

SUMMARY
Fragile X syndrome (FXS) is the most common form of inherited intellectual disability. Previous studies have implicated mGlu5 in the pathogenesis of the disease, but a crucial unanswered question is whether pharmacological mGlu5 inhibition is able to reverse an already established FXS phenotype in mammals. Here we have used the novel, potent, and selective mGlu5 inhibitor CTEP to address this issue in the Fmr1 knockout mouse. Acute CTEP treatment corrects elevated hippocampal long-term depression, protein synthesis, and audiogenic seizures. Chronic treatment that inhibits mGlu5 within a receptor occupancy range of 81% ± 4% rescues cognitive deficits, auditory hypersensitivity, aberrant dendritic spine density, overactive ERK and mTOR signaling, and partially corrects macroorchidism. This study shows that a comprehensive phenotype correction in FXS is possible with pharmacological intervention starting in young adulthood, after development of the phenotype. It is of great interest how these findings may translate into ongoing clinical research testing mGlu5 inhibitors in FXS patients.
INTRODUCTION
Fragile X syndrome (FXS) is a monogenic developmental disorder associated with a complex neuropsychiatric phenotype (Hagerman et al., 2009). FXS is caused by mutations in the fragile X mental retardation 1 (FMR1) gene, triggering partial or complete gene silencing and partial or complete lack of the fragile X mental retardation protein (FMRP) (Oostra and Willemsen, 2003).
It has been proposed that exaggerated consequences of mGlu5-mediated signaling in the absence of FMRP play a causal role in FXS (Bear et al., 2004). This theory is strongly supported by the finding that genetic reduction of mGlu5 expression is sufficient to correct a broad range of phenotypes in the Fmr1 knockout (KO) mouse (Dölen et al., 2007). Additionally, a number of pharmacological studies have shown that short-acting mGlu5 inhibitors, such as MPEP and fenobam, can ameliorate fragile X phenotypes in several evolutionarily distant animal models (see Krueger and Bear, 2011, for review).
Although these studies support the mGlu theory, they do not address the very important conceptual question of whether mammalian fragile X phenotypes can be prevented or reversed with late-onset mGlu5 inhibition. A failure to correct mutant phenotypes with treatment starting after symptom onset would suggest a missed critical period and indicate that fragile X syndrome is a terminally differentiated phenotype of altered brain development. On the other hand, amelioration of phenotypes with late treatment would support the notion that many problems are due to an ongoing imbalance in synaptic signaling, which can be substantially improved once the normal balance is restored. The genetic rescue experiments to date have not addressed this question because they are germline manipulations present in utero. Neither have the pharmacological experiments to date been able to address this question in mammals, because they have relied on compounds with a short duration of action. Experiments with acute drug treatment cannot explore the full therapeutic potential of mGlu5 antagonists in view of the chronic and developmental nature of FXS.
In the current study, we used a new pharmacological tool, CTEP, a selective, orally bioavailable, and long-acting mGlu5 inhibitor (Lindemann et al., 2011) to test whether chronic pharmacological mGlu5 inhibition can reverse FXS phenotypes in a fully developed brain. We chose to start treatment at an age of 4–5 weeks, when the mouse brain development is anatomically complete but highly plastic and when all FXS phenotypes relevant for the study are established. Our results show that chronic treatment of young adult Fmr1 KO mice with an mGlu5 inhibitor rescues a broad range of phenotypes, including learning and memory deficits, hyperreactivity to sensory stimuli, elevated locomotor activity, and increased dendritic spine density in the cortex. Our data also reveal correction of elevated sensitivity to epilepsy, excessive protein synthesis, long-term depression (LTD), activity of signaling pathways, and an amelioration of macroorchidism. Taken together, the data suggest beneficial effects in a wide range of symptoms and a disease-modifying potential for mGlu5 inhibitors in FXS.
RESULTS
CTEP Enables Chronic Pharmacological Inhibition of mGlu5 in Mice
CTEP is a novel potent, selective, and orally bioavailable mGlu5 inhibitor with a unique long half-life of approximately 18 hr in mice (Figure 1A) (Lindemann et al., 2011). In vivo receptor occupancy measurements with the tracer [3H]-ABP688 (Hintermann et al., 2007) revealed 50% mGlu5 occupancy (EC50) by CTEP concentrations in plasma and brain of 12.1 ng/ml and 75.0 ng/g, respectively (Figure 1B). A regimen of one dose of 2 mg/kg CTEP per os (p.o.) per 48 hr achieved uninterrupted mGlu5 occupancy. The minimal (trough level) drug exposure reached after 2 weeks of treatment was 98 ± 14 ng/ml in plasma and 215 ± 28 ng/g in brain (Figure 1C), corresponding to an estimated mean receptor occupancy level of 81%, with a peak to trough range of 85%–77% (Figure 1D).
Figure 1. CTEP Corrects Protein Synthesis and LTD and Is Suitable for Chronic Dosing in Fmr1 KO Mice.
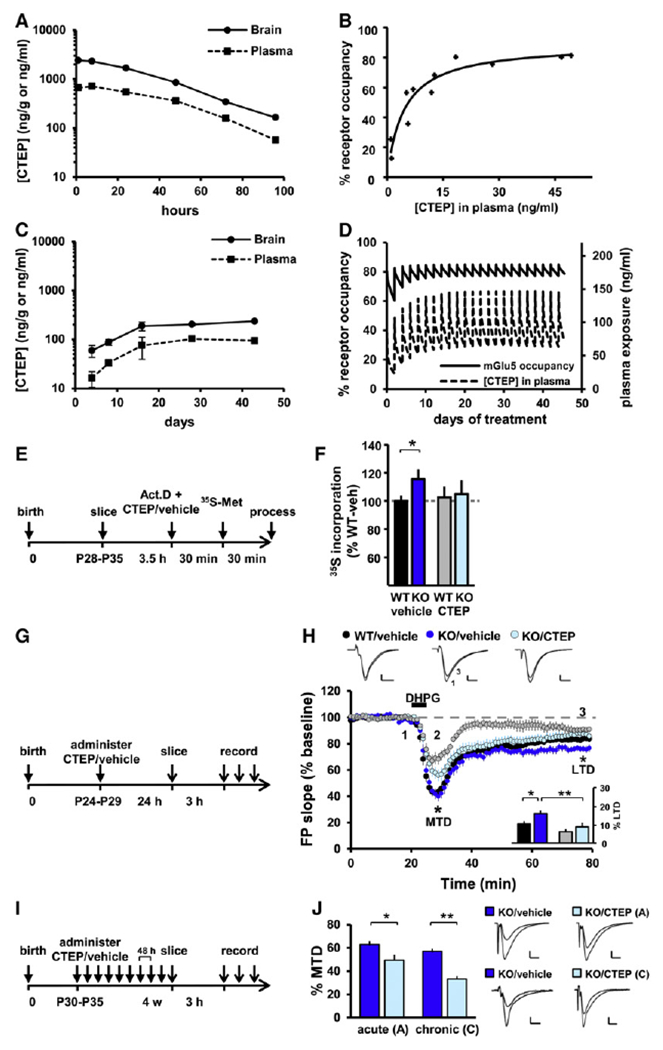
(A) Drug exposure after a single oral dose of CTEP at 4.5 mg/kg; two mice per time point. (B) mGlu5 receptor occupancy in vivo as a function of drug exposure; 11 mice. (C) Drug-exposure monitoring during chronic dosing at 2 mg/kg per 48 hr p.o. Samples were collected 48 hr after each drug administration and thus reveal the minimal levels of drug exposure; two to four mice per time point. (D) Simulation of mGlu5 receptor occupancy during the course of 6 week chronic treatment at 2 mg/kg per 48 hr p.o. This dosing regimen achieved sustained receptor occupancy of 81% ± 4%. (E) Timeline of the protein synthesis assay measuring 35[S]-Met/Cys incorporation in hippocampus sections in vitro. (F) Protein synthesis rate in WT and Fmr1 KO slices in presence or absence of CTEP in the bath; mean ± SEM of 9–11 animals per group with two slices per animal and drug treatment; *p < 0.05. (G) Timeline of acute s.c. treatment with CTEP or vehicle 24 hr before dissection and LTD induction. (H) Gp1 mGlu LTD was enhanced in Fmr1 KO and was rescued by a single CTEP treatment. Two-way ANOVA revealed a significant effect of genotype (p < 0.05) and treatment (p < 0.01), but no interaction. Inset: post hoc tests showed a significant LTD enhancement in KO/vehicle versus WT/vehicle slices (*p < 0.05), a significant rescue by CTEP (**p < 0.01), and no significant effect of CTEP in WT slices (p = 0.14). (I) Timeline of chronic treatment schedule (2 mg/kg/48 hr p.o) beginning at the age of 4–5 weeks. (J) The maximal transient depression (MTD) induced by DHPG was significantly reduced by both acute (*p < 0.05) and chronic (**p < 0.01) CTEP treatment in KO slices, showing that the drug efficacy is maintained throughout chronic treatment. For (H) and (J), mean ± SEM of 14–18 slices per condition.
CTEP Corrects Excessive Protein Synthesis in the Hippocampus of Fmr1 KO Mice
FMRP binds hundreds of mRNAs in vivo and represses their translation (Darnell et al., 2011). Accordingly, at the core of FXS pathophysiology is an elevated rate of protein synthesis (Qin et al., 2005; Dölen et al., 2007; Osterweil et al., 2010). This phenotype was confirmed by measuring [35S]-methionine/cysteine incorporation in acute hippocampal slices (Fmr1 KO: 115% ± 7% of wild-type [WT]; p < 0.05; Figures 1E and 1F). As previously shown with MPEP (Osterweil et al., 2010), bath application of CTEP (10 μM) corrected the elevated protein synthesis rate in Fmr1 KO hippocampal slices (KO/CTEP: 104.9% ± 10% of WT/vehicle) with no significant effects in WT slices.
Correction of Elevated mGlu-LTD in the Hippocampus after In Vivo CTEP Administration
Fmr1 KO mice show an elevated group 1 (Gp1) mGlu-dependent long-term depression (Huber et al., 2002) which can be corrected by genetic reduction of mGlu5 expression levels (Dölen et al., 2007), but not by bath application of MPEP (Volk et al., 2006). We therefore determined whether in vivo administration of CTEP could reduce the elevated LTD ex vivo in the Fmr1 KO hippocampus to WT levels. WT and KO animals (postnatal day 25–30) received a single dose of CTEP (2 mg/kg, subcutaneous [s.c.]) or vehicle 24 hr prior to euthanasia and hippocampal slice preparation. We found that Gp1 mGlu-mediated hippocampal LTD was elevated in vehicle-treated Fmr1 KO mice compared to WT (WT/vehicle: 84.6% ± 2.4%; KO/vehicle: 76.1% ± 2.5%; p < 0.05; Figures 1G and 1H) and was normalized by a single dose of CTEP (KO/vehicle versus KO/CTEP: 86.9% ± 3.3%; p < 0.01).
CTEP treatment also reduced the maximum transient depression (MTD) to DHPG, which represents an electrophysiological readout of Gp1 mGlu activation. After 4 weeks of chronic dosing, MTD was strongly suppressed by CTEP (KO/vehicle: 57.1% ± 2.2%; KO/CTEP: 33.2% ± 2.6%; p < 0.01; Figures 1I and 1J), even more so than after a single dose (KO/vehicle: 62.9% ± 3.0% versus KO/CTEP: 49.4% ± 4.6%; p < 0.05), showing that the drug efficacy is maintained throughout chronic treatment.
Correction of Learning and Memory Deficits
Cognitive impairment is a core symptom in FXS. We confirmed that Fmr1 KO mice exhibit deficits in inhibitory avoidance (IA) (Figure 2). Vehicle-treated Fmr1 KO mice showed significantly reduced latencies to enter the dark compartment compared to vehicle-treated WT littermates 6 hr after conditioning and during all extinction trials (6 hr, p = 0.0186; 24 hr, p = 0.0095; 48 hr, p = 0.0582; Figures 2B–2D). There was no difference in the pain threshold between Fmr1 KO and WT mice (Figure 2E).
Figure 2. Pharmacological Correction of Learning and Memory Deficit and Hypersensitivity to Auditory Stimuli in Fmr1 KO Mice.
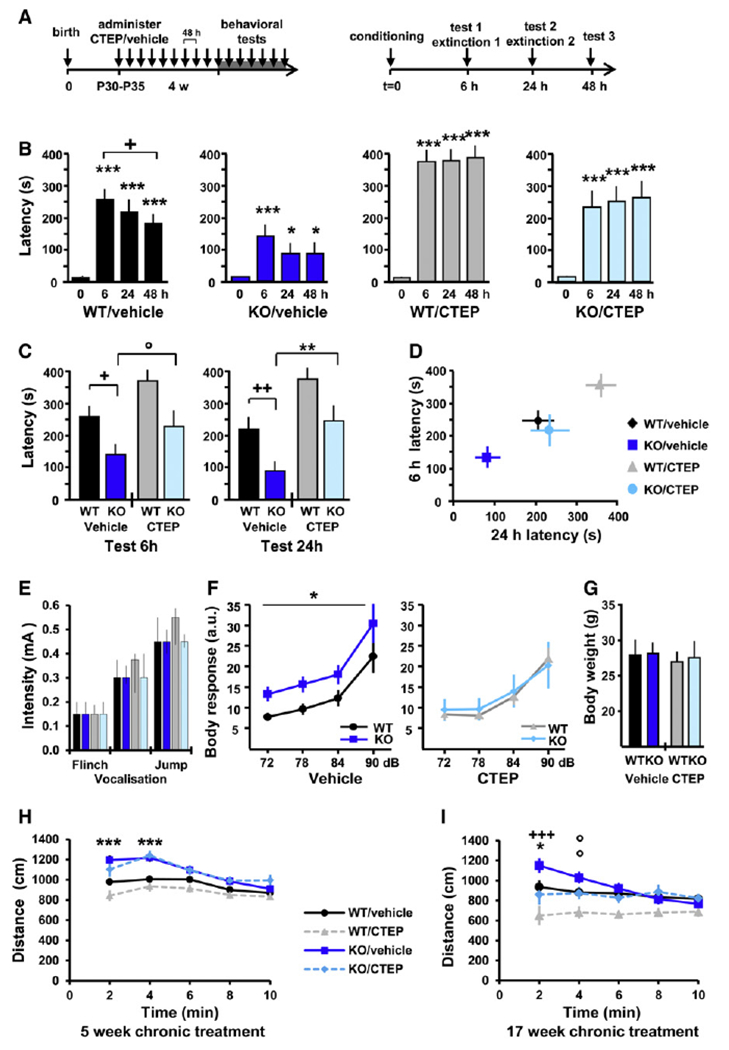
(A) Timeline of chronic dosing prior to behavioral evaluation and the inhibitory avoidance and extinction (IAE) tests. (B) All experimental groups exhibited a significant increase in latency following the conditioning session (different from t = 0; *p < 0.05; **p < 0.01; ***p < 0.001), and WT/vehicle animals also showed significant memory extinction (different from t = 6 hr; +p < 0.05). (C) Comparison of latency across experimental groups at 6 and 24 hr test sessions; KO/vehicle versus WT/vehicle: *p < 0.05, **p < 0.01; KO/CTEP versus KO/vehicle: °p < 0.1, ++p < 0.01. (D) Multivariate analysis of latency at 6 versus 24 hr; the learning deficit observed in KO/vehicle mice was fully compensated by treatment, and the effect of treatment was similar in WT and Fmr1 KO mice. For (B)–(D), mean ± SEM of 15–16 mice per group. (E) Pain threshold: vehicle- and CTEP-treated Fmr1 KO and WT mice showed no differences in response to electrical foot shocks. Mean ± interquartile range, with six mice per group. (F) Sensitivity to auditory stimuli: mice were exposed to short auditory stimuli at 72 (+6), 78 (+12), 84 (+18), and 90 (+24) dB over a white background noise at 66 dB, and the whole-body startle response was recorded. Genotype effect: *p < 0.05; treatment effect: p < 0.05; mean ± SEM of 13–16 mice per group, with eight presentations of each sound intensity. (G) Body weight: there was no significant difference in body weight between the experimental groups on the day of the whole-body startle response experiment; mean ± SD of 13–16 mice per group. (H and I) Locomotor activity in the open field: Fmr1 KO mice exhibited elevated novelty-induced activity compared to WT mice at the age of 2 months (H) and 5 months (I). Correction of the hyperactivity phenotype was observed after 17 weeks (I), but not after 5 weeks (H), of chronic CTEP treatment. KO/vehicle versus WT/vehicle: °p < 0.1, *p < 0.05, ***p < 0.001; KO/CTEP versus KO/vehicle: +p < 0.05, ++p < 0.01; mean ± SEM of 16–17 (H) and 13–15 (I) mice per group.
Chronic treatment fully rescued the learning and memory deficit in the IA paradigm, with CTEP-treated Fmr1 KO mice exhibiting latencies to enter the dark compartment similar to vehicle-treated WT mice at all test sessions. Correspondingly, CTEP-treated Fmr1 KO mice exhibited significantly more avoidance than vehicle-treated Fmr1 KO mice (6 hr, p = 0.0817; 24 hr, p = 0.0016; 48 hr, p = 0.0007).
Correction of Hypersensitivity to Sensory Stimuli
FXS patients frequently present a hypersensitivity to sensory stimuli (Miller et al., 1999), mirrored in Fmr1 KO mice by a hypersensitivity to low-intensity auditory stimuli (Nielsen et al., 2002). The whole-body startle response to short auditory stimuli of moderate intensity (6–24 dB over background) was measured in chronically treated Fmr1 KO and WT mice. The elevated startle response of Fmr1 KO mice compared to WT mice was fully corrected by chronic CTEP treatment (genotype effect: p = 0.029; treatment effect: p = 0.035; Figure 2F). Treatment with CTEP had no effect on the response of WT animals. There was no potential bias between the experimental groups due to body weight (Figure 2G).
Correction of Elevated Locomotor Activity
Hyperactivity is frequently observed in FXS patients, a symptom that is reproduced in Fmr1 KO mice (The Dutch-Belgian Fragile X Consortium, 1994). In the open-field test, vehicle-treated Fmr1 KO mice exhibited elevated novelty-induced locomotor activity compared to vehicle-treated WT mice at the age of 2 and 5 months (2 months, p < 0.001; 5 months, p = 0.014; Figures 2H and 2I). The increased locomotor activity was corrected after 17 weeks (treatment effect: p = 0.009; KO/CTEP versus KO/vehicle at 2 min, p < 0.001; 4 min, p = 0.06; Figure 2I), but not after 5 weeks (Figure 2H), of chronic CTEP treatment.
Correction of Increased Susceptibility to Audiogenic Seizures
FXS patients have increased rates of epilepsy, and this is reflected in Fmr1 KO mice by an increased susceptibility to audiogenic seizures (AGS) (Musumeci et al., 1999, 2000). Drug-naive Fmr1 KO mice presented an elevated seizure response to intense auditory stimuli (120 dB) compared to WT littermates on both C57BL/6 and FVB genetic backgrounds. This hypersensitivity to AGS was fully corrected by a single dose of CTEP administrated 4 hr before testing (Table 1). These results are consistent with the previously reported anticonvulsant activity of other mGlu5 antagonists in Fmr1 KO mice (Qiu et al., 2009; Yan et al., 2005).
Table 1.
Pharmacological Rescue of Elevated Susceptibility for Audiogenic Seizure
| Wild Running | Seizure | Death | Incidence | |
|---|---|---|---|---|
| FVB | ||||
| WT/vehicle | 1/13 | 1/13 | 0/13 | 8% |
| WT/CTEP | 0/12 | 0/12 | 0/12 | 0% |
| KO/vehicle | 12/17 | 10/17 | 5/17 | 71%* |
| KO/CTEP | 5/16 | 6/16 | 0/16 | 31%** |
| C57BL/6 | ||||
| WT/vehicle | 1/14 | 0/14 | 0/14 | 7% |
| WT/CTEP | 0/12 | 0/12 | 0/12 | 0% |
| KO/vehicle | 8/19 | 3/19 | 2/19 | 42%* |
| KO/CTEP | 1/15 | 1/15 | 0/15 | 7%** |
Fmr1 KO mice display an elevated susceptibility to audiogenic seizure in both C57BL/6 and FVB genetic backgrounds, which was rescued by acute administration of CTEP (4 hr before testing, 2 mg/kg, s.c. or p.o.). The tables present for each experimental group the number of mice exhibiting wild running and/or jumping, tonic-clonic seizures, or death compared to the total number of animals tested. Pairwise group comparisons with the Fisher’s exact test: different from WT/vehicle,
p < 0.05; different from KO-vehicle,
p < 0.05.
Correction of the Dendritic Spine Phenotype in the Visual Cortex
Increased dendritic spine density was reported in postmortem analysis of FXS patient brain tissue (Irwin et al., 2001) and can be observed in Fmr1 KO mice (Galvez and Greenough, 2005). Vehicle-treated Fmr1 KO animals showed a significantly higher spine density in pyramidal neurons of the binocular visual cortex compared to vehicle-treated WT animals in basal, but not apical, dendrites (KO/vehicle versus WT/vehicle: segments 50 μm, p = 0.029; 75 μm, p = 0.030; Figures 3A–3C). Chronic treatment with CTEP corrected this phenotype, reducing spine density in Fmr1 KO animals to WT levels. In basal dendrites, spine density in CTEP-treated KO animals was significantly lower than vehicle-treated KO animals (25 μm, p = 0.009; 50 μm, p = 0.002; 75 μm, p = 0.022). In WT animals, CTEP treatment had no significant effect on the spine density.
Figure 3. Pharmacological Correction of Elevated Dendritic Spine Density and Altered Intracellular Signaling In Vivo and Partial Correction of Macroorchidism.
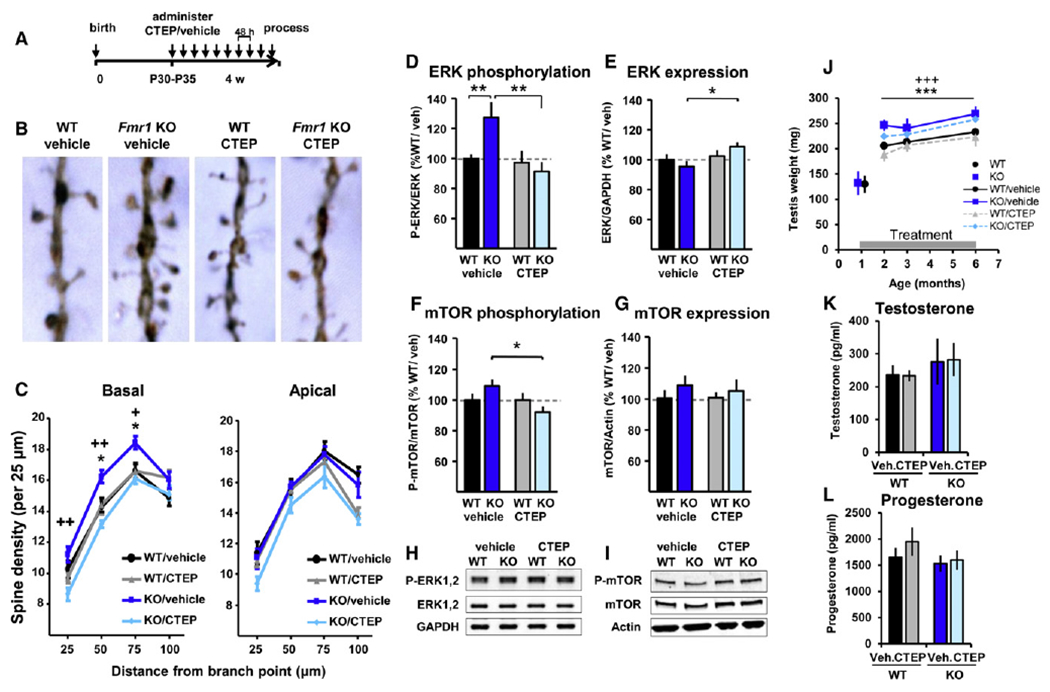
(A) Timeline of chronic dosing for morphological and biochemical analyses. (B) Representative images of Golgi-stained neurons in the primary visual cortex; each photograph represents a 10-μm-long spine segment. (C) Spine density was increased in basal, but not apical, dendrites of KO/vehicle compared to WT/vehicle littermates (*p < 0.05) and normalized in chronically treated KO animals (KO/CTEP versus KO/vehicle: +p < 0.05, ++p < 0.01). Mean ± SEM of ten mice per group, with three dendrites on three different neurons counted per animal. (D–I) Quantification of phosphorylation and expression levels of ERK and mTOR in cortical extracts collected from chronically treated animals. (D) Elevated ERK1,2 phosphorylation (Thr202/Tyr204) in KO/vehicle mice was corrected in chronically treated KO mice (**p < 0.01). (E) Treatment also increased ERK expression levels in KO animals (*p < 0.05). (F) Similarly, mTOR phosphorylation (Ser2481) levels were significantly decreased in chronically CTEP-treated mice compared to vehicle-treated Fmr1 KO mice (*p < 0.05). (G) mTOR expression levels were not altered in Fmr1 KO mice and were not altered upon chronic treatment. For (F) and (G), mean ± SEM of 11 mice per group and triplicate measurements. (H and I) Typical western blot results. (J) Testis weight (Table S2). Adult Fmr1 KO mice presented an increased testis weight compared to WT mice (genotype effect: ***p < 0.001), which was partially corrected (~40% correction) upon chronic treatment (treatment effect: +++p < 0.001). There was no significant genotype × treatment interaction. Mean ± SD of 9–12 mice per age and per group. (K and L) Testosterone and progesterone levels were determined in the plasma of animals subjected to 17 weeks of chronic treatment. For both hormones, the levels were similar in WT and Fmr1 KO animals and were not affected by treatment. Mean ± SEM of 7–10 mice per group and duplicate measurements.
Correction of Abnormal Intracellular Signaling in the Cerebral Cortex
The ERK and mTOR signaling pathways have been implicated in the coupling of mGlu5 to the synaptic protein synthesis machinery (Banko et al., 2006; Gallagher et al., 2004). The basal activity levels of ERK and mTOR in the cortex of mice chronically treated with CTEP and vehicle were analyzed by semiquantitative phosphospecific western blots. The phosphorylation of ERK1,2 at Thr202 and Tyr204 and the autophosphorylation of mTOR at Ser2481 correspond to the active forms of these kinases (Dalby et al., 1998; Soliman et al., 2010).
The ERK phosphorylation level was significantly higher in the cortex of vehicle-treated Fmr1 KO animals compared to vehicle-treated WT littermates (KO/vehicle: 122.9% ± 9.3% of WT/vehicle; p = 0.010; Figures 3D and 3H). Chronic treatment with CTEP specifically reduced the elevated ERK activity in Fmr1 KO cortex (KO/CTEP: 89.5% ± 6.5% of WT/vehicle; KO/CTEP versus KO/vehicle; p = 0.0012) with no effect on ERK activity in WT cortex. Chronic CTEP treatment also triggered a modest increase of the total ERK expression level in Fmr1 KO mice compared to vehicle-treated KO animals (KO/CTEP: 109.0% ± 6.5%; KO/vehicle: 95.1% ± 6.0%; p = 0.013; Figure 3E).
The mTOR phosphorylation level was nonsignificantly increased in vehicle-treated Fmr1 KO animals compared to vehicle-treated WT littermates (KO/vehicle: 109.1% ± 5.0% of WT/vehicle; p = 0.13; Figure 3F). Chronic CTEP treatment significantly reduced the mTOR phosphorylation level specifically in Fmr1 KO mice and not in WT animals (KO/CTEP: 92.0% ± 4.6% of WT/vehicle; KO/CTEP versus KO/vehicle; p = 0.006). mTOR expression levels were similar in WT and KO animals and were unchanged by treatment (Figure 3G).
Partial Correction of Macroorchidism upon Chronic Treatment
The postadolescent macroorchidism observed in FXS patients is reflected in elevated testis weight in Fmr1 KO mice (The Dutch-Belgian Fragile X Consortium, 1994). Testis weight was monitored starting with drug-naive 5-week-old mice throughout 17 weeks of chronic treatment with CTEP and vehicle. Fmr1 KO mice presented significantly increased testis weight compared to WT animals at all adult ages (effect size: +32.8 mg, p < 0.001; Figure 3J; see Table S2 available online), which was partially corrected upon chronic treatment (effect size: −13.5 mg, p < 0.001). No significant differences in plasma levels of testosterone (Figure 3K) and progesterone (Figure 3L) were observed between genotypes and treatment groups.
Absence of Chronic Treatment Effect on Motor Coordination and General Fitness of the Animals
Chronic treatment was well tolerated by the animals independent of the genotype. There was a minimal reduction in body weight gain (Figure S1A) and a modest decrease in body temperature of 0.5°C on average (Figure S1B) in animals receiving chronic CTEP treatment compared to vehicle in both genotypes. Chronic drug treatment for 4 weeks had no effect on the rotarod performance (Figure S1C). A small but significantly reduced grip strength in vehicle-treated Fmr1 KO compared to WT mice and in CTEP-treated mice of both genotypes compared to vehicle-treated WT mice was observed (Figure S1D). A modified version of the Irwin battery of simple neurological and observational measures (Irwin, 1968) did not reveal any noticeable alteration in the general fitness of the animals resulting from the mutation or the treatment (Table S1).
DISCUSSION
This study assessed the therapeutic potential of chronic pharmacological mGlu5 inhibition in a mouse model of FXS, with treatment starting in young adulthood. The study became possible with the discovery of the novel mGlu5 inhibitor CTEP (Lindemann et al., 2011), enabling continuous mGlu5 inhibition with a receptor occupancy of ca. 81% ± 4% (Figure 1). Acute treatment with CTEP rescued elevated protein synthesis in hippocampal slices, and single-dose administration in vivo normalized LTD ex vivo and suppressed the audiogenic seizure phenotype. Four weeks of chronic CTEP treatment starting at the age of 5 weeks reversed the learning and memory deficit in the inhibitory avoidance test (Figure 2), the hypersensitivity to auditory stimuli, the increased dendritic spine density in the primary visual cortex (Figure 3), and the elevated ERK and mTOR activities in the cortex of Fmr1 KO mice. Chronic CTEP treatment for 17 weeks also corrected elevated locomotor activity (Figures 2H and 2I) and partially reversed macroorchidism (Figure 3J) without affecting testosterone and progesterone plasma levels (Figures 3K and 3L). For some measures (e.g., elevated protein synthesis, auditory hypersensitivity, basal dendrite spine density, and ERK phosphorylation), the corrective effects of CTEP were specific for Fmr1 KO mice, whereas for others (e.g., LTD, inhibitory avoidance, and locomotor activity) CTEP treatment also had a proportional effect on WT mice. Regardless, CTEP treatment moved fragile X phenotypes closer to the untreated WT situation for all these measures. The important and therapeutically relevant conclusion is that a broad spectrum of FXS phenotypes—biochemical, structural, and behavioral—can be improved with treatment onset in early adulthood in mammals.
Our results are in good agreement with the comprehensive phenotypic rescue obtained by genetic reduction of mGlu5 expression levels (Dölen et al., 2007). A limitation of the genetic approach, however, was that mGlu5 expression levels were reduced at the earliest stage of embryonic development and thus may prevent the development of phenotypes rather than correct them. With respect to pharmacological mGlu5 inhibition, a study by Su et al. (2011) reported a rescue of increased dendritic spine density in cortical neurons in vivo by 2 weeks of MPEP administration when treatment started at birth, but not when treatment started in 6-week-old animals. All other experiments reporting correction of the increased spine density phenotype with mGlu5 antagonists (MPEP, fenobam, and AFQ056) were limited to in vitro experiments on primary cultured neurons (de Vrij et al., 2008; Levenga et al., 2011). In contrast to the results of Su et al. (2011), our data show that starting treatment immediately after birth is not a requirement; instead, chronic treatment starting in young adulthood can reverse an established phenotype. The difference in the outcomes of these experiments could be due to the duration of treatment (2 versus 4 weeks), continuous or fluctuating receptor inhibition achieved with long versus short half-life molecules (CTEP and MPEP), respectively, as well as the targeted mGlu5 receptor occupancy range, which is not available for previous studies.
Previous studies have noted impaired inhibitory avoidance acquisition and exaggerated extinction in the Fmr1 KO mice (Yuskaitis et al., 2010; Dölen et al., 2007). Consistent with findings in Fmr1 KO (Dölen et al., 2007) and Grm5 KO (Xu et al., 2009) mice, chronic mGlu5 inhibition retarded memory extinction. We were surprised to discover, however, that long-term CTEP treatment also increased acquisition in both genotypes. We speculate that metaplasticity after chronic partial mGlu5 inhibition promotes the synaptic modifications that accompany inhibitory avoidance acquisition (Whitlock et al., 2006).
FXS patients frequently present a hypersensitivity to sensory stimuli (Hagerman, 1996) and a deficit in the prepulse inhibition (PPI) of the startle response (Frankland et al., 2004). In Fmr1 KO mice, correction of the increased PPI by acute MPEP administration could be demonstrated based on eye-blink response (de Vrij et al., 2008), but not by measuring whole-body startle response (Thomas et al., 2012). The interpretation of these PPI results in mice is confounded, because Fmr1 KO compared to WT mice show a reduced whole-body startle in response to loud (>110 dB) auditory stimuli but an elevated whole-body startle response to low-intensity auditory stimuli (<90 dB) (Nielsen et al., 2002). On this background, we studied the elevated whole-body startle response in Fmr1 KO compared to WT mice to low-intensity stimuli, which was fully corrected by chronic CTEP treatment (Figure 2F).
To better understand the molecular underpinning of the treatment effects, we studied ERK and mTOR phosphorylation in the cortex of adult animals after chronic CTEP treatment. ERK is an important component of the signaling cascade downstream of Gp1 mGlu receptors, and ERK inhibition is sufficient to normalize the elevated protein synthesis rate in Fmr1 KO hippocampus sections and to suppress seizures (Chuang et al., 2005; Osterweil et al., 2010). Like ERK, mTOR is an important regulator of protein synthesis and also modulates Gp1 mGlu-dependent hippocampal LTD (Hou and Klann, 2004). In Fmr1 KO mice, the level of mTOR activity is elevated in some preparations and unresponsive to mGlu1/5 activation (Osterweil et al., 2010; Sharma et al., 2010). These observations suggest that the normalization of ERK and mTOR activity in Fmr1 KO mice by chronic CTEP treatment is likely an integral part of the cellular mechanism through which mGlu5 inhibitors correct the altered hippocampal LTD, elevated AGS susceptibility, and deficient learning and memory in FXS.
Taken together, our data provide evidence for the potential of mGlu5 inhibitors to correct a broad range of complex behavioral, cellular, and neuroanatomical phenotypes closely related to patients’ symptoms in Fmr1 KO mice. The data further reveal that a pharmacological correction of key FXS phenotypes can be achieved with treatment beginning in young adulthood after anatomically completed brain development. The breadth of phenotypes addressed and the degree of normalization by mGlu5 inhibition supports the expectation that mGlu5 inhibitors might have the ability to change the developmental trajectory of FXS patients and thus could hold the potential for disease modification. Currently, several mGlu5 inhibitors are under clinical examination in FXS (RO4917523, F. Hoffmann-La Roche; AFQ056, Novartis; STX107, Seaside Therapeutics). It will be of great interest to see whether the clinical phenotype can be addressed in a similar broad fashion and with a similar magnitude as suggested by the preclinical data.
EXPERIMENTAL PROCEDURES
Animals
Fmr1 KO mice (The Dutch-Belgian Fragile X Consortium, 1994) were initially obtained from The Jackson Laboratory and were maintained on congenic C57BL/6J and FVB genetic backgrounds, respectively. All animal work was approved by local veterinary authorities. All experiments were conducted with experimenters blind to genotype and drug treatment.
Drug Treatment, Characterization of CTEP Pharmacological Properties, Metabolic Labeling, Electrophysiology, Inhibitory Avoidance, Audiogenic Seizure, Golgi Analysis, and Western Blot Analysis
Methods were identical to the ones described previously (Dölen et al., 2007; Lindemann et al., 2011; Osterweil et al., 2010). Full method descriptions are provided in Supplemental Experimental Procedures.
Whole-Body Startle Response to Auditory Stimuli, Locomotor Activity, Neurological Assessment, Motor Coordination and Grip-Strength Test, Testis Weight, and Hormone Levels
For method descriptions, see Supplemental Experimental Procedures.
Statistics
Data were analyzed with two-way analysis of variance (ANOVA) with genotype and treatment as independent factors and repeated measures as covariate when appropriate. Post hoc tests were used to compare groups only if the global analysis indicated a statistically significant (p < 0.05) main effect or a significant interaction. A post hoc Bonferroni test was applied to LTD data, and a protected post hoc Fisher’s test was used for all other experiments. Testis weight was analyzed with a three-way ANOVA with genotype, treatment and age as independent factors, and the corresponding effect sizes are reported. AGS experiments were analyzed with nonparametric statistics for small sample size (Fisher’s exact test).
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank Neil Parrott for the modeling of mGlu5 receptor occupancy; Christophe Fischer and Catherine Diener for hormone measurements; Gerhard Hoffmann, Thomas Thelly, Christophe Flament, and Daniela Doppler for analyzing CTEP exposure; Michael Honer, Edilio Borroni, Patricia Glaentzlin, and Celine Sutter for in vivo binding experiments; and Marco Celio and coworkers at Frimorfo for Golgi-Cox analysis of dendritic spines. We would like to thank Anita Albientz (animal breeding and genotyping), Marie Haman (inhibitory avoidance extinction, locomotor activity, and neurological assessment), and Daniel Rüher and Antonio Ricci (CTEP synthesis) for their excellent technical assistance. We would like to further thank Luca Santarelli and Anrivan Ghosh for their continued support of the project. M.F.B. discloses a financial interest in Seaside Therapeutics. A.M., T.M.B., L.O., J.G.W., G.J., and L.L. are full-time employees of F. Hoffmann-La Roche. Research was partially supported by a grant from the NICHD (M.F.B, principal investigator) and a fellowship from the Autism Science Foundation (M.S.). A.M. and M.S. performed experiments, wrote the manuscript, and participated in the study design. L.L. and M.F.B. designed and directed the study and wrote the manuscript. T.M.B. and L.O. performed experiments. G.J. contributed CTEP. J.G.W. and W.S. contributed to the writing of the manuscript. The following experiments were performed at F. Hoffmann-La Roche (A.M., T.M.B., L.O., W.S., J.G.W., G.J., and L.L.): CTEP pharmacokinetic and receptor occupancy studies and modeling, hormone measurements, pharmacological rescue of inhibitory avoidance extinction deficit, elevated locomotor activity, hypersensitivity to auditory stimuli, elevated synaptic spine density, ERK/mTOR signaling alterations, and macroorchidism. The following experiments were performed at the Picower Institute for Learning and Memory, MIT (M.S. and M.F.B.): rescue of elevated audiogenic seizure sensitivity and elevated hippocampal LTD.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes one figure, two tables, and Supplemental Experimental Procedures and can be found with this article online at doi:10.1016/j.neuron.2012.03.009.
REFERENCES
- Banko JL, Hou L, Poulin F, Sonenberg N, and Klann E (2006). Regulation of eukaryotic initiation factor 4E by converging signaling pathways during metabotropic glutamate receptor-dependent long-term depression. J. Neurosci 26, 2167–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Huber KM, and Warren ST (2004). The mGluR theory of fragile X mental retardation. Trends Neurosci. 27, 370–377. [DOI] [PubMed] [Google Scholar]
- Chuang SC, Zhao W, Bauchwitz R, Yan Q, Bianchi R, and Wong RK (2005). Prolonged epileptiform discharges induced by altered group I metabotropic glutamate receptor-mediated synaptic responses in hippocampal slices of a fragile X mouse model. J. Neurosci 25, 8048–8055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalby KN, Morrice N, Caudwell FB, Avruch J, and Cohen P (1998). Identification of regulatory phosphorylation sites in mitogen-activated protein kinase (MAPK)-activated protein kinase-1a/p90rsk that are inducible by MAPK. J. Biol. Chem 273, 1496–1505. [DOI] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, et al. (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vrij FM, Levenga J, van der Linde HC, Koekkoek SK, De Zeeuw CI, Nelson DL, Oostra BA, and Willemsen R (2008). Rescue of behavioral phenotype and neuronal protrusion morphology in Fmr1 KO mice. Neurobiol. Dis 31, 127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dölen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, and Bear MF (2007). Correction of fragile X syndrome in mice. Neuron 56, 955–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankland PW, Wang Y, Rosner B, Shimizu T, Balleine BW, Dykens EM, Ornitz EM, and Silva AJ (2004). Sensorimotor gating abnormalities in young males with fragile X syndrome and Fmr1-knockout mice. Mol. Psychiatry 9, 417–425. [DOI] [PubMed] [Google Scholar]
- Gallagher SM, Daly CA, Bear MF, and Huber KM (2004). Extracellular signal-regulated protein kinase activation is required for metabotropic glutamate receptor-dependent long-term depression in hippocampal area CA1. J. Neurosci 24, 4859–4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvez R, and Greenough WT (2005). Sequence of abnormal dendritic spine development in primary somatosensory cortex of a mouse model of the fragile X mental retardation syndrome. Am. J. Med. Genet. A 135, 155–160. [DOI] [PubMed] [Google Scholar]
- Hagerman RJ (1996). Physical and behavioral phenotype. In Fragile X Syndrome: Diagnosis, Treatment and Research, Second Edition, Hargeman RJ and Cronister A, eds. (Baltimore, MD: The John Hopkins University Press; ), pp. 3–109. [Google Scholar]
- Hagerman RJ, Berry-Kravis E, Kaufmann WE, Ono MY, Tartaglia N, Lachiewicz A, Kronk R, Delahunty C, Hessl D, Visootsak J, et al. (2009). Advances in the treatment of fragile X syndrome. Pediatrics 123, 378–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hintermann S, Vranesic I, Allgeier H, Brülisauer A, Hoyer D, Lemaire M, Moenius T, Urwyler S, Whitebread S, Gasparini F, and Auberson YP (2007). ABP688, a novel selective and high affinity ligand for the labeling of mGlu5 receptors: identification, in vitro pharmacology, pharmacokinetic and biodistribution studies. Bioorg. Med. Chem 15, 903–914. [DOI] [PubMed] [Google Scholar]
- Hou L, and Klann E (2004). Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J. Neurosci 24, 6352–6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, and Bear MF (2002). Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. USA 99, 7746–7750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin S (1968). Comprehensive observational assessment: Ia. A systematic, quantitative procedure for assessing the behavioral and physiologic state of the mouse. Psychopharmacology (Berl.) 13, 222–257. [DOI] [PubMed] [Google Scholar]
- Irwin SA, Patel B, Idupulapati M, Harris JB, Crisostomo RA, Larsen BP, Kooy F, Willems PJ, Cras P, Kozlowski PB, et al. (2001). Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am. J. Med. Genet 98, 161–167. [DOI] [PubMed] [Google Scholar]
- Krueger DD, and Bear MF (2011).Toward fulfilling the promise of molecular medicine in fragile X syndrome. Annu. Rev. Med 62, 411–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenga J, Hayashi S, de Vrij FM, Koekkoek SK, van der Linde HC, Nieuwenhuizen I, Song C, Buijsen RA, Pop AS, Gomezmancilla B, et al. (2011). AFQ056, a new mGluR5 antagonist for treatment of fragile X syndrome. Neurobiol. Dis 42, 311–317. [DOI] [PubMed] [Google Scholar]
- Lindemann L, Jaeschke G, Michalon A, Vieira E, Honer M, Spooren W, Porter R, Hartung T, Kolczewski S, Büttelmann B, et al. (2011). CTEP: a novel, potent, long-acting, and orally bioavailable metabotropic glutamate receptor 5 inhibitor. J. Pharmacol. Exp. Ther 339, 474–486. [DOI] [PubMed] [Google Scholar]
- Miller LJ, McIntosh DN, McGrath J, Shyu V, Lampe M, Taylor AK, Tassone F, Neitzel K, Stackhouse T, and Hagerman RJ (1999). Electrodermal responses to sensory stimuli in individuals with fragile X syndrome: a preliminary report. Am. J. Med. Genet 83, 268–279. [PubMed] [Google Scholar]
- Musumeci SA, Hagerman RJ, Ferri R, Bosco P, Dalla Bernardina B, Tassinari CA, De Sarro GB, and Elia M (1999). Epilepsy and EEG findings in males with fragile X syndrome. Epilepsia 40, 1092–1099. [DOI] [PubMed] [Google Scholar]
- Musumeci SA, Bosco P, Calabrese G, Bakker C, De Sarro GB, Elia M, Ferri R, and Oostra BA (2000). Audiogenic seizures susceptibility in transgenic mice with fragile X syndrome. Epilepsia 41, 19–23. [DOI] [PubMed] [Google Scholar]
- Nielsen DM, Derber WJ, McClellan DA, and Crnic LS (2002). Alterations in the auditory startle response in Fmr1 targeted mutant mouse models of fragile X syndrome. Brain Res. 927, 8–17. [DOI] [PubMed] [Google Scholar]
- Oostra BA, and Willemsen R (2003). A fragile balance: FMR1 expression levels. Hum. Mol. Genet 12 (Spec No 2), R249–R257. [DOI] [PubMed] [Google Scholar]
- Osterweil EK, Krueger DD, Reinhold K, and Bear MF (2010). Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J. Neurosci 30, 15616–15627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin M, Kang J, Burlin TV, Jiang C, and Smith CB (2005). Postadolescent changes in regional cerebral protein synthesis: an in vivo study in the FMR1 null mouse. J. Neurosci 25, 5087–5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu LF, Lu TJ, Hu XL, Yi YH, Liao WP, and Xiong ZQ (2009). Limbic epileptogenesis in a mouse model of fragile X syndrome. Cereb. Cortex 19, 1504–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Hoeffer CA, Takayasu Y, Miyawaki T, McBride SM, Klann E, and Zukin RS (2010). Dysregulation of mTOR signaling in fragile X syndrome. J. Neurosci 30, 694–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soliman GA, Acosta-Jaquez HA, Dunlop EA, Ekim B, Maj NE, Tee AR, and Fingar DC (2010). mTOR Ser-2481 autophosphorylation monitors mTORC-specific catalytic activity and clarifies rapamycin mechanism of action. J. Biol. Chem 285, 7866–7879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su T, Fan HX, Jiang T, Sun WW, Den WY, Gao MM, Chen SQ, Zhao QH, and Yi YH (2011). Early continuous inhibition of group 1 mGlu signaling partially rescues dendritic spine abnormalities in the Fmr1 knockout mouse model for fragile X syndrome. Psychopharmacology (Berl.) 215, 291–300. [DOI] [PubMed] [Google Scholar]
- The Dutch-Belgian Fragile X Consortium, Bakker CE, Verheij C, Willemsen R, van der Helm R, Oerlemans F, Vermey M, Bygrave A, Hoogeveen T, Oostra BA, Reyniers E, et al. (1994). Fmr1 knockout mice: a model to study fragile X mental retardation. Cell 78, 23–33. [PubMed] [Google Scholar]
- Thomas AM, Bui N, Perkins JR, Yuva-Paylor LA, and Paylor R (2012). Group I metabotropic glutamate receptor antagonists alter select behaviors in a mouse model for fragile X syndrome. Psychopharmacology (Berl.) 219, 47–58. [DOI] [PubMed] [Google Scholar]
- Volk LJ, Daly CA, and Huber KM (2006). Differential roles for group 1 mGluR subtypes in induction and expression of chemically induced hippocampal long-term depression. J. Neurophysiol 95, 2427–2438. [DOI] [PubMed] [Google Scholar]
- Whitlock JR, Heynen AJ, Shuler MG, and Bear MF (2006). Learning induces long-term potentiation in the hippocampus. Science 313, 1093–1097. [DOI] [PubMed] [Google Scholar]
- Xu J, Zhu Y, Contractor A, and Heinemann SF (2009). mGluR5 has a critical role in inhibitory learning. J. Neurosci 29, 3676–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan QJ, Rammal M, Tranfaglia M, and Bauchwitz RP (2005). Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology 49, 1053–1066. [DOI] [PubMed] [Google Scholar]
- Yuskaitis CJ, Mines MA, King MK, Sweatt JD, Miller CA, and Jope RS (2010). Lithium ameliorates altered glycogen synthase kinase-3 and behavior in a mouse model of fragile X syndrome. Biochem. Pharmacol 79, 632–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.