

Abstract
The hallmark of tumorigenesis is the successful circumvention of cell death regulation for achieving unlimited replication and immortality. Ferroptosis is a newly identified type of cell death dependent on lipid peroxidation which differs from classical programmed cell death in terms of morphology, physiology and biochemistry. The broad spectrum of injury and tumor tolerance are the main reasons for radiotherapy and chemotherapy failure. The effective rate of tumor immunotherapy as a new treatment method is less than 30%. Ferroptosis can be seen in radiotherapy, chemotherapy, and tumor immunotherapy; therefore, ferroptosis activation may be a potential strategy to overcome the drug resistance mechanism of traditional cancer treatments. In this review, the characteristics and causes of cell death by lipid peroxidation in ferroptosis are briefly described. In addition, the three metabolic regulations of ferroptosis and its crosstalk with classical signaling pathways are summarized. Collectively, these findings suggest the vital role of ferroptosis in immunotherapy based on the interaction of ferroptosis with tumor immunotherapy, chemotherapy and radiotherapy, thus, indicating the remarkable potential of ferroptosis in cancer treatment.
Keywords: cancer, ferroptosis, immunotherapy
List of Abbreviations
- 4‐HNE
4‐Hydroxy‐2‐nonanal
- AA
arachidonic acid
- ACC
acetyl‐coenzyme A carboxylase
- ACSL4
acyl‐CoA synthetase long‐chain family member 4
- AdA
epinephrine
- AIFM2
apoptosis‐inducing factor mitochondrial‐related 2
- ALOX15
arachidonic acid 15‐lipoxygenase
- ARE
antioxidant response element
- Atg5
autophagy‐related 5
- BH4
tetrahydrobiopterin
- CDDP
cisplatin
- CoQ10
ubiquinone
- CoQ10H2
panthenol
- CRC
colorectal cancer
- DAMP
damage‐related molecular pattern
- DFO
deferoxamine mesylate
- DHO
dihydroorotic acid
- DHODH
dihydroorotate dehydrogenase
- DMT1
divalent metal transporter
- DPP4
dipeptidyl peptidase‐4
- ECAD
E‐cadherin
- EGFR‐TKI
epidermal growth factor receptor‐tyrosine kinase inhibitor
- ETC
electron transfer chain
- FDA
U.S. Food & Drug Administration
- FIN56
ferroptosis inducer derived from CIL56
- FPN1
ferroportin
- FSP1
ferroptosis inhibitor protein 1
- GCH1
GTP cyclohydrolase 1
- GCLC
glutamate‐cysteine ligase catalytic subunit
- GPX
glutathione peroxidase
- GPX4
glutathione peroxidase 4
- GSH
glutathione
- H2O2
hydrogen peroxide
- HCAR1
lactate receptor
- HCC
hepatocellular carcinoma
- HGSOC
high‐grade serous ovarian cancer
- HMGB1
high‐mobility group protein B1
- HMGCR
3‐hydroxy‐3‐methyl‐glutaryl‐CoA reductase
- HO‐1
heme oxygenase‐1
- JAK
Janus kinase
- LIP
labile iron pool
- LOX
lipoxygenase
- LOX
lipoxygenase
- LPCAT3
lysophosphatidylcholine acyltransferase 3
- MCT1
lactate transporter
- MDA
malondialdehyde
- MtROS
mitochondrial reactive oxygen species
- MUFA
monounsaturated fatty acid
- NCOA4
nuclear receptor co‐activator 4
- NK
natural killer cell
- NOX
NADPH oxidase
- NRF2
NFE2‐related factor 2
- OA
orotic acid
- PARP‐1
poly polymerase‐1
- PC
phosphatidylcholine
- PCD
programmed cell death
- PD‐1
programmed cell death protein 1
- PE
phosphatidylethanolamine
- PEBP1
phosphatidylethanolamine binding protein 1
- PI3K
phosphatidylinositol 3‐kinase
- PL
piperine
- PRX
peroxidase
- PUFA
polyunsaturated fatty acid
- Rb
retinoblastoma
- RIP1
receptor‐interacting protein kinase 1
- RIP3
receptor‐interacting protein kinase 3
- ROS
reactive oxygen species
- RSL3
oncogenic‐RAS‐selective lethal compounds
- SCD1
stearoyl‐CoA desaturase 1
- SOD1
superoxide dismutase
- SREBP1
sterol regulatory element‐binding protein 1
- SREBP2
sterol regulatory element binding protein‐2
- SSZ
sulfadiazine
- STAT1
signal transducer activator of transcription 1
- STEAP3
Six‐transmembrane epithelial antigen of the prostate 3
- TAM
tumor‐associated macrophage
- TCA
tricarboxylic acid
- TEAD4
TEA domain transcription factor 4
- TF
transferrin
- TFR1
transferrin receptor
- TFRC
transferrin receptor
- TNFα
tumor necrosis factor α
- VDAC2/3
voltage‐dependent anion channel 2/3
- YAP
Yes‐Associated Protein
- α‐KG
α‐ketoglutarate
- β‐CD
β‐cyclodextrin
- γ‐Glu‐AAs
γ‐glutamyl peptides
1. BACKGROUND
Programmed cell death (PCD) is the basis of embryogenesis, tissue homeostasis, immune response, and other processes, and is essential for normal development, homeostasis, and prevention of hyperproliferative diseases such as cancer [1]. Various types of programmed cell death such as apoptosis, pyroptosis, parthanatos, necroptosis, and ferroptosis have been identified by researchers.
Apoptosis is a form of cell death during development, homeostasis, infection, and pathogenesis. It is conserved in the evolutionary process. Therefore, the regulation of death in mammalian cells is caused by the core steps of caspase activation [2]. In addition, new cell death mechanisms, such as pyroptosis, parthanatos and necroptosis, are gradually being discovered and improved. Pyroptosis is a type of PCD that critically depends on the formation of plasma membrane pores by members of the gasdermin protein family, often (but not always), as a result of inflammatory caspase activation [3]. In pyroptosis, the cells swell and form pores before rupture, causing the cell membrane to lose its integrity, releasing the cellular contents and leading to inflammation, accompanied by nuclear shrinkage and DNA breaks [4]. Parthanatos is based on poly polymerase‐1 (PARP‐1) which activates a type of PCD that is unrelated to caspase. It plays a vital role in a variety of neurological diseases. Parthanatos is characterized by chromatin condensation which produces large amounts of DNA fragments [5]. Necroptosis involves the activation of receptor‐interacting protein kinase 1 or 3 (RIP1 or RIP3) [6], with the accumulation of ATP and reactive oxygen species (ROS), calcium overload, mitochondrial permeability transition, opening of pores, increased cell volume, organelle swelling, plasma membrane rupture, and subsequent leakage of cell contents [7, 8, 9]. Cancer cells can be regulated by inducing necroptosis; however, the above assumption is extremely limited owing to the acquired or intrinsic resistance of cancer cells to necroptosis [10].
Erastin and RSL3(oncogenic‐RAS‐selective lethal compounds) are two different inducers of ferroptosis which are selectively cytotoxic to carcinogenic RAS mutant cell lines. RSL‐induced cell death is related to increased ROS production, which is inhibited by reduced cellular iron ingestion or chelation of iron. Accordingly, cell death caused by the accumulation of iron‐dependent cellular ROS leading to the breakdown of cell redox homeostasis is called ferroptosis [11, 12, 13]. Under transmission electron microscopy, erastin‐treated HRAS mutants do not undergo apoptosis (chromatin condensation and marginalization), necrosis (cytoplasm and organelle swelling and plasma membrane rupture) or autophagy (the formation of double‐membrane closed vesicles) related to the classic morphological characteristics; however, the mitochondria show an increase in membrane density [13, 14]. In short, ferroptosis and known cell death have different phenotypic characteristics, biochemical characteristics, and regulatory factors (Table 1) [15, 25].
TABLE 1.
The main feature of ferroptosis, apoptosis, pyroptosis, parthanatos and necroptosis
| Type of programmed cell death | Morphological feature | Regulator | Biochemical feature | ROS function | Reference |
|---|---|---|---|---|---|
| Ferroptosis | The cell membrane did not rupture and blisters. Mitochondrial crests are reduced or disappeared, and the outer mitochondrial membrane ruptures. Normal nuclear size and chromatin. | NOX, GPX4, DHODH, FSP1, BH4, GOT1, NRF2 |
Lipids, iron and ROS promote ferroptosis GPX4, FSP1 and DHODH synergistically inhibit ferroptosis |
Promote lipid peroxidation, which may further induce 4‐hydroxy‐2‐nonanal production, leading to cell membrane rupture and ferroptosis | [13, 15‐18] |
| Apoptosis | Plasma membrane blebbing; cellular and nuclear volume reduction. Nuclear fragmentation and chromatin condensation | p53, Bax, Bak, Bcl‐ 2, Bcl‐XL |
DNA breaks in the junction of nucleosomes Activation of caspases Phosphatidylserine shift |
Excessive ROS induces autophagy and maintains cell homeostasis by degrading damaged mitochondria | [2, 19‐22] |
| Pyroptosis | The cells swelled to form protrusions. The cell membrane forms pores, nucleus shrinks and DNA breaks. | CASP1, CASP4, CASP5, GSDMD | Activate caspase‐1 and promote the production of inflammatory cytokines IL‐1β and IL‐18. GSDMD is the key executor of pyrolysis | ROS activate the classical pyroptosis pathway dependent on NLRP 3 inflammasome to activate Caspase‐1 | [3, 4, 22] |
| Parthanatos | Chromatin condensation, which produces a large amount of DNA fragments | PARP1, AIFM1 | PARP1 activation produces a large amount of PAR,, independent of caspases | Excessive ROS triggers parthanatos | [5, 21, 23] |
| Necroptosis | Rupture of plasma membrane.organelle swelling. Moderate chromatin condensation | RIP1/3, MLKL |
Activation of RIP1, RIP3,and MLKL PARP1 hyperactivation |
RIPK1 autophosphorylation, RIPK3 recruitment, and necrosome formation | [6, 22, 24, 25] |
Abbreviations: NOX, NADPH oxidase; GPX4, glutathione peroxidase 4; DHODH, dihydroorotate dehydrogenase; FSP1, ferroptosis inhibitor protein 1; BH4, tetrahydrobiopterin; GOT1, aspartate aminotransaminase; NRF2, NFE2‐related factor 2; CASP1, Caspase‐1;CASP4, Caspase‐4; CASP5, Caspase‐5; GSDMD, Gasdermin‐D; PARP1, Poly(ADP‐ribose) polymerase‐1; AIFM1, apoptosis‐inducing factor mitochondria‐associated 1; RIP1/3, Receptor‐interacting protein 1/3; MLKL, mixed‐lineage kinase domain‐like protein; ROS, reactive oxygen species; GPX4, glutathione peroxidase 4; FSP1, ferroptosis inhibitor protein 1; DHODH, dihydroorotate dehydrogenase; IL‐1β, interleukin‐1beta; IL‐18, Interleukin 18; GSDMD, Gasdermin‐D; RIP1, Receptor‐interacting protein 1; RIP3, Receptor‐interacting protein 3; MLKL, mixed‐lineage kinase domain‐like protein; NLRP 3, NACHT, LRR, and PYD domain‐containing protein (NLRP) 3.
Many studies have confirmed that ferroptosis plays an important role in cancer biology. Dysregulated ferroptosis has been shown to be involved in cancer, neurodegeneration, tissue damage, inflammation, and infection [26]. The metabolic plasticity of cancer cells provides interesting insights into the mechanism by which metabolic reconnection plays a key role in cancer persistence. In some cases, this metabolic reprogramming is related to acquired sensitivity to ferroptosis; hence, targeting ferroptosis is expected to enhance the efficacy of immunotherapy, representing the potential physiological functions of ferroptosis in tumor suppression and immune surveillance [27]; however, the potential importance of ferroptosis in cancer therapy has not been fully emphasized.
Here, we review the discovery of ferroptosis and the molecular details of its underlying mechanism. The metabolic regulation of ferroptosis and its interaction with the classic signaling pathways involved in cell survival are also discussed. In addition, this review focuses on the availability of functional ferroptosis for tumor treatment and its response to regulate the survival and metastasis of tumor cells by regulating the three metabolic pathways to achieve ferroptosis therapy. This review summarizes the general biomarkers of ferroptosis and the application and mining of exosomes for cancer treatment.
2. THE OCCURRENCE OF FERROPTOSIS
Lipid peroxidation is a positive feedback chain reaction driven by free radicals; ROS, including superoxide (O2·), peroxide (H2O2 and ROOH), and free radicals (HO·and RO·) initiate the oxidation of polyunsaturated fatty acids (Figure 1) [28], which is called oxidative stress [29].
FIGURE 1.
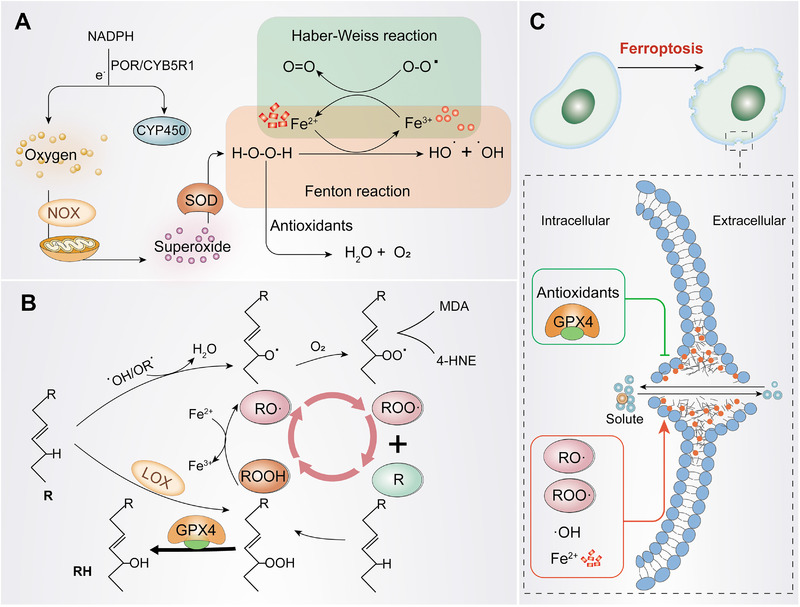
The occurrence of ferroptosis. (A) Generation of OH· and O2·. Oxygen produces O2· under the action of NADPH or NOX and mitochondria. Subsequently, O2· is converted into H2O2 under the action of SOD. H2O2 forms HO· in the presence of iron ions, which deprive PUFA of hydrogen atoms, causing lipid peroxidation. (B) Lipid peroxidation cycle and its termination. HO· or RO· catalyzes R to form RO·, RO· reacts quickly with oxygen to form ROO·, then extracts hydrogen from another R to produce new RO· and ROOH (positive feedback chain reaction). GPX4 can be used as an antioxidant to reduce ROOH to R to stop the peroxidation reaction. (C) The occurrence of ferroptosis. ROO· will decompose to produce MDA and 4‐HNE, then form covalent adducts with macromolecules such as protein, DNA, and lipids, or crosslink and inactivate proteins that promote ferroptosis, thereby promoting cell membrane rupture and ferroptosis. Abbreviations: NOX, NADPH oxidase; O2, superoxide radicals; SOD, peroxidase; HO·, hydroxyl radicals; RO·, lipid free radicals; ROO·, lipid peroxy‐free radicals; ROOH, lipids hydroperoxide; MDA, malondialdehyde; POR, NADPH‐cytochrome P450 reductase; CYB5R1, NADH‐cytochrome b5 reductase; CYP450, cytochrome P450; NOX, NADPH oxidase; SOD, superoxide dismutase; GPX4, glutathione peroxidase 4; 4‐HNE, 4‐Hydroxy‐2‐nonanal
The generation of hydroxyl‐free radicals (HO∙) and peroxy‐free radicals (RO∙) is a prerequisite for initiating lipid peroxidation. Oxygen competitively deprives the electrons of Nicotinamide Adenine Dinucleotide phosphate (NADPH) accepted by oxidoreductase NADPH‐cytochrome P450 reductase (POR), and NADH‐cytochrome b5 reductase (CYB5R1) on the endoplasmic reticulum is converted into superoxide (O2 –) [30, 31]. The electron transport chain of mitochondria and NADPH oxidase (NOX) [29] also produce superoxide. Recently, it has been demonstrated that P53 blocks the ability of dipeptidyl peptidase‐4 (DPP4) to form NOX1‐DPP4 complexes with NOX1 in the nucleus, reducing ROS production to inhibit ferroptosis [32], indicating that NOX plays a critical role in ferroptosis. Superoxide groups are highly active free radicals that can be eliminated by superoxide dismutase (SOD1), catalyzed and decomposed into hydrogen peroxide (H2O2) and water (H2O) with reduced reactivity [29, 33]. Hydrogen peroxide forms hydroxyl radicals (HO·) under the catalytic action of Fe2+, which is the basis of free radical lipid peroxidation, called the Fenton reaction [33, 34]. HO is generated through the redox cycle of the Fenton reaction, forming a closed‐loop cycle through the Haber‐Weiss reaction (Figure 1A) [15, 35]. Hydrogen peroxide can be reduced to water and oxygen through the synergistic action of glutathione peroxidase (GPX), peroxidase (PRX) and catalase [29]. Hydroxyl radicals (HO) and hydroperoxides (ROO) are the two most widespread ROS that affect lipids [15, 36], which may compromise the integrity of lysosomal membranes during oxidative stress, thereby, affecting cell survival [37].
Oxidative stress‐induced lipid peroxidation is the basis for ferroptosis. Lipid peroxides are produced by the oxidative attack of ROS on polyunsaturated fatty acids (PUFAs) (Figure 1B) [38]. The first step is the production of RO [34], followed by the creation of a new RO· (reproduction) [34, 39, 40]. The final step is to stop the reaction with antioxidants [41]. Enzymatic lipid peroxidation is mediated by the activity of the lipoxygenase (LOX) family [42], and phosphatidylethanolamine binding protein 1 (PEBP1), a scaffold protein of LOX15, which directly promotes ferroptosis [43]. glutathione peroxidase 4(GPX4) is the only GPX that can reduce hydroperoxide in the membrane, reducing ROOH to R, and interrupting the lipid auto‐oxidation reaction [35].
Accumulation of phospholipid hydroperoxides in cellular membranes is recognized as the hallmark and rate‐limiting step of ferroptosis; however, the exact mechanism leading to the downstream ferroptosis of lipid peroxidation remains elusive. 4‐Hydroxy‐2‐nonanal (4‐HNEs) is the final product of malondialdehyde (MDA) produced by lipid peroxidation which may form covalent adducts with biological macromolecules to reduce membrane integrity and crosslink and inactivate proteins, thereby, promoting cell membrane rupture and ferroptosis [15, 16]. Molecular dynamics models hypothesized that the continuous extensive oxidation and consumption of PUFAs can lead to membrane thinning and local membrane curvature as much as possible, driving the oxidant into the membrane, and the oxidant further damages the integrity of the membrane. This process makes the membrane increasingly sparse and curved. The vicious circle of the membrane eventually destroyed the stability of the membrane. Therefore, without GPX4, uncontrolled positive feedback may occur, forming a protein pore similar to that observed in necrosis and sagging, which leads to pores and micellization [44, 45]. The opening of the plasma membrane pores allows solutes to exchange with the external environment, leading to cell swelling and death (Figure 1C) [31].
3. FERROPTOSIS METABOLIC PATHWAY
Ferroptosis is caused by lipid peroxidation due to the accumulation of ROS, which exceeds the redox content of glutathione (GSH) and GPX4 [46]. Iron, lipids and ROS play an irreplaceable role in cell survival. However, excessive dependence is a double‐edged sword. These three maintain normal body function in a steady‐state and strike a deadly blow to cells when metabolic disorders occur. The complex biological processes involved in ferroptosis are induced by the imbalance of antioxidants, iron, and lipid dynamics (Figure 2); however, their role and contribution to the occurrence and metastasis of cancer remain unclear.
FIGURE 2.
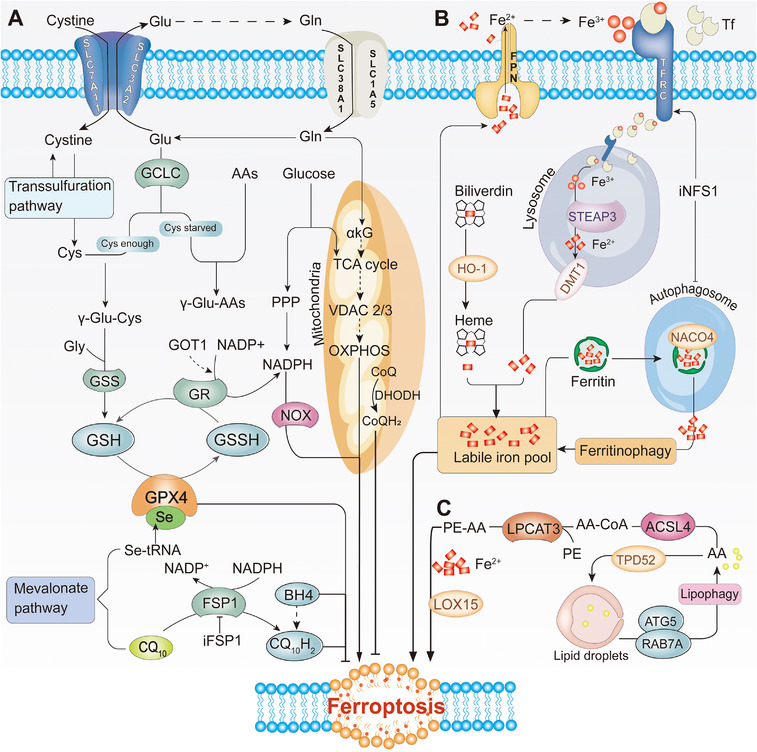
Ferroptosis metabolic pathway. (A) Antioxidant metabolism. GPX4 and CoQ10 are two parallel pathways of antioxidant metabolism. GPX4 exerts an antioxidant function through the metabolism of GSH. CoQ10 is reduced to CQ10H2 under the action of FSP1, inhibiting lipid peroxidation by capturing lipid peroxidation free radicals. (B) Iron metabolism. Iron binds to TF and is released into the cytoplasm by TFRC. Ferritin releases a large amount of iron‐by‐iron autophagy mediated by NACO4. Heme releases iron under the catalysis of HO‐1. FPN can transport iron from cells to the blood. (C) Lipid metabolism signaling pathway. AA/AdA is converted into polyunsaturated fatty acids (PE‐AA) by ACSL4 and LPCAT3. The balance between lipid synthesis, storage, and degradation can also manipulate the ferroptosis process. Acetyl‐CoA can also produce PUFA under the action of ACC synthase, immediately produce a large amount of ROS under the action of Fe2+ and LOX15. Abbreviations: GPX4, glutathione peroxidase 4; CoQ10, ubiquinone; FSP1, ferroptosis inhibitor protein 1; TF, transferrin; TFRC, transferrin receptor 1; HO‐1, heme oxygenase‐1; NFS1, cysteine desulfurase 1; ACSL4, acyl‐CoA synthetase 4; AA, arachidonic acid; AdA, epinephrine; LPCAT3, lysophosphatidylcholine acyltransferase 3; ATG5, autophagy‐related 5; BACO4, ###; BH4, tetrahydrobiopterin; Cys, Cysteine; DMT1, divalent metal transporter; FPN, ferroportin; GCLC, glutamate‐cysteine ligase catalytic subunit; Gln, Glutamine; Glu, Glutamate; GR, gluathione reductase; GSH, glutathione; GSS, Glutathione synthetase; GSSH, glutathione counterpart; iFSP1, Inhibit FSP1; iNFS1, Inhibit NFS1; LOX15, lipoxygenase 15; NADPH, nicotinamide adenine dinucleotide phosphate; NOX, NADPH oxidase; PE, Phosphatidylethanolamine; RAB7A, a small GTPase of the Rab family; SLC3A2, solute carrier family 3 member 2; SLC7A11, cystine/glutamate antiporter solute carrier family 7 member 11; STEAP3, Six‐transmembrane epithelial antigen of the prostate 3
3.1. Antioxidant metabolism
The balance of antioxidant metabolism is dynamically maintained mainly by GPX4, coenzyme CQ10, and dihydroorotate dehydrogenase (DHODH), all of which contain a metabolite molecule that exists in a chemically reduced and oxidized state that can inhibit ferroptosis (Figure 2A). GPX4 is a selenoprotein that reduces hydroperoxide in the cell membrane and uses GSH as an essential cofactor [35]. Glutamine‐cysteine synthase activity, cysteine concentration, and GSH feedback inhibition directly regulate GSH synthesis, thereby, affecting the enzymatic activity of GPX4 [47, 48, 49]. System XC and the trans‐sulfurization pathway are the two primary sources of cysteine.
The cystine/glutamate transport complex XC, comprising of heterodimers SLC7A11 (xCT) and SLC3A2 (4F2hc), takes up cystine (cystine) in a 1:1 form and excretes intracellular glutamate [13, 50, 51]. Researchers have established that SLC7A11 is the key hub for regulating “iron overload ferroptosis” [52], and activating SLC7A11 expression in cardiomyocytes can create “dead” resurrection [53]. Many factors regulate the expression of XC in the system, including targeted reduction of SLC7A11 levels, inhibition of SLC7A11 degradation and promotion of SLC7A11 expression; indicating that the XC system can determine the tolerance of ferroptosis (Table 2) [54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101].
TABLE 2.
Substances that regulate ferroptosis
| Category | Molecule | Target | Mechanism | Reference |
|---|---|---|---|---|
|
Antioxidant metabolism |
(1S, 3R)‐RSL3 | GPX4 | Covalently binds and directly inhibits GPX4 | [54] |
| Erastin | GSH, XC, VCAD2/3 | Inhibit the formation of GSH, inhibit the uptake of cystine, and induce ROS | [13, 54, 55] | |
| TFAP2c/ Sp1 | GPX4 | Enhance the expression of GPX4 | [56] | |
| Afelin A | GPX4 | Directly reduce the protein level and activity of GPX4 | [57] | |
| FIN56 | GPX4,MVA | Direct degradation of GPX4 and consumption of CoQ10 | [58, 59] | |
| HSPA5 | GPX4 | Binds and protects GPX4 protein from degradation | [60] | |
| Abramycin | GPX4 | Direct inhibition of GPX4 | [61] | |
| nitrile‐oxide electrophiles | GPX4 | Selectively target the active site of GPX4 | [62] | |
| FINO2 | GPX4 | Indirectly inhibit the function of GPX4 enzyme | [63] | |
| BSO | GSH | Inhibit the activity of GSH synthase | [64] | |
| miRNA‐18a | GCL | Inhibit the activity of glutathione‐cysteine ligase | [65] | |
| Cysteinase | Cysteine | Excessive consumption of cysteine, induces pancreatic cancer cell ferroptosis | [66] | |
| Estatin | SLC7A5 | Reduce glutamate intake | [67] | |
| CDO1 | GSH | Competitively restrict cysteine synthesis of GSH | [68] | |
| CARS | Transsulfurization pathway | Knockout CARS induce the transsulfur pathway and inhibit cystine deprivation | [69] | |
| P53 | SLC7A11, DPP4 | Inhibit SLC7A11 transcription, block DPP4 and reduce induction of ROS | [70] | |
| DPP4 | NOX1 | Interacts with NOX1 to form NOX1‐DPP4 complex | [32] | |
| ATF3 | XC | Targeted reduction of SLC7A11 mRNA and protein levels | [71] | |
| BECN1 | SLC7A11 | Binds and inhibits SLC7A11 | [72] | |
| BAP1 | XC | Epigenetic modification inhibits transcription of SLC7A11 | [73] | |
| ATF4 | XC | Combines with SLC7A11 promoter and activates its expression | [74] | |
| mTORC1 | SLC7A11 | Degrade SLC7A11 in the lysosome | [75] | |
| OTUB1 | XC | Directly interact with SLC7A11 to prevent its degradation | [76] | |
| NRF2 | FTH, OH‐1, GPX4, SCL7A11,GCLC | Both promote iron death and inhibit ferroptosis | [77, 78] | |
| BH4 | CoQ10 | Potentially promote the production of CoQ10 | [79] | |
| DHODH | CoQH2 | Reduces CoQ to CoQH2, protects quality from oxidative damage | [80, 81] | |
| ARF | NRF2 | Inhibit NRF2 transcription and activate SLC7A11 | [78] | |
| miRNA ‐137 | SLC1A5 | Reduce glutamine intake | [82] | |
| Myc | Glutamine transporter | Induce glutamine intake and promote glutamine decomposition | [83] | |
| p53 S47 | GLS2 | Inhibit the decomposition of glutamine | [84] | |
| FSP1 | CoQ10 | Catalyze the regeneration of CoQ10H2 | [85, 86] | |
| SREBP1 | SCD1 | Forms BECN1‐SLC7A11 complex, inhibits ferroptosis | [87] | |
| SCD1 | Saturated fatty acid, CoQ10 | Convert saturated fatty acids into monounsaturated fatty acids | [88] | |
| Iron metabolism | NCOA4 | FTH1 | Ferritin phagocytosis | [89, 90] |
| HO‐1 | Heme | Catalyzes the release of iron from heme | [57, 91] | |
| Hepcidin | FPN1 | Decrease cell iron output | [92] | |
| NFS1 | TFRC, FPN1 | Increase iron levels in cells | [55, 93, 94] | |
| Deferoxamine | Iron | Chelate free iron, leading to depletion of iron storage | [95] | |
| GOT1 | Aspartic acid | Knockdown of GOT1 increases the unstable iron pool | [96] | |
|
Lipid metabolism |
SAT1 | LOX15 | Induce LOX15 expression to increase | [97] |
| PEBP1 | LOX15 | As a scaffold protein of LOX15, promote LOX15 expression | [43] | |
| ACC | Acyl‐CoA | Synthetic ferroptosis substrate polyunsaturated fatty acid | [59, 98] | |
| iPLA2β | Unknow | Hydrolyzed oxidized phospholipid activity decreased | [99] | |
| Hippo pathway | ACSL4 | Inhibit the expression of ACSL4 | [100] | |
| MCT1 | ACSL4 | Inhibit the expression of ACSL4 | [88] | |
| AGPS | PUFA‐ePLs | Catalyze the synthesis of ether lipids,increase the susceptibility to ferroptosis | [101] |
Abbreviations: GPX4, glutathione peroxidase 4; ACSL4, acyl‐CoA synthetase long‐chain family member 4; CoQ10, ubiquinone; CoQH2, ubiquinol; FPN1, ferroportin; GCLC, glutamate‐cysteine ligase catalytic subunit; FTH1, ferritin heavy chain 1; GCL, glutamate cysteine ligase; GLS2, glutaminase 2; GPX4, ###; MVA, Mevalonate pathway; GSH, glutathione; VCAD2/3, voltage‐dependent anion channel 2/3; LOX15, lipoxygenase 15; NOX1, NADPH oxidase 1; NRF2, NFE2‐related factor 2; SCD1, stearoyl‐CoA desaturase 1; SLC1A5, Alanine, serine, cysteine‐preferring transporter 2 (ASCT2; SLC1A5); SLC7A11, cystine/glutamate antiporter solute carrier family 7 member 11; TFRC, transferrin receptor; XC, cystine‐glutamate reverse transporter.
Glutamate is another important raw material required for the synthesis of GHS, and its uptake mainly depends on SLC38A1 and SLC1A5 [102]. GCLC(glutamate‐cysteine ligase catalytic subunit) catalyzes the first step in glutathione synthesis through the linkage of cysteine and glutamate. However, when cysteine is insufficient, GCLC promotes the synthesis of γ‐glutamyl peptides (γ‐Glu‐AAs), eliminating glutamate, and preventing ferroptosis [103]. Overall, both the biosynthesis/uptake of selenocysteine [56, 104] and the synthesis of GSH [65, 67] affect the homeostasis of GPX4, indicating that GPX4 plays a pivotal role in regulating ferroptosis.
Mitochondrial metabolism is the main source of cellular ROS, and the decomposition of glutamine, the raw material of GSH, is thought to regulate ferroptosis by providing α‐ketoglutarate (α‐KG) in the mitochondrial tricarboxylic acid (TCA) cycle [93, 105]. Inhibition of the mitochondrial TCA cycle, knockout of mitochondrial voltage‐dependent anion channel 2/3 (VDAC2/3), or suppression of electron transfer chain (ETC) reduces lipid peroxide accumulation and ferroptosis, indicating that ferroptosis involves abnormal ROS production [11, 55, 106]. Blocking the mitochondria strongly inhibits ferroptosis; however, when GPX4 is pharmacologically inhibited, ferroptosis can be performed independently of mitochondria. When cysteine is inadequate, mitochondrial metabolism promotes rapid depletion of glutathione and subsequent lipid ROS production and ferroptosis [106], suggesting that the role of mitochondria in ferroptosis may be related to the upstream and downstream; however, the exact role of mitochondria in ferroptosis remains unclear.
Recent studies have revealed that FSP1‐CoQ is an antioxidant system that is parallel to the GPX4 pathway and acts only on GPX4‐depleted cells. Ferroptosis inhibitor protein 1 (FSP1), formerly known as apoptosis‐inducing factor mitochondrial‐related 2 (AIFM2), was identified as a ferroptosis inhibitor. FSP1 is recruited to the plasma membrane through N‐terminal myristoylation as an oxidoreductase, reduces ubiquinone (CoQ10) which is another product of mevalonate metabolism to the lipophilic free radical scavenger panthenol (CoQ10H2), which limits the accumulation of lipid ROS in the membrane without GPX4 [85, 86]. Isopentenyl pyrophosphate, a metabolite of mevalonate, is essential for producing selenoproteins, GPX4 and ubiquinone. Another product, squalene synthase, is a regulator of oxidation levels and is used to accumulate ROS to induce ferroptosis [58]. Regarding the involvement of squalene synthase in the mevalonate pathway, a rare lymphoma cell lacks an enzyme involved in cholesterol synthesis. Cells without this enzyme will accumulate squalene and the accumulation of squalene can increase the antioxidant capacity of a cell, thereby, providing benefits to cancer cells by a phenomenon known as an anti‐ferroptotic property [107]. The specific inducers of ferroptosis, ferroptosis inducer derived from CIL56 (FIN56) [58] and statins [108], can inhibit the CoQ10 and mevalonate pathways to mediate cell ferroptosis. In addition to this pathway, a recent study identified another GPX4‐independent ferroptosis blocking pathway involving GTP cyclohydrolase 1 (GCH1), which is a rate‐limiting step in the production of tetrahydrobiopterin (BH4). BH4 inhibits ferroptosis by facilitating the formation of CoQ10 and blocking the peroxidation of specific lipids [79].
FSP1 inhibits ferroptosis by producing ubiquinol but its activity is limited to the cell membrane. Thus, it is still unclear whether the mitochondria use a similar mechanism to produce ubiquinol to repair the oxidative damage caused by mitochondrial membrane lipids. The latest research proposes a third protection system: DHODH, an enzyme located on the outer surface of the inner mitochondrial membrane and is responsible for oxidizing dihydroorotic acid (DHO) in the inner mitochondrial membrane to orotic acid (OA). DHODH reduces CoQ to CoQH2 [80, 81]. Therefore, DHODH is a protective mitochondrial lipid system that protects against oxidative damage. The following three systems have unique subcellular locations: GPX4 in the cytoplasm and mitochondria, FSP1 on the plasma membrane, and DHODH on the mitochondria. Damage to one system will force cells to rely more on the other, while simultaneous loss of all three protective systems will trigger ferroptosis induced by lipid peroxidation. The above results indicate the existence of three parallel antioxidant systems in cells, namely GPX4, FSP1‐CoQ, and DHODH, which precisely regulate the metabolism of cellular antioxidant substances to prevent ferroptosis.
3.2. Iron metabolism
As one of the “bioelements,” iron in an oxygen‐rich environment can generate ROS through the Fenton reaction with different types of phospholipid hydroperoxides and lipid (fatty acid) hydroperoxides [28]. Fe3+ circulating in the blood is transported to the cells by binding to transferrin (TF) and through endocytosis mediated by the transferrin receptor (TFRC). After Fe3+ is taken into cells, it is reduced to Fe2+ by the action of metal reductase Six‐transmembrane epithelial antigen of the prostate 3(STEAP3) on the ferrosomes. The divalent metal transporter (DMT1) mediates the release of Fe2+ from ferrosomes to the labile iron pool (LIP) medium (Figure 2B) [53, 67, 109, 110]. Recent studies have revealed that transferrin in the liver inhibits liver damage, fibrosis, and cirrhosis by regulating ferroptosis [111], and the iron homeostasis pathway driven by sterol regulatory element binding protein‐2 (SREBP2) contributes to cancer progression, drug resistance and metastasis [112]; indicating that iron homeostasis is essential for organ survival.
Most iron can be stored in ferritin or heme. Ferritin includes two subunits: FTL (light chain) and FTH1 (heavy chain) [113]. The nuclear receptor co‐activator 4 (NCOA4) directly binds to FTH1 to form the ferritin complex targeting lysosomes for “ferritinophagy,” promoting the degradation of ferritin and increasing intracellular free iron during iron deficiency [89, 90]. As another common protein containing iron in cells, Fe2+ released by heme under the catalysis of heme oxygenase‐1 (HO‐1) accumulates in cardiomyocytes and induces ferroptosis [114] which may be activated by NRF2 and BAY [57, 91]. Ferroportin (FPN1) is the only cellular efflux channel for iron which is degraded by binding to hepcidin secreted by the liver, resulting in a decrease in cellular iron output (Figure 2B) [92]. However, most cells do not have an effective mechanism to export iron, resulting in an increase in LIP levels when the amount of iron exceeds the storage capacity [46]. Regardless of the pathway, the increase in intracellular free iron increases the induction of ferroptosis by LIP.
3.3. Lipid metabolism
Lipid production is the basis of survival and lipid peroxidation is a biomarker of ferroptosis [115]. Lipid peroxidation in the process of ferroptosis requires acyl‐CoA synthetase long‐chain family member 4 (ACSL4), lysophosphatidylcholine acyltransferase 3 (LPCAT3), and arachidonic acid 15‐lipoxygenase (ALOX15) (Figure 2C). Quantitative lipidomics has found that arachidonic acid (AA) and epinephrine (AdA), which are phosphatidylethanolamines (PEs), are necessary for ferroptosis [116]. In addition, it has recently been found that enhancing TPD52‐dependent lipid storage or blocking ATG5, and RAB7A‐dependent lipid degradation both prevent RSL3‐induced lipid peroxidation and subsequent ferroptosis, indicating that the balance between lipid synthesis, storage, and degradation can be manipulated by ferroptosis [117]. ACSL4 expression is regulated by the Hippo signaling pathways [100], radiotherapy [118], and the lactic microenvironment [88]. Located in the endoplasmic reticulum, LPCAT3 is mainly expressed in the metabolic tissues such as liver, fat, and pancreas [119], by specifically inserting acyl groups in phosphatidylcholine (PC) and PE‐CoA produces PE‐AA [120]. Lack of LPCAT3 can lead to a significant decrease in cell membrane AA levels and accumulation of cytoplasmic lipid droplets, which leads to lethality and intestinal cell damage in mice [120], indicating that LPCAT3 is essential for cell survival. Another interesting study recently defined the saturation of ether lipids as a determinant of ferroptosis. It was also found that in the differentiation of cardiomyocytes and nerve cells, increased levels of polyunsaturated ether phospholipids (PUFA‐ePLs) are accompanied by increased sensitivity to ferroptosis, and selective downregulation of highly unsaturated PUFA‐ePLs levels may be an important approach for cells to escape ferroptosis [101]. The above findings are of great importance to the rational design and use of ferroptosis‐targeted drugs in disease treatment.
Lipoxygenases (LOXs) can oxidize PUFAs into their corresponding hydroperoxides. In mammalian cells, linoleic acid and AA are the most abundant polyunsaturated fatty acid substrates in LOXs [42, 119]. PUFAs are the lipids that are most prone to peroxidation during ferroptosis. Therefore, pretreatment of cells with PUFAs containing heavy hydrogen isotope deuterium at the peroxidation site (D‐PUFA) can prevent PUFA oxidation and block ferroptosis [121]. Acetyl‐coenzyme A carboxylase (ACC) is an enzyme involved in the fatty acid synthesis and directly synthesizes PUFAs from acyl‐CoA. PE‐AA is a polyunsaturated fatty acid PUFA, which can be used in LOX15 and Fe2+ catalytic production of oxidized phospholipid ox‐PE, contributes to the development of ferroptosis [59, 98]. The mutation of the key protein iPLA2β for phospholipid remodeling leads to a decrease in the activity of its hydrolyzed oxidized phospholipid ox‐PE, leading to the accumulation of oxidized phospholipids in dopaminergic neurons [99]. Evidence has shown that antioxidant metabolism, iron levels and lipid metabolism play an extremely important role in hypertrophy, and steady‐state changes on either side induce ferroptosis (Table 2). However, when and how lipid peroxidation regulates ferroptosis and non‐ferroptotic cell death in different ways remain to be discovered.
4. CROSSTALK BETWEEN FERROPTOSIS AND CLASSIC CELL SIGNALING PATHWAYS IN CANCER
As previously mentioned, induced by lipid peroxidation, ferroptosis controls many processes in a cell type‐and context‐specific manner; however, the role of ferroptosis in the airframe remains elusive. Many studies have revealed that the synergistic regulation of ferroptosis by antioxidant, iron, and lipid metabolism affects the occurrence and development of specific types of cancer cells (Figure 2). However, the crosstalk between ferroptosis and other signaling pathways is not well understood. This review discusses the crosstalk between ferroptosis and other classic cell signaling pathways in cancer to provide cancer immunotherapy targets (Figure 3).
FIGURE 3.
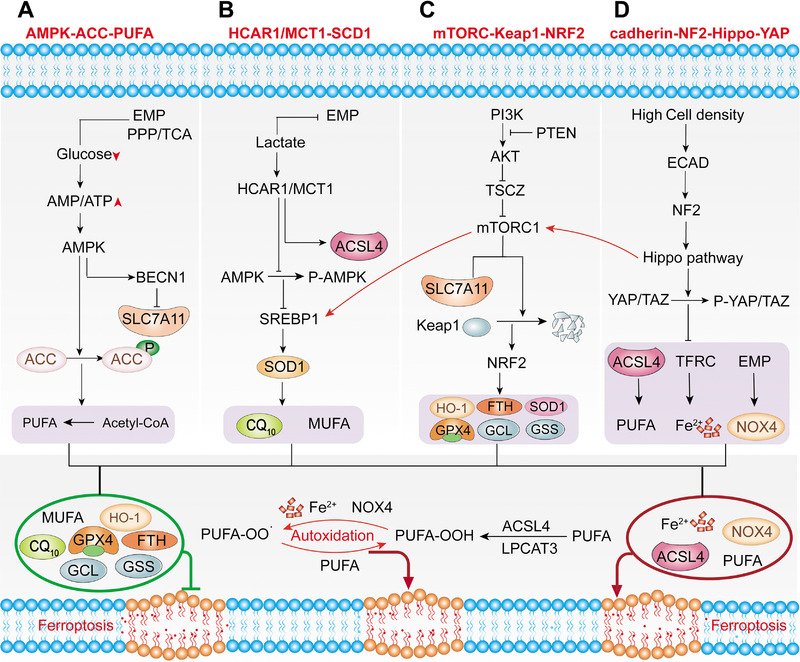
Crosstalk between ferroptosis and classic signaling pathways. (A) AMPK‐ACC‐PUFA pathway. EMP, PPP and TCA cycles all require glucose as a substrate. When glucose is lacking, insufficient intracellular energy metabolism will activate AMPK, reducing the synthesis of PUFA by inhibiting ACC. (B) HCAR/MCT1‐SREBP1‐SCD1 pathway. Lactate enhances cell lipid synthesis by inhibiting glycolysis. Lactate induces the expression of HCAR1, MCT1 and inhibiting ferroptosis by directly inhibit ACSL4. Moreover, lactate promotes the phosphorylation of AMPK, which inhibits the ability of SREBP1 to promote SCD1, which directly inhibit ferroptosis by promoting the production of CQ10 and MUFA. (C) MTORC1‐Keap1‐NRF2 pathway. PI3K‐AKT‐mTORC1 signaling pathway activation will upregulate SREBP1 and SCD1, promoting MUFA production to resist ferroptosis. In addition to degrading SLC7A11, mTORC1 also promotes the accumulation of NRF2 by phosphorylation of p62, and indirectly regulates NRF2 target genes. mTORC2 directly regulates the phosphorylation of SLC7A11. (D) Cadherin‐NE2‐Hippo‐YAP pathway. At high cell density, ECAD will send a signal to the Hippo signaling pathway through NF2, inhibiting the ferroptosis process by promoting the phosphorylation of YAP and TZA. The green box indicates inhibition of ferroptosis, and the red box indicates the inducer of ferroptosis. Abbreviations: HCAR1, lactate receptor; MCT1, lactate transporter; SREBP1, sterol regulatory element‐binding protein 1; SCD1, stearoyl‐CoA desaturase 1; MUFA, monounsaturated fatty acid; ACC, acetyl‐coenzyme A carboxylase; ACSL4, acyl‐CoA synthetase long‐chain family member 4; AMP, adenosine monophosphate; AMPK, AMP‐activated protein kinase; ATP, adenosine triphosphate; BECN1, beclin 1; EMP, Embden‐Meyerhof‐Parnas pathway; FTH, ferritin heavy chain; GCL, glutamate cysteine ligase; GPX4, glutathione peroxidase 4; GSS, glutathione synthetase; LPCAT3, lysophosphatidylcholine acyltransferase 3; NF2, neurofibromatosis type 2; NOX4, NADPH oxidase 4; NRF2, NFE2‐related factor 2; PI3K, phosphatidylinositol 3‐kinase; PTEN, phosphatase and tensin homolog; PUFA, polyunsaturated fatty acids; SLC7A11, cystine/glutamate antiporter solute carrier family 7 member 11; SOD1, superoxide dismutase; TAZ, PDZ‐binding motif; TCA, tricarboxylic acid; TSC, tuberous sclerosis; YAP, yes‐associated protein
4.1. Energy metabolism pathways
Recent studies have indicated that energy metabolism pathways are involved in the regulation of ferroptosis. Because of the Warburg effect [122], cancer cells are extremely dependent on glucose and have low productivity. Insufficient energy metabolism in the cell leads to a decrease in ATP content and activates AMPK, an important hub that senses and regulates the balance of cell energy metabolism. ACC1 and ACC2 are two enzymes that catalyze the synthesis of malonyl‐CoA from acetyl‐CoA, promoting fatty acid synthesis and inhibiting fatty acid oxidation. AMPK inhibits ACC to reduce the synthesis of PUFAs, ultimately inhibiting ferroptosis [123]. The above results indicate that the AMPK‐ACC‐PUFA pathway activated by energy metabolism reduces the synthesis of polyunsaturated fatty acid PUFAs, thereby inhibiting ferroptosis (Figure 3A). AMPK phosphorylation also mediates the phosphorylation of BECN1, the main component of the phosphatidylinositol 3‐kinase (PI3K) complex, which promotes BECN1‐SLC7A11 complex formation, system XC inhibition, and subsequent ferroptosis [72]. These results indicate that tumor cells avoid the occurrence of ferroptosis to a certain extent through metabolic reprogramming. Inhibition of AMPK‐ACC‐PUFA signaling may produce antitumor effects by targeting this pathway in different cell types of the tumor and its niche, by enhancing the efficacy of immunotherapies and potentiating chemotherapy treatments. However, therapeutic interference with AMPK‐ACC‐PUFA signaling may also result in unexpected adverse effects; therefore, these inhibitors should be used with care.
4.2. HCAR1/MCT1‐SREBP1‐SCD1 pathway
In addition to forming a microenvironment that is prone to tumor metastasis, the lactate produced by tumor metabolism may also inhibit the ferroptosis of tumor cells through the HCAR1/MCT1‐SREBP1‐SCD1 pathway, providing potential for its metastasis and development (Figure 3B). Lactate produced by tumors enhances lipid synthesis by inhibiting glycolysis Moreover, lactate induces the expression of HCAR1 (lactate receptor) and MCT1 (lactate transporter). MCT1‐mediated lactate uptake promotes the production of ATP in hepatocellular carcinoma (HCC) cells, increasing the AMP/ATP ratio and phosphorylating AMPK, leading to the upregulation of sterol regulatory element‐binding protein 1 (SREBP1) [88, 124]. SREBP1 is a transcription factor that regulates the expression of multiple lipid synthesis‐related genes, including stearoyl‐CoA desaturase 1 (SCD1), which converts saturated fatty acids into monounsaturated fatty acids (MUFAs), thereby, inhibiting ferroptosis [87]. Lipidomic testing found that the knockdown of MCT1 upregulates the expression of ACSL4, reduces the production of phospholipids containing MUFA, and downregulates the expression of another ferroptosis inhibitor, CoQ10, downstream of SCD1 [88]. The above results indicate that lactate induces the formation of monounsaturated fatty acids through the HCAR1/MCT1‐SREBP1‐SCD1 pathway which helps liver cancer cells to resist lipid peroxidation induced by oxidative stress.
4.3. PI3K‐AKT‐mTOR pathway
Regardless of the energy stress and lactate signaling pathway activated by tumor metabolism, the master growth regulator of the cell, mTORC1, and another different mTOR kinase complex, mTORC2, have also been shown to be involved in the regulation of ferroptosis (Figure 3C). Many cancers require mTORC1 to maintain their ability to regulate protein synthesis and cell growth, coordinating nutrition and energy supply, to ensure that they undergo unlimited cell division, whereas mTORC2 mainly promotes cell proliferation and survival through phosphorylation of AKT [125]. Both mTORC1 and mTORC2 are involved in ferroptosis. mTORC2 interacts with SLC7A11 to directly phosphorylate serine at position 26 of SLC7A11, inhibiting its transporter activity [126]. High cell density activates LATS1/2 kinase in the Hippo pathway, subsequently phosphorylating a component of mTORC1, leading to inactivation of mTORC1 and preventing mTORC1 from degrading SLC7A11 in the lysosome [75]. Activated PI3K‐AKT‐mTOR is one of the most mutated signaling pathways in human cancers, and activation of the PI3K‐AKT‐mTORC1 signaling pathway ensures that cancer cells resist ferroptosis through SREBP1‐mediated MUFA production [87].
Excessive metabolism and proliferation burden lead to higher oxidative stress in cancer cells. NFE2‐related factor 2 (NRF2) is a transcription factor that integrates cell stress signals and directs various transcription elements [127]. However, during periods of increased oxidative stress or mutations in KEAP1, CUL3 or NRF2, NRF2 is no longer ubiquitinated and degraded, enabling translated NRF2 to be transported to the nucleus to activate the transcription of genes containing antioxidant response elements (ARE) [128, 129]. In addition, mTORC1 promotes the binding of p62 and Keap1 by phosphorylating p62, leading to the degradation of Keap1 and accumulation of NRF2 [87]. Many antioxidant defense proteins involved in iron and ROS metabolism, including SOD1, GPX4, glutathione synthesis enzymes (GCL and GSS), HO‐1 and FTH1, are all induced by NRF2 transcription [128, 129, 130]. Therefore, the PI3K‐AKT‐mTORC1 signaling pathway plays a critical role in maintaining cellular redox homeostasis through NRF2‐mediated signal transduction and SREBP1/SCD1‐mediated MUFA synthesis (Figure 3C).
4.4. Cadherin‐NE2‐Hippo‐YAP pathway
Apart from the mTORC1 pathway which controls tumor cell growth, Hippo controls organ size by regulating cell proliferation and apoptosis, and E‐cadherin, maintains cell polarity and is also involved in ferroptosis. The Hippo pathway is a kinase chain composed of a series of highly conserved protein kinases and transcription factors [131]. E‐cadherin (ECAD) is an important regulator of inter‐cell interconnection in epithelial cells and is positively correlated with cell density [132]. Knockout of the Hippo element or overexpression of YAP inhibits the reduction of cell proliferation caused by the binding of E‐cadherin to the cell surface. The E‐cadherin/catenin complex acts as an upstream regulator of the Hippo signaling pathway in mammalian cells and inhibits the activity of YAP in the nucleus, directly regulating the Hippo pathway [133].
Recent studies have also revealed that neighboring cells can also activate the NF2 and Hippo signaling pathways through ECAD‐mediated interactions, thereby, inhibiting the ferroptosis of tumor cells (Figure 3D). TEA domain transcription factor 4 (TEAD4), which interacts with Yes‐associated protein (YAP) and activates its function, binds to the promoter regions of the ferroptosis markers TFRC and ACSL4. This effect was inhibited by the phosphorylation of YAP. Therefore, an increase in cell density leads to a decrease in the expression of TFRC and ACSL4. This phenomenon can be reversed by knocking out ECAD, NF2 or inhibiting the phosphorylation of YAP, thereby promoting ferroptosis [100]. In addition, LATS1/2 is a key component of Hippo and inhibits mTORC1 by phosphorylating Ser606 of Raptor, an important component of mTORC1, realizing the interaction between the Hippo signaling pathway and mTORC1 [75]. The above results indicate that the intercellular mechanism may be a vital defense function of cells which resists oxidative stress and ferroptosis.
Unlimited growth is a major feature of tumors, accompanied by massive consumption of energy, “self‐toxic” density, and violent biochemical metabolism. Thus, ferroptosis was significantly induced in tumor cells. However, in the natural state, few tumor cells lose the ability to develop infiltration due to ferroptosis, and there must be a special mechanism to antagonize ferroptosis. Some studies have indicated that the classic energy metabolism pathways, density effects and biochemical reaction metabolites in tumor cells interact closely with ferroptosis and significantly inhibit cell ferroptosis. Therefore, inhibition of ferroptosis may be the result of tumor cells achieving a large amount of growth. In summary, a better understanding of the crosstalk between ferroptosis and classic cancer signaling pathways could help developing new compounds to target specific ferroptosis activation mechanisms or particular effectors, increasing selectivity and decreasing the clinical drawbacks of current inhibitors.
5. FERROPTOSIS AND ANTITUMOR TREATMENT
Current treatments for cancer cannot effectively cope with the resistance of cancer cells to existing chemotherapy drugs. Many studies have demonstrated that ferroptosis plays a vital role in killing tumor cells and inhibiting tumor growth (Figure 4). Ferroptosis has been identified as a cause of non‐small cell lung cancer [134], breast cancer [135], pancreatic cancer [136], HCC, and other tumor‐causing cell deaths. Consequently, targeted induction of ferroptosis may be a new cancer treatment strategy.
FIGURE 4.
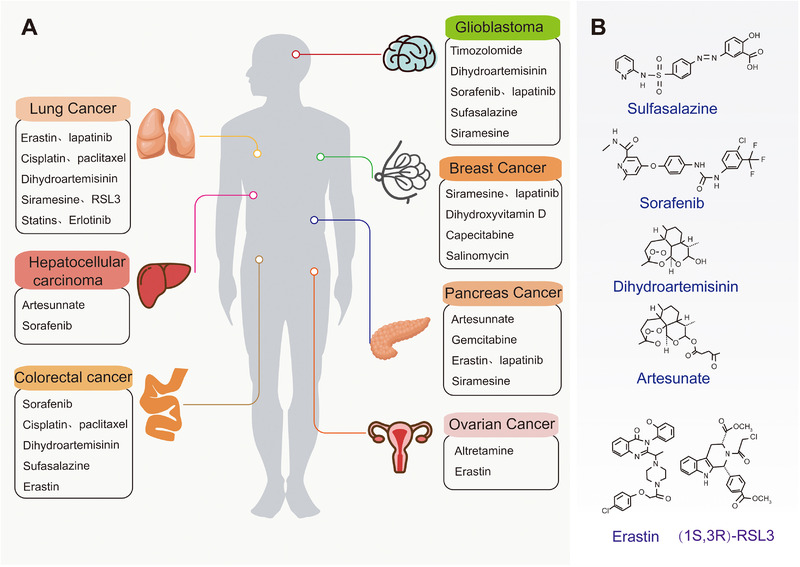
Ferroptosis and drug treatment of tumor. (A) Classic cancer and corresponding treatment drugs of ferroptosis. (B) Small molecules used in ferroptosis therapy to treat cancer
Lung cancer is a malignant tumor with the fastest increase in morbidity and mortality rates. It is the greatest threat to the health and life of the population. Three main drug mechanisms target the induction of ferroptosis in lung cancer cells. The first is to target the inhibition of the XC system to limit the uptake of cystine and induce the accumulation of ROS, such as the small‐molecule compound erastin [13, 137] and the FDA (U.S. Food & Drug Administration)‐approved immunosuppressant sulfasalazine [47]. Second, ferroptosis can be induced in lung cancer by increasing the level of free iron, typically cisplatin [138], changing the iron regulatory protein/iron response element ratio to regulate iron homeostasis, dihydroartemisinin [139], and NFS1, which tricks lung cancer cells to absorb and release large amounts of iron [94]. Research has also indicated that drug combination therapy has great application potential. Erastin makes lung cancer cells more sensitive to cisplatin, in turn triggering ferroptosis [134], and may enhance the therapeutic effect and improve the survival rate of lung cancer patients treated with epidermal growth factor receptor‐tyrosine kinase inhibitors (EGFR‐TKIs) [140].
Glioblastoma is a very aggressive brain tumor that often recurs and causes death even after surgery and radiotherapy [141]. Sulfasalazine inhibits cysteine uptake through XC, leading to ferroptosis in glioma cells Temozolomide induces ferroptosis by activating NRF2/ATF4 at the mRNA and protein levels to induce the expression of SLC7A11 and increase the activity of cystathionine gamma‐lyase in the trans‐sulfur pathway [142]. Even at low concentrations, the combination of aspirin and sorafenib leads to XC inhibition, GSH depletion, and ROS accumulation in cancer cells, simultaneously enhancing the cytotoxicity of cisplatin to drug‐resistant cells by inhibiting XC and DNA damage [143]. Therefore, ferroptosis also has potential applications in the management of treatment‐resistant malignant tumors.
HCC (hepatocellular carcinoma) is the most common primary hepatocellular carcinoma, with the fastest increase in the incidence of all cancers. Sorafenib was originally thought to inhibit a variety of oncogenic kinases that induce ferroptosis in HCC. This effect is significantly inhibited by deferoxamine which depletes intracellular iron storage [95]. A recent study indicated that inhibition of metallothionein‐1G enhanced the anticancer activity of sorafenib in vitro and tumor xenograft models, knocking out MT‐1G by RNA interference enhanced ferroptosis caused by sorafenib [144]. Loss of retinoblastoma (Rb) protein function is a hallmark of HCC development. HCC cells with reduced Rb levels increase cell death induction two to three times when exposed to sorafenib, indicating that Rb inhibits sorafenib treatment of ferroptosis [145], which may be the reason for the acquired resistance to sorafenib in HCC patients.
Colorectal cancer (CRC) is the third most common malignant tumor [146]. Sulfasalazine effectively depleted GSH in CRC cell lines and enhanced the anticancer activity of cisplatin, resulting in significant accumulation of ROS and growth inhibition of CRC, indicating that sulfasalazine is a relatively non‐toxic drug targeting the cystine transporter. Moreover, its combination with CDDP(cisplatin) may have an effective therapeutic effect on CRC [147]. RSL3‐treated CRC cells accelerate ROS production by activating transferrin and reducing GPX4 levels [148].
Pancreatic cancer is one of the deadliest cancer types with strong chemical resistance and has a clinical mortality rate close to 95%; the occurrence of aggressive metastasis and resistance to conventional chemotherapy is considered a major challenge for treatment [149]. Sulfasalazine is an anti‐inflammatory drug with a strong XC inhibitory effect which can significantly reduce cysteine uptake and glutathione levels of human MIA, Pa, Ca‐2 and PANC‐1 pancreatic cancer cells [150]. Combining sulfadiazine (SSZ) and piperine (PL) treatment can significantly increase ROS levels in pancreatic cancer and induce cytotoxic ferroptosis. This effect is eliminated by ferroptosis inhibitors and deferoxamine [136]. Artesunate is an antimalarial drug that induces iron‐and ROS‐dependent cell killing in pancreatic cancer cell lines with K‐Ras mutations and wild‐type, indicating that artesunnate is a specific inducer of pancreatic cancer cell ferroptosis [151].
Ovarian cancer is a lethal malignant tumor but no major progress has been made in the treatment of ovarian cancer for more than 30 years. The cells of patients with high‐grade serous ovarian cancer (HGSOC) are accompanied by a decrease in FPN expression and an increase in the expression of transferrin receptor (TFR1) in the iron efflux pump, leading to excessive iron accumulation in the cell and enhancement of iron dependence. Consequently, the increase in iron efflux and erastin inhibits the metastasis and proliferation of ovarian cancer, the effects of which are inhibited by Fer‐1 and Deferoxamine mesylate (DFO) [152]. In addition to killing pancreatic cancer cells, artesunnate can also induce HGSOC ferroptosis to exert antitumor effects [153], and combining siramesine and lapatinib has been shown to synergistically inducing breast cancer ferroptosis by increasing the levels of transferrin and ROS, the effect of which is significantly inhibited by the iron chelator deferoxamine [135]. Similarly, CDO1 overexpression reduces GSH levels in breast cancer cells, leading to the accumulation of high levels of ROS and aggravating ferroptosis [154], indicating that small‐molecule drugs targeting different metabolic axes of ferroptosis have great potential for clinical treatment.
As a large number of clinical drugs are modified or combined with small‐molecule inhibitors, their specificities may be different (Figure 4). There are also drugs that only target specific cancers, and these may be special drugs that have been selected by researchers through screening in cancer models. For ease of understanding, ferroptosis‐associated anticancer drugs are also listed here (Table 3) [155, 156, 157, 158, 159, 160, 161], along with their targets and functions. It should be noted that the characteristics of the tumor itself determine the type and effect of drugs in relation to ferroptosis. For example, iron‐rich tumors (such as HCC and breast cancer) or reactive oxygen‐rich tumors (lung cancer) are particularly sensitive to drugs that promote ferroptosis. In this case, the use of ferroptosis‐targeted drugs can achieve a better curative effect. Second, the selection of specific drugs largely depends on the degree of expression of drug targets in tumor cells. Different ferroptosis‐regulatory genes have different gene expression levels in different tumors. For example, certain types of tumor cells (lung cancer) overexpress SLC7A11, and thus, the ferroptosis inducer sulfasalazine which targets SLC7A11 may be effective against them. However, although other ferroptosis‐related drugs (such as lapatinib) can also induce ferroptosis, they cannot directly target SLC7A11, so their effects in the treatment of lung cancer are not satisfactory. It is also interesting to note that traditional drugs have been repurposed to target ferroptosis treatment. For example, sulfasalazine, which was originally used to treat colitis and arthritis, was found to promote ferroptosis by inhibiting cysteine uptake and GPX4 synthesis. This reminds us that the function of drugs may expand with more scientific exploration. It is worth mentioning that many nano‐drugs have great potential in specifically inducing ferroptosis in tumor cells, and can be simply classified according to the nanomaterials and coating materials used (Table 4) [161, 162, 163, 164, 165, 166, 167, 168, 169, 170, 171, 172, 173, 174, 175, 176, 177, 178]. However, not all nanomedicines are listed because they have fewer clinical applications compared with conventional drugs.
TABLE 3.
Applications of ferroptosis in cancer
| Compound/Drug | Cancer type | Target | Function | Reference |
|---|---|---|---|---|
| Sulfasalazine | Lung cancer, Glioblastoma, Colorectal cancer, Pancreatic cancer, Prostate cancer | SLC7A11 | Inhibit cysteine uptake and GPX4 synthesis, promote ferroptosis | [47, 128, 147, 150] |
| Cisplatin | Lung cancer, Colorectal cancer | GSH | Induce Lipid peroxidation, increase the level of free iron, | [134, 138] |
| Dihydroartemisinin | Lung cancer, Glioblastoma, Colorectal cancer, Breast cancer | Ferritinophagy | Induce Iron metabolism | [139] |
| Statins | Lung cancer | GPX4 | Block the synthesis of GPX4 | [140] |
| Gefitinib | Lung cancer | ROS | Inhibition of EGFR‐ERK/AKT by gefitinib, reduces ROS | [155] |
| Sorafenib |
Glioblastoma, Hepatocellular carcinoma, Liver Cancer, Colorectal cancer |
SLC7A11 |
Induce lipid peroxidation, accumulation of ROS to promote ferroptosis |
[95, 143, 144] |
| Lapatinib |
Breast Cancer, Glioblastoma |
Fe | Induces ferroptosis by elevating the intracellular iron level | [156] |
| Temozolomide | Glioblastoma | NRF2/ATF4 | Induce the expression of SLC7A11 | [142] |
| Artesunnate |
Glioblastoma, Pancreatic cancer, Ovarian cancer |
Ferritin | Enhance lysosomal degradation of ferritin | [151, 153, 157] |
| Paclitaxel | Colorectal cancer | P53, SLC7A11 | Lipid peroxidation | [158] |
| Siramesine | Breast cancer | Iron | increases iron | [135] |
| Gemcitabine | Pancreatic cancer | GPX4 | Inhibit lipid peroxidation | [159] |
| Tanshinone | Pancreatic cancer | STAT3 | Regulating ferroptosis through lysosome pathway | [160] |
| Cotylenin A | Pancreatic cancer | ROS | Promoting ROS production | [161] |
Abbreviations: SLC7A11, cystine/glutamate antiporter solute carrier family 7 member 11; GSH, glutathione; GPX4, glutathione peroxidase 4; ROS, reactive oxygen species; ATF4, Activating transcription factor 4; NRF2, NFE2‐related factor 2; STAT3, signal transducer and activator of transcription 3; ROS, reactive oxygen species; EGFR, epidermal growth factor receptor; ERK, extracellular signal‐regulated kinase; AKT, protein kinase B.
TABLE 4.
Recently reported nanomaterial‐induced ferroptosis‐related nanoparticles for cancer therapy applications
| Nanoparticle | Material type | Encapsulation | Mechanism | Reference |
|---|---|---|---|---|
| Ferumoxytol | Polysaccharide | Fe2O3 | Fe3+ increasing and ROS overloading | [162] |
| FePt | Black phosphorus, metal | Fe | Promote Fenton reaction | [163] |
| RSL3@COF‐Fc | COFs | RSL3 |
Promote Fenton reaction, GPX4 inhibition, Lipid peroxidation |
[164] |
| FePt@MnO@DSPE | Metal nanoparticles | Fe2+, Mn2+ | Promote Fenton reaction | [165] |
| UPDA‐PEG@Fe2+/3+ | Coordination polymer | Fe2+, Fe3+ | Promote Fenton reaction | [166] |
| DS@MA‐LS | Liposome | Doxorubicin and Sorafenib | Increase ROS and SLC7A11 inhibition | [167] |
| FaPEG‐MnMSN | Mesoporous silica | Sorafenib | Promote Fenton reaction, SLC7A11 inhibition and GSH depletion | [168] |
| MoS2, WS2 | Metal sulfide | None | GPX4 inhibition, Oxygen free radical generation, Lipid peroxidation | [169] |
| TA‐Fe/ART@ZIF | MOFs | Artemisinin, Fe2+ | Promote Fenton reaction | [170] |
| RSL3@mPEG‐PLys‐AA | Polymer | RSL3 | Promote Fenton reaction, GPX4 inhibition, Lipid peroxidation | [171] |
| SRF@Hb‐Ce6 | Protein | Sorafenib | GPX4 inhibition | [172] |
| LDL‐DHA | Self‐assembled nanoparticles | Omega‐3 fatty acid, docosahexaenoic acid | GSH depletion, GPX4 inhibition, Lipid peroxidation | [173] |
| UCNP@LP(Azo‐CA4) | Upconversion material | Azobenzene combretastatin A4 | Promote Fenton reaction | [174] |
| BNP@R | Phenylboronate ester | RSL‐3 | Induces the secretion of IFN‐γ and inhibits GPX4 | [175] |
| Erastin@FA‐exo | Exosome | Erastin | GPX4 inhibition and ROS overloading | [176] |
| MnO2@HMCu2‐xS | Nanocomposites | Mn2+,rapamycin | GSH exhaustion and ROS overloading | [177] |
| Ascorbate plus nanocarrier loading Fe3+ and RSL3 | Lipid‐coated calcium phosphate | Ascorbate, iron, RSL3 |
Iron overload, GPX4 inhibitions, ROS overgeneration |
[178] |
Abbreviations: COF, covalent organic frameworks; MOF, metal–organic frameworks; RSL3, oncogenic‐RAS‐selective lethal compounds; ROS, reactive oxygen species; GPX4, glutathione peroxidase 4; SLC7A11, cystine/glutamate antiporter solute carrier family 7 member 11; IFN‐γ, interferon‐gamma.
To summarize, drugs related to ferroptosis mainly exert their clinical functions in the following ways: (1) inhibition of the XC‐glutathione/GPX4 axis by regulating antioxidants; (2) regulating the p62‐Keap1‐NRF2 pathway and the downstream antioxidant gene expression of NRF2; and (3) activation of the iron axis to trigger ferroptosis by regulating the functions of lysosomes, ferritin, transferrin, and iron autophagosomes. Previous research provides a comprehensive review of the drugs that induce ferroptosis and lays the foundation for future clinical treatment of cancer.
6. FERROPTOSIS AND TUMOR IMMUNOTHERAPY
Traditional tumor treatment methods, including chemotherapy and radiotherapy, are not effective in tumor treatment because of the broad spectrum of their targets and tumor cell tolerance. The response rate of tumor immunotherapy to many tumors is still limited; consequently, cancer treatment still needs development, and recent research has revealed that ferroptosis has a certain degree of crosstalk with radiotherapy and the immune system, combined with the fragility of tumor cell metabolism and the difference in sensitivity brought about by different cell states [155, 156, 157, 158, 159, 160, 161, 162, 163, 164, 165, 166, 167, 168, 169, 170, 171, 172, 173, 174, 175, 176, 177, 178, 179]. Therefore, this review proposes six potential methods for the treatment of cancer, including the combination of ferroptosis and tumor immune checkpoint inhibitor therapy; using ferroptosis to activate tumor immune cells and reverse resistance to radiotherapy; targeting tumor metabolic features and specifically inducing ferroptosis; using ferroptosis to induce innate transformation of anti‐cancer macrophages; inducing damage‐related molecular pattern generation and targeting plasticity in cancer cell differentiation.(Figure 5).
FIGURE 5.
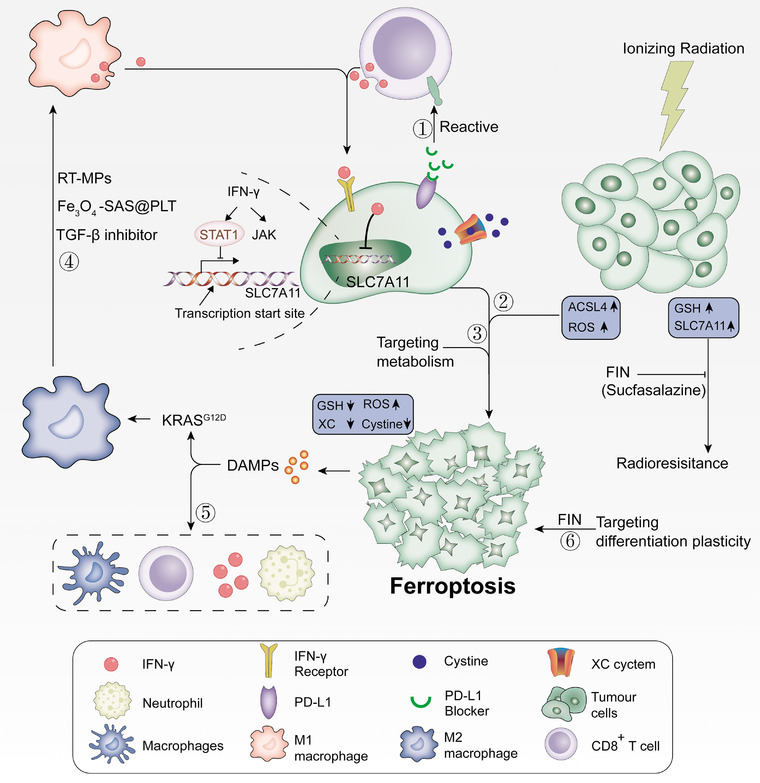
Ferroptosis and tumor immunotherapy. CD8+ T cells inhibit tumor cell cystine uptake by downregulating SLC3A2 and SLC7A11 by releasing IFNγ, and at the same time assisting immune checkpoint inhibitor PD‐L1, which can synergistically enhance T cell‐mediated anti‐tumor immunity and induce tumor cell ferroptosis. Ferroptosis is dependent on glucose and glutamine, and selectively targeting tumor cells by hindering metabolism has a bright future. The DAMPs released in vitro by cancer cells undergoing ferroptosis can induce the maturation of dendritic cells, cross‐induction of CD8+ T cells, production of IFN‐γ, and production of M2 macrophages. Subsequently, it activates adaptability in the tumor microenvironment, forming a positive feedback of the immune response. Radiotherapy produces a large amount of ROS by upregulating ACSL4 and inactivating SLC7A11 or GPX4 with ferroptosis inducers (FINs) at the same time, which makes radiation‐resistant cancer cells sensitive to radiotherapy, reversing the resistance to radiotherapy. Different differentiation stages of cancer cells have different sensitivities to ferroptosis, and targeted therapy can be implemented according to the differentiation state of the sensitive stage of ferroptosis induced by the differentiation plasticity of cancer cells. Abbreviations: RT‐MP, irradiated tumor cell‐released microparticles; ACSL4, acyl‐CoA synthetase long‐chain family member 4; DAMP, damage‐related molecular patterns; FIN, ferroptosis‐inducer; GPX4, glutathione peroxidase 4; GSH, glutathione; IFNγ, interferon‐gamma; PD‐L1, programmed death 1 ligand; ROS, reactive oxygen species; SLC3A2, solute carrier family 3 member 2; SLC7A11, cystine/glutamate antiporter solute carrier family 7 member 11; TGF‐β, transforming growth factor‐beta
6.1. Tumor immune checkpoint inhibitor therapy
Traditional NK(natural killer cell) cells and innate lymphoid cells interact with cancer cells through perforin, TNF family death receptors (such as TRAIL), and immune checkpoint receptors (such as programmed cell death protein 1 [PD‐1]) [180]. PD‐1 is lacking on resting T cells, while high levels of PD‐1 expression are detected in tumor‐infiltrating T cells, indicating that immune‐induced tumor PD‐L1 expression may be an adaptive drug resistance mechanism for tumor cells to respond to immune challenges [181, 182, 183]. Studies have revealed that interferon has the dual function of activating and suppressing the immune system. Interferon signals in cancer cells and immune cells oppose each other to establish a regulatory relationship that limits adaptability and natural immune killing. Therefore, blocking PD‐1 inhibitors in breast, melanoma, and CRC cells can block the interferon signal (IFNγ) and enhance the abundance of CD8+ T cells, and ultimately activate the immune system to attack cancer cells [184]. However, due to the complexity of the tumor microenvironment and insufficient activation of the host immune system, the systemic antitumor response rate of immune checkpoint blocking therapy for many cancers is still limited (Figure 5) [185, 186]. Ferroptosis‐related lipid peroxides can be used as an identifying signal to promote the recognition, phagocytosis and processing of tumor antigens by dendritic cells, and present tumor‐associated antigens to CD8+ T lymphocytes, activating cytotoxic T lymphocytes to enhance tumor immunotherapy. Selective enrichment of RSL‐3‐containing nanoparticles in tumor tissues can induce the immunogenic death of tumor cells, initiate antitumor immune responses and induce cytotoxic T lymphocytes to secrete IFN‐γ. More importantly, IFN‐γ and RSL‐3 can block the lipid peroxide repair pathway and increase the accumulation of lipid peroxide in tumor cells. After further treatment with immune checkpoints, the infiltration of cytotoxic T lymphocytes in tumor tissues increased by four times, effectively inhibiting tumor growth and metastasis [175]. This shows that tumor immune checkpoint inhibitor therapy based on tumor cell ferroptosis is expected to provide a new strategy for improving ferroptosis‐mediated tumor immunotherapy.
6.2. Activating tumor immune cells and reversing radiotherapy resistance
Immunotherapy is a method of treating cancer by generating or enhancing the immune response to cancer [187]. CD8+ T cells activated by immunotherapy are traditionally thought to be through (i) perforating granzyme and (ii) Fas‐FasL induces tumor cell death [188]. Studies have also established that Janus kinase (JAK) and signal transducer activator of transcription 1 (STAT1) mediate IFNγ signal activation and regulate immune responses [189]. Further research found that CD8+ T cell‐derived IFN‐γ increased the binding of STAT1 to the transcription start site of SLC7A11 and inhibited its transcription. Therefore, the lack of STAT1 in tumor cells eliminates IFN‐γ‐mediated downregulation of SLC7A11 and simultaneously reverses RSL3‐induced lipid peroxidation and ferroptosis [190]. Erastin and RSL3 promote ferroptosis in cancer cells, except in human and mouse CD8+ T cells [191], suggesting that tumor cells and T cells may have different susceptibilities to ferroptosis inducers. When mice are treated with immune checkpoint inhibitors and ferroptosis inducers, they can produce a strong immune response and promote ferroptosis against tumors [190].
Radiotherapy was once considered to be one of the most vital tumor treatment methods [191, 192]. However, radiation resistance is still the main factor leading to the failure of radiation therapy, accompanied by tumor recurrence and metastasis; therefore, it is used in combination with specific chemical drugs to sensitize patients to radiotherapy. Studies have indicated that ferroptosis may crosstalk with radiotherapy. For example, radiotherapy can cause mutations in the KEAP1 gene, which upregulates the expression of SLC7A11, subsequently triggering cell resistance to ferroptosis and radiotherapy [118]. In addition, the combination of radiotherapy and immunotherapy increases cell sensitivity to ferroptosis, and cytoplasmic radiation instead of nuclear radiation has a synergistic effect with ferroptosis inducers. These treatments promote cell death by increasing sugar absorption and inducing peroxidation, rather than the typical DNA‐damaging effect [193].
Recent studies have indicated that effector T cells and radiotherapy interact through ferroptosis to promote tumor clearance. CD8+ T cells activated by immunotherapy increase lipid peroxidation and ferroptosis through IFNγ, the latter directly sensitizing tumor cells to radiation therapy [194]. Radiotherapy activates the expression of telangiectasia mutant gene ATM to inhibit the expression of SLC7A11, limiting the uptake of tumor cystine, reducing glutathione, increasing the level of lipid peroxidation, and mediating tumor cell ferroptosis [194]. In addition, radiotherapy produces a large amount of ROS and reduces the level of ferroptosis marker 4‐HNE; combining FINs to inhibit SLC7A11 or GPX4 significantly enhances the efficacy of radiotherapy and reverses radiation resistance [118], indicating that radiotherapy combined with ferroptosis‐inducer FINs and immune checkpoint inhibitors may be a new strategy to eliminate radiotherapy resistance (Figure 5). It is noteworthy that ferroptosis is related to the adverse events of radiation, such as pulmonary fibrosis and granulocyte‐macrophage hematopoietic progenitor cell death [195, 196].
6.3. Targeting tumor metabolism addiction
Maintaining the Warburg effect of tumor metabolism requires higher glucose uptake and metabolic activity, making tumor cells heavily dependent on antioxidant mechanisms; therefore, they are more susceptible to oxidative stress [122]. Moreover, tumor cells consume glucose and glutamine at a higher rate than normal cells, which is called glucose and glutamine addiction. New treatment methods may selectively target tumor cells by blocking their metabolism [197]. For example, the metabolic pressure of mitochondria increases the ROS level of T cells under hypoxia, causing severe T cell dysfunction and exhaustion, thereby, reducing the internal ROS of T cells and alleviating the hypoxic environment of tumor cells effectively blocks T cell immune failure and achieves a synergistic anticancer effect with tumor immunotherapy [198].
Cancer cells with high SLC7A11 expression are more sensitive to glutamine starvation and exhibit glucose‐PPP dependence. Using glucose transporter inhibitors to limit the glucose in SLC7A11 highly expressing cancer cells leads to NADPH depletion and redox system breakdown, causing intracellular cystine accumulation and ferroptosis [199]. Aspartic acid undergoes transamination with GOT1(aspartate aminotransaminase) to produce oxaloacetate, which undergoes downstream biochemical reactions to produce NADPH. NADPH is used to support redox balance in cancer cell proliferation. Knockdown of GOT1 damages the oxidative phosphorylation of the mitochondria, prevents mitochondrial metabolism, increases the unstable iron pool, and increases cell sensitivity to ferroptosis, thereby triggering ferroptosis [96]. Furthermore, the combination of tryptophan depletion and immune checkpoint blockade synergistically enhanced T cell‐mediated antitumor immunity and induced tumor cell ferroptosis [190]. In conclusion, the treatment methods for the characteristics of cancer cell metabolism may include: (1) directly blocking the activity of the SLC7A11 cystine transporter to inhibit cystine absorption, ultimately inducing lipid peroxidation and ferroptosis; (2) inhibiting glucose uptake to target the glucose dependence of SLC7A11 highly expressing cancer cells, leading to rapid cell death caused by nutrient deficiency; (3) in glutamine ‐dependent cancer cells, the growth of cancer cells is inhibited by using glutaminase inhibitors.
6.4. Innate transformation of anticancer macrophages
Recent studies have established that ferroptosis is directly immunogenic or triggers an inflammatory response to other forms of regulatory necrosis in the tumor microenvironment [49]. Tumor‐associated macrophages (TAMs) can exhibit different activation states based on the local microenvironment stimuli. The M1 macrophage phenotype leads to tumor regression and inhibits tumor growth and occurrence [200]. M2 macrophages secrete pro‐angiogenic factors and immunosuppressive cytokines to effectively inhibit the effector functions of T cells and M1 macrophages [201].
The above results indicate that TAMs play an important role in promoting tumor growth and metastasis, and inhibiting the antitumor immune response. Therefore, it is conceivable that the development of anticancer macrophage innate transformation therapy, directly targeting intracellular signaling pathways to increase the ratio of M1/M2, may be another direction for tumor therapy. For example, the protein, KRASG12D, released from pancreatic cancer cells undergoing ferroptosis, is further taken up by macrophages through a specific receptor for hyper‐glycosylation end products, ultimately inducing STAT3‐dependent fatty acid oxidation and driving the formation of M2 macrophages [66]. In addition to sulfadiazine‐loaded magnetic nanoparticles (Fe3O4‐SAS@PLT) [186], combined therapy with microparticles released from tumor cells and radiotherapy with PD‐1 antibody and TGF‐β inhibitor [202], repolarizes M2‐TAMS to the antitumor M1‐TAMS, regulating the antitumor interaction between tumor cells and TAMS [203]. Additionally, the combined treatment of oxygen‐proliferative photosensitizer (PS‐PDT) [204] and strong ferroptosis‐inducer leads to an increase in lipid peroxidation, IFN‐γ secretion, and immunity reshaping of the TME [172]. In addition, M1 macrophages are easier to activate and produce IFN‐γ than M2 macrophages [205] whereas PD‐1 antibody and TGF‐β inhibitors synergistically enhance the immune response in the TME, leading to M1 polarization, and the increase in H2O2 levels in macrophages triggers a Fenton reaction with ions released from the core of the nanoparticle, which subsequently contributes to tumor cell ferroptosis [206]. In summary, anticancer macrophage innate transformation combined with ferroptosis therapy may have broad application prospects.
6.5. Clever use of damage‐related molecular patterns (DAMPs)
Necrotic cancer cells release DAMPs in vitro which bind to receptors in various immune cells to induce the maturation of dendritic cells and the cross‐induction of CD8+ T cells, accompanied by the production of IFN‐γ, subsequently activating the adaptive immune system in the tumor microenvironment and mediating antitumor immunity [207, 208, 209]. In the process of tumorigenesis, ferroptosis has the dual effect of promoting and inhibiting tumors, which mainly depends on the release of DAMPs in the tumor microenvironment. Cell death caused by ferroptosis is characterized by the release of restricted intracellular components, including DAMPs. Once released outside the cell, these components acquire immunostimulatory properties and act as adjuvants, thereby, activating the innate and adaptive immune system by binding to pattern recognition receptors [210]. For example, in a model of cardiac injury, ferroptotic cells attract neutrophils in a TLR4‐TRIF‐dependent manner, which indicates that DAMPs are released when ferroptotic cell death triggers TLR4 signals [211]. These results indicate that cells undergoing ferroptosis secrete factors that strongly activate the innate immune system, as shown in several pathological and genetic models of kidney and brain ferroptosis. Phagocytosis of bone marrow‐derived dendritic cells is activated by three key DAMPs (CRT, HMGB1, and ATP) in cells undergoing ferroptosis [204]. Neutrophils are also recruited through the TLR4/TRIF/1 IFN or Wnt signaling pathways induced by cancer cells undergoing ferroptosis, which are important regulators and potential therapeutic targets against cancer progression [210]. In addition, DAMP, high‐mobility group protein B1 (HMGB1), plays a crucial role in DNA regulation, such as DNA repair, transcription, and replication [212]. Both type I and type II ferroptosis activators, including erastin, sorafenib, RSL3, and FIN56, can induce the release of HMGB1 [213], which in turn interacts with the immune system and regulates cancer treatment, and plays a vital role in the success of the therapy [214]. Therefore, it is speculated that the accumulation of DAMPs can trigger tissue inflammation and regulate ferroptosis in a spontaneous cascade [215], and the delicate use of DAMPs can activate positive immune feedback to inhibit tumor growth.
6.6. Targeting cancer cell differentiation plasticity
There is different drug resistance in cancer cell lines in different growth and differentiation states. For instance, melanoma can be divided into four subtypes, each of which has a specific sensitivity to ferroptosis induction which targets differentiation plasticity to improve the target and provides a treatment method for immunotherapy efficacy [216]. Tumor necrosis factor α (TNFα) and interferon γ cause the dedifferentiation of melanoma cells. Compared with the vector or cytokine control, cell death increased after Erastin or RSL3 combination treatment, which verified that the failure differentiation subtypes were the most sensitive and melanocyte subtypes had the strongest drug resistance [216].
The hypermesenchymal cell status observed in human cancer cell lines is related to the resistance of different cancer lineages to multiple treatment modalities. This treatment‐resistant hyper‐mesenchymal cell status relies on a drug‐targetable lipid peroxidase pathway, characterized by increased lipid peroxidase activity and promotion of polyunsaturated lipid synthesis [108]. Two different types of compounds are closely related to the selective induction of cell death in epithelial cancer cell lines in a high‐mesenchymal state. The first type includes RSL3, ML210 and ML162, which are known to induce ferroptosis. The second category is statins that inhibit 3‐hydroxy‐3‐methyl‐glutaryl‐CoA reductase (HMGCR). The inhibitory effect of statins on HMG‐CoA reductase also consumes CoQ10 and mevalonate pathways, mediating its selectivity for high mesenchymal cancer cells [108]. Therefore, targeting the differentiation plasticity of cancer cell lines and adjusting their different cell states to improve targeting and immunotherapy may be a potential cancer treatment method.
Ferroptosis is a new therapy that depends on iron‐mediated lipids. Several studies have shown that ferroptosis is closely related to chemotherapy and radiotherapy [60, 217]. However, the connection between ferroptosis and cancer immunotherapy is speculative at best because the available evidence is based on very few well‐conducted studies, and only few ferroptosis‐related genes have been subjected to prognostic analyses [218, 219]; moreover, these analyses are unreliable because these genes possess many physiological functions beyond ferroptosis.
Nevertheless, when one treatment strategy is used alone, its systemic antitumor response rate for multiple cancers is limited. Thus, considering the characteristics of cancer cell metabolism and differentiation, we propose that a combination of the abovementioned six ferroptosis‐related methods will be of great significance in cancer treatment.
7. CONCLUSIONS
ROS accumulation exceeding the redox content maintained by the phospholipid catalase in the cell mediates the occurrence of ferroptosis [13]. Previous studies on ferroptosis are based on the use of pharmacological inducers/inhibitors to affect lipid oxidation, iron metabolism, or lipid metabolism, and there is a lack of specific biomarkers for ferroptosis; thus, it is uncertain whether ferroptosis always occurs in cancer, and the corresponding clinical therapies for ferroptosis are still inadequate.
The effectiveness of anticancer drugs to kill cancer cells is inhibited by the non‐targetability of oncoproteins and the necessity of cancer essential genes for normal cells [59]. Therefore, precision medicine based on ferroptosis needs to identify the biomarker of ferroptosis for each cancer patient to achieve subsequent molecular targeted precision therapy. In vitro experiments have shown that cell viability, SLC7A11, ACSL4 and ROS levels [98, 220, 221], the final product of AA and larger PUFA decomposition, 4‐HNE [15, 16, 118], the ER stress response gene CHAC1 which is a downstream target of the eIF2α‐ATF4 pathway [137], prostaglandin peroxidase synthase PTGS2 [54, 210], NADPH abundance [58], and lipid droplet‐associated protein HILPDA activated by hypoxia‐inducible factor HIF‐2α, are biomarkers for predicting tumor cell ferroptosis [222]; however, some of the current fluorescence‐based techniques for lipid peroxide detection accurately and precisely measure cellular lipid peroxides. Still, further research on ferroptosis biomarkers will greatly promote the treatment of related diseases.
Recent studies have shown that metabolic reprogramming is involved in the regulation of ferroptosis, which indicates that tumor cells avoid the occurrence of ferroptosis to a certain extent through metabolic reprogramming. Inhibition of AMPK‐ACC‐PUFA signaling may produce antitumor effects by targeting this pathway in different types of tumors and its niche [123], thereby, enhancing the efficacy of immunotherapies and potentiating chemotherapy treatments. Ferroptosis inhibitors potently increase neuronal reprogramming by inhibiting lipid peroxidation during fate conversion [223] which suggests that ferroptosis is also associated with cell reprogramming. In some cases, this metabolic reprogramming has been linked to an acquired sensitivity to ferroptosis [60, 61]; therefore, regulation of the metabolic pathways controlling ferroptosis might reshape the tumor niche, opening up new opportunities to treat therapy‐insensitive tumors [62]. Furthermore, defining the metabolic mechanisms governing iron and lipid turnover might lead to the discovery of novel therapeutic strategies.
Autophagy plays a vital role in determining the survival of cells under various stresses, and it contributes to ferroptosis through degradation of cellular iron storage proteins (a process known as ferritinophagy), causing cellular iron homeostasis [63]. Moreover, intracellular ferrous iron and lipid peroxidation levels decrease following knockdown of Atg5 (autophagy‐related 5) and Atg7, limiting erastin‐induced ferroptosis [64]. Therefore, ferroptosis is an autophagic cell death. Whether oxidative stress induces autophagy that is dependent on KRASG12D protein [66], the core clock protein ARNT that facilitates ferroptosis induction [69], or mitochondrial reactive oxygen species (MtROS), regulation of iron homeostasis [78] still has a complex relationship with ferroptosis. Taken together, autophagy is of great significance to ferroptosis, but the complex relationship between the two, whether it is interdependence or mutual antagonism, needs to be reexamined.
Combining exosome technology and nano‐drug development to increase free iron or oxides in cells is one of the interesting ways to achieve ferroptosis therapy. Examples of new regimens for the treatment of breast cancer include grafting heparin (NLC/H), DOX, dimer with β‐cyclodextrin (β‐CD) iron, and TGF‐β receptor inhibitor (SB431542) co‐loaded with modified nanoparticles, which can effectively increase the level of intracellular ROS to activate the ferroptosis pathway [82]. Furthermore, exosomes may be able to optimize drug delivery. Various Pt‐free iron oxide nanoparticles are constructed by coating polyethyleneimine and polyethylene glycol. They are transported through the exosome channel in the acidic environment of cancer through absorption and degradation, followed by the release of intracellular iron, enhancing the Fenton response and ROS production, and leading to tumor cell ferroptosis [83]. However, drug‐resistant cells can induce the expression of glutaric acid protein prominin‐2 that regulates lipid motility, thereby stimulating the formation of ferritin‐containing exosomes, transporting iron from the cells, and promoting ferroptosis resistance through iron transport in breast epithelial cells and breast cancer cells [84]. Moreover, drug‐induced increases in intracellular glutathione concentration mediate the drug resistance of tumor cells [97]. Therefore, the double‐edged sword of exosome use in ferroptosis therapy requires more in‐depth research.
Tumor cell metastasis poses a challenge for targeted therapies. For example, tumor cells migrate to lymph nodes to avoid ferroptosis caused by high levels of oxidative stress and iron overload in blood vessels. Moreover, the lymph nodes are rich in lipids, which improves the ability of tumor cells to metabolize fatty acids and use the ACSL3 enzyme to coat a layer of oleic acid molecular protective film on their surface [224]. Thus, when they re‐enter the bloodstream, the amount and density of unsaturated fatty acids in tumor cells are low, implying that the use of ferroptosis to limit tumor metastasis is still unknown.
Recent studies have greatly advanced the knowledge on ferroptosis, such as the negative effect of ferroptosis on tumorigenesis and its potential use in tumor therapy [225]. Moreover, research has revealed that the depletion of GPX4 or SLC7A11 not only causes ferroptosis, but also other types of cell death [226]. The defense mechanism of DHODH‐mediated mitochondrial ferroptosis has also been reported [80]. In addition, the lack of GPX4 enhances cellular lipid peroxidation, specifically inhibiting the cGAS‐STING pathway, which may be a direct connection between GPX4 and immunity [227]. Combination of ferroptosis‐mediated immunotherapies with image‐guided therapy may be a promising method. For example, iron‐chelated semiconductor multi‐composite nanoparticles are used in photothermal ferroptosis treatment under the guidance of photoacoustic imaging [228]. With high temporal and spatial accuracies, easy control, noninvasive property, and minimal side effects, photodynamic therapy is also an attractive method for image‐guided cancer cell ablation [229]. However, further studies are required to explore more inducers of ferroptosis (which may be more useful than inhibitors), clarify the synergistic or antagonistic relationship between ferroptosis and other types of cell death, and develop combined therapies of ferroptosis with exosomes, nanomedicines, and tumor immunotherapies. Detailed information regarding ferroptosis will be gradually discovered in the near future, with an in‐depth understanding of the occurrence, development, and treatment of cancer.
DECLARATIONS
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable
CONSENT FOR PUBLICATION
Not applicable
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
FUNDING
This work was supported by a special program from the Ministry of Science and Technology of China (2016YFA0502500), the Chinese National Natural Science Funds (U20A201376, 31925013, 82041009, 31871405, and 31701232), Jiangsu National Science Foundation (BK20180043) and the Science and Technology Plan Project of Suzhou (SYS2019020).
AUTHORS' CONTRIBUTIONS
L.Z.(Lei Zhao), Xiaoxue Zhou, Feng Xie, and L.Z.(Lei Zhang) conceived and drafted the manuscript, drew the figures, and summarized the tables. L.Z.(Lei Zhao), Xiaoxue Zhou, Feng Xie, L.Z.(Lei Zhang), Fangfang Zhou, and L.Z.(Long Zhang) discussed the concepts of the manuscript. Haiyan Yan, Jun Huang, Chong Zhang and Jun Chen provided valuable suggestion. L.Z.(Long Zhang) and Fangfang Zhou approved the submission of the manuscript L.Z.(Lei Zhao), Xiaoxue Zhou, Feng Xie, and L.Z.(Lei Zhang) contributed equally to this work.
ACKNOWLEDGMENTS
Not applicable.
Zhao L, Zhou X, Xie F, Zhang L, Yan H, Huang J, et al. Ferroptosis in cancer and cancer immunotherapy. Cancer Commun. 2022;42:88–116. 10.1002/cac2.12250
Contributor Information
Chong Zhang, Email: zhangchong@zucc.edu.cn.
Fangfang Zhou, Email: zhoufangfang@suda.edu.cn.
Jun Chen, Email: zjdxcj@126.com.
Long Zhang, Email: zhanglong.2003@tsinghua.org.cn.
DATA AVAILABILITY STATEMENT
Not applicable
REFERENCES
- 1. Allocati N, Masulli M, Di Ilio C, De Laurenzi V. Die for the community: an overview of programmed cell death in bacteria. Cell Death Dis. 2015;6(1):e1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. 2011;147(4):742‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25(3):486‐541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7(2):99‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang Y, Dawson VL, Dawson TM. Poly(ADP‐ribose) signals to mitochondrial AIF: a key event in parthanatos. Exp Neurol. 2009;218(2):193‐202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11(10):700‐14. [DOI] [PubMed] [Google Scholar]
- 7. Yuan J, Kroemer G. Alternative cell death mechanisms in development and beyond. Genes Dev. 2010;24(23):2592‐602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Christofferson DE, Yuan J. Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol. 2010;22(2):263‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vanden Berghe T, Linkermann A, Jouan‐Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non‐apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15(2):135‐47. [DOI] [PubMed] [Google Scholar]
- 10. Su Z, Yang Z, Xie L, DeWitt JP, Chen Y. Cancer therapy in the necroptosis era. Cell Death Differ. 2016;23(5):748‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron‐dependent, nonapoptotic cell death in oncogenic‐RAS‐harboring cancer cells. Chem Biol. 2008;15(3):234‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype‐selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3(3):285‐96. [DOI] [PubMed] [Google Scholar]
- 13. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron‐dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shaw AT, Winslow MM, Magendantz M, Ouyang C, Dowdle J, Subramanian A, et al. Selective killing of K‐ras mutant cancer cells by small molecule inducers of oxidative stress. Proc Natl Acad Sci U S A. 2011;108(21):8773‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ayala A, Muñoz MF, Argüelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4‐hydroxy‐2‐nonenal. Oxid Med Cell Longev. 2014;2014:360438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhong H, Yin H. Role of lipid peroxidation derived 4‐hydroxynonenal (4‐HNE) in cancer: focusing on mitochondria. Redox Biol. 2015;4:193‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23(3):369‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yu H, Guo P, Xie X, Wang Y, Chen G. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J Cell Mol Med. 2017;21(4):648‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guo QQ, Wang SS, Zhang SS, Xu HD, Li XM, Guan Y, et al. ATM‐CHK2‐Beclin 1 axis promotes autophagy to maintain ROS homeostasis under oxidative stress. Embo J. 2020;39(10):e103111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16(1):3‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res. 2019;29(5):347‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sendler M, Mayerle J, Lerch MM. Necrosis, Apoptosis, Necroptosis, Pyroptosis: It Matters How Acinar Cells Die During Pancreatitis. Cell Mol Gastroenterol Hepatol. 2016;2(4):407‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ma D, Lu B, Feng C, Wang C, Wang Y, Luo T, et al. Deoxypodophyllotoxin triggers parthanatos in glioma cells via induction of excessive ROS. Cancer Lett. 2016;371(2):194‐204. [DOI] [PubMed] [Google Scholar]
- 24. Schenk B, Fulda S. Reactive oxygen species regulate Smac mimetic/TNFα‐induced necroptotic signaling and cell death. Oncogene. 2015;34(47):5796‐806. [DOI] [PubMed] [Google Scholar]
- 25. Zhang Y, Su SS, Zhao S, Yang Z, Zhong CQ, Chen X, et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun. 2017;8:14329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31(2):107‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22(4):266‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014;10(1):9‐17. [DOI] [PubMed] [Google Scholar]
- 29. Sullivan LB, Chandel NS. Mitochondrial reactive oxygen species and cancer. Cancer and Metab. 2014;2(1):17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yan B, Ai Y, Sun Q, Ma Y, Cao Y, Wang J, et al. Membrane Damage during Ferroptosis Is Caused by Oxidation of Phospholipids Catalyzed by the Oxidoreductases POR and CYB5R1. Mol Cell. 2021;81(2):355‐69. e10. [DOI] [PubMed] [Google Scholar]
- 31. Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W, et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol. 2020;16(3):302‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, et al. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep. 2017;20(7):1692‐704. [DOI] [PubMed] [Google Scholar]
- 33. Papa S, Martino PL, Capitanio G, Gaballo A, De Rasmo D, Signorile A, et al. The oxidative phosphorylation system in mammalian mitochondria. Adv Exp Med Biol. 2012;942:3‐37. [DOI] [PubMed] [Google Scholar]
- 34. Cheng Z, Li Y. What is responsible for the initiating chemistry of iron‐mediated lipid peroxidation: an update. Chem Rev. 2007;107(3):748‐66. [DOI] [PubMed] [Google Scholar]
- 35. Brigelius‐Flohé R, Maiorino M. Glutathione peroxidases. Biochim Biophys Acta. 2013;1830(5):3289‐303. [DOI] [PubMed] [Google Scholar]
- 36. Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell. 2019;35(6):830‐49. [DOI] [PubMed] [Google Scholar]
- 37. Terman A, Kurz T. Lysosomal iron, iron chelation, and cell death. Antioxid Redox Signal. 2013;18(8):888‐98. [DOI] [PubMed] [Google Scholar]
- 38. Conrad M, Kagan VE, Bayir H, Pagnussat GC, Head B, Traber MG, et al. Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev. 2018;32(9‐10):602‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yin H, Xu L, Porter NA. Free radical lipid peroxidation: mechanisms and analysis. Chem Rev. 2011;111(10):5944‐72. [DOI] [PubMed] [Google Scholar]
- 40. Imai H, Nakagawa Y. Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radic Biol Med. 2003;34(2):145‐69. [DOI] [PubMed] [Google Scholar]
- 41. Feng H, Stockwell BR. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018;16(5):e2006203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Haeggström JZ, Funk CD. Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chem Rev. 2011;111(10):5866‐98. [DOI] [PubMed] [Google Scholar]
- 43. Wenzel SE, Tyurina YY, Zhao J, St Croix CM, Dar HH, Mao G, et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell. 2017;171(3):628‐41. e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, et al. Caspase‐11 cleaves gasdermin D for non‐canonical inflammasome signalling. Nature. 2015;526(7575):666‐71. [DOI] [PubMed] [Google Scholar]
- 45. Agmon E, Solon J, Bassereau P, Stockwell BR. Modeling the effects of lipid peroxidation during ferroptosis on membrane properties. Sci Rep. 2018;8(1):5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Qiu Y, Cao Y, Cao W, Jia Y, Lu N. The Application of Ferroptosis in Diseases. Pharmacol Res. 2020;159:104919. [DOI] [PubMed] [Google Scholar]
- 47. Guan J, Lo M, Dockery P, Mahon S, Karp CM, Buckley AR, et al. The xc‐ cystine/glutamate antiporter as a potential therapeutic target for small‐cell lung cancer: use of sulfasalazine. Cancer Chemother Pharmacol. 2009;64(3):463‐72. [DOI] [PubMed] [Google Scholar]
- 48. Hatem E, El Banna N, Huang ME. Multifaceted Roles of Glutathione and Glutathione‐Based Systems in Carcinogenesis and Anticancer Drug Resistance. Antioxid Redox Signal. 2017;27(15):1217‐34. [DOI] [PubMed] [Google Scholar]
- 49. Bebber CM, Müller F, Prieto Clemente L, Weber J, von Karstedt S. Ferroptosis in Cancer Cell Biology. Cancers (Basel). 2020;12(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ishii T, Sugita Y, Bannai S. Regulation of glutathione levels in mouse spleen lymphocytes by transport of cysteine. J Cell Physiol. 1987;133(2):330‐6. [DOI] [PubMed] [Google Scholar]
- 51. Conrad M, Sato H. The oxidative stress‐inducible cystine/glutamate antiporter, system x (c) (‐) : cystine supplier and beyond. Amino Acids. 2012;42(1):231‐46. [DOI] [PubMed] [Google Scholar]
- 52. Wang H, An P, Xie E, Wu Q, Fang X, Gao H, et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology. 2017;66(2):449‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fang X, Cai Z, Wang H, Han D, Cheng Q, Zhang P, et al. Loss of Cardiac Ferritin H Facilitates Cardiomyopathy via Slc7a11‐Mediated Ferroptosis. Circ Res. 2020;127(4):486‐501. [DOI] [PubMed] [Google Scholar]
- 54. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1‐2):317‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, et al. RAS‐RAF‐MEK‐dependent oxidative cell death involving voltage‐dependent anion channels. Nature. 2007;447(7146):864‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Alim I, Caulfield JT, Chen Y, Swarup V, Geschwind DH, Ivanova E, et al. Selenium Drives a Transcriptional Adaptive Program to Block Ferroptosis and Treat Stroke. Cell. 2019;177(5):1262‐79. e25. [DOI] [PubMed] [Google Scholar]
- 57. Hassannia B, Wiernicki B, Ingold I, Qu F, Van Herck S, Tyurina YY, et al. Nano‐targeted induction of dual ferroptotic mechanisms eradicates high‐risk neuroblastoma. J Clin Invest. 2018;128(8):3341‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shimada K, Hayano M, Pagano NC, Stockwell BR. Cell‐Line Selectivity Improves the Predictive Power of Pharmacogenomic Analyses and Helps Identify NADPH as Biomarker for Ferroptosis Sensitivity. Cell Chem Biol. 2016;23(2):225‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. 2016;12(7):497‐503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Friedmann Angeli JP, Krysko DV, Conrad M. Ferroptosis at the crossroads of cancer‐acquired drug resistance and immune evasion. Nat Rev Cancer. 2019;19(7):405‐14. [DOI] [PubMed] [Google Scholar]
- 61. Koppula P, Zhuang L, Gan B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell. 2021;12(8):599‐620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wu Y, Zhang S, Gong X, Tam S, Xiao D, Liu S, et al. The epigenetic regulators and metabolic changes in ferroptosis‐associated cancer progression. Mol Cancer. 2020;19(1):39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26(9):1021‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ, 3rd , et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12(8):1425‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Anderton B, Camarda R, Balakrishnan S, Balakrishnan A, Kohnz RA, Lim L, et al. MYC‐driven inhibition of the glutamate‐cysteine ligase promotes glutathione depletion in liver cancer. EMBO Rep. 2017;18(4):569‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Dai E, Han L, Liu J, Xie Y, Kroemer G, Klionsky DJ, et al. Autophagy‐dependent ferroptosis drives tumor‐associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy. 2020;16(11):2069‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhou B, Liu J, Kang R, Klionsky DJ, Kroemer G, Tang D. Ferroptosis is a type of autophagy‐dependent cell death. Semin Cancer Biol. 2020;66:89‐100. [DOI] [PubMed] [Google Scholar]
- 68. Hao S, Yu J, He W, Huang Q, Zhao Y, Liang B, et al. Cysteine Dioxygenase 1 Mediates Erastin‐Induced Ferroptosis in Human Gastric Cancer Cells. Neoplasia. 2017;19(12):1022‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yang M, Chen P, Liu J, Zhu S, Kroemer G, Klionsky DJ, et al. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci Adv. 2019;5(7):eaaw2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53‐mediated activity during tumour suppression. Nature. 2015;520(7545):57‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wang L, Liu Y, Du T, Yang H, Lei L, Guo M, et al. ATF3 promotes erastin‐induced ferroptosis by suppressing system Xc(). Cell Death Differ. 2020;27(2):662‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, et al. AMPK‐Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System X(c)(‐) Activity. Curr Biol. 2018;28(15):2388‐99. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhang Y, Shi J, Liu X, Feng L, Gong Z, Koppula P, et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol. 2018;20(10):1181‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chen D, Fan Z, Rauh M, Buchfelder M, Eyupoglu IY, Savaskan N. ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT‐dependent manner. Oncogene. 2017;36(40):5593‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Gan W, Dai X, Dai X, Xie J, Yin S, Zhu J, et al. LATS suppresses mTORC1 activity to directly coordinate Hippo and mTORC1 pathways in growth control. Nat Cell Biol. 2020;22(2):246‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Liu T, Jiang L, Tavana O, Gu W. The Deubiquitylase OTUB1 Mediates Ferroptosis via Stabilization of SLC7A11. Cancer Res. 2019;79(8):1913‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ye P, Mimura J, Okada T, Sato H, Liu T, Maruyama A, et al. Nrf2‐ and ATF4‐dependent upregulation of xCT modulates the sensitivity of T24 bladder carcinoma cells to proteasome inhibition. Mol Cell Biol. 2014;34(18):3421‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wei S, Qiu T, Yao X, Wang N, Jiang L, Jia X, et al. Arsenic induces pancreatic dysfunction and ferroptosis via mitochondrial ROS‐autophagy‐lysosomal pathway. J Hazard Mater. 2020;384:121390. [DOI] [PubMed] [Google Scholar]
- 79. Soula M, Weber RA, Zilka O, Alwaseem H, La K, Yen F, et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol. 2020;16(12):1351‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. DHODH‐mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593(7860):586‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Garcia‐Bermudez J, Birsoy K. A mitochondrial gatekeeper that helps cells escape death by ferroptosis. Nature. 2021;593(7860):514‐5. [DOI] [PubMed] [Google Scholar]
- 82. Zhang J, Yang J, Zuo T, Ma S, Xokrat N, Hu Z, et al. Heparanase‐driven sequential released nanoparticles for ferroptosis and tumor microenvironment modulations synergism in breast cancer therapy. Biomaterials. 2021;266:120429. [DOI] [PubMed] [Google Scholar]
- 83. Ma P, Xiao H, Yu C, Liu J, Cheng Z, Song H, et al. Enhanced Cisplatin Chemotherapy by Iron Oxide Nanocarrier‐Mediated Generation of Highly Toxic Reactive Oxygen Species. Nano Lett. 2017;17(2):928‐37. [DOI] [PubMed] [Google Scholar]
- 84. Brown CW, Amante JJ, Chhoy P, Elaimy AL, Liu H, Zhu LJ, et al. Prominin2 Drives Ferroptosis Resistance by Stimulating Iron Export. Dev Cell. 2019;51(5):575‐86. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione‐independent ferroptosis suppressor. Nature. 2019;575(7784):693‐8. [DOI] [PubMed] [Google Scholar]
- 86. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575(7784):688‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Yi J, Zhu J, Wu J, Thompson CB, Jiang X. Oncogenic activation of PI3K‐AKT‐mTOR signaling suppresses ferroptosis via SREBP‐mediated lipogenesis. Proc Natl Acad Sci U S A. 2020;117(49):31189‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zhao Y, Li M, Yao X, Fei Y, Lin Z, Li Z, et al. HCAR1/MCT1 Regulates Tumor Ferroptosis through the Lactate‐Mediated AMPK‐SCD1 Activity and Its Therapeutic Implications. Cell Rep. 2020;33(10):108487. [DOI] [PubMed] [Google Scholar]
- 89. Dowdle WE, Nyfeler B, Nagel J, Elling RA, Liu S, Triantafellow E, et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol. 2014;16(11):1069‐79. [DOI] [PubMed] [Google Scholar]
- 90. Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509(7498):105‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Chang LC, Chiang SK, Chen SE, Yu YL, Chou RH, Chang WC. Heme oxygenase‐1 mediates BAY 11‐7085 induced ferroptosis. Cancer Lett. 2018;416:124‐37. [DOI] [PubMed] [Google Scholar]
- 92. Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306(5704):2090‐3. [DOI] [PubMed] [Google Scholar]
- 93. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol Cell. 2015;59(2):298‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Alvarez SW, Sviderskiy VO, Terzi EM, Papagiannakopoulos T, Moreira AL, Adams S, et al. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature. 2017;551(7682):639‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Louandre C, Ezzoukhry Z, Godin C, Barbare JC, Mazière JC, Chauffert B, et al. Iron‐dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int J Cancer. 2013;133(7):1732‐42. [DOI] [PubMed] [Google Scholar]
- 96. Kremer DM, Nelson BS, Lin L, Yarosz EL, Halbrook CJ, Kerk SA, et al. GOT1 inhibition promotes pancreatic cancer cell death by ferroptosis. Nat Commun. 2021;12(1):4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Mohell N, Alfredsson J, Fransson Å, Uustalu M, Byström S, Gullbo J, et al. APR‐246 overcomes resistance to cisplatin and doxorubicin in ovarian cancer cells. Cell Death Dis. 2015;6(6):e1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Yuan H, Li X, Zhang X, Kang R, Tang D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun. 2016;478(3):1338‐43. [DOI] [PubMed] [Google Scholar]
- 99. Sun WY, Tyurin VA, Mikulska‐Ruminska K, Shrivastava IH, Anthonymuthu TS, Zhai YJ, et al. Phospholipase iPLA(2)β averts ferroptosis by eliminating a redox lipid death signal. Nat Chem Biol. 2021;17(4):465‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR, et al. Intercellular interaction dictates cancer cell ferroptosis via NF2‐YAP signalling. Nature. 2019;572(7769):402‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Zou Y, Henry WS, Ricq EL, Graham ET, Phadnis VV, Maretich P, et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature. 2020;585(7826):603‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. McGivan JD, Bungard CI. The transport of glutamine into mammalian cells. Front Biosci. 2007;12:874‐82. [DOI] [PubMed] [Google Scholar]
- 103. Kang YP, Mockabee‐Macias A, Jiang C, Falzone A, Prieto‐Farigua N, Stone E, et al. Non‐canonical Glutamate‐Cysteine Ligase Activity Protects against Ferroptosis. Cell Metab. 2021;33(1):174‐89. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide‐Induced Ferroptosis. Cell. 2018;172(3):409‐22. e21. [DOI] [PubMed] [Google Scholar]
- 105. Gao M, Jiang X. To eat or not to eat‐the metabolic flavor of ferroptosis. Curr Opin Cell Biol. 2018;51:58‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, et al. Role of Mitochondria in Ferroptosis. Mol Cell. 2019;73(2):354‐63. e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Garcia‐Bermudez J, Baudrier L, Bayraktar EC, Shen Y, La K, Guarecuco R, et al. Squalene accumulation in cholesterol auxotrophic lymphomas prevents oxidative cell death. Nature. 2019;567(7746):118‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore‐Ludlow B, et al. Dependency of a therapy‐resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547(7664):453‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Muñoz M, García‐Erce JA, Remacha AF. Disorders of iron metabolism. Part 1: molecular basis of iron homoeostasis. J Clin Pathol. 2011;64(4):281‐6. [DOI] [PubMed] [Google Scholar]
- 110. Yanatori I, Kishi F. DMT1 and iron transport. Free Radic Biol Med. 2019;133:55‐63. [DOI] [PubMed] [Google Scholar]
- 111. Yu Y, Jiang L, Wang H, Shen Z, Cheng Q, Zhang P, et al. Hepatic transferrin plays a role in systemic iron homeostasis and liver ferroptosis. Blood. 2020;136(6):726‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Hong X, Roh W, Sullivan RJ, Wong KHK, Wittner BS, Guo H, et al. The Lipogenic Regulator SREBP2 Induces Transferrin in Circulating Melanoma Cells and Suppresses Ferroptosis. Cancer Discov. 2021;11(3):678‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Bradley JM, Le Brun NE, Moore GR. Ferritins: furnishing proteins with iron. J Biol Inorg Chem. 2016;21(1):13‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Fang X, Wang H, Han D, Xie E, Yang X, Wei J, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. 2019;116(7):2672‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Tang Z, Xu Z, Zhu X, Zhang J. New insights into molecules and pathways of cancer metabolism and therapeutic implications. Cancer Commun (Lond). 2021;41(1):16‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13(1):81‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Bai Y, Meng L, Han L, Jia Y, Zhao Y, Gao H, et al. Lipid storage and lipophagy regulates ferroptosis. Biochem Biophys Res Commun. 2019;508(4):997‐1003. [DOI] [PubMed] [Google Scholar]
- 118. Lei G, Zhang Y, Koppula P, Liu X, Zhang J, Lin SH, et al. The role of ferroptosis in ionizing radiation‐induced cell death and tumor suppression. Cell Res. 2020;30(2):146‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Zhao Y, Chen YQ, Bonacci TM, Bredt DS, Li S, Bensch WR, et al. Identification and characterization of a major liver lysophosphatidylcholine acyltransferase. J Biol Chem. 2008;283(13):8258‐65. [DOI] [PubMed] [Google Scholar]
- 120. Hashidate‐Yoshida T, Harayama T, Hishikawa D, Morimoto R, Hamano F, Tokuoka SM, et al. Fatty acid remodeling by LPCAT3 enriches arachidonate in phospholipid membranes and regulates triglyceride transport. Elife. 2015;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. 2016;113(34):E4966‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Warburg O, Minami S. Versuche an Überlebendem Carcinom‐gewebe. Klinische Wochenschrift. 1923;2(17):776‐7. [Google Scholar]
- 123. Lee H, Zandkarimi F, Zhang Y, Meena JK, Kim J, Zhuang L, et al. Energy‐stress‐mediated AMPK activation inhibits ferroptosis. Nat Cell Biol. 2020;22(2):225‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Bertolio R, Napoletano F, Mano M, Maurer‐Stroh S, Fantuz M, Zannini A, et al. Sterol regulatory element binding protein 1 couples mechanical cues and lipid metabolism. Nat Commun. 2019;10(1):1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Kim J, Guan KL. mTOR as a central hub of nutrient signalling and cell growth. Nat Cell Biol. 2019;21(1):63‐71. [DOI] [PubMed] [Google Scholar]
- 126. Gu Y, Albuquerque CP, Braas D, Zhang W, Villa GR, Bi J, et al. mTORC2 Regulates Amino Acid Metabolism in Cancer by Phosphorylation of the Cystine‐Glutamate Antiporter xCT. Mol Cell. 2017;67(1):128‐38. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Sporn MB, Liby KT. NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer. 2012;12(8):564‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the p62‐Keap1‐NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63(1):173‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Anandhan A, Dodson M, Schmidlin CJ, Liu P, Zhang DD. Breakdown of an Ironclad Defense System: The Critical Role of NRF2 in Mediating Ferroptosis. Cell Chem Biol. 2020;27(4):436‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Dreger H, Westphal K, Weller A, Baumann G, Stangl V, Meiners S, et al. Nrf2‐dependent upregulation of antioxidative enzymes: a novel pathway for proteasome inhibitor‐mediated cardioprotection. Cardiovasc Res. 2009;83(2):354‐61. [DOI] [PubMed] [Google Scholar]
- 131. Zhao B, Lei QY, Guan KL. The Hippo‐YAP pathway: new connections between regulation of organ size and cancer. Curr Opin Cell Biol. 2008;20(6):638‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. van Roy F, Berx G. The cell‐cell adhesion molecule E‐cadherin. Cell Mol Life Sci. 2008;65(23):3756‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Kim NG, Koh E, Chen X, Gumbiner BM. E‐cadherin mediates contact inhibition of proliferation through Hippo signaling‐pathway components. Proc Natl Acad Sci U S A. 2011;108(29):11930‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Guo J, Xu B, Han Q, Zhou H, Xia Y, Gong C, et al. Ferroptosis: A Novel Anti‐tumor Action for Cisplatin. Cancer Res Treat. 2018;50(2):445‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Ma S, Henson ES, Chen Y, Gibson SB. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016;7(7):e2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Yamaguchi Y, Kasukabe T, Kumakura S. Piperlongumine rapidly induces the death of human pancreatic cancer cells mainly through the induction of ferroptosis. Int J Oncol. 2018;52(3):1011‐22. [DOI] [PubMed] [Google Scholar]
- 137. Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine‐glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 2014;3:e02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Zhang X, Sui S, Wang L, Li H, Zhang L, Xu S, et al. Inhibition of tumor propellant glutathione peroxidase 4 induces ferroptosis in cancer cells and enhances anticancer effect of cisplatin. J Cell Physiol. 2020;235(4):3425‐37. [DOI] [PubMed] [Google Scholar]
- 139. Chen GQ, Benthani FA, Wu J, Liang D, Bian ZX, Jiang X. Artemisinin compounds sensitize cancer cells to ferroptosis by regulating iron homeostasis. Cell Death Differ. 2020;27(1):242‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Hung MS, Chen IC, Lee CP, Huang RJ, Chen PC, Tsai YH, et al. Statin improves survival in patients with EGFR‐TKI lung cancer: A nationwide population‐based study. PLoS One. 2017;12(2):e0171137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Xu T, Ding W, Ji X, Ao X, Liu Y, Yu W, et al. Molecular mechanisms of ferroptosis and its role in cancer therapy. J Cell Mol Med. 2019;23(8):4900‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Chen L, Li X, Liu L, Yu B, Xue Y, Liu Y. Erastin sensitizes glioblastoma cells to temozolomide by restraining xCT and cystathionine‐γ‐lyase function. Oncol Rep. 2015;33(3):1465‐74. [DOI] [PubMed] [Google Scholar]
- 143. Roh JL, Kim EH, Jang H, Shin D. Aspirin plus sorafenib potentiates cisplatin cytotoxicity in resistant head and neck cancer cells through xCT inhibition. Free Radic Biol Med. 2017;104:1‐9. [DOI] [PubMed] [Google Scholar]
- 144. Sun X, Niu X, Chen R, He W, Chen D, Kang R, et al. Metallothionein‐1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology. 2016;64(2):488‐500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Louandre C, Marcq I, Bouhlal H, Lachaier E, Godin C, Saidak Z, et al. The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett. 2015;356(2 Pt B):971‐7. [DOI] [PubMed] [Google Scholar]
- 146. Fearon ER. Molecular genetics of colorectal cancer. Ann N Y Acad Sci. 2010;768(1):101‐10. [DOI] [PubMed] [Google Scholar]
- 147. Ma MZ, Chen G, Wang P, Lu WH, Zhu CF, Song M, et al. Xc‐ inhibitor sulfasalazine sensitizes colorectal cancer to cisplatin by a GSH‐dependent mechanism. Cancer Lett. 2015;368(1):88‐96. [DOI] [PubMed] [Google Scholar]
- 148. Sui X, Zhang R, Liu S, Duan T, Zhai L, Zhang M, et al. RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front Pharmacol. 2018;9:1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Burris HA, 3rd , Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, et al. Improvements in survival and clinical benefit with gemcitabine as first‐line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15(6):2403‐13. [DOI] [PubMed] [Google Scholar]
- 150. Lo M, Ling V, Low C, Wang YZ, Gout PW. Potential use of the anti‐inflammatory drug, sulfasalazine, for targeted therapy of pancreatic cancer. Curr Oncol. 2010;17(3):9‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Eling N, Reuter L, Hazin J, Hamacher‐Brady A, Brady NR. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience. 2015;2(5):517‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Basuli D, Tesfay L, Deng Z, Paul B, Yamamoto Y, Ning G, et al. Iron addiction: a novel therapeutic target in ovarian cancer. Oncogene. 2017;36(29):4089‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Roh JL, Kim EH, Jang HJ, Park JY, Shin D. Induction of ferroptotic cell death for overcoming cisplatin resistance of head and neck cancer. Cancer Lett. 2016;381(1):96‐103. [DOI] [PubMed] [Google Scholar]
- 154. Hasegawa M, Takahashi H, Rajabi H, Alam M, Suzuki Y, Yin L, et al. Functional interactions of the cystine/glutamate antiporter, CD44v and MUC1‐C oncoprotein in triple‐negative breast cancer cells. Oncotarget. 2016;7(11):11756‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Yamaguchi H, Hsu JL, Chen CT, Wang YN, Hsu MC, Chang SS, et al. Caspase‐independent cell death is involved in the negative effect of EGF receptor inhibitors on cisplatin in non‐small cell lung cancer cells. Clin Cancer Res. 2013;19(4):845‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Ma S, Dielschneider RF, Henson ES, Xiao W, Choquette TR, Blankstein AR, et al. Ferroptosis and autophagy induced cell death occur independently after siramesine and lapatinib treatment in breast cancer cells. PLoS One. 2017;12(8):e0182921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Yang ND, Tan SH, Ng S, Shi Y, Zhou J, Tan KS, et al. Artesunate induces cell death in human cancer cells via enhancing lysosomal function and lysosomal degradation of ferritin. J Biol Chem. 2014;289(48):33425‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Lv C, Qu H, Zhu W, Xu K, Xu A, Jia B, et al. Low‐Dose Paclitaxel Inhibits Tumor Cell Growth by Regulating Glutaminolysis in Colorectal Carcinoma Cells. Front Pharmacol. 2017;8:244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Zhu S, Zhang Q, Sun X, Zeh HJ 3rd, Lotze MT, Kang R, et al. HSPA5 Regulates Ferroptotic Cell Death in Cancer Cells. Cancer Res. 2017;77(8):2064‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Gao H, Bai Y, Jia Y, Zhao Y, Kang R, Tang D, et al. Ferroptosis is a lysosomal cell death process. Biochem Biophys Res Commun. 2018;503(3):1550‐6. [DOI] [PubMed] [Google Scholar]
- 161. Kasukabe T, Honma Y, Okabe‐Kado J, Higuchi Y, Kato N, Kumakura S. Combined treatment with cotylenin A and phenethyl isothiocyanate induces strong antitumor activity mainly through the induction of ferroptotic cell death in human pancreatic cancer cells. Oncol Rep. 2016;36(2):968‐76. [DOI] [PubMed] [Google Scholar]
- 162. Zanganeh S, Hutter G, Spitler R, Lenkov O, Mahmoudi M, Shaw A, et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro‐inflammatory macrophage polarization in tumour tissues. Nat Nanotechnol. 2016;11(11):986‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Yao X, Yang B, Wang S, Dai Z, Zhang D, Zheng X, et al. A novel multifunctional FePt/BP nanoplatform for synergistic photothermal/photodynamic/chemodynamic cancer therapies and photothermally‐enhanced immunotherapy. J Mater Chem B. 2020;8(35):8010‐21. [DOI] [PubMed] [Google Scholar]
- 164. Zhou LL, Guan Q, Li WY, Zhang Z, Li YA, Dong YB. A Ferrocene‐Functionalized Covalent Organic Framework for Enhancing Chemodynamic Therapy via Redox Dyshomeostasis. Small. 2021;17(32):e2101368. [DOI] [PubMed] [Google Scholar]
- 165. Yang B, Liu Q, Yao X, Zhang D, Dai Z, Cui P, et al. FePt@MnO‐Based Nanotheranostic Platform with Acidity‐Triggered Dual‐Ions Release for Enhanced MR Imaging‐Guided Ferroptosis Chemodynamic Therapy. ACS Appl Mater Interfaces. 2019;11(42):38395‐404. [DOI] [PubMed] [Google Scholar]
- 166. Chen L, Lin Z, Liu L, Zhang X, Shi W, Ge D, et al. Fe(2+)/Fe(3+) Ions Chelated with Ultrasmall Polydopamine Nanoparticles Induce Ferroptosis for Cancer Therapy. ACS Biomater Sci Eng. 2019;5(9):4861‐9. [DOI] [PubMed] [Google Scholar]
- 167. Kou L, Sun R, Jiang X, Lin X, Huang H, Bao S, et al. Tumor Microenvironment‐Responsive, Multistaged Liposome Induces Apoptosis and Ferroptosis by Amplifying Oxidative Stress for Enhanced Cancer Therapy. ACS Appl Mater Interfaces. 2020;12(27):30031‐43. [DOI] [PubMed] [Google Scholar]
- 168. Tang H, Li C, Zhang Y, Zheng H, Cheng Y, Zhu J, et al. Targeted Manganese doped silica nano GSH‐cleaner for treatment of Liver Cancer by destroying the intracellular redox homeostasis. Theranostics. 2020;10(21):9865‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Xu S, Zheng H, Ma R, Wu D, Pan Y, Yin C, et al. Vacancies on 2D transition metal dichalcogenides elicit ferroptotic cell death. Nat Commun. 2020;11(1):3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Li Z, Wu X, Wang W, Gai C, Zhang W, Li W, et al. Fe(II) and Tannic Acid‐Cloaked MOF as Carrier of Artemisinin for Supply of Ferrous Ions to Enhance Treatment of Triple‐Negative Breast Cancer. Nanoscale Res Lett. 2021;16(1):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Gao M, Deng J, Liu F, Fan A, Wang Y, Wu H, et al. Triggered ferroptotic polymer micelles for reversing multidrug resistance to chemotherapy. Biomaterials. 2019;223:119486. [DOI] [PubMed] [Google Scholar]
- 172. Xu T, Ma Y, Yuan Q, Hu H, Hu X, Qian Z, et al. Enhanced Ferroptosis by Oxygen‐Boosted Phototherapy Based on a 2‐in‐1 Nanoplatform of Ferrous Hemoglobin for Tumor Synergistic Therapy. ACS Nano. 2020;14(3):3414‐25. [DOI] [PubMed] [Google Scholar]
- 173. Ou W, Mulik RS, Anwar A, McDonald JG, He X, Corbin IR. Low‐density lipoprotein docosahexaenoic acid nanoparticles induce ferroptotic cell death in hepatocellular carcinoma. Free Radic Biol Med. 2017;112:597‐607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Zhu J, Dai P, Liu F, Li Y, Qin Y, Yang Q, et al. Upconverting Nanocarriers Enable Triggered Microtubule Inhibition and Concurrent Ferroptosis Induction for Selective Treatment of Triple‐Negative Breast Cancer. Nano Lett. 2020;20(9):6235‐45. [DOI] [PubMed] [Google Scholar]
- 175. Song R, Li T, Ye J, Sun F, Hou B, Saeed M, et al. Acidity‐Activatable Dynamic Nanoparticles Boosting Ferroptotic Cell Death for Immunotherapy of Cancer. Adv Mater. 2021;33(31):e2101155. [DOI] [PubMed] [Google Scholar]
- 176. Yu M, Gai C, Li Z, Ding D, Zheng J, Zhang W, et al. Targeted exosome‐encapsulated erastin induced ferroptosis in triple negative breast cancer cells. Cancer Sci. 2019;110(10):3173‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. An P, Gao Z, Sun K, Gu D, Wu H, You C, et al. Photothermal‐Enhanced Inactivation of Glutathione Peroxidase for Ferroptosis Sensitized by an Autophagy Promotor. ACS Appl Mater Interfaces. 2019;11(46):42988‐97. [DOI] [PubMed] [Google Scholar]
- 178. An Y, Zhu J, Liu F, Deng J, Meng X, Liu G, et al. Boosting the Ferroptotic Antitumor Efficacy via Site‐Specific Amplification of Tailored Lipid Peroxidation. ACS Appl Mater Interfaces. 2019;11(33):29655‐66. [DOI] [PubMed] [Google Scholar]
- 179. Wang Y, Wang M, Wu HX, Xu RH. Advancing to the era of cancer immunotherapy. Cancer Commun (Lond). 2021;41(9):803‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Gao Y, Souza‐Fonseca‐Guimaraes F, Bald T, Ng SS, Young A, Ngiow SF, et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat Immunol. 2017;18(9):1004‐15. [DOI] [PubMed] [Google Scholar]
- 181. Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol. 2016;13(6):394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Zou W, Wolchok JD, Chen L. PD‐L1 (B7‐H1) and PD‐1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8(328):328rv4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med. 2018;24(10):1550‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Benci JL, Johnson LR, Choa R, Xu Y, Qiu J, Zhou Z, et al. Opposing Functions of Interferon Coordinate Adaptive and Innate Immune Responses to Cancer Immune Checkpoint Blockade. Cell. 2019;178(4):933‐48. e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Dong MB, Wang G, Chow RD, Ye L, Zhu L, Dai X, et al. Systematic Immunotherapy Target Discovery Using Genome‐Scale In Vivo CRISPR Screens in CD8 T Cells. Cell. 2019;178(5):1189‐204. e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Jiang Q, Wang K, Zhang X, Ouyang B, Liu H, Pang Z, et al. Platelet Membrane‐Camouflaged Magnetic Nanoparticles for Ferroptosis‐Enhanced Cancer Immunotherapy. Small. 2020;16(22):e2001704. [DOI] [PubMed] [Google Scholar]
- 187. Coley WB. The Treatment of Inoperable Sarcoma by Bacterial Toxins (the Mixed Toxins of the Streptococcus erysipelas and the Bacillus prodigiosus). Proc R Soc Med. 1910;3(Surg Sect):1. [PMC free article] [PubMed] [Google Scholar]
- 188. Nguyen LT, Ohashi PS. Clinical blockade of PD1 and LAG3–potential mechanisms of action. Nat Rev Immunol. 2015;15(1):45‐56. [DOI] [PubMed] [Google Scholar]
- 189. Wang W, Kryczek I, Dostál L, Lin H, Tan L, Zhao L, et al. Effector T Cells Abrogate Stroma‐Mediated Chemoresistance in Ovarian Cancer. Cell. 2016;165(5):1092‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569(7755):270‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Delaney G, Jacob S, Featherstone C, Barton M. The role of radiotherapy in cancer treatment: estimating optimal utilization from a review of evidence‐based clinical guidelines. Cancer. 2005;104(6):1129‐37. [DOI] [PubMed] [Google Scholar]
- 192. Baidoo KE, Yong K, Brechbiel MW. Molecular pathways: targeted α‐particle radiation therapy. Clin Cancer Res. 2013;19(3):530‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Ye LF, Chaudhary KR, Zandkarimi F, Harken AD, Kinslow CJ, Upadhyayula PS, et al. Radiation‐Induced Lipid Peroxidation Triggers Ferroptosis and Synergizes with Ferroptosis Inducers. ACS Chem Biol. 2020;15(2):469‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Lang X, Green MD, Wang W, Yu J, Choi JE, Jiang L, et al. Radiotherapy and Immunotherapy Promote Tumoral Lipid Oxidation and Ferroptosis via Synergistic Repression of SLC7A11. Cancer Discov. 2019;9(12):1673‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Li X, Duan L, Yuan S, Zhuang X, Qiao T, He J. Ferroptosis inhibitor alleviates Radiation‐induced lung fibrosis (RILF) via down‐regulation of TGF‐β1. J Inflamm (Lond). 2019;16:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Zhang X, Xing X, Liu H, Feng J, Tian M, Chang S, et al. Ionizing radiation induces ferroptosis in granulocyte‐macrophage hematopoietic progenitor cells of murine bone marrow. Int J Radiat Biol. 2020;96(5):584‐95. [DOI] [PubMed] [Google Scholar]
- 197. Gwangwa MV, Joubert AM, Visagie MH. Crosstalk between the Warburg effect, redox regulation and autophagy induction in tumourigenesis. Cell Mol Biol Lett. 2018;23:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Scharping NE, Rivadeneira DB, Menk AV, Vignali PDA, Ford BR, Rittenhouse NL, et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat Immunol. 2021;22(2):205‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Liu X, Olszewski K, Zhang Y, Lim EW, Shi J, Zhang X, et al. Cystine transporter regulation of pentose phosphate pathway dependency and disulfide stress exposes a targetable metabolic vulnerability in cancer. Nat Cell Biol. 2020;22(4):476‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Watkins SK, Egilmez NK, Suttles J, Stout RD. IL‐12 rapidly alters the functional profile of tumor‐associated and tumor‐infiltrating macrophages in vitro and in vivo. J Immunol. 2007;178(3):1357‐62. [DOI] [PubMed] [Google Scholar]
- 201. Nucera S, Biziato D, De Palma M. The interplay between macrophages and angiogenesis in development, tissue injury and regeneration. Int J Dev Biol. 2011;55(4‐5):495‐503. [DOI] [PubMed] [Google Scholar]
- 202. Wan C, Sun Y, Tian Y, Lu L, Dai X, Meng J, et al. Irradiated tumor cell‐derived microparticles mediate tumor eradication via cell killing and immune reprogramming. Sci Adv. 2020;6(13):eaay9789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Sindrilaru A, Peters T, Wieschalka S, Baican C, Baican A, Peter H, et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J Clin Invest. 2011;121(3):985‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Turubanova VD, Balalaeva IV, Mishchenko TA, Catanzaro E, Alzeibak R, Peskova NN, et al. Immunogenic cell death induced by a new photodynamic therapy based on photosens and photodithazine. J Immunother Cancer. 2019;7(1):350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M‐1/M‐2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000;164(12):6166‐73. [DOI] [PubMed] [Google Scholar]
- 206. Zhang F, Li F, Lu GH, Nie W, Zhang L, Lv Y, et al. Engineering Magnetosomes for Ferroptosis/Immunomodulation Synergism in Cancer. ACS Nano. 2019;13(5):5662‐73. [DOI] [PubMed] [Google Scholar]
- 207. Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F, et al. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci U S A. 2014;111(47):16836‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Aaes TL, Kaczmarek A, Delvaeye T, De Craene B, De Koker S, Heyndrickx L, et al. Vaccination with Necroptotic Cancer Cells Induces Efficient Anti‐tumor Immunity. Cell Rep. 2016;15(2):274‐87. [DOI] [PubMed] [Google Scholar]
- 209. Tang R, Xu J, Zhang B, Liu J, Liang C, Hua J, et al. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J Hematol Oncol. 2020;13(1):110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Proneth B, Conrad M. Ferroptosis and necroinflammation, a yet poorly explored link. Cell Death Differ. 2019;26(1):14‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Li W, Feng G, Gauthier JM, Lokshina I, Higashikubo R, Evans S, et al. Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J Clin Invest. 2019;129(6):2293‐304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, et al. HMG‐1 as a late mediator of endotoxin lethality in mice. Science. 1999;285(5425):248‐51. [DOI] [PubMed] [Google Scholar]
- 213. Wen Q, Liu J, Kang R, Zhou B, Tang D. The release and activity of HMGB1 in ferroptosis. Biochem Biophys Res Commun. 2019;510(2):278‐83. [DOI] [PubMed] [Google Scholar]
- 214. Garg AD, Krysko DV, Vandenabeele P, Agostinis P. Hypericin‐based photodynamic therapy induces surface exposure of damage‐associated molecular patterns like HSP70 and calreticulin. Cancer Immunol Immunother. 2012;61(2):215‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Linkermann A, Stockwell BR, Krautwald S, Anders HJ. Regulated cell death and inflammation: an auto‐amplification loop causes organ failure. Nat Rev Immunol. 2014;14(11):759‐67. [DOI] [PubMed] [Google Scholar]
- 216. Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, et al. Multi‐stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug‐Induced Iron‐Dependent Oxidative Stress. Cancer Cell. 2018;33(5):890‐904. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Chen X, Kang R, Kroemer G, Tang D. Ferroptosis in infection, inflammation, and immunity. J Exp Med. 2021;218(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol. 2021;18(5):280‐96. [DOI] [PubMed] [Google Scholar]
- 219. Wang H, Cheng Y, Mao C, Liu S, Xiao D, Huang J, et al. Emerging mechanisms and targeted therapy of ferroptosis in cancer. Mol Ther. 2021;29(7):2185‐208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Su Y, Zhao B, Zhou L, Zhang Z, Shen Y, Lv H, et al. Ferroptosis, a novel pharmacological mechanism of anti‐cancer drugs. Cancer Lett. 2020;483:127‐36. [DOI] [PubMed] [Google Scholar]
- 221. Liu DS, Duong CP, Haupt S, Montgomery KG, House CM, Azar WJ, et al. Inhibiting the system x(C)(‐)/glutathione axis selectively targets cancers with mutant‐p53 accumulation. Nat Commun. 2017;8:14844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, et al. A GPX4‐dependent cancer cell state underlies the clear‐cell morphology and confers sensitivity to ferroptosis. Nat Commun. 2019;10(1):1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Gascón S, Murenu E, Masserdotti G, Ortega F, Russo GL, Petrik D, et al. Identification and Successful Negotiation of a Metabolic Checkpoint in Direct Neuronal Reprogramming. Cell Stem Cell. 2016;18(3):396‐409. [DOI] [PubMed] [Google Scholar]
- 224. Ubellacker JM, Tasdogan A, Ramesh V, Shen B, Mitchell EC, Martin‐Sandoval MS, et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature. 2020;585(7823):113‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Dai E, Han L, Liu J, Xie Y, Zeh HJ, Kang R, et al. Ferroptotic damage promotes pancreatic tumorigenesis through a TMEM173/STING‐dependent DNA sensor pathway. Nat Commun. 2020;11(1):6339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Canli Ö, Alankuş YB, Grootjans S, Vegi N, Hültner L, Hoppe PS, et al. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood. 2016;127(1):139‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Jia M, Qin D, Zhao C, Chai L, Yu Z, Wang W, et al. Redox homeostasis maintained by GPX4 facilitates STING activation. Nat Immunol. 2020;21(7):727‐35. [DOI] [PubMed] [Google Scholar]
- 228. He S, Jiang Y, Li J, Pu K. Semiconducting Polycomplex Nanoparticles for Photothermal Ferrotherapy of Cancer. Angew Chem Int Ed Engl. 2020;59(26):10633‐8. [DOI] [PubMed] [Google Scholar]
- 229. Wang Y, Xu S, Shi L, Teh C, Qi G, Liu B. Cancer‐Cell‐Activated in situ Synthesis of Mitochondria‐Targeting AIE Photosensitizer for Precise Photodynamic Therapy. Angew Chem Int Ed Engl. 2021;60(27):14945‐53. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable