

Abstract
Cyclic tetrapeptide histone deacetylase inhibitors represent a promising class of antiplasmodial agents that epigenetically disrupt a wide range of cellular processes in Plasmodium falciparum. Unfortunately, certain limitations, including reversible killing effects (plasmodistatic) and host cell toxicity, have played a role in preventing these inhibitors from entering into clinical use as antimalarials. In this study, we present a series of cyclic tetrapeptide analogs derived primarily from the fungus Wardomyces dimerus that inhibit P. falciparum with low nanomolar potency and high selectivity. This cyclic tetrapeptide scaffold was diversified further via semi-synthesis, leading to the identification of several key structural changes that positively impacted the selectivity, potency, and in vitro killing rate profiles of these compounds. We confirmed their effectiveness as HDAC inhibitors through the inhibition of PfHDAC1 catalytic activity, in silico modeling, and the hyperacetylation of histone H4. Additional analysis revealed the in vitro inhibition of the most active epoxide-containing analog was plasmodistatic, and reversible once removed after 24 or 48 hours. In contrast, one of the new diacetyloxy semi-synthetic analogs, CTP-NPDG 19, displayed a rapid and irreversible action against the parasite once removed (plasmodicidal) after 24 hours of compound exposure.
Keywords: HDAC, cyclic tetrapeptide, Plasmodium, PfHDAC1, malaria, natural products
Graphical Abstract
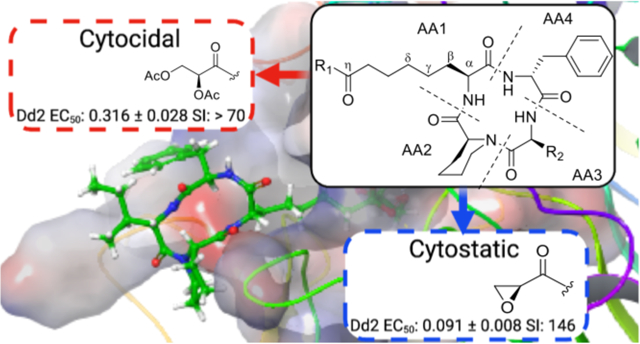
Malaria remains a constant threat to both global health and economic stability, with over 229 million new infections and 409,000 deaths reported worldwide in 20191. Plasmodium, the apicomplexan parasite responsible for malaria, has a long history of evading frontline clinical agents, with numerous cases of drug resistance now documented2–6. As a consequence, the steady progress made by frontline treatment with artemisinin combination therapies (ACTs) is threatened by the development of drug resistance in Southeast Asia and Rwanda4, 6. Equally alarming is the independent emergence of the resistance conferring C580Y mutation of pfk13 in parasite samples taken from Guyana, demonstrating the concurrent evolution of resistance with spatial isolation5. These developments necessitate the identification of new antimalarial inhibitors with distinct targets for which there is no pre-existing resistance.
Histone deacetylase (HDAC) inhibitors, which have garnered widespread attention as anti-cancer drugs, have demonstrated promise as a candidate class of antimalarials. In eukaryotes, HDAC enzymes act in coordination with histone acetyl transferases (HATS) to alter the acetylation state of certain amino acid residues on histone proteins7. While histone acetylation has an activating effect on transcription, histone deacetylation results in the condensation of chromatin and concomitant suppression of gene expression. This form of epigenetic regulation is prominent in all stages of the Plasmodium life cycle, and is critical to parasite stress response and thought to contribute to the transcriptional regulation of drug-resistance7–9. HDAC enzymes can be divided into four classes of Zn2+ dependent enzymes and the class of NAD+ dependent sirtuins. Plasmodium falciparum, the most lethal malaria causing species that infects humans, has been found to possess one class I HDAC enzyme (PfHDAC1, PF3D7_0925700), two class II HDAC enzymes (PF14_069 and PF10_0078), and two sirtuins (PF3D7_1328800 and PF3D7_1451400). The sirtuins (PfSir2A and PfSir2B) have been found to be non-essential, and are primarily involved in the regulation of var gene expression responsible for antigenic variation, making them less appealing as drug targets9. Of the Zn2+ dependent plasmodial HDACs, PfHDAC1 is the most highly conserved across all species, in agreement with its essential nature10 .
The potential of HDAC inhibitors as antiplasmodials has been validated through the repurposing of FDA approved inhibitors that exhibit nanomolar activity in both P. falciparum and Plasmodium vivax10–13. Two chemotypes that have received considerable attention are hydroxamic acids and cyclic tetrapeptides (Figure 1).
Figure 1.
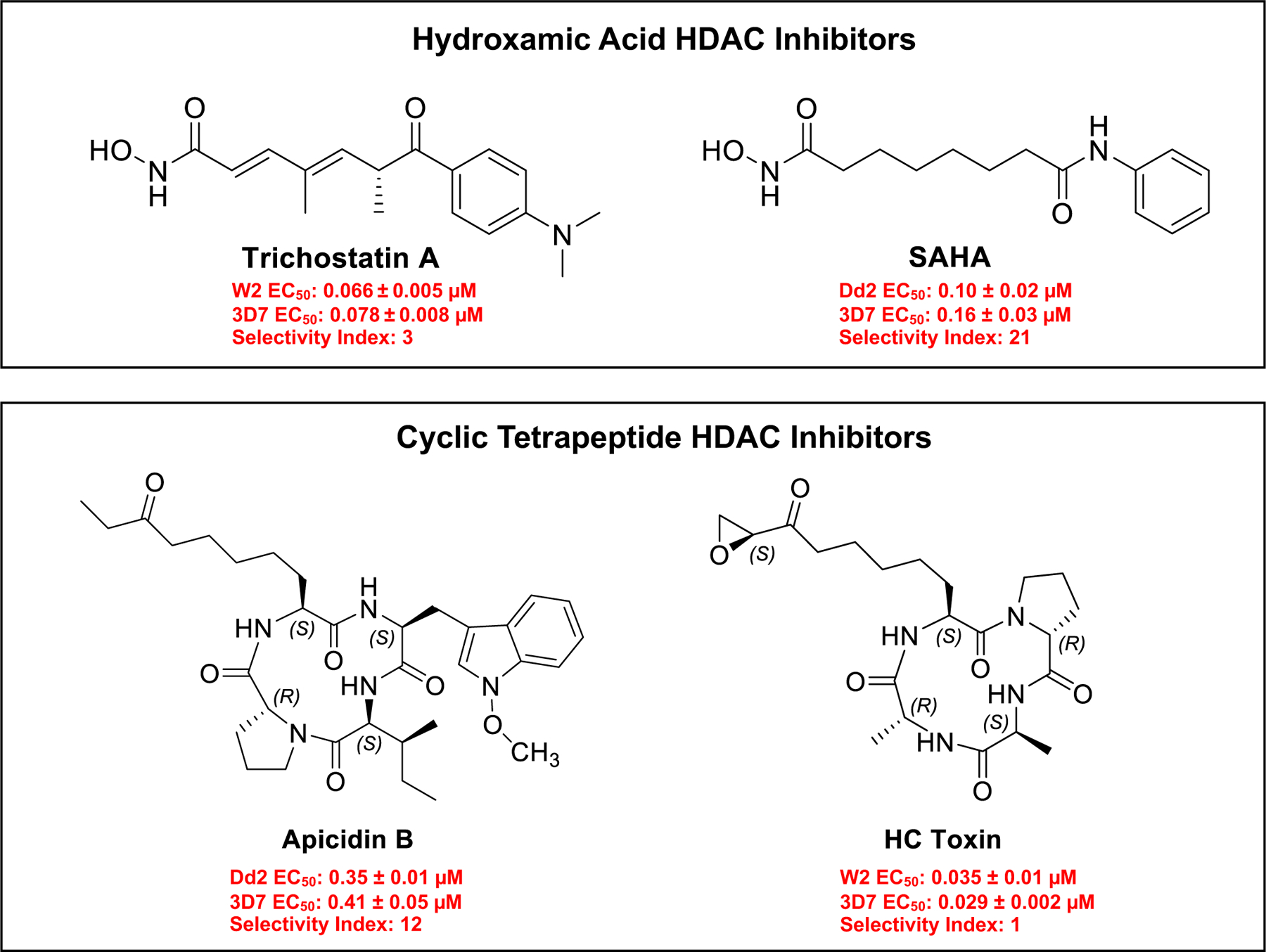
Activity of common hydroxamic acid and cyclic tetrapeptide HDAC inhibitors. Selectivity index (SI) for Apicidin B and SAHA based on HepG2 EC50 / Dd2 and 3D7 EC50 Average. Values for TSA and HC Toxin from reference 7, and SI based on 3D7 and W2 EC50 average. Results are expressed as means from triplicate experiments +/− SEM.
Common hydroxamic acid HDAC inhibitors include compounds such as trichostatin A (TSA) and suberanilohydroxamic acid (SAHA) (also known as vorinostat) with P. falciparum 3D7 EC50 0.078 ± 0.0087 μM and 0.16 ± 0.03 μM, respectively (Supporting Table 1). Hydroxamic acids have a high affinity for Zn2+, and are typically pan-HDAC inhibitors capable of targeting multiple classes of HDAC enzymes14. In addition to exhibiting potent inhibition against standard drug resistant strains, hydroxamic acids have also shown activity against multidrug resistant clinical isolates of both P. falciparum and P. vivax, as well as prevent schizont maturation in P. vivax15. Due to their potent activities, good bioavailability, and ease of preparation, these compounds have found favor with researchers looking to develop HDAC inhibitors as antimalarials. Unfortunately, some of these analogs have been found to offer only plasmodistatic effects when tested against Plasmodium berghei in vivo, meaning the parasite quickly rebounded once the drug was removed16. The transient inhibitory nature of these compounds has been demonstrated at the transcriptional level as well in P. falciparum parasites following treatment with the HDAC inhibitor TSA. While TSA caused a dramatic deregulation of genes associated with the parasites intraerythrocytic life cycle, the majority of these effects were reversible in under 30 min8. Compound toxicity has historically been a concern with hydroxamic acid derivates; although steady progress has been made to produce more selective analogs15, 17. Modifications to these hydroxamic acid scaffolds has resulted in inhibitors that exhibit a slow binding to class I HDAC complexes, and improved selectivity18.
Among the reported cyclic tetrapeptides with antiplasmodial activities are HC toxin and apicidin B, with 3D7 EC50 values of 0.029 ± 0.002 μM and 0.41 ± 0.05 μM, respectively. An interest in apicidin has persisted over time, due in part, to its broad antiprotozoal activity13. In P. falciparum, apicidin has been validated as an inhibitor of class I plasmodial HDACs19. P. falciparum contains just the single class I HDAC (PfHDAC1), which is highly conserved, and shows a moderate sequence similarity (61–62%) to human HDACs10, 20 . Unfortunately, apicidin B and its analogs exhibit limited selectivity for the parasite, as compared to mammalian cell (Supporting Table 2). Additionally, in vivo treatment was found to reduce parasitemia, but was not curative13, 21. To overcome the deficiencies of these HDAC inhibitory scaffolds, compounds with greater selectivity and permanent killing effects against the parasite (plasmodicidal) are needed, preferably in under 24 h.
In this study we investigated a series of 29 natural and semi-synthetic cyclic tetrapeptide HDAC inhibitors identified through our screening efforts of fungus-derived natural product scaffolds. Several of these inhibitors exhibited potent in vitro activity against multidrug-resistant Dd2 and drug-sensitive 3D7 P. falciparum strains, high selectivity, and low nanomolar inhibition of PfHDAC1. We provide additional evidence that these compounds act as HDAC inhibitors through the rapid hyperacetylation of histone H4, generally accepted as a canonical signature of HDAC inhibition in Plasmodium8 . Additionally, our results indicated that the natural product 3 exhibited a plasmodistatic and reversible effect, whereas the semi-synthetic analog 26 demonstrated plasmodicidal action against the parasite. These findings provide insight into the different chemotypes relevant to the reversibility and selectivity of antiplasmodial activity among cyclic tetrapeptide HDAC inhibitors, and their potential as antimalarial therapeutics.
RESULTS AND DISCUSSION:
Phenotypic screening identifies known and unknown cyclic tetrapeptides with antiplasmodial potency:
A SYBR Green I-based fluorescence assay was used to screen a library of 759 purified natural products from the University of Oklahoma, Natural Product Discovery Group, at a fixed concentration (1 μM) using the multidrug resistant P. falciparum strain, Dd2. Compounds that demonstrated > 50% inhibition of parasite growth were prioritized, and concentration-response curves were generated to determine their EC50 values. Two of the compounds detected as hits were the fungus-derived cyclic tetrapeptides chlamydocin (compound 1, Dd2 EC50: 0.031 ± 0.007 μM) and 1-alaninechlamydocin (compound 2, Dd2 EC50: 0.010 ± 0.001 μM) (Table 1). Chlamydocin and its analogues were previously explored for their cancer cell inhibitory effects22–23; however, this is the first report of antiplasmodial activity associated with these natural products. A counter screen was then performed to assess the selectivity of compounds 1 and 2 for P. falciparum parasites versus the human HepG2 cell line. The results revealed both compounds were relatively toxic to HepG2 cells, with some variation (EC50 values of 0.53 ± 0.007 μM and 0.038 ± 0.006 μM for chlamydocin and 1-alaninechlamydocin, respectively). Additional analogs were later identified via a separate bioassay guided dereplication of Lasionectria hilhorsti, compounds 3–5 (Table 1). The activity of these analogs provides additional examples of the relative toxicity of this stereoisomeric scaffold. The isolation and identification of these compounds are provided in the Supporting Information (Supporting Figures 1–6, Supporting Tables 3–9). We identified compound 3 to be the natural product FR235222 (synonym: WF 27082B), a cyclic tetrapeptide previously reported from a Acremonium sp24. Compound 3 was reported to cause class I HDAC inhibition in Toxoplasma gondii, and exhibited low nanomolar antiplasmodial inhibition in Dd2 and 3D725. Our testing confirmed similar bioactivity at Dd2 EC50: 0.033 ± 0.007, and significant toxicity in HepG2, EC50: 0.26 ± 0.06 (Table 1). Compound 3 and its analogue 4 (WF 27082F), have also been reported for their activities against human class I HDACs and T-cell proliferation26. We found compound 4, and the remaining analogue, compound 5, demonstrated low nanomolar potencies against P. falciparum Dd2 with EC50 values of 0.024 ± 0.005 and EC50 0.062 ± 0.01, respectively (Table 1). Toxicity against human HepG2 cells varied with an EC50 of 0.15 ± 0.04 for 4, and 0.88 ± 0.05 for compound 5. While all 5 analogs show significant activity and toxicity, some generalized trends can be identified. For R1, analogs with an epoxide or alcohol group showed comparable levels of activity; while the ethyl analog (compound 5) showed a 2–6 fold increase in EC50 comparatively. Compound 5 also had a higher HepG2 EC50, suggesting activity and toxicity increases concurrently and non-specifically. The impact of a hydrogen to methyl substitution at R2 was either negligible, or confounded given the sample size. In general, the substitution of a methyl group on R3 to a less sterically hindered hydrogen resulted in both higher potency, and increased cytotoxicity, as with compounds 2 vs 1 and 3 or 5 vs 4. Despite the toxicity, the degree of variation in the selectivity exhibited by the natural products led us to surmise that achieving greater levels of selectivity might be possible through further exploration of this cyclic tetrapeptide scaffold.
Table 1.
Antimalarial Effects of SRSS Cyclic Tetrapeptides.

| |||||||
|---|---|---|---|---|---|---|---|
| ID | R1 | R2 | R3 | Dd2 EC50 (μM) | 3D7 EC50 (μM) | HepG2 EC50 (μM) | SI |
| Chlamydocin (1) |
|
H | CH3 | 0.031 ± 0.007 | - | 0.53 ± 0.007 | 17 |
| 1-Alaninechlamydocin (2) |
|
H | H | 0.010 ± 0.001 | 0.010 ± 0.001 | 0.038 ± 0.006 | 4 |
| WF 20782B (3) |
|
CH3 | CH3 | 0.033 ± 0.007 | 0.050 ± 0.002 | 0.26 ± 0.06 | 6 |
| WF 27082F (4) |
|
CH3 | H | 0.024 ± 0.005 | 0.026 ± 0.001 | 0.15 ± 0.04 | 6 |
| Deoxy-WF 20782B (5) |

|
CH3 | CH3 | 0.062 ± 0.01 | 0.092 ± 0.001 | 0.88 ± 0.05 | 11 |
Results are expressed as means from triplicate experiments +/− SE. Selectivity Index (SI) = HepG2 EC50 / (: Dd2 EC50, 3D7 EC50)
Identification of cyclic tetrapeptide variants with potent antiplasmodial activity and improved selectivity:
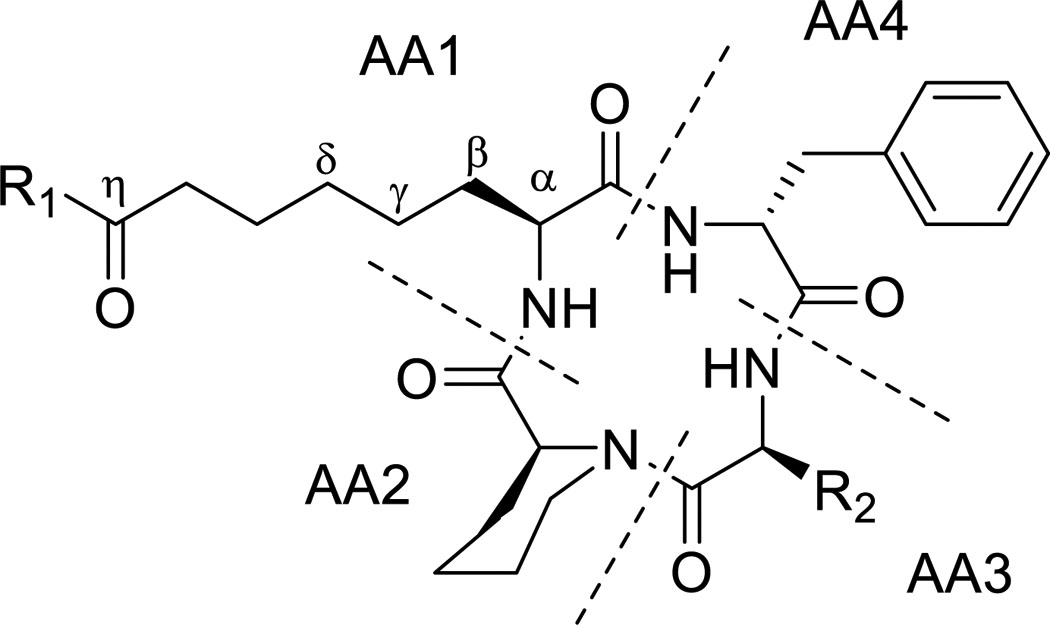
To further determine whether cyclic tetrapeptide scaffolds could be identified that exhibited increased selectivity toward inhibiting Plasmodium spp., a search of the University of Oklahoma fungal library was conducted, which led to the identification of a Wardomyces dimerus isolate that produced a large number of natural products containing the target scaffold, including the known compounds 6 (PF1070A), 7 (NA1309), and 11 (WF-3161)27–28. Purification efforts resulted in to the identification of 10 previously unreported cyclic tetrapeptide analogues (compounds 8–10, 12–18). Their structures were subsequently determined based on interpretation of data from a combination of mass spectrometry, NMR, additional spectroscopic methods (e.g., X-ray), and chemical (e.g., Marfey’s and GITC methods) experiments. These data, along with summaries of the data interpretation, are provided in the Supporting Information (Supporting Figures 1–6, Supporting Tables 3–9). Notably, our structure determination studies included efforts to independently analyze the absolute configurations of the epoxyketone moieties in the AA1 residues of 6, 11, and 16; as well as assess the absolute configurations of the analogous alcohol-bearing stereogenic centers of the naturally-occurring analogues. Those endeavors resulted in the generation of 11 semisynthetic products (compounds 19-29) that afforded opportunities to test how alterations of the oxidation states of the carbon atoms comprising the AA1 epoxyketone system impacted antiplasmodial activity and selectivity. The activities of the cyclic tetrapeptides are summarized in Table 2.
Table 2.
Antimalarial Effects of SSSR Cyclic Tetrapeptides.
| ||||||
|---|---|---|---|---|---|---|
| ID | R1 | R2 | Dd2 EC50 (μM) | 3D7 EC50 (μM) | HepG2 EC50 (μM) | SI |
| PF 1070A (6) |
|
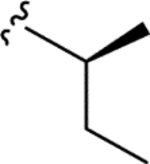
|
0.091 ± 0.008 | 0.069 ± 0.003 | 12 ± 1 | 150 |
| NA 1309 (7) |
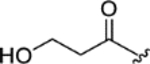
|
|
> 5.0 | > 5.0 | > 25 | - |
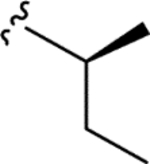
| CTP-NPDG 1 (8) |
|
|
> 5.0 | > 5.0 | > 25 | - |
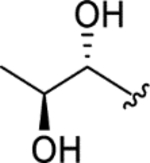
| CTP-NPDG 2 (9) |

|

|
2.5 ± 0.05 | 3.5 ± 0.3 | > 25 | > 8 |
| CTP-NPDG 3 (10) |
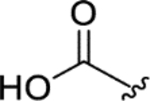
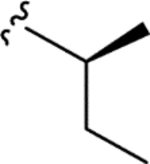
|
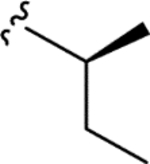
|
> 5.0 | > 5.0 | > 25 | - |
| WF-3161 (11) |
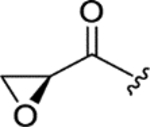
|
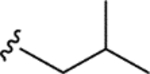
|
0.12 ± 0.011 | 0.13 ± 0.04 | 22 ± 0.9 | 176 |
| CTP-NPDG 4 (12) |

|

|
> 5.0 | > 5.0 | > 25 | - |
| CTP-NPDG 5 (13) |
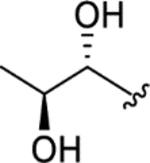
|
|
> 5.0 | > 5.0 | > 25 | - |
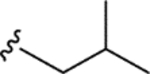
| CTP-NPDG 6 (14) |
|
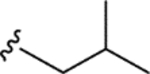
|
1.7 ± 0.09 | 2.3 ± 0.1 | > 25 | > 13 |
| CTP-NPDG 7 (15) |
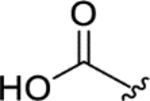
|
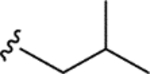
|
1.6 ± 0.05 | 2.4 ± 0.1 | > 25 | > 13 |
| CTP-NPDG 8 (16) |
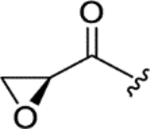
|

|
0.24 ± 0.02 | 0.35 ± 0.07 | > 25 | > 85 |
| CTP-NPDG 9 (17) |
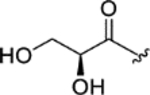
|

|
3.3 ± 0.3 | 4.2 ± 0.2 | > 25 | > 7 |
| CTP-NPDG 10 (18) |
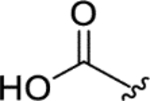
|
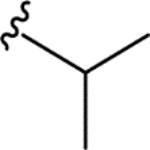
|
> 5.0 | > 5.0 | > 25 | - |
| CTP-NPDG 11 (19) |
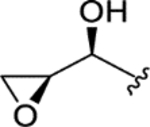
|
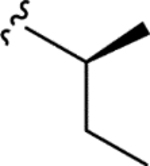
|
> 5.0 | > 5.0 | > 25 | - |
| CTP-NPDG 12 (20) |
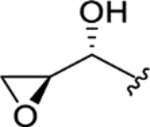
|
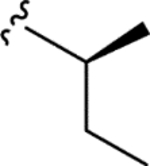
|
> 5.0 | > 5.0 | > 25 | - |
| CTP-NPDG 13 (21) |
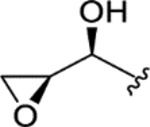
|
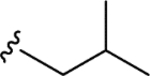
|
2.2 ± 0.1 | 3.0 ± 0.4 | > 25 | > 10 |
| CTP-NPDG 14 (22) |
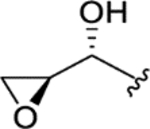
|
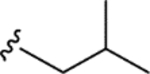
|
> 5.0 | > 5.0 | > 25 | - |
| CTP-NPDG 15 (23) |
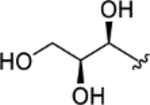
|
|
> 5.0 | > 5.0 | > 25 | - |
| CTP-NPDG 16 (24) |
|
|
> 5.0 | > 5.0 | > 25 | - |
| CTP-NPDG 17 (25) |
|

|
> 5.0 | 3.6 ± 0.1 | > 25 | > 7* |
| CTP-NPDG 18 (26) |
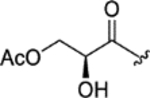
|
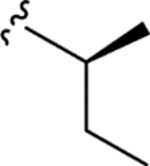
|
1.5 ± 0.1 | 1.9 ± 0.3 | > 25 | > 15 |
| CTP-NPDG 19 (27) |
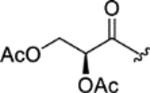
|

|
0.32 ± 0.03 | 0.40 ± 0.08 | > 25 | > 70 |
| CTP-NPDG 20 (28) |

|

|
3.2 ± 0.2 | 2.6 ± 0.3 | > 25 | > 9 |
| CTP-NPDG 21 (29) |
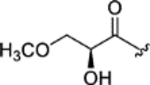
|

|
1.3 ± 0.05 | 1.7 ± 0.009 | > 25 | > 17 |
Results are expressed as means from triplicate experiments ± SEM. Selectivity index (SI) = HepG2 EC50 / (Dd2 E50, 3D7 EC50 average) *Selectivity of 25 based on 3D7 EC50 only.
The data generated for the 29-total cyclic tetrapeptides revealed several trends and sheds new insight into potential avenues for developing this chemical scaffold for its antiplasmodial effects. First, the cyclic tetrapeptide core could be modified to accept an appreciable spectrum of changes in the relative order, alpha-carbon configurations, and identities of the lipophilic AA2-AA4 residues. Those changes included the interchange of Pro and pipecolate (Pip) residues, incorporation of either D- or L-Phe residues, and the substitution of Leu/Iso/Val residues. Second, these structural changes often resulted in fluctuations in not just potency, but also in selectivity. For example, altering the cyclic tetrapeptide macrocycle from Pro (AA2) – L-phenylalanine (AA 3) – Ala/Aib (AA4) (e.g., chlamydocin) to Pip (AA2) – Leu/Iso/Val (AA3) – D-phenylalanine (AA4) resulted in compounds with > 10-fold improvements in selectivity against P. falciparum versus human cells. Third, the AA1 side-chain supported a surprisingly wide range of functionalization options at its terminus; albeit, some of the changes resulted in losses in activity. For example, a carbonyl was required in most cases at the seventh carbon (i.e., η-carbon) atom of AA1 for antiplasmodial activity to be retained. However, an exception was noted with epoxy alcohol 21, which retained modest activity, while epimeric 22 was inactive. The structures containing a η-keto group, accompanied by a vicinal diol or derivatized diol moieties, proved more nuanced in terms of antiplasmodial effects. Whereas most of these compounds retained only modest antiplasmodial activities, a surprising activity against both P. falciparum strains (Dd2 EC50: 0.32 ± 0.03 μM and 3D7 EC50: 0.40 ± 0.08 μM), it exhibited a high level of selectivity versus HepG2 cells (SI > 70). exception was noted for the Structurally, the natural and semi-synthetic analogues can be organized into groups based on variations occurring at two sites with the compounds: the aliphatic residues incorporated into the cyclotetrapeptide scaffold (i.e., R1: isoleucine, leucine, or valine) and the type of functional groups present among C29-C31 (R2). In general, changing R1 from a Val to a Leu increased in vitro antiparasitic activity with the trend exemplified by compound pairs 18 (Dd2 EC50: > 5.0 μM) versus 15 (Dd2 EC50: 1.6 ± 0.05 μM), 17 (Dd2 EC50: 3.3 ± 0.3 μM) versus 14 (Dd2 EC50: 1.7 ± 0.09 μM), and 16 (Dd2 EC50: 0.24 ± 0.02 μM) versus 11 (Dd2 EC50: 0.12 ± 0.01 μM). There was also a notable decrease in antiplasmodial activity when one of the methyl groups in leucine residues was shifted to generate an isoleucine, as evident based on comparisons of the activities for compounds 9 (Dd2 EC50: 2.5 ± 0.05 μM) versus 14 (Dd2 EC50: 1.7 ± 0.09 μM), 10 (Dd2 EC50: > 5.0 μM) versus 15 (Dd2 EC50: 1.6 ± 0.05 μM), 19 (Dd2 EC50: > 5.0 μM) versus 21 (Dd2 EC50: 2.2 ± 0.1 μM), and 28 (Dd2 EC50: 3.2 ± 0.03 μM) versus 29 (Dd2 EC50: 1.3 ± 0.05 μM). In studies of broad spectrum HDAC inhibitors such as SAHA, even modest changes to the ‘cap’ regions of those compounds were found to alter interaction around the L1 loop of human HDAC 6 (D460-P484) and cause substantial changes in their enzyme affinities29–30. Our results mirror those effects showing that modifications to this region may alter isoform selectivity, offering the potential to further reducing mammalian cell toxicity18.
As seen with modifications to R2, changes that reduce the reactivity of the Zn2+ binding ketone greatly reduced, or even abolished, antiplasmodial activity. This agrees with published reports demonstrating the zinc binding group is crucial to HDAC catalytic activity7. Reports based on analogous HDAC inhibitors, and class I human HDACs 6 and 8, indicate that this Zn2+ binding ketone is hydrolyzed, forming a geminal-diolate that is stabilized via intermolecular bonding with the Zn2+ and the surrounding catalytic amino acid residues29, 31. In summary, R2 groups composed of cooccurring ketone and epoxide groups showed the greatest antiplasmodial potency, followed by compounds bearing vicinal alcohols at the alpha and beta carbons. In comparison, compounds in which the carbonyl had been reduced tended to be inactive at the concentrations tested, with the exception of compound 21, which showed modest inhibition of Dd2 (EC50 of 2.2 ± 0.1 μM). Possible trends involving other functional group combinations proved more difficult to discern; however, sufficient data were available to enable recognition that R2 analogues bearing acetyloxy or methoxy groups retained activity, with the diacetoxy compound 27 showing good antiplasmodial potency (EC50 of 0.32 ± 0.03 μM against Dd2) and a high level of selectivity (SI > 70). Most of the compounds exhibited little or no cytotoxicity towards HepG2 cells at the concentrations tested, with compound 6 and 11 standing as the only molecules producing HepG2 EC50 values of 12 ± 1, and 22 ± 0.9 μM, respectively. This limited cytotoxicity could be due to insolubility, however several of the toxic analogs showed a nearly identical predicted log P, log D, and log S scores compared to non-toxic analogs, suggesting solubility and lipophilicity may be comparable. A full table of the predicted log P, log D and log S scores for compounds 1-29, SAHA, and Apicidins B and C can be seen in Supporting Table 9.
Natural and semi-synthetic Wardomyces-derived cyclic tetrapeptides inhibit PfHDAC1 catalytic activity:
Based on the activity observed for the chlamydocin analogues against class I HDACs, we sought to validate the HDAC inhibition of these new tetrapeptides by testing them against class I plasmodial HDAC, PfHDAC1, utilizing a recombinant enzyme23, 32. As listed in Table 3, of the 24 Wardomyces-derived natural and semi-synthetic cyclic tetrapeptides, 19 of the compounds showed inhibitory activities against the enzyme with IC50 values < 10 μM, and several showed activities in a low nanomolar range (i.e., compounds 6, 11, 14–16, 18, 21, 25–27). These findings confirm that cyclic tetrapeptides are capable of preventing deacetylation by PfHDAC1 in situ and support its role as the likely in vitro target for these compounds. All in situ activities for these cyclic tetrapeptides are summarized in Table 3.
Table 3.
Enzyme activity against PfHDAC1. All concentrations reported in micromolar (μM).
| ID | PfHDAC1 IC50 (μM) |
|---|---|
| PF 1070A (6) | 0.0011 ± 0.001 |
| NA 1309 (7) | 6.3 ± 0.8 |
| CTP-NPDG 1 (8) | > 10 |
| CTP-NPDG 2 (9) | 3.2 ± 0.6 |
| CTP-NPDG 3 (10) | 7.0 ± 0.5 |
| WF-3161 (11) | 0.0020 ± 0.002 |
| CTP-NPDG 4(12) | > 10 |
| CTP-NPDG 5 (13) | > 10 |
| CTP-NPDG 6 (14) | 0.68 ± 0.1 |
| CTP-NPDG 7 (15) | 0.70 ± 0.09 |
| CTP-NPDG 8(16) | 0.015 ± 0.003 |
| CTP-NPDG 9 (17) | 1.1 ± 0.1 |
| CTP-NPDG 10 (18) | 1.0 ± 0.08 |
| CTP-NPDG 11 (19) | 1.6 ± 0.1 |
| CTP-NPDG 12 (20) | 1.1 ± 0.2 |
| CTP-NPDG 13 (21) | 0.14 ± 0.03 |
| CTP-NPDG 14 (22) | 1.4 ± 0.02 |
| CTP-NPDG 15 (23) | > 10 |
| CTP-NPDG 16 (24) | > 10 |
| CTP-NPDG 17 (25) | 0.66 ± 0.03 |
| CTP-NPDG 18 (26) | 0.47 ± 0.1 |
| CTP-NPDG 19 (27) | 0.20 ± 0.02 |
| CTP-NPDG 20 (28) | 1.2 ± 0.2 |
| CTP-NPDG 21 (29) | 1.3 ± 0.1 |
Notes: Results expressed as means from duplicate experiments +/− SEM.
Linear regression was used to further probe the relationship of enzyme inhibition and in vitro EC50 in Dd2 and 3D7 activities of the cyclic tetrapeptides. This analysis revealed that inhibition of in vitro parasite growth strongly correlated with enzyme inhibition (p < 0.0001), with more potent antiplasmodial analogs showing increased inhibition of PfHDAC1 (Figure 2). For example, compounds 6 and 11 demonstrated IC50 values of 0.0011 ± 0.001 and 0.0020 ± 0.002 μM against PfHDAC1, and in vitro Dd2 EC50 values of 0.091 ± 0.008 and 0.12 ± 0.01 μM, respectively. These distinct measurements of potency were on par, or better, than the FDA approved hydroxamic acid-based inhibitor SAHA, which inhibited PfHDAC with an IC50 of 0.092 ± 0.02 and an in vitro Dd2 EC50 of 0.10 ± 0.02 (Supporting Table 1). Focusing on the R2 group, the PfHDAC1 inhibitory activity results appeared to follow a similar trend to the in vitro P. falciparum inhibition data, with epoxide containing ketones exhibiting the greatest potency, followed by the vicinal diols, simple alcohols, and the reduced ketones. . To account for potential physiochemical differences between these analogs, a linear regression was also performed on the average in vitro pEC50 and the predicted chemical parameters log S and log D (Supporting Figure 7A–B). No correlation was found based on a Pearson two-tailed test. When these parameters were also compared to the predicted chemical parameters log S and log D, no significant correlation was found overall, however some intergroup significance was seen (Supporting Figure 7C–D). There was a significant relationship between the enzyme pIC50 of the reduced ketones and the log S (p = 0.0297), and the enzyme pIC50 of the epoxides and the log D (p = 0.0336). While the later should be considered cautiously due to the small group size (only 3 epoxides), the relationship between the predicted aqueous solubility (log S) and the enzyme activity of the reduced ketones may explain why the correlation between the enzyme and in vitro activity for these analogs falls slightly outside the 99% confidence interval.
Figure 2.

Linear regression of enzyme and in vitro inhibition values. Average in vitro pEC50s calculated as the negative log of the 3D7 and Dd2 average when converted to molar. Average enzyme pCI50 calculated as the negative log of pfHDAC1 activity when converted to molar. Dotted lines represent the 99% confidence interval. Color key represents classification of R2 group. Correlation as calculated by two-tailed Pearson test p < 0.0001. For purposes of analysis, if no IC50 of EC50 value below the maximum concentration tested was available, whole integers were substituted for greater than values (> 5 changed to 5, > 10 changed to 10).
Semi-Synthetic Diacetyloxy Analogue 27 Exhibits a Plasmodicidal Killing Profile:
To further explore the activities of the selective cyclic tetrapeptides, we examined the plasmodial killing rate profiles for two of the most active inhibitors, compounds 6 and 27 (compound 16 was also screened, but exhibited an near identical killing profile to 6, as can be seen in Supporting Figure 8). For these tests, asynchronous P. falciparum Dd2 parasites were exposed to a 10 × EC50 concentrations of the inhibitors for fixed periods of time and the remaining compound removed via washing and replacement with fresh medium. Samples were collected every 24 h for flow cytometric analysis with SYBR Green I and Mitotracker Deep Red FM (MTR) to assess levels of parasitemia and parasite viability. The epoxide containing analogue, compound 6, produced a plasmodistatic effect (Figure 3). In both 24 and 48 h incubations, parasitemia levels rebounded after 3 days; in addition, the fraction of MTR− parasites remained low throughout the time course, reaching the DMSO control average by day 5 with a 24 h exposure (Figure 3A–3B) or day 6 with a 48-h exposure (Figure 3C–3D). As previously discussed, reversibility is a known concern for class I HDAC inhibitors8. However, compound 27 exhibited a pronounced irreversible impairment to the parasite after 24 h, and prevented the recovery of parasitemia throughout the entirety of this time course study (6 days). Also distinct from compound 6, the fraction of MTR− parasites consistently remained above that of the DMSO vehicle control following incubation with compound 27. Taken together, these results indicate that compound 27 produces a plasmodicidal response in P. falciparum distinct from previously reported cyclic tetrapeptides.
Figure 3.
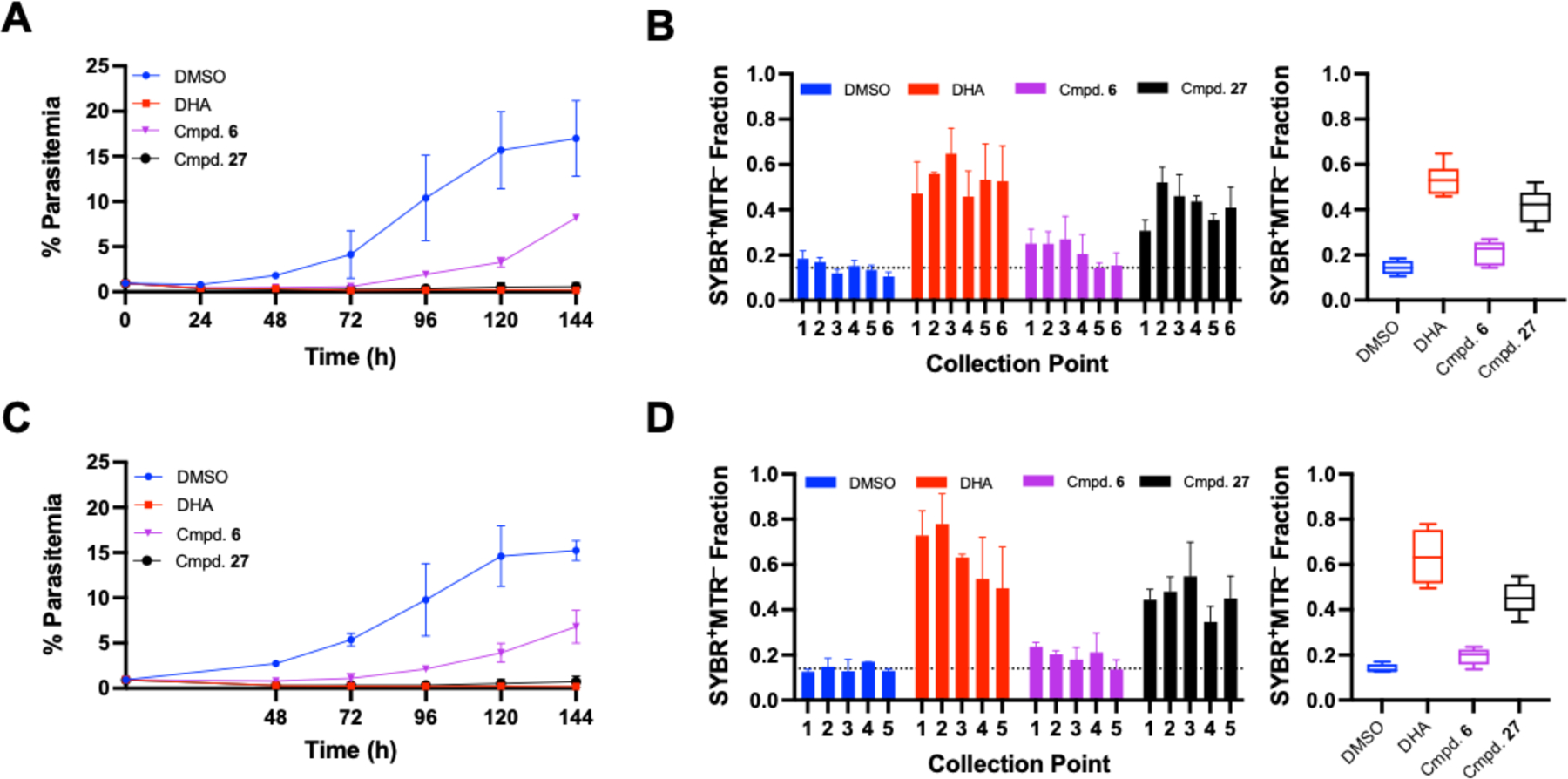
Rate of killing of compounds 6 and 27. Parasites were treated for 24 (A-B) or 48 h (C-D) prior to compound wash off. Flow cytometry with SYBR Green I and Mitotracker Deep Red FM (MTR) was performed after compound wash-off and every 24 h after for 6 days (100,000 events per sample) to track parasitemia (A, C). Parasites (SYBR+) were gated as either MTR+/MTR− and the results are expressed as the fraction of mitotracker positive parasites at each collection point, with the variance between all points expressed via box and whisker plot. Results expressed as means from duplicate experiments +/− SEM.
Molecular modeling illustrates binding of active compounds to PfHDAC1:
To further explore the binding of compounds 6 and 27 within the catalytic pocket of HDAC, we used an in silico modeling approach to generate the inhibitor-enzyme complex (Supporting Figures 9 and 10). Although the crystal structure of PfHDAC1 and the other plasmodial HDAC enzymes have not been reported, the structures of human class I HDAC analogues are well documented; and due to the extensive sequence homologies, these proteins have been used as the starting points for computational studies of the plasmodial isoforms12, 30, 33. The hydrolyzed forms of both inhibitors were used, based on mechanistic studies on class I HDACs 6 and 8 that found that the ketone carbon of related cyclic tetrapeptide HDAC inhibitors must be hydrolyzed to form a geminal-diolate29, 31. Additionally, crystal structure data (PDB: 5VI6) of HDAC8 complexed with close analog trapoxin A (PDB: 5VI6), indicates the usually reactive epoxide remains intact during enzyme binding31.
In the course of this study, we discovered that 27 exhibited a poor fit with human class I HDAC 8 due to steric hinderance from the bulky acetyloxy groups. Likewise, analogues containing a single acetyloxy moiety, such as 25 or 26, exhibited poor fits with the catalytic site of the enzyme, based on modeling. These issues might have arisen from structural differences between the human and plasmodial HDAC isoforms. Based on the reported findings of other antiplasmodial HDAC inhibitor screenings, and homology modeling with Schistosoma mansoni, parasite isoforms such as PfHDAC1 may be more able to tolerate bulky substituents34. Additionally, compound 27 may convert to a more accessible form, such as the deacetylated analog, compound 9. We therefore modeled both compound 6, and compound 9, to provide additional insight. As indicated by the modeling results, both compounds can form a gem diol that interacts with Zn2+, HIS 142, and TYR 306 (Supporting Figures 9 and 10). The docking results vary based on their potential hydrophilic interactions, and the direct binding of the ‘cap’ region with ASP101 and HIS180. As determined by the docking software, compound 6 binding has an improved lipophilic contact score, and compound 9 binding resulted in a better gliding and overall docking score.
Compound 27 may convert to compound 9 in vitro:
As discussed, the model limitations for compound 27 raised the possibility that the acetyloxy groups in compound 27 hydrolyze under assay conditions to generate compound 9, as illustrated in Figure 4. Compound 9 however, is significantly less active both in vitro (Dd2 EC50: 2.5 ± 0.05) and in situ against the PfHDAC1 enzyme (IC50: 3.2 ± 0.6). Ester groups are often added in pro-drug design to protect the hydrogen bonding groups and increase lipophilicity35. Similarly, the presence of the ester groups in compound 27 may increase the activity from compound 9 by improving the chemical stability at varying pH, and increasing the compounds resistance to hydrolysis, resulting in slower degradation and thus higher concentrations of the active component at any given time. To explore whether compound 27 converted to 9, and if that transformation could account for the increased activity of 27, both compounds were subjected to culture conditions for 2 and 6 h, followed by LCMS detection.
Figure 4.
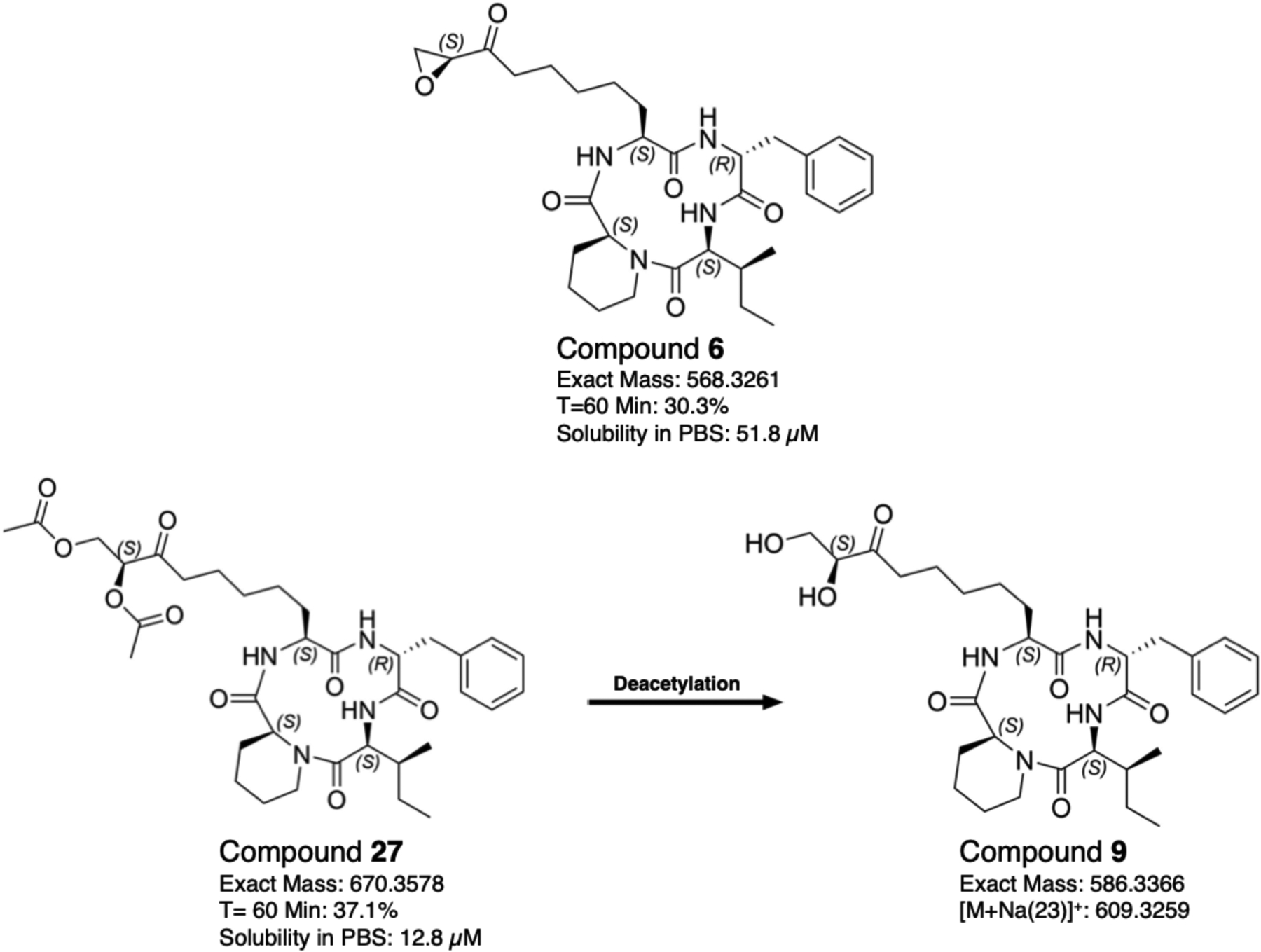
Structure and properties of compounds 6, 27, and potential active metabolite 9. The microsomal stability of compounds 6 and 27 is reported as the percent of compound remaining after a 1 h incubation with the corresponding murine liver enzymes. Solubility studies were performed at room temperature in PBS, pH 7.4.
The resulting data revealed that levels of compound 27 diminished below detection limits after just 2 h, with a majority of 27 converted to 9 (data not shown). This loss of compound 27 was observed in all test conditions regardless of whether cells were present in the culture medium. Since compound 27 was being depleted in the cultures, we shifted to focusing our analysis to focus on the relative levels of compounds 9 (deemed the ‘active’ form of compound 27) in the medium and cells. Interestingly, the relative levels of compound 9 detected in uninfected and infected red blood cells (RBCs) was higher in several scenarios when compound 27 was used to treat cells, compared to when compound 9 was directly added (Supporting Figure 11A–B). This pattern was most pronounced in the supernatant and cells of infected RBCs after 6 hours of incubation (Supporting Figure 11C–D). These results mimicked what would be expected if compound 27 served as a ‘delayed-release’ prodrug-like capacity in which the diacetyloxy-group-containing compound served as a reservoir for producing active compound 9. While these results hint at an intriguing path to the design of a selective plasmodicidal HDAC inhibitor, we cannot rule out a number of alternative possibilities such as compound 27 having better membrane penetration or cellular partitioning, or engagement with a different cellular target.
Natural and semi-synthetic Wardomyces cyclic tetrapeptides cause hyperacetylation of histone H4 in Plasmodium culture:
To further examine the effects of cyclic tetrapeptides on blocking histone deacetylation in Plasmodium parasites, we selected a polyclonal anti-acetyl-histone H4 antibody designed to recognize acetylated lysines 5, 8, 12, and 16 on histone H4. This H4 antibody was chosen since the hyperacetylation of histone H4 is generally recognized as a canonical signature of HDAC inhibition in Plasmodium8. In addition, an increase in histone H4 acetylation levels for all life cycle stages was observed in P. falciparum 3D7 culture following treatment with the similar cyclic tetrapeptide apicidin35. Synchronous 3D7 trophozoite stage cultures were exposed to 5× the EC50 concentration of compounds 6 and 27, along with the less selective inhibitor, compound 2. For comparative purposes, the compounds SAHA and chloroquine (CQ) were also tested. A Western blot of the collected lysate shows similar levels of histone H4 hyperacetylation following treatment with the HDAC inhibitors (p < 0.01 for compound 6, and p < 0.05 for SAHA, compound 27, and compound 2) (Figure 5A–B).
Figure 5.
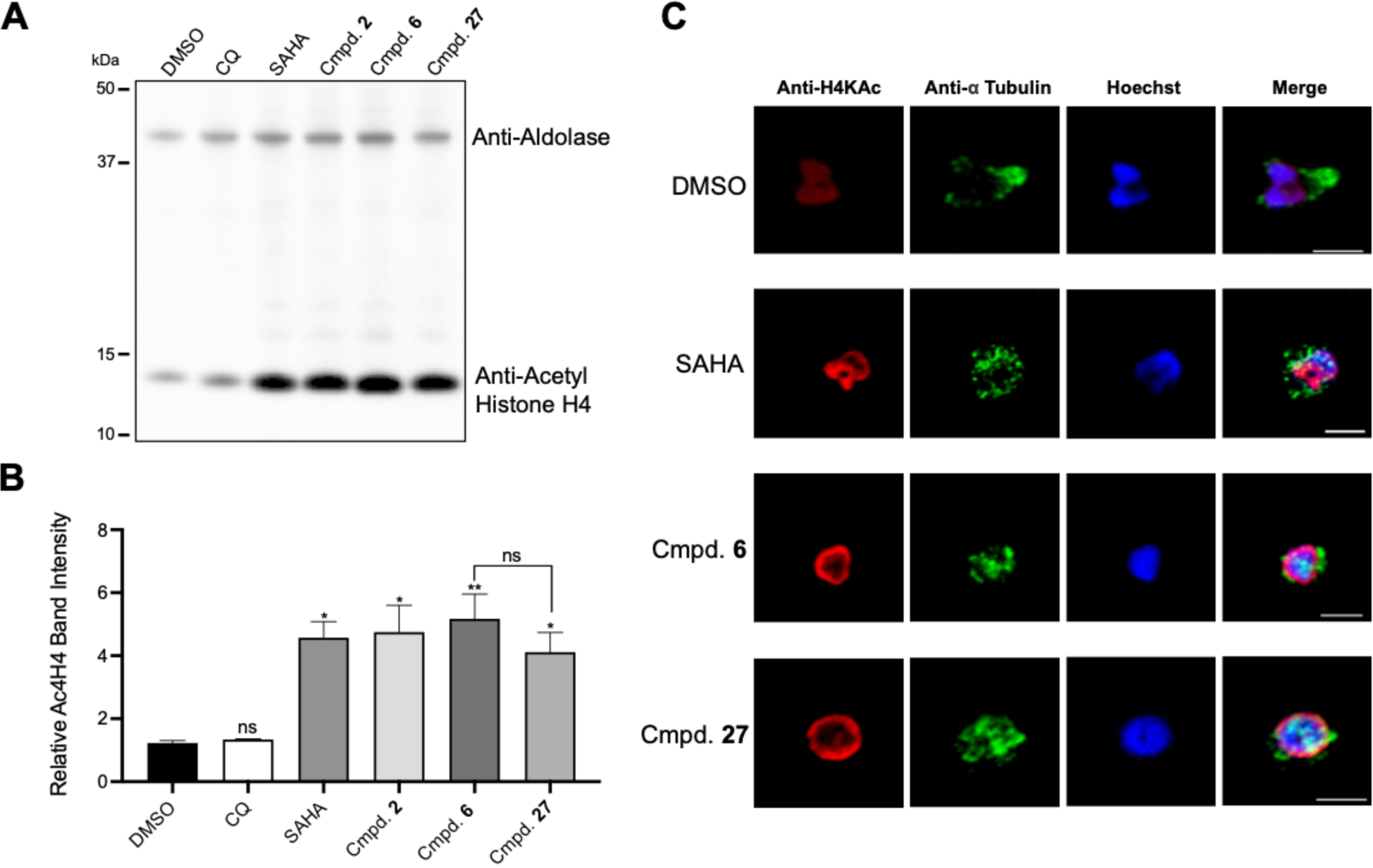
(A, B) Hyperactylation of HDAC inhibitors after 4 h treatment with 5x EC50. (B) Average band intensity of 3 western blots normalized to aldolase. Significance seen between DMSO and SAHA (p < 0.05), DMSO and cmpd. 2 (p < 0.05), DMSO and cmpd. 27 (p < 0.05), and DMSO and cmpd. 6 (p < 0.01). (C) Parasites treated in ring and collected in troph prior to incubation in chambered slides. Probing preformed with anti-acetyl histone H4 antibody in addition to anti-alpha tubulin and Hoechst.
We next performed an immunofluorescence assay (IFA) with nucleic marker Hoechst and anti-acetyl histone H4, to better visualize the state of histone 4 acetylation in cells. Again, synchronous cultures were treated with 5x EC50 of SAHA, or compounds 6 or 27 (Figure 5C). Anti-alpha tubulin was used to evaluate any change in the microtubular structure following the compound exposure. Treatment with HDAC inhibitors resulted in a noticeable increase in histone H4 acetylation localized to the nuclear periphery. Previous research indicated these regions of hyperacetylation likely correspond to the euchromatin being activitely transcribed and regulated8, 36–37. Compared to the diffuse acetylation signal in the vehicle control, these results suggest potential chromatin reorganization in response to HDAC inhibition. This is consistent with the developing model of epigenetic regulation in eukaryotes that indicates genes under strong activation/suppression localize to the perimeter of the nucleus38; and chromatin reorganization in eukaryotes has been well documented in response to HDAC inhibition with the compound TSA37. We also noticed reorganization of microtubule cytoskeleton following exposure to HDAC inhibitors.
IFA studies comparing the individual H4 histone acetylations in Plasmodium found that H3K9Ac, H4K8Ac, H4K5Ac, and H4Ac4 showed varying degrees of polarization to the nuclear periphery at distinct points in the intraerythrocytic lifecycle. While H4Ac4 and H3K9Ac signal maintained a horseshoe-like distribution to the nuclear perimeter from ring stage onward, H4K5Ac became increasingly polarized as the cell cycle progressed36. In contrast, signal specific to histone H4K8Ac which is known to be one of the most dynamic H4 makers, became more irregular as the life cycle progressed into trophozoite, reaching a diffuse nuclear pattern36. Of all four H4 acetylations, H4K8ac is suspected to be the rate limited step, and is most sensitive to HDAC inhibitors8.
Compound 27 blocks cell cycle progression more rapidly:
To further examine the effects of cyclic tetrapeptides 6 and 27, we performed a stage specific inhibition assay (SSA) with synchronous culture, using SAHA as a non-cyclic tetrapeptide HDAC inhibitor control. Cultures were incubated with a 5 × EC50 concentration of inhibitors added every 12 hours post invasion (HPI), followed by analysis of samples every 12 hours, until the start of a second life cycle (54 HPI). The results indicate that while the parasites were affected by HDAC inhibitor treatment at all stages prior to late schizogony, compound addition preceding the trophozoite stage (18–30 HPI) exhibited the greatest degree of inhibition (Figure 6). This supports the view of HDAC inhibition as the mechanism of action, since HDAC activity peaks in trophozoite but has been shown to be impacted by inhibitor addition at all stages8, 35. When compared to compound 6 or SAHA, compound 27 appears to arrest growth at an earlier stage of the intraerythrocytic developmental cycle after 6 and 18 HPI treatments.
Figure 6.
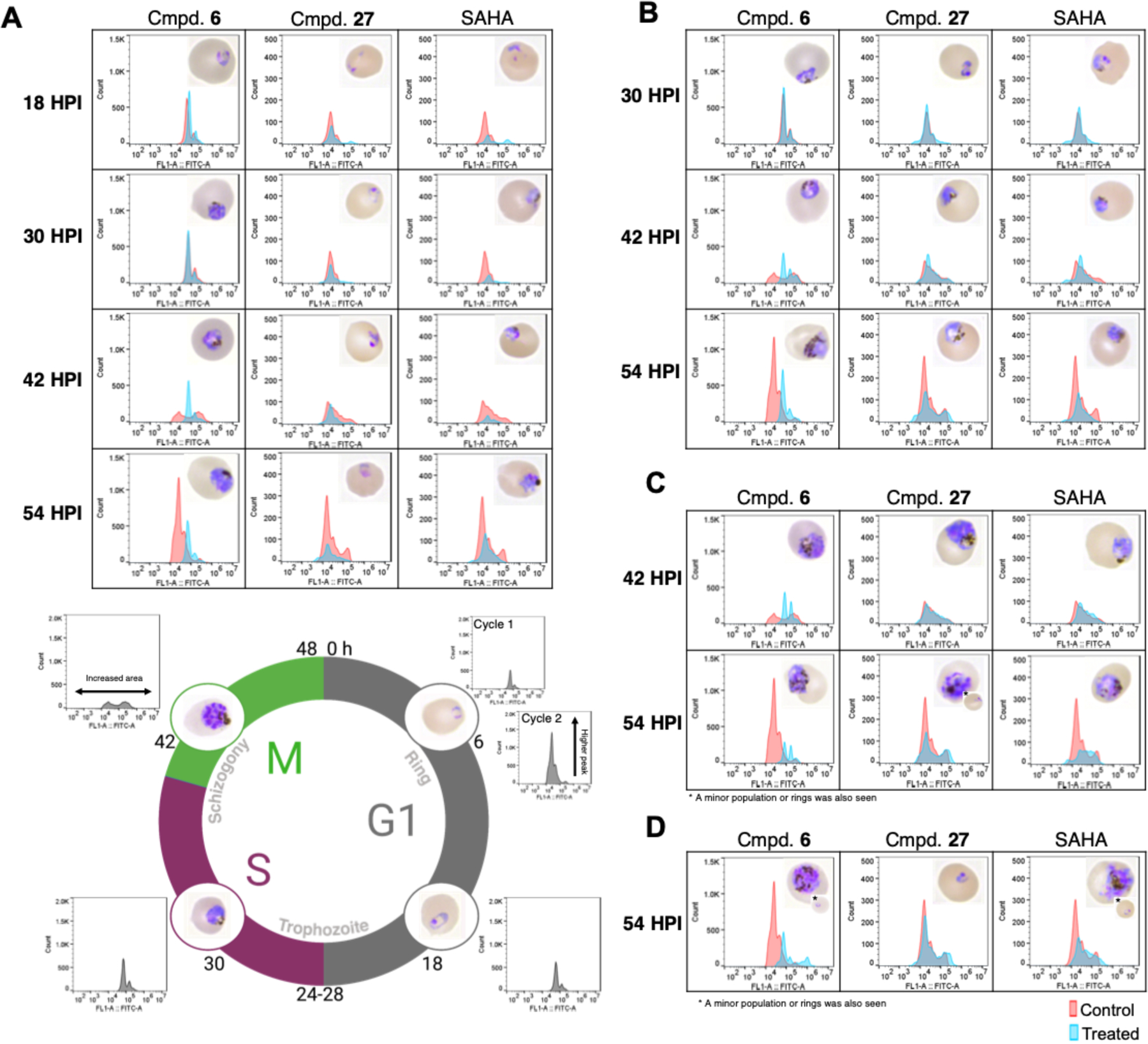
Stage specific inhibition following inhibitor treatment at 5 × EC50 in synchronous Dd2 parasites at select timepoints. Parasites monitored every 12 h until second cycle (54 HPI) using YOYO-1 and Geimsa thin smears. (A) Treatment at 6 HPI with compounds 6, 27, or SAHA. Life cycle graph representative of intraerythrocytic stage in the DMSO vehicle control at selected times of 6, 18, 30, 42, and 54 HPI. (B) Treatment at 18 HPI. (C) Treatment at 30 HPI. (D) Treatment at 42 HPI. Images and flow for each timepoint shown taken simultaneous from the same replicate. Total results representative of 3 independent replicates, with the displayed results chosen from replicate 1 or 2.
Exposure to any of the HDAC inhibitors at 6 HPI caused cell cycle arrest prior to nuclear division, with no shift in fluorescence in the FITC channel associated with DNA replication at 42 HPI (Figure 6A). Morphologically, parasites treated at 6 HPI in the SAHA and compound 6 conditions resemble late trophozoite, while the compound 27 treated parasites resemble rings or early trophozoites. There is a distinct increase in size that occurs after compound 6 addition, and to a lesser degree following exposure to SAHA. These size increases were not apparent when parasites were exposed to compound 27. Parasites treated at 18 HPI showed inhibition in all treatment conditions. However, compound 27 treated parasites again appeared smaller; in this case, they were morphologically more consistent with SAHA treated parasites (Figure 6B). With 30 HPI treatment, all HDAC inhibitors appeared to arrest growth in schizogony; although with compound 27, a minor percentage of parasites were able to undergo reinvasion, resulting in rings in the next developmental cycle (Figure 6C). Treatment at 42 HPI either had no apparent effect (compound 27), or parasites were partially inhibited at schizogony with some minor reinvasion of rings (SAHA and compound 6) (Figure 6D). Taken together, these results are consistent with a faster rate of killing for compound 27 compared to compound 6; and a potential shift towards early life cycle stage inhibition.
Conclusion:
This study led to the identification of 21 new cyclic tetrapeptides that inhibit plasmodial HDAC activity, resulting in pronounced antiplasmodial effects. More importantly, several of the compounds exhibit improved selectivity over the previously reported cyclic tetrapeptides. Based on enzymatic studies, antiplasmodial activity was strongly correlated with inhibition of class I HDAC, PfHDAC1. While the epoxide analog compound 6 showed a plasmodistatic killing profile, diacetyl compound 27 had a plasmodicidal effect on the parasite that may in fact be due to a bioactive metabolite. Both compounds cause the in vivo hyperacetylation of histone H4, and chromatin remodeling compared to the control, consistent with HDAC inhibition as the mechanism of action. Despite these similarities, compound 27 appears to show a stronger preference for early life stage activity, which could contribute to its superior killing profile. While the possibility that compound 27 has additional off target effects cannot be ignored, it is clear strong HDAC inhibition is occurring, based on the extremely potent in situ enzyme activity and matching acetylation profile to compound 6. Together, these results may potentially shed light on the complex specificity of cyclic tetrapeptide inhibitors in P. falciparum, and provide a roadmap to future selective HDAC inhibitor development.
METHODS
Culture Conditions:
Parasite culture maintenance followed a protocol by Trager and Jensen with slight modifications39. Parasite lines were grown in RPMI 1640 supplemented with 25 mM HEPES pH 7.4, 26 mM NaHCO3, 2% dextrose, 15 mg/L hypoxanthine, 25 mg/L gentamycin, and 0.5% Albumax II in human A+ erythrocytes, hematocrit 4%. Incubator conditions were kept constant at 37°C and 95% air, 5% CO2. Human HepG2 culture maintenance followed ATCC guidelines at the time of the experiment40. Media was DMEM, supplemented with 10% FBS, 1% Antibiotic/Antimycotic, 0.3% NaHCO3, 5 mM HEPEs pH 7.4, and 2 mM L-Glutamine as needed.
Phenotypic Antiplasmodial Screening:
To determine the antiplasmodial EC50 in 3D7 and Dd2 lines, a SYBR Green I fluorescence-based assay was employed. Serial dilutions of fungal compounds in diluted DMSO were added to microtiter plates with asynchronous parasites, diluted to 1% parasitemia and 1% hematocrit for a final DMSO percentage under 0.25%. Parasites were incubated for 72 h at standard growth conditions prior to freezing at −80°C. Plates were subsequently thawed and incubated with 1X SYBR Green in a lysis buffer composed of 20 mM Tris-HCL, 0.08% saponin, 5 mM EDTA, and 0.8% Triton X-100 for 45 min at room temperature. The resulting fluorescent signal was collected at excitation 485 nm, emission 530 nm, on a Synergy Neo2 multi-mode reader (BioTek, Winsooki, VT). Values were normalized to chloroquine and vehicle controls, and dose response curves were generated in CDD Vault.
Stage Specificity Assay:
SSA was performed as previously described with some modifications41. In brief, Dd2 culture was tightly synchronized using a combination of MACS column (Miltenyi Biotec, Auburn, CA)42 and treatment with 5% sorbitol43. Culture was then diluted to 1% parasitemia, 2% hematocrit, and compound was added at 6, 18, 30, or 42 HPI, with samples collected every 12 h for each condition until the second life cycle (54 HPI). Stage development was compared with Giemsa smearing for all conditions and time points tested. For flow cytometry, samples were fixed in 4% paraformaldehyde and incubated for 30 min at 37°C. Fixed samples were then washed in PBS 3x, and stored at 4°C prior to reading. Prepared samples were resuspended in permeabilization solution (0.25% Triton X-100) and mixed at 500 RPM for 10 min on horizontal plate shaker. Samples were then centrifuged at 900 × g for 2 min and resuspended in RNAse solution (50 μg/mL RNAse). RNAse treatment lasted for 3 h at 37°C before staining with YOYO-1 (500 nM final concentration). Signal acquisition was performed with a CytoFLEX S (Beckman Coulter, Brea, CA) at excitation wavelength 488 nm, with 500,000 events collected per sample. Parasite population was gated using uninfected RBC control with FlowJo version 10. Gated histograms from treated culture overlaid on top of vehicle treated culture for graphical comparison.
Human Cell Cytotoxicity:
HepG2 hepatocarcinoma cells were used in an MTS (3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) based assay to determine compound cytotoxicity. 2,250 cells per well were seeded onto a 384-micotiter plate and incubated for 24 h at 37°C, 5% CO2, prior to compound addition. Treated cells were then incubated for an additional 48 h. Control wells were incubated with 5% Triton X-100 for 5 min prior to MTS addition; after which, plates were incubated for 4 h at 37°C before reading absorbance at 490 nm with a Synergy Neo2 multi-mode reader (BioTek, Winsooki, VT). Dose response curves generated in CDD Vault (Burlingame, CA).
Enzymatic assays:
PfHDAC1 enzymatic assays were performed following a modified version of the manufacturer’s (BPS Bioscience, San Diego, CA) protocol. The assay reaction included 0.018 – 0.048 μg of recombinant PfHDAC1 per reaction based on enzyme activity of the given batch (Cat # 50063), 25 μM of fluorogenic HDAC substrate 3 (Cat # 50037), assay buffer (Cat # 50031), and assay developer (BPS # 50030). Reactions were incubated in 384-well plates at 37°C for 2 h with inhibitor dilutions using no-enzyme and no-inhibitor controls. Assay developer was then added to stop the reaction, and plates were incubated for 30 min at room temperature protected from light. Plates were then read excitation 360 nm, emission 450 nm on a BioTek Synergy neo2 (Winooski, VT). IC50 curves and linear regression analysis generated using GraphPad Prism, version 8.
Microsomal Stability Assay:
Microsomal stability was determined by Analiza (Cleveland, OH) using microsomes collected from Menoxtech, Male (CD-1), LN:1710069. Prior to the assay, samples were diluted to 20% DMSO/80% acetonitrile. The microsomal solution (0.63 mg/mL protein in 100 mM KPO4 with 1.3 mM EDTA) was prewarmed to 37°C prior to compound addition at 0.1 mM. The reaction solution was dispensed into assay plates for T = 0, T = 60 min, or T = 60 min - NADPH conditions. For T = 0, the reaction was quenched using cold (4°C) MeOH and a NADPH regeneration solution was added. For T = 60, NADPH was added to the reaction solution and the plate was sealed and incubated for 1 h at 37°C, prior to quenching with cold MeOH. This process was repeated for the T = 60, -NADPH condition, with LC-MS grade H2O added in place of NADPH. Plates were then sealed, vortexed, and centrifuged at 3000 × RPM for 15 min at 4°C. Supernatants were then analyzed using LC-TOFMS and the % compound remaining was calculated as T = 60 min / T = 0 min.
Kinetic Solubility Assay:
Solubility studies were performed by Analiza using standard HPLC-UV analysis. In brief, compound was diluted to a final concentration of 200 μM, in a final DMSO concentration of 2%, into 1X PBS, 7.4. Compound was subsequently added to Millipore solubility plates containing a 0.45 μM polycarbonate filter membrane and plates were sealed and incubated for 24 h at room temperature. Samples were then vacuum filtered and filtrate was collected for analysis by HPLC using an Agilent 1290 (Santa Clara, CA). Peaks were plotted against a calibration curve, and quantified using linear regression with Agilent OpenLab Intelligent Reporting Software.
LC-MS Incubation Study:
Starting compounds 9, or 27, were added in triplicate to the following culture conditions at 2.5 μM; culture media alone (RPMI 1640 supplemented with 25 mM HEPES pH 7.4, 26 mM NaHCO3, 2% dextrose, 15 mg/L hypoxanthine, 25 mg/L gentamycin, and 0.5% Albumax II), culture media with human A+ erythrocytes (4% hematocrit), and culture media with erythrocytes inoculated with 5% Dd2, asynchronous parasites. After compound addition, culture was kept at standard in vitro growth conditions of 37°C and 5% CO2. After 2 or 6 h incubations, the supernatant portion of each sample was collected and plated for later analysis by LCMS (Shimadzu, Kyoto, Japan); the corresponding cell portion was then collected in PBS. In infected conditions, an additional separation of the parasite population was performed by lysis with 0.02% saponin as described elsewhere44. The lysed cell supernatant was collected for analysis and the corresponding parasite pellet was collected in PBS. All samples were then centrifuged and frozen at −80°C to facilitate additional cell lysis prior to LCMS preparation.
For LCMS, samples were thawed and extracted with a 9:1 MeOH: Water solution prior to sonication for 1 h. The evaporation solvents were then resuspended in MeOH at equal concentrations based on culture proportion. The samples from the media supernatant were resuspended at 100 mg/mL, while the smaller cell and parasite portions were all resuspended at 25 mg/mL. An equal volume of each sample was then injected for LCMS analysis using a C18 column and 10–100% acetonitrile in water with 0.1% formic acid for 13 min. The relative intensity of the peak associated with compound 6 was then tracked in all conditions. Given the mathematical relationship between concentration (area, A) and intensity (height, H) where A = H2, the peak intensity for media supernatant samples was divided by 2 to account for the 4 × higher concentration. Data was then analyzed in GraphPad Prism version 8.
Molecular Docking:
The computational molecular docking experiments were performed using the Schrödinger Small-Molecule Drug Discovery Suite (Maestro®, 2018–4)45. The crystal structure (5VI6) was downloaded from PDB and optimized using the Protein Preparation Wizard following the standard protocol. Water molecules were removed. The ligand structures were optimized using the Ligand Preparation Wizard (LigPrep). Energy minimization of the ligands was conducted using the OPLS3 force field, and the ionization states were generated at pH 7.0 using Epik. The structure of compounds 6 and 29 were drawn using Maestro based on the X-ray structure of compound 6 we had obtained. Docking experiments were carried out using the Glide SP mode. The metal constraint was kept on during docking and any ligand atom was allowed to perform the metal coordination.
Immunoblot analysis:
3D7 parasite culture was incubated with 5 × EC50 of compound for 4 h at 37°C. Parasites were collected from infected RBC for the hyperacetylation assay using a 0.1% saponin lysis as described46. Cell-free parasite lysate was then prepared by treating the collected parasite pellet with 100 μL of M-PER (Thermo Scientific, Waltham, MA) containing 1 μL HALT protease inhibitor cocktail (Thermo Scientific, Waltham, MA) per 1 mg of pellet. Fifteen μg of lysate was resolved on a 10% Bis-Tris SDS_PAGE for immunoblot blot analysis using primary antibodies with ratios 1:2,500 anti-acetyl-histone H4 (MilliporeSigma, Burlington, MA, Cat # 06–598) and 1:40,000 anti-Plasmodium aldolase (Abcam, Cambridge, U.K., Cat # ab207494), with anti-rabbit secondary antibodies at 1:20,000 (Promega, Madison, WI, Cat # W401B). Immunoblot signals were developed by addition of SuperSignal west pico chemiluminescent substrate (Thermo Scientific, Waltham, MA, Cat # 34580). Relative density was determined using Bio-Rad Image Lab software normalized to aldolase loading control. Statistical analysis with one-way ANOVA performed in GraphPad Prism version 8.
Immunofluorescence assay:
Immunofluorescence assay performed using a modified protocol by Mehnert et al47. Lak-Tek II (Millipore Sigma, St. Louis, MO) 4 well chambered slides were coated with 5 mg/mL Concanavalin A and incubated for 20 min at 37°C. Wells were washed twice with pre-warmed PBS before the addition of 150 μL treated parasite culture, pre-washed twice with PBS. Chamber slides were then incubated for 10 min at 37°C. Wells were washed twice with PBS, then incubated overnight in 400 μL treated culture media to improve resolution. Wells were then rinsed in PBS and fixed with 4% paraformaldehyde. Wells were washed with PBS and then permeabilized with 0.1% Triton® X-100 for 15 min on horizontal shaker followed by three more washes in PBS prior to incubation with 0.1 mg/mL NaBH4 to quench free aldehydes. After two subsequent PBS washes, wells were blocked in 3% BSA overnight. Blocking with primary antibodies 1:1,000 anti-acetyl-histone H4 (MilliporeSigma, Burlington, MA, Cat # 06–598), 1:500 anti-alpha tubulin (Sigma-Aldrich, St. Louis, MO., Cat # T5168) was done for 2.5 h followed by three 5 min washes in 0.5% Tween® 20. Blocking with secondary fluorophores 1:1,000 Alex 555, and 1:1,000 Alexa 488, and Hoechst was performed for 1 h protected from light. Wells were washed three times with 0.5% Tween® 20 then once with PBS before being resuspended in 300 μL PBS.
Killing rate:
Rate of killing assays were performed on asynchronous Dd2 culture starting at ~1% parasitemia, 4% hematocrit. 10 × EC50 of inhibitor or a DMSO vehicle control was added for either 24, or 48 h prior to removal. Culture aliquots were collected at the time of wash off, and once every 24 h following for Geimsa thin smears and flow cytometry. For flow cytometric analysis, culture samples were incubated for 30 min at room temp with 1:1,000 SYBR Green I and 0.6 μM Mitotracker Deep red FM, and 100,000 events were collected using a CytoFLEX S (Beckman Coulter, Brea, CA). DHA, no treatment, no Mitotracker, and uninfected RBC controls were used for gating. The fraction of parasites dead was determined by taking Mitotracker positive, SYBR positive / total SYBR positive events. Data was analyzed using FlowJo version 10.
Supplementary Material
ACKNOWLEDGEMENTS
Support for this research was provided by NIH grants R21AI143052 and R01AI154777 to D. C. and R. H. C. We thank Natalia Mojica Santos and Raphaella Paes for their assistance screening the Lasionectria hilhorsti analogs.
ABBREVIATIONS.
- HDAC
histone deacetylase
- P. falciparum
Plasmodium falciparum
- SI
selectivity index
- SAR
structure activity relationship
- PfHDAC1
Plasmodial HDAC 1
- ACT
artemisinin combination therapy
- FDA
Food and Drug Administration
- HAT
histone acetyl transferase
- TSA
Trichostatin A
- SAHA
suberoylanilide hydroxamic acid
- HepG2
human hepatocellular carcinoma cell line
- Dd2
multidrug resistant P. falciparum line
- 3D7
Chloroquine sensitive P. falciparum line
- MIA PacA-2
human pancreatic cancer cell line
- EC50
50 percent effective concentration
- EC90
90 percent effective concentration
- IC50
50 percent inhibitory concentration
- MTS
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt
- μM
micromolar
- nM
nanomolar
- SSA
stage specific assay
- HPI
hours post invasion
- DMSO
dimethylsulfoxide
- HeLa
human cervical cancer cell line
- DHA
dihydroartemisinin
- T. gondii
Toxoplasma gondii
- ZBG
Zinc binding group
- CQ
Chloroquine
Footnotes
Supporting Information.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsinfecdis.1c00341
General information: Extraction and purification of source fungus (W. dimerus); Structure elucidation of natural compounds 6-18 with 1D (1H and 13C) and 2D NMR (HSQC, HMBC, COSY and ROESY), ECD and HRESIMS spectrum, chemical reaction analysis (Marfey’s, GITC and Mosher’s methods), and single crystal X-ray diffraction analysis of compound 6; Chemical synthesis methods and 1D (1H and 13C) and 2D NMR (HSQC, HMBC and COSY) and HRESIMS spectrum of synthetic compounds 19-29; Tables for antimalarial effects of additional HDAC inhibitors
The authors declare no competing financial interests.
REFERENCES.
- 1.World Health Organization, G. M. P. World malaria report 2020; 2020.
- 2.WHO Artemisinin resistance and artemisinin-based combination therapy efficacy World Health Organization: 2019. [Google Scholar]
- 3.Straimer J; Gnadig NF; Witkowski B; Amaratunga C; Duru V; Ramadani AP; Dacheux M; Khim N; Zhang L; Lam S; Gregory PD; Urnov FD; Mercereau-Puijalon O; Benoit-Vical F; Fairhurst RM; Menard D; Fidock DA, Drug resistance. K13-propeller mutations confer artemisinin resistance in Plasmodium falciparum clinical isolates. Science 2015, 347 (6220), 428–431. DOI: 10.1126/science.1260867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ouji M; Augereau JM; Paloque L; Benoit-Vical F, Plasmodium falciparum resistance to artemisinin-based combination therapies: A sword of Damocles in the path toward malaria elimination. Parasite 2018, 25, 24–36. DOI: 10.1051/parasite/2018021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mathieu LC; Cox H; Early AM; Mok S; Lazrek Y; Paquet JC; Ade MP; Lucchi NW; Grant Q; Udhayakumar V; Alexandre JS; Demar M; Ringwald P; Neafsey DE; Fidock DA; Musset L, Local emergence in Amazonia of Plasmodium falciparum k13 C580Y mutants associated with in vitro artemisinin resistance. Elife 2020, 9, 1–21. DOI: 10.7554/eLife.51015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Uwimana A; Legrand E; Stokes BH; Ndikumana JM; Warsame M; Umulisa N; Ngamije D; Munyaneza T; Mazarati JB; Munguti K; Campagne P; Criscuolo A; Ariey F; Murindahabi M; Ringwald P; Fidock DA; Mbituyumuremyi A; Menard D, Emergence and clonal expansion of in vitro artemisinin-resistant Plasmodium falciparum kelch13 R561H mutant parasites in Rwanda. Nat Med 2020, 26 (10), 1602–1608. DOI: 10.1038/s41591-020-1005-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coetzee N; von Gruning H; Opperman D; van der Watt M; Reader J; Birkholtz LM, Epigenetic inhibitors target multiple stages of Plasmodium falciparum parasites. Sci Rep 2020, 10 (1), 2355–2366. DOI: 10.1038/s41598-020-59298-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gupta AP; Zhu L; Tripathi J; Kucharski M; Patra A; Bozdech Z, Histone 4 lysine 8 acetylation regulates proliferation and host-pathogen interaction in Plasmodium falciparum. Epigenetics Chromatin 2017, 10 (1), 40–57. DOI: 10.1186/s13072-017-0147-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deshmukh AS; Srivastava S; Dhar SK, Plasmodium falciparum: epigenetic control of var gene regulation and disease. Subcell Biochem 2013, 61, 659–682. DOI: 10.1007/978-94-007-4525-4_28. [DOI] [PubMed] [Google Scholar]
- 10.Kanyal A; Rawat M; Gurung P; Choubey D; Anamika K; Karmodiya K, Genome-wide survey and phylogenetic analysis of histone acetyltransferases and histone deacetylases of Plasmodium falciparum. FEBS J 2018, 285 (10), 1767–1782. DOI: 10.1111/febs.14376. [DOI] [PubMed] [Google Scholar]
- 11.Diedrich D; Stenzel K; Hesping E; Antonova-Koch Y; Gebru T; Duffy S; Fisher G; Scholer A; Meister S; Kurz T; Avery VM; Winzeler EA; Held J; Andrews KT; Hansen FK, One-pot, multi-component synthesis and structure-activity relationships of peptoid-based histone deacetylase (HDAC) inhibitors targeting malaria parasites. Eur J Med Chem 2018, 158, 801–813. DOI: 10.1016/j.ejmech.2018.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Traore MDM; Zwick V; Simoes-Pires CA; Nurisso A; Issa M; Cuendet M; Maynadier M; Wein S; Vial H; Jamet H; Wong YS, Hydroxyl Ketone-Based Histone Deacetylase Inhibitors To Gain Insight into Class I HDAC Selectivity versus That of HDAC6. ACS Omega 2017, 2 (4), 1550–1562. DOI: 10.1021/acsomega.6b00481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Darkin-Rattray SJ; Gurnett AM; Myers RW; Dulski PM; Crumley TM; Allocco JJ; Cannova C; Meinke PT; Colletti SL; Bednarek MA; Singh SB; Goetz MA; Dombrowski AW; Polishook JD; Schmatz DM, Apicidin: a novel antiprotozoal agent that inhibits parasite histone deacetylase. Proceedings of the National Academy of Sciences of the United States of America 1996, 93 (23), 13143–7. DOI: 10.1073/pnas.93.23.13143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giannini G; Battistuzzi G; Vignola D, Hydroxamic acid based histone deacetylase inhibitors with confirmed activity against the malaria parasite. Bioorg Med Chem Lett 2015, 25 (3), 459–61. DOI: 10.1016/j.bmcl.2014.12.051. [DOI] [PubMed] [Google Scholar]
- 15.Marfurt J; Chalfein F; Prayoga P; Wabiser F; Kenangalem E; Piera KA; Fairlie DP; Tjitra E; Anstey NM; Andrews KT; Price RN, Ex vivo activity of histone deacetylase inhibitors against multidrug-resistant clinical isolates of Plasmodium falciparum and P. vivax. Antimicrob Agents Chemother 2011, 55 (3), 961–6. DOI: 10.1128/AAC.01220-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andrews KT; Walduck A; Kelso MJ; Fairlie DP; Saul A; Parsons PG, Anti-malarial effect of histone deacetylation inhibitors and mammalian tumour cytodifferentiating agents. Int J Parasitol 2000, 30 (6), 761–8. DOI: 10.1016/s0020-7519(00)00043-6. [DOI] [PubMed] [Google Scholar]
- 17.Kahnberg P; Lucke AJ; Glenn MP; Boyle GM; Tyndall JD; Parsons PG; Fairlie DP, Design, synthesis, potency, and cytoselectivity of anticancer agents derived by parallel synthesis from alpha-aminosuberic acid. J Med Chem 2006, 49 (26), 7611–22. DOI: 10.1021/jm050214x. [DOI] [PubMed] [Google Scholar]
- 18.Bantscheff M; Hopf C; Savitski MM; Dittmann A; Grandi P; Michon AM; Schlegl J; Abraham Y; Becher I; Bergamini G; Boesche M; Delling M; Dumpelfeld B; Eberhard D; Huthmacher C; Mathieson T; Poeckel D; Reader V; Strunk K; Sweetman G; Kruse U; Neubauer G; Ramsden NG; Drewes G, Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat Biotechnol 2011, 29 (3), 255–65. DOI: 10.1038/nbt.1759. [DOI] [PubMed] [Google Scholar]
- 19.Duffy MF; Selvarajah SA; Josling GA; Petter M, Epigenetic regulation of the Plasmodium falciparum genome. Brief Funct Genomics 2014, 13 (3), 203–16. DOI: 10.1093/bfgp/elt047. [DOI] [PubMed] [Google Scholar]
- 20.Melesina J; Robaa D; Pierce RJ; Romier C; Sippl W, Homology modeling of parasite histone deacetylases to guide the structure-based design of selective inhibitors. J Mol Graph Model 2015, 62, 342–361. DOI: 10.1016/j.jmgm.2015.10.006. [DOI] [PubMed] [Google Scholar]
- 21.Chua MJ; Arnold MS; Xu W; Lancelot J; Lamotte S; Spath GF; Prina E; Pierce RJ; Fairlie DP; Skinner-Adams TS; Andrews KT, Effect of clinically approved HDAC inhibitors on Plasmodium, Leishmania and Schistosoma parasite growth. Int J Parasitol Drugs Drug Resist 2017, 7 (1), 42–50. DOI: 10.1016/j.ijpddr.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Du L; Risinger AL; King JB; Powell DR; Cichewicz RH, A potent HDAC inhibitor, 1-alaninechlamydocin, from a Tolypocladium sp. induces G2/M cell cycle arrest and apoptosis in MIA PaCa-2 cells. J Nat Prod 2014, 77 (7), 1753–7. DOI: 10.1021/np500387h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang S; Li X; Wei Y; Xiu Z; Nishino N, Discovery of potent HDAC inhibitors based on chlamydocin with inhibitory effects on cell migration. ChemMedChem 2014, 9 (3), 627–37. DOI: 10.1002/cmdc.201300372. [DOI] [PubMed] [Google Scholar]
- 24.Mori H; Abe F; Furukawa S; Furukawa S; Sakai F; Hino M; Fujii T, FR235222, a fungal metabolite, is a novel immunosuppressant that inhibits mammalian histone deacetylase (HDAC) II. Biological activities in animal models. J Antibiot (Tokyo) 2003, 56 (2), 80–6. [PubMed] [Google Scholar]
- 25.Bougdour A; Maubon D; Baldacci P; Ortet P; Bastien O; Bouillon A; Barale JC; Pelloux H; Menard R; Hakimi MA, Drug inhibition of HDAC3 and epigenetic control of differentiation in Apicomplexa parasites. J Exp Med 2009, 206 (4), 953–66. DOI: 10.1084/jem.20082826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sasamura S; Sakamoto K; Takagaki S; Yamada T; Takase S; Mori H; Fujii T; Hino M; Hashimoto M, AS1387392, a novel immunosuppressive cyclic tetrapeptide compound with inhibitory activity against mammalian histone deacetylase. J Antibiot (Tokyo) 2010, 63 (11), 633–6. DOI: 10.1038/ja.2010.51. [DOI] [PubMed] [Google Scholar]
- 27.Asahi I; Miura N; Yamabe Y; Toyoda H; Imura N; Koyama M; Naganuma A, PF1070A, a novel and potent inducer of the synthesis of metallothionein. Biochemistry 1999, 38 (32), 10415–10423. [DOI] [PubMed] [Google Scholar]
- 28.Kawai M; Pottorf RS; Rich DH, Structure and solution conformation of the cytostatic cyclic tetrapeptide WF-3161, cyclo[L-leucyl-L-pipecolyl-L-(2-amino-8-oxo-9, 10-epoxydecanoyl)-D-phenylalanyl]. J Med Chem 1986, 29 (11), 2409–11. DOI: 10.1021/jm00161a046. [DOI] [PubMed] [Google Scholar]
- 29.Hai Y; Christianson DW, Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat Chem Biol 2016, 12 (9), 741–7. DOI: 10.1038/nchembio.2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vickers CJ; Olsen CA; Leman LJ; Ghadiri MR, Discovery of HDAC Inhibitors That Lack an Active Site Zn(2+)-Binding Functional Group. ACS Med Chem Lett 2012, 3 (6), 505–8. DOI: 10.1021/ml300081u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Porter NJ; Christianson DW, Binding of the Microbial Cyclic Tetrapeptide Trapoxin A to the Class I Histone Deacetylase HDAC8. ACS Chem Biol 2017, 12 (9), 2281–2286. DOI: 10.1021/acschembio.7b00330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim B; Hong J, An overview of naturally occurring histone deacetylase inhibitors. Curr Top Med Chem 2015, 14 (24), 2759–82. DOI: 10.2174/1568026615666141208105614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hsu KC; Liu CY; Lin TE; Hsieh JH; Sung TY; Tseng HJ; Yang JM; Huang WJ, Novel Class IIa-Selective Histone Deacetylase Inhibitors Discovered Using an in Silico Virtual Screening Approach. Sci Rep 2017, 7 (1), 3228–3241. DOI: 10.1038/s41598-017-03417-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hansen FK; Sumanadasa SD; Stenzel K; Duffy S; Meister S; Marek L; Schmetter R; Kuna K; Hamacher A; Mordmuller B; Kassack MU; Winzeler EA; Avery VM; Andrews KT; Kurz T, Discovery of HDAC inhibitors with potent activity against multiple malaria parasite life cycle stages. Eur J Med Chem 2014, 82, 204–13. DOI: 10.1016/j.ejmech.2014.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chaal BK; Gupta AP; Wastuwidyaningtyas BD; Luah YH; Bozdech Z, Histone deacetylases play a major role in the transcriptional regulation of the Plasmodium falciparum life cycle. PLoS Pathog 2010, 6 (1), e1000737–50. DOI: 10.1371/journal.ppat.1000737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luah YH; Chaal BK; Ong EZ; Bozdech Z, A moonlighting function of Plasmodium falciparum histone 3, mono-methylated at lysine 9? PLoS One 2010, 5 (4), e10252–61. DOI: 10.1371/journal.pone.0010252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cowell IG; Papageorgiou N; Padget K; Watters GP; Austin CA, Histone deacetylase inhibition redistributes topoisomerase IIbeta from heterochromatin to euchromatin. Nucleus 2011, 2 (1), 61–71. DOI: 10.4161/nucl.2.1.14194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taddei A, Active genes at the nuclear pore complex. Curr Opin Cell Biol 2007, 19 (3), 305–10. DOI: 10.1016/j.ceb.2007.04.012. [DOI] [PubMed] [Google Scholar]
- 39.Trager W; Jensen JB, Human malaria parasites in continuous culture. Science 1976, 193 (4254), 673–5. [DOI] [PubMed] [Google Scholar]
- 40.ATCC American Type Culture Collection https://www.atcc.org/products/all/HB-8065.aspx#culturemethod (accessed 7 August 2018).
- 41.Wright AE; Collins JE; Roberts B; Roberts JC; Winder PL; Reed JK; Diaz MC; Pomponi SA; Chakrabarti D, Antiplasmodial Compounds from Deep-Water Marine Invertebrates. Mar Drugs 2021, 19 (4), 179–190. DOI: 10.3390/md19040179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mata-Cantero L; Lafuente MJ; Sanz L; Rodriguez MS, Magnetic isolation of Plasmodium falciparum schizonts iRBCs to generate a high parasitaemia and synchronized in vitro culture. Malar J 2014, 13, 112–121. DOI: 10.1186/1475-2875-13-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lambros C; Vanderberg JP, Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol 1979, 65 (3), 418–20. [PubMed] [Google Scholar]
- 44.Allman EL; Painter HJ; Samra J; Carrasquilla M; Llinas M, Metabolomic Profiling of the Malaria Box Reveals Antimalarial Target Pathways. Antimicrob Agents Chemother 2016, 60 (11), 6635–6649. DOI: 10.1128/AAC.01224-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sastry GM; Adzhigirey M; Day T; Annabhimoju R; Sherman W, Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. Journal of computer-aided molecular design 2013, 27 (3), 221–234. [DOI] [PubMed] [Google Scholar]
- 46.Hasenkamp S; Wong EH; Horrocks P, An improved single-step lysis protocol to measure luciferase bioluminescence in Plasmodium falciparum. Malar J 2012, 11, 42–49. DOI: 10.1186/1475-2875-11-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mehnert AK; Simon CS; Guizetti J, Immunofluorescence staining protocol for STED nanoscopy of Plasmodium-infected red blood cells. Mol Biochem Parasitol 2019, 229, 47–52. DOI: 10.1016/j.molbiopara.2019.02.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.