

Abstract
Renal interstitial fibrosis (RIF) is an incurable pathological lesion in chronic kidney diseases. Pericyte activation is the major pathological characteristic of RIF. Fibroblast and macrophage activation are also involved in RIF. Studies have revealed that core fucosylation (CF), an important post-translational modification of proteins, plays a key role in pericyte activation and RIF by regulating multiple profibrotic signaling pathways as a hub-like target. Here, we reveal that mesenchymal stem cell (MSC)-derived exosomes reside specifically in the injured kidney and deliver microRNA (miR)-34c-5p to reduce cellular activation and RIF by inhibiting CF. Furthermore, we showed that the CD81-epidermal growth factor receptor (EGFR) ligand-receptor complex aids the entry of exosomal miR-34c-5p into pericytes, fibroblasts, and macrophages. Altogether, our findings reveal a novel role of MSC-derived exosomes in inhibiting multicellular activation via CF and provide a potential intervention strategy for renal fibrosis.
Keywords: mesenchymal stem cells, exosomes, pericyte, core fucosylation, renal interstitial fibrosis, microRNA-34c-5p
Graphical abstract
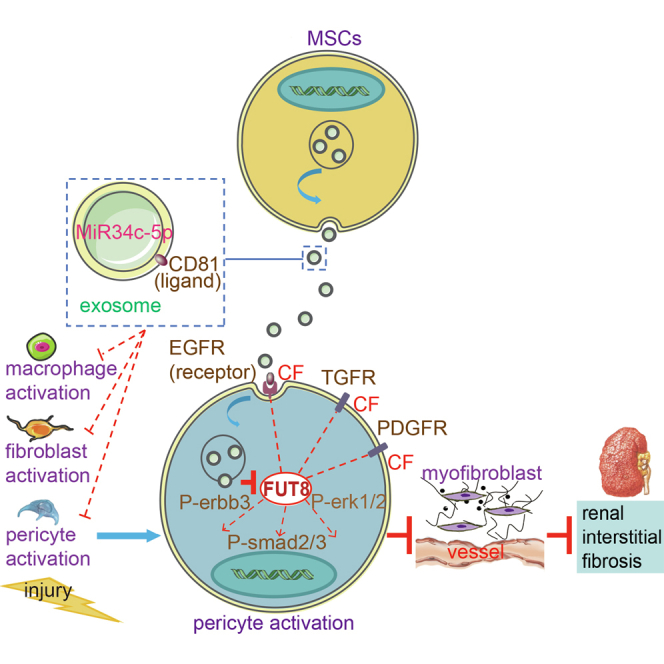
Myofibroblast proliferation, one of the major events in renal interstitial fibrosis (RIF), relies on cellular activation. However, the underlying mechanisms are poorly understood. Lin et al. report that microRNA-34c-5p from mesenchymal stem cell exosomes inhibits the core fucosylation needed for cellular activation, indicating a possible RIF treatment strategy.
Introduction
Persistent and unremitting renal interstitial fibrosis (RIF) ultimately leads to end-stage renal diseases, which are fatal diseases that force patients to undergo dialysis for life or to receive kidney transplants. Unfortunately, there has been no effective method for halting RIF.1 A progressive increase in myofibroblasts in the renal interstitium is well recognized as the main culprit in RIF.2 Meanwhile, the destruction of renal interstitial microvessels during RIF has recently become a matter of increasing concern.3
Myofibroblasts are of multicellular origin. Pericytes are the main source of myofibroblasts; fibroblasts and macrophages are other sources.4 Pericytes are perivascular cells attached to the abluminal surface of the renal interstitial microvessels responsible for stabilizing vascular walls. Upon activation, pericytes detach from the vascular basement membrane, and are prone to instability, ultimately leading to vessel rarefaction.5 In addition, Duffield and LeBleu have shown that pericyte-myofibroblast transition is the major source of myofibroblasts in RIF. These studies suggest that pericytes can be identified as a culprit obviously involved in both the microvessel rarefaction and myofibroblast proliferation in RIF.2,6 Fibroblast and macrophage transition into myofibroblasts is also involved in RIF.4 Accordingly, the inhibition of pericyte, fibroblast, and macrophage activation is worth exploiting to develop a potential effective therapeutic method against RIF. However, RIF is a complex process involving multiple cells; the effect of single-cell or single-signaling-pathway inhibition may be unsatisfactory.
The overactivation of multiple cross-linked profibrotic signaling pathways is the basis for cellular activation.7, 8, 9 However, how the overactivation of these pathways can be suppressed has not been explored. As post-translational modification of proteins is essential for their functions, it would be more reasonable to target a common modification site of these pathway proteins rather than interfering with the protein molecules individually.10
We have found that core fucosylation (CF), which is specifically catalyzed by α1,6-fucosyltransferase (FUT8), is a key post-translational modification of multiple profibrotic proteins.11 We postulate that CF might be a promising therapeutic hub-like target for multiple relevant cells and multiple signaling pathways of RIF. Previously, we found that RIF was reduced in FUT8 knockdown mice,12 but the gene modification method could not be applied clinically.
Cell-based therapies involving mesenchymal stem cells (MSCs) have emerged as promising options for kidney diseases.13, 14, 15 Although MSCs have beneficial effects against RIF, their clinical application faces many challenges, including ethics, safety, tumorigenesis, immunosuppression, and multidirectional differentiation.16 Therefore, we used MSC-derived exosomes instead of MSCs as a candidate regulator of CF.
Here, we present evidence that MSC-derived exosomes reside specifically in the injured kidney, assisted by the CD81-epidermal growth factor receptor (EGFR) complex located on the membrane and by fusion into pericytes, fibroblasts, and macrophages, subsequently delivering microRNA (miR)-34c-5p into the cells to inhibit the CF of several important profibrotic proteins, resulting in the amelioration of multicellular activation and renal fibrosis.
Results
MSCs inhibited CF and pericyte activation in RIF
Our previous studies showed that CF and its unique catalytic enzyme FUT8 were elevated and acted as the culprits in pericyte activation and RIF.11 To investigate whether MSCs can inhibit CF during pericyte activation, we co-cultured activated pericytes with MSCs in Transwell chambers in vitro. As CF is specifically recognized by Lens culinaris lectin (LCA),7,17,18 we detected LCA to evaluate the CF levels. We found that MSCs markedly suppressed FUT8 expression and activity (Figures 1A and 1B), resulting in LCA downregulation (Figure 1C). Meanwhile, MSCs significantly decreased α-SMA expression and pericyte transition into spindle-like myofibroblasts (Figures 1D and 1E).
Figure 1.

MSCs inhibited CF and pericyte activation in RIF
(A) FUT8 levels were assessed using western blotting. The bottom panel shows the quantification. Scale bar, 50 μm. Data are the mean ± SD (n = 3). Control, pericytes without TGFβ1 stimulation. TGFβ1+MSCs, 48 h after TGFβ1 stimulation of pericytes, MSCs were added immediately after the TGFβ1 stimulation of pericytes. (B) FUT8 activity examined by HPLC. Data are the mean ± SD (n = 3); S indicates the peptide substrate; P is the fucosylation product. (C) Representative images of PDGFRβ (green) and LCA (red) staining, and α-SMA (green) and LCA (red) staining. The bottom panel shows the quantification. Scale bar, 75 μm. Data are the mean ± SD (n = 3). (D) Representative morphological alterations in pericytes. Scale bar, 200 μm. (E) Representative images of PDGFRβ (green) and α-SMA (red) staining. The right panel shows the quantification. Scale bar, 75 μm. Data are the mean ± SD (n = 3). α-SMA levels were assessed using western blotting. The right panel shows the quantification. Scale bar, 50 μm. Data are the mean ± SD (n = 3). TGFβ1+MSCs, 48-h TGFβ1–stimulated pericytes plus MSCs. (F) Representative images of PDGFRβ (green) and LCA (red) staining, and α-SMA (green) and LCA (red) staining. The bottom panel shows the quantification. Scale bar, 75 μm. Data are the mean ± SD (n = 6). Control, C57 mice without UUO surgery. UUO+MSCs, 7 days after mice had been injected with MSCs via the tail vein immediately after UUO surgery. (G) Representative images of PDGFRβ (green) and CD31 (red) staining, and PDGFRβ (green) and α-SMA (red) staining. The bottom panel shows the quantification. Scale bar, 75 μm. Data are the mean ± SD (n = 6). The arrows represent the pericytes leaving the endothelium. (H) Representative images of Masson's trichrome and PAS staining of UUO mouse kidney sections. The bottom panel shows the quantification. Scale bar, 200 μm. Data are the mean ± SD (n = 6).
Next, we assessed whether MSCs inhibit CF in the unilateral ureteral obstruction (UUO) kidney during renal fibrosis. MSCs (1 × 106) were injected into mouse tail vein immediately after UUO operation. The MSCs markedly reduced LCA (Figure 1F) and markedly attenuated both pericyte detachment from endothelial cells and following transition to myofibroblasts at 7 days after UUO (Figure 1G). The pathological results showed that RIF was significantly reduced (Figure 1H). We found that MSCs markedly suppressed FUT8 activity (Figure S1A). We detected the distribution of MSCs in the mice by injecting MSCs/red fluorescent protein (RFP) via the tail vein into PDGFRβ-enhanced green fluorescent protein (EGFP) fluorescent reporter mice, whose pericytes emit green autofluorescence. However, MSCs were not seen in UUO mouse kidneys (Figures S1B and S1C). The above in vitro and in vivo data suggest that the benefit of MSCs against renal fibrosis in UUO mice might follow the paracrine mode rather than a direct and contact effect.
MSCs inhibited pericyte activation and RIF mainly through secreting exosomes
Many studies have indicated that the therapeutic effects of MSCs are largely mediated by paracrine factors, including exosomes.19,20 Accordingly, we investigated whether the therapeutic effects depend on exosomes derived from MSCs. Exosomes were collected from the supernatant of MSCs cultured for 24 h in serum-free medium (concentration, 3 μg/mL) and were identified by electron microscopy, nanoparticle tracking analysis, and western blotting (Figure 2A). Then, we performed a tracer experiment to detect exosome distribution. Exosomes were labeled by PKH67 fluorescence. In the MSC conditioned medium (CM; the culture medium), exosome secretion peaked at 24 h (Figure 2B). In vitro imaging of MSCs co-cultured with TGFβ1-stimulated pericytes in Transwell chambers showed that the fluorescently labeled exosomes were visible in the pericytes at 24 h, and peaked at 48 h. In the absence of TGFβ1, we observed that exosomes entered the pericytes at 0 h, 24 h, 48 h, and only a small amount of exosomes entered the pericytes at 48 h (Figure 2C). Exosome distribution after injection into UUO mouse tail vein was then traced using an ex vivo imaging system. After exosomes had been injected into the tail vein, the dynamic fluorescence tracer demonstrated that they were distributed in the liver, lung, kidneys, brain, spleen, and heart at 6 h of UUO, and were mainly accumulated in the liver, lung, and kidneys at 12 h of UUO. The exosomes began to accumulate in the obstructed kidney at 24 h of UUO, and peaked in the obstructed kidney at 72 h of UUO. There remained a small amount of exosomes in the obstructed kidney at 168 h of UUO, and they were nearly undetectable at 240 h of UUO. However, only a few exosomes were detectable in the contralateral kidney at 24 h and 72 h of UUO (Figure 2D).21,22 In addition, there was no obvious pathological change in the heart, liver, spleen, lung, or brain (Figure S2A). These results suggest that MSC-derived exosomes may be candidate therapeutic tools due to their special targeting ability. Next, we observed the efficacy of the exosomes.
Figure 2.

Exosomes accumulated in activated pericytes and obstructed kidney both in vitro and in vivo
(A) (Left) Representative electron micrograph of exosomes. Scale bar, 100 μm. (Middle) Nanoparticle tracking analysis. (Right) CD63 and CD81 levels were assessed using western blotting. Scale bar, 50 μm (n = 3). (B) The exosome content of MSC-CM was detected by luciferase reporter. Data are the mean ± SD (n = 3). (C) Representative images of PKH67 (green) and PDGFRβ (red) staining in vitro. The right panel shows the quantification. Scale bar, 75 μm. Control, pericytes without TGFβ1 stimulation. 0 h, 24 h, 48 h, pericytes without TGFβ1 stimulation for 0 h, 24 h, 48 h. (D) Ex vivo UUO mouse imaging at 0 h, 6 h, 12 h, 24 h, 72 h, 168 h, and 240 h.
We then prepared MSCs, MSC-CM, exosomes extracted from CM (Exo), and exosome-free MSC-CM [CM(−)Exo], and compared their effects on CF and pericyte activation. Notably, MSCs, exosomes, and CM all suppressed FUT8 protein overexpression and activity effectively and synchronously with LCA elevation in the pericytes following 48-h incubation with TGFβ1 (Figures 3A–3C). MSCs, exosomes, and CM alleviated the number of pericytes transitioning to myofibroblasts. However, CM(−)Exo did not exhibit the above inhibitory effects (Figures 3D and 3E).
Figure 3.

MSCs inhibited pericyte activation and RIF mainly through secreting exosomes
(A) FUT8 levels were assessed using western blotting. The bottom panel shows the quantification. Scale bar, 50 μm. Data are the mean ± SD (n = 3). Control, pericytes without TGFβ1 stimulation. TGFβ1+MSCs, 48-h TGFβ1-stimulated pericytes plus MSCs; TGFβ1+Exo, 48-h TGFβ1-stimulated pericytes plus exosomes; TGFβ1+CM, 48-h TGFβ1-stimulated pericytes plus CM; TGFβ1+CM(−)Exo, 48-h TGFβ1-stimulated pericytes plus CM(−)Exo. (B) FUT8 activity examined by HPLC. Data are the mean ± SD (n = 3); S indicates the peptide substrate; P is the fucosylation product. (C) Representative images of PDGFRβ (green) and LCA (red) staining, and α-SMA (green) and LCA (red) staining. The right panel shows the quantification. Scale bar, 75 μm. Data are the mean ± SD (n = 3). (D) Representative morphological alterations in pericytes. Scale bar, 200 μm. (E) Representative images of PDGFRβ (green) and α-SMA (red) staining. The bottom panel shows the quantification. Scale bar, 75 μm. α-SMA levels were assessed using western blotting. The right panel shows the quantification. Scale bar, 50 μm. Data are the mean ± SD (n = 6). (F) Representative images of PDGFRβ (green) and LCA (red) staining, and α-SMA (green) and LCA (red) staining. The bottom right panel shows the quantification. Scale bar, 75 μm. Data are the mean ± SD (n = 6). Control, C57 mice without UUO surgery. UUO+MSCs, 7 days after MSC injection into UUO mouse tail vein; UUO+Exo, 7 days after Exo injection into UUO mouse tail vein; UUO+CM, 7 days after CM injection into UUO mouse tail vein; UUO+CM(−)Exo, 7 days after CM(−)Exo injection into UUO mouse tail vein. (G) Representative images of PDGFRβ (green) and CD31 (red) staining, and PDGFRβ (green) and α-SMA (red) staining. The bottom panel shows the quantification. Scale bar, 75 μm. Data are the mean ± SD (n = 6). (H) Representative images of Masson's trichrome and PAS staining of UUO mouse kidney sections. The bottom panel shows the quantification. Scale bar, 200 μm. Data are the mean ± SD (n = 6).
Then we investigated the effects of the exosomes on UUO mice. LCA levels were decreased following the injection of exosomes, CM, and MSCs (Figure 3F). The degree of pericyte separation from vascular endothelial cells and α-SMA expression levels were decreased by exosomes, MSCs, and CM, but not by CM(−)Exo (Figure 3G). Both CM and exosome injection alleviated RIF, similar to that of MSCs, but CM(−)Exo had no such effects (Figure 3H). Notably, MSCs, exosomes, and CM all suppressed FUT8 activity effectively (Figure S2B). Our data show that CM, exosomes, and MSCs had similar inhibitory effects on pericyte activation and CF, while the exosome-free CM was ineffective in vitro or in vivo. These findings suggest that MSCs inhibit pericyte activation mainly through exosomes in UUO mice. However, whether the role of MSCs and exosomes is dependent on CF inhibition is not known.
MSC-derived exosome inhibition of pericyte activation was dependent on CF
To further investigate whether the effects of MSCs and exosomes were dependent on CF inhibition, we performed a FUT8 rescue experiment. RT-PCR, immunofluorescence, and western blotting were used. FUT8 plasmid was transfected into the pericytes (Figure 4A). Following the transfection, FUT8 and LCA expression was increased, the degree of pericyte-myofibroblast transition was aggravated, and the curative effect of MSCs and exosomes was significantly reversed (Figures 4B–4F). These data indicate that FUT8 overexpression disabled the efficacy of the MSCs and exosomes. These results reveal a novel mechanism of MSC-derived exosomes for inhibiting pericyte activation.
Figure 4.

MSC-derived exosomes inhibited pericyte activation mainly by regulating CF
(A) (Left) RT-PCR, (middle) representative images of LCA (red) staining, (right) FUT8 levels assessed by western blotting. The bottom panel shows the quantification. Data are the mean ± SD (n = 3). (B) FUT8 level was assessed by western blotting. The bottom panel shows the quantification. Scale bar, 50 μm. Data are the mean ± SD (n = 3). (C) FUT8 activity tested by HPLC. Data are the mean ± SD (n = 3); S indicates the peptide substrate; P is the fucosylation product. (D) Representative images of PDGFRβ (green) and LCA (red) staining, and α-SMA (green) and LCA (red) staining. The bottom panel shows the quantification. Scale bar, 75 μm. Data are the mean ± SD (n = 3). (E) Representative morphological alterations in pericytes. Scale bar, 200 μm. (F) Representative images of PDGFRβ (green) and α-SMA (red) staining. The bottom panel shows the quantification. Scale bar, 75 μm. Data are the mean ± SD (n = 3). α-SMA level was assessed by western blotting. The bottom panel shows the quantification. Scale bar, 50 μm. Data are the mean ± SD (n = 3).
MSC-derived exosomes inhibited the CF of multiple proteins, resulting in profibrotic signaling pathway inactivation
The TGFβ-SMAD2/3, PDGF-ERK1/2, and EGFR-ERBB3 pathways have been recognized as key classical profibrotic signaling pathways in renal pericytes,11,23 and these three proteins are modified by CF.24 Further, BMP7-SMAD6/7, a typical antifibrotic signaling pathway that competitively inhibits SMAD2/3, has been recognized as an important anti-fibrotic signaling pathway in renal fibrosis,25, 26, 27 so we investigated the effect of the exosomes on these pathways. Lectin blotting and immunofluorescence analysis showed that the addition of exosomes decreased the CF of TGFβR, PDGFRβ, and EGFR without affecting protein expression levels, and decreased the phosphorylation of SMAD2/3, ERK1/2, and ERBB3 in the pericytes following 48-h incubation with TGFβ1 (Figures 5A–5D). On the contrary, lectin blotting showed that CF did not modify the BMP7 receptor; BMP7 protein levels did not change, and its downstream SMAD6/7 increased significantly after the addition of the exosomes. However, the addition of exosome-free CM did not have any of the above effects (Figure 5E). Our study is the first to show that the unique beneficial effects of MSC-derived exosomes on renoprotection depend on CF regulation, which leads to the inactivation of several fibrogenic pathways, while the anti-fibrogenic signaling pathway is activated.
Figure 5.

Exosomes inhibited the CF of multiple proteins, resulting in the inactivation of these signaling pathways
(A) TGFβR1, PDGFRβ levels in total cell lysates were assessed by western blotting. Immunoprecipitated TGFβR1, PDGFRβ proteins were analyzed by lectin blotting. TGFβR1, PDGFRβ were immunoprecipitated from whole-cell lysates with the corresponding antibodies, and the blots were probed with LCA. SMAD2/3, phosphorylated (P)-SMAD2/3, ERK1/2, and P-ERK1/2 levels were assessed using western blotting. Total cell lysates underwent immunoblotting. Representative data are shown. The bottom panel shows the quantification. Data are the mean ± SD (n = 3). (B) Representative images of EGFR (green) and LCA (red) staining in vitro. The right panel shows the quantification. Scale bar, 75 μm. Data are the mean ± SD (n = 3). (C) Representative images of EGFR (green) and LCA (red) staining in vivo. The bottom panel shows the quantification. Scale bar, 75 μm. Data are the mean ± SD (n = 6). (D) EGFR levels in total cell lysates were assessed by western blotting. Immunoprecipitated EGFR protein was analyzed by lectin blotting. EGFR was immunoprecipitated from whole-cell lysates with the corresponding antibodies, and the blots were probed with LCA. ERBB3 and P-ERBB3 levels were assessed using western blotting. Total cell lysates underwent immunoblotting. Representative data are shown. The right panel shows the quantification. Data are the mean ± SD (n = 3). (E) BMP level in total cell lysates was assessed by western blotting. Immunoprecipitated BMP protein was analyzed by lectin blotting. BMP was immunoprecipitated from whole-cell lysates with the corresponding antibodies, and the blots were probed with LCA. SMAD6/7 level was assessed using western blotting. Total cell lysates underwent immunoblotting. Representative data are shown. The right panel shows the quantification. Data are the mean ± SD (n = 3).
MSC-derived exosomes downregulated CF by delivering miR-34c-5p
MSC-derived exosomes perform biological functions by releasing microRNAs (miRNAs).28, 29, 30 To explore the mechanism of the role of MSC-derived exosomes, MSCs were cultured for 0 h, 24 h, and 48 h without serum and the exosomes were isolated for miRNA omics analysis. We detected 176 miRNAs with different expression changes at the three timepoints. We overlapped these miRNAs with 71 miRNAs from the miRDB miRNA library that specifically regulate FUT8, and screened out only one miRNA (i.e., miR-34c-5p). The heatmap showed significant differential expression of miR-34c-5p in the 24-h group compared with the 0-h group (Figure 6A). Real-time PCR (RT-PCR) detection showed that miR-34c-5p expression was decreased significantly after UUO 7 d, and returned to control levels after exosome treatment in the mouse kidneys (Figure S3A). Further, we verified whether FUT8 is regulated by miR-34c-5p by luciferase assay. The data indicated that miR-34c-5p markedly reduced the luciferase activity of the wild-type (WT) FUT8 reporter plasmid compared with the control (Figure 6B), which confirmed that miR-34c-5p could specifically interact with FUT8.
Figure 6.

Exosomes downregulated CF by delivering miR-34c-5p during pericyte activation and RIF
(A) (Left) Heatmap of exosomal miRNA and (right) histogram of miR-34c-5p at 0 h, 24 h, and 48 h. Data are the mean ± SD (n = 3). (B) Luciferase reaction between FUT8 and miR-34c-5p. Data are the mean ± SD (n = 3). (C) Representative morphological alterations in pericytes. Scale bar, 200 μm. Also shown are representative images of PDGFRβ (green) and α-SMA (red) staining, and α-SMA (green) and LCA (red) staining in vitro. The right panel shows the quantification. Scale bar, 75 μm. Data are the mean ± SD (n = 3). (D) Representative images of Masson's trichrome and PAS staining of UUO mouse kidney sections. Scale bar, 200 μm. Also shown are representative images of PDGFRβ (green) and α-SMA (red) staining, and α-SMA (green) and LCA (red) staining in vivo. The right panel shows the quantification. Scale bar, 75 μm. Data are the mean ± SD (n = 6).
Then, miR-34c-5p mimics and inhibitor encoding GFP were commercially constructed (GenePharma). The synthetic mimic or inhibitor was transfected into MSCs, and the exosomes were collected from the supernatant. Flow cytometry detected the FAM-labeled miR-34c-5p mimic and inhibitor, and RT-PCR showed that FUT8 was dramatically reduced after miR-34c-5p mimic transfection and was increased after miR-34c-5p inhibitor transfection in the MSC and exosome groups (Figure S3B). These data indicate the successful generation of miR-34c-5p mimic and inhibitor. The miR-34c-5p mimic greatly decreased pericyte detachment and pericyte transition to myofibroblasts following 48-h incubation with TGFβ1, and downregulated α-SMA expression and CF (Figure 6C). The inhibitory effect of miR-34c-5p in the UUO mouse kidney during RIF was then assessed. Injection of miR-34c-5p mimic markedly decreased tubulointerstitial damage and fibrosis at 7 days after UUO, and downregulated α-SMA expression and CF (Figure 6D). These data show that the inhibitory effect of exosomes on CF and pericyte activation is mainly dependent on miR-34c-5p. We performed a tracer experiment to elucidate the delivery of miR-34c-5p. Fluorescent fam-labeled miR-34c-5p was transfected into MSCs, and the fluorescence-labeled miR-34c-5p in the CM was illustrated using the luciferase marker. TGFβ1-stimulated pericytes were cultured with the CM in Transwell chambers, and the CM was injected into the mice via the tail vein. The fluorescence-labeled miR-34c-5p was observed in the pericytes and obstructed kidney (Figure S3C). MiR-34c-5p was also observed in the lung, liver, and spleen after separate injection of the miR-34c-5p mimic(fam) without packaging with exosomes. These data suggest that miR-34c-5p has no targeting capability without the exosome-packaging effect (Figure S3D). We confirmed that exosomes regulate CF to inhibit pericyte activation and RIF mainly by delivering miR-34c-5p from MSCs to pericytes. As the UUO model was a model of postrenal acute obstruction, we supplemented another chronic model of prerenal ischemia-reperfusion (IR), and found that miR-34c-5p could also inhibit renal injury (Figures S4A and S4B).
CD81-EGFR complex formation aided miR-34c-5p entry into pericytes to downregulate CF
However, how miR-34c-5p moves from exosomes to pericytes is still unclear. Recent discoveries have highlighted the fact that exosomes mediate crosstalk between different cell types by miRNA delivery to specific recipient cell types via receptor-mediated binding.31,32 We screened out 1,708 pericyte proteins and 1,687 exosome proteins using proteomic analysis. Eighty-four pericyte-exosome interacting membrane proteins were identified by examining protein-protein interaction using STRING software. A protein-protein interaction network was drawn to provide a framework for visualizing receptor-mediated binding. As shown in the network, larger nodes indicated more protein interactions, suggesting that the protein is more important in the network. The nodes were largest for EGFR protein. Among the proteins closest to EGFR, the nodes were largest for CD81. Exosome and pericyte proteomics screened out the membrane ligand and target receptor: CD81-EGFR. We hypothesized that the EGFR-CD81 complex might play an important role in mediating miR-34c-5p delivery from exosomes to pericytes (Figure 7A).
Figure 7.

CD81–EGFR complex formation aided miR-34c-5p entry into pericytes to downregulate CF
(A) (Top) Exosome and pericyte proteomics; (bottom) protein interaction network. (B) Computer simulation of CD81-EGFR molecular docking. (C) Representative images of EGFR (green) and CD81 (red) staining both in vitro (n = 3) and in vivo (n = 6). The right panel shows the quantification. Scale bar, 75 μm. Data are the mean ± SD. (D) Representative images of α-SMA (green), LCA (red), and miR-34c-5p (fam) staining in vitro. The right panel shows the quantification. Scale bar, 75 μm. (E) Representative images of α-SMA (green), LCA (red), and miR-34c-5p (fam) staining in vivo. The right panel shows the quantification. Scale bar, 75 μm. Data are the mean ± SD (n = 6).
Then, we constructed the spatial 3D structures of a full-size EGFR and CD81 using computer-based techniques. Interactions between EGFR and the CD81 extracellular domain estimated using a molecular docking approach aided the identification of three sites for EGFR binding to CD81. The CD81 extracellular region binds to the EGFR ligand-binding region to form a complex (Figure 7B), and FUT8 does not bind in the EGFR ligand-binding region and does not affect CD81-EGFR complex formation (Figure S5A). Immunofluorescence staining aimed at verifying CD81-EGFR complex formation showed decreased EGFR fluorescence after exosome treatment. In view of the structure and function of proteins and their interaction results, the possible reason is that CD81 binds to the EGFR ligand-binding region mainly by hydrogen bonds and hydrophobic action, which may affect the conformation of amino acid residues and decrease EGFR fluorescence intensity. The 3D fluorescence showed the binding of exogenous CD81 to EGFR (Figure 7C). These data provide evidence for the coupling between EGFR and CD81.
To study the function of the CD81-EGFR complex in the process of pericyte activation and renal fibrosis, GFP-encoding CD81 and EGFR short hairpin RNA (shRNA) recombinant adenovirus vectors were commercially constructed (GenePharma) (Figure S5B). Adenoviral transfections were performed on MSCs, pericytes, and mice through tail vein injection. RT-PCR and flow cytometry showed that CD81 and EGFR were dramatically reduced after CD81 shRNA and EGFR shRNA transfection (Figure S5C).
We also used rhodamine-labeled LCA (LCA-tetramethylrhodamine isothiocyanate [TRITC]) to investigate whether the CD81-EGFR complex aids miR-34c-5p entry into pericytes to inhibit CF. Following the inhibition of CD81 or EGFR, exosomes could not inhibit CF or pericyte activation, and we could barely detect miR-34c-5p in the pericytes (Figures 7D, 7E, and S5D). The data show that CD81-EGFR complex formation aids miR-34c-5p entry into pericytes.
CD81-EGFR complex formation aided miR-34c-5p entry into fibroblasts and macrophages to downregulate CF and inhibit cellular activation
We found that fibroblasts(vimentin) and macrophages(F4/80) gradually transitioned into myofibroblasts at 1, 3, and 7 days of UUO, and CF levels were increased (Figure 8A). Following exosome and miR-34c-5p treatment, fibroblast and macrophage activation was decreased, and CF levels were decreased (Figure 8B).
Figure 8.

CD81–EGFR complex formation aided miR-34c-5p entry into fibroblasts and macrophages to downregulate CF and inhibit cellular activation
(A and B) The top two rows show representative images of vimentin (green) and α-SMA (red) staining, and F4/80 (green) and α-SMA (red) staining; bottom two rows show representative images of vimentin (green) and LCA (red) staining, and F4/80 (green) and LCA (red) staining. The bottom panel shows the quantification. Scale bar, 75 μm. Data are the mean ± SD (n = 6). Vimentin represents fibroblasts; F4/80 represents macrophages. (C) Representative images of EGFR (green), vimentin (red), and EGFR (green) and F4/80 (red) staining. The bottom panel shows the quantification. Scale bar, 75 μm. Data are the mean ± SD (n = 6). (D) Representative images of miR-34c-5p (fam) (green), vimentin (red), and miR-34c-5p (fam) (green) and F4/80 (red) staining. The bottom panel shows the quantification. Scale bar, 75 μm. Data are the mean ± SD (n = 6).
Is CD81-EGFR interaction also involved in miR-34c-5p delivery in fibroblasts and macrophages? We found that both fibroblasts and macrophages expressed EGFR. Following the inhibition of CD81 or EGFR, we found only a small amount of miR-34c-5p in the fibroblasts and macrophages (Figures 8C and 8D). These data show that the CD81-EGFR complex aids miR-34c-5p entry into fibroblasts and macrophages.
Discussion
MSCs can repair kidney injury effectively and reduce RIF progression,14,19,33, 34, 35, 36, 37 but the mechanism warrants further exploration. In the present study, we found that MSCs inhibit both CF and pericyte activation, while FUT8 overexpression disabled the efficacy of the MSCs. The data demonstrate that MSCs reduced pericyte activation and RIF mainly by regulating CF. To the best of our knowledge, this is the first report of the ability of MSCs to antagonize pericyte activation by regulating post-translational modification of proteins.
MSCs function through local colonization or in a paracrine manner.38 However, the occurrence of local colonization is in the minority.36 Previously, we found that although MSCs injected through mouse tail vein decreased kidney injury, they mainly accumulated in the lungs instead of the injured kidney.39 Here, our data show that MSCs in the upper Transwell chamber reduced pericyte activation in the bottom chamber with no contact, and exogenous red fluorescent-labeled MSCs did not accumulate in the obstructed kidney. We deduced that the benefits of MSCs were not due to colonization but mainly arose in a paracrine manner.
MSC exosome secretion can contribute greatly to the effect of MSCs, such as regulating the development of individual organisms, cell activation, and repairing damaged tissues.20,40, 41, 42, 43 Previously, we explored a new method for exosome enrichment based on polyethylene glycol (PEG) secondary precipitation, which was simple and efficient and did not affect exosome activity.44
Exosome chemotaxis and agglomeration from the liver to the obstructed kidney can take place, reducing the possibility of engulfment and degradation of exosomes in the obstructed kidney.8,45, 46, 47 We speculate that this may be the reason for the long existence of exosomes in the injured kidney. The specific mechanism remains to be further studied. Meanwhile, only a few exosomes in the contralateral kidney were detectable at 24 h,72 h of UUO, which is similar to the findings in acute kidney injury.21,22 Here, we also monitored other organs, i.e., the heart, liver, spleen, lung, and brain, and observed no pathological change when the exosomes were injected via the tail vein. These results suggest that exosomes may be desirable therapeutic tools with good targeting ability. The possible mechanism is that MSCs may be recruited by receptor-mediated interaction48; exosomes bear the same membrane receptors as MSCs, and may accumulate at the site of injury via similar mechanisms. However, the specific colonization mechanism and long-term efficacy require further investigation.
Our data show that the CM, exosomes, and MSCs had similar inhibitory effects on pericyte activation and CF, while the exosome-free CM was ineffective both in vitro and in vivo. FUT8 overexpression disabled the efficacy of the exosomes. The data demonstrate that exosomes reduce pericyte activation and RIF mainly by regulating CF.
Interestingly, we found that the therapeutic effect of MSCs was slightly better than that of exosomes, but was almost equivalent to that of CM. We postulated that there were other cytokines in the CM, and exosomes were virtually the main therapeutic component of the CM. The MSCs exerted inhibitory effects mainly through exosome secretion.
We hypothesized that the inhibitory effects of the exosomes on CF may inactivate multiple profibrotic pathways simultaneously, which may account for the therapeutic benefit of MSCs and exosomes. The activation of multiple signaling pathways is the initiative basis of pericyte activation and RIF. Regarding the important fibrogenic signaling pathways, we investigated the EGFR-ERBB3, TGFβR-SMAD2/3, and PDGFR-ERK1/2 pathways. The MSCs and exosomes did not affect the expression of membrane receptors in these signaling pathways, while both membrane receptor CF and phosphorylation of the downstream response elements were decreased remarkably. This inhibitory regulation of the fibrogenic signaling pathways may account for the therapeutic benefit of MSCs and exosomes in pericyte activation and RIF. Several studies have reported the inefficiency of single-pathway blockade in mitigating fibrosis.7,9,11 The compensation of other fibrogenic pathways or the relative inadequacy of anti-fibrosis pathways might account for the inefficiency of single-pathway blockade. Then, we investigated the reported anti-fibrogenic BMP7-SMAD6/7 signaling pathways, where BMP7 expression (not regulated by CF) was not affected by TGFβ1 or the exosomes. The interesting finding is that the BMP7-SMAD6/7 and TGF-β-SMAD2/3 signaling pathways may antagonize each other.42 Whether there is another CF-independent mechanism is worth further study. Theoretically, the anti-fibrogenic function of BMP7-SMAD6/7 remained unaffected.49 These data suggest that exosomes have inhibitory effects specifically for CF-regulated signaling pathways. Intervention for the hub-like control of multiple signaling pathways, such as CF in the present study, may be a more effective and specific strategy for inhibiting cellular activation and organ fibrosis.
MSC-derived exosomes are extracellular vesicles and can deliver functionally active materials such as proteins, miRNAs, and non-coding RNAs.28,30,31,50, 51, 52 Exosomes can inhibit renal tubular epithelial cells and endothelial cell damage to play a role in renal tissue injury repair via miRNAs28,30; however, this has not been reported in pericytes. Therefore, we speculated that some miRNAs contained in exosomes might be involved in pericyte activation. In our preliminary experiment, we observed that the treatment effect of exosomes secreted by serum-free stem cells for 24 h was significant compared with that for 48 h, and that exosome secretion peaked at 24 h. After preliminary screening of candidates through omics statistical analysis, miRNAs with peak secretion at 24 h and statistically significant changes were likely to be candidates and play a major therapeutic role. We used the exosome transcriptome and miRNA library regulation of FUT8 to screen out miR-34c-5p.52 MiR-34c-5p was significantly decreased in UUO at 7 days, and returned to control levels after exosome treatment. The supplementation of exogenous miR-34c-5p inhibited FUT8 expression, thereby inhibiting cellular activation and RIF. Accordingly, we used a fluorescent reporter enzyme reaction to verify that FUT8 is a substrate of miR-34c-5p. We confirmed that exosomes regulate CF to inhibit pericyte activation and RIF mainly by delivering miR-34c-5p from MSCs to the pericytes.
There are two types of exosome signaling: membrane fusion and endocytosis. Ligands-receptors form an endocytic complex to aid miRNA entry into recipient cells.31,32,40 Here, exosome and pericyte proteomics screened out the membrane ligand and target receptor: CD81-EGFR. CD81 is a member of a four-transmembrane protein that binds to fusion receptor cells.53 Here, we used computer simulation, 3D fluorescence imaging, and blockade experiments to confirm that exosomes delivered miR-34c-5p via CD81-EGFR complexes to regulate CF and pericyte activation in RIF.
Following exosome or miR-34c-5p treatment, fibroblast and macrophage activation was decreased; interestingly, CF levels were also decreased. MiR-34c-5p entered the fibroblasts and macrophages mainly in the manner of CD81-EGFR. Following the inhibition of CD81 or EGFR, we found only a small amount of miR-34c-5p in the fibroblast and macrophage, so there might be other ways that assist miR-34c-5p to enter the fibroblast and macrophage. This is the first time that CF expression changes in fibroblasts and macrophages have been found, and it also suggests that CF not only acts as a multi-target signal pathway but also plays an important role in multiple relevant cells. Further, this is also the first report that MSC exosomal miR-34c-5p can inhibit fibroblast and macrophage activation.
In summary, we reveal the process of MSCs acting on renal pericyte, fibroblast, and macrophage activation. MSC-derived exosomes deliver miR-34c-5p into pericytes, fibroblasts, and macrophages through the CD81-EGFR complex. Then, miR-34c-5p downregulates CF to inhibit multiple signaling pathways, and consequently ameliorates multiple cellular activations and RIF. With CF as the target, MSC-derived exosomes exert a widespread effect on multiple cells and signaling pathways. We reveal a novel therapeutic mechanism of MSCs on RIF, which provides a basis for clinical application.
Materials and methods
MSC culture
C57BL/6 mouse bone marrow-derived MSCs/RFP were obtained commercially (Cyagen Biosciences, Sunnyvale, CA). The culture was initiated following the manufacturer's instructions. The MSCs were placed in 25-cm2 culture flasks and cultured in MSC growth medium (Cyagen Biosciences) at 37°C under 5% CO2 and 90% humidity. The medium was changed every 3 days. Passage 6–8 MSCs were used for the experiments. MSC/RFP identification was confirmed commercially (Cyagen Biosciences). Following previously described methods,37 fluorescence-activated cell sorting (Beckman Coulter, Indianapolis, IN) was used to examine the representative markers of MSCs (CD45, CD90 [BD Biosciences, San Diego, CA], CD11a, CD54 [AbD Serotec, Oxford, UK]), and MSC multilineage differentiation was examined under adipogenic and osteogenic differentiation conditions, and conformed to the characteristics of MSCs.
Preparation of MSC exosomes, CM, and exosome-free CM
The exosomes were separated by an internationally recognized overspeed centrifugation method.44 MSCs cultured for 24 h in serum-free medium were removed from the incubator and observed under a microscope. The supernatant was collected and centrifuged (500 × g, 20 min). After centrifugation, the supernatant was placed in an ultrafiltration tube and centrifuged (5,000 × g, 100 min) to condense the culture volume by 30 times to prepare CM for injection, which was then aliquoted and stored at a density of 1 × 106 cells. The same ultracentrifugation method was used for producing the exosome-free CM: MSC supernatant medium collected for 24 h was centrifuged in a centrifuge tube (500 × g, 20 min); after centrifugation, the supernatant was placed in a new centrifuge tube and centrifuged again (10,000 × g, 20 min). The supernatant was then centrifuged again in an ultracentrifuge tube (100,000 × g, 180 min). The supernatants were collected as exosome-free CM. The pellet in the ultracentrifuge tube was resuspended in phosphate-buffered saline (PBS) and transferred to a new ultracentrifuge tube (100,000 × g, 180 min). The supernatant was discarded and the pelleted exosomes were resuspended in a small amount of PBS.
Animal care and UUO mouse models
PDGFR-EGFP mice were obtained from Cyagen Biosciences; C57BL/6 mice were obtained from the Dalian Medical University Laboratory Animal Center and were housed at a constant room temperature with a 12-h light/dark cycle. Standard rodent chow and water were provided ad libitum. The animal experiments were conducted in accordance with the regulations established by the Institutional Committee for the Care and Use of Laboratory Animals and were approved by the Dalian Medical University Laboratory Animal Center; all efforts were made to minimize suffering. All surgical procedures were conducted by a single surgeon under aseptic conditions in the Laboratory Animal Unit. The mice were anesthetized with an intraperitoneal injection of freshly prepared 10% chloral hydrate. A midline incision was made in the abdominal wall, and the left ureter was isolated and ligated using a 4-0 silk suture at two points along its length. The wound was closed with a silk suture, and the mice were returned to their cages. The mice were humanely sacrificed on days 1, 3, and 7 (n = 6), and the kidneys were harvested for analysis.
Acute kidney IR mouse model
C57BL/6 mice were anesthetized with an intraperitoneal injection of freshly prepared 10% chloral hydrate. The left renal pedicle was clamped with an atraumatic vascular clip for 40 min (Roboz Surgical Instrument Co., Germany) through a flank incision, and the left kidney turned purple subsequent to clamping. The clamps were removed after 40 min to start the reperfusion, and the left kidney reverted to red within approximately 10 s. The muscle layer and skin were closed with 4-0 silk sutures.
Mouse tail vein injection of MSCs, MSC-derived exosomes, CM, and exosome-free CM
After the UUO mouse model had been constructed, the mice were divided into UUO, MSC, exosome, CM, and exosome-free CM groups. The mice from each group were fixed on the fixture. The tail was immersed in water at 40°C for 40 s, and the tail hair was scraped with a blade to expose the tail vein. Using a 1-mL syringe, the reagent was slowly injected into the tail at about 30°C for about 1 mm. After the injection, the injection site was pressed until the bleeding had stopped. We injected 1 × 106 MSCs or exosomes (100 μg) per mouse.
Renal histopathological staining
The mouse kidneys were fixed for 24 h in 10% formalin, removed, and dehydrated in alcohol series: 70%, 80%, and 95% alcohol solution for 30 min each; 100% alcohol solution for 30 min twice, and then cleared for 20 min in chloroform. The kidney tissue was then placed in a paraffin solution for 25 min for paraffin embedding. Using a microtome, the kidney tissue was cut into 1-μm thick paraffin sections on glass slides, and placed in a 60°C incubator for 5 h. Then, the specimens were dewaxed in xylene I and II for 20 min each, followed by immersion in absolute ethanol I and II for 10 min each. Subsequently, the specimens were washed in 95%, 90%, 80%, and 70% alcohol for 5 min each. Masson pathological staining was performed as follows: the nuclei were stained with Weigert's iron hematoxylin for 5 min, differentiated for a few seconds using 1% hydrochloric acid alcohol, and rinsed in tap water for 2 min, returning to blue. Then, the specimens were stained using Ponceau-acid fuchsin for 6–14 min, and rinsed with distilled water before 4-min phosphomolybdic acid treatment, 6-min aniline blue counterstaining, and 1-min 1% glacial acetic acid treatment. Finally, the specimens were placed in 95% alcohol I and II, absolute ethanol I and II, and then xylene I and II for 5 min each, dehydrated and cleared, and sealed using neutral gum. For periodic acid-Schiff (PAS) staining, the specimens were treated using 1% periodate oxide for 5–10 min, stained with Schiff's solution for 10–30 min, and rinsed in running water. This was followed by hematoxylin staining for 2–5 min, treatment with 1% hydrochloric acid alcohol, washing with tap water, treatment with 1% ammonia back to blue, and washing with tap water. Finally, the specimens were placed in 95% alcohol I and II, absolute ethanol I and II, and xylene I and II each for 5 min, dehydrated and cleared, and sealed in neutral gum.
The extent of tubulointerstitial damage (including the number of inflammatory cells, renal tubule expansion/atrophy, and proliferation of renal interstitial fibrous tissue) was evaluated in each group of stained sections. There were 0–3 levels of damage: 0 indicated normal (0 points); 1 indicated <25% fibrosis (1 point); 2 indicated 25–50% fibrosis (2 points); and 3 indicated >50% fibrosis (3 points). Three fields per slice were randomly selected under ×200 magnification, and the results were expressed as the mean ± standard deviation (SD). All pathological sections were evaluated independently by three investigators in a double-blinded manner.
Pericyte isolation and the establishment of a pericyte activation model
Here, we used a model we have described previously,9 with some modifications. The kidney was diced and incubated with Liberase (0.5 mg/mL, Roche, Mannheim, Germany) and DNase (100 U/mL, Roche) in Hank's buffered salt solution for 45 min at 37°C. After centrifugation, the cells were resuspended in Hank's buffered salt solution, and filtered (40 μm). Then, impurities were removed using 42% Percoll solution, and the cell layer was collected. Pericytes were purified by isolating PDGFR-β+ cells using magnetic-activated cell sorting (Miltenyi Biotec, Gladbach, Germany), and cultured in pericyte medium supplemented with 2% fetal bovine serum, 1% pericyte growth supplement, and 1% penicillin/streptomycin (ScienCell, Carlsbad, CA) at 37°C in a 5% CO2 atmosphere with 90% humidity. The medium was changed every 3 days. Passage 1 or 2 cells were used for the experiments. Pericyte activation was induced using 10 ng/mL recombinant TGFβ1.
Immunofluorescence (kidney tissue and pericytes)
Freshly harvested mouse kidney tissues were fixed in 4% paraformaldehyde (PFA) for 24 h. The tissues were successively dehydrated in 30%, 20%, and 10% sucrose solutions for 1 h each, and embedded in optimal cutting temperature compound (Tissue-Tek, Sakura Finetek, Tokyo, Japan). Next, 4-μm cryosections were collected on Superfrost Plus glass slides, rinsed with PBS, and permeabilized with 1% Triton X-100 for 5 min. Then, the sections were blocked with blocking buffer (Vector Laboratories, Burlingame, CA) for 1 h. The sections were incubated with primary antibodies against PDGFRβ (1:100; ab32570; Abcam, Cambridge, MA), CD31 (1:100; ab24590; Abcam), α-SMA (1:100; ab205718; Abcam), CD81 (1:100; D5O2Q; Cell Signaling Technology, Danvers, MA), EGFR (1:100; ab52894; Abcam), PKH67 (1:100; UR52303; Umibio, Shanghai, China), vimentin (1:100; ab92547; Abcam), F4/80 (1:100, ab6640; Abcam), and LCA (1:1,000; LCA, b-1045; Vector Laboratories) for 1 h at room temperature, and then incubated overnight at 4°C. Then, the sections were incubated with secondary antibodies for 1 h at room temperature. Subsequently, the sections were mounted with fluorescent mounting medium with DAPI (Abcam). Each section was observed under a confocal laser scanning microscope (Leica SP8, Leica, Wetzlar, Germany) at ×400 magnification, if necessary. The primary antibodies were omitted for the negative controls.
The pericytes were fixed in freshly prepared 4% PFA for 10 min at room temperature, washed three times with PBS, and each coverslip was incubated in 1% BSA. Then, the cells were incubated with the same primary antibodies as above for 1 h at room temperature. After washing in PBS, the cells were incubated with secondary antibodies conjugated with fluorescein isothiocyanate (FITC) or Cy3 for 1 h at room temperature in a darkened humidified chamber. Finally, the preparations were washed with PBS and mounted in fluorescent mounting medium with DAPI (Abcam). Images were acquired using a confocal laser scanning microscope (Leica SP8) at ×400 magnification. The primary antibodies were omitted for the negative controls.
Western blotting
Cells were collected and lysed in radioimmunoprecipitation assay (RIPA) buffer (1% Triton X-100, 150 mmol/L NaCl, 1 mmol/L EGTA, 50 mmol/L Tris-HCl, 0.1% sodium dodecyl sulfate [SDS], 1% sodium deoxycholate, phenylmethylsulfonyl fluoride [PMSF]) on ice for 20 min. Protein concentrations were determined with a bicinchoninic acid protein assay kit (Thermo, Rockford, IL). Proteins (equal amounts, 10 μg/lane) were separated on 6%–10% SDS-PAGE (polyacrylamide gel electrophoresis) and electrotransferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad, Hercules, CA) at 400 mA for 2 h. The membranes were then incubated in blocking buffer (PBS) with 0.05% Tween 20 and 0.1% BSA for 1 h at room temperature, and then incubated overnight at 4°C with primary antibodies against α-SMA (1:500; ab205718; Abcam), FUT8 (1:500; ab19874; Abcam), TGFβR (1:500; ab31013; Abcam), PDGFRβ (1:500; ab32570; Abcam), SMAD2/3 (1:500; ab202445; Abcam), ERK1/2 (1:500; ab184699; Abcam), phosphorylated (P)-SMAD2/3 (1:500; ab272332; Abcam), P-ERK1/2 (1:500; ab214362; Abcam), SMAD6/7 (1:500; ap52036; Abcepta, Beijing, China), BMP7 (1:500; ab129156; Abcam), EGFR (1:500; ab52894; Abcam), ERBB3 (1:500; D22C5; Cell Signaling Technology), P-ERBB3 (1:500; d1b5; Cell Signaling Technology), CD63 (1:500; ab217345; Abcam), and CD81 (1:500; ab109201; Abcam). The membranes were incubated with secondary antibodies for 1 h at room temperature. The blots were washed three times in 1× PBS-Tween 20 solution, and incubated for 1 min with electrochemiluminescence (ECL) reagents (Amersham, Pittsburgh, PA). Proteins were visualized with Super RX-N film (Fujifilm Corporation, Tokyo, Japan). Anti-GAPDH antibody was used as the loading control to normalize the protein levels detected.
Immunoprecipitation
Cell lysates were centrifuged (12,000 rpm for 20 min at 4°C), and the supernatant was collected and precleared using Protein G PLUS-Agarose (Santa Cruz Biotechnology, Santa Cruz, CA). The cell lysates (500 μg) were then incubated with 2 μg of antibody against TGFβR, PDGFRβ, EGFR, and BMP7 for 2 h at 4°C on a rocker platform (30 rocks/min). Protein-antibody complexes were collected with 20 μL Protein G PLUS-Agarose on a rocker platform (30 rocks/min) overnight at 4°C. The immunoprecipitates were washed three times with lysis buffer. Equal amounts (10 μg/lane) of proteins underwent 12% SDS-PAGE for lectin blotting (described below). The primary antibodies were omitted for the negative controls.
Lectin blotting
Immunoprecipitated EGFR BMP7, TGFβR, and PDGFRβ were separated using 12% SDS-PAGE and electrotransferred to PVDF membranes. The membranes were blocked with 5% BSA (w/v) in Tris-buffered saline containing 0.05% Tween 20 (TBST) overnight at 4°C, and then incubated for 1 h at 23°C with TBST containing LCA-Biotin (b-1045, Vector Laboratories), which preferentially recognizes Fuc-1,6 GlcNAc. The blots were washed three times with 1× PBS-Tween solution and incubated for 1 min with ECL reagents (Amersham). The lectin-reactive proteins were visualized using Super RX-N film (Fujifilm Corporation).
Enzyme activity detection
FUT8 activity was measured using a previously described method.54 Cell lysates (5 μg) as the enzyme source were added to assay buffer (200 mM 2-(N-morpholino)ethanesulfonic acid hydrate [MES], 1% Triton X-100) supplemented with donor (500 μM GDP-L-fucose) and substrate (50 μM GnGn-Asn-4-(2-pyridylamine) butylamine [PABA]). The mixture was incubated at 37°C for 4 h, and the reaction was stopped by heating at 100°C for 5 min. The reaction solution was then centrifuged at 12,000 × g for 10 min, and 10 μL of reaction products underwent high-performance liquid chromatography (HPLC) with a fluorescence detector (Waters Corporation, Milford, MA) at excitation and emission wavelengths of 320 nm and 400 nm, respectively.
Ex vivo animal imaging
Exosomes were labeled with PKH67. The UUO obstruction model was established in mice. PKH67-labeled exosomes were injected via the tail vein, and the in vivo biodistributions were imaged at 0 h, 6 h, 12 h, 24 h, 72 h, 168 h, and 240 h with a FCZAA8-E1JHAA imaging system (Bruker, Bremen, Germany) (λex/λem = 466/504 nm). The amount of exosomes was 100 μg per mouse via tail vein.
DNA and RNA transfection
GP-transfect-Mate (GenePharma, Suzhou, China) was placed at room temperature before being mixed gently. Serum-free medium (50 μL) was added into a 1.5-mL sterile centrifuge tube, and the transfection reagents were added, gently pipette-mixed, and stood at room temperature for 5 min. At the same time, 50 μL of serum-free medium was added into another 1.5-mL sterile centrifuge tube, and the DNA/RNA oligo was added, and pipette-mixed gently. The GP-transfect-Mate medium mixture was added to the DNA/RNA oligo medium mixture at room temperature for 5 min. The mixture was lightly mixed with a transfer gun and transfected immediately after 15 min at room temperature. DNA/RNA expression was detected after a 24–72-h transfection of 100 μL of compound.
Inhibition of CD81 and EGFR expression
Adenoviruses carrying CD81 shRNA and EGFR shRNA were constructed commercially (GenePharma, Suzhou, China). The shRNA-carrying adenoviruses were administered to the mice via tail vein injection of 1 × 106 plaque-forming units (PFU) before anesthesia for UUO surgery. The shRNAs were designed for knockdown in pericytes in vitro. To confirm the knockdown, we extracted RNA from the cells and assessed CD81 and EGFR expression using reverse transcriptase-PCR (RT-PCR) according to the manufacturer's protocol.
PCR
We purchased and reproduced PDGFRβ-EGFP fluorescent reporter mice from Cyagen Biosciences, and verified the mice by PCR (the molecular size of the product is 260 bp; the molecular size of the internal reference is 413 bp).
RT-PCR
RNA was extracted from the cells, and the RNA concentration was measured. Then, the genomic DNA was removed, and the cDNA was reverse transcribed using a Verso cDNA Synthesis Kit (Thermo Fisher Scientific) using EvaGreen qPCR Supermix (Solis BioDyne, Shanghai, China), followed by PCR. The forward (F) and reverse (R) PCR primer sequences were as follows: FUT8, F 5′-TTCCTGGCGTTGGATTAT-3′, R 5′-TTGGACAGTTCTCGGCTA-3′; CD81, F 5′-TGGCTTCCTGGGCTGCTA-3′, R 5′-GCGATCTGGTCCTTGTTGA-3′; EGFR, F 5′-CATCTCCGAAAGCCAACA-3′, R 5′-CGACGGTCCTCCAAGTAG-3′. MiR-34c-5p, AGGCAGTGTAGTTAGCTGATTGC.
Flow cytometry
Cells were fixed with 0.05% glutaraldehyde (Sigma-Aldrich, St. Louis, MO) for 10 min at room temperature, and then permeabilized with 0.1% Triton X-100 (Life Technologies, Carlsbad, CA) for 5 min at room temperature. The cells were transfected with fluorescent protein/miRNA via a transfection kit (GenePharma), then washed with PBS three times to prepare single-cell suspensions. The fluorescence signals were collected from the suspensions.
Protein extraction and digestion
For protein extraction, 4% 1-dodecyl-3-methylimidazolium chloride ([C12 mim]Cl) (m/v, containing 1% v/v protease inhibitor cocktail) was added to the cell pellet, followed by sonication at 10% amplitude for 5 s on and 10 s off, with a total working time of 3 min. The extracted proteins were denatured by incubation at 95°C for 5 min. The insoluble debris was removed by centrifugation at 16,000 × g for 10 min. The supernatant was collected, and the protein concentration was determined using a bicinchoninic acid (BCA) quantification kit (Beyotime Biotechnology, Shanghai, China).
Proteins were reduced in 20 mM dithiothreitol at 95°C for 5 min, and then alkylated in 40 mM iodoacetamide at room temperature in the dark for 30 min. The proteins were then transferred to 10-kDa filter devices and washed with 8 M urea by centrifugation (14,000 × g) three times. The concentrates were washed with 100 mM triethylammonium bicarbonate (TEAB) three times. Thereafter, the concentrates were diluted with 100 μL of 100 mM TEAB containing 1 μg trypsin, and incubated at 37°C for 12 h.
TMT 10-plex labeling
Isobaric labeling was conducted according to the instructions of the tandem mass tag (TMT) kit (Thermo Fisher Scientific, Erlangen, Germany). For the TMT 10-plex labeling experiment, 10 samples were labeled with 10 channels (127N, 128N, 129N, 130N, 131N, 127C, 128C, 129C, 130C, 131C). TMT reagents (0.4 mg) were dissolved in anhydrous acetonitrile (41 μL) and added to 100 μg of peptides (dissolved in 60 μL of 100 mM TEAB) to achieve a final acetonitrile concentration of approximately 30% (v/v). After 60-min reaction at room temperature, 16 μL of 5% (w/v) hydroxylamine was added to each tube, and mixed for 20 min. The aliquots were then combined for fractionation.
High-pH RPLC fractionation
To increase the depth of protein identification, peptide fractionation was performed with high-pH reverse-phase liquid chromatography (RPLC). Combined peptides were separated and collected using an Agilent 2100 HPLC system (Santa Clara, CA) with a high pH-stable RP column (4.6 mm × 150 mm, 5 μm, 100 Å, Durashell, Agela Technology, Torrance, CA) at a flow rate of 0.3 mL/min using a gradient of 5%–45% solvent B over 55 min (solvent A, 30 mM ammonium acetate, pH 10; solvent B, acetonitrile, 30 mM ammonium acetate, pH 10). A total of 50 fractions (0.5 mL) were collected from 6 min to 55 min, and the fractions with equal collection time intervals (6 min) were pooled. Finally, six pooled fractions were lyophilized in a SpeedVac Concentrator (Thermo Fisher Scientific, Waltham, MA).
Proteomic analysis
Nanoflow RPLC was performed on an EASY-nLC 1000 system coupled online to an Orbitrap Fusion Lumos mass spectrometer equipped with a nano-electrospray ion source (Thermo Fisher Scientific, Erlangen, Germany). Mobile-phase buffer A comprised 2% acetonitrile and 98% H2O (0.1% formic acid), and buffer B comprised 98% acetonitrile and 2% H2O (0.1% formic acid). The tryptic peptides were dissolved in buffer A and loaded onto a 15-cm analytical column (internal diameter [ID], 150 μm) packed with ReproSil-Pur C18-AQ 1.9-μm beads (Ammerbuch-Entringen, Germany). The peptides were separated at a flow rate of 600 nL/min with a linear gradient of 8%–36% mobile-phase B within 78 min, followed by a linear increase from 36% to 50% mobile-phase B within 8 min, and 50%–95% mobile B within 1 min, followed by 8 min of washing. The spray voltage was set at 2,900 V in positive ion mode, and the ion transfer tube temperature was set at 320°C. Data-dependent acquisition was performed using Xcalibur software (Thermo Fisher Scientific, Erlangen, Germany) in profile spectrum data mode.
Database searching of MS data
All raw data were analyzed using MaxQuant 1.5.1.0 against mouse reference proteomes (https://www.uniprot.org, version 2019/06, 17,331 sequences). Carbamidomethyl cysteine was searched as a fixed modification. Oxidized methionine and protein N-terminal acetylation were set as variable modifications. The enzyme specificity was set as trypsin. The maximum missing cleavage site was set as 2. The tolerances of the first search and the main search for peptides were set at 20 ppm and 4.5 ppm, respectively. The minimal peptide length was set at seven. The false discovery rates (FDRs) for peptides, proteins, and sites were all <0.01.
Statistical analysis
Data are presented as the means ± SDs. Multiple comparisons of parametric data were performed using one-way analysis of variance (ANOVA). Nonparametric data were compared with the Mann-Whitney U test to identify differences between groups. Pearson's correlation analysis was calculated to determine the associations, and p < 0.05 was considered to indicate statistical significance. All statistical analyses were performed using SPSS version 21.0 (IBM, Inc., Armonk, NY).
Data availability
ProteomeXchange member repository, accession code: PXD022358. Reviewer login information: https://www.iprox.org/page/SSV024.html;url=1604542455529Sw7W. Password: riyQ. Sequence Read Archive (SRA) database: PRJNA674943. Dalian Medical University Animal experimental ethical inspection number: 8177032062.
Acknowledgments
This research was supported by grants from the Natural Science Foundation of China (NSFC, nos. 81530021, 81470996, and 81770694); the Science and Technology Talent Training Plan of the First Affiliated Hospital of Dalian Medical University (no. 2015YC002); Liaoning province Xingliao Talents Plan project (no. XLYC1908029).
Author contributions
X.H., W.W., Q.T., X.D., B.Q. Z.L., and L.L. performed the animal experiments, PCR, immunohistochemistry, immunofluorescence, and western blotting experiments and collected and analyzed the data. X.H., A.L., L.Z., Z.S., and W.Y. performed the cell-based experiments and collected and analyzed the data. H.L., N.W., X.H., N.S., and N.Y. designed the study, interpreted the results, wrote the paper, revised the paper, and discussed the analyses. All authors have read and approved the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.10.012.
Contributor Information
Nan Wang, Email: pyran78@163.com.
Hongli Lin, Email: linhongli@vip.163.com.
Supplemental information
References
- 1.Zhang L., Wang F., Wang L., Wang W., Liu B., Liu J., Chen M., He Q., Liao Y., Yu X., et al. Prevalence of chronic kidney disease in China: a cross-sectional survey. Lancet. 2012;379:815–822. doi: 10.1016/S0140-6736(12)60033-6. [DOI] [PubMed] [Google Scholar]
- 2.LeBleu V.S., Taduri G., O'Connell J., Teng Y., Cooke V.G., Woda C., Sugimoto H., Kalluri R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013;19:1047–1053. doi: 10.1038/nm.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DiRocco D.P., Kobayashi A., Taketo M.M., McMahon A.P., Humphreys B.D. Wnt4/β-catenin signaling in medullary kidney myofibroblasts. J. Am. Soc. Nephrol. 2013;24:1399–1412. doi: 10.1681/ASN.2012050512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuppe C., Ibrahim M.M., Kranz J., Zhang X., Ziegler S., Perales-Patón J., Jansen J., Reimer K.C., Smith J.R., Dobie R., et al. Decoding myofibroblast origins in human kidney fibrosis. Nature. 2021;589:281–286. doi: 10.1038/s41586-020-2941-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawakami T., Mimura I., Shoji K., Tanaka T., Nangaku M. Hypoxia and fibrosis in chronic kidney disease: crossing at pericytes. Kidney Int. Suppl. 2014;4:107–112. doi: 10.1038/kisup.2014.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duffield J.S. Cellular and molecular mechanisms in kidney fibrosis. J. Clin. Invest. 2014;124:2296–2306. doi: 10.1172/JCI72267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen N., Lin H., Wu T., Wang D., Wang W., Xie H., Zhang J., Feng Z. Inhibition of TGF-β1-receptor posttranslational core fucosylation attenuates rat renal interstitial fibrosis. Kidney Int. 2013;84:64–77. doi: 10.1038/ki.2013.82. [DOI] [PubMed] [Google Scholar]
- 8.Wang N., Shao Y., Mei Y., Zhang L., Li Q., Li D., Shi S., Hong Q., Lin H., Chen X. Novel mechanism for mesenchymal stem cells in attenuating peritoneal adhesion: accumulating in the lung and secreting tumor necrosis factor α-stimulating gene-6. Stem Cell Res. Ther. 2012;3:51. doi: 10.1186/scrt142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Y.T., Chang F.C., Wu C.F., Chou Y.H., Hsu H.L., Chiang W.C., Shen J., Chen Y.M., Wu K.D., Tsai T.J., et al. Platelet-derived growth factor receptor signaling activates pericyte-myofibroblast transition in obstructive and post-ischemic kidney fibrosis. Kidney Int. 2011;80:1170–1181. doi: 10.1038/ki.2011.208. [DOI] [PubMed] [Google Scholar]
- 10.García I., Arenas-Alfonseca L., Moreno I., Gotor C., Romero L.C. HCN regulates cellular processes through posttranslational modification of proteins by S-cyanylation. Plant Physiol. 2019;179:107–123. doi: 10.1104/pp.18.01083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang N., Deng Y., Liu A., Shen N., Wang W., Du X., Tang Q., Li S., Odeh Z., Wu T., et al. Novel mechanism of the pericyte-myofibroblast transition in renal interstitial fibrosis: core fucosylation regulation. Sci. Rep. 2017;7:16914. doi: 10.1038/s41598-017-17193-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fang M., Kang L., Wang X., Guo X., Wang W., Qin B., Du X., Tang Q., Lin H. Inhibition of core fucosylation limits progression of diabetic kidney disease. Biochem. Biophys. Res. Commun. 2019;520:612–618. doi: 10.1016/j.bbrc.2019.10.037. [DOI] [PubMed] [Google Scholar]
- 13.Chuang H.M., Shih T.E., Lu K.Y., Tsai S.F., Harn H.J., Ho L.I. Mesenchymal stem cell therapy of pulmonary fibrosis: improvement with target combination. Cell Transpl. 2018;27:1581–1587. doi: 10.1177/0963689718787501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bianchi F., Sala E., Donadei C., Capelli I., La Manna G. Potential advantages of acute kidney injury management by mesenchymal stem cells. World J. Stem Cells. 2014;6:644–650. doi: 10.4252/wjsc.v6.i5.644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lou G., Chen Z., Zheng M., Liu Y. Mesenchymal stem cell-derived exosomes as a new therapeutic strategy for liver diseases. Exp. Mol. Med. 2017;49:e346. doi: 10.1038/emm.2017.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mishra V.K., Shih H.H., Parveen F., Lenzen D., Ito E., Chan T.F., Ke L.Y. Identifying the therapeutic significance of mesenchymal stem cells. Cells. 2020;9:1145. doi: 10.3390/cells9051145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ihara H., Ikeda Y., Taniguchi N. Reaction mechanism and substrate specificity for nucleotide sugar of mammalian alpha1,6-fucosyltransferase–a large-scale preparation and characterization of recombinant human FUT8. Glycobiology. 2006;16:333–342. doi: 10.1093/glycob/cwj068. [DOI] [PubMed] [Google Scholar]
- 18.Ihara H., Ikeda Y., Toma S., Wang X., Suzuki T., Gu J., Miyoshi E., Tsukihara T., Honke K., Matsumoto A., et al. Crystal structure of mammalian alpha1,6-fucosyltransferase, FUT8. Glycobiology. 2007;17:455–466. doi: 10.1093/glycob/cwl079. [DOI] [PubMed] [Google Scholar]
- 19.He J., Wang Y., Sun S.S., Yu M., Wang C., Pei X., Zhu B., Wu J., Zhao W. Bone marrow stem cells-derived microvesicles protect against renal injury in the mouse remnant kidney model. Nephrology (Carlton) 2012;17:493–500. doi: 10.1111/j.1440-1797.2012.01589.x. [DOI] [PubMed] [Google Scholar]
- 20.Wang Y., Yu D., Liu Z., Zhou F., Dai J., Wu B., Zhou J., Heng B.C., Zou X.H., Ouyang H., et al. Exosomes from embryonic mesenchymal stem cells alleviate osteoarthritis through balancing synthesis and degradation of cartilage extracellular matrix. Stem Cell Res. Ther. 2017;8:189. doi: 10.1186/s13287-017-0632-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bruno S., Grange C., Deregibus M.C., Calogero R.A., Saviozzi S., Collino F., Morando L., Busca A., Falda M., Bussolati B., et al. Mesenchymal stem cell-derived microvesicles protect against acute tubular injury. J. Am. Soc. Nephrol. 2009;20:1053–1067. doi: 10.1681/ASN.2008070798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grange C., Tapparo M., Bruno S., Chatterjee D., Quesenberry P.J., Tetta C., Camussi G. Biodistribution of mesenchymal stem cell-derived extracellular vesicles in a model of acute kidney injury monitored by optical imaging. Int. J. Mol. Med. 2014;33:1055–1063. doi: 10.3892/ijmm.2014.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nolan-Stevaux O., Truitt M.C., Pahler J.C., Olson P., Guinto C., Lee D.C., Hanahan D. Differential contribution to neuroendocrine tumorigenesis of parallel egfr signaling in cancer cells and pericytes. Genes Cancer. 2010;1:125–141. doi: 10.1177/1947601909358722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ying W., Hao Y., Zhang Y., Peng W., Qin E., Cai Y., Wei K., Wang J., Chang G., Sun W., et al. Proteomic analysis on structural proteins of severe acute respiratory syndrome coronavirus. Proteomics. 2004;4:492–504. doi: 10.1002/pmic.200300676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zou G.L., Zuo S., Lu S., Hu R.H., Lu Y.Y., Yang J., Deng K.S., Wu Y.T., Mu M., Zhu J.J., et al. Bone morphogenetic protein-7 represses hepatic stellate cell activation and liver fibrosis via regulation of TGF-β/Smad signaling pathway. World J. Gastroenterol. 2019;25:4222–4234. doi: 10.3748/wjg.v25.i30.4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu H., Zhang W., Lilly B. Evaluation of Notch3 deficiency in diabetes-induced pericyte loss in the retina. J. Vasc. Res. 2018;55:308–318. doi: 10.1159/000493151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuan K., Shamskhou E.A., Orcholski M.E., Nathan A., Reddy S., Honda H., Mani V., Zeng Y., Ozen M.O., Wang L., et al. Loss of endothelium-derived Wnt5a is associated with reduced pericyte recruitment and small vessel loss in pulmonary arterial hypertension. Circulation. 2019;139:1710–1724. doi: 10.1161/CIRCULATIONAHA.118.037642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Valadi H., Ekström K., Bossios A., Sjöstrand M., Lee J.J., Lötvall J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007;9:654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 29.Qu Y., Zhang Q., Cai X., Li F., Ma Z., Xu M., Lu L. Exosomes derived from miR-181-5p-modified adipose-derived mesenchymal stem cells prevent liver fibrosis via autophagy activation. J. Cell. Mol. Med. 2017;21:2491–2502. doi: 10.1111/jcmm.13170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang B., Yao K., Huuskes B.M., Shen H.H., Zhuang J., Godson C., Brennan E.P., Wilkinson-Berka J.L., Wise A.F., Ricardo S.D. Mesenchymal stem cells deliver exogenous microRNA-let7c via exosomes to attenuate renal fibrosis. Mol. Ther. 2016;24:1290–1301. doi: 10.1038/mt.2016.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ohno S., Takanashi M., Sudo K., Ueda S., Ishikawa A., Matsuyama N., Fujita K., Mizutani T., Ohgi T., Ochiya T., et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol. Ther. 2013;21:185–191. doi: 10.1038/mt.2012.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Diao J., Pantua H., Ngu H., Komuves L., Diehl L., Schaefer G., Kapadia S.B. Hepatitis C virus induces epidermal growth factor receptor activation via CD81 binding for viral internalization and entry. J. Virol. 2012;86:10935–10949. doi: 10.1128/JVI.00750-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.da Silva A.F., Silva K.A., Reis L.A., Teixeira V.P., Schor N. Bone marrow-derived mesenchymal stem cells and their conditioned medium attenuate fibrosis in an irreversible model of unilateral ureteral obstruction. Cell Transpl. 2015;24:2657–2666. doi: 10.3727/096368915X687534. [DOI] [PubMed] [Google Scholar]
- 34.Liu B., Ding F., Hu D., Zhou Y., Long C., Shen L., Zhang Y., Zhang D., Wei G. Human umbilical cord mesenchymal stem cell conditioned medium attenuates renal fibrosis by reducing inflammation and epithelial-to-mesenchymal transition via the TLR4/NF-κB signaling pathway in vivo and in vitro. Stem Cell Res. Ther. 2018;9:7. doi: 10.1186/s13287-017-0760-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sung P.H., Chang C.L., Tsai T.H., Chang L.T., Leu S., Chen Y.L., Yang C.C., Chua S., Yeh K.H., Chai H.T., et al. Apoptotic adipose-derived mesenchymal stem cell therapy protects against lung and kidney injury in sepsis syndrome caused by cecal ligation puncture in rats. Stem Cell Res. Ther. 2013;4:155. doi: 10.1186/scrt385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duffield J.S., Park K.M., Hsiao L.L., Kelley V.R., Scadden D.T., Ichimura T., Bonventre J.V. Restoration of tubular epithelial cells during repair of the postischemic kidney occurs independently of bone marrow-derived stem cells. J. Clin. Invest. 2005;115:1743–1755. doi: 10.1172/JCI22593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li D., Wang N., Zhang L., Hanyu Z., Xueyuan B., Fu B., Shaoyuan C., Zhang W., Xuefeng S., Li R., et al. Mesenchymal stem cells protect podocytes from apoptosis induced by high glucose via secretion of epithelial growth factor. Stem Cell Res. Ther. 2013;4:103. doi: 10.1186/scrt314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kinnaird T., Stabile E., Burnett M.S., Epstein S.E. Bone-marrow-derived cells for enhancing collateral development: mechanisms, animal data, and initial clinical experiences. Circ. Res. 2004;95:354–363. doi: 10.1161/01.RES.0000137878.26174.66. [DOI] [PubMed] [Google Scholar]
- 39.Hu J., Zhang L., Wang N., Ding R., Cui S., Zhu F., Xie Y., Sun X., Wu D., Hong Q., et al. Mesenchymal stem cells attenuate ischemic acute kidney injury by inducing regulatory T cells through splenocyte interactions. Kidney Int. 2013;84:521–531. doi: 10.1038/ki.2013.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Camussi G., Deregibus M.C., Bruno S., Cantaluppi V., Biancone L. Exosomes/microvesicles as a mechanism of cell-to-cell communication. Kidney Int. 2010;78:838–848. doi: 10.1038/ki.2010.278. [DOI] [PubMed] [Google Scholar]
- 41.Marote A., Teixeira F.G., Mendes-Pinheiro B., Salgado A.J. MSCs-derived exosomes: cell-secreted nanovesicles with regenerative potential. Front. Pharmacol. 2016;7:231. doi: 10.3389/fphar.2016.00231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nargesi A.A., Lerman L.O., Eirin A. Mesenchymal stem cell-derived extracellular vesicles for renal repair. Curr. Gene Ther. 2017;17:29–42. doi: 10.2174/1566523217666170412110724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu K., Li H.Y., Yang K., Wu J.L., Cai X.W., Zhou Y., Li C.Q. Exosomes as potential alternatives to stem cell therapy for intervertebral disc degeneration: in-vitro study on exosomes in interaction of nucleus pulposus cells and bone marrow mesenchymal stem cells. Stem Cell Res. Ther. 2017;8:108. doi: 10.1186/s13287-017-0563-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weng Y., Sui Z., Shan Y., Hu Y., Chen Y., Zhang L., Zhang Y. Effective isolation of exosomes with polyethylene glycol from cell culture supernatant for in-depth proteome profiling. Analyst. 2016;141:4640–4646. doi: 10.1039/c6an00892e. [DOI] [PubMed] [Google Scholar]
- 45.Abello J., Nguyen T.D.T., Marasini R., Aryal S., Weiss M.L. Biodistribution of gadolinium- and near infrared-labeled human umbilical cord mesenchymal stromal cell-derived exosomes in tumor bearing mice. Theranostics. 2019;9:2325–2345. doi: 10.7150/thno.30030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu Y., Bai L., Guo K., Jia Y., Zhang K., Liu Q., Wang P., Wang X. Focused ultrasound-augmented targeting delivery of nanosonosensitizers from homogenous exosomes for enhanced sonodynamic cancer therapy. Theranostics. 2019;9:5261–5281. doi: 10.7150/thno.33183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Royo F., Cossio U., Ruiz de Angulo A., Llop J., Falcon-Perez J.M. Modification of the glycosylation of extracellular vesicles alters their biodistribution in mice. Nanoscale. 2019;11:1531–1537. doi: 10.1039/c8nr03900c. [DOI] [PubMed] [Google Scholar]
- 48.Herrera M.B., Bussolati B., Bruno S., Morando L., Mauriello-Romanazzi G., Sanavio F., Stamenkovic I., Biancone L., Camussi G. Exogenous mesenchymal stem cells localize to the kidney by means of CD44 following acute tubular injury. Kidney Int. 2007;72:430–441. doi: 10.1038/sj.ki.5002334. [DOI] [PubMed] [Google Scholar]
- 49.Caja L., Tzavlaki K., Dadras M.S., Tan E.J., Hatem G., Maturi N.P., Morén A., Wik L., Watanabe Y., Savary K., et al. Snail regulates BMP and TGFβ pathways to control the differentiation status of glioma-initiating cells. Oncogene. 2018;37:2515–2531. doi: 10.1038/s41388-018-0136-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baglio S.R., Rooijers K., Koppers-Lalic D., Verweij F.J., Pérez Lanzón M., Zini N., Naaijkens B., Perut F., Niessen H.W., Baldini N., et al. Human bone marrow- and adipose-mesenchymal stem cells secrete exosomes enriched in distinctive miRNA and tRNA species. Stem Cell Res. Ther. 2015;6:127. doi: 10.1186/s13287-015-0116-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quan Y., Wang Z., Gong L., Peng X., Richard M.A., Zhang J., Fornage M., Alcorn J.L., Wang D. Exosome miR-371b-5p promotes proliferation of lung alveolar progenitor type II cells by using PTEN to orchestrate the PI3K/Akt signaling. Stem Cell Res. Ther. 2017;8:138. doi: 10.1186/s13287-017-0586-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morizane R., Fujii S., Monkawa T., Hiratsuka K., Yamaguchi S., Homma K., Itoh H. miR-34c attenuates epithelial-mesenchymal transition and kidney fibrosis with ureteral obstruction. Sci. Rep. 2014;4:4578. doi: 10.1038/srep04578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sharma N.R., Mateu G., Dreux M., Grakoui A., Cosset F.L., Melikyan G.B. Hepatitis C virus is primed by CD81 protein for low pH-dependent fusion. J. Biol. Chem. 2011;286:30361–30376. doi: 10.1074/jbc.M111.263350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liang W., Mao S., Sun S., Li M., Li Z., Yu R., Ma T., Gu J., Zhang J., Taniguchi N., et al. Core fucosylation of the T cell receptor is required for T cell activation. Front. Immunol. 2018;9:78. doi: 10.3389/fimmu.2018.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ProteomeXchange member repository, accession code: PXD022358. Reviewer login information: https://www.iprox.org/page/SSV024.html;url=1604542455529Sw7W. Password: riyQ. Sequence Read Archive (SRA) database: PRJNA674943. Dalian Medical University Animal experimental ethical inspection number: 8177032062.