

Abstract
Pharmacologic targeting of components of the MAPK/ERK pathway in differentiated thyroid carcinoma (DTC) is often limited due to the development of adaptive resistance. However, the detailed mechanism of MEK inhibitor (MEKi) resistance is not fully understood. Here, MEKi-resistant models were constructed successfully, in which multiple receptor tyrosine kinases (RTKs) signaling pathways and Src-homology 2 domain-containing phosphatase 2 (SHP2) were activated in MEKi-resistant cells. Given the physiological role of SHP2 as the downstream target of many RTKs, we first found blockade of SHP2 enhanced the sensitivity to MEKi in constructed MEKi-resistant models. Interestingly, we also found that compared with MEKi treatment alone, MEKi in combination with an SHP2 inhibitor markedly suppressed the reactivation of the MEK/ERK pathway; thus, the addition of the SHP2 inhibitor significantly improved the antitumor effects of MEKi. The synergistic suppression of DTC upon treatment with both inhibitors was further confirmed in xenograft models and transgenic models. Thus, our data suggest that RTKs activation leads to reactivation of the MAPK pathway and resistance to MEKi in DTC, which is reversed by SHP2 blockade. As a novel active inhibitor of SHP2, SHP099 in combination with MEKi is a promising therapeutic approach for advanced DTC and MEKi-resistant one.
Keywords: Differentiated thyroid carcinoma, MEK inhibitor resistance, RTK, SHP2, combination strategy
Introduction
Despite the promising overall prognosis of differentiated thyroid cancer (DTC), 5-10% of patients continue to suffer from advanced and treatment-refractory disease after standard therapy [1-3]. Once progression occurs, surgery, external beam radiation, watchful waiting and experimental trials are sequentially performed, and these usually have only marginal survival benefits [4]. Patients with advanced DTC only have a 10% survival rate, in contrast to the approximately 98% survival rate of patients with indolent DTC [5,6]. Hence, there is an urgent need to develop novel reliable treatment strategies for advanced DTC.
Molecular-targeted therapy for advanced DTC has received sustained attention due to its promising therapeutic effects on advanced DTC. MAPK/ERK signaling is the most commonly affected pathway in DTC and is widely associated with cell proliferation, differentiation, gene transcription and immune escape [7,8]. Impairment of ERK signaling is the primary strategy for targeted therapy, and it is mainly achieved at the levels of BRAF and MEK [7,9]. Supported by encouraging preclinical data, the MEK inhibitor (MEKi) selumetinib has been approved for the treatment of advanced DTC [10]. However, consistent with other targeted drugs, there are still two limitations related to the clinical application of MEKi. First and most importantly, the occurrence of MEKi resistance is common. Despite initial promising results, a prolonged response to MEKi is infrequently seen [7]. Furthermore, the clinical data show that MEKi has less effect on thyroid cancer cells that lack the BRAFV600E mutation [11,12]. Therefore, exploring combination strategies that can abolish intrinsic resistance and extend the application range is of great clinical value [13,14].
Src homology 2 (SH2) domain-containing tyrosine phosphatase-2 (SHP2), a nonreceptor protein tyrosine phosphatase encoded by the protein tyrosine phosphatase nonreceptor type 11 (PTPN11) gene in humans, is widely expressed in various tumor cells and tissues, such as breast, liver and lung cancer, leukemia and neuroblastoma tissues [15-20]. SHP2 contains a protein tyrosine phosphatase catalytic (PTP) domain, a proline-rich motif, a C-terminal tail that includes two tyrosine phosphorylation sites (Y580 and Y542) and two SH2 domains. As a common downstream target of multiple cytokines, growth factors and other extracellular stimuli, SHP2 manipulates various important cellular life activities, including cell proliferation, activation, migration and differentiation, and is therefore considered a novel therapeutic target [21,22]. It has been shown that SHP2 expression is enhanced in thyroid carcinoma tissues compared with normal thyroid tissues, associated with thyroid cancer metastasis, suggesting that SHP2 may be useful in evaluating prognostic outcomes [23,24]. However, there have been few studies on the mechanisms involved in SHP2 function in thyroid carcinoma at present. Moreover, the role of SHP2 in drug resistance in thyroid carcinoma remains unknown.
In the present study, we constructed drug-resistant DTC cell clones through continuously stimulating with a MEKi (AZD6244). RNA sequencing (RNA-seq) was performed to identify resistance-related genes and pathways. We found that multiple RTKs were activated in MEKi-resistant models and that p-ERK was reactivated in DTC cell lines. As a common downstream signal transductor for multiple RTKs, SHP2 was found to be activated in resistant cell lines. Inhibition of SHP2 enhanced the susceptibility of MEKi-resistant DTC models to MEKi. Furthermore, the combination strategy (MEKi and SHP2i) showed superior antitumor activity than SHP099 or AZD6244 alone in four DTC cell lines (K1, BCPAP, KTC-1 and TPC-1) harboring the BRAF mutation or RET activation. We verified the above mechanism using DTC a xenograft model. Compared with other models, transgenic animal model is thought to be able to better simulate the process of human tumorigenesis and development than other models since its mature immune system, and thus have greater advantages for the study of tumorigenesis mechanisms and tumor-targeted therapy. The combined strategy showed significantly superior antitumor effects than the individual treatments in the transgenic mouse model, which further demonstrated the potential therapeutic value of the combined strategy for DTC.
Materials and methods
Cells and drugs
The K1 (BRAF mutation) and BCPAP (BRAF mutation) cell lines were purchased from Guangzhou Cellcook Biotech Co. (Guangzhou, China). The TPC-1 (RET activation) and KTC-1 (BRAF mutation) cell lines were purchased from the Chinese Academy of Science (Shanghai, China). All cell lines were identified by short tandem repeat (STR) analysis and cultured in RPMI 1640 (Gibco) supplemented with 10% fetal bovine serum, penicillin/streptomycin (5,000 units/mL, Gibco) and l-glutamine (2 mM, Gibco). The passage number of the cells used for the experiments was approximately 20-30. All cell lines were tested for mycoplasma contamination. AZD6244 (Selleck Chemicals) and SHP099 (a potent, selective, orally available SHP2 inhibitor; Selleck Chemicals) were used in the present study.
RNA-sequence (RNA-Seq) analysis
RNA was isolated from the DTC cell line BCPAP and the MEKi-resistant model BCPAP-R. The samples were then processed for RNA-sequencing using the NuGen Ovation Human FFPE RNA-Seq Multiplex System. Total RNA quality was determined (bioanalyzer), rRNA was depleted with RiboMinus, multiplexed paired-end libraries were prepared with Illumina TruSeq, and sequencing was performed with an Illumina HiSeq instrument. The quality of the sequencing was determined by running FastQC. GSEA was performed to identify the enriched pathways and activated network modules. All of the data have been deposited in the Dryad database with access URL: https://datadryad.org/stash/share/RuLPFKPoSPLWlYUstqt2awejpLGkoJGMC-qxsS561Ug.
Plasmid and cell transfection
A lentivirus containing the SHP2 shRNA plasmid was purchased from GeneChem Co. (GV298, Shanghai, China). The shRNA construct for PTPN11 was inserted into the U6-MCS-Ubiquitin-Cherry-IRES-puromycin vector. The shRNA targeting sequence for human PTPN11 are listed in Table 1. Cells were seeded in 6-well plates at a density of 50000 cells/well. For transfection, the cells were cultured in RPMI 1640 comprising 10% fetal bovine serum and lentivirus vectors for 48 h. After that, the cells were transferred to RPMI 1640 comprising 10% fetal bovine serum and puromycin (4 μM) for further screening. Transfection was confirmed by western blot analysis.
Table 1.
The shRNA targeting sequence
| Sequence | |
|---|---|
| shRNA | 5’-gcTGAAATAGAAAGCAGAGTT-3’ |
Small interfering RNAs (siRNA) and cell transfection
Small interfering RNAs (siRNA) of SHP2 were obtained from Qiagen. Transfections were performed using Lipofectamine TM 3000 (Invitrogen) following the manufacturer’s instructions. The siRNA sequences are listed in Table 2. Cells were seeded in 6-well plates at a density of 50000 cells/well. 2 hours after the complete medium was replaced with serum-free 1640 medium, a serum-free 1640 premix containing 5 ul siRNA and 3 ul lipo3000 was added to each well. After 4-6 h culture in the cell incubator, the supernatant was discarded and replaced with complete medium. After 24-48 h, RNA and cell precipitation were collected for subsequent experiments.
Table 2.
The siRNA sequences
| Sense | Antisense | |
|---|---|---|
| siRNA | 5’-UUAGACUUGCCGUCAUUGCUC-3’ | 5’-GCAAUGACGGCAAGUCUAAAG-3’ |
Cell viability assay
Cells were seeded in a 96-well plate at a density of 500 cells per well. After incubation for 24 hours, the cells were treated with DMSO, SHP099 (10 µM), AZD6244 (1 µM), or the combination of the inhibitors (Comb, 10 µM SHP099 and 1 µM AZD6244) at various concentrations. After the indicated period of time, the cells were incubated with CCK8 substrates (5 mg/mL) for two hours. The optical density was measured at 450 nm.
Colony formation assay
Cells were seeded in a 24-well plate at a density of 200 cells per well. After 24 hours, the cells were cultured with DMSO, SHP099 (10 µM), AZD6244 (1 µM), or the combination of the inhibitors (Comb, 10 µM SHP099 and 1 µM AZD6244) for two weeks. Then, the cells were stained with 0.05% crystal violet before being imaged.
Cell cycle analysis
The cells treated as described above were fixed in cold 70% ethanol and incubated at 4°C overnight. After incubation, the cells were resuspended in PBS supplemented with 60 µg/mL RNase A and 25 µg/mL propidium iodide (PI) for 15 minutes in the dark at room temperature. Then, the samples were analyzed using a FACSCalibur flow cytometer (BD Biosciences).
Western blot analysis and RTK arrays
Cells treated as described above were lysed in modified RIPA buffer comprising 1% PMSF. Equal amounts of total protein were resolved by SDS-PAGE and transferred onto PVDF membranes (Millipore). Then, the membranes were immunoblotted overnight with primary antibodies. The primary antibodies used for the western blot were p-MEK (Cell Signaling Technology, 2338; 1:1000), MEK (Cell Signaling Technology, 4694; 1:1000), p-ERK (Cell Signaling Technology, 4370; 1:1000), ERK (Cell Signaling Technology, 4695; 1:1000), p-SHP2 (Abcam, ab62322; 1:1000), SHP2 (Abcam, ab32083; 1:1000) and GAPDH (Cell Signaling Technology, 5174; 1:40000). Human phospho-RTK arrays were purchased from R&D Systems (ARY001B) and were used according to the manufacturer’s guidelines.
Animal experiments
All animal experiments were approved by the Tianjin Medical University Cancer Institute and the Hospital Animal Care and Use Committee and performed according to the IACUC protocol. To establish a drug resistance model in vivo, ten mice with DTC xenografts of similar volumes received oral doses of 20 mg/kg AZD6244 once daily. After 15 or 40 days, representative xenografts from the two groups were picked for human phospho-RTK arrays. The remaining xenografts from the mice treated for 40 days were cut into equal sections and transplanted into other mice. After seven days, the transplanted animals were randomly assigned to the same four groups and treated as described above. At the indicated time points, the animals were sacrificed, and the tumors were excised for further analysis.
To confirm the combination strategy in vivo, thyroid cancer cell line xenografts were established through subcutaneous injection of 1×105 cells into 4-week-old male NSG mice. When the tumor volume reached approximately 15 mm3, the animals were randomly assigned to four groups: the control (Ctrl) group, which was orally treated with DMSO, q.d.; the SHP099 group, which was orally treated with 50 mg/kg SHP099, q.o.d; the AZD6244 group, which received daily oral doses of 20 mg/kg AZD6244; and the Comb group, which was treated with the combination therapy. A transgenic mouse model of spontaneous DTC was established as previously described. Cre recombinase was overexpressed in TPO-Cre mice under the control of the human thyroid peroxidase (TPO) gene promoter. Activated BRAFV600E was expressed at physiological levels in the BrafCA mice. Endogenous Braf alleles were replaced by BRAFV600E by crossing BrafCA mice with TPO-Cre mice to produce a set of double transgene-positive offspring that develop DTC around the age of 4-5 weeks [25]. In our study, the mice spontaneously developed DTC at 6-12 weeks of age. According to their weight, six-week-old TPO-Cre BrafCA mice were randomly assigned to four groups: the control (Ctrl) group, which was orally treated with DMSO, q.d.; the SHP099 group, which was orally treated with 50 mg/kg SHP099, q.o.d; the AZD6244 group, which received daily oral doses of 20 mg/kg AZD6244; and the Comb group, which was treated with the combination therapy.
Immunohistochemistry (IHC)
To measure the expression levels of p-ERK and Ki67 in the animal models, xenografts were collected, fixed in 4% paraformaldehyde (PFA) overnight, and embedded in optimum cutting temperature compound (OCT). IHC was performed according to standard protocols. The primary antibodies used for the IHC assays were p-ERK (Cell Signaling Technology, 4370) and Ki67 (Cell Signaling Technology, 9027). The stained slides were independently examined by two pathologists who were blinded to the treatment information. Hematoxylin and eosin staining was performed by the Department of Pathology of Tianjin Medical University Cancer Institute and Hospital. The expression levels of p-ERK and Ki67 were quantified to determine histological scores (histoscores). The staining intensity in labeled cells was scored from weak to strong as follows: 0 points (nuclei exhibited no yellow granular bodies); 1 point (nuclei exhibited light yellow granular bodies); 2 points (nuclei exhibited brown yellow granular bodies); and 3 points (nuclei exhibited dark brown yellow granular bodies). The positive cells and total cells were counted with ImageJ software. Histoscores was calculated according to the following formula: Histoscore = staining intensity × (positive cell count/total cell count) ×100%.
RT-PCR
RT-PCR assays were performed as previously described [21]. Briefly, total RNA was extracted from cells using a RNeasy Mini Kit (Qiagen). cDNA synthesis was conducted using a Transcriptor First Strand cDNA Synthesis Kit (Takara) according to the manufacturer’s instructions. PCR was performed with FastStart Universal SYBR Green Master Mix (Takara) on an ABI ViiA7 system. The primers are listed in Table 3.
Table 3.
The primers of RT-PCR assays
| Forward | Reverse | |
|---|---|---|
| ETV1 | CTTAGCCGTTCACTCCGCTAT | TCTGTCTTCAGCAGTGGACG |
| ETV4 | GCCCATTTCATTGCCTGGAC | TACACGTAACGCTCACCAGC |
| ETV5 | TAGAACCGGAAGAGGTTGCTC | TTATCCGGGAAAGCCATGGAG |
| FOSL1 | CAGCCCAGCAGAAGTTCCA | ACTGAGGGTAGGTCAGAGGC |
Statistical analysis
The data are presented as the mean ± SD of three independent experiments. Statistical analysis was performed using SPSS (IBM Corporation, Armonk, NY, USA) and GraphPad Prism 8.0 software (La Jolla, CA, USA). T-tests and ANOVA were used to determine the statistical significance (*P<0.05, **P<0.01, ***P<0.001, compared with the Ctrl group, @P<0.05, @@P<0.01, @@@P<0.001, compared with the Comb group) of differences between the groups treated with different inhibitors treated and the control.
Results
SHP2 is a potential therapeutic target for MEKi-resistant DTC cell lines
To verify the intrinsic mechanism of MEKi resistance, resistant models were constructed in vitro utilizing BCPAP and TPC-1 cells. The cells were cultured with increasing concentrations of AZD6244 until drug-resistant clones termed BCPAP-R and TPC-1-R were more than 10 times less sensitive to AZD6244 than their parental lines (Figure S1A). To confirm the resistance of BCPAP-R and TPC-1-R cells, we performed a colony formation assay and found that the MEKi-resistant cell lines were not significantly inhibited after treatment with AZD6244, unlike the primary cell lines, which were significantly inhibited by AZD6244 (P<0.01 for both BCPAP and TPC-1 cells) (Figure S1B). The above indicated that cell lines developed acquired resistance under the constant stimulation of MEKi. Then we performed RNA-sequencing analysis of BCPAP (N=3) and BCPAP-R cells (N=3), and RNA-seq identified 4,270 mRNAs with significant changes in BCPAP-R cells relative to BCPAP cells (Figure S2A and S2B). Based on the total identified mRNAs, GSEA revealed that the mRNAs of RTKs pathways (including VEGFA, VEGF, IGF1 and EGFR) were significantly enriched in BCPAP-R cells (Figure 1A and 1B). SHP2 is a positive RTK downstream signal transducer that was recently regarded as a novel target for RTKs-driven cancers [22]. As a marker of SHP2 downstream of RTK signaling [26], p-SHP2 expression in both primary and MEKi-resistant cell lines were determined by western blot analysis. Compared with that in the parental cell lines, p-SHP2 expression in BCPAP-R and TPC-1-R cells was significantly upregulated (Figure 1C). At the same time, p-MEK and p-ERK expression was upregulated continuously in the resistant cell lines compared with the parent cell lines (Figure S3).
Figure 1.
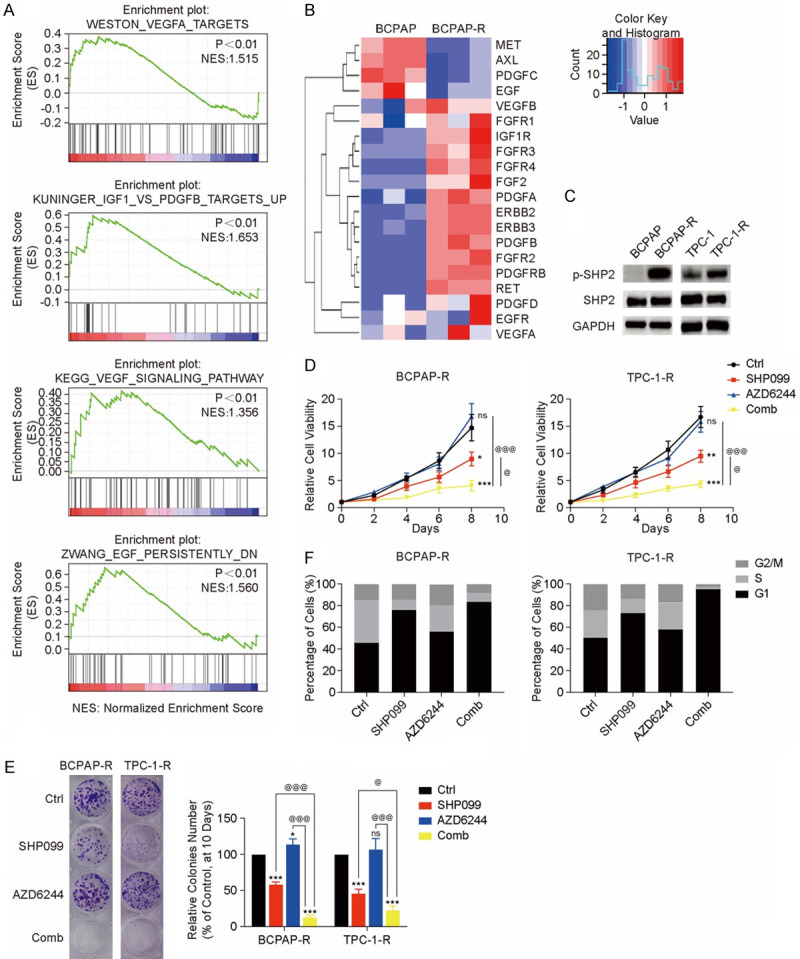
SHP2 is a potential therapeutic target for MEKi-resistant DTC cell lines. (A) GSEA of all identified mRNAs. mRNA expression of RTKs significantly increased in BCPAP-R cells. (B) Heatmap of RTKs that were upregulated, with the scale showing fold changes. (C) SHP2 was significantly activated in the BCPAP-R and TPC-1-R cell lines. Two MEKi-resistant models were treated with DMSO (Ctrl), SHP099 (10 µM), AZD6244 (1 µM), or both drugs (Comb). (D) and (E) The cell viability (D) and colony formation (E) assays were performed on days 2-8 and 10, respectively; *P<0.05, **P<0.01, ***P<0.001, compared with the Ctrl group, @P<0.05, @@P<0.01, @@@P<0.001, compared with Comb group, ANOVA. All experiments were repeated independently at least three times, and the data are presented as the mean ± SD. The data showed that the combination of SHP099 and AZD6244 more significantly inhibited proliferation in the MEKi-resistant model than the other treatments. (F) The combination of SHP099 and AZD6244 significantly induced G1 phase arrest in the BCPAP-R and TPC-1-R cell lines. Cell cycle distribution was assessed by flow cytometry. The percentage of cells in the G1, S and G2/M phases was plotted.
To verify whether SHP2 contributes to MEKi resistance, two MEKi-resistant cell lines (BCPAP-R and TPC-1-R cells) were treated with DMSO (Ctrl), SHP099, AZD6244, or the combination of SHP099 and AZD6244 (Comb). All examined MEKi-resistant cell lines demonstrated susceptibility to SHP099 with suppressed cell colony formation and viability (all P<0.05) (Figure 1D and 1E). Cell cycle analysis revealed that SHP099 arrested MEKi-resistant cell lines at G1/S phase (Figure 1F). Coadministration with AZD6244 resulted in an increase in efficacy with additive synergistic effects. SHP099 resensitized BCPAP-R and TPC-1-R cells to AZD6244, and the combination strategy suppressed cell colony formation, growth, and cell cycle progression more effectively than either SHP099 or AZD6244 alone (all P<0.05) (Figure 1D-F). These data indicated that RTK-induced ERK reactivation was a dominant mechanism of the MEKi-resistant phenotype, which was significantly impaired by the SHP2 inhibitor SHP099.
The combination strategy suppresses MEKi-resistant tumor growth in vivo
To test the effectiveness of the combined strategy in a MEKi-resistant murine model, mice with K1 xenografts were established and treated as indicated in the flow chart (Figure 2A). Ten mice with DTC xenografts of similar volumes were divided into two groups and received oral doses of AZD6244 (20 mg/kg, q.d.) for 15 or 40 days. During the 40 days of AZD6244 treatment, the tumor growth rate was suppressed for approximately 25 days, and then the tumors rapidly grew for approximately 25-40 days (Figure S4). Representative xenografts treated with AZD6244 for 15 or 40 days were selected for the phospho-RTK array, which revealed that multiple RTKs were consistently activated after 40 days of MEKi treatment (all P<0.05) (Figure 2B). Consistent with our results in vitro, above data revealed the reactivated RTKs in DTC after long-term treatment with AZD6244 in vivo.
Figure 2.
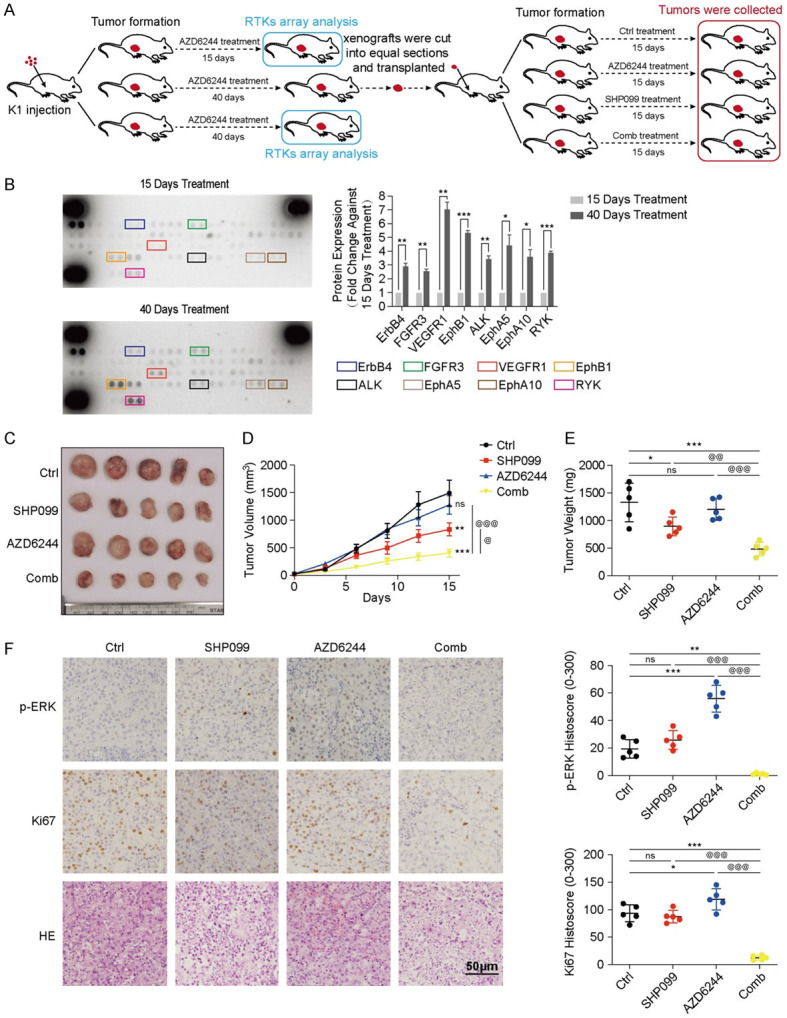
SHP099 abolished MEKi resistance in a MEKi-resistant murine model. A. Treatment protocol for mice with K1 xenografts. B. Phospho-RTK arrays revealed the upregulation of multiple activated RTKs in the tumor tissue of mice after 40 days of AZD6244 treatment compared with 15 days of treatment. *P<0.05, **P<0.01, ***P<0.001, two-tailed t-test. C. Images of the dissected tumors obtained from xenograft mice in the Ctrl group (n=5), SHP099 group (n=5), AZD6244 group (n=5) and Comb group (n=5). D. The growth curve of the xenografts after treatment with DMSO (Ctrl), SHP099 (50 mg/kg, q.o.d), AZD6244 (20 mg/kg, q.d.) and both drugs (Comb: 20 mg/kg AZD6244, q.d.; 50 mg/kg SHP099, q.o.d). The volumes of all transplanted xenografts were measured every five days until the 15th day. E. The weights of the dissected tumors from transplanted mice were analyzed after treatment for 15 days; n=5, per group, *P<0.05, **P<0.01, ***P<0.001, compared with the Ctrl group; @P<0.05, @@P<0.01, @@@P<0.001, compared with the Comb group, ANOVA. F. Representative images and QuPath quantitation of p-ERK and Ki67 immunostaining in the xenografts.
To analyze the efficacy of the combined strategy for RTK-activated xenografts, the remains of xenografts treated with MEKi for 40 days were cut into equal sections and transplanted into other mice. After seven days, the transplanted mice were randomly assigned to four groups and treated with DMSO (Ctrl), SHP099, AZD6244 or the combination of both drugs (Comb) for 15 days. AZD6244 had minimal effects on the RTK-activated tumors, demonstrating that the tumor models had partial resistance to the MEKi. The increased susceptibility of MEKi-resistant tumors to SHP099 indicated that activated RTKs played a major role in DTC cell proliferation. Of note, the combination of SHP099 and AZD6244 caused more effective tumor shrinkage than a single agent (all P<0.05) (Figure 2C-E), demonstrating that MEKi resistance development depended on SHP2. As revealed by the IHC assay (Figure 2F), the group treated with the combination of SHP099 and AZD6244 had lower p-ERK expression and fewer Ki67-positive cells than the groups treated with single agents (all P<0.05). Stable weight, normal behavior and health were observed in the MEKi-resistant models after combination treatment (Figure S5). These data suggested that the combination of SHP2 and MEK inhibition continued to be effective in suppressing tumor growth in MEKi-resistant models in vivo.
Suppression of SHP2 abrogates RTK-induced MAPK/ERK pathway rebound
After treatment with MAPK inhibitors, ERK signaling rebound induced by RTKs can develop in various cancer models [26-29]. To examine whether ERK rebound existed in the DTCs after treatment with MEKi, p-ERK expression was detected after 0, 1, 24 and 48 hours of AZD6244 treatment. Western blot analysis revealed that p-ERK expression initially declined after one hour and then rapidly rebounded to baseline levels or to an even higher level at 48 hours, indicating that the reactivation of p-ERK rapidly appeared during 48 hours MEKi treatment (Figure 3A). To verify the relationship between ERK signaling rebound and activated RTKs in DTCs, a human phospho-RTK array was performed to determine the RTK activation status. As expected, the phosphorylation levels of several RTKs were increased in K1 after MEKi treatment (all P<0.05) (Figure 3B). The expression of p-SHP2, which was manipulated by activated RTKs, was significantly increased (P<0.001) in all four cell lines treated with AZD6244 (Figure 3C). Hence, it was hypothesized that therapies targeting both MEK and SHP2 could effectively abolish ERK signaling reactivation.
Figure 3.
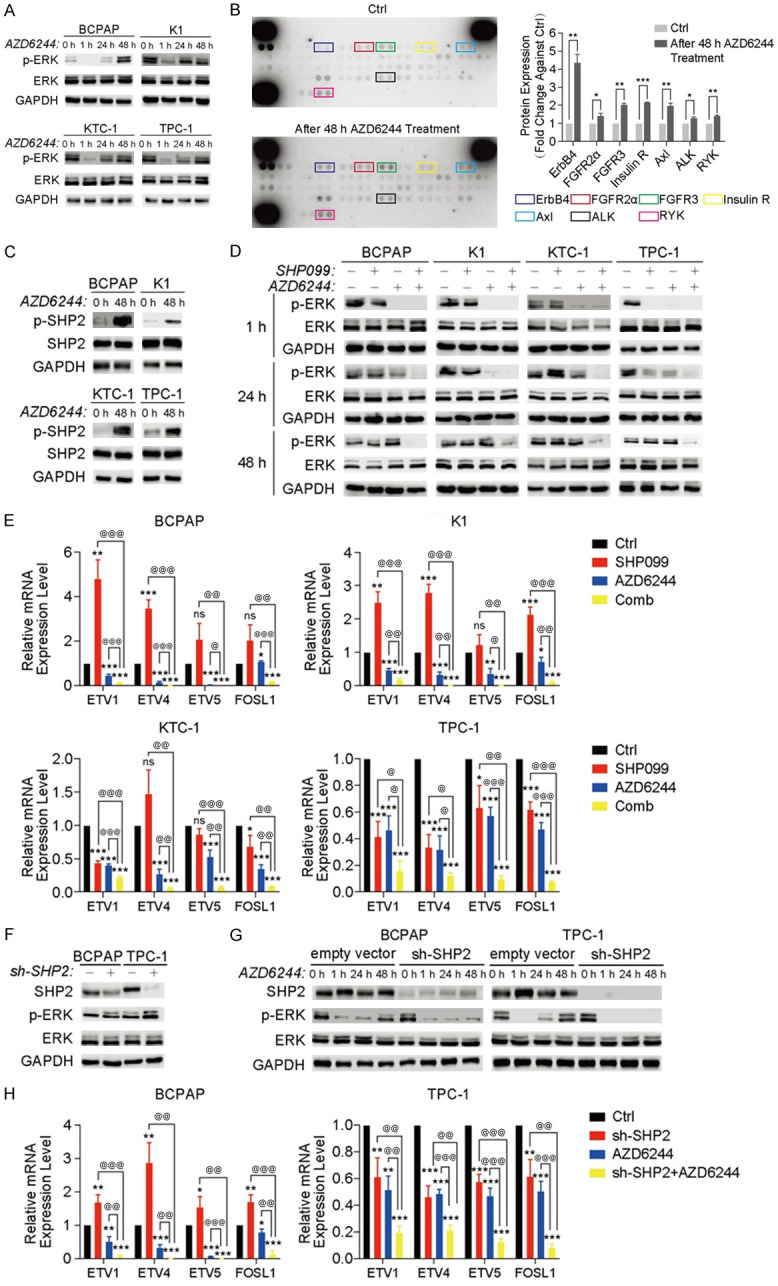
SHP099 abrogated the RTK-induced rebound of the ERK pathway in DTC cell lines after 48 h of MEKi treatment. A. Western blot analysis revealed ERK reactivation in 4 DTC cell lines after 48 h of AZD6244 (1 µM) treatment. Representative images from one experiment are shown. B. The phospho-RTK arrays revealed that multiple RTKs were activated in the DTC cell line K1 after 48 h (1 µM) of AZD6244 treatment. *P<0.05, **P<0.01, ***P<0.001, two-tailed t-test. C. Western blot analysis revealed the SHP2 activation level in DTC cell lines treated with or without AZD6244 for 48 h. D. The ERK activation level was analyzed by western blotting in four DTC cell lines treated with DMSO (Ctrl), SHP099 (10 µM), AZD6244 (1 µM), or both drugs (Comb) within 48 h. E. The expression of ERK-dependent genes (ETV1, ETV4, ETV5, and FOSL1) was assessed by qRT-PCR in 4 DTC cell lines that were treated as indicated. *P<0.05, **P<0.01, ***P<0.001, compared with the Ctrl group, @P<0.05, @@P<0.01, @@@P<0.001, compared with the Comb group, two-tailed t-test. F and G. Knocking down SHP2 expression exhibited a similar effect as SHP099 treatment in BCPAP and TPC-1 cell lines. F. Western blot analysis of SHP2 expression after knockdown of SHP2 expression in BCPAP and TPC-1 cell lines. G. Knocking down SHP2 expression similarly abolished ERK rebound after 48 h of MEKi treatment. H. The combination of AZD6244 (1 µM) and SHP2 knockdown suppressed the expression of ERK-dependent genes (ETV1, ETV4 and ETV5, and FOSL1). All data are presented as the mean ± SD of three independent experiments.
For further confirmation, four DTC cell lines were treated with DMSO, SHP099 alone, AZD6244 alone, or the combination of SHP099 and AZD6244, and the change in p-ERK expression within 48 hours was measured. Compared with SHP099 or AZD6244 alone, the combination of SHP099 and AZD6244 persistently inhibited p-ERK expression (Figure 3D). As indicated by ETV1, ETV4, ETV5, and FOSL1 mRNA levels, ERK-dependent transcription was also significantly inhibited (all P<0.05) by the combination strategy [30] (Figure 3E). In addition, the combination of MEKi and SHP2 knockdown had similar effects as SHP099/MEKi treatment, indicating that SHP099 is “on-target” in DTC cell lines (Figure 3F-H). Taken together, these data demonstrated that the combination of SHP099 and MEKi could more effectively suppress p-ERK expression than SHP099 or AZD6244 alone and that dual SHP2/MEK inhibition caused these observed effects.
SHP2 inhibition combined with AZD6244 inhibits cell progression in DTC in vitro
To determine whether the combination SHP2/MEK inhibition was more effective in DTC cell lines, colony formation and viability (CCK8) assays were performed (Figure 4A and 4B). As a single agent, SHP099 had a variable effect on colony formation in DTC cell lines with different genetic backgrounds. A minimal effect was detected in three BRAF mutation cell lines (which was consistent with previous studies), while a marked effect (all P<0.01) was observed in BRAF wild-type cells TPC-1 harboring the RET fusion mutation. However, unlike after SHP099 or AZD6244 treatment alone, few or no detectable colonies were found in all DTC cells after treatment with the combination of SHP099 and AZD6244 (all P<0.001). Similar effects were observed in the cell viability assays. The combination treatment resulted in intense growth inhibition (P<0.01) in all DTC cells.
Figure 4.

The combination of SHP099 and AZD6244 exhibited synergistic effects in DTC cells with various genetic backgrounds. (A) and (B) Four DTC cell lines were treated with DMSO (ctrl), SHP099 (10 µM), AZD6244 (1 µM), or both drugs (Comb). The colony formation (A) and cell viability (B) assays were performed on days 10 and 0-8, respectively. *P<0.05, **P<0.01, ***P<0.001, compared with the Ctrl group, @P<0.05, @@P<0.01, @@@P<0.001, compared with the Comb group, ANOVA. All data are presented as the mean ± SD of three independent experiments. Compared with the single agent, the combination of SHP099 and AZD6244 exhibited a more significant inhibitory effect on the progression of the four DTC cell lines. (C) The combination of SHP099 and AZD6244 significantly induced G1 phase arrest in the four DTC cell lines. Cell cycle distribution was assessed by flow cytometry. The percentage of cells in the G1, S and G2/M phases was plotted. (D-F) Colony formation assays (D), cell viability assays (E) and cell cycle distribution analysis (F) revealed that knockdown of SHP2 expression exhibited a similar effect as SHP099 treatment after combination with AZD6244.
Given the effect of the combination of SHP099 and AZD6244 on DTC cell lines, it was then determined whether the combination treatment suppressed cell proliferation by regulating cell cycle progression (Figure 4C). The combination of SHP099 and AZD6244 arrested DTC cell lines in G1/S phase. Similar to the previous study, SHP2 knockdown had similar effects as SHP099/MEKi treatment, suggesting that SHP099 is “on-target” in DTC cell lines (Figures 4D-F and S6). These data suggested that the combination of SHP2/MEK inhibition could powerfully inhibit DTC cell proliferation in vitro.
The combination of SHP099 and AZD6244 is a promising therapeutic approach in vivo
To confirm our results in models in vivo, mice carrying K1 or TPC-1 xenografts were utilized to examine the antitumor activity of MEK/SHP2 inhibition. Compared with mice treated with SHP099 or AZD6244 treatment alone, mice in the combination group had markedly reduced tumor volumes and weight (all P<0.05) (Figure 5A-C) and exhibited more effective suppression of p-ERK levels (all P<0.01) (Figure 5D). Xenografts isolated from combination-treated mice had fewer proliferating cells (all P<0.01) than those derived from other mice, as measured by Ki67 staining (Figure 5D). The effects of a single agent were similar to the above in vitro results. After treatment with the combination strategy, the mice had a stable weight, appeared healthy and exhibited normal behavior, and this effect was observed in all mice carrying K1 or TPC-1 xenografts (Figure S7A and S7B).
Figure 5.
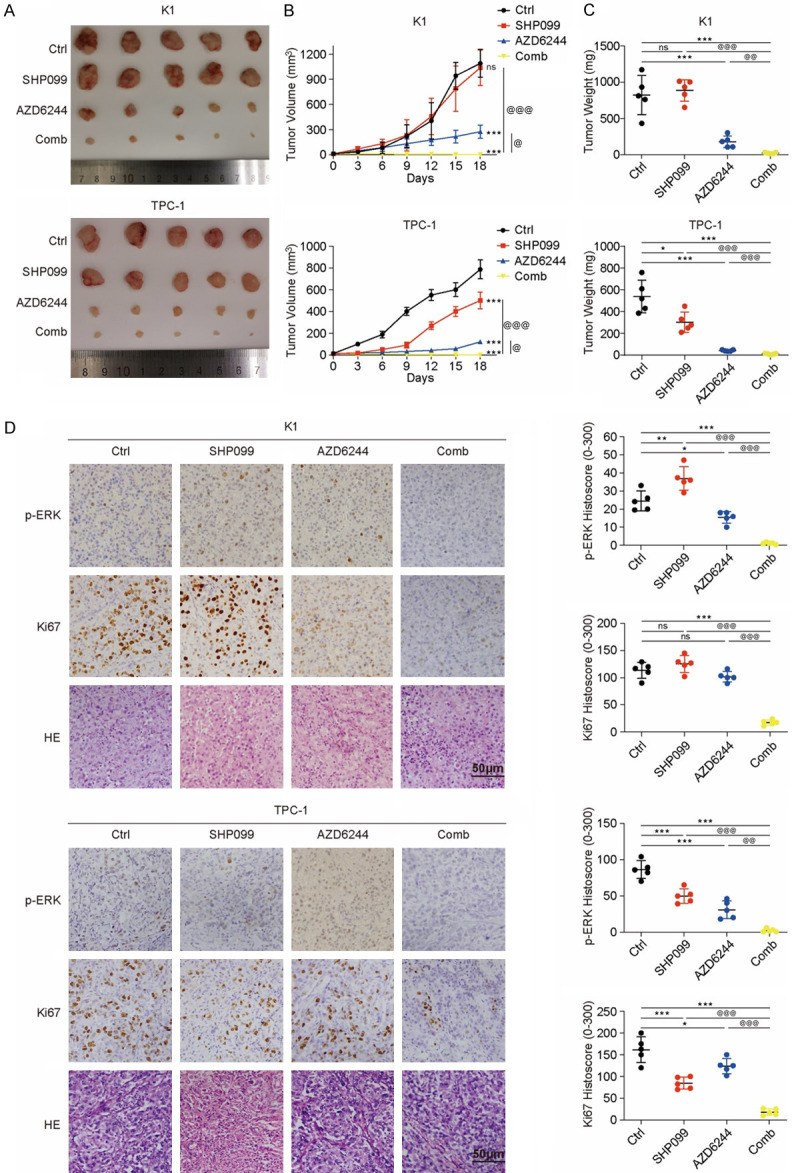
The combination of SHP099 and AZD6244 is a promising therapeutic approach for advanced DTC in vivo. A. Images of tumors from NSG mice injected with K1 and TPC-1 cells. B. The growth curves of K1 and TPC-1 xenografts after treatment with DMSO (Ctrl), SHP099, AZD6244 and both drugs (Comb). All xenograft tumor tissue volumes were measured every three days until the 18th day. C. The weight of the xenografts was analyzed after treatment for 18 days; n=5, per group, *P<0.05, **P<0.01, ***P<0.001, compared with the Ctrl group, @P<0.05, @@P<0.01, @@@P<0.001, compared with the Comb group, ANOVA. D. Representative images and QuPath quantitation of p-ERK and Ki67 immunostaining in xenografts described above. *P<0.05, **P<0.01, ***P<0.001, compared with the Ctrl group, @P<0.05, @@P<0.01, @@@P<0.001, compared with the Comb group, ANOVA.
We further confirmed our results in a transgenic murine model of spontaneous thyroid cancer (Figure 6A). Mice treated with SHP099 or AZD6244 treatment alone exhibited a reduction in tumor volume to approximately 40-50% of the initial volume, although the combination of SHP099 and AZD6244 produced greater reductions in tumor volume and weight than either therapy alone (all P<0.05) (Figure 6B and 6C). IHC assays revealed that the group treated with the combination of SHP099 and AZD6244 had lower p-ERK expression (all P<0.05) and fewer Ki67-positive cells than the groups treated with the individual agents (all P<0.001) (Figure 6D). Stable weight, normal behavior and health were observed in the transgenic models after combination treatment (Figure S7C). These data suggested that the combination of SHP2/MEK inhibition could serve as a powerful therapeutic approach for DTC, in which single-agent targeted therapeutics usually have a limited effect.
Figure 6.

The combined strategy was effective in transgenic murine models of spontaneous thyroid cancer. A. Representative images of mouse thyroid tumors. B. Images of tumors from transgenic murine models. C. The weights of the mouse thyroid tumors were analyzed after treatment for 18 days; n=5, per group, *P<0.05, **P<0.01, ***P<0.001, compared with the Ctrl group, @P<0.05, @@P<0.01, @@@P<0.001, compared with the Comb group, ANOVA. D. Representative images and QuPath quantitation of p-ERK and Ki67 immunostaining of mouse thyroid tumors described above. *P<0.05, **P<0.01, ***P<0.001, compared with the Ctrl group, @P<0.05, @@P<0.01, @@@P<0.001, compared with the Comb group, ANOVA.
Discussion
Though the study and application of MAPK inhibitors have profoundly changed the treatment prospects and prognoses of many malignant tumors in recent years, acquired resistance to MAPK-targeted therapy is almost inevitable. Detailed studies have explored the mechanism of tumor resistance to MAPK inhibitors in various cancer types. Adaptive resistance to sorafenib is acquired in hepatocellular carcinoma (HCC) cells through the reactivation of multiple RTKs, such as EGFR, AXL, and EPH receptor A2, leading to RAS/MEK/ERK and AKT reactivation [17]. Another study proved that partial reprogramming of melanoma cells induces dedifferentiation and adaptation to MAPK inhibitors [31]. As an important factor in the MAPK signaling pathway, MEK has been targeted for inhibition to treat a variety of malignant tumors. Furthermore, drug resistance to MEK inhibitors has also been studied. In addition to RTK reactivation, tumors evade long-term MEK blockade therapies via various resistance mechanisms [32], such as BRAF gene amplification in colorectal cancers harboring BRAFV600E [33], increased formation of Raf-1/B-Raf dimers in melanoma cells [34], and enhanced activation of the PI3K/AKT pathway in melanoma cells [35]. However, while inhibiting the MAPK signaling pathway is a major strategy for targeting thyroid cancer, studies on MAPK inhibitor resistance in DTC remain rare and weak. Considering the limited effective strategies for treating advanced DTC and the different genetic backgrounds of DTC compared with other malignancies, it is particularly important to explore the mechanism of DTC resistance to MEK inhibitors.
Upregulation of the expression of multiple RTKs was revealed by RNA-seq in an MEKi-resistant model in vitro and identified by an RTK assay in MEKi-resistant models in vivo. SHP2, which is activated by RTKs, interacts with Ras, participating in signal transduction and activation of the MAPK pathway to promote tumor progression [36]. Higher SHP2 activity was found in all resistant clones, which indicated that SHP2 can be activated by RTKs and plays a major role in MEKi resistance. The combination of AZD6244 and SHP099 significantly blocked tumor growth and ERK activation in all MEKi-resistant models, demonstrating that DTC models with either BRAFV600E mutation or RET activation acquire MEKi resistance through activated RTKs, and this mainly depends on SHP2.
The combined strategy of SHP2 inhibition and MEK inhibition has been confirmed to be effective in a variety of tumor models, such as pancreatic ductal adenocarcinoma (PDAC), non-small cell lung cancer and malignant peripheral nerve sheath tumor (MPNST) [21,37,38]. Moreover, the combined strategies associated with SHP2 inhibitors and other MAPK inhibitors have also been reported to be promising and effective. The combination of SHP099 and sorafenib has been proven to be a very promising and safe treatment [17] in hepatocellular carcinoma (HCC) by blocking downstream SHP2 activation by RTK reactivation and impeding RTK-driven reactivation of the ERK and AKT pathways [17]. SHP2 inhibition can also enhance systemic antitumor effects in the anti-PD-1-resistant model of non-small cell lung cancer [39,40]. The above studies highlight the promising clinical value of SHP2 inhibition for targeted therapy for various advanced cancers. In our study, we not only confirmed the efficacy of the combination strategy in DTC but also demonstrated that the combination of SHP099 and AZD6244 may be a promising treatment approach for MEKi-resistant DTC. Considering SHP2/MEK inhibition effectively suppressed tumor progression in all tested DTC models both in vivo and in vitro, the combination of SHP099 and AZD6244 is a promising therapeutic approach for advanced DTC and MEKi-resistant one. A schematic model summarizing the mechanism of the SHP099/AZD6244 combination strategy in DTC is presented in Figure S8.
In our research, SHP2 inhibitor failed to inhibit DTC cell lines harboring BRAFV600E mutation. However, after the long-term MEKi treatment, instead of BRAF mutation, activated RTKs played the dominant role in ERK pathway activation in DTC cell lines which had developed acquired resistance to MEKi. Hence, different from the parental cells, SHP2 inhibitor can significantly suppress the growth in MEKi-resistant cell lines.
Different from DTC cell lines harboring BRAFV600E mutation including BCPAP, K1, KTC-1, the TPC-1 cell line harbored activated RET, which is also a kind of RTK [41]. As the downstream signal of RTKs (including RET), the inhibition of SHP2 had certain therapeutic effects to TPC-1 cell line. The effect of SHP2 inhibitor in immunodeficiency mice model was consistent with that observed in vitro. Nevertheless, SHP2 inhibitor can also partially inhibit the progression of DTC in a transgenic murine model of spontaneous thyroid cancer harboring BRAFV600E mutation, revealing that distinct immune microenvironment influenced the efficacy of SHP2 inhibitor, which was proven by other studies. Above results suggest that the efficacy of SHP2 inhibitors may depend on the activated signaling pathways in tumor cells and the tumor microenvironment. However, the combination strategy effectively inhibited tumor progression in all tested DTC models, which indicates that the combination strategy is effective, widely applicable and of potential clinical value.
RET is an RTK involved in the development of DTC and medullary thyroid carcinoma (MTC) [42,43]. RET/PTC fusions can be present in papillary thyroid carcinoma (PTC) which is an important classification of DTC, and their prevalence can reach 50-70% in pediatric patients and in patients that have experienced radioiodine exposure [44]. In contrast with the DTC cell line harboring the BRAFV600E mutation, high SHP2 activity and SHP099 sensitivity were found in TPC-1, a DTC cell line harboring a RET gain-of-function fusion. A similar trend also occurred in TT (Figure S9A and S9B), an MTC cell line harboring the constitutive activation RETC634W mutation [45]. The above data suggested that SHP099 significantly suppresses RET activation-driven DTC progression, although SHP2/MEK blockade produced greater inhibition of tumor development.
The present study has several limitations. RAS is the direct downstream signal for SHP2, and the GTPase activity in different RAS mutants is positively correlated with sensitivity to SHP099. Certain RAS mutations (G13D and Q61X) have lower intrinsic GTPase activity than other mutations, thereby showing emerging resistance to this combination [37,46,47]. Our study failed to evaluate the GTPase activity in different DTC models. And, the effect of the combination of SHP099 and AZD6244 in DTC cell lines harboring RAS mutations. This was limited by the low mutation rate of RAS in DTC [48] and the well-known relationship between RAS mutations and the effect of SHP099 [20,37,46]. In addition, the impact of the combination strategy on the microenvironment of DTC could not be evaluated. Both SHP099 and AZD6244 have been reported to be correlated with the activation of various immune cells [42,49,50]. Further studies are needed to determine the synergistic effect of SHP099 and AZD6244 in shaping the microenvironment of DTC.
In conclusion, SHP2 blockade abolished ERK rebound-induced MEKi resistance, resulting in persistent growth inhibition. Compared with the use of SHP099 or AZD6244 alone, the combination strategy can more effectively overcome MEKi-resistance due to activation of MAPK pathway in DTC. The combination of the SHP2 inhibitor SHP099 and AZD6244 may be a promising and effective therapeutic strategy for advanced DTC and MEKi-resistant one.
Acknowledgements
This work was partially supported by grants from the National Natural Science Foundation of China (Grant No. 81872169, 82172821 and 82103386), Tianjin Key Research and Development Program Science and Technology Support Key Projects (Grant No. 17YFZCSY00690), Tianjin Municipal Science and Technology Project (Grant No. 19JCYBJC27400) and Beijing-Tianjin-Hebei Basic Research Cooperation Project (Grant No. 20JCZXJC00120).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 2.Lim H, Devesa SS, Sosa JA, Check D, Kitahara CM. Trends in thyroid cancer incidence and mortality in the United States, 1974-2013. JAMA. 2017;317:1338–1348. doi: 10.1001/jama.2017.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cronin KA, Lake AJ, Scott S, Sherman RL, Noone AM, Howlader N, Henley SJ, Anderson RN, Firth AU, Ma J, Kohler BA, Jemal A. Annual report to the nation on the status of cancer, part I: national cancer statistics. Cancer. 2018;124:2785–2800. doi: 10.1002/cncr.31551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lirov R, Worden FP, Cohen MS. The treatment of advanced thyroid cancer in the age of novel targeted therapies. Drugs. 2017;77:733–745. doi: 10.1007/s40265-017-0733-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.American Thyroid Association (ATA) Guidelines Taskforce on Thyroid Nodules, Differentiated Thyroid Cancer. Cooper DS, Doherty GM, Haugen BR, Kloos RT, Lee SL, Mandel SJ, Mazzaferri EL, McIver B, Pacini F, Schlumberger M, Sherman SI, Steward DL, Tuttle RM. Revised American Thyroid Association management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid. 2009;19:1167–1214. doi: 10.1089/thy.2009.0110. [DOI] [PubMed] [Google Scholar]
- 6.Durante C, Haddy N, Baudin E, Leboulleux S, Hartl D, Travagli JP, Caillou B, Ricard M, Lumbroso JD, De Vathaire F, Schlumberger M. Long-term outcome of 444 patients with distant metastases from papillary and follicular thyroid carcinoma: benefits and limits of radioiodine therapy. J Clin Endocrinol Metab. 2006;91:2892–2899. doi: 10.1210/jc.2005-2838. [DOI] [PubMed] [Google Scholar]
- 7.Zaballos MA, Acuna-Ruiz A, Morante M, Crespo P, Santisteban P. Regulators of the RAS-ERK pathway as therapeutic targets in thyroid cancer. Endocr Relat Cancer. 2019;26:R319–R344. doi: 10.1530/ERC-19-0098. [DOI] [PubMed] [Google Scholar]
- 8.Varricchi G, Loffredo S, Marone G, Modestino L, Fallahi P, Ferrari SM, de Paulis A, Antonelli A, Galdiero MR. The immune landscape of thyroid cancer in the context of immune checkpoint inhibition. Int J Mol Sci. 2019;20:3934. doi: 10.3390/ijms20163934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Savoia P, Fava P, Casoni F, Cremona O. Targeting the ERK signaling pathway in melanoma. Int J Mol Sci. 2019;20:1483. doi: 10.3390/ijms20061483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Naoum GE, Morkos M, Kim B, Arafat W. Novel targeted therapies and immunotherapy for advanced thyroid cancers. Mol Cancer. 2018;17:51. doi: 10.1186/s12943-018-0786-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ball DW, Jin N, Rosen DM, Dackiw A, Sidransky D, Xing M, Nelkin BD. Selective growth inhibition in BRAF mutant thyroid cancer by the mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244. J Clin Endocrinol Metab. 2007;92:4712–4718. doi: 10.1210/jc.2007-1184. [DOI] [PubMed] [Google Scholar]
- 12.Hayes DN, Lucas AS, Tanvetyanon T, Krzyzanowska MK, Chung CH, Murphy BA, Gilbert J, Mehra R, Moore DT, Sheikh A, Hoskins J, Hayward MC, Zhao N, O’Connor W, Weck KE, Cohen RB, Cohen EE. Phase II efficacy and pharmacogenomic study of selumetinib (AZD6244; ARRY-142886) in iodine-131 refractory papillary thyroid carcinoma with or without follicular elements. Clin Cancer Res. 2012;18:2056–2065. doi: 10.1158/1078-0432.CCR-11-0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herr R, Halbach S, Heizmann M, Busch H, Boerries M, Brummer T. BRAF inhibition upregulates a variety of receptor tyrosine kinases and their downstream effector Gab2 in colorectal cancer cell lines. Oncogene. 2018;37:1576–1593. doi: 10.1038/s41388-017-0063-5. [DOI] [PubMed] [Google Scholar]
- 14.Ponz-Sarvise M, Corbo V, Tiriac H, Engle DD, Frese KK, Oni TE, Hwang CI, Ohlund D, Chio IIC, Baker LA, Filippini D, Wright K, Bapiro TE, Huang P, Smith P, Yu KH, Jodrell DI, Park Y, Tuveson DA. Identification of resistance pathways specific to malignancy using organoid models of pancreatic cancer. Clin Cancer Res. 2019;25:6742–6755. doi: 10.1158/1078-0432.CCR-19-1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ostman A, Hellberg C, Bohmer FD. Protein-tyrosine phosphatases and cancer. Nat Rev Cancer. 2006;6:307–320. doi: 10.1038/nrc1837. [DOI] [PubMed] [Google Scholar]
- 16.Aceto N, Sausgruber N, Brinkhaus H, Gaidatzis D, Martiny-Baron G, Mazzarol G, Confalonieri S, Quarto M, Hu G, Balwierz PJ, Pachkov M, Elledge SJ, van Nimwegen E, Stadler MB, Bentires-Alj M. Tyrosine phosphatase SHP2 promotes breast cancer progression and maintains tumor-initiating cells via activation of key transcription factors and a positive feedback signaling loop. Nat Med. 2012;18:529–537. doi: 10.1038/nm.2645. [DOI] [PubMed] [Google Scholar]
- 17.Leung CON, Tong M, Chung KPS, Zhou L, Che N, Tang KH, Ding J, Lau EYT, Ng IOL, Ma S, Lee TKW. Overriding adaptive resistance to sorafenib through combination therapy with Src homology 2 domain-containing phosphatase 2 blockade in hepatocellular carcinoma. Hepatology. 2020;72:155–168. doi: 10.1002/hep.30989. [DOI] [PubMed] [Google Scholar]
- 18.Xia L, Wen L, Wang S. SHP2 inhibition benefits epidermal growth factor receptor mutated non-small cell lung cancer therapy. Mini Rev Med Chem. 2020;21:1314–1321. doi: 10.2174/1389557520666201127104104. [DOI] [PubMed] [Google Scholar]
- 19.Dong L, Yu WM, Zheng H, Loh ML, Bunting ST, Pauly M, Huang G, Zhou M, Broxmeyer HE, Scadden DT, Qu CK. Leukaemogenic effects of Ptpn11 activating mutations in the stem cell microenvironment. Nature. 2016;539:304–308. doi: 10.1038/nature20131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Valencia-Sama I, Ladumor Y, Kee L, Adderley T, Christopher G, Robinson CM, Kano Y, Ohh M, Irwin MS. NRAS status determines sensitivity to SHP2 inhibitor combination therapies targeting the RAS-MAPK pathway in neuroblastoma. Cancer Res. 2020;80:3413–3423. doi: 10.1158/0008-5472.CAN-19-3822. [DOI] [PubMed] [Google Scholar]
- 21.Fedele C, Ran H, Diskin B, Wei W, Jen J, Geer MJ, Araki K, Ozerdem U, Simeone DM, Miller G, Neel BG, Tang KH. SHP2 inhibition prevents adaptive resistance to MEK inhibitors in multiple cancer models. Cancer Discov. 2018;8:1237–1249. doi: 10.1158/2159-8290.CD-18-0444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang J, Zhang F, Niu R. Functions of Shp2 in cancer. J Cell Mol Med. 2015;19:2075–2083. doi: 10.1111/jcmm.12618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu ZQ, Ma R, Zhang CM, Li J, Li L, Hu ZT, Gao QI, Li WM. Expression and clinical significance of tyrosine phosphatase SHP2 in thyroid carcinoma. Oncol Lett. 2015;10:1507–1512. doi: 10.3892/ol.2015.3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cao J, Huang YQ, Jiao S, Lan XB, Ge MH. Clinicopathological and prognostic significance of SHP2 and Hook1 expression in patients with thyroid carcinoma. Hum Pathol. 2018;81:105–112. doi: 10.1016/j.humpath.2018.06.016. [DOI] [PubMed] [Google Scholar]
- 25.Zhi J, Zhang P, Zhang W, Ruan X, Tian M, Guo S, Zhang W, Zheng X, Zhao L, Gao M. Inhibition of BRAF sensitizes thyroid carcinoma to immunotherapy by enhancing tsMHCII-mediated immune recognition. J Clin Endocrinol Metab. 2021;106:91–107. doi: 10.1210/clinem/dgaa656. [DOI] [PubMed] [Google Scholar]
- 26.Ahmed TA, Adamopoulos C, Karoulia Z, Wu X, Sachidanandam R, Aaronson SA, Poulikakos PI. SHP2 drives adaptive resistance to ERK signaling inhibition in molecularly defined subsets of ERK-dependent tumors. Cell Rep. 2019;26:65–78. e5. doi: 10.1016/j.celrep.2018.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amodio V, Yaeger R, Arcella P, Cancelliere C, Lamba S, Lorenzato A, Arena S, Montone M, Mussolin B, Bian Y, Whaley A, Pinnelli M, Murciano-Goroff YR, Vakiani E, Valeri N, Liao WL, Bhalkikar A, Thyparambil S, Zhao HY, de Stanchina E, Marsoni S, Siena S, Bertotti A, Trusolino L, Li BT, Rosen N, Di Nicolantonio F, Bardelli A, Misale S. EGFR blockade reverts resistance to KRAS(G12C) inhibition in colorectal cancer. Cancer Discov. 2020;10:1129–1139. doi: 10.1158/2159-8290.CD-20-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosell R, Karachaliou N, Morales-Espinosa D, Costa C, Molina MA, Sansano I, Gasco A, Viteri S, Massuti B, Wei J, Gonzalez Cao M, Martinez Bueno A. Adaptive resistance to targeted therapies in cancer. Transl Lung Cancer Res. 2013;2:152–159. doi: 10.3978/j.issn.2218-6751.2012.12.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi H, Kong X, Ribas A, Lo RS. Combinatorial treatments that overcome PDGFRbeta-driven resistance of melanoma cells to V600EB-RAF inhibition. Cancer Res. 2011;71:5067–5074. doi: 10.1158/0008-5472.CAN-11-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pratilas CA, Taylor BS, Ye Q, Viale A, Sander C, Solit DB, Rosen N. (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc Natl Acad Sci U S A. 2009;106:4519–4524. doi: 10.1073/pnas.0900780106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Granados K, Huser L, Federico A, Sachindra S, Wolff G, Hielscher T, Novak D, Madrigal-Gamboa V, Sun Q, Vierthaler M, Larribere L, Umansky V, Utikal J. T-type calcium channel inhibition restores sensitivity to MAPK inhibitors in de-differentiated and adaptive melanoma cells. Br J Cancer. 2020;122:1023–1036. doi: 10.1038/s41416-020-0751-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lake D, Correa SA, Muller J. Negative feedback regulation of the ERK1/2 MAPK pathway. Cell Mol Life Sci. 2016;73:4397–4413. doi: 10.1007/s00018-016-2297-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corcoran RB, Dias-Santagata D, Bergethon K, Iafrate AJ, Settleman J, Engelman JA. BRAF gene amplification can promote acquired resistance to MEK inhibitors in cancer cells harboring the BRAF V600E mutation. Sci Signal. 2010;3:ra84. doi: 10.1126/scisignal.2001148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lito P, Pratilas CA, Joseph EW, Tadi M, Halilovic E, Zubrowski M, Huang A, Wong WL, Callahan MK, Merghoub T, Wolchok JD, de Stanchina E, Chandarlapaty S, Poulikakos PI, Fagin JA, Rosen N. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell. 2012;22:668–682. doi: 10.1016/j.ccr.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Turke AB, Song Y, Costa C, Cook R, Arteaga CL, Asara JM, Engelman JA. MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors. Cancer Res. 2012;72:3228–3237. doi: 10.1158/0008-5472.CAN-11-3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang WQ, Lin Q, Zhuang X, Cai LL, Ruan RS, Lu ZX, Tzeng CM. Structure, function, and pathogenesis of SHP2 in developmental disorders and tumorigenesis. Curr Cancer Drug Targets. 2014;14:567–588. doi: 10.2174/1568009614666140717105001. [DOI] [PubMed] [Google Scholar]
- 37.Mainardi S, Mulero-Sanchez A, Prahallad A, Germano G, Bosma A, Krimpenfort P, Lieftink C, Steinberg JD, de Wit N, Goncalves-Ribeiro S, Nadal E, Bardelli A, Villanueva A, Bernards R. SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat Med. 2018;24:961–967. doi: 10.1038/s41591-018-0023-9. [DOI] [PubMed] [Google Scholar]
- 38.Wang J, Pollard K, Allen AN, Tomar T, Pijnenburg D, Yao Z, Rodriguez FJ, Pratilas CA. Combined inhibition of SHP2 and MEK is effective in models of NF1-deficient malignant peripheral nerve sheath tumors. Cancer Res. 2020;80:5367–5379. doi: 10.1158/0008-5472.CAN-20-1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fan Z, Tian Y, Chen Z, Liu L, Zhou Q, He J, Coleman J, Dong C, Li N, Huang J, Xu C, Zhang Z, Gao S, Zhou P, Ding K, Chen L. Blocking interaction between SHP2 and PD-1 denotes a novel opportunity for developing PD-1 inhibitors. EMBO Mol Med. 2020;12:e11571. doi: 10.15252/emmm.201911571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen D, Barsoumian HB, Yang L, Younes AI, Verma V, Hu Y, Menon H, Wasley M, Masropour F, Mosaffa S, Ozgen T, Klein K, Cortez MA, Welsh JW. SHP-2 and PD-L1 inhibition combined with radiotherapy enhances systemic antitumor effects in an anti-PD-1-resistant model of non-small cell lung cancer. Cancer Immunol Res. 2020;8:883–894. doi: 10.1158/2326-6066.CIR-19-0744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li J, Shang G, Chen YJ, Brautigam CA, Liou J, Zhang X, Bai XC. Cryo-EM analyses reveal the common mechanism and diversification in the activation of RET by different ligands. Elife. 2019;8:e47650. doi: 10.7554/eLife.47650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haroon Al Rasheed MR, Xu B. Molecular alterations in thyroid carcinoma. Surg Pathol Clin. 2019;12:921–930. doi: 10.1016/j.path.2019.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Groot JW, Links TP, Plukker JT, Lips CJ, Hofstra RM. RET as a diagnostic and therapeutic target in sporadic and hereditary endocrine tumors. Endocr Rev. 2006;27:535–560. doi: 10.1210/er.2006-0017. [DOI] [PubMed] [Google Scholar]
- 44.Zhu Z, Ciampi R, Nikiforova MN, Gandhi M, Nikiforov YE. Prevalence of RET/PTC rearrangements in thyroid papillary carcinomas: effects of the detection methods and genetic heterogeneity. J Clin Endocrinol Metab. 2006;91:3603–3610. doi: 10.1210/jc.2006-1006. [DOI] [PubMed] [Google Scholar]
- 45.Santoro M, Carlomagno F. Central role of RET in thyroid cancer. Cold Spring Harb Perspect Biol. 2013;5:a009233. doi: 10.1101/cshperspect.a009233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ruess DA, Heynen GJ, Ciecielski KJ, Ai J, Berninger A, Kabacaoglu D, Görgülü K, Dantes Z, Wörmann SM, Diakopoulos KN, Karpathaki AF, Kowalska M, Kaya-Aksoy E, Song L, van der Laan EAZ, López-Alberca MP, Nazaré M, Reichert M, Saur D, Erkan MM, Hopt UT, Sainz B Jr, Birchmeier W, Schmid RM, Lesina M, Algül H. Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat Med. 2018;24:954–960. doi: 10.1038/s41591-018-0024-8. [DOI] [PubMed] [Google Scholar]
- 47.Nichols RJ, Haderk F, Stahlhut C, Schulze CJ, Hemmati G, Wildes D, Tzitzilonis C, Mordec K, Marquez A, Romero J, Hsieh T, Zaman A, Olivas V, McCoach C, Blakely CM, Wang Z, Kiss G, Koltun ES, Gill AL, Singh M, Goldsmith MA, Smith JAM, Bivona TG. RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat Cell Biol. 2018;20:1064–1073. doi: 10.1038/s41556-018-0169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Landa I, Pozdeyev N, Korch C, Marlow LA, Smallridge RC, Copland JA, Henderson YC, Lai SY, Clayman GL, Onoda N, Tan AC, Garcia-Rendueles MER, Knauf JA, Haugen BR, Fagin JA, Schweppe RE. Comprehensive genetic characterization of human thyroid cancer cell lines: a validated panel for preclinical studies. Clin Cancer Res. 2019;25:3141–3151. doi: 10.1158/1078-0432.CCR-18-2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Durante C, Puxeddu E, Ferretti E, Morisi R, Moretti S, Bruno R, Barbi F, Avenia N, Scipioni A, Verrienti A, Tosi E, Cavaliere A, Gulino A, Filetti S, Russo D. BRAF mutations in papillary thyroid carcinomas inhibit genes involved in iodine metabolism. J Clin Endocrinol Metab. 2007;92:2840–2843. doi: 10.1210/jc.2006-2707. [DOI] [PubMed] [Google Scholar]
- 50.Liu Q, Qu J, Zhao M, Xu Q, Sun Y. Targeting SHP2 as a promising strategy for cancer immunotherapy. Pharmacol Res. 2020;152:104595. doi: 10.1016/j.phrs.2019.104595. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.