the cytotoxic activity of each CAR T construct against Nalm-6-PD-L1 cells at various effector-to-target (E:T) ratios. In contrast to the differentiation pattern observed above, 19PTBBz and 19PBBz demonstrated similarly higher killing efficiency compared with 19TBBz and 19GBBz at a low E:T ratio (0.1:1), suggesting that PD-1 signaling, but not TIGIT, has a major impact on the short-term cytotoxicity of CAR T cells (Figure 4D). Overall, the results from our in vitro comparisons suggest that inhibitory PD-1 and TIGIT signaling have non-redundant effects on the differentiation, proliferation, and effector functions of CAR T cells; thus, simultaneous downregulation of these two ICRs may synergistically benefit their antitumor activity.
Downregulation of PD-1 and TIGIT distinctly reprograms the transcriptomic profiles of CAR T cells
To better understand the functional characteristics of ICR downregulation, we performed bulk RNA sequencing (RNA-seq) on CAR T cells generated from two independent donors. First, we analyzed 19GBBz CAR T cells harvested after repeated stimulation and found significant changes in their transcriptomic profiles (Figure S16A–S16C). Specifically, the expression of genes encoding inhibitory receptors (PD-1, TIGIT, LAG-3, TIM-3, and CD244), exhaustion-related transcription factors (PRDM1, TOX2, EOMES, and EGR2/3), and chemokines (XCL1, CCL3, and CLL4) was significantly increased, whereas the expression of genes encoding naive/central memory-associated markers and transcription factors (BCL6, IL7R, TCF7, LEF1, SELL, CD27, and CCR7), alongside those encoding proteins associated with glucose metabolism (HK2, PFKFPB4, PDK1, GYS1, and GBE1), was decreased (Figure S16D). A gene set enrichment analysis (GSEA) also revealed that CAR T cells exhibited a more differentiated and exhausted state after repeated stimulation (Figure S16E).49,50
Next, we compared the gene expression profiles of 19GBBz, 19PBBz, 19TBBz, and 19PTBBz CAR T cells after second stimulation with Nalm-6-PD-L1 cells. A Pearson’s correlation analysis revealed significant similarity between the two TIGIT-downregulated CAR T cells 19TBBz and 19PTBBz relative to 19GBBz and 19PBBz, indicating that the high degree of transcriptomic reprogramming of CAR T cells is mainly regulated by TIGIT rather than PD-1 downregulation (Figure 5A). We then analyzed the hierarchically clustered heatmap of differentially expressed genes in each group of CAR T cells (Figure 5B). Notably, the PD-1-downregulated CAR T cells, 19PBBz and 19PTBBz, showed similar increases in the number of transcripts for effector and proliferation-related molecules (IL2, MKI67, FASLG, and TNFSF10). In contrast, transcriptional features related to the differentiation and exhaustion status of T cells were largely shared between 19TBBz and 19PTBBz CAR T cells but were clearly distinct from those of 19GBBz and 19PBBz CAR T cells. In general, 19TBBz and 19PTBBz CAR T cells exhibited decreased expression of inhibitory receptor (LAG3 and CD244) and chemokine genes (XCL1, XCL2, and CCL3) and higher expression of naive/central memory-phenotype (IL7R, BCL6, and CD27) and active glucose metabolism genes (HK2, PFKFB4, and PDK1), indicating a less differentiated/exhausted state (Figure 5C). GSEA also revealed the downregulation of exhaustion-related genes and the upregulation of naive/memory-related genes in 19PTBBz CAR T cells compared with 19PBBz (Figure 5D). These RNA-seq results are consistent with our in vitro functional studies and further support our hypothesis that the downregulation of PD-1 enhances short-term effector function in 19PTBBz CAR T cells, while the downregulation of TIGIT is primarily responsible for maintaining a less differentiated/exhausted state.
Figure 5.

RNA-seq analysis uncovers the distinct roles of PD-1 and TIGIT downregulation
Second-stimulated 19GBBz, 19PBBz, 19TBBz, and 19PTBBz cells were prepared as shown in Figure S16 (A) for RNA-seq. (A) Pearson’s correlation analysis of the transcriptomic profiles and (B) hierarchical clustering of differentially expressed genes from second-stimulated 19GBBz, 19PBBz, 19TBBz, and 19PTBBz cells derived from two donors (FDR q ≤ 0.1). (C) Heatmap of selected genes associated with T cell function in second-stimulated 19GBBz, 19PBBz, 19TBBz, and 19PTBBz cells. Asterisks represent the statistical significance as measured by q value. (D) Normalized enrichment scores (NESs) of significantly enriched gene sets associated with phenotypic and functional T cell signatures in second-stimulated 19GBBz, 19PBBz, 19TBBz, and 19PTBBz cells as determined by GSEA analysis. For all gene sets, FDR q ≤ 0.03 unless otherwise indicated. ∗q < 0.05, ∗∗q < 0.01, ∗∗∗q < 0.001, ∗∗∗∗q < 0.0001.
In vivo efficacy of patient-derived, clinical-grade PD-1/TIGIT-downregulated CAR T cells
To assess the feasibility of the clinical translation of PD-1/TIGIT dual-downregulated CD19-targeting CAR T cells, we established a large-scale CAR T manufacturing protocol using a semi-automated closed system (Figure S17A). In this protocol, we used the modified lentiviral vectors 19PBBz-nt and 19PTBBz-nt in which the ΔLNGFR tag, which was inserted for purification of CAR T, was removed from the original constructs. For comparison, we also generated 19BBz-nt without an shRNA cassette (Figure S17B). Consistent with the results obtained above, healthy donor-derived 19PTBBz-nt CAR T cells outperformed 19BBz-nt and 19PBBz-nt in our Nalm-6-PD-L1 leukemia model (Figure S17C). Next, following the same protocol, we manufactured 19BBz-nt and 19PTBBz-nt CAR T from diffuse large B cell lymphoma (DLBCL) patient-derived T cells (Figure S18). After 5 days of ex vivo culture, CAR T cells were successfully manufactured with a similar transduction efficiency (26.8% ± 11.44% for 19PTBBz-nt and 32.3% ± 11.07% for 19BBz-nt, respectively; Figure S18A), CD4/CD8 ratio, and differentiation profiles (Figures S18B and S18C). Cell growth was also comparable as determined by absolute cell count and glucose consumption (Figures S18D and S18E). However, robust downregulation of PD-1 and TIGIT was observed only in the CAR+ population of the 19PTBBz-nt group (Figure S18F). Both groups showed persistent Nalm-6-PD-L1 tumor clearance in all mice treated with a dose of 1 × 106 CAR T cells. However, mice treated with 19BBz-nt experienced tumor relapse at a dose of 0.5 × 106 CAR T cells and almost lost tumor control at a dose 0.25x106. In contrast, 19PTBBz-nt CAR T cells effectively suppressed tumor relapse even at a dose of 0.25x106 CAR T cells (Figures 6A and 6B). Mouse survival was also significantly improved by treatment with 19PTBBz-nt CAR T cells compared to 19BBz-nt at a dose of 0.25 × 106 CAR T cells (Figure 6C), a finding that may be explained by the difference in the persistence (Figure 6D) of CAR T cells in vivo at a dose of 0.5 × 106. Consistent with our in vitro RNA-seq data, 19PTBBz-nt cells harvested from mice on day 14 were less differentiated than 19BBz-nt cells, as determined by the ratio between TCM and TEM populations (Figure 6E). We also analyzed the ex vivo functionality of CAR T cells isolated on day 43 and found superior cytotoxicity of 19PTBBz-nt cells compared to 19BBz-nt cells (Figure 6F). Interestingly, although 19PTBBz-nt cells demonstrated superior proliferation and persistent activity in vivo, the serum levels of granulocyte-macrophage colony-stimulating factor (GM-CSF) at the early time point (3 days after CAR T infusion) were comparable to that of 19BBz-nt cells (Figure 6G), alleviating potential concerns about adverse side effects, such as cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS) associated with CAR T therapy.51,52
Figure 6.

Clinical-scale manufactured CAR T cells with PD-1/TIGIT downregulation showed a superior in vivo functionality against leukemia and lymphoma tumor models
(A and B) NSG mice were injected intravenously with 1 × 106 Nalm-6-GL-PD-L1 cells. 5 days after tumor growth, mock, 19BBz-nt, and 19PTBBz-nt cells were intravenously injected at the indicated doses. Tumor burden was monitored based on the bioluminescence intensity from the IVIS imaging system. Data are from n = 4 mock mice and n = 6 mice for all CAR T cell treatment groups. (C) Kaplan-Meier survival analysis with log-rank test comparing the CAR T cell-treated mice at 0.25 × 106 dose from (A) and (B). (D) The number of CAR T cells in the blood and spleen was determined 43 days after CAR T cell injection at a 0.5 × 106 dose. Data are the mean ± SD from six mice per group. (E) Differentiation state of T cells in the spleen of mice 14 days after 0.5 × 106 CAR T infusion. The differentiation state of CD4+ and CD8+ CAR T cells was determined by flow cytometry for CCR7 and CD45RA expression. Data are the mean ± SD from 19BBz-nt-treated mice (n = 6) and 19PTBBz-nt-treated mice (n = 6). (F) IncuCyte-based cytotoxicity kinetics of CAR T cells in the spleen of mice 43 days after CAR T cell injection against Nalm-6-GL-PD-L1 cells at a 1:3 E:T ratio. Data are the mean ± SD from two mice per group performed in duplicate. (G) NSG mice were injected intravenously with 1 × 106 Nalm-6-GL-PD-L1 leukemia cells. 5 days later, 1 × 106 19BBz-nt or 19PTBBz-nt cells were injected intravenously. 3 days after CAR T cell infusion, a plasma was obtained from blood and used to measure GM-CSF levels. Data are from n = 2 (mock) and n = 4 (19BBz-nt and 19PTBBz) mice. Data are mean ± SD from each group. (H and I) NOG mice were injected subcutaneously on the right flank with 5 × 106 Raji-GL-PD-L1 lymphoma cells. When the mean tumor volume reached approximately 100 mm3, CAR T cells were injected intravenously at the indicated doses. Tumor burden was monitored based on the bioluminescence intensity from the IVIS imaging system. Statistical analysis for (D), (E), and (F) by an unpaired two-tailed t test and for (G) by a one-way ANOVA. ∗∗p < 0.01, ∗∗∗p < 0.0001, ∗∗∗∗p < 0.0001; ns, not significant.
Lastly, we evaluated the in vivo efficacy of 19PTBBz-nt and 19BBz-nt in a subcutaneous Raji-PD-L1 lymphoma model and confirmed the improved antitumor activity of 19PTBBz-nt CAR T cells over 19BBz-nt across different doses (4 × 106, 2 × 106, and 1 × 106 CAR T cells; Figures 6H and 6I; Figure S19). Collectively, these results show that our cell-intrinsic PD-1/TIGIT dual-downregulation strategy, which is readily applicable to a clinical-grade CAR T manufacturing process, may provide an effective approach for enhancing the in vivo efficacy of CAR T cells by averting immune checkpoint-mediated dysfunction.
Discussion
Because inhibitory checkpoint signaling is involved in the suboptimal efficacy of CAR T cells, we applied an shRNA-based gene-silencing approach to achieve sustained downregulation of checkpoint receptors. While CRISPR-Cas9-mediated gene knockout could alternatively be used,53 in a mouse model of chronic viral infection, the genetic absence of PD-1 in antigen-specific CD8 T cells significantly reduces long-term T cell survival, likely owing to chronic overstimulation.54 Similarly, complete deficiency in thymocyte selection-associated high mobility group box (TOX) or eomesodermin (EOMES) results in a more rapid decline of antigen-specific T cells in mouse models of chronic viral infection and cancer.55, 56, 57 However, deletion of only one allele of TOX or EOMES was shown to rescue the persistence of antigen-specific T cells, resulting in superior disease control compared with wild-type T cells.56,57 Furthermore, homozygous or heterozygous deletion of PD-1 failed to suppress the malignant transformation of T cells in mouse lymphoma models, suggesting a risk in using PD-1 knockout T cells for therapeutic purposes.58 Thus, partial inhibition of exhaustion-associated factors in engineered T cells may not only be sufficient, but also beneficial for optimal therapeutic outcomes. In addition, our approach to integrating shRNA cassettes into lentiviral vectors is readily applicable to currently established protocols for commercial CAR T manufacturing.
We showed that cell-intrinsic PD-1 downregulation endowed 41BB-based CD19-targeting CAR T cells with resistance to the inhibitory PD-1/PDL-1 signaling axis, and the resulting 41BB-based CAR T cells exerted more persistent in vitro and in vivo activity compared with their CD28-based counterpart. Interestingly, this is consistent with the results from a comparison of conventional 41BB- and CD28-based CAR T cells, wherein the CD28 domain within the CAR induces more robust signaling than the 41BB domain, leading to early T cell exhaustion.59, 60, 61 Indeed, in our comparison of cytotoxicity between 19PBBz versus 19P28z, a difference was observed only in the restimulation setting but not in the freshly prepared CAR T samples.
Next, to assess the effects of dual ICR downregulation, we compared the combination of PD-1 with TIM-3, LAG-3, TIGIT, and CTLA-4, which have been demonstrated to have synergistic effects in several preclinical models and/or clinical trials as antibody-based blockade targets when combined with anti-PD-1 antibodies.20,23,24,62, 63, 64 However, among the four different combinations, only PD-1/TIGIT enhanced the antitumor activity of CD19-targeting CAR T cells compared with single downregulation of PD-1. We cannot currently rule out the possibility that these results are attributable to limitations in the mouse xenograft model used. Although we confirmed that our Nalm-6-GL-PD-L1 cell line expresses galectin-9, HLA-DR, and CD112, which are the respective ligands for TIM-3, LAG-3, and TIGIT, B7 molecules (CD80 and CD86), the ligand for CTLA-4, was not detected. Thus, although endogenous B7 molecules expressed on mouse antigen-presenting cells (APCs) can cross-react with human CTLA-4,65,66 it is possible that using different tumor models expressing human B7 molecules may lead to different outcomes with respect to the combination of PD-1 and CTLA-4 downregulation.
Nonetheless, it is also plausible that our results reflect a biological feature of CAR T cells that distinguishes them from endogenous tumor-reactive T cells. For example, anti-CTLA-4 antibodies are known to exert their effects primarily by rescuing the costimulatory CD28/B7 axis between naive T cells and activated dendritic cells (DCs) within the priming site,16 which may not be relevant for CAR T cells with a 41BB costimulatory domain that has undergone ex vivo stimulation by anti-CD3 and anti-CD28 antibodies. Similarly, HLA-DR, the ligand for LAG-3, is also highly expressed on APCs, and it has been shown that the therapeutic effect of LAG-3 blockade is mediated by enhancing T cell activation by these cells,63 which may also not be relevant for CAR T cells.
Our results may further be explained by mechanistic differences between our cell-intrinsic downregulation approach and systemic antibody treatment. A recent clinical study showed that combination therapy using anti-PD-1 and anti-CTLA-4 antibodies induced different therapeutic effects depending on the treatment sequence.23 It is therefore conceivable that each antibody used in the combination therapy exerts a synergistic effect by engaging distinct T cell populations undergoing different activation and differentiation stages, rather than engaging the same T cells simultaneously.
In vitro functional assays and RNA-seq analysis revealed that downregulation of PD-1 enhances effector function whereas downregulation of TIGIT is primarily responsible for the acquisition of a less differentiated/exhausted phenotype. There have been several reports that inhibition of PD-1 signaling enhances the effector function of T cells.16,26,28,34,67,68 Interestingly, in our experimental conditions, PD-1 downregulation did not affect the transcription of the soluble effectors IFN-γ, granzyme B, and perforin 1, but did promote the transcription of the membrane-bound death ligands TNF superfamily member 10 (TNFSF10) and Fas ligand (FASLG). It was recently reported that genetic disruption of TNFSF10 or FASLG in CAR T cells significantly impairs cytotoxic activity and induces the progressive dysfunction of CAR T cells.69
In the case of TIGIT, its inhibitory effects can be mediated through its intracellular ITIM domain or through competition with the costimulatory receptor CD226.46,70 It also has been reported that TIGIT regulates immune responses by modulating DCs and regulatory T cells (Tregs).70,71 In addition, a recent study showed that treatment with an agonistic antibody targeting glucocorticoid-induced tumor necrosis factor receptor-related protein (GITR) in combination with a PD-1-blocking antibody generated early memory CD8 T cells with high proliferative capacity, resulting in a synergistic regression of tumors dependent on CD226 signaling.72 This finding is in line with our results that highlights the role of CD226 in TIGIT blockade73 and strongly suggests that one of its principal mechanisms of actions can be the modulation of the differentiation and memory status of tumor-reactive T cells.
In recent clinical trials, combination therapy using anti-PD-L1 and anti-TIGIT blocking antibodies has shown promising results in terms of both efficacy and safety, raising expectations for this particular combination of checkpoint receptors. To the best of our knowledge, our study is the first to report that downregulation of these two ICRs in a cell-intrinsic fashion synergistically enhances the antitumor activity of CAR T cells. Our study also provides a mechanistic rationale for the synergy, laying the groundwork for more rigorous future single cell-level analyses and confirmation through alternative blockade strategies such as knockout or antibody treatment. Based on these promising preclinical results, phase 1/2 clinical trials of PD-1/TIGIT KD CD19 CAR T cells are in progress in adult patients with relapsed and refractory DLBCL (ClinicalTrials.gov: NCT04836507). During these studies, it will be important to carefully monitor the potential for increased risk of adverse events such as CRS and ICANS. Moving forward, whether this combination can be applied to other engineered T cell platforms, such as TCR T therapy, as well as to CAR T cells targeting solid tumor antigens, is a subject of considerable interest that is currently under investigation in preclinical models.
Materials and methods
Plasmid construction
To construct the lentiviral transfer vector encoding the CD19-specific CAR, the anti-CD19 scFv (FMC63) was fused by overlapping PCR to the CD8α spacer and transmembrane domains, the 4-1BB (CD137) or CD28 costimulatory domains, and the CD3ζ signaling domain. shRNA expression cassettes were added upstream to the pLV-EF-1α-ΔLNGFR-P2A-CD19 CAR vector in the antisense direction under the RNA Pol III promoter (mU6, hU6, or hH1). For dual immune checkpoint inhibition, two shRNA-expressing modules, respectively controlled by the mU6 and hU6 promoters, were cloned-in facing each other upstream of the of the EF-1α promoter. Immune checkpoint targeting small interfering RNA (siRNA) or shRNA sequences are listed in Table S1.
Cell lines and culture conditions
Nalm-6-GL cells were kindly provided by R. Kochenderfer (National Cancer Institute). K562 cells were purchased from the American Type Culture Collection (ATCC). Raji-PD-L1 cells were purchased from InvivoGen (USA). K562 cells were engineered to express human CD19 by lentiviral transduction. Nalm-6-GL or K562-CD19 cells were transduced to express human PD-L1 (Sino Biological; HG10084-UT cDNA subcloned into a lentiviral vector) to generate Nalm-6-GL-PD-L1 and K562-CD19-PD-L1 cells. Raji-PD-L1 cells were transduced to express firefly luciferase-green fluorescent protein (ffluc/GFP) to generate Raji-GL-PD-L1 cells. Nalm-6-GL-PD-L1 cells were transduced with human CD80 (human CD80 open reading frame [ORF] referring to GenBank: NM_005191.4 was cloned to lentiviral vector) to generate Nalm-6-GL-PD-L1-CD80. Lenti-X 293T packaging cells were obtained from Takara Bio (Japan). Raji, K562, and Nalm-6 cell lines were cultured in RPMI 1640 medium (Gibco) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Gibco), 2 mM l-glutamine (Gibco), and 1% penicillin/streptomycin (Gibco) in a humidified incubator with a 5% CO2 atmosphere at 37°C. Lenti-X 293T cells were cultured Dulbecco’s modified Eagle’s medium (Gibco) supplemented with 10% heat-inactivated FBS, 2 mM l-glutamine, 0.1 mM non-essential amino acids (Gibco), 1 mM sodium pyruvate (Gibco), and 1% penicillin/streptomycin. Cell line authentication was performed by Korea Cell Line Bank based on criteria established by the International Cell Line Authentication Committee. None of the cell lines used in this research is included in the commonly misidentified cell lines registry. Mycoplasma contamination tests were conducted by the Laboratory Animal Resources Center of the Korea Research Institute of Bioscience and Biotechnology before use in in vivo experiments and results were negative.
Generation of human CAR T cells
Human peripheral blood mononuclear cells (PBMCs) were obtained from healthy adult donors at the Seoul National University Hospital (SNUH) using protocols approved by the Institutional Review Board (IRB no. H-1607-155-778). Lenti-X 293T cells were seeded at 6.5 × 106 cells per dish in poly-d-lysine-coated 100-mm dishes 3 days prior to transduction. After 24 h, cells were co-transfected with 7.5 μg of lentiviral transfer plasmid, 4.5 μg of pMDG.1 encoding a vesicular stomatitis virus glycoprotein G (VSV-G) envelope, 6 μg of pRSV-Rev encoding Rev, and 6 μg of pMDLg/pRRE encoding Gag/Pol using Lipofectamine 2000 according to the manufacturer’s instructions. Lentiviral supernatants were collected 40 h following transfection, removed of cell debris by centrifugation at 1,600 rpm for 5 min, and immediately used to transduce T cells. PBMCs were collected from whole blood samples of healthy donors using SepMate tubes (STEMCELL Technologies, Canada) in accordance with the manufacturer’s instructions. The PBMCs were stimulated with 4 μg/mL plate-bound anti-CD3 antibody (clone OKT3; Bio X Cell), 2 μg/mL soluble anti-CD28 antibody (clone CD28.2; Bio X Cell), and 300 IU/mL human recombinant IL-2 (BMI Korea, Republic of Korea) in complete T cell medium containing 90% RPMI 1640 supplemented with 10% heat-inactivated FBS, 0.1 mM non-essential amino acids, 2 mM GlutaMAX (Gibco, USA), 1 mM sodium pyruvate, and 0.05 mM 2-mercaptoethanol (Gibco, USA). Two days after stimulation, activated T cells were transduced with lentiviral supernatants and 10 μg/mL protamine sulfate followed by centrifugation at 1,000 × g for 90 min at 32°C and further incubated at 37°C. 24 h later, the lentiviral supernatants were removed and the transduced T cells were expanded in the conditions described above. Fresh medium was added on day 3. The percentage of transduced T cells was evaluated by ΔLNGFR expression on day 4 after transduction. ΔLNGFR-positive transduced T cells were isolated using a human CD271 MicroBead kit (catalog no. 130-099-023, Miltenyi Biotec) according to the manufacturer’s instructions. CAR+ ΔLNGFR+ T cells were maintained in complete T cell medium with 300 IU/mL recombinant human (rh)IL-2.
Flow cytometry
All flow cytometry experiments were performed with the LSRFortessa X-20 cytometer (BD Biosciences, USA) and analyzed with FlowJo software (Tree Star, USA). For cell surface staining, 2 × 105 cells were stained with antibodies resuspended in 100 μL of fluorescence-activated cell sorting (FACS) buffer (1% BSA in Dulbecco’s phosphate-buffered saline [DPBS]) for 20 min at 4°C in the dark. Cells were washed with 2 mL of FACS buffer, resuspended, and analyzed. For intracellular staining of CTLA-4, T cells were fixed/permeabilized using 200 μL of BD Cytofix/Cytoperm solution (BD Biosciences, USA) for 20 min at 4°C. Cells were washed with 2 mL of intracellular staining buffer (1% BSA, 0.1% sodium azide, and 0.1% saponin in DPBS) and then stained with antibodies. Intracellular staining of IL-2 followed the same protocol as that for CTLA-4 but included the addition of 1 μg/mL GolgiPlug (BD Biosciences) to the culture media 12 h prior to sample preparation. Viable cells were determined with Fixable Viability Dye eFluor 780 (Thermo Fisher Scientific, USA). The CD19-CAR moiety was labeled by Alexa Fluor 647 (AF647)-conjugated anti-mouse F(ab′)2 antibody (Jackson ImmunoResearch) or biotin-conjugated rhCD19-Fc (ACRO Biosystems, USA) with AF647-conjugated streptavidin (BioLegend, USA). ΔLNGFR was labeled with allophycocyanin (APC)-conjugated LNGFR antibody (clone ME20.4-1.H4, Miltenyi Biotec, Germany). All flow cytometry analyses were assessed using the following antibody clones: from BioLegend: CD4-Brilliant Violet (BV)605 (clone OKT4), CD8-APC or phycoerythrin (PE) (clone SK1), CCR7-PE (clone G034H7), CD45RO-PerCp (peridinin-chlorophyll-protein)-Cy5.5 (clone UCHL1), CTLA-4-PE (clone BNI3), LAG3-PE (clone 7H2C65), CD226-PE (clone 11A8), CD155-PE (clone SKII.4), CD112-PE (clone TX31), CD19-APC (clone HIB19), PD-L1-APC (clone 29E.2A3), CD80-PE (clone 2D10), HLA-DR-PE (clone L243), Galectin-9-PerCp-Cy5.5 (clone 9M1-3), and IL-2-PE (clone MQ1-17H12); from BD Biosciences: CCR7-BV421 (clone 150503) and CD45RA-fluorescein isothiocyanate (FITC) (clone HI100); from Thermo Fisher Scientific: PD-1-PE (clone J105) and TIGIT-PE (clone MBSA43); from R&D Systems: TIM3-PE (clone 344823).
Repeated antigen stimulation
For the first stimulation, 1 × 106 CAR T cells were co-cultured with gamma-irradiated Nalm-6-GL-PD-L1-CD80 or K562-CD19-PD-L1 at a 1:3 E:T ratio on day 0 for 6 days. For the second stimulation, CAR T cells were harvested and numerated with a Countess II automated cell counter before re-seeding (1 × 106 cells) with the same target cells at a 1:3 E:T ratio for another 6 days before further cell counting.
PD-1/PD-L1-dependent proliferation assay
To determine the impact of the PD-L1/PD-1 axis on their proliferation, 1 × 106 CAR T cells were co-cultured with gamma-irradiated K562-CD19 or K562-CD19-PD-L1 at a 1:3 E:T ratio for 7 days and counted with a Countess II automated cell counter (Thermo Fisher Scientific, USA).
CD226 blocking assay
To evaluate the contribution of CD226 signaling to the proliferative activity of 19PTBBz cells, day first-stimulated CAR T cells were co-cultured with gamma-irradiated target cells at a 1:3 E:T ratio in the presence of 10 μg/mL plate-bound human CD226 blocking monoclonal antibody (clone DX11, Abcam, UK). Ultra-LEAF purified mouse IgG1 (clone MOPC-21, BioLegend, USA) was used as for isotype control.
Quantitation of cytokine levels
3 × 105 CAR T cells were cultured with 9 × 105 gamma-irradiated target cells in 2 mL of complete T cell medium without rhIL-2 in 24-well plates. After 48 h of coculture, supernatants were collected.
A cytometric bead array (CBA) assay was performed to measure the IFN-γ level using a human Th1/Th2 cytokine kit II (BD Biosciences, USA). An ELISA assay was conducted to measure the GM-CSF level using a GM-CSF Quantikine ELISA kit (R&D Systems, USA) in the culture supernatant following the manufacturer’s instructions. Plasma was harvested from the peripheral blood of mice by centrifugation for 10 min at 1,500 × g at room temperature. Plasma was stored at −80°C until analysis with a GM-CSF Quantikine ELISA kit.
Cytotoxicity assays
To evaluate the cytotoxic activity of CAR T cells in the presence or absence of PD-L1, 1 × 105 Nalm-6-GL or Nalm-6-GL-PD-L1 cells were co-cultured with CAR T cells at a 1:1, 1:0.3, or 0.1:1 E:T ratio in 250 μL of complete T cell medium in 96-well flat-bottom plates for up to 72 h. Triplicate wells were used for each CAR T group. GFP fluorescence intensity was detected every 2 h using the IncuCyte S3 live-cell analysis system (Sartorius, Germany). Total integrated GFP intensity per well was used as a quantitative measure of viable target cells. Values of total integrated GFP intensity were normalized to the GFP intensity of the starting point.
In vivo xenograft models
All animal testing procedures described herein were approved by the Korea Advanced Institute of Science and Technology (KAIST) and Osong Medical Innovation Foundation Laboratory Animal Center Institutional Animal Care and Use Committee and were performed according to national and institutional guidelines. 6- to 8-week-old NSG mice were obtained from The Jackson Laboratory and bred in the KAIST Laboratory Animal Resource Center under a protocol approved by the KAIST Institutional Animal Care and Use Committee. 6- to 8-week-old female NOG mice were obtained from Koatech (Republic of Korea). 6- to 8-week-old NSG mice were administered with 1 × 106 Nalm-6-GL or Nalm-6-GL-PD-L1 cells by tail vein injection. At 5 days post-injection, the indicated dose of CAR T cells was injected intravenously. Tumor burden was measured after intraperitoneal injection of luciferin (PerkinElmer) per the manufacturer’s instructions and subsequent bioluminescence imaging using the IVIS Lumina X5 imaging system. Bioluminescence values were analyzed using the Living Image software (PerkinElmer). Tumor growth was measured weekly. To evaluate the in vivo efficacy of CAR T cells against a subcutaneous tumor model, 5 × 106 Raji-GL-PD-L1 lymphoma cells were subcutaneously injected into the right flank of NOG mice. When the mean tumor volume reached approximately 100 mm3 (measured with a caliper), CAR T cells were intravenously injected into the mice at the indicated doses. Tumor growth was determined weekly by bioluminescence imaging. Absolute CAR T cell counts in the peripheral blood of mice were determined using CountBright absolute counting beads (Thermo Fisher Scientific, USA) according to the manufacturer’s instructions. Splenocytes were obtained by physical dissociation of spleen on a cell strainer. Splenocytes were pelleted and red blood cells were lysed with the addition of red blood cell lysis solution (Miltenyi Biotec, 130-094-183), subsequently washed, filtered on a cell strainer, and collected in a tube. Splenocytes were resuspended in FACS buffer with Fc blocking reagent; antibodies were added and washed off after the incubation time.
Bulk RNA-seq
First-stimulated CAR T cells (2 × 107) were prepared by culturing over gamma-irradiated Nalm-6-PD-L1 cells for 48 h prior to LNGFR isolation and freezing for cDNA preparation. Second-stimulated CAR T cells were prepared by culturing over gamma-irradiated Nalm-6-PD-L1-CD80 cells for 6 days followed by harvesting and seeding 2 × 107 cells over gamma-irradiated Nalm-6-PD-L1 cells for 48 h, LNGFR isolation, and freezing for cDNA preparation. Total RNA was isolated using the NucleoSpin RNA XS kit (Macherey-Nagel, Germany) according to the manufacturer’s instructions. RNA quality was determined with the Agilent 4200 TapeStation (RNA integrity number [RIN] value >9). RNA-seq libraries were constructed using the Illumina TruSeq stranded mRNA LP kit. Briefly, mRNA was purified by oligo(dT) beads and fragmented through enzymatic reaction. After fragmentation, cDNA was generated through reverse transcription. The cDNA libraries were constructed and followed by the 100-bp paired-end mode on the DNBSEQ-400 platform. External RNA Controls Consortium (ERCC) RNA spike-in mixes (Thermo Fisher Scientific, 4456740) were included for quality assurance. Libraries were sequenced on a MGISEQ-2000. The quality of all paired-end reads was analyzed using FastQC software, and the per base sequence quality of all sample was above Q30. RNA-seq analysis was carried out via the Galaxy platform (https://usegalaxy.org/). FASTQ data were mapped to the reference genome hg19 using HISAT2 v2.1.0 software. The number of reads per annotated gene was then computed from the mapped reads using the featureCounts v1.6.4 software. The R package limma with voom method v3.38.3 was used to normalize all datasets and analyze differential expression between groups. Pearson’s correlation matrices and hierarchical clustering plots of differentially expressed genes (false discovery rate [FDR] < 0.1) were generated with the Instant Clue software. The heatmap of selected genes associated with T cell function was generated using GraphPad Prism 8. Each row represents the Z score of normalized gene expression values for the selected genes. GSEA was performed using a pre-ranked file generated by t-statistic on curated gene sets from the Broad Institute Molecular Signature Database. The RNA-seq data have been deposited in the Gene Expression Omnibus (GEO) and are available under the accession no. GEO: GSE158676.
Statistical analysis
Statistical analyses were determined by a Student’s t test (two-tailed, unpaired), one-way ANOVA, or two-way ANOVA. Percent survival was estimated using the Kaplan-Meier method, and statistical significance was calculated by the log-rank test. All statistical analyses were conducted using GraphPad Prism 8 (GraphPad). For all analyses, a p value <0.05 was considered statistically significant.
Acknowledgments
This work was supported by Curocell Inc., the Osong Medical Innovation Foundation funded by the Ministry of Health & Welfare (2016M3A9D9945471, HO18C0005), the National Research Foundation of Korea (NRF) (2017R1A6A3A11031455), the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), the Ministry of Health & Welfare (HI20C0043), and by the KAIST Global Centre for Open Research with Enterprise (GCORE) grant funded from the Ministry of Science and ICT (N11190028).
Author contributions
C.H.K., Y.-H.L., and H.C.K. designed the experiments and wrote the manuscript. Y.-H.L., H.J.L., Y.L., S.K.N., J.S.Y.M., X.W., and O.S. performed the experiments and analyzed the data. C.H. analyzed the data and wrote the manuscript. W.S.K., S.J.K., Y.K., and I.J. designed the experiments and analyzed the data.
Declaration of interests
C.H.K., Y.-H.L., H.J.L., and Y.L. are inventors on a patent that was filed based on these works. C.H.K. is a co-founder of and holds equity in Curocell Inc. Y.K. is a consultant at Curocell Inc. H.C.K., Y.-H.L., and H.J.L. are employees at Curocell Inc. The remaining authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.10.004.
Contributor Information
Young-Ho Lee, Email: yhlee@curocellbtx.com.
Chan Hyuk Kim, Email: kimchanhyuk@kaist.ac.kr.
Supplemental information
References
- 1.Guedan S., Ruella M., June C.H. Emerging cellular therapies for cancer. Annu. Rev. Immunol. 2019;37:145–171. doi: 10.1146/annurev-immunol-042718-041407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maude S.L., Laetsch T.W., Buechner J., Rives S., Boyer M., Bittencourt H., Bader P., Verneris M.R., Stefanski H.E., Myers G.D., et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 2018;378:439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park J.H., Rivière I., Gonen M., Wang X., Sénéchal B., Curran K.J., Sauter C., Wang Y., Santomasso B., Mead E., et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N. Engl. J. Med. 2018;378:449–459. doi: 10.1056/NEJMoa1709919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schuster S.J., Bishop M.R., Tam C.S., Waller E.K., Borchmann P., McGuirk J.P., Jäger U., Jaglowski S., Andreadis C., Westin J.R., et al. JULIET Investigators Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med. 2019;380:45–56. doi: 10.1056/NEJMoa1804980. [DOI] [PubMed] [Google Scholar]
- 5.Locke F.L., Ghobadi A., Jacobson C.A., Miklos D.B., Lekakis L.J., Oluwole O.O., Lin Y., Braunschweig I., Hill B.T., Timmerman J.M., et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019;20:31–42. doi: 10.1016/S1470-2045(18)30864-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grupp S.A., Kalos M., Barrett D., Aplenc R., Porter D.L., Rheingold S.R., Teachey D.T., Chew A., Hauck B., Wright J.F., et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cappell K.M., Sherry R.M., Yang J.C., Goff S.L., Vanasse D.A., McIntyre L., Rosenberg S.A., Kochenderfer J.N. Long-term follow-up of anti-CD19 chimeric antigen receptor T-cell therapy. J. Clin. Oncol. 2020;38:3805–3815. doi: 10.1200/JCO.20.01467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Porter D.L., Hwang W.T., Frey N.V., Lacey S.F., Shaw P.A., Loren A.W., Bagg A., Marcucci K.T., Shen A., Gonzalez V., et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015;7:303ra139. doi: 10.1126/scitranslmed.aac5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Majzner R.G., Mackall C.L. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 2018;8:1219–1226. doi: 10.1158/2159-8290.CD-18-0442. [DOI] [PubMed] [Google Scholar]
- 10.Mueller K.T., Maude S.L., Porter D.L., Frey N., Wood P., Han X., Waldron E., Chakraborty A., Awasthi R., Levine B.L., et al. Cellular kinetics of CTL019 in relapsed/refractory B-cell acute lymphoblastic leukemia and chronic lymphocytic leukemia. Blood. 2017;130:2317–2325. doi: 10.1182/blood-2017-06-786129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maude S.L., Frey N., Shaw P.A., Aplenc R., Barrett D.M., Bunin N.J., Chew A., Gonzalez V.E., Zheng Z., Lacey S.F., et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fraietta J.A., Lacey S.F., Orlando E.J., Pruteanu-Malinici I., Gohil M., Lundh S., Boesteanu A.C., Wang Y., O’Connor R.S., Hwang W.T., et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018;24:563–571. doi: 10.1038/s41591-018-0010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pardoll D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keir M.E., Butte M.J., Freeman G.J., Sharpe A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hashimoto M., Kamphorst A.O., Im S.J., Kissick H.T., Pillai R.N., Ramalingam S.S., Araki K., Ahmed R. CD8 T cell exhaustion in chronic infection and cancer: Opportunities for interventions. Annu. Rev. Med. 2018;69:301–318. doi: 10.1146/annurev-med-012017-043208. [DOI] [PubMed] [Google Scholar]
- 16.Wei S.C., Duffy C.R., Allison J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8:1069–1086. doi: 10.1158/2159-8290.CD-18-0367. [DOI] [PubMed] [Google Scholar]
- 17.Riley J.L. PD-1 signaling in primary T cells. Immunol. Rev. 2009;229:114–125. doi: 10.1111/j.1600-065X.2009.00767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Waldman A.D., Fritz J.M., Lenardo M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020;20:651–668. doi: 10.1038/s41577-020-0306-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gide T.N., Wilmott J.S., Scolyer R.A., Long G.V. Primary and acquired resistance to immune checkpoint inhibitors in metastatic melanoma. Clin. Cancer Res. 2018;24:1260–1270. doi: 10.1158/1078-0432.CCR-17-2267. [DOI] [PubMed] [Google Scholar]
- 20.Koyama S., Akbay E.A., Li Y.Y., Herter-Sprie G.S., Buczkowski K.A., Richards W.G., Gandhi L., Redig A.J., Rodig S.J., Asahina H., et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016;7:10501. doi: 10.1038/ncomms10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Curran M.A., Montalvo W., Yagita H., Allison J.P. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. USA. 2010;107:4275–4280. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Valsecchi M.E. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 2015;373:1270. doi: 10.1056/NEJMc1509660. [DOI] [PubMed] [Google Scholar]
- 23.Weber J.S., Gibney G., Sullivan R.J., Sosman J.A., Slingluff C.L., Jr., Lawrence D.P., Logan T.F., Schuchter L.M., Nair S., Fecher L., et al. Sequential administration of nivolumab and ipilimumab with a planned switch in patients with advanced melanoma (CheckMate 064): An open-label, randomised, phase 2 trial. Lancet Oncol. 2016;17:943–955. doi: 10.1016/S1470-2045(16)30126-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Woo S.R., Turnis M.E., Goldberg M.V., Bankoti J., Selby M., Nirschl C.J., Bettini M.L., Gravano D.M., Vogel P., Liu C.L., et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72:917–927. doi: 10.1158/0008-5472.CAN-11-1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fourcade J., Sun Z., Pagliano O., Chauvin J.M., Sander C., Janjic B., Tarhini A.A., Tawbi H.A., Kirkwood J.M., Moschos S., et al. PD-1 and Tim-3 regulate the expansion of tumor antigen-specific CD8+ T cells induced by melanoma vaccines. Cancer Res. 2014;74:1045–1055. doi: 10.1158/0008-5472.CAN-13-2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.John L.B., Devaud C., Duong C.P., Yong C.S., Beavis P.A., Haynes N.M., Chow M.T., Smyth M.J., Kershaw M.H., Darcy P.K. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin. Cancer Res. 2013;19:5636–5646. doi: 10.1158/1078-0432.CCR-13-0458. [DOI] [PubMed] [Google Scholar]
- 27.Cherkassky L., Morello A., Villena-Vargas J., Feng Y., Dimitrov D.S., Jones D.R., Sadelain M., Adusumilli P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Invest. 2016;126:3130–3144. doi: 10.1172/JCI83092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chong E.A., Melenhorst J.J., Lacey S.F., Ambrose D.E., Gonzalez V., Levine B.L., June C.H., Schuster S.J. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: Refueling the CAR. Blood. 2017;129:1039–1041. doi: 10.1182/blood-2016-09-738245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rafiq S., Yeku O.O., Jackson H.J., Purdon T.J., van Leeuwen D.G., Drakes D.J., Song M., Miele M.M., Li Z., Wang P., et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018;36:847–856. doi: 10.1038/nbt.4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kloss C.C., Lee J., Zhang A., Chen F., Melenhorst J.J., Lacey S.F., Maus M.V., Fraietta J.A., Zhao Y., June C.H. Dominant-negative TGF-β receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol. Ther. 2018;26:1855–1866. doi: 10.1016/j.ymthe.2018.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu X., Ranganathan R., Jiang S., Fang C., Sun J., Kim S., Newick K., Lo A., June C.H., Zhao Y., Moon E.K. A chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR T cells in advanced solid tumors. Cancer Res. 2016;76:1578–1590. doi: 10.1158/0008-5472.CAN-15-2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shin J.H., Park H.B., Oh Y.M., Lim D.P., Lee J.E., Seo H.H., Lee S.J., Eom H.S., Kim I.H., Lee S.H., Choi K. Positive conversion of negative signaling of CTLA4 potentiates antitumor efficacy of adoptive T-cell therapy in murine tumor models. Blood. 2012;119:5678–5687. doi: 10.1182/blood-2011-09-380519. [DOI] [PubMed] [Google Scholar]
- 33.Hoogi S., Eisenberg V., Mayer S., Shamul A., Barliya T., Cohen C.J. A TIGIT-based chimeric co-stimulatory switch receptor improves T-cell anti-tumor function. J. Immunother. Cancer. 2019;7:243. doi: 10.1186/s40425-019-0721-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rupp L.J., Schumann K., Roybal K.T., Gate R.E., Ye C.J., Lim W.A., Marson A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep. 2017;7:737. doi: 10.1038/s41598-017-00462-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu W., Zi Z., Jin Y., Li G., Shao K., Cai Q., Ma X., Wei F. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol. Immunother. 2019;68:365–377. doi: 10.1007/s00262-018-2281-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ren J., Zhang X., Liu X., Fang C., Jiang S., June C.H., Zhao Y. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget. 2017;8:17002–17011. doi: 10.18632/oncotarget.15218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choi B.D., Yu X., Castano A.P., Darr H., Henderson D.B., Bouffard A.A., Larson R.C., Scarfò I., Bailey S.R., Gerhard G.M., et al. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J. Immunother. Cancer. 2019;7:304. doi: 10.1186/s40425-019-0806-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Y., Zhang X., Cheng C., Mu W., Liu X., Li N., Wei X., Liu X., Xia C., Wang H. CRISPR-Cas9 mediated LAG-3 disruption in CAR-T cells. Front. Med. 2017;11:554–562. doi: 10.1007/s11684-017-0543-6. [DOI] [PubMed] [Google Scholar]
- 39.Wei J., Luo C., Wang Y., Guo Y., Dai H., Tong C., Ti D., Wu Z., Han W. PD-1 silencing impairs the anti-tumor function of chimeric antigen receptor modified T cells by inhibiting proliferation activity. J. Immunother. Cancer. 2019;7:209. doi: 10.1186/s40425-019-0685-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zou F., Lu L., Liu J., Xia B., Zhang W., Hu Q., Liu W., Zhang Y., Lin Y., Jing S., et al. Engineered triple inhibitory receptor resistance improves anti-tumor CAR-T cell performance via CD56. Nat. Commun. 2019;10:4109. doi: 10.1038/s41467-019-11893-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.An D.S., Qin F.X., Auyeung V.C., Mao S.H., Kung S.K., Baltimore D., Chen I.S. Optimization and functional effects of stable short hairpin RNA expression in primary human lymphocytes via lentiviral vectors. Mol. Ther. 2006;14:494–504. doi: 10.1016/j.ymthe.2006.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hui E., Cheung J., Zhu J., Su X., Taylor M.J., Wallweber H.A., Sasmal D.K., Huang J., Kim J.M., Mellman I., Vale R.D. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. 2017;355:1428–1433. doi: 10.1126/science.aaf1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Josefsson S.E., Beiske K., Blaker Y.N., Førsund M.S., Holte H., Østenstad B., Kimby E., Köksal H., Wälchli S., Bai B., et al. TIGIT and PD-1 mark intratumoral T cells with reduced effector function in B-cell non-Hodgkin lymphoma. Cancer Immunol. Res. 2019;7:355–362. doi: 10.1158/2326-6066.CIR-18-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang R.-Y., Francois A., McGray A.R., Miliotto A., Odunsi K. Compensatory upregulation of PD-1, LAG-3, and CTLA-4 limits the efficacy of single-agent checkpoint blockade in metastatic ovarian cancer. OncoImmunology. 2016;6:e1249561. doi: 10.1080/2162402X.2016.1249561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blake S.J., Dougall W.C., Miles J.J., Teng M.W., Smyth M.J. Molecular pathways: Targeting CD96 and TIGIT for cancer immunotherapy. Clin. Cancer Res. 2016;22:5183–5188. doi: 10.1158/1078-0432.CCR-16-0933. [DOI] [PubMed] [Google Scholar]
- 46.Lozano E., Dominguez-Villar M., Kuchroo V., Hafler D.A. The TIGIT/CD226 axis regulates human T cell function. J. Immunol. 2012;188:3869–3875. doi: 10.4049/jimmunol.1103627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gattinoni L., Lugli E., Ji Y., Pos Z., Paulos C.M., Quigley M.F., Almeida J.R., Gostick E., Yu Z., Carpenito C., et al. A human memory T cell subset with stem cell-like properties. Nat. Med. 2011;17:1290–1297. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sommermeyer D., Hudecek M., Kosasih P.L., Gogishvili T., Maloney D.G., Turtle C.J., Riddell S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia. 2016;30:492–500. doi: 10.1038/leu.2015.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Feucht J., Sun J., Eyquem J., Ho Y.-J., Zhao Z., Leibold J., Dobrin A., Cabriolu A., Hamieh M., Sadelain M. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat. Med. 2019;25:82–88. doi: 10.1038/s41591-018-0290-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miller B.C., Sen D.R., Al Abosy R., Bi K., Virkud Y.V., LaFleur M.W., Yates K.B., Lako A., Felt K., Naik G.S., et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019;20:326–336. doi: 10.1038/s41590-019-0312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barker C.A., Kim S.K., Budhu S., Matsoukas K., Daniyan A.F., D’Angelo S.P. Cytokine release syndrome after radiation therapy: Case report and review of the literature. J. Immunother. Cancer. 2018;6:1–14. doi: 10.1186/s40425-017-0311-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sterner R.M., Sakemura R., Cox M.J., Yang N., Khadka R.H., Forsman C.L., Hansen M.J., Jin F., Ayasoufi K., Hefazi M., et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood. 2019;133:697–709. doi: 10.1182/blood-2018-10-881722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Depil S., Duchateau P., Grupp S.A., Mufti G., Poirot L. ‘Off-the-shelf’ allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020;19:185–199. doi: 10.1038/s41573-019-0051-2. [DOI] [PubMed] [Google Scholar]
- 54.Odorizzi P.M., Pauken K.E., Paley M.A., Sharpe A., Wherry E.J. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J. Exp. Med. 2015;212:1125–1137. doi: 10.1084/jem.20142237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khan O., Giles J.R., McDonald S., Manne S., Ngiow S.F., Patel K.P., Werner M.T., Huang A.C., Alexander K.A., Wu J.E., et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature. 2019;571:211–218. doi: 10.1038/s41586-019-1325-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Scott A.C., Dündar F., Zumbo P., Chandran S.S., Klebanoff C.A., Shakiba M., Trivedi P., Menocal L., Appleby H., Camara S., et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature. 2019;571:270–274. doi: 10.1038/s41586-019-1324-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li J., He Y., Hao J., Ni L., Dong C. High levels of Eomes promote exhaustion of anti-tumor CD8+ T cells. Front. Immunol. 2018;9:2981. doi: 10.3389/fimmu.2018.02981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wartewig T., Kurgyis Z., Keppler S., Pechloff K., Hameister E., Öllinger R., Maresch R., Buch T., Steiger K., Winter C., et al. PD-1 is a haploinsufficient suppressor of T cell lymphomagenesis. Nature. 2017;552:121–125. doi: 10.1038/nature24649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Philipson B.I., O’Connor R.S., May M.J., June C.H., Albelda S.M., Milone M.C. 4-1BB costimulation promotes CAR T cell survival through noncanonical NF-κB signaling. Sci. Signal. 2020;13:eaay8248. doi: 10.1126/scisignal.aay8248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Salter A.I., Ivey R.G., Kennedy J.J., Voillet V., Rajan A., Alderman E.J., Voytovich U.J., Lin C., Sommermeyer D., Liu L., et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci. Signal. 2018;11:eaat6753. doi: 10.1126/scisignal.aat6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Long A.H., Haso W.M., Shern J.F., Wanhainen K.M., Murgai M., Ingaramo M., Smith J.P., Walker A.J., Kohler M.E., Venkateshwara V.R., et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015;21:581–590. doi: 10.1038/nm.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sakuishi K., Apetoh L., Sullivan J.M., Blazar B.R., Kuchroo V.K., Anderson A.C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 2010;207:2187–2194. doi: 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lichtenegger F.S., Rothe M., Schnorfeil F.M., Deiser K., Krupka C., Augsberger C., Schlüter M., Neitz J., Subklewe M. Targeting LAG-3 and PD-1 to enhance T cell activation by antigen-presenting cells. Front. Immunol. 2018;9:385. doi: 10.3389/fimmu.2018.00385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hodi F.S., Chiarion-Sileni V., Gonzalez R., Grob J.J., Rutkowski P., Cowey C.L., Lao C.D., Schadendorf D., Wagstaff J., Dummer R., et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018;19:1480–1492. doi: 10.1016/S1470-2045(18)30700-9. [DOI] [PubMed] [Google Scholar]
- 65.Blazar B.R., Taylor P.A., Linsley P.S., Vallera D.A. In vivo blockade of CD28/CTLA4: B7/BB1 interaction with CTLA4-Ig reduces lethal murine graft-versus-host disease across the major histocompatibility complex barrier in mice. Blood. 1994;83:3815–3825. [PubMed] [Google Scholar]
- 66.Vasu C., Wang A., Gorla S.R., Kaithamana S., Prabhakar B.S., Holterman M.J. CD80 and CD86 C domains play an important role in receptor binding and co-stimulatory properties. Int. Immunol. 2003;15:167–175. doi: 10.1093/intimm/dxg017. [DOI] [PubMed] [Google Scholar]
- 67.Rota G., Niogret C., Dang A.T., Barros C.R., Fonta N.P., Alfei F., Morgado L., Zehn D., Birchmeier W., Vivier E., Guarda G. Shp-2 is dispensable for establishing T cell exhaustion and for PD-1 signaling in vivo. Cell Rep. 2018;23:39–49. doi: 10.1016/j.celrep.2018.03.026. [DOI] [PubMed] [Google Scholar]
- 68.Buchbinder E.I., Desai A. CTLA-4 and PD-1 pathways: Similarities, differences, and implications of their inhibition. Am. J. Clin. Oncol. 2016;39:98–106. doi: 10.1097/COC.0000000000000239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Singh N., Lee Y.G., Shestova O., Ravikumar P., Hayer K.E., Hong S.J., Lu X.M., Pajarillo R., Agarwal S., Kuramitsu S., et al. Impaired death receptor signaling in leukemia causes antigen-independent resistance by inducing CAR T-cell dysfunction. Cancer Discov. 2020;10:552–567. doi: 10.1158/2159-8290.CD-19-0813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yu X., Harden K., Gonzalez L.C., Francesco M., Chiang E., Irving B., Tom I., Ivelja S., Refino C.J., Clark H., et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009;10:48–57. doi: 10.1038/ni.1674. [DOI] [PubMed] [Google Scholar]
- 71.Kurtulus S., Sakuishi K., Ngiow S.F., Joller N., Tan D.J., Teng M.W., Smyth M.J., Kuchroo V.K., Anderson A.C. TIGIT predominantly regulates the immune response via regulatory T cells. J. Clin. Invest. 2015;125:4053–4062. doi: 10.1172/JCI81187. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 72.Wang B., Zhang W., Jankovic V., Golubov J., Poon P., Oswald E.M., Gurer C., Wei J., Ramos I., Wu Q., et al. Combination cancer immunotherapy targeting PD-1 and GITR can rescue CD8+ T cell dysfunction and maintain memory phenotype. Sci. Immunol. 2018;3:eaat7061. doi: 10.1126/sciimmunol.aat7061. [DOI] [PubMed] [Google Scholar]
- 73.Jin H.S., Ko M., Choi D.S., Kim J.H., Lee D.H., Kang S.H., Kim I., Lee H.J., Choi E.K., Kim K.P., et al. CD226hiCD8+ T cells are a prerequisite for anti-TIGIT immunotherapy. Cancer Immunol. Res. 2020;8:912–925. doi: 10.1158/2326-6066.CIR-19-0877. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.