

Abstract
Background
In recent years, the availability of high throughput technologies, establishment of large molecular patient data repositories, and advancement in computing power and storage have allowed elucidation of complex mechanisms implicated in therapeutic response in cancer patients. The breadth and depth of such data, alongside experimental noise and missing values, requires a sophisticated human-machine interaction that would allow effective learning from complex data and accurate forecasting of future outcomes, ideally embedded in the core of machine learning design.
Objective
In this review, we will discuss machine learning techniques utilized for modeling of treatment response in cancer, including Random Forests, support vector machines, neural networks, and linear and logistic regression. We will overview their mathematical foundations and discuss their limitations and alternative approaches in light of their application to therapeutic response modeling in cancer.
Conclusion
We hypothesize that the increase in the number of patient profiles and potential temporal monitoring of patient data will define even more complex techniques, such as deep learning and causal analysis, as central players in therapeutic response modeling.
Keywords: Therapeutic response, therapeutic resistance, machine learning, cancer, prediction, data repositories
1. INTRODUCTION
In recent years, the availability of high throughput technologies, the establishment of large molecular patient data repositories such as TCGA [1], SU2C [2], TARGET [3], etc., and advancement in computing power and storage [4, 5] have allowed elucidation of complex mechanisms implicated in cancer progression and therapeutic response [2, 6-15], building a foundation for the development of personalized medicine and precision therapeutics. Such molecular data, spanning clinical information, human genome, epigenome, and transcriptome, is referred to as Big Data and, if utilized effectively, holds a promise to make individualized predictions of therapeutic response directly at diagnosis and in real time [7, 13, 16, 17], enhancing clinical decision making and improving patient outcomes.
The volume and depth of such data, alongside experimental noise and missing values, requires a sophisticated human-machine interaction that would allow effective learning from complex data and accurate predictions of the future outcomes based on the learning experiences even in the presence of noise, ideally embedded in the core of machine learning (ML) design. In 1950, “Turing test” evaluated machine's ability to exhibit intelligent behavior equivalent to a human [18]. Following its success, machine learning officially originated in 1956, when John McCarthy organized the infamous Dartmouth Conference, coining the term artificial intelligence [19] (i.e., the ability of a computer to perform learning and reasoning similar to the human mind) and in 1959, when Andrew Samuel introduced the term machine learning (i.e., “field of study that gives computers the ability to learn without being explicitly programmed”) [20]. After the success of the Dartmouth conference, in 1958, Frank Rosenblatt introduced the first neural network (i.e., perceptron) [21], followed by Widrow and Hoff in 1960, who developed a single layer neural network (known as ADALINE) and a multilayer neural network MADALINE - a three-layered (input, hidden and output layers) feed forward neural network, with ADALINE units in their hidden and output layers [22, 23], applied to detect binary patterns and eliminate echo from phone lines, respectively. The machine learning experienced further expansion throughout 60’s via works by Hunt et al. [24] in symbolic learning, Nilsson [25] in statistical methods and Rosenblatt [26] in neural networks, laying the solid foundation for the field.
After the initial bricks for the field were laid out, late 1960s welcomed significant enhancement in ML. Some of the iconic algorithms introduced during that time included the nearest neighbor algorithm [27], k-means clustering [28], and cross-validation technique [29]. To improve the neural network accuracy, in 1974, Werbos first described neural network specific back propagation [30], which was then implemented in 1982, leading to a surge in the interest for the field in the years to follow. In 1979, Fukushima introduced neocognitron, a hierarchical multilayered neural network, which was for the first time capable of performing multilayer network training/learning to recognize patterns. In 1982, Hopfield proposed the idea of building a bidirectional network, which later became popularly known as Hopfield network [31], one of the first types of recurrent neural networks. Following these discoveries, in 1983, Hinton and Sejnowski introduced Botlzmann machine, which was stochastic in nature and could be utilized to determine optimal solution (by optimizing the weights in the network) for the associated problem [32]. The earlier discovery of neocognitron by Fukushima in 1979 inspired the development of convolutional neural networks (a type of deep neural network utilized for image processing at the time) in late 80’s to 90’s, including LeNet-1, LeNet-4, and LeNet-5 [33-36].
Alongside these developments, several groups significantly contributed to the field, laying the foundation for theoretical machine learning, including work by Vapnik and Chervonenkis [37] (VC) in 1971, which introduced the concept of VC dimension, a measure of capacity for a classifier to accurately classify data points in a sample, where VC dimension along with training error was utilized to compute the upper bound of the test error. Following this, Valiant in 1984 introduced a probably approximately correct (PAC) learning model, where a model was learned by applying an approximation function [38]. Furthermore, several mathematical methods have been effectively adopted into the ML field to improve its accuracy and precision, including Fisher’s Linear Discriminant Analysis [39], Naive Bayes [40], Least squares [41], Markov Chains [42], etc. The 80s and 90s also witnessed massive development in broad areas of ML, including classification and regression decision trees [43, 44], and boosting techniques [45].
Late 90s and the beginning of the 21st century further contributed to significant advances in machine learning. In fact, 90s introduced advanced algorithms such as support vector machines (SVM) [46], Random Forests [47], bagging technique [48], least absolute shrinkage and selection operator (LASSO) [49], etc., whereas the 21st century witnessed a surge in popularity of algorithms for deep (representation) learning due to the exceptionally good performance of AlexNet on the ImageNet image recognition task [50]. Some of the algorithms introduced since AlexNet included ResNet [51], U-net [52], Google Brain [53], DeepFace [54] etc., revolutionizing the field and creating an arsenal of computational tools to analyze real-life data, efficiently dealing with noise, missing values, and data sparsity.
With high-throughput patient molecular data becoming accessible came the true manifestation of machine learning, with its effective applications in making decisions that can affect patient lives, undoubtedly including its significant-impact utilization in cancer therapeutic response. While relatively recent in its application to treatment response in cancer, machine learning has already established itself as a major player in predictive therapeutic modeling, with significant promise for high impact on patients’ lives and clinical decision making. In particular, most recent applications in this field have included utilization of Random Forests to predict response to chemotherapy in oral squamous cell carcinoma patients [55], support vector machines to predict response to chemotherapy across 19 cancer types available in TCGA [56], and regression-based modeling to predict response to first generation androgen-deprivation therapy in prostate cancer [6], among others [8, 57-62]. This review will focus on the machine learning algorithms that have already been utilized to successfully predict therapeutic response in cancer and will describe mathematical and statistical foundations of their implementation, discuss their limitations and advantages over other methods, and explore future avenues to enhance personalized treatment predictions and precision therapeutics.
2. DATA SOURCES FOR PREDICTING THERAPEUTIC RESPONSE
Predictive modeling of therapeutic response aims to learn relationships between two essential components: predictor variables and response variables and then subsequently utilize predictor variables to predict therapeutic response. Further, predictor variables recapitulate clinical and molecular patient characteristics, where clinical data involves age, gender, race, demographics, initial disease aggressiveness, accompanied treatments, etc., and molecular data includes gene expression, alternative splicing, mutations, epigenomic changes, etc., and is obtained from biopsies, tumor-removing surgery, or blood/urine samples. At the same time, response variables recapitulate treatment-related disease progression, which for example, includes time to treatment failure (e.g., where treatment failure can be defined as detection of minimal residual disease, change in blood markers, tumor re-occurrence, local or distant metastasis, cancer-related death, etc.) or an indication if treatment response was good or poor (often defined for a specific time frame, for example within 6 months, 1-year, or 5-year period).
In recent years, advancements in high throughput technologies have significantly increased the availability of clinical and molecular data in cancer therapeutic response experimental systems. Yet, interpretability and compatibility of different in vitro and in vivo models with human samples have been a long-standing problem, especially for advancing predictive modeling of therapeutic response. In fact, it has been reported that these systems differ in their ability to capture genomic and transcriptomic features of the primary tumors of patients [63], including their microenvironment [64]. Thus, in this review, we specifically focus on data sources derived from therapeutic administration to patients (Fig. 1, Table 1). Examples of such resources include (i) The Tumor Genome Atlas (TCGA) database [1]; (ii) Stand Up To Cancer (SU2C) East Coast project [2, 9, 65, 66]; (iii) Stand Up To Cancer (SU2C) West Coast project [67-69]; (iv) PROstate Cancer Medically Optimized Genome Enhanced ThErapy (PROMOTE) [70]; (v) Cancer Genome Characterization Initiative (CGCI) [71]; (vi) Therapeutically Applicable Research To Generate Effective Treatments (TARGET) [3,72-74]; (vii) Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) database [75]; alongside cohorts from GEO repository, such as (viii) GSE6532 [76]; (ix) GSE1379 [77]; (x) GSE1456 [78]; (xi) GSE78870 [79]; (xii) GSE41994 [80] etc. Some of these resources have already been utilized to study therapeutic response using non- machine learning approaches, including work of (i) Abida et al. [9], which utilized Whole Exome Sequencing data from SU2C East Coast prostate cancer cohort to identify alterations in TP53, RB1 and AR as associated with resistance to androgen receptor signaling inhibitors (ARSI) in metastatic castration-resistant prostate cancer patients; (ii) Epsi et al. [8], which integrated RNA Sequencing and DNA Methylation data from TCGA to identify pathways that govern chemotherapy response in lung adenocarcinoma; and (iii) Oshi et al. [81], which utilized RNA Sequencing data from METABRIC to identify E2F pathway as a predictive marker governing response to neoadjuvant chemotherapy in ER+/HER2- breast cancer.
Fig. (1).

Schematic representation of the workflow for predictive modeling of therapeutic response: Patient clinical information alongside molecular profiles (e.g., Epigenomic alterations, DNA alterations, RNA alterations) are utilized as input to machine learning for predictive modeling of patient therapeutic response. (A higher resolution / colour version of this figure is available in the electronic copy of the article).
Table 1.
Description of data sources for therapeutic response. Detailed description of data sources for predictor variables (e.g., RNA sequencing, DNA methylation, etc.) and response variables (e.g., treatment response etc.).
| Data Sources | Data Types | Cancer Types | Response Variables | Sources |
|---|---|---|---|---|
| TCGA [1] | DNA Methylation | 33 cancer types (including Lung, Breast, Colon, Prostate, etc.) | Overall survival, Disease progression, Treatment response | Genomics Data Commons (GDC) (https://portal.gdc.cancer.gov/) |
| RNA Sequencing | ||||
| miRNA Sequencing | ||||
| Whole Exome Sequencing | ||||
| ATAC Sequencing | ||||
| Genotyping Array | ||||
| SU2C East Coast [9, 65, 66, 82] | RNA Sequencing | Prostate cancer, Pancreatic cancer, Lung cancer | Overall survival, Treatment response | dbGaP phs000915.v2.p2 |
| Whole Exome Sequencing | ||||
| Single Nucleotide Variation | ||||
| SU2C West Coast [67-69] | Bisulfite Sequencing | Prostate cancer, Pancreatic cancer | Treatment response | Genomics Data Commons (GDC) (https://portal.gdc.cancer.gov/projects/WCDT-MCRPC) |
| RNA Sequencing | ||||
| Whole Genome Sequencing | dbGap phs001648.v2.p1 | |||
| PROMOTE [70] | RNA Sequencing | Prostate cancer | Treatment response | dbGaP phs001141.v1.p1 |
| Whole Exome Sequencing | ||||
| Single Nucleotide Polymorphism | ||||
| Cancer Genome Characterization Initiative (CGCI) [71] | RNA Sequencing | Cervical cancer | Overall survival, Disease progression, Treatment response | Genomics Data Commons (GDC) (https://portal.gdc.cancer.gov/projects/CGCI-HTMCP-CC) |
| miRNA Sequencing | ||||
| Whole Genome Sequencing | ||||
| Targeted Sequencing | ||||
| TARGET [3, 72-74] | RNA Sequencing | Acute myeloid leukemia, Acute lymphoblastic leukemia, Neuroblastoma, kidney, Osteosarcoma, Rhabdoid tumor, Wills tumor, Clear cell sarcoma | Overall survival, Treatment response | Genomics Data Commons (GDC) https://portal.gdc.cancer.gov/ |
| miRNA Sequencing | ||||
| Whole Exome Sequencing | ||||
| Whole Genome Sequencing | ||||
| Genotyping Array | ||||
| METABRIC [75] | Copy Number Variation | Breast cancer | Overall survival, Disease specific survival, Treatment response | https://www.synapse.org/#!Synapse:syn1688369/wiki/27311 |
| mRNA Expression (Illumina HT 12 arrays) | ||||
| GSE6532 [76] | mRNA Expression (Affymetrix) | Breast cancer | Treatment response | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE6532 |
| GSE1379 [77] | mRNA Expression (Arcturus 22k human oligonucleotide microarray) | Breast cancer | Treatment response | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE1379 |
| GSE1456 [78] | mRNA Expression (Affymetrix) | Breast cancer | Treatment response | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE1456 |
| GSE78870 [79] | miRNA Expression (TaqMan microRNA Low-Density Array pools A and B version 2.0) | Breast cancer | Treatment response | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE78870 |
| GSE41994 [80] | mRNA Expression (Agilent_ human_DiscoverPrint_15746) | Breast cancer | Treatment response | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE41994 |
3. MACHINE LEARNING FOR TREATMENT RESPONSE: RATIONALE AND STUDY DESIGN
Since the ultimate goal of machine learning in therapeutic predictive modeling is to learn features (i.e., inputs/predictor variables) associated with treatment response (i.e., called outcomes, outputs/response variables, or labels in classical machine learning) and then utilize this knowledge to predict future therapeutic response for new incoming patients, supervised learning (i.e., where outputs are known as ground truth and are actively utilized in the learning process) has earned its solid place in the state-of-the-art therapeutic response modeling. In fact, while unsupervised learning (e.g., k-means [28], Principal Component Analysis [83], etc.) has been widely applied in cancer-related research, it only discovers associations among input variables and does not utilize their relationship to the outputs. On the other hand, supervised learning (e.g., decision tree [84], Random Forests [85], support vector machines [46], regression-based models [86], etc.) utilizes outputs as ground truth and learns relationships between input and output variables so that the final model can be used to predict the outputs for a new set of inputs (e.g., in new patients).
Generally speaking, supervised learning estimates a function f that maps input variable/s X (i.e., predictor) to output variable/s Y (i.e., outcome/response variables), so that,
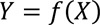
As mentioned above, in predictive modeling of therapeutic response, predictor variables could include clinical patient data (i.e., age, gender, race, initial disease aggressiveness, etc) and molecular data (i.e., gene expression, mutations, epigenomic changes such as DNA methylation, etc) obtained from biopsies, tumor-removing surgery, blood or urine samples, etc. Outcomes/response variables include time to treatment failure (e.g., defined as tumor re-occurrence, local or distant metastasis, or cancer-related death etc) or simply an indication, if treatment response was good or poor (defined using a specific clinical test or time-related threshold, such as a 1-year or a 5-year relapse or survival) [2, 9]. Depending on the type of outcome/response data, supervised learning can either utilize (a) regression model (i.e., output data is continuous, such as time to treatment failure) or (b) classification model (i.e., output data is categorical, such as good or poor response).
In a clinical setting, supervised learning tailored for predictive modeling of therapeutic response utilizes the following three steps: training (i.e., the model is learned/trained), testing (i.e., evaluating the ability of the model to predict outcomes), and forecasting (i.e., outcomes are predicted for new incoming cases) (Fig. 2). To successfully implement the first two steps, supervised learning divides available data into training and test sets (usually training set constitutes 2/3rd and test set 1/3rd of the available data). Training data is utilized to learn the model (function f), while test data is utilized to test the ability of such model to effectively predict outputs. In training step, inputs and outputs (labels) are known to the model and their relationships are actively learned (Fig. 2, Left), while in the test step, the outputs are hidden on purpose and are only uncovered at the end in order to evaluate if the predictions were correct (Fig. 2, Middle). The culmination of such model training and testing results in the third, most important step in clinical decision making - forecasting - predicting outputs/labels for new incoming patients (Fig. 2, Right). If such predictions are later proven to be accurate, these additional data are utilized to re-train and improve the original model.
Fig. (2).

Schematic representation of steps in supervised machine learning to build a predictive model for classification problem. Left: Build/learn the model using training patient cohort. Cross-validation is employed to reduce model variance and improve robustness. Middle: Test and evaluate the model’s performance using the test patient cohort. Right: Forecasting/predicting outcomes for new incoming patients, with subsequent model re-training/improvement. (A higher resolution / colour version of this figure is available in the electronic copy of the article).
One of the essential sub-steps in the training step of supervised learning is cross-validation. Cross-validation allows to mitigate overfitting (where the model can perform well by chance due to the nature of the training set selected) and evaluate how the model is expected to perform on the unseen data. This technique is also utilized to tune parameters when necessary (e.g., for supervised learning methods that require parameter estimation to define f, called parametric models, e.g., linear regression). To achieve this, the training set is divided into k folds/subsets (as for example in Fig. 2, Left, k = 5), so that one of the subsets is kept on-hold and the model is trained on the k-1 subsets. Once trained, the subset on-hold is used to evaluate (i.e., validate) model’s expected accuracy using Mean Squared Error (i.e., the average of the sum of squared difference between actual response and predicted response, MSE), which reflects how far our predictions are from the actual output values. The process is repeated k times, combining MSEs for all folds, followed by averaging it over k, which results in the estimation of cross- validation error. This error is used to evaluate how the constructed model is expected to perform on unseen data or (when parameter tuning) which parameters result in the lowest error and should be selected for optimal model performance.
As a part of supervised learning, the machine learning field has adopted two main methods on how to learn/estimate parameters from training data for prediction purposes: frequentist and conditionalist (i.e., Bayesian) [87]. Frequentists’ viewpoint estimates a parameter that is a constant and assume no prior knowledge for this process [88]. In Bayesian viewpoint, a parameter is viewed as a variable with its own distribution (set of values), utilized to make predictions with degrees of certainty, and prior knowledge is considered for this process [88]. The main difference between these viewpoints is in the way they measure uncertainly in parameter estimation [89]. When frequentist methods obtain a point estimate of the parameters, they do not assign probabilities to possible parameter values. To measure uncertainty, they rely on confidence intervals, where at least 95% of estimated confidence intervals (from enough population samples) are expected to include the true value of the parameter [90]. At the same time, when Bayesian methods estimate a full posterior distribution over the parameters (or point estimates that maximize the posterior distribution), this allows them to get uncertainty of the estimate by integrating the full posterior distribution [91]. In large, utilization of any of these approaches depends on the philosophy, type of prediction we want to achieve (point estimate or probability of potential values) and data availability of appropriate data (i.e., where we have prior knowledge that can be used in the modeling process) [92]. Classical examples of supervised machine learning models that utilize a frequentist approach include logistic and linear regression [93, 94] and those that utilize the Bayesian approach include Bayesian Neural Networks [95], Markov Chain Monte Carlo [96], Bayesian linear regression [97], etc.
These general principles of supervised learning design are utilized as essential building blocks by different machine learning algorithms for predictive modeling of therapeutic response, including tree-based methods (e.g., decision trees and Random Forests), support vector machines, artificial neural networks, and classical regression-based models (e.g., linear regression and logistic regression). Here, we will discuss their mathematical foundations, advantages, disadvantages, and clinical applications, specifically in modeling therapeutic response in cancer patients.
4. SURVEY OF MACHINE LEARNING IN TREATMENT RESPONSE MODELING
4.1. Random Forests
Random Forests is a collection of decision trees [47, 84, 98-101], which have been highly popular in healthcare and medical research due to their interpretability and decision- making capability. A typical decision tree consists of root node, inner nodes, and leaf nodes, all connected by tree branches (Fig. 3). In a decision tree, features/inputs are utilized for each tree split (represented by the root node and internal nodes), allowing to make a decision about the output categorization (outputs or “decisions” are stored at the leaf nodes). For example, in a classical classification example (Fig. 3A), in a dataset with n = 10 patients (i.e., four patients with good response and six patients with poor response) and M = 3 features (i.e., gene A, B, and C), expression level θb of gene B is selected at the root as the most important feature to best split/classify the patients (four patients for the left branch with the expression level of gene B ≤ θb and six patients for the right branch with the expression level of gene B > θb ).
Fig. (3).

Training and testing of a decision tree; (A) Decision tree built using 3 genes A, B and C for 6 poor responders (patients in orange) and 4 good responders (patients in green). At a single expression threshold, entropy gain for each gene is calculated and gene with highest entropy gain is selected as a node for splitting. For example, entropy gain for each gene is calculated (Table, left) and gene B is selected as it has the highest entropy gain (highlighted in red) for the root node. The root node is denoted by a diamond and the intermediate nodes are denoted by a circle. At threshold θb, gene B splits (black arrows) the training set such that 6 patients have gene B expression less than or equal to threshold θb and the 4 patients have expression above threshold θb. All 4 patients with gene B expression less than or equal to threshold θb belong to the poor responder category and, therefore, are represented in leaf node (light blue rectangle). (B) The predictive ability of the decision tree model is evaluated using a test set. (A higher resolution / colour version of this figure is available in the electronic copy of the article).
In general, to select the most important feature at each node split, a decision tree evaluates all provided features and calculates a so-called node purity, which for example, can be estimated by minimizing the residual sum of squares (for regression models), Gini Index or entropy (for classification models). Entropy (E), which conceptually measures the randomness associated with the outcome at each node, is calculated as:
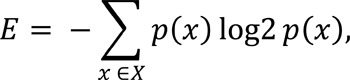
where p(x) is the probability of a category X (i.e., patients with poor or good treatment response) in the training set. It is calculated for each available feature at each node split (starting from the root), so that a feature with the highest entropy gain (compared to the entropy for the entire set) is selected at each split, as described in Fig. (3A) (where expression levels of gene B are selected for a root node split due to its highest gain in entropy - for simplicity, we assume a single expression threshold available for each gene). This principle is employed at each node split until all the samples have been classified or until a certain threshold set by the user or estimated by a tuning parameter is reached (we will touch on Random Forests’ parameters that can be tuned later). Once built, such decision tree is utilized to either make predictions for out-of-bag patients or forecasting for new patients (Fig. 3B).
While a single decision tree is prone to overfitting, an ensemble of decision trees, known as Random Forests, has been widely utilized to increase prediction accuracy [101, 102]. In particular, to reduce variance and increase model robustness, Random Forests utilizes several important techniques, including (i) bootstrapping (where patients are sub- sampled with replacement multiple times and each sub-sample is utilized to build a decision tree) (Fig. 4, top); (ii) feature sub-sampling (only a specific number of features are selected for each tree split) (Fig. 4, middle); (iii) bagging (where the output of sample and feature sub-sampling is integrated and averaged for predictive purposes) (Fig. 4, bottom). Bootstrapping employs sampling with replacement, producing a bagged subset (n bagged patients, sampled with replacement from a patients’ set of size n) and an out-of-bag subset (similar to hold-on cross-validation subset in Fig. 2). On average, during bootstrapping 2/3rd of the training set is utilized to build a bagged subset and 1/3rdof the training set for out-of-bag subset. Each kth round of bootstrapping produces a decision tree, resulting in k decision trees overall (Fig. 4, middle).
Fig. (4).

Schematics for Random Forests. Random Forests has three unique features (i) bootstrapping (top), feature selection (middle) and bagging (bottom). First Random Forests performs bootstrapping whereby n random patients are chosen with replacement from a patient set of size n. This process is continued k times, producing k different patient subsets. Each patient subset (known as bootstrap sample) is used to build a decision tree. Patients considered for building a tree are known as bagged patients and patients that are left out of the bootstrap samples are known as out-of bag samples. Next, to build a tree, at each split, out of total M features available, a subset of m features is considered. This process is known as feature selection. Finally, after building k decision trees, Random Forests performs a procedure called bagging whereby each patient from out of bag subsets is validated across trees that did not use that specific patient while building their trees. The final output by the Random Forests is the prediction from each tree is recorded and the vote is selected as the final output of the model, known as average bagging. Finally, to evaluate the predictive ability of the Random Forests, out of bag error is calculated. (A higher resolution / colour version of this figure is available in the electronic copy of the article).
To ensure that all decision trees in the Random Forests are uncorrelated, each tree split feature sub-sampling is employed. If a total number of features is M, it is recommended that features selected for classification lie within the range of
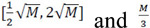 features are selected for the regression model (Fig. 4, middle). Finally, bagging utilizes outputs from bootstrapping and feature sub-sampling so that each sample from the out-of-bag subsets (from each bootstrap round) is validated using decision trees built without utilizing this specific sample. After predictions are made for each sample/patient, bagging utilizes a majority vote to make a final prediction, used to calculate Mean Squared Error or classification error (average misclassifications) (Fig. 4, bottom), thus minimizing model variance. To control for bias- variance trade-off, important parameters in Random Forests to consider and thus tune are the number of trees, tree depth (or number of samples at the leaf nodes), number of features at each tree split etc.
features are selected for the regression model (Fig. 4, middle). Finally, bagging utilizes outputs from bootstrapping and feature sub-sampling so that each sample from the out-of-bag subsets (from each bootstrap round) is validated using decision trees built without utilizing this specific sample. After predictions are made for each sample/patient, bagging utilizes a majority vote to make a final prediction, used to calculate Mean Squared Error or classification error (average misclassifications) (Fig. 4, bottom), thus minimizing model variance. To control for bias- variance trade-off, important parameters in Random Forests to consider and thus tune are the number of trees, tree depth (or number of samples at the leaf nodes), number of features at each tree split etc.
One of the clinically relevant and most widely used outputs in Random Forests is feature importance, which is often used to evaluate which clinical or molecular determinant/s are most important for predicting therapeutic response. It is calculated using the average of the total decrease in Gini Index/ entropy for each feature across all trees (for classification model) or the average of the total decrease in residual sum of squares across all trees (for regression models). Yet, when evaluating feature importance, one should be careful about the presence of collinear features. While not affecting model performance per se, they can reduce the importance of one another and could be easily misinterpreted in the clinical setting.
Due to its robustness and ability to perform well even in moderate-sized datasets, Random Forests has been actively utilized for predictive modeling of treatment response in cancer patients [55, 103-122]. In a classic example by Tsuji et al. [59], Random Forests was implemented to identify gene expression markers to stratify patients based on their response to mFOLFOX therapy in colorectal cancer. A total of 83 patients with colorectal cancer without prior treatment were enrolled and received mFOLFOX6 treatment after sample collection. Out of 83 samples, 54 samples (2/3rd of 83) were selected for training purposes and 29 (1/3rd of 83) for testing. Gene expression profiles (i.e., 17,920 probes) were used as inputs/features. Response to the therapy (outcomes/labels) was assessed through computer tomography (for the appearance of lesions) and evaluated after 4 cycles of the treatment. The multi-layered analysis identified 14 most important genes, which successfully predicted 12 out of 15 (80%) patients with good response and 13 out of 14 (92.8%) patients with poor response in the test set, establishing Random Forests as a robust, reliable method for therapeutic response modeling.
4.2. Support Vector Machines
Support vector machines or SVMs [46, 123, 124] are popularly used for binary classification problems (yet their recent extensions can handle multi-class [125, 126] and regression modeling [127, 128]). Conceptually, SVM is a generalization of the optimal separating (i.e., maximal margin) classifier and support vector (i.e., soft margin) classifier, with the advantage of allowing for misclassified samples and non-linear class boundaries. The main objective of SVM is to identify an optimal hyperplane which would effectively separate classes from each other (e.g., poor responders and good responders). The SVM hyperplane is defined in a way such that the distance between the separating hyperplane and training data observations is maximized (such distance is also known as a margin) (Fig. 5). One can think about the hyperplane as the widest/maximal ribbon that can fit between the two classes (this is classically known as a maximal margin classifier, Fig. 5A). Yet, an advancement over the maximal margin classifier - support vector classifier - allows a margin to be “soft” and have some observations inside a margin or even have some observations (i.e., mismatches) on a wrong part of the hyperplane, having at most epsilon deviation from the hyperplane (Fig. 5B). In support vector classifier, samples that lie directly on the margin are known as support vectors as they “support” the hyperplane (only these observations affect the hyperplane and if they move, the hyperplane would move as well). It is interesting that SVM classification is only based on a small number of observations (i.e., support vectors) and is robust to the observations that are far from the hyperplane/margin. The size of the margin (and the corresponding support vectors) is a parameter to optimize in SVM.
Fig. (5).

Support vector machines determine a hyperplane that can separate patients into two classes. (A) SVM identifies a hyperplane such that it can maximize the margin and no patients lie inside the margin. (B) SVM can also identify hyperplane where patients lie inside the margin. This is known as soft margin. (A higher resolution / colour version of this figure is available in the electronic copy of the article).
A unique and valuable characteristic of SVM in addition to utilizing a support vector classifier is that it works not only with linear but also with non-linear observations. In order to accommodate non-linear boundaries between the classes, SVM enlarges the feature space through kernels (widely used non-linear kernels include polynomial [129], radial [130], and hyperbolic tangent kernels [131]). However, utilization of kernels could be computationally expensive, as it turns optimization involved in SVM in a quadratic programming problem [132-134]. This might cause a computational challenge, especially as data depth and breadth increase, as is the case with Big Data [135-141].
The mathematical way to define a hyperplane (which is M-1 dimensional) is,
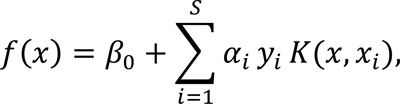
where β0 is the intercept, S is the number of support vectors, αi is the Lagrange multiplier, yi is the class label for a support vector i so that y1…ys are in {-1,1} (where 1 represents one class/good response and -1 the other class/poor response), K(x, xi) is a kernel function, and xi is a feature vector of size M for a support vector i. One can think of hyperplane as an entity that divides M-1 dimensional space into two parts, so that all points/samples with ƒ(x) > 0 lie to one side of the hyperplane and points/samples with ƒ(x) < 0 lie to the other side of the hyperplane [141, 142].
Once SVM classifier is built, the samples to be evaluated/predicted are subjected to ƒ(x) and their class is predicted/assigned based on the sign of the ƒ(x) (i.e., if it is positive, the sample is assigned to class 1 and if it is negative, to class -1). Interestingly, the magnitude of ƒ(x) can suggest how far the observation is from the hyperplane and thus how confident we are in assigning a class membership [143] (i.e., the further away from the hyperplane a sample is, the more confident we are in its predicted membership).
Given its flexibility in allowing mismatches and ability to work with non-linear relationships, SVM have been widely utilized for predictive modeling of treatment response in cancer patients in the last decade [144-164]. One of the bright examples is the work of Huang et al. [60], which developed an open sourced SVM to predict drug response to seven chemotherapeutic drugs using gene expression data across 60 human cancer cell lines. To increase performance accuracy and reduce the number of features (especially important for SVM and discussed later in the Limitations and alternative approaches section), they utilized recursive feature elimination (RFE) approach. The model was tested on 273 ovarian cancer patients and showed significant predictive ability, when compared to previous reports. In addition, the same group later demonstrated that utilization of the SVM-RFE model (i.e., SVM model along with recursive feature elimination approach) when employed on 152 patients with different cancers from TCGA produced predictions of treatment response to gemcitabine and 5-flurouracil with high accuracy > 80% [56].
4.3. Artificial Neural Networks
Artificial neural network (ANN) is an algorithm inspired by the biological neural network of the human brain and has been widely utilized in pattern recognition and image processing [165]. Generally, ANN consists of three parts: one input layer, multiple hidden layers, and one output layer (Fig. 6A). The hidden layers allow for processing of the data that are not linearly separable and if more than one hidden layer is present, the neural network is commonly known as a deep neural network. Inputs to the input layer are predictors (e.g., molecular or clinical features), which are then assigned weights that either amplify or dampen the inputs thus indicating input significance. Value for each predictor (e.g., expression level for a gene) multiplied by its weight (called weighted nodes) along with a bias (which also has its own weight) are summed up in a summation function (also known as Net input function) (Fig. 6B). The output of the summation function is then sent to an activation function which is an important step of the ANN as it directly affects its output, accuracy, convergence, and computational efficiency. Activation function can be as simple as a binary step function (i.e., based on a threshold, determines if a neuron is activated or repressed) or account for non-linear relationships and data complexity utilizing sigmoid, hyperbolic tangent, rectified linear unit, soft-max, swish functions, etc [166-169].
Fig. (6).

Schematics of Artificial Neural Network (ANN). (A) An example of deep neural network which has three components: input layer, 2 hidden layers, and output layer. (B) Each of the predictors in input layer (red and yellow stars) is multiplied by its corresponding weights (black balls) and along with bias is input to net input function (also known as summation function). The output of the summation function is passed through the activation function (i.e., stepwise function), to determine the output of ANN. (A higher resolution / colour version of this figure is available in the electronic copy of the article).
The objective of the training step in ANN is to find the best/optimal set of weights for inputs and bias to solve a specific problem (i.e., treatment response prediction). This is often implemented as a backpropagation [169], where weights for input and bias are optimized to minimize the difference between the actual and the predicted output values (e.g., measured as sum of squared errors or entropy), although this solution is not always global. To control for bias-variance trade-off, the model could be tuned for the number of units in hidden layer and amount of weight decay.
ANN has been utilized by several groups to study treatment response in cancer [170-175]. One of the bright examples is the study of Tadayyon et al. [61], which built an artificial neural network classifier based on quantitative ultrasound imaging to predict response to neoadjuvant chemotherapy for 100 breast cancer patients. The ANN classifier could predict response to the treatment with an accuracy of 96 ± 6%.
4.4. Linear and Logistic Regression
Linear and logistic regressions have earned their historical foundational role in statistical inference and learning and have been widely utilized in treatment response modeling in the recent decade [115, 120, 176-183].
Linear regression estimates linear relationship between input and output variables and fits a so-called regression line (Fig. 7A) in a way so that the sum of the squares of the distances between the line and the data points (i.e., residuals) is minimized. In mathematical terms, function f for a regression line can be re written as:
Fig. (7).

Schematics for linear and logistic regression. (A) Simple linear regression between gene expression (input/predictor variable) and time to treatment response (output/dependent variable) is shown. The regression determines the regression line with slope β 1 and β 0 y-intercept . (B) Logistic regression between gene expression (predictor/independent variable) and response to treatment (response/dependent variable). At a threshold (p=0.5), if the probability of belonging to a specific class is p ≥ 0.5, patients belong to a class (i.e., poor responders) and if p < 0.5, patients belong to another class (i.e., good responders). (A higher resolution / colour version of this figure is available in the electronic copy of the article).
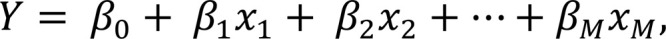
where M is the number of input variables/predictors, β 0 is the y-intercept and β1, β2, ... β M are the slope coefficients for input variables x1, x2, ... xM (reflecting how much each predictor affects the outcome Y). If only one input/predictor variable is present, it is referred to as a simple linear regression and when more than one input/predictor variable is present, it is referred to as a multiple (or multivariable) linear regression. One of the significant extensions of linear regression is Cox proportional hazards modeling, particularly important in modeling therapeutic response, where the outcomes are represented by treatment-related survival time: time to treatment failure or time to latest follow-up (i.e., for censored patients).
In logistic regression, the output is a binary variable (i.e., class membership) and if p is the probability of belonging to a specific output class (e.g., good or poor response), then f takes the following form:
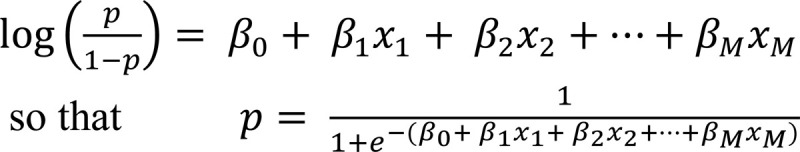
For example, if the probability threshold is p = 0.5, patients with probability p ≥ 0.5 are classified as poor responders and p < 0.5 as good responders (Fig. 7B).
Due to their interpretability and wide dissemination, linear and logistic regression have been widely utilized to model treatment response in cancer [115, 120, 178-183]. For example, Jahani et al. [62], analyzed DCA-MR images of 132 locally advanced breast cancer patients after being treated with neoadjuvant chemotherapy. Voxel-wise changes in morphologic, kinetic, and structural features were quantified using image registration technique. Strength of identified features in determining pathological complete response was evaluated using logistic regression analysis first on a baseline model which included age, race, hormone receptor status, and tumor volume as explanatory variables. Following this, voxel-wise features were added to the baseline model and were shown to improve early prediction of response to neoadjuvant chemotherapy in locally advanced breast cancer patients.
Recently, a series of regression-based methods have been utilized for integration of different data types in predictive therapeutic response modeling. In particular, in Panja et al. [6], linear regression-based analysis was employed to elucidate relationships between epigenomic (i.e., DNA methylation) and transcriptomic (i.e., gene expression) determinants of response to first generation androgen-deprivation therapy in prostate cancer. To specifically study primary resistance, localized primary prostate cancer tumors (at radical prostatectomy) from The Cancer Genome Atlas (TCGA-PRAD) patient cohort, not receiving any treatment prior to sample collection, but treated with adjuvant (post-operative) androgen deprivation therapy, were specifically selected. Linear regression analysis between DNA methylation sites (independent variable) and gene expression of the site-harboring genes (dependent variable) identified 5 site-gene pairs with functional importance in therapeutic response. These markers were shown to differentiate patients at risk of resistance to androgen deprivation therapy in prostate cancer with 90% accuracy and were demonstrated to be active in patients that failed androgen-deprivation with metastatic disease.
In Epsi et al. [8], and Rahem et al. [17], molecular determinants of therapeutic response were evaluated not as single independent entities, but as groups of genes connected by their biological function - biological pathways. These studies utilized logistic regression-based methods and Cox proportional hazard modeling to establish relationship between activity levels of biological pathways (used as features) and therapeutic response to carboplatin + paclitaxel in lung adenocarcinoma [8] and to tamoxifen in breast cancer [17]. Identified pathway markers were shown to accurately stratify patients at risk of resistance across multiple independent patient cohorts (82%-94% accuracy) and have been shown to outperform non-pathway-based methods.
5. LIMITATIONS AND ALTERNATIVE APP- ROACHES
As more clinical and molecular data from cancer patients become available for computational use, machine learning is becoming a backbone for predictive modeling of treatment response. Yet, some of the limitations inherent to its design needs special attention, especially when applied to therapeutic response modeling.
Big Data provides the necessary breadth and depth for the elucidation of complex mechanisms that govern treatment response, yet since its single determinants are used as features/inputs in a machine learning setting, their magnitude can easily overwhelm the system, resulting in overfitting. In fact, it is recommended that the number of features should be significantly less compared to the number of samples/patients M<<n. Given that the thousands of molecular features are routinely profiled using high-throughput technologies, it would require even more patient samples and might not be feasible. To overcome this limitation, various feature selection techniques have been proposed and utilized, including (i) wrapper methods [184-186], which evaluates all possible combination of features to identify optimal set of features that maximize model performance, where commonly used wrapper methods include forward [187], backward [188], stepwise selection [189], simulated annealing [190], genetic algorithms [191], etc.; (ii) filter methods [192-195], which evaluate relevance of predictors outside of the training model (i.e., usually features are evaluated individually), where commonly used filter methods include correlation [196], information theory [197], rough set theory [198], distance measures [199], etc.; (iii) hybrid methods [200-202], which identify features using a combination of both filter and wrapper methods, with most popular being F-score and Supported Sequential Forward Search (FSSFS) method [203], which utilizes F-score (i.e., filter method) to first preprocess and identify a subset of features which is then subjected to supported sequential forward search (i.e., a wrapper method) to identify the final list of features; and (iv) embedded methods [204-206], where feature selection is a part of model selection process, including L1 - regularization based methods such as Least Absolute Shrinkage and Selection Operator (LASSO) [49], which is a regularized linear regression model that penalizes all features equivalently, shrinking unimportant ones (i.e., features which are unlikely to impact response variable) to zero. Apart from LASSO, another commonly used embedded method for feature selection is Smoothly Clipped Absolute Deviation Penalty (SCAD) [207], which penalizes both important and unimportant features, shrinking unimportant features to zero whereas having a lesser impact on important features compared to LASSO. Besides computational methods, feature selection can also be performed through feature masking based on domain knowledge, where users can utilize their domain knowledge to facilitate feature selection. A classic example of such feature selection was described by Yan et al. [208], incorporating prior knowledge of staining pattern to identify texture based features that can help quantify cellular phenotype.
It is possible to pre-select features even prior to feature selection, as is referred to as feature screening, such as (i) Sure Independence Screening (SIS) [209], which determines the association between each predictor and response variable through correlation analysis to determine the important features; (ii) Sure Independence Ranking and Screening (SIRS) [210], which utilizes expectation of squared correlation between a predictor and an indicator function of the response variable to determine a minimum number of important features; (iii) Distance Correlation Sure Independence Screening (DC-SIS) method [211] which screens features based on their distance correlation with response variable (by computing distance between simultaneous observations of each predictor, and as well as simultaneous observations of response variable), etc.
The large number of predictors can also lead to the substantial presence of non-informative features. While this can be easily overcome with some machine learning algorithms (e.g., Random Forests), it might substantially affect the performance of other methods such as multiple linear and logistic regression, SVM and neural networks. One of the solutions is to filter features based on their data cross-integration or biological relevance (e.g., biological pathways, like in Epsi et al. [8] and Rahem et al. [17]) and for their association with therapeutic response ahead of time. Additional advantage in reducing the feature space to informative features only is in the fact that fewer corresponding model terms/parameters need to be optimized, thus improving model performance.
Furthermore, the presence of multiple co-occurring molecular features or a correlation between clinical and molecular features (often observed in therapeutic response data) could lead to feature co-linearity, which can substantially interfere with model performance (e.g., in neural networks and SVM) and could substantially affect its interpretation (e.g., Random Forests’ feature importance is not interpretable in cases of feature co-linearity). To overcome these limitations, in addition to feature selection techniques described above, it is recommended to test for feature co-linearity ahead of time and keep the most important representative feature or the most biologically relevant feature from the group, “eliminating” non-important features. Alternatively, co-linear features could be represented as a group and utilized in the analysis as one entity.
While high throughput techniques to generate Big Data have brought significant advantages to our understanding of cancer progression and therapeutic response, they could be prone to experimental noise or missing values [212]. While some machine learning algorithms are relatively immune to noise or missing data (e.g., Random Forests), others will suffer in terms of their model performance. To address this problem, in the last two decades, several methods have been developed to deal with noise in the data [213-215], including robust regression methods such as M-estimation, S-estimation and MM-estimation [216, 217] and domain knowledge (e.g., pathology expertise) [218]. M-estimation minimizes a function of residuals to estimate coefficients for a regression model, in the presence of outliers (i.e., noise), specifically in response variables [219], yet not taking into account outliers from predictor variables [219]. Thus to overcome this limitation, S-estimation was developed, which modified the residual function of M-estimation by introducing the standard deviation of residuals, being able to handle more diverse sources of noise [220]. However, S-estimation has a major drawback as it requires a large number of samples to accurately estimate coefficients for regression model (i.e., has low efficiency) [220]. Therefore, to compensate for the efficiency and at the same time to have a model which can consider outliers from both predictor and response variables, MM-estimation, a combination of M- and S-estimation, was introduced [220].
At the same time, missing data can substantially affect model performance and accuracy of prediction [221] and can be tackled with (i) expectation-maximization (EM) algorithms [222-224], utilized to estimate missing data from expected complete data by maximizing a likelihood function; (ii) matrix completion-based methods such as simple, complex optimization program [225, 226], which compute a complete low rank matrix from a matrix with missing data by minimizing the nuclear norm.
Furthermore, even though molecular Big Data has produced a lot of features (i.e., M is large), the available datasets for therapeutic response modeling still offer cohorts of relatively small sizes (i.e., n is smaller than M), thus limiting possible machine learning applicability and performance. This is especially important for methods that requires estimation of parameters for each hidden layer (thus the number of parameters is further amplified) such as neural networks, while other methods perform relatively well even in moderate-sized patient cohorts (i.e., linear and logistic regression, Random Forests, etc.). Finally, while linear relationships are the most natural way to start data explorations, molecular Big Data’s complexity and its association with therapeutic response often require non-linear solutions. In such settings, machine learning methods that account for such relationships are preferred, such as Random Forests, SVM with non-linear kernels, or neural nets.
6. DISCUSSION
Recent advancements in Big Data high throughput technology hold a promise to move the field of therapeutic predictive modeling fast forward. Techniques such as CRISPR, ChIP-Seq, HI-C etc. have been widely utilized in cancer research [227-229], with great potential to be effectively expanded to predicting treatment response. One of the most promising shifting paradigms, which has revolutionized cancer research in recent years, is single-cell sequencing [230]. Not only such technique is utilized to analyze complexities of biological systems at single cell level, it also reflects tumor heterogeneity [231-233], clonality [234, 235], and epithelial-stromal interactions [236, 237], opening doors to better precision therapeutics and in-depth monitoring of treatment response, perfectly suited for complex machine learning tasks [238].
While such advances have significantly improved the treatment response investigation, several challenges in the field of therapeutic monitoring remain to be thoroughly addressed. First of all, access to available molecular data in the public domain pose significant challenges when rapid predictions need to be made or results reproduced/validated [239]. Furthermore, the access to facilities and cost of the tumor molecular profiling at the time of biopsy and surgery remain substantial obstacles for many patients and institutions [240] and pose a substantial challenge for subsequent effective application of predictions from multi-omic integrative machine learning techniques [241]. Moreover, this challenge is further amplified if such samples need to be obtained repeatedly, for treatment monitoring [242]. One of the ways to overcome this problem and effectively monitor disease and treatment progression is through utilizing liquid biopsies, a rapid non-invasive technique, which can analyze cancer cells from tumors circulating in the blood [243] and can be applied repeatedly. Such technique has been widely utilized by the cancer community [244, 245] and holds a promise for effective therapeutic monitoring and analyses, providing plethora of data for effective machine learning utilization and accurate predictions.
As the therapeutic monitoring becomes more accessible and molecular datasets become larger (i.e., n increase), we foresee the utilization of more advanced machine learning techniques, which require sufficient number of samples for their optimal performance. One such example is deep learning [57, 246]. The advantage of deep learning is in its ability to capture the biological complexities at a more granular level compared to other machine learning algorithms. One of the algorithms widely utilized for deep learning is deep neural networks (i.e., neural networks with multiple hidden layers), where its additional hidden layers allow for a “deeper” learning. Given the complexity of mechanisms and molecular cross-talks implicated in therapeutic response, deep learning is ideally suited to elucidate mechanisms and markers of therapeutic response, yet in large-sized patient cohorts.
Even though deep learning might offer an elucidation of more complex deep relationships in the data, it often suffers from output interpretability [247, 248], when knowledge about the prediction is essential for a well-informed decision [249, 250]. In deep neural network, tracing which variables are combined to make the prediction could become too complex and hides conditions at which the models can fail (i.e., black box model) [251]. Several alternative solutions have been proposed to overcome this problem, where a complex model is followed by the subsequent explanatory model [252, 253], yet not fully providing an accurate representation [252].
Another example of machine learning algorithms ideally suited for therapeutic modeling is causal methods [254-256]. Causal methods look for causal rather than accidental associations among data points, essential in identifying mechanisms underlying treatment response and novel therapeutic targets. Causal models and analysis have already been used in a clinical setting, such as establishing a causal relationship between lower lipid levels in the body and higher bone mineral density [257], in epidemiology [258], or in cancer progression [259]. Yet, the absolute beauty of causal analysis is obtained with time series data, established by Kleinberg et al. [254, 256, 260], and later applied to cancer progression using cross-sectional data by Ramazzotti et al., [261]. As the availability of time-series monitoring data for therapeutic response in cancer patients is underway, its pressing need, importance, and interpretability will undoubtedly benefit from causal analysis.
We foresee that future utilization of currently utilized approaches for predictive modeling alongside causal analysis, as machine learning paradigm for modeling of therapeutic response, will not only overcome limitations of finding simple association relationships, but will also provide outputs easily interpretable by the clinicians and pave a road to interpretable precision therapeutics.
CONCLUSION
Over the last decade, there has been a significant increase in the utilization of machine learning in predictive modeling of treatment response in cancer patients. In this review, we have discussed machine learning algorithms currently utilized for this purpose, their mathematical foundations, and specific applications in a practical setting. Volume and heterogeneity of Big Data in therapeutic modeling allows for elucidation of complex mechanisms implicated in treatment response, yet requires special considerations due to the large number of unfiltered determinants/features it provides. We have discussed these limitations and approaches to overcome them. We conclude that as patient datasets become larger and better characterized, we foresee effective utilization of deep learning and causal analysis in therapeutic modeling in cancer patients, paving a road to interpretable precise outcomes.
ACKNOWLEDGEMENTS
Declared none.
LIST OF ABBREVIATIONS
- ML
Machine Learning
- DNA
Deoxyribonucleic Acid
- RNA
Ribonucleic Acid
- SVM
Support Vector Machine
- ANN
Artificial Neural Network
- TCGA
The Cancer Genome Atlas
- PRAD
Prostate Adenocarcinoma
- ICGC
International Cancer Genome Consortium
- RFE
Recursive Feature Elimination
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
This work was supported by New Jersey Commission on Cancer Research (DCHS20PPC028) and Rutgers School of Health Professions, USA, start-up funds.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Tomczak K., Czerwińska P., Wiznerowicz M. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp. Oncol. (Pozn.) 2015;19(1A):A68–A77. doi: 10.5114/wo.2014.47136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barrett M.T., Deiotte R., Lenkiewicz E., Malasi S., Holley T., Evers L., Posner R.G., Jones T., Han H., Sausen M., Velculescu V.E., Drebin J., O’Dwyer P., Jameson G., Ramanathan R.K., Von Hoff D.D. Clinical study of genomic drivers in pancreatic ductal adenocarcinoma. Br. J. Cancer. 2017;117(4):572–582. doi: 10.1038/bjc.2017.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.TARGET N. Therapeutically applicable research to generate effective treatments. Available from: https://ocg.cancer.gov/programs/target . [Google Scholar]
- 4.Vamathevan J., Clark D., Czodrowski P., Dunham I., Ferran E., Lee G., Li B., Madabhushi A., Shah P., Spitzer M., Zhao S. Applications of machine learning in drug discovery and development. Nat. Rev. Drug Discov. 2019;18(6):463–477. doi: 10.1038/s41573-019-0024-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Santoro G., Turvani G., Graziano M. New logic-in-memory paradigms: an architectural and technological perspective. Micromachines (Basel) 2019;10(6):368. doi: 10.3390/mi10060368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Panja S., Hayati S., Epsi N.J., Parrott J.S., Mitrofanova A. Integrative (epi) genomic analysis to predict response to androgen-deprivation therapy in prostate cancer. EBioMedicine. 2018;31:110–121. doi: 10.1016/j.ebiom.2018.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dutta A., Panja S., Virk R.K., Kim J.Y., Zott R., Cremers S., Golombos D.M., Liu D., Mosquera J.M., Mostaghel E.A., Barbieri C.E., Mitrofanova A., Abate-Shen C. Co-clinical analysis of a genetically engineered mouse model and human prostate cancer reveals significance of NKX3.1 expression for response to 5α-reductase inhibition. Eur. Urol. 2017;72(4):499–506. doi: 10.1016/j.eururo.2017.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Epsi N.J., Panja S., Pine S.R., Mitrofanova A. pathCHEMO, a generalizable computational framework uncovers molecular pathways of chemoresistance in lung adenocarcinoma. Commun. Biol. 2019;2:334. doi: 10.1038/s42003-019-0572-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abida W., Cyrta J., Heller G., Prandi D., Armenia J., Coleman I., Cieslik M., Benelli M., Robinson D., Van Allen E.M., Sboner A., Fedrizzi T., Mosquera J.M., Robinson B.D., De Sarkar N., Kunju L.P., Tomlins S., Wu Y.M., Nava Rodrigues D., Loda M., Gopalan A., Reuter V.E., Pritchard C.C., Mateo J., Bianchini D., Miranda S., Carreira S., Rescigno P., Filipenko J., Vinson J., Montgomery R.B., Beltran H., Heath E.I., Scher H.I., Kantoff P.W., Taplin M-E., Schultz N., deBono J.S., Demichelis F., Nelson P.S., Rubin M.A., Chinnaiyan A.M., Sawyers C.L. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA. 2019;116(23):11428–11436. doi: 10.1073/pnas.1902651116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kahles A., Lehmann K.-V., Toussaint N. C., Hüser M., Stark S. G., Sachsenberg T., Stegle O., Kohlbacher O., Sander C., Caesar-Johnson S. J. Comprehensive analysis of alternative splicing across tumors from 8,705 patients. Cancer Cell. 2018;34(2):211–224. doi: 10.1016/j.ccell.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cancer Genome Atlas Research Network. The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015;163(4):1011–1025. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aytes A., Giacobbe A., Mitrofanova A., Ruggero K., Cyrta J., Arriaga J., Palomero L., Farran-Matas S., Rubin M.A., Shen M.M., Califano A., Abate-Shen C. NSD2 is a conserved driver of metastatic prostate cancer progression. Nat. Commun. 2018;9(1):5201. doi: 10.1038/s41467-018-07511-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aytes A., Mitrofanova A., Lefebvre C., Alvarez M.J., Castillo- Martin M., Zheng T., Eastham J.A., Gopalan A., Pienta K.J., Shen M.M., Califano A., Abate-Shen C. Cross-species regulatory network analysis identifies a synergistic interaction between FOXM1 and CENPF that drives prostate cancer malignancy. Cancer Cell. 2014;25(5):638–651. doi: 10.1016/j.ccr.2014.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Le Magnen C., Virk R.K., Dutta A., Kim J.Y., Panja S., Lopez-Bujanda Z.A., Califano A., Drake C.G., Mitrofanova A., Abate-Shen C. Cooperation of loss of NKX3.1 and inflammation in prostate cancer initiation. Dis. Model. Mech. 2018;11(11):dmm035139. doi: 10.1242/dmm.035139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dillon L.W., Hayati S., Roloff G.W., Tunc I., Pirooznia M., Mitrofanova A., Hourigan C.S. Targeted RNA-sequencing for the quantification of measurable residual disease in acute myeloid leukemia. Haematologica. 2019;104(2):297–304. doi: 10.3324/haematol.2018.203133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duffy M.J. Predictive markers in breast and other cancers: a review. Clin. Chem. 2005;51(3):494–503. doi: 10.1373/clinchem.2004.046227. [DOI] [PubMed] [Google Scholar]
- 17.Rahem SM, Epsi NJ, Coffman FD, Mitrofanova A. Genome-wide analysis of therapeutic response uncovers molecular pathways governing tamoxifen resistance in ER+ breast cancer. EBioMedicine. 2020;61:103047. doi: 10.1016/j.ebiom.2020.103047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turing A.M.I. Computing machinery and intelligence. Mind. 1950;LIX(236):433–460. doi: 10.1093/mind/LIX.236.433. [DOI] [Google Scholar]
- 19.McCarthy J., Minsky M.L., Rochester N., Shannon C.E. A proposal for the dartmouth summer research project on artificial intelligence, August 31, 1955. AI Mag. 2006;27(4):12–12. [Google Scholar]
- 20.Samuel A.L. Some studies in machine learning using the game of checkers. IBM J. Res. Develop. 1959;3(3):210–229. doi: 10.1147/rd.33.0210. [DOI] [Google Scholar]
- 21.Rosenblatt F. The perceptron: a probabilistic model for information storage and organization in the brain. Psychol. Rev. 1958;65(6):386–408. doi: 10.1037/h0042519. [DOI] [PubMed] [Google Scholar]
- 22.Widrow B., Hoff M.E. Adaptive switching circuits. Stanford Univ. Ca Stanford Electronics Labs; 1960. [DOI] [Google Scholar]
- 23. Widrow, B. Generalization and Information Storage in Networks of Adaline Neurons. In: Self-Organizing Systems, Spartan Books, M.D. Yovits, G.T. Jacobi and G.D. Goldstein, Eds., Washington DC, 1962, pp. 435-461. [Google Scholar]
- 24.Hunt E.B., Marin J., Stone P.J. Experiments in induction. Academic Press. New York: 1966. [Google Scholar]
- 25.Nilsson N. Learning machines. McGraw-Hill; New York: 1965. p. 19652. [Google Scholar]
- 26. Rosenblatt, F. Principles of Neurodynamics: Perceptions and the Theory of Brain Mechanism. Spartan Books: Washington, DC, 1962. [Google Scholar]
- 27.Cover T., Hart P. Nearest neighbor pattern classification. IEEE Trans. Inf. Theory. 1967;13(1):21–27. doi: 10.1109/TIT.1967.1053964. [DOI] [Google Scholar]
- 28.MacQueen J. In: Some methods for classification and analysis of multivariate observations.; Proceedings of the fifth Berkeley symposium on mathematical statistics and probability; Oakland, CA, USA, . 1967. pp. 281–297. [Google Scholar]
- 29.Mosteller F., Tukey J.W. Data analysis, including statistics. Collected Works of John W. Tukey: Graphics. 1988;5:123. [Google Scholar]
- 30. Werbos, P. Beyond Regression: New Tools for Prediction and Analysis in the Behavioral Sciences. Ph.D. Thesis, Harvard University, Cambridge, 1974. [Google Scholar]
- 31.Hopfield J.J. Neural networks and physical systems with emergent collective computational abilities. Proc. Natl. Acad. Sci. USA. 1982;79(8):2554–2558. doi: 10.1073/pnas.79.8.2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hinton G.E., Sejnowski T.J. Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition. Citeseer; In: Optimal perceptual inference. 1983. [Google Scholar]
- 33.LeCun Y., Bottou L., Bengio Y., Haffner P. Gradient-based learning applied to document recognition. Proc. IEEE. 1998;86(11):2278–2324. doi: 10.1109/5.726791. [DOI] [Google Scholar]
- 34.LeCun Y., Jackel L., Bottou L., Cortes C., Denker J.S., Drucker H., Guyon I., Muller U.A., Sackinger E., Simard P. Learning algorithms for classification: A comparison on handwritten digit recognition. Neural Networks. 1995;261:276. [Google Scholar]
- 35.LeCun Y., Boser B.E., Denker J.S., Henderson D., Howard R.E., Hubbard W.E., Jackel L.D. In: Handwritten digit recognition with a back-propagation network. Adv. Neural Inf. Process. Syst. 1990:pp. 396–404. [Google Scholar]
- 36.LeCun Y., Boser B., Denker J.S., Henderson D., Howard R.E., Hubbard W., Jackel L.D. Backpropagation applied to handwritten zip code recognition. Neural Comput. 1989;1(4):541–551. doi: 10.1162/neco.1989.1.4.541. [DOI] [Google Scholar]
- 37.Vapnik V.N., Chervonenkis A.Y. Measures of complexity. Springer; 2015. On the uniform convergence of relative frequencies of events to their probabilities. In: ; pp. 11–30. [DOI] [Google Scholar]
- 38.Valiant L.G. A theory of the learnable. Commun. ACM. 1984;27(11):1134–1142. doi: 10.1145/1968.1972. [DOI] [Google Scholar]
- 39.Fisher R.A. The use of multiple measurements in taxonomic problems. Ann. Eugen. 1936;7(2):179–188. doi: 10.1111/j.1469-1809.1936.tb02137.x. [DOI] [Google Scholar]
- 40.Bayes T. LII. An essay towards solving a problem in the doctrine of chances. By the late Rev. Mr. Bayes, FRS communicated by Mr. Price, in a letter to John Canton, AMFR S. Philos. Trans. R. Soc. Lond. 1763;53:370–418. [Google Scholar]
- 41.Legendre A.M. Nouvelles méthodes pour la détermination des orbites des comètes. F. Didot: Paris; 1805. p. 1752-1833. [Google Scholar]
- 42.Markov A.A. Extension of the law of large numbers to dependent quantities. Izv. Fiz.-Matem. Obsch. Kazan Univ. 1906;15:135–156. [Google Scholar]
- 43.Quinlan J.R. Induction of decision trees. Mach. Learn. 1986;1(1):81–106. doi: 10.1007/BF00116251. [DOI] [Google Scholar]
- 44.Breiman L., Friedman J., Stone C.J., Olshen R.A. Classification and regression trees. CRC press; 1984. [Google Scholar]
- 45.Schapire R.E. The strength of weak learnability. Mach. Learn. 1990;5(2):197–227. doi: 10.1007/BF00116037. [DOI] [Google Scholar]
- 46.Cortes C., Vapnik V. Support-vector networks. Mach. Learn. 1995;20(3):273–297. doi: 10.1007/BF00994018. [DOI] [Google Scholar]
- 47.Breiman L. Random forests. Mach. Learn. 2001;45(1):5–32. doi: 10.1023/A:1010933404324. [DOI] [Google Scholar]
- 48.Breiman L. Bagging predictors. Mach. Learn. 1996;24(2):123–140. doi: 10.1007/BF00058655. [DOI] [Google Scholar]
- 49.Tibshirani R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. B. 1996;58(1):267–288. doi: 10.1111/j.2517-6161.1996.tb02080.x. [DOI] [Google Scholar]
- 50.Krizhevsky A., Sutskever I., Hinton G.E. In: Imagenet classification with deep convolutional neural networks. Advances in Neural Information Processing. System. 2012:pp. 1097–1105. [Google Scholar]
- 51.Targ S., Almeida D., Lyman K. Resnet in resnet: Generalizing residual architectures. arXiv preprint. 2016.
- 52.Ronneberger O., Fischer P., Brox T. In: U-net: Convolutional networks for biomedical image segmentation.; International Conference on Medical Image Computing and Computer-assisted Intervention; 2015. pp. 234–241. [DOI] [Google Scholar]
- 53.Abadi M., Barham P., Chen J., Chen Z., Davis A., Dean J., Devin M., Ghemawat S., Irving G., Isard M. In: Google Brain. Tensorflow: A system for large-scale machine learning.; Proceedings of the 12th USENIX Symposium on Operating Systems Design and Implementation (OSDI’16); 2016. pp. 265–284. [Google Scholar]
- 54.Taigman Y., Yang M., Ranzato M.A., Wolf L. In: Deepface: Closing the gap to human-level performance in face verification.; Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition; 2014. pp. 1701–1708. [DOI] [Google Scholar]
- 55.Schomberg J. Identification of targetable pathways in oral cancer patients via random forest and chemical informatics. Cancer Inform. 2019;18:1176935119889911. doi: 10.1177/1176935119889911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang C., Clayton E.A., Matyunina L.V., McDonald L.D., Benigno B.B., Vannberg F., McDonald J.F. Machine learning predicts individual cancer patient responses to therapeutic drugs with high accuracy. Sci. Rep. 2018;8(1):16444. doi: 10.1038/s41598-018-34753-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu Y., Hosny A., Zeleznik R., Parmar C., Coroller T., Franco I., Mak R.H., Aerts H.J.W.L. deep learning predicts lung cancer treatment response from serial medical imaging. Clin. Cancer Res. 2019;25(11):3266–3275. doi: 10.1158/1078-0432.CCR-18-2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Falgreen S., Dybkær K., Young K.H., Xu-Monette Z.Y., El- Galaly T.C., Laursen M.B., Bødker J.S., Kjeldsen M.K., Schmitz A., Nyegaard M., Johnsen H.E., Bøgsted M. Predicting response to multidrug regimens in cancer patients using cell line experiments and regularised regression models. BMC Cancer. 2015;15:235–235. doi: 10.1186/s12885-015-1237-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsuji S., Midorikawa Y., Takahashi T., Yagi K., Takayama T., Yoshida K., Sugiyama Y., Aburatani H. Potential responders to FOLFOX therapy for colorectal cancer by Random Forests analysis. Br. J. Cancer. 2012;106(1):126–132. doi: 10.1038/bjc.2011.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang C., Mezencev R., McDonald J.F., Vannberg F. Open source machine-learning algorithms for the prediction of optimal cancer drug therapies. PLoS One. 2017;12(10):e0186906. doi: 10.1371/journal.pone.0186906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tadayyon H., Gangeh M., Sannachi L., Trudeau M., Pritchard K., Ghandi S., Eisen A., Look-Hong N., Holloway C., Wright F., Rakovitch E., Vesprini D., Tran W.T., Curpen B., Czarnota G. A priori prediction of breast tumour response to chemotherapy using quantitative ultrasound imaging and artificial neural networks. Oncotarget. 2019;10(39):3910–3923. doi: 10.18632/oncotarget.26996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jahani N., Cohen E., Hsieh M-K., Weinstein S.P., Pantalone L., Hylton N., Newitt D., Davatzikos C., Kontos D. Prediction of treatment response to neoadjuvant chemotherapy for breast cancer via early changes in tumor heterogeneity captured by DCE-MRI registration. Sci. Rep. 2019;9(1):12114. doi: 10.1038/s41598-019-48465-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu K., Chen B., Aran D., Charalel J., Yau C., Wolf D.M., van ’t Veer L.J., Butte A.J., Goldstein T., Sirota M. Comprehensive transcriptomic analysis of cell lines as models of primary tumors across 22 tumor types. Nat. Commun. 2019;10(1):3574. doi: 10.1038/s41467-019-11415-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Niu N., Wang L. In vitro human cell line models to predict clinical response to anticancer drugs. Pharmacogenomics. 2015;16(3):273–285. doi: 10.2217/pgs.14.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Blackford A.L., Canto M.I., Klein A.P., Hruban R.H., Goggins M. Recent trends in the incidence and survival of stage 1A pancreatic cancer: A surveillance, epidemiology, and end results analysis. J. Natl. Cancer Inst. 2020;112(11):1162–1169. doi: 10.1093/jnci/djaa004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Skoulidis F., Goldberg M.E., Greenawalt D.M., Hellmann M.D., Awad M.M., Gainor J.F., Schrock A.B., Hartmaier R.J., Trabucco S.E., Gay L., Edwards R., Bufill J.A., Sharma N., Ou S.-H.I, Peled N., Spigel D.R., Rizvi H., Aguilar E.J., Carter B.W, Erasmus J., Halpenny D.F., Wistuba I.I., Miller V.A., Frampton G.M., Wolchok J.D., Shaw A.T., Jänne P.A., Stephens P.J., Rudin C.M., Geese W.J., Albacker L.A., Heymach J.V. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 2018;8(7):822. doi: 10.1158/2159-8290.CD-18-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Quigley D.A., Dang H.X., Zhao S.G., Lloyd P., Aggarwal R., Alumkal J.J., Foye A., Kothari V., Perry M.D., Bailey A.M., Playdle D., Barnard T.J., Zhang L., Zhang J., Youngren J.F., Cieslik M.P., Parolia A., Beer T.M., Thomas G., Chi K.N., Gleave M., Lack N.A., Zoubeidi A., Reiter R.E., Rettig M.B., Witte O., Ryan C.J., Fong L., Kim W., Friedlander T., Chou J., Li H., Das R., Li H., Moussavi-Baygi R., Goodarzi H., Gilbert L.A., Lara P.N., Jr, Evans C.P., Goldstein T.C., Stuart J.M., Tomlins S.A., Spratt D.E., Cheetham R.K., Cheng D.T., Farh K., Gehring J.S., Hakenberg J., Liao A., Febbo P.G., Shon J., Sickler B., Batzoglou S., Knudsen K.E., He H.H., Huang J., Wyatt A.W., Dehm S.M., Ashworth A., Chinnaiyan A.M., Maher C.A., Small E.J., Feng F.Y. Genomic hallmarks and structural variation in metastatic prostate cancer. Cell. 2018;174(3):758–769.e9. doi: 10.1016/j.cell.2018.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Aggarwal R., Huang J., Alumkal J.J., Zhang L., Feng F.Y., Thomas G.V., Weinstein A.S., Friedl V., Zhang C., Witte O.N., Lloyd P., Gleave M., Evans C.P., Youngren J., Beer T.M., Rettig M., Wong C.K., True L., Foye A., Playdle D., Ryan C.J., Lara P., Chi K.N., Uzunangelov V., Sokolov A., Newton Y., Beltran H., Demichelis F., Rubin M.A., Stuart J.M., Small E.J. Clinical and genomic characterization of treatment-emergent small-cell neuroendocrine prostate cancer: a multi-institutional prospective study. J. Clin. Oncol. 2018;36(24):2492–2503. doi: 10.1200/JCO.2017.77.6880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Highlander S.K., Chung V., Alistar A.T., Borazanci E., Jameson G., Pearson T., Cantafio N.A., Hoff D.D.V. Abstract B24: Gastrointestinal microbiome changes in stage IV pancreatic cancer patients treated with pembrolizumab with or without paricalcitol on the Stand Up to Cancer (SU2C) Pancreas Catalyst Trial. Cancer Res. 2019;79(24) Suppl.:B24. [Google Scholar]
- 70.Wang L., Dehm S.M., Hillman D.W., Sicotte H., Tan W., Gormley M., Bhargava V., Jimenez R., Xie F., Yin P., Qin S., Quevedo F., Costello B.A., Pitot H.C., Ho T., Bryce A.H., Ye Z., Li Y., Eiken P., Vedell P.T., Barman P., McMenomy B.P., Atwell T.D., Carlson R.E., Ellingson M., Eckloff B.W., Qin R., Ou F., Hart S.N., Huang H., Jen J., Wieben E.D., Kalari K.R., Weinshilboum R.M., Wang L., Kohli M. A prospective genome-wide study of prostate cancer metastases reveals association of wnt pathway activation and increased cell cycle proliferation with primary resistance to abiraterone acetate-prednisone. Ann. Oncol. 2018;29(2):352–360. doi: 10.1093/annonc/mdx689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gagliardi A., Porter V.L., Zong Z., Bowlby R., Titmuss E., Namirembe C., Griner N.B., Petrello H., Bowen J., Chan S.K., Culibrk L., Darragh T.M., Stoler M.H., Wright T.C., Gesuwan P., Dyer M.A., Ma Y., Mungall K.L., Jones S.J.M., Nakisige C., Novik K., Orem J., Origa M., Gastier-Foster J.M., Yarchoan R., Casper C., Mills G.B., Rader J.S., Ojesina A.I., Gerhard D.S., Mungall A.J., Marra M.A. Analysis of Ugandan cervical carcinomas identifies human papillomavirus clade-specific epigenome and transcriptome landscapes. Nat. Genet. 2020;52(8):800–810. doi: 10.1038/s41588-020-0673-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mullighan C.G., Su X., Zhang J., Radtke I., Phillips L.A., Miller C.B., Ma J., Liu W., Cheng C., Schulman B.A., Harvey R.C., Chen I.M., Clifford R.J., Carroll W.L., Reaman G., Bowman W.P., Devidas M., Gerhard D.S., Yang W., Relling M.V., Shurtleff S.A., Campana D., Borowitz M.J., Pui C.H., Smith M., Hunger S.P., Willman C.L., Downing J.R. Children’s Oncology Group. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N. Engl. J. Med. 2009;360(5):470–480. doi: 10.1056/NEJMoa0808253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pugh T.J., Morozova O., Attiyeh E.F., Asgharzadeh S., Wei J.S., Auclair D., Carter S.L., Cibulskis K., Hanna M., Kiezun A., Kim J., Lawrence M.S., Lichenstein L., McKenna A., Pedamallu C.S., Ramos A.H., Shefler E., Sivachenko A., Sougnez C., Stewart C., Ally A., Birol I., Chiu R., Corbett R.D., Hirst M., Jackman S.D., Kamoh B., Khodabakshi A.H., Krzywinski M., Lo A., Moore R.A., Mungall K.L., Qian J., Tam A., Thiessen N., Zhao Y., Cole K.A., Diamond M., Diskin S.J., Mosse Y.P., Wood A.C., Ji L., Sposto R., Badgett T., London W.B., Moyer Y., Gastier-Foster J.M., Smith M.A., Guidry Auvil J.M., Gerhard D.S., Hogarty M.D., Jones S.J., Lander E.S., Gabriel S.B., Getz G., Seeger R.C., Khan J., Marra M.A., Meyerson M., Maris J.M. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 2013;45(3):279–284. doi: 10.1038/ng.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Eleveld T.F., Oldridge D.A., Bernard V., Koster J., Colmet Daage L., Diskin S.J., Schild L., Bentahar N.B., Bellini A., Chicard M., Lapouble E., Combaret V., Legoix-Né P., Michon J., Pugh T.J., Hart L.S., Rader J., Attiyeh E.F., Wei J.S., Zhang S., Naranjo A., Gastier-Foster J.M., Hogarty M.D., Asgharzadeh S., Smith M.A., Guidry Auvil J.M., Watkins T.B., Zwijnenburg D.A., Ebus M.E., van Sluis P., Hakkert A., van Wezel E., van der Schoot C.E., Westerhout E.M., Schulte J.H., Tytgat G.A., Dolman M.E., Janoueix-Lerosey I., Gerhard D.S., Caron H.N., Delattre O., Khan J., Versteeg R., Schleiermacher G., Molenaar J.J., Maris J.M. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat. Genet. 2015;47(8):864–871. doi: 10.1038/ng.3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Curtis C., Shah S.P., Chin S-F., Turashvili G., Rueda O.M., Dunning M.J., Speed D., Lynch A.G., Samarajiwa S., Yuan Y., Gräf S., Ha G., Haffari G., Bashashati A., Russell R., McKinney S., Langerød A., Green A., Provenzano E., Wishart G., Pinder S., Watson P., Markowetz F., Murphy L., Ellis I., Purushotham A., Børresen-Dale A.L., Brenton J.D., Tavaré S., Caldas C., Aparicio S. METABRIC Group. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486(7403):346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Loi S., Haibe-Kains B., Desmedt C., Lallemand F., Tutt A.M., Gillet C., Ellis P., Harris A., Bergh J., Foekens J.A., Klijn J.G., Larsimont D., Buyse M., Bontempi G., Delorenzi M., Piccart M.J., Sotiriou C. Definition of clinically distinct molecular subtypes in estrogen receptor-positive breast carcinomas through genomic grade. J. Clin. Oncol. 2007;25(10):1239–1246. doi: 10.1200/JCO.2006.07.1522. [DOI] [PubMed] [Google Scholar]
- 77.Ma X.J., Wang Z., Ryan P.D., Isakoff S.J., Barmettler A., Fuller A., Muir B., Mohapatra G., Salunga R., Tuggle J.T., Tran Y., Tran D., Tassin A., Amon P., Wang W., Wang W., Enright E., Stecker K., Estepa-Sabal E., Smith B., Younger J., Balis U., Michaelson J., Bhan A., Habin K., Baer T.M., Brugge J., Haber D.A., Erlander M.G., Sgroi D.C. A two-gene expression ratio predicts clinical outcome in breast cancer patients treated with tamoxifen. Cancer Cell. 2004;5(6):607–616. doi: 10.1016/j.ccr.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 78.Pawitan Y., Bjöhle J., Amler L., Borg A.L., Egyhazi S., Hall P., Han X., Holmberg L., Huang F., Klaar S., Liu E.T., Miller L., Nordgren H., Ploner A., Sandelin K., Shaw P.M., Smeds J., Skoog L., Wedrén S., Bergh J. Gene expression profiling spares early breast cancer patients from adjuvant therapy: derived and validated in two population-based cohorts. Breast Cancer Res. 2005;7(6):R953–R964. doi: 10.1186/bcr1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ramirez-Ardila D.E., Ruigrok-Ritstier K., Helmijr J.C., Look M.P., van Laere S., Dirix L., Berns E.M.J.J., Jansen M.P.H.M. LRG1 mRNA expression in breast cancer associates with PIK3CA genotype and with aromatase inhibitor therapy outcome. Mol. Oncol. 2016;10(8):1363–1373. doi: 10.1016/j.molonc.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jansen M.P., Knijnenburg T., Reijm E.A., Simon I., Kerkhoven R., Droog M., Velds A., van Laere S., Dirix L., Alexi X., Foekens J.A., Wessels L., Linn S.C., Berns E.M., Zwart W. Hallmarks of aromatase inhibitor drug resistance revealed by epigenetic profiling in breast cancer. Cancer Res. 2013;73(22):6632–6641. doi: 10.1158/0008-5472.CAN-13-0704. [DOI] [PubMed] [Google Scholar]
- 81.Oshi M., Tokumaru Y., Katsuta E., Yan L., Endo I., Takabe K. E2F cell cycle pathway score as a predictive biomarker of ER+/HER2- breast cancer response to neoadjuvant chemotherapy. J. Clin. Oncol. 2020;38(15):e12593–e12593. [Google Scholar]
- 82.Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, Colin C.; Attard, G.; Beltran, H.; Abida, W.; Bradley, Robert K.; Vinson, J.; Cao, X.; Vats, P.; Kunju, Lakshmi P.; Hussain, M.; Feng, Felix Y.; Tomlins, Scott A.; Cooney, K.A.; Smith, D.C.; Brennan, C.; Siddiqui, J.; Mehra, R.; Chen, Y.; Rathkopf, D.E.; Morris, M.J.; Solomon, S.B.; Durack, J.C.; Reuter, V.E.; Gopalan, A.; Gao, J.; Loda, M.; Lis, R.T.; Bowden, M.; Balk, S.P.; Gaviola, G.; Sougnez, C.; Gupta, M.; Yu, E.Y.; Mostaghel, Elahe A.; Cheng, Heather H.; Mulcahy, H.; True, Lawrence D.; Plymate, Stephen R.; Dvinge, H.; Ferraldeschi, R.; Flohr, P.; Miranda, S.; Zafeiriou, Z.; Tunariu, N.; Mateo, J.; Perez-Lopez, R.; Demichelis, F.; Robinson, Brian D.; Schiffman, M.; Nanus, David M.; Tagawa, Scott T.; Sigaras, A.; Eng, Kenneth W.; Elemento, O.; Sboner, A.; Heath, E.I.; Scher, H.I.; Pienta, K.J.; Kantoff, P.; de Bono, J.S.; Rubin, M.A.; Nelson, P.S.; Garraway, L.A.; Sawyers, C.L.; Chinnaiyan, A.M. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161(5):1215–1228. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pearson K. LIII. On lines and planes of closest fit to systems of points in space. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1901;2(11):559–572. doi: 10.1080/14786440109462720. [DOI] [Google Scholar]
- 84.Azar A.T., El-Metwally S.M. Decision tree classifiers for automated medical diagnosis. Neural Comput. Appl. 2013;23(7):2387–2403. doi: 10.1007/s00521-012-1196-7. [DOI] [Google Scholar]
- 85.Liaw A., Wiener M. Classification and regression by randomForest. R News. 2002;2(3):18–22. [Google Scholar]
- 86.Liu C., Li B., Vorobeychik Y., Oprea A. In: Robust linear regression against training data poisoning.; Proceedings of the 10th ACM Workshop on Artificial Intelligence and Security; 2017. pp. 91–102. [DOI] [Google Scholar]
- 87.Guyon I., Saffari A., Dror G., Cawley G. Model selection: Beyond the bayesian/frequentist divide. J. Mach. Learn. Res. 2010;11(1):61–87. [Google Scholar]
- 88.Perkins J., Wang D. A comparison of Bayesian and frequentist statistics as applied in a simple repeated measures example. J. Mod. Appl. Stat. Methods. 2004;3(1):24. doi: 10.22237/jmasm/1083371040. [DOI] [Google Scholar]
- 89.Bickel P., Lehmann E. Selected Works of EL Lehmann. Springer; 2012. Frequentist interpretation of probability. pp. 1083–1085. [DOI] [Google Scholar]
- 90.Bzdok D., Altman N., Krzywinski M. Statistics versus machine learning. Nat. Methods. 2018;15(4):233–234. doi: 10.1038/nmeth.4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Vilares I., Kording K. Bayesian models: the structure of the world, uncertainty, behavior, and the brain. Ann. N. Y. Acad. Sci. 2011;1224(1):22–39. doi: 10.1111/j.1749-6632.2011.05965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hackenberger B.K. Bayes or not Bayes, is this the question? Croat. Med. J. 2019;60(1):50–52. doi: 10.3325/cmj.2019.60.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tolles J., Meurer W.J. Logistic regression: relating patient characteristics to outcomes. JAMA. 2016;316(5):533–534. doi: 10.1001/jama.2016.7653. [DOI] [PubMed] [Google Scholar]
- 94.Seal H.L. Studies in the history of probability and statistics. XV. The historical development of the Gauss linear model. Biometrika. 1967;54(1):1–24. [PubMed] [Google Scholar]
- 95.Bishop C.M. Bayesian neural networks. J. Braz. Comput. Soc. 1997, 4(1), Available from: https://www.scielo.br/j/jbcos/a/ NYV8LvmFH7pWpC77KZJQwSP/?lang=en#top . [Google Scholar]
- 96.Hayes B. First links in the Markov chain. Am. Sci. 2013;101(2):252. doi: 10.1511/2013.101.92. [DOI] [Google Scholar]
- 97.Box G.E., Tiao G.C. Bayesian inference in statistical analysis. John Wiley & Sons, Vol. 40, 2011. [Google Scholar]
- 98.Tanner L., Schreiber M., Low J.G.H., Ong A., Tolfvenstam T., Lai Y.L., Ng L.C., Leo Y.S., Thi Puong L., Vasudevan S.G., Simmons C.P., Hibberd M.L., Ooi E.E. Decision tree algorithms predict the diagnosis and outcome of dengue fever in the early phase of illness. PLoS Negl. Trop. Dis. 2008;2(3):e196–e196. doi: 10.1371/journal.pntd.0000196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Barach P., Levashenko V., Zaitseva E. Fuzzy Decision Trees in Medical Decision Making Support Systems.; Proceedings of the International Symposium on Human Factors and Ergonomics in Health Care; 2019. pp. 37–42. [DOI] [Google Scholar]
- 100.Valdes G., Luna J.M., Eaton E., Simone C.B., II, Ungar L.H., Solberg T.D. MediBoost: a patient stratification tool for interpretable decision making in the era of precision medicine. Sci. Rep. 2016;6(1):37854. doi: 10.1038/srep37854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Horiguchi K., Toi M., Horiguchi S., Sugimoto M., Naito Y., Hayashi Y., Ueno T., Ohno S., Funata N., Kuroi K., Tomita M., Eishi Y. Predictive value of CD24 and CD44 for neoadjuvant chemotherapy response and prognosis in primary breast cancer patients. J. Med. Dent. Sci. 2010;57(2):165–175. [PubMed] [Google Scholar]
- 102.Kureshi N., Abidi S.S., Blouin C. A predictive model for personalized therapeutic interventions in non-small cell lung cancer. IEEE J. Biomed. Health Inform. 2016;20(1):424–431. doi: 10.1109/JBHI.2014.2377517. [DOI] [PubMed] [Google Scholar]
- 103.Desbordes P., Ruan S., Modzelewski R., Pineau P., Vauclin S., Gouel P., Michel P., Di Fiore F., Vera P., Gardin I. Predictive value of initial FDG-PET features for treatment response and survival in esophageal cancer patients treated with chemo-radiation therapy using a random forest classifier. PLoS One. 2017;12(3):e0173208. doi: 10.1371/journal.pone.0173208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shi M., He J. SNRFCB: sub-network based random forest classifier for predicting chemotherapy benefit on survival for cancer treatment. Mol. Biosyst. 2016;12(4):1214–1223. doi: 10.1039/C5MB00399G. [DOI] [PubMed] [Google Scholar]
- 105.Rahman R., Matlock K., Ghosh S., Pal R. Heterogeneity aware random forest for drug sensitivity prediction. Sci. Rep. 2017;7(1):11347. doi: 10.1038/s41598-017-11665-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gardin I., Fdhila M., Desbordes P., Smadja J., Lebtahi R., Dieudonne A. Predictive value of dosimetry indices for treatment response in liver cancer patients treated with yttrium 90 microspheres using a random forest algorithm. J. Nucl. Med. 2017;58(Suppl. 1):197–197. [Google Scholar]
- 107.Nguyen L., Naulaerts S., Bomane A., Bruna A., Ghislat G., Ballester P.J. Machine learning models to predict in vivo drug response via optimal dimensionality reduction of tumour molecular profiles. bioRxiv. 2018 [Google Scholar]
- 108.Tabib S., Larocque D. Non-parametric individual treatment effect estimation for survival data with random forests. Bioinformatics. 2020;36(2):629–636. doi: 10.1093/bioinformatics/btz602. [DOI] [PubMed] [Google Scholar]
- 109.Jeong S.Y., Kim W., Byun B.H., Kong C.B., Song W.S., Lim I., Lim S.M., Woo S.K. Prediction of chemotherapy response of osteosarcoma using baseline 18F-FDG textural features machine learning approaches with PCA. Contrast Media Mol. Imaging. 2019;2019:3515080. doi: 10.1155/2019/3515080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gonçalves-Ribeiro S., Sanz-Pamplona R., Vidal A., Sanjuan X., Guillen Díaz-Maroto N., Soriano A., Guardiola J., Albert N., Martínez-Villacampa M., López I., Santos C., Serra- Musach J., Salazar R., Capellà G., Villanueva A., Molleví D.G. Prediction of pathological response to neoadjuvant treatment in rectal cancer with a two-protein immunohistochemical score derived from stromal gene-profiling. Ann. Oncol. 2017;28(9):2160–2168. doi: 10.1093/annonc/mdx293. [DOI] [PubMed] [Google Scholar]
- 111.Sørlie T., Perou C.M., Fan C., Geisler S., Aas T., Nobel A., Anker G., Akslen L.A., Botstein D., Børresen-Dale A-L., Lønning P.E. Gene expression profiles do not consistently predict the clinical treatment response in locally advanced breast cancer. Mol. Cancer Ther. 2006;5(11):2914–2918. doi: 10.1158/1535-7163.MCT-06-0126. [DOI] [PubMed] [Google Scholar]
- 112.Lu M., Sadiq S., Feaster D.J., Ishwaran H. Estimating individual treatment effect in observational data using random forest methods. J. Comput. Graph. Stat. 2018;27(1):209–219. doi: 10.1080/10618600.2017.1356325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gan Z., Zou Q., Lin Y., Xu Z., Huang Z., Chen Z., Lv Y. Identification of a 13-gene-based classifier as a potential biomarker to predict the effects of fluorouracil-based chemotherapy in colorectal cancer. Oncol. Lett. 2019;17(6):5057–5063. doi: 10.3892/ol.2019.10159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mani S., Chen Y., Arlinghaus L.R., Li X., Chakravarthy A.B., Bhave S.R., Welch E.B., Levy M.A., Yankeelov T.E. Early prediction of the response of breast tumors to neoadjuvant chemotherapy using quantitative MRI and machine learning. AMIA Annu. Symp. Proc. 2011;2011:868–877. [PMC free article] [PubMed] [Google Scholar]
- 115.Midorikawa Y., Tsuji S., Takayama T., Aburatani H. Genomic approach towards personalized anticancer drug therapy. Pharmacogenomics. 2012;13(2):191–199. doi: 10.2217/pgs.11.157. [DOI] [PubMed] [Google Scholar]
- 116.Daemen A., Griffith O.L., Heiser L.M., Wang N.J., Enache O.M., Sanborn Z., Pepin F., Durinck S., Korkola J.E., Griffith M., Hur J.S., Huh N., Chung J., Cope L., Fackler M.J., Umbricht C., Sukumar S., Seth P., Sukhatme V.P., Jakkula L.R., Lu Y., Mills G.B., Cho R.J., Collisson E.A., van’t Veer L.J., Spellman P.T., Gray J.W. Modeling precision treatment of breast cancer. Genome Biol. 2013;14(10):R110. doi: 10.1186/gb-2013-14-10-r110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Menden M.P., Iorio F., Garnett M., McDermott U., Benes C.H., Ballester P.J., Saez-Rodriguez J. Machine learning prediction of cancer cell sensitivity to drugs based on genomic and chemical properties. PLoS One. 2013;8(4):e61318–e61318. doi: 10.1371/journal.pone.0061318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Stetson L.C., Pearl T., Chen Y., Barnholtz-Sloan J.S. Computational identification of multi-omic correlates of anticancer therapeutic response. BMC Genomics. 2014;15(Suppl. 7):S2. doi: 10.1186/1471-2164-15-S7-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dean J.A., Wong K.H., Welsh L.C., Jones A.B., Schick U., Newbold K.L., Bhide S.A., Harrington K.J., Nutting C.M., Gulliford S.L. Normal tissue complication probability (NTCP) modelling using spatial dose metrics and machine learning methods for severe acute oral mucositis resulting from head and neck radiotherapy. Radiother. Oncol. 2016;120(1):21–27. doi: 10.1016/j.radonc.2016.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Abajian A., Murali N., Savic L.J., Laage-Gaupp F.M., Nezami N., Duncan J.S., Schlachter T., Lin M., Geschwind J.F., Chapiro J. Predicting treatment response to intra-arterial therapies for hepatocellular carcinoma with the use of supervised machine learning-an artificial intelligence concept. J. Vasc. Interv. Radiol. 2018;29(6):850–857.e1. doi: 10.1016/j.jvir.2018.01.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lind A.P., Anderson P.C. Predicting drug activity against cancer cells by random forest models based on minimal genomic information and chemical properties. PLoS One. 2019;14(7):e0219774. doi: 10.1371/journal.pone.0219774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shen W-C., Chen S-W., Wu K-C., Lee P-Y., Feng C-L., Hsieh T-C., Yen K-Y., Kao C-H. Predicting pathological complete response in rectal cancer after chemoradiotherapy with a random forest using 18F-fluorodeoxyglucose positron emission tomography and computed tomography radiomics. Ann. Transl. Med. 2020;8(5):207. doi: 10.21037/atm.2020.01.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Vapnik V. Estimation of Dependences Based on Empirical Data: Springer Series in Statistics (Springer Series in Statistics). Springer-Verlag; 1982. [Google Scholar]
- 124.Duda R.O., Hart P.E., Stork D.G. Pattern classification. John Wiley & Sons; 2012. [Google Scholar]
- 125.Hsu C.W., Lin C.J. A comparison of methods for multiclass support vector machines. IEEE Trans. Neural Netw. 2002;13(2):415–425. doi: 10.1109/72.991427. [DOI] [PubMed] [Google Scholar]
- 126.Duan K-B., Keerthi S.S. In: An Empirical Study, Multiple Classifier Systems. Oza N.C., Polikar R., Kittler J., Roli F., editors. Springer; Berlin, Heidelberg: 2005. In: Which Is the Best Multiclass SVM Method? pp. 278–285. [Google Scholar]
- 127.Drucker H., Burges C.J., Kaufman L., Smola A.J., Vapnik V. In: Support vector regression machines. Adv. Neural Inf. Process. Syst. 1997:155–161. [Google Scholar]
- 128.Ho C-H., Lin C-J. Large-scale linear support vector regression. J. Mach. Learn. Res. 2012;13(1):3323–3348. [Google Scholar]
- 129.dos Santos E.M., Gomes H.M. In: A comparative study of polynomial kernel svm applied to appearance-based object recognition.; International Workshop on Support Vector Machines; 2002. pp. 408–418. [DOI] [Google Scholar]
- 130.Ye Z., Li H. In: Based on Radial Basis Kernel function of Support Vector Machines for speaker recognition.; 2012 5th International Congress on Image and Signal Processing.; 16-18 Oct, ; 2012. pp. 1584–1587. [Google Scholar]
- 131.Kuhn M., Johnson K. Applied predictive modeling. Springer; 2013. p. 26. [DOI] [Google Scholar]
- 132.Bottou L., Lin C.-J. Support vector machine solvers. Large scale kernel machines. 2007;3(1):301–320. [Google Scholar]
- 133.González-Mendoza M., Hernández-Gress N., Titli A. Quadratic Optimization Fine Tuning for the Learning Phase of SVM. In: Ramos F.F., Larios R.V., Unger H., editors. Advanced Distributed Systems. Springer; Berlin, Heidelberg: 2005. pp. 347–357. [Google Scholar]
- 134.Scheinberg K. An efficient implementation of an active set method for SVMs. J. Mach. Learn. Res. 2006;7:2237–2257. [Google Scholar]
- 135.Ben-Hur A., Ong C.S., Sonnenburg S., Schölkopf B., Rätsch G. Support vector machines and kernels for computational biology. PLOS Comput. Biol. 2008;4(10):e1000173–e1000173. doi: 10.1371/journal.pcbi.1000173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Camps-Valls G., Bruzzone L. Kernel-based methods for hyperspectral image classification. IEEE Trans. Geosci. Remote Sens. 2005;43(6):1351–1362. doi: 10.1109/TGRS.2005.846154. [DOI] [Google Scholar]
- 137.Haasdonk B. Feature space interpretation of SVMs with indefinite kernels. IEEE Trans. Pattern Anal. Mach. Intell. 2005;27(4):482–492. doi: 10.1109/TPAMI.2005.78. [DOI] [PubMed] [Google Scholar]
- 138.Frénay B., Verleysen M. Using SVMs with randomised feature spaces: an extreme learning approach. ESANN; 2010. [Google Scholar]
- 139.Sonnenburg S., Rätsch G., Schäfer C., Schölkopf B. Large scale multiple kernel learning. J. Mach. Learn. Res. 2006;7:1531–1565. [Google Scholar]
- 140.Leslie C.S., Eskin E., Cohen A., Weston J., Noble W.S. Mismatch string kernels for discriminative protein classification. Bioinformatics. 2004;20(4):467–476. doi: 10.1093/bioinformatics/btg431. [DOI] [PubMed] [Google Scholar]
- 141.Cervantes J., Garcia-Lamont F., Rodríguez-Mazahua L., Lopez A. A comprehensive survey on support vector machine classification: Applications, challenges and trends. Neurocomputing. 2020;408:189–215. doi: 10.1016/j.neucom.2019.10.118. [DOI] [Google Scholar]
- 142.AACR Project GENIE Consortium. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017;7(8):818–831. doi: 10.1158/2159-8290.CD-17-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.James G., Witten D., Hastie T., Tibshirani R. An introduction to statistical learning. Springer; 2013. p. 112. [DOI] [Google Scholar]
- 144.Sannachi L., Gangeh M., Tadayyon H., Gandhi S., Wright F.C., Slodkowska E., Curpen B., Sadeghi-Naini A., Tran W., Czarnota G.J. Breast cancer treatment response monitoring using quantitative ultrasound and texture analysis: comparative analysis of analytical models. Transl. Oncol. 2019;12(10):1271–1281. doi: 10.1016/j.tranon.2019.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Klement R. J., Allgäuer M., Appold S., Dieckmann K., Ernst I., Ganswindt U., Holy R., Nestle U., Nevinny-Stickel M., Semrau S., Sterzing F., Wittig A., Andratschke N., Guckenberger M. Support vector machine-based prediction of local tumor control after stereotactic body radiation therapy for early-stage non-small cell lung cancer. Int. J. Radiation Oncol. Biol. Phys. 2014;88(3):732–738. doi: 10.1016/j.ijrobp.2013.11.216. [DOI] [PubMed] [Google Scholar]
- 146.Sobhani N., Generali D., Roviello G. PAK6-associated support vector machine classifier: a new way to evaluate response and survival of gastric cancer treated by 5-FU/oxaliplatin chemotherapy. EBioMedicine. 2017;22:18–19. doi: 10.1016/j.ebiom.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lu T-P., Kuo K-T., Chen C-H., Chang M-C., Lin H-P., Hu Y-H., Chiang Y-C., Cheng W-F., Chen C-A. Developing a prognostic gene panel of epithelial ovarian cancer patients by a machine learning model. Cancers (Basel) 2019;11(2):10.3390. doi: 10.3390/cancers11020270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Mucaki E.J., Zhao J.Z.L., Lizotte D., Rogan P.K. Predicting response to platin chemotherapy agents with biochemically-inspired machine learning. bioRxiv. 2018;2018:4. doi: 10.1038/s41392-018-0034-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wu S., Jiao Y., Zhang Y., Ren X., Li P., Yu Q., Zhang Q., Wang Q., Fu S. Imaging-based individualized response prediction of carbon ion radiotherapy for prostate cancer patients. Cancer Manag. Res. 2019;11:9121–9131. doi: 10.2147/CMAR.S214020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Dorman S.N., Baranova K., Knoll J.H.M., Urquhart B.L., Mariani G., Carcangiu M.L., Rogan P.K. Genomic signatures for paclitaxel and gemcitabine resistance in breast cancer derived by machine learning. Mol. Oncol. 2016;10(1):85–100. doi: 10.1016/j.molonc.2015.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ahuja K., Rather G.M., Lin Z., Sui J., Xie P., Le T., Bertino J.R., Javanmard M. Toward point-of-care assessment of patient response: a portable tool for rapidly assessing cancer drug efficacy using multifrequency impedance cytometry and supervised machine learning. Microsyst. Nanoeng. 2019;5(1):34. doi: 10.1038/s41378-019-0073-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Polano M., Chierici M., Dal Bo M., Gentilini D., Di Cintio F., Baboci L., Gibbs D.L., Furlanello C., Toffoli G. A pan-cancer approach to predict responsiveness to immune checkpoint inhibitors by machine learning. Cancers (Basel) 2019;11(10):1562. doi: 10.3390/cancers11101562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Mucaki E.J., Baranova K., Pham H.Q., Rezaeian I., Angelov D., Ngom A., Rueda L., Rogan P.K. Predicting outcomes of hormone and chemotherapy in the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) Study by biochemically-inspired machine learning. F1000 Res. 2016;5(2124):2124. doi: 10.12688/f1000research.9417.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Shayesteh S.P., Alikhassi A., Fard Esfahani A., Miraie M., Geramifar P., Bitarafan-Rajabi A., Haddad P. Neo-adjuvant chemoradiotherapy response prediction using MRI based ensemble learning method in rectal cancer patients. Phys. Med. 2019;62:111–119. doi: 10.1016/j.ejmp.2019.03.013. [DOI] [PubMed] [Google Scholar]
- 155.Cheng C., Komljenovic D., Pan L., Dimitrakopoulou-Strauss A., Strauss L., Bäuerle T. Evaluation of treatment response of cilengitide in an experimental model of breast cancer bone metastasis using dynamic PET with 18F-FDG. Hell. J. Nucl. Med. 2011;14(1):15–20. [PubMed] [Google Scholar]
- 156.Land W. H., Margolis D., Gottlieb R., Krupinski E. A., Yang J. Y. Improving CT prediction of treatment response in patients with metastatic colorectal carcinoma using statistical learning theory. BMC genomics. 2010;11(Suppl 3):S15–S15. doi: 10.1186/1471-2164-11-S3-S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Han M., Dai J., Zhang Y., Lin Q., Jiang M., Xu X., Liu Q., Jia J. Support vector machines coupled with proteomics approaches for detecting biomarkers predicting chemotherapy resistance in small cell lung cancer. Oncol. Rep. 2012;28(6):2233–2238. doi: 10.3892/or.2012.2037. [DOI] [PubMed] [Google Scholar]
- 158.Zhang J., Jia J., Zhu F., Ma X., Han B., Wei X., Tan C., Jiang Y., Chen Y. Analysis of bypass signaling in EGFR pathway and profiling of bypass genes for predicting response to anticancer EGFR tyrosine kinase inhibitors. Mol. Biosyst. 2012;8(10):2645–2656. doi: 10.1039/c2mb25165e. [DOI] [PubMed] [Google Scholar]
- 159.Imani F., Boada F.E., Lieberman F.S., Davis D.K., Mountz J.M. Molecular and metabolic pattern classification for detection of brain glioma progression. Eur. J. Radiol. 2014;83(2):e100–e105. doi: 10.1016/j.ejrad.2013.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Jiang Y., Xie J., Han Z., Liu W., Xi S., Huang L., Huang W., Lin T., Zhao L., Hu Y., Yu J., Zhang Q., Li T., Cai S., Li G. Immunomarker support vector machine classifier for prediction of gastric cancer survival and adjuvant chemotherapeutic benefit. Clin. Cancer Res. 2018;24(22):5574–5584. doi: 10.1158/1078-0432.CCR-18-0848. [DOI] [PubMed] [Google Scholar]
- 161.Dong Z., Zhang N., Li C., Wang H., Fang Y., Wang J., Zheng X. Anticancer drug sensitivity prediction in cell lines from baseline gene expression through recursive feature selection. BMC Cancer. 2015;15:489. doi: 10.1186/s12885-015-1492-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Choy C.T., Wong C.H., Chan S.L. Embedding of genes using cancer gene expression data: biological relevance and potential application on biomarker discovery. Front. Genet. 2019;9:682. doi: 10.3389/fgene.2018.00682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hazai E., Hazai I., Ragueneau-Majlessi I., Chung S.P., Bikadi Z., Mao Q. Predicting substrates of the human breast cancer resistance protein using a support vector machine method. BMC Bioinformatics. 2013;14:130. doi: 10.1186/1471-2105-14-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kourou K., Exarchos T.P., Exarchos K.P., Karamouzis M.V., Fotiadis D.I. Machine learning applications in cancer prognosis and prediction. Comput. Struct. Biotechnol. J. 2014;13:8–17. doi: 10.1016/j.csbj.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Neal R.M. Connectionist learning of belief networks. Artif. Intell. 1992;56(1):71–113. doi: 10.1016/0004-3702(92)90065-6. [DOI] [Google Scholar]
- 166.LeCun Y., Bengio Y., Hinton G. Deep learning. Nature. 2015;521(7553):436–444. doi: 10.1038/nature14539. [DOI] [PubMed] [Google Scholar]
- 167.Nair V., Hinton G.E. Rectified linear units improve restricted boltzmann machines. ICML; 2010. [Google Scholar]
- 168.Badrinarayanan V., Kendall A., Cipolla R. Segnet: A deep convolutional encoder-decoder architecture for image segmentation. IEEE Trans. Pattern Anal. Mach. Intell. 2017;39(12):2481–2495. doi: 10.1109/TPAMI.2016.2644615. [DOI] [PubMed] [Google Scholar]
- 169.Rumelhart D.E., Hinton G.E., Williams R.J. Learning representations by back-propagating errors. Nature. 1986;323(6088):533–536. doi: 10.1038/323533a0. [DOI] [Google Scholar]
- 170.Chen Y-C., Chang Y-C., Ke W-C., Chiu H-W. Cancer adjuvant chemotherapy strategic classification by artificial neural network with gene expression data: An example for non-small cell lung cancer. J. Biomed. Inform. 2015;56:1–7. doi: 10.1016/j.jbi.2015.05.006. [DOI] [PubMed] [Google Scholar]
- 171.Nasief H., Zheng C., Schott D., Hall W., Tsai S., Erickson B., Allen Li X. A machine learning based delta-radiomics process for early prediction of treatment response of pancreatic cancer. NPJ Precision Oncol. 2019;3(1):25. doi: 10.1038/s41698-019-0096-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Aghaei F., Tan M., Hollingsworth A.B., Qian W., Liu H., Zheng B. Computer-aided breast MR image feature analysis for prediction of tumor response to chemotherapy. Med. Phys. 2015;42(11):6520–6528. doi: 10.1118/1.4933198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Bilsland A.E., Pugliese A., Liu Y., Revie J., Burns S., McCormick C., Cairney C.J., Bower J., Drysdale M., Narita M., Sadaie M., Keith W.N. Identification of a selective G1-phase benzimidazolone inhibitor by a senescence-targeted virtual screen using artificial neural networks. Neoplasia. 2015;17(9):704–715. doi: 10.1016/j.neo.2015.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Gulliford S.L., Webb S., Rowbottom C.G., Corne D.W., Dearnaley D.P. Use of artificial neural networks to predict biological outcomes for patients receiving radical radiotherapy of the prostate. Radiother. Oncol. 2004;71(1):3–12. doi: 10.1016/j.radonc.2003.03.001. [DOI] [PubMed] [Google Scholar]
- 175.Murphy G.P., Snow P., Simmons S.J., Tjoa B.A., Rogers M.K., Brandt J., Healy C.G., Bolton W.E., Rodbold D. Use of artificial neural networks in evaluating prognostic factors determining the response to dendritic cells pulsed with PSMA peptides in prostate cancer patients. Prostate. 2000;42(1):67–72. doi: 10.1002/(SICI)1097-0045(20000101)42:1<67::AID-PROS8>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 176.Gore S.D., Fenaux P., Santini V., Bennett J.M., Silverman L.R., Seymour J.F., Hellström-Lindberg E., Swern A.S., Beach C.L., List A.F. A multivariate analysis of the relationship between response and survival among patients with higher-risk myelodysplastic syndromes treated within azacitidine or conventional care regimens in the randomized AZA-001 trial. Haematologica. 2013;98(7):1067–1072. doi: 10.3324/haematol.2012.074831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Lamarca A., Barriuso J., Kulke M., Borbath I., Lenz H-J., Raoul J.L., Meropol N.J., Lombard-Bohas C., Posey J., Faivre S., Raymond E., Valle J.W. Determination of an optimal response cut-off able to predict progression-free survival in patients with well-differentiated advanced pancreatic neuroendocrine tumours treated with sunitinib: an alternative to the current RECIST-defined response. Br. J. Cancer. 2018;118(2):181–188. doi: 10.1038/bjc.2017.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Tran W.T., Gangeh M.J., Sannachi L., Chin L., Watkins E., Bruni S.G., Rastegar R.F., Curpen B., Trudeau M., Gandhi S., Yaffe M., Slodkowska E., Childs C., Sadeghi-Naini A., Czarnota G.J. Predicting breast cancer response to neoadjuvant chemotherapy using pretreatment diffuse optical spectroscopic texture analysis. Br. J. Cancer. 2017;116(10):1329–1339. doi: 10.1038/bjc.2017.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Devasia T., Dewaraja Y., Roberson P., Schipper M. Logistic regression models for accurate prediction of tumor control using radiobiologic dose metrics in Y-90 radioembolization. J. Nucl. Med. 2019;60(Suppl. 1):120. [Google Scholar]
- 180.Song B-N., Kim S-K., Mun J-Y., Choi Y-D., Leem S-H., Chu I-S. Identification of an immunotherapy-responsive molecular subtype of bladder cancer. EBioMedicine. 2019;50:238–245. doi: 10.1016/j.ebiom.2019.10.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Xu Y., Dong Q., Li F., Xu Y., Hu C., Wang J., Shang D., Zheng X., Yang H., Zhang C., Shao M., Meng M., Xiong Z., Li X., Zhang Y. Identifying subpathway signatures for individualized anticancer drug response by integrating multi-omics data. J. Transl. Med. 2019;17(1):255. doi: 10.1186/s12967-019-2010-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Bhojwani D., Kang H., Menezes R.X., Yang W., Sather H., Moskowitz N.P., Min D-J., Potter J.W., Harvey R., Hunger S.P., Seibel N., Raetz E.A., Pieters R., Horstmann M.A., Relling M.V., den Boer M.L., Willman C.L., Carroll W.L. Children’s Oncology Group Study; Dutch Childhood Oncology Group; German Cooperative Study Group for Childhood Acute Lymphoblastic Leukemia. Gene expression signatures predictive of early response and outcome in high-risk childhood acute lymphoblastic leukemia: A Children’s Oncology Group Study [corrected]. J. Clin. Oncol. 2008;26(27):4376–4384. doi: 10.1200/JCO.2007.14.4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Wang Q., He Z., Chen Y. Comprehensive analysis reveals a 4-gene signature in predicting response to Temozolomide in low-grade glioma patients. Cancer Control: J. Moffitt Cancer Center. 2019;26(1):1073274819855118. doi: 10.1177/1073274819855118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Caruana R., Freitag D. Machine Learning Proceedings 1994. Elsevier; 1994. Greedy attribute selection. pp. 28–36. [DOI] [Google Scholar]
- 185.Dy J.G., Brodley C.E. Feature subset selection and order identification for unsupervised learning, ICML. Citeseer; 2000. pp. 247–254. [Google Scholar]
- 186.Kohavi R., John G.H. Wrappers for feature subset selection. Artif. Intell. 1997;97(1-2):273–324. doi: 10.1016/S0004-3702(97)00043-X. [DOI] [Google Scholar]
- 187.Whitney A.W. A direct method of nonparametric measurement selection. IEEE Trans. Comput. 1971;100(9):1100–1103. doi: 10.1109/T-C.1971.223410. [DOI] [Google Scholar]
- 188.Marill T., Green D. On the effectiveness of receptors in recognition systems. IEEE Trans. Inf. Theory. 1963;9(1):11–17. doi: 10.1109/TIT.1963.1057810. [DOI] [Google Scholar]
- 189.Yongkang K., Seungyeoun L., Min-Seok K., Ahrum N., Yonghwan C., Sung Gon Y., Junghyun N., Sangjo H., Meejoo K., Sun Whe K., Jin-Young J., Yikwon K., Youngsoo K., Taesung P. In: Developing cancer prediction model based on stepwise selection by AUC measure for proteomics data.; 2015 IEEE International Conference on Bioinformatics and Biomedicine (BIBM); 2015. pp. 1345–1350. [DOI] [Google Scholar]
- 190.Mafarja M.M., Mirjalili S. Hybrid whale optimization algorithm with simulated annealing for feature selection. Neurocomputing. 2017;260:302–312. doi: 10.1016/j.neucom.2017.04.053. [DOI] [Google Scholar]
- 191.Frohlich H., Chapelle O., Scholkopf B. Proceedings, 15th IEEE International Conference on Tools with Artificial Intelligence. IEEE; 2003. In: Feature selection for support vector machines by means of genetic algorithm, pp. 142–148. [DOI] [Google Scholar]
- 192.Dash M., Choi K., Scheuermann P., Liu H. In: Feature selection for clustering-a filter solution.; 2002 IEEE International Conference on Data Mining; 2002. pp. 115–122. [DOI] [Google Scholar]
- 193. Hall, M.A. Correlation-based feature selection of discrete and numeric class machine learning. ICML '00: Proceedings of the Seventeenth International Conference on Machine Learning, June 2000, pp. 359-366. [Google Scholar]
- 194.Liu H., Setiono R. A probabilistic approach to feature selection-a filter solution, ICML. Citeseer; 1996. pp. 319–327. [Google Scholar]
- 195.Yu L., Liu H. In: Feature selection for high-dimensional data: A fast correlation-based filter solution.; Proceedings of the 20th international conference on machine learning (ICML-03); 2003. pp. 856–863. [Google Scholar]
- 196. Hall, M.A. Correlation-based feature selection for machine learning. 1999, Ph.D. Thesis, University of Waikato, New Zealand, 1974, p. 178. [Google Scholar]
- 197.Kumar G., Kumar K. An information theoretic approach for feature selection. Secur. Commun. Netw. 2012;5(2):178–185. doi: 10.1002/sec.303. [DOI] [Google Scholar]
- 198.Anaraki J.R., Eftekhari M. In: Rough set based feature selection: A Review.; 5th Conference on Information and Knowledge Technology.; 28-30 May, ; 2013. pp. 301–306. [Google Scholar]
- 199.Yu Q., Jiang S-J., Wang R-C., Wang H-Y. A feature selection approach based on a similarity measure for software defect prediction. Front. Inform. Technol. Electronic Eng. 2017;18(11):1744–1753. doi: 10.1631/FITEE.1601322. [DOI] [Google Scholar]
- 200.Renders J.M., Flasse S.P. Hybrid methods using genetic algorithms for global optimization. IEEE Trans. Syst. Man Cybern. B Cybern. 1996;26(2):243–258. doi: 10.1109/3477.485836. [DOI] [PubMed] [Google Scholar]
- 201.Wang S., Wei Y., Li D., Zhang W., Li W. Fourth International Conference on Fuzzy Systems and Knowledge Discovery (FSKD 2007) IEEE; 2007. In: A hybrid method of feature selection for Chinese text sentiment classification. pp. 435–439. [DOI] [Google Scholar]
- 202.Hsu H-H., Hsieh C-W., Lu M-D. Hybrid feature selection by combining filters and wrappers. Expert Syst. Appl. 2011;38(7):8144–8150. doi: 10.1016/j.eswa.2010.12.156. [DOI] [Google Scholar]
- 203.Lee M-C. Using support vector machine with a hybrid feature selection method to the stock trend prediction. Expert Syst. Appl. 2009;36(8):10896–10904. doi: 10.1016/j.eswa.2009.02.038. [DOI] [Google Scholar]
- 204.Lal T.N., Chapelle O., Weston J., Elisseeff A. Feature extraction. Springer; 2006. Embedded methods. pp. 137–165. [DOI] [Google Scholar]
- 205.Wang S., Tang J., Liu H. In: Embedded unsupervised feature selection.; Twenty-ninth AAAI conference on artificial intelligence.; 2015. [Google Scholar]
- 206.Maldonado S., López J. Dealing with high-dimensional class-imbalanced datasets: Embedded feature selection for SVM classification. Appl. Soft Comput. 2018;67:94–105. doi: 10.1016/j.asoc.2018.02.051. [DOI] [Google Scholar]
- 207.Fan J., Li R. Variable selection via nonconcave penalized likelihood and its oracle properties. J. Am. Stat. Assoc. 2001;96(456):1348–1360. doi: 10.1198/016214501753382273. [DOI] [Google Scholar]
- 208.Yan D., Wang P., Knudsen B.S., Linden M., Randolph T.W. Statistical methods for tissue array images - algorithmic scoring and co-training. Ann. Appl. Stat. 2012;6(3):1280–1305. doi: 10.1214/12-AOAS543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Zhang H.H. Discussion of "sure independence screening for ultra-high dimensional feature space. J. R. Stat. Soc. Series B Stat. Methodol. 2008;70(5):903–903. doi: 10.1111/j.1467-9868.2008.00674.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Zhu L., Li L., Li R., Zhu L. Model-free feature screening for ultrahigh dimensional data. J. Am. Stat. Assoc. 2011;106(496):1464–1475. doi: 10.1198/jasa.2011.tm10563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Li R., Zhong W., Zhu L. Feature screening via distance correlation learning. J. Am. Stat. Assoc. 2012;107(499):1129–1139. doi: 10.1080/01621459.2012.695654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Bohannan Z.S., Mitrofanova A. Calling variants in the clinic: informed variant calling decisions based on biological, clinical, and laboratory variables. Comput. Struct. Biotechnol. J. 2019;17:561–569. doi: 10.1016/j.csbj.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Gupta S., Gupta A. Dealing with noise problem in machine learning data-sets: A systematic review. Procedia Comput. Sci. 2019;161:466–474. doi: 10.1016/j.procs.2019.11.146. [DOI] [Google Scholar]
- 214.Nie Y., Yu J. Mining breast cancer genes with a network based noise-tolerant approach. BMC Syst. Biol. 2013;7(1):49. doi: 10.1186/1752-0509-7-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Guo Z., Zhang T., Li X., Wang Q., Xu J., Yu H., Zhu J., Wang H., Wang C., Topol E.J., Wang Q., Rao S. Towards precise classification of cancers based on robust gene functional expression profiles. BMC Bioinformatics. 2005;6:58–58. doi: 10.1186/1471-2105-6-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Alma Ö.G. Comparison of robust regression methods in linear regression. Int. J. Contemp. Math. Sciences. 2011;6(9):409–421. [Google Scholar]
- 217.Almetwally E., Almongy H. Comparison between m estimation, s estimation, and mm estimation methods of robust estimation with application and simulation. Int. J. Math. Archive. 2018;9(11):1–9. [Google Scholar]
- 218.Yan D., Randolph T.W., Zou J., Gong P. Incorporating deep features in the analysis of tissue microarray images. arXiv preprint. 2018. [DOI] [PMC free article] [PubMed]
- 219.Huber P.J. Robust regression: Asymptotics, conjectures and monte carlo. Ann. Stat. 1973;1(5):799–821. doi: 10.1214/aos/1176342503. [DOI] [Google Scholar]
- 220.Susanti Y., Pratiwi H. M estimation, S estimation, and MM estimation in robust regression. Int. J. Pure Appl. Math. 2014;91(3):349–360. doi: 10.12732/ijpam.v91i3.7. [DOI] [Google Scholar]
- 221.Kang H. The prevention and handling of the missing data. Korean J. Anesthesiol. 2013;64(5):402–406. doi: 10.4097/kjae.2013.64.5.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Dempster A.P., Laird N.M., Rubin D.B. Maximum likelihood from incomplete data via the EM Algorithm. J. R. Stat. Soc. B. 1977;39(1):1–22. doi: 10.1111/j.2517-6161.1977.tb01600.x. [DOI] [Google Scholar]
- 223.Little R., Rubin D. Statistical Analysis with Missing Data. Wiley, New York; 2002. A taxonomy of missing-data methods. p. 19. [Google Scholar]
- 224.Kaplan E.L., Meier P. Nonparametric estimation from incomplete observations. J. Am. Stat. Assoc. 1958;53(282):457–481. doi: 10.1080/01621459.1958.10501452. [DOI] [Google Scholar]
- 225.Candès E.J., Recht B. Exact matrix completion via convex optimization. Found. Comput. Math. 2009;9(6):717. doi: 10.1007/s10208-009-9045-5. [DOI] [Google Scholar]
- 226.Recht B. A simpler approach to matrix completion. J. Mach. Learn. Res. 2011;12(12):3413–3430. [Google Scholar]
- 227.Ben-Hamo R., Jacob Berger A., Gavert N., Miller M., Pines G., Oren R., Pikarsky E., Benes C.H., Neuman T., Zwang Y., Efroni S., Getz G., Straussman R. Predicting and affecting response to cancer therapy based on pathway-level biomarkers. Nat. Commun. 2020;11(1):3296. doi: 10.1038/s41467-020-17090-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Kobunai T., Matsuoka K., Takechi T. ChIP-seq analysis to explore DNA replication profile in Trifluridine-treated human colorectal cancer cells in vitro. Anticancer Res. 2019;39(7):3565–3570. doi: 10.21873/anticanres.13502. [DOI] [PubMed] [Google Scholar]
- 229.Chen H., Li C., Peng X., Zhou Z., Weinstein J.N., Caesar-Johnson S.J., Demchok J.A., Felau I., Kasapi M., Ferguson M.L. A pan-cancer analysis of enhancer expression in nearly 9000 patient samples. Cell. 2018;173(2):386–399. doi: 10.1016/j.cell.2018.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Mitra A.K., Mukherjee U.K., Harding T., Jang J.S., Stessman H., Li Y., Abyzov A., Jen J., Kumar S., Rajkumar V., Van Ness B. Single-cell analysis of targeted transcriptome predicts drug sensitivity of single cells within human myeloma tumors. Leukemia. 2016;30(5):1094–1102. doi: 10.1038/leu.2015.361. [DOI] [PubMed] [Google Scholar]
- 231.Levitin H.M., Yuan J., Sims P.A. Single-cell transcriptomic analysis of tumor heterogeneity. Trends Cancer. 2018;4(4):264–268. doi: 10.1016/j.trecan.2018.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.González-Silva L., Quevedo L., Varela I. Tumor functional heterogeneity unraveled by scRNA-seq technologies. Trends Cancer. 2020;6(1):13–19. doi: 10.1016/j.trecan.2019.11.010. [DOI] [PubMed] [Google Scholar]
- 233.Ren X., Kang B., Zhang Z. Understanding tumor ecosystems by single-cell sequencing: promises and limitations. Genome Biol. 2018;19(1):211. doi: 10.1186/s13059-018-1593-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Singh M., Al-Eryani G., Carswell S., Ferguson J.M., Blackburn J., Barton K., Roden D., Luciani F., Giang Phan T., Junankar S., Jackson K., Goodnow C.C., Smith M.A., Swarbrick A. High-throughput targeted long-read single cell sequencing reveals the clonal and transcriptional landscape of lymphocytes. Nat. Commun. 2019;10(1):3120. doi: 10.1038/s41467-019-11049-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Meir Z., Mukamel Z., Chomsky E., Lifshitz A., Tanay A. Single-cell analysis of clonal maintenance of transcriptional and epigenetic states in cancer cells. Nat. Genet. 2020;52(7):709–718. doi: 10.1038/s41588-020-0645-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Chen J., Lau B.T., Andor N., Grimes S.M., Handy C., Wood-Bouwens C., Ji H.P. Single-cell transcriptome analysis identifies distinct cell types and niche signaling in a primary gastric organoid model. Sci. Rep. 2019;9(1):4536. doi: 10.1038/s41598-019-40809-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Puram S. V., Tirosh I., Parikh A. S., Patel A. P., Yizhak K., Gillespie S., Rodman C., Luo C. L., Mroz E. A., Emerick K. S. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell. 2017;171(7):1611–1624. doi: 10.1016/j.cell.2017.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Liu R., Zhang G., Yang Z. Towards rapid prediction of drug-resistant cancer cell phenotypes: single cell mass spectrometry combined with machine learning. Chem. Commun. (Camb.) 2019;55(5):616–619. doi: 10.1039/C8CC08296K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Cagan R., Meyer P. Rethinking cancer: current challenges and opportunities in cancer research. Dis. Model. Mech. 2017;10(4):349–352. doi: 10.1242/dmm.030007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Arnedos M., Soria J-C., Andre F., Tursz T. Personalized treatments of cancer patients: a reality in daily practice, a costly dream or a shared vision of the future from the oncology community? Cancer Treat. Rev. 2014;40(10):1192–1198. doi: 10.1016/j.ctrv.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 241.Pinu F.R., Beale D.J., Paten A.M., Kouremenos K., Swarup S., Schirra H.J., Wishart D. Systems biology and multi-omics integration: Viewpoints from the metabolomics research community. Metabolites. 2019;9(4):76. doi: 10.3390/metabo9040076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Zugazagoitia J., Guedes C., Ponce S., Ferrer I., Molina-Pinelo S., Paz-Ares L. Current challenges in cancer treatment. Clin. Ther. 2016;38(7):1551–1566. doi: 10.1016/j.clinthera.2016.03.026. [DOI] [PubMed] [Google Scholar]
- 243.Palmirotta R., Lovero D., Cafforio P., Felici C., Mannavola F., Pellè E., Quaresmini D., Tucci M., Silvestris F. Liquid biopsy of cancer: a multimodal diagnostic tool in clinical oncology. Ther. Adv. Med. Oncol. 2018;10:1758835918794630. doi: 10.1177/1758835918794630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Mattox A. K., Bettegowda C., Zhou S., Papadopoulos N., Kinzler K.W., Vogelstein B. Applications of liquid biopsies for cancer. Sci. Translat. Med. 2019;11(507):10.1126. doi: 10.1126/scitranslmed.aay1984. [DOI] [PubMed] [Google Scholar]
- 245.Shohdy K.S., West H.J. Circulating tumor DNA testing-liquid biopsy of a cancer. JAMA Oncol. 2020;6(5):792–792. doi: 10.1001/jamaoncol.2020.0346. [DOI] [PubMed] [Google Scholar]
- 246.Sakellaropoulos T., Vougas K., Narang S., Koinis F., Kotsinas A., Polyzos A., Moss T.J., Piha-Paul S., Zhou H., Kardala E., Damianidou E., Alexopoulos L.G., Aifantis I., Townsend P.A., Panayiotidis M.I., Sfikakis P., Bartek J., Fitzgerald R.C., Thanos D., Mills Shaw K.R., Petty R., Tsirigos A., Gorgoulis V.G. a deep learning framework for predicting response to therapy in cancer. Cell Rep. 2019;29(11):3367–3373.e4. doi: 10.1016/j.celrep.2019.11.017. [DOI] [PubMed] [Google Scholar]
- 247.Chakraborty S. In Interpretability of deep learning models: A survey of results.; 2017 IEEE SmartWorld, Ubiquitous Intelligence Computing, Advanced Trusted Computed, Scalable Computing Communications, Cloud Big Data Computing, Internet of People and Smart City Innovation (SmartWorld/SCALCOM/UIC/ATC/CBDCom/IOP/SCI),; 4-8 Aug . 2017. pp. 1–6. [Google Scholar]
- 248.Montavon G., Samek W., Müller K-R. Methods for interpreting and understanding deep neural networks. Digit. Signal Process. 2018;73:1–15. doi: 10.1016/j.dsp.2017.10.011. [DOI] [Google Scholar]
- 249.Ahmad M.A., Eckert C., Teredesai A. In: Interpretable machine learning in healthcare.; Proceedings of the 2018 ACM international conference on bioinformatics, computational biology, and health informatics; 2018. pp. 559–560. [Google Scholar]
- 250.Stiglic G., Kocbek P., Fijacko N., Zitnik M., Verbert K., Cilar L. Interpretability of machine learning-based prediction models in healthcare. WIREs Data Mining Knowledge Discov. 2020;10(5):1379. doi: 10.1002/widm.1379. [DOI] [Google Scholar]
- 251.Noack A., Ahern I., Dou D., Li B. Does interpretability of neural networks imply adversarial robustness? arXiv preprint. 2019.
- 252.Rudin C. Stop explaining black box machine learning models for high stakes decisions and use interpretable models instead. Nature Mach. Intelligence. 2019;1(5):206–215. doi: 10.1038/s42256-019-0048-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Gilpin L.H., Bau D., Yuan B.Z., Bajwa A., Specter M., Kagal L. In: Explaining Explanations: An Overview of Interpretability of Machine Learning.; 2018 IEEE 5th International Conference on Data Science and Advanced Analytics (DSAA); 1-3 Oct ; 2018. pp. 80–89. [Google Scholar]
- 254.Kleinberg S., Hripcsak G. A review of causal inference for biomedical informatics. J. Biomed. Inform. 2011;44(6):1102–1112. doi: 10.1016/j.jbi.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Cooper G.F., Bahar I., Becich M.J., Benos P.V., Berg J., Espino J.U., Glymour C., Jacobson R.C., Kienholz M., Lee A.V., Lu X., Scheines R. Center for Causal Discovery team. The center for causal discovery of biomedical knowledge from big data. J. Am. Med. Inform. Assoc. 2015;22(6):1132–1136. doi: 10.1093/jamia/ocv059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Kleinberg S., Mishra B. The temporal logic of causal structures.; Proceedings of the Twenty-Fifth Conference on Uncertainty in Artificial Intelligence; Montreal, Quebec, Canada . 2009. pp. 303–312. [Google Scholar]
- 257.Cherny S.S., Freidin M.B., Williams F.M.K., Livshits G. The analysis of causal relationships between blood lipid levels and BMD. PLoS One. 2019;14(2):e0212464. doi: 10.1371/journal.pone.0212464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Vineis P., Kriebel D. Causal models in epidemiology: past inheritance and genetic future. Environ. Health. 2006;5(1):21. doi: 10.1186/1476-069X-5-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Olde Loohuis L., Caravagna G., Graudenzi A., Ramazzotti D., Mauri G., Antoniotti M., Mishra B. Inferring tree causal models of cancer progression with probability raising. PLoS One. 2014;9(10):8358. doi: 10.1371/journal.pone.0108358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Zheng M., Claassen J., Kleinberg S. Automated Identification of Causal Moderators in Time-Series Data. In: Proceedings of 2018 ACM SIGKDD Workshop on Causal Disocvery, PMLR. Proceedings of Machine Learning Research. 2018;92:pp. 4–22. [PMC free article] [PubMed] [Google Scholar]
- 261.Ramazzotti D., Caravagna G., Olde Loohuis L., Graudenzi A., Korsunsky I., Mauri G., Antoniotti M., Mishra B. CAPRI: efficient inference of cancer progression models from cross-sectional data. Bioinformatics. 2015;31(18):3016–3026. doi: 10.1093/bioinformatics/btv296. [DOI] [PubMed] [Google Scholar]