

Abstract
Treatment for metastatic colorectal cancer (mCRC) is focused on prolonging survival and maintaining quality of life. It is important to establish prognostic and predictive markers to avoid extended, ineffective treatment. The aim of the present study was, by a novel approach, to analyze the association between cell-free (cf)DNA levels and outcome in patients receiving systemic therapy for incurable mCRC. The study was a prospective non-interventional biomarker study for patients receiving standard of systemic treatment for mCRC. Patients with mCRC, who, according to standard guidelines, were considered for treatment with EGFR inhibitors, were included. The cfDNA levels in consecutive plasma samples were measured by a direct fluorescence assay. The study included 47 patients. Blood samples were available at baseline (n=47); prior to the third treatment cycle (n=31); the first (n=33), second (n=22) and third response evaluation during treatment (n=17); and at progression (n=22). The disease control rate was 42 and 91% in patients with high (≥75th percentile of baseline cfDNA levels) and low cfDNA levels (<75th percentile of baseline cfDNA levels), respectively (P<0.001). Median progression-free survival (PFS) was 3.8 and 8.5 months in patients with high and low cfDNA levels, respectively (hazard ratio=3.03, 95% CI 1.46-6.29, P<0.01). Median overall survival (OS) was 5.0 and 26.6 months in patients with high and low cfDNA levels, respectively (hazard ratio=3.48, 95% CI 1.44-8.44, P<0.01). In the multivariate analysis, baseline cfDNA levels remained a significant predictor of PFS and OS. In conclusion, cfDNA is a promising prognostic tool in the personalized treatment of mCRC. cfDNA levels were estimated by a simple, rapid and inexpensive method (OPTIPAL II: ClinicalTrials.gov identifier no. NCT03750175; registered November 21, 2018).
Keywords: cell-free DNA, biomarker, colorectal adenocarcinoma, metastatic colorectal cancer, systemic therapy
Introduction
Colorectal cancer (CRC) is the third most common cancer type worldwide and the second leading cause of global cancer-related deaths (1). Approximately 20% of patients with CRC exhibit metastasis at the time of diagnosis and ~50% eventually develop metastatic disease (2). The majority of patients with metastatic CRC (mCRC) cannot be cured. For patients not eligible for metastasis-directed therapy, the treatment strategy for mCRC is palliative focusing on prolonging survival and maintaining quality of life (3). It is important to establish prognostic and predictive markers to detect early treatment failure and avoid prolonged, ineffective therapy.
It has been known for decades that small fragments of cell-free DNA (cfDNA) are present in the circulation (4) with elevated levels in cancer patients (5). A proportion of cfDNA detected in cancer patients is derived from cancer cells, so-called circulating tumor DNA (ctDNA) (6). At present, the research landscape is focusing on investigating the clinical value of tumor-specific mutations in ctDNA, rather than on the value of cfDNA. Compared to ctDNA, the measurement of cfDNA is applicable to all cancer types and not dependent on the presence of specific mutations. It has been demonstrated that cfDNA levels in plasma have a prognostic and predictive value in patients with mCRC, but the methods used to measure cfDNA are complex, time-consuming and expensive, which limits their clinical utility (7-11).
Direct fluorescent assay (DFA), first described by Goldshtein et al (12) in 2009 and refined by Boysen et al (13) in 2018, is able to quantify cfDNA in plasma or serum. This method is simple, rapid and inexpensive. The method has proved feasible in patients with locally advanced rectal cancer (14), mCRC (15,16) and squamous cell carcinoma of the anus undergoing chemoradiotherapy (17).
The aim of the present study was to measure the levels of cfDNA by DFA in patients with mCRC prior to palliative systemic therapy, during treatment and at progression, and to determine their possible association with patient characteristics, treatment response and survival. It was hypothesized that in patients with mCRC, high cfDNA levels were associated with advanced disease and shorter survival compared to low cfDNA levels.
Materials and methods
Study design
The OPTIPAL II study was designed to prospectively enroll patients with mCRC prior to the start of systemic palliative treatment. The study was a non-interventional biomarker study for patients receiving standard systemic treatment for mCRC. The study was explorative and designed to include ~50 patients for analysis. The examined variable in OPTIPAL II was ‘circulating free DNA’. The term ‘circulating free DNA’ covers both measurements of total cfDNA and measurements of the proportion of cfDNA originating from tumor cells (ctDNA). In the present study, the cfDNA results are presented.
Patients
Patients with mCRC, who, according to standard guidelines, were considered for treatment with EGFR inhibitors, were included in a prospective biomarker study (OPTIPAL II: ClinicalTrials.gov identifier no. NCT03750175) at the Department of Oncology, Aarhus University Hospital (Aarhus, Denmark). The OPTIPAL II trial was designed to evaluate the feasibility of ctDNA for anti-EGFR therapy selection and the primary outcome data will be presented in a separate publication. The level of total DNA measurement and correlation to outcome was among the prespecified secondary endpoints.
The key inclusion criteria were as follows: Histopathologically verified mCRC, indication for systemic palliative treatment with standard anti-EGFR monoclonal antibodies, fit for therapy with EGFR inhibition and age ≥18 years. The exclusion criteria were a World Health Organization (WHO) performance status >2, significant other cancer disease within 5 years of inclusion, conditions precluding sampling during therapy and treatment breaks.
In the case of plasma RAS/RAF wild-type status [determined by droplet digital (dd)PCR], treatment consisted of chemotherapy and cetuximab/panitumumab. Patients with plasma RAS/RAF mutation (determined by ddPCR) were treated with chemotherapy.
Imaging response was determined based on CT scans of the chest, abdomen and pelvis performed at baseline, after every fourth cycle of treatment and thereafter every third month during follow-up until progression, death or end of follow-up, whichever came first. Progression-free survival (PFS) was measured from the date of inclusion to progression according to RECIST version 1.1(18), death or censoring, whichever came first. Overall survival (OS) was calculated from the date of inclusion to the date of death from any cause. Patients still alive were censored at the last known date alive. Disease control (DC) was defined as stable disease (SD), partial response (PR) or complete response (CR) as the best response according to RECIST version 1.1(18).
For comparison, blood samples were available from 94 fully anonymized presumed healthy individuals from the Lolland-Falster Health Study (NCT02482896), as previously described (13). All healthy donors gave written informed consent and the study was approved by Region Zealand's Ethical Committee on Health Research (approval no. SJ-421).
Total circulating DNA analysis
Blood samples were collected prospectively at baseline; prior to the third treatment cycle; and the first, second and third response evaluation during treatment. The last sample was drawn at the documented time of progression. A total of 30 ml was drawn at each time-point.
Plasma samples were obtained in EDTA tubes and after 30 min, they were centrifuged at 1,200 x g for 10 min at 21˚C and then stored at -80˚C. cfDNA levels were detected directly in the plasma samples by a modified version (13) of the method described by Goldshtein et al (12). In brief, 40 µl of plasma was added to 160 µl PBS containing DMSO (1:8) and SYBR® Gold Nucleic Acid Gel Stain (1:8,000; Invitrogen; Thermo Fisher Scientific, Inc.). Samples were analyzed in a black 96-well plate (Pio-Plex Pro Flat Bottom Plates; Bio-Rad Laboratories, Inc.). Fluorescence was measured with the 96-well fluorometer (Infinite F200 PRO; Tecan Group) at an emission wavelength of 535 nm and an excitation wavelength of 485 nm.
Standards for DNA were prepared from Human Control Genomic DNA (Thermo Fisher Scientific, Inc.) diluted in PBS containing 10% bovine serum albumin (Sigma-Aldrich; Merck KGaA). The concentration of cfDNA in the plasma samples was calculated from a standard curve.
The final concentration of each sample was calculated as the mean of four measurements. Outliers were removed according to Dixon's q-test if the standard deviation (SD) exceeded 10% if the removal of one value was able to bring the SD <10%; otherwise, the sample was discarded if SD >15%.
The samples were analyzed blinded to clinical parameters and study endpoints.
Statistical analysis
Categorized variables were expressed as counts and proportions and continuous variables were expressed as median values and ranges. The Mann-Whitney U-test was applied to examine the association between patient characteristics and cfDNA levels. Fisher's exact test was applied to compare categorical variables. Wilcoxon's signed-rank test was used for comparison of nonparametric paired samples. Values for cfDNA levels, lactate dehydrogenase (LDH) levels and tumor burden were not normally distributed; therefore, natural log transformation was performed to achieve a normal distribution (confirmed with histograms and QQ-plots) prior to analyses of the associations with a linear regression model.
In a normal reference cohort, the mean cfDNA level was 0.54 ng/µl as measured by the DFA (13), which was used for cut-off point analysis as described below. PFS and OS were estimated by Kaplan-Meier curves and differences between groups with an unadjusted univariate Cox proportional hazards model. All analysis was by intention to treat. The prognostic value of all baseline characteristics was analyzed. Multivariate analysis was performed using the Cox proportional hazards model. Variables with P<0.05 by univariate analysis were included in the multivariate analysis. cfDNA was included as a continuous variable.
All reported P-values were two-sided and P<0.05 was considered to indicate statistical significance. Effect sizes were indicated by 95% confidence intervals (95% CI). Statistical analyses were performed using STATA/IC16.0 (StataCorp LLC).
The study is reported in accordance with the REMARK guideline (19).
Results
Patient characteristics
Between 2018 and 2020, a total of 48 patients were included, but one patient was excluded due to failed analysis of the baseline sample. Blood samples were collected between 2018 and 2020. All baseline characteristics collected are presented in Table I. The median age was 65 years (range, 37-76 years) and the majority of patients were male (57%). The WHO performance status was 0-1 in all patients except three patients with a WHO performance status of 2. Of all primary tumors, 28 (60%) were left-sided. In one-third of the patients (n=15), the primary tumor was in situ at inclusion. Furthermore, 50% of the patients (n=24) were diagnosed with liver involvement. The majority of patients were included prior to first-line therapy (n=45) and two prior to second- and third-line treatment, respectively.
Table I.
Baseline characteristics of patients (n=47) and association with cfDNA levels at baseline.
| Characteristic | Value | cfDNA, ng/µl [median (95% CI)] | P-valuea |
|---|---|---|---|
| All patients | 47(100) | ||
| Age, years | |||
| Median (interquartile range) | 65 (60-70) | ||
| ≤65 | 24(51) | 0.81 (0.61-0.97) | 0.32 |
| >65 | 23(49) | 0.66 (0.55-0.89) | |
| Sex | 0.64 | ||
| Male | 27(57) | 0.73 (0.61-0.92) | |
| Female | 20(43) | 0.74 (0.57-1.05) | |
| WHO PSb | 0.07 | ||
| 0, 1 | 43(93) | 0.73 (0.61-0.86) | |
| 2 | 3(7) | 1.18 (0.81-2.85c) | |
| Location primary tumor | 0.04 | ||
| Colon | 30(64) | 0.83 (0.61-1.01) | |
| Rectum | 17(36) | 0.65 (0.54-0.81) | |
| Sidedness primary tumor | 0.02 | ||
| Right | 19(40) | 0.87 (0.61-1.20) | |
| Left | 28(60) | 0.66 (0.55-0.80) | |
| Resection status primary tumor | 0.04 | ||
| Resected | 32(68) | 0.63 (0.57-0.77) | |
| Not resected | 15(32) | 0.93 (0.70-1.04) | |
| Number of lines of previous anticancer therapies | 0.07 | ||
| 0 | 45(96) | 0.73 (0.61-0.86) | |
| ≥1 | 2(4) | 1.20 (1.08-1.32c) | |
| Time of metastases | 0.19 | ||
| Synchronous | 26(55) | 0.86 (0.63-0.95) | |
| Metachronous | 21(45) | 0.61 (0.54-0.81) | |
| Number of metastatic sites | 0.24 | ||
| 1 site | 20(43) | 0.73 (0.56-0.85) | |
| >1 site | 27(57) | 0.77 (0.61-0.96) | |
| Metastatic location | |||
| Non-liver | 23(49) | 0.61 (0.55-0.75) | 0.01d |
| Liver (+ other sites) | 24(51) | 0.94 (0.66-1.11) | |
| Non-lung | 27(57) | 0.76 (0.58-0.87) | 0.32e |
| Lung (+ other sites) | 20(43) | 0.66 (0.57-1.21) | |
| Tissue mutation statusf | 0.60 | ||
| RAS/BRAF wild-type | 8(17) | 0.75 (0.49-1.26) | |
| RAS/BRAF mutation | 38(83) | 0.70 (0.59-0.87) | |
| LDHg | <0.01 | ||
| <ULN | 23(56) | 0.66 (0.59-0.79) | |
| >ULN | 18(44) | 0.98 (0.88-1.27) |
Values are expressed as n (%) unless otherwise specified.
aMann-Whitney U-test;
bOne missing value for PS;
cUpper confidence limit held at maximum of sample;
d‘Non-liver’ vs. ‘Liver (+ other sites)’;
e‘Non-lung’ vs. ‘Lung (+ other sites)’;
fOne patient did not have their tumor tissue analyzed due to insufficient material;
gSix missing values for LDH. cfDNA, cell-free DNA; WHO, World Health Organization; PS, performance status; LDH, lactate dehydrogenase; ULN, upper limit of normal (>205 U/l).
A total of 37 patients were treated with FOLFIRI as the study treatment with the addition of anti-EGFR in 18 cases. Furthermore, three patients had contraindications to 5-fluorouracil and were treated with irinotecan in combination with anti-EGFR. Moreover, one patient was treated with CAPOX. A total of six patients deteriorated prior to receiving the first cycle of treatment and were not treated. At the end of the follow-up, disease progression had occurred in 38 and 21 patients had died. No patients were lost to follow-up. The median follow-up time was 12.8 months (range, 0.2-28.4 months) and the median PFS and OS were 8.0 months (95% CI 5.7-8.7) and 21.4 months (95% CI 12.8-not reached), respectively.
None of the patients achieved a CR during treatment, but one patient underwent successful salvage surgery after major tumor regression during chemotherapy. A PR was observed in 11 patients and SD in 24 patients. Early disease progression occurred in 10 patients; including four patients who experienced progressive disease at the first radiographic response evaluation and six patients where radiographic response evaluation was not possible due to symptomatic deterioration.
Baseline levels of cfDNA
The total cfDNA levels were measured by DFA in all baseline samples, with a mean value of 0.84 ng/µl (95% CI 0.71-0.98 ng/µl) and a median level of 0.73 ng/µl, respectively. The baseline levels of cfDNA in the study cohort were significantly higher compared to the cfDNA levels of a previously analyzed normal cohort of 94 presumed healthy individuals with a median level of 0.52 ng/µl and mean level of 0.54 ng/µl (95% CI 0.50-0.59, P<0.001) (13).
At baseline, the median cfDNA level was higher in patients with right-sided primary tumor (0.87 ng/µl, 95% CI 0.61-1.20) compared to patients with left-sided primary tumor (0.66 ng/µl, 95% CI 0.55-0.80, P=0.02). The same applied to patients with primary tumor in situ (0.93 ng/µl, 95% CI 0.70-1.04) compared to patients with resected primary tumor (0.63 ng/µl, 95% CI 0.57-0.77, P=0.04). Furthermore, the median cfDNA level was higher in patients with LDH > upper limit of normal (ULN; >205 U/l) (0.66 ng/µl, 95% CI 0.59-0.79) compared to patients with LDH<ULN (0.98 ng/µl, 95% CI 0.88-1.27, P<0.01) and in patients with liver involvement (0.94 ng/µl, 95% CI 0.66-1.11) compared to patients without liver involvement (0.61 ng/µl, 95% CI 0.55-0.75, P=0.01) (Table I and Fig. 1).
Figure 1.
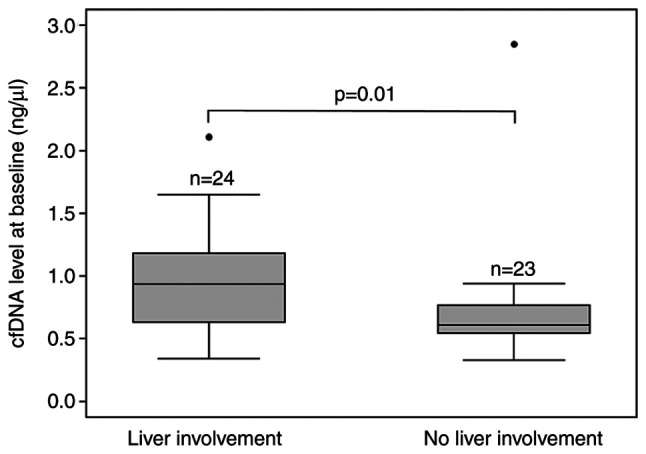
Association of baseline plasma cfDNA levels with liver involvement in patients with metastatic colorectal cancer (n=47). Box and whisker plot with 25th, 50th and 75th percentiles, upper and lower adjacent values and outliers (dots) of cfDNA levels. Statistically significant differences are presented as P-values calculated using the Mann-Whitney U-test. cfDNA, cell-free DNA.
A low positive correlation was determined between tumor burden measured as the sum of diameters (mm) of target lesions according to RECIST version 1.1(19) and cfDNA levels at baseline (R2=0.17, adjusted R2=0.15, P=0.01, n=36; Fig. 2). LDH and cfDNA levels at baseline had a moderate positive correlation (R2=0.45, adjusted R2=0.43, P<0.01, n=40; Fig. S1).
Figure 2.
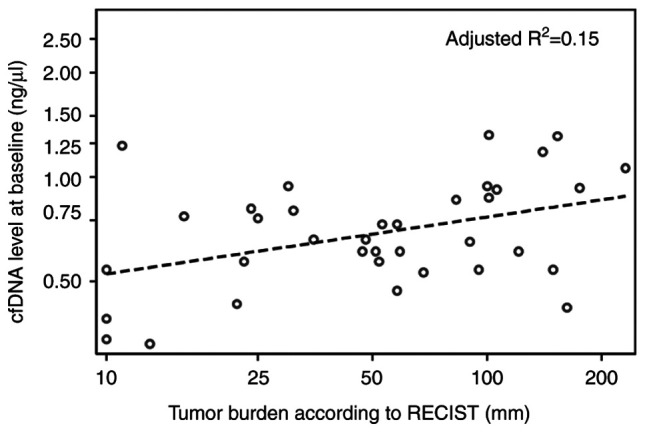
Relation between tumor burden according to RECIST version 1.1 (mm) and baseline plasma cfDNA levels (ng/µl) evaluated using a linear regression model for all patients (n=36). cfDNA, cell-free DNA.
Prognostic value of baseline cfDNA levels
To determine the most appropriate cutoff, the study population was divided into four groups according to internal cohort quartiles of cfDNA levels at baseline. This exploratory approach revealed that patients with the highest level (the upper quartile group) had shorter median PFS and OS compared to the other quartile groups (data not shown). Consequently, the study population was dichotomized according to the 75th percentile of cfDNA levels at baseline. High cfDNA levels were associated with unfavorable outcome. The median PFS was 3.8 months in patients with high cfDNA levels compared to 8.5 months in those with lower cfDNA levels (hazard ratio=3.03, 95% CI 1.46-6.29, P<0.01, n=47). The Median OS was 5.0 months in patients with high cfDNA levels and 26.6 months in patients with low cfDNA levels (hazard ratio=3.48, 95% CI 1.44-8.44, P<0.01, n=47; Fig. 3).
Figure 3.

Kaplan-Meier curves indicating the association between plasma cfDNA levels (ng/µl) and PFS and OS in 47 patients with metastatic colorectal cancer. Patients were dichotomized according to 75th percentile of cfDNA levels at baseline. Low cfDNA, <75th percentile; high cfDNA, ≥75th percentile. Tick marks indicate censored data. n.r., not reached; CI, confidence interval; cfDNA, cell-free DNA; PFS, progression-free survival; OS, overall survival.
According to the univariate analysis, baseline cfDNA levels, number of lines of previous anticancer therapy and liver involvement were associated with PFS. Baseline cfDNA levels, WHO performance status, number of lines of previous anticancer therapy and LDH levels were associated with OS. On multivariate analysis, the baseline cfDNA levels remained a significant predictor of PFS and OS (Table SI).
Baseline cfDNA levels and correlation with tumor response
In patients with PR, SD and progressive disease as the best response, the baseline cfDNA levels were 0.73, 0.70 and 1.05 ng/µl, respectively. The median baseline cfDNA level was significantly lower in patients who achieved DC (0.73 ng/µl) compared to those who progressed early (1.05 ng/µl, P=0.04) (Table SII). When dichotomizing the total cohort according to the 75th percentile of cfDNA levels at baseline, the DC rate was significantly higher in the group with low cfDNA levels (91%) compared to the group with high cfDNA levels (42%, P<0.001; Table II).
Table II.
Baseline cfDNA levels and tumor response according to RECIST version 1.1.
| Best response | Total (n=45)a | <75th percentile (n=33) | ≥75th percentile (n=12) |
|---|---|---|---|
| DC (PR+SD) | 35(78) | 30(91) | 5 (42%) |
| PD | 10(22) | 3(9) | 7 (58%) |
P=0.001 according to Fisher's exact test. The total cohort (n=47) was dichotomized acccording to the 75th percentile of cfDNA levels at baseline.
aFor two patients, the first response evaluation had not yet been performed. cfDNA, cell-free DNA; DC, disease control; PR, partial response; SD, stable disease; PD, progressive disease.
Prognostic value of cfDNA dynamics during treatment
Blood samples were available from patients at baseline (n=47); prior to the third treatment cycle (n=31); the first (n=33), second (n=22) and third response evaluation during treatment (n=17); and at progression (n=22). At baseline (pretreatment), the median cfDNA level of 0.73 ng/µl was significantly higher compared to the median levels during treatment of 0.55, 0.56, 0.44 and 0.48 ng/µl at the third treatment cycle and first, second and third response evaluation during treatment, respectively (Fig. 4A).
Figure 4.

Median plasma cfDNA levels at various time-points: Baseline; just before third treatment cycle; first, second and third response evaluation; and progression in (A) patients with metastatic colorectal cancer (n=47) and (B) a subgroup of patients with liver involvement (n=24). Box and whisker plots with 25, 50 and 75 percentiles, upper and lower adjacent values and outliers (dots) of cfDNA levels. P-values were calculated using the Mann-Whitney U-test. *P<0.01. cfDNA, cell-free DNA.
In the normal reference cohort, the mean cfDNA level was 0.54 ng/µl as measured by DFA. In brief, it may be hypothesized that levels >0.54 ng/µl are caused by the tumor's contribution to total cfDNA levels. It was further hypothesized that ctDNA is eliminated when cfDNA levels decrease to ≤0.54 ng/µl during treatment. Thus, using 0.54 ng/µl as a cut-off, it was evaluated whether a change in cfDNA during treatment is predictive of outcomes.
Patients with cfDNA levels ≤0.54 ng/µl at both baseline and at the first response evaluation demonstrated the longest median PFS (10.0 months) and patients with cfDNA levels ≤0.54 ng/µl at baseline with an increase to >0.54 ng/µl at the first response evaluation had the shortest median PFS (4.0 months). The remaining patients with high levels at baseline (>0.54 ng/µl), which either remained high or fell below the cut-off, had a median PFS of 8.4 months (P=0.03, comparing PFS in the four groups described above by log-rank test). No significant association between the change in cfDNA levels and OS (P=0.09) or DC rate (P=0.66) was obtained. Of note, only 33 patients were included in these analyses.
Enhanced effect of total cfDNA in patients with liver involvement
A total of 24 patients had liver involvement at inclusion. This subgroup of patients had significantly higher cfDNA levels at baseline (0.94 ng/µl) compared to the study population as a whole. In addition, there appeared to be a larger decrease in the cfDNA level after initiation of treatment in the group of patients with liver involvement (Fig. 4A and B); however, this was not significant due to the small number of patients (data not shown) and no conclusion may be drawn from the current dataset. During treatment, cfDNA levels in the subgroup with liver involvement were comparable to the cfDNA levels in the whole study population (Fig. 4A and B).
In the subgroup of patients with liver involvement, there was a moderate correlation between the tumor burden measured as the sum of diameters (mm) of target lesions according to RECIST version 1.1(18) and cfDNA levels at baseline (R2=0.42, adjusted R2=0.38, P=0.01, n=18; Fig. 5).
Figure 5.

Relation between tumor burden according to RECIST version 1.1 (mm) and baseline plasma cfDNA levels (ng/µl) evaluated using a linear regression model for patients with liver involvement (n=18). cfDNA, cell-free DNA.
When this subgroup was dichotomized according to the 75th percentile of cfDNA levels at baseline, it was confirmed that high cfDNA levels were associated with unfavorable outcome. The median PFS was 1.6 months in patients with high cfDNA levels compared to 7.7 months in those with lower cfDNA levels (hazard ratio=2.04, 95% CI 0.77-5.43, P=0.15, n=24). The median OS was 3.3 months in patients with high cfDNA levels and 21.7 months in patients with low cfDNA levels (hazard ratio=3.27, 95% CI 1.03-10.37, P=0.04, n=24; Fig. 6).
Figure 6.

Kaplan-Meier curves indicating the association between plasma cfDNA levels (ng/µl) and PFS and OS in 24 patients with metastatic colorectal cancer with liver involvement. Patients were dichotomized according to 75th percentile of cfDNA levels at baseline. Low cfDNA, <75th percentile; high cfDNA, ≥75th percentile. Tick marks indicate censored data. n.r., not reached; CI, confidence interval; cfDNA, cell-free DNA; PFS, progression-free survival; OS, overall survival.
Discussion
In the present study, a potential clinical utility of cfDNA was demonstrated, quantified by an assay applied directly to the biological sample (plasma), for patients with mCRC receiving standard of care systemic treatment. The present study was performed for the purpose of testing the clinical utility of cfDNA levels monitored by DFA during palliative systemic treatment for mCRC. When analyzing the total level of cfDNA, the analysis is not limited to patients with detectable mutations in the blood (e.g., RAS or BRAF mutations). Quantification of cfDNA levels may be performed for all patients regardless of geno- or phenotype, thus being a potential biomarker for the majority of cancer patients. Previously, cfDNA measurements by DFA have proven valuable as a prognostic and predictive marker in patients with liver metastasis of CRC treated with hepatic arterial infusion (16) and chemoembolization (15). To the best of our knowledge, the present study was the first to examine cfDNA levels by DFA during palliative systemic treatment for mCRC. The method proved feasible and the major findings were in agreement with the existing literature concerning cfDNA levels and mCRC.
In the present study, RECIST was used as a simplified pseudo-marker for tumor burden. A more precise marker of the full tumor burden may be tumor volume defined by positron emission tomography (PET)-CT. Nygaard et al (20) evaluated the correlation of the disease volume determined by PET-CT and the cfDNA level. In accordance with the present results, Nygaard et al (20) obtained a weak correlation supporting the assumption that cfDNA levels reflect both tumor burden and biological mechanisms, giving a complex picture of the disease.
In the present study, the baseline level of cfDNA was related to outcomes in terms of response, PFS and OS. High cfDNA levels at baseline correlated with shorter PFS, OS and lower DC rate, which is in line with previous studies (7-11). In both uni- and multivariate analysis, high cfDNA levels demonstrated sustained prognostic value. The results of the multivariate analyses should be interpreted with caution due to the low numbers of participants and events and should be confirmed in larger-scale studies.
Not only baseline cfDNA levels had a prognostic value. Also compared measurements of cfDNA levels at baseline and at the first response evaluation had a correlation with PFS. Patients who maintained a cfDNA level equal to those in healthy individuals had the longest median PFS. Patients with an increasing cfDNA level from baseline to first response evaluation had the shortest median PFS. This should be interpreted with caution. The small sample size and variations of cfDNA do not allow for any solid conclusions to the utility of dynamics during therapy, but merely a hypothesis description, since the present study was the first report of this marker in this setting. In general, an externally and pre-defined cut-off based on healthy individuals increases the clinical utility, by allowing for reproducibility and comparison with other cohorts. The results indicated that early changes in cfDNA levels during systemic therapy may contribute to the early identification of treatment effects. Similarly, Thomsen et al (21) reported a potential correlation between the initial dynamics in methylated ctDNA and early prediction of treatment benefit during first-line systemic treatment for mCRC.
In accordance with the present results, several previous studies have demonstrated higher cfDNA levels in patients with liver involvement compared to those without (7,22,23). Hence, the subgroup of patients with liver involvement appears to have a different biology when it comes to cfDNA and therefore, this subgroup was evaluated independently. In addition to higher baseline cfDNA levels, a tendency to a steeper decrease in cfDNA was observed when initiating treatment. Otherwise, the subgroup was comparable to the study group as a whole. Baseline cfDNA levels and early changes in cfDNA levels were correlated with outcome. These results should be interpreted with caution due to the low number of patients. In future studies, it would be relevant to further investigate patients with liver involvement in comparison to patients without any liver involvement.
Carcinoembryonic antigen (CEA) is a well-known and widely used prognostic marker in mCRC. CEA measurements are not available in the present study. The correlation between CEA and cfDNA was previously analyzed by our group using ddPCR methods, suggesting that cfDNA appeared to provide a much stronger prognostic value than CEA on multivariate analysis (22,24). It was previously reported that LDH levels correlate with cfDNA levels (12,22) and the results of the present study were similar. In accordance with the present study, a previous study by our group (22) reported that the LDH level is not an independent prognosticator of survival.
The major limitation of the present study was the relatively low sample size, and naturally, the present data call for validation in external cohorts. Furthermore, measurements of cfDNA levels by DFA may be slightly influenced by comorbidity, as previously reported (7), and are hence not absolutely cancer-specific. Finally, with the current method, it is not possible to adjust for contamination by DNA from lymphocytes as described in relation to PCR-based techniques (25). It would have been relevant to perform an age-matched comparison to the normal cohort, but this was not possible, since no information regarding age was available for the healthy control group. These factors are important to acknowledge, particularly in early-stage disease, where the contribution from normal cells weighs higher.
Although the current focus of research on liquid biopsies is on the more time- and resource-demanding ctDNA analyses, it is of utmost importance not to overlook potential clinically relevant information of cfDNA, which may be obtained from the feasible and simple approach of the present study.
In conclusion, quantification of cfDNA is a promising prognostic tool when personalizing the treatment of mCRC and the current study presented a simple, rapid and inexpensive method to determine cfDNA levels. Future research should evaluate the value of cfDNA determined by DFA, e.g., in a randomized clinical trial evaluating treatment guided by cfDNA levels against standard of care in mCRC or other cancer types.
Supplementary Material
Acknowledgements
The content of the present study was presented at the Danish Society for Clinical Oncology meeting (online event) on April 22, 2021 (abstract no. 1).
Funding Statement
Funding: This project was funded by grants from The Danish Cancer Society (Kræftens Bekæmpelse; grant nos. R99-A6323-14-S25 and R269-A15652), Novo Nordisk Foundation (grant no. 9297), Health Research Foundation of Central Denmark Region (grant no. A1602) and Aase and Ejner Danielsen Foundation (grant no. 10-002001). The funding sources had no influence on the study design, data collection, analysis and interpretation of data and neither took part in the writing of the report nor in the decision to submit the article for publication.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Authors' contributions
KLGS designed the outline of the study. LBC, IGL, KLGS, AKB, NP and BSS collaborated in the acquisition of data. KLGS and LC analyzed and interpreted the data. BSS, NP and AKB participated in analyzing and interpreting of the data. LBC wrote the manuscript. KLGS, AKB, NP, BSS and IGL revised the manuscript critically for important intellectual content. KLGS, NP and LBC confirm the authenticity of all raw data. All authors have read and agreed to the published version of the manuscript.
Ethics approval and consent to participate
The study was approved by Regional Committees on Health Research Ethics for Central Denmark Region (June 12, 2017; no. 1-10-72-111-17) and Danish Data Protection Agency (May 2, 2017; no. 1-16-02-153-17). Both oral and written informed consent was obtained from all participants for inclusion in the study, also specifically for the collection of blood samples prior to, during and after therapy (as described in the methods section) and for the use of the samples in scientific research.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.van der Geest LG, Lam-Boer J, Koopman M, Verhoef C, Elferink MA, de Wilt JH. Nationwide trends in incidence, treatment and survival of colorectal cancer patients with synchronous metastases. Clin Exp Metastasis. 2015;32:457–465. doi: 10.1007/s10585-015-9719-0. [DOI] [PubMed] [Google Scholar]
- 3.Van Cutsem E, Cervantes A, Nordlinger B, Arnold D. Metastatic colorectal cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2014;25 (Suppl 3):iii1–iii9. doi: 10.1093/annonc/mdu260. ESMO Guidelines Working Group. [DOI] [PubMed] [Google Scholar]
- 4.Mandel P, Metais P. Les acides nucléiques du plasma sanguin chez l'homme. C R Seances Soc Biol Fil. 1948;142:241–243. [PubMed] [Google Scholar]
- 5.Leon SA, Shapiro B, Sklaroff DM, Yaros MJ. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977;37:646–650. [PubMed] [Google Scholar]
- 6.Stroun M, Anker P, Maurice P, Lyautey J, Lederrey C, Beljanski M. Neoplastic characteristics of the DNA found in the plasma of cancer patients. Oncology. 1989;46:318–322. doi: 10.1159/000226740. [DOI] [PubMed] [Google Scholar]
- 7.Spindler KL, Appelt AL, Pallisgaard N, Andersen RF, Brandslund I, Jakobsen A. Cell-free DNA in healthy individuals, noncancerous disease and strong prognostic value in colorectal cancer. Int J Cancer. 2014;135:2984–2991. doi: 10.1002/ijc.28946. [DOI] [PubMed] [Google Scholar]
- 8.Spindler KL, Pallisgaard N, Andersen RF, Brandslund I, Jakobsen A. Circulating free DNA as biomarker and source for mutation detection in metastatic colorectal cancer. PLoS One. 2015;10(e0108247) doi: 10.1371/journal.pone.0108247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spindler KL, Pallisgaard N, Vogelius I, Jakobsen A. Quantitative cell-free DNA, KRAS, and BRAF mutations in plasma from patients with metastatic colorectal cancer during treatment with cetuximab and irinotecan. Clin Cancer Res. 2012;18:1177–1185. doi: 10.1158/1078-0432.CCR-11-0564. [DOI] [PubMed] [Google Scholar]
- 10.Spindler KL, Pallisgaard N, Andersen RF, Jakobsen A. Changes in mutational status during third-line treatment for metastatic colorectal cancer - results of consecutive measurement of cell free DNA, KRAS and BRAF in the plasma. Int J Cancer. 2014;135:2215–2222. doi: 10.1002/ijc.28863. [DOI] [PubMed] [Google Scholar]
- 11.Spindler KG, Boysen AK, Pallisgård N, Johansen JS, Tabernero J, Sørensen MM, Jensen BV, Hansen TF, Sefrioui D, Andersen RF, et al. Cell-Free DNA in metastatic colorectal cancer: A systematic review and meta-analysis. Oncologist. 2017;22:1049–1055. doi: 10.1634/theoncologist.2016-0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldshtein H, Hausmann MJ, Douvdevani A. A rapid direct fluorescent assay for cell-free DNA quantification in biological fluids. Ann Clin Biochem. 2009;46(Pt 6):488–494. doi: 10.1258/acb.2009.009002. [DOI] [PubMed] [Google Scholar]
- 13.Boysen AK, Sørensen BS, Lefevre AC, Abrantes R, Johansen JS, Jensen BV, Schou JV, Larsen FO, Nielsen D, Taflin H, et al. Methodological development and biological observations of cell free DNA with a simple direct fluorescent assay in colorectal cancer. Clin Chim Acta. 2018;487:107–111. doi: 10.1016/j.cca.2018.09.029. [DOI] [PubMed] [Google Scholar]
- 14.Schou JV, Larsen FO, Sørensen BS, Abrantes R, Boysen AK, Johansen JS, Jensen BV, Nielsen DL, Spindler KL. Circulating cell-free DNA as predictor of treatment failure after neoadjuvant chemo-radiotherapy before surgery in patients with locally advanced rectal cancer. Ann Oncol. 2018;29:610–615. doi: 10.1093/annonc/mdx778. [DOI] [PubMed] [Google Scholar]
- 15.Boysen AK, Jensen M, Nielsen DT, Mortensen FV, Sørensen BS, Jensen AR, Spindler KL. Cell-free DNA and chemoembolization in patients with liver metastases from colorectal cancer. Oncol Lett. 2018;16:2654–2660. doi: 10.3892/ol.2018.8925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boysen AK, Schou JV, Jensen BV, Nielsen D, Sørensen BS, Johansen JS, Spindler KG. Prognostic and predictive value of circulating DNA for hepatic arterial infusion of chemotherapy for patients with colorectal cancer liver metastases. Mol Clin Oncol. 2020;13(77) doi: 10.3892/mco.2020.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lefèvre AC, Kronborg C, Sørensen BS, Krag SRP, Serup-Hansen E, Spindler KG. Measurement of circulating free DNA in squamous cell carcinoma of the anus and relation to risk factors and recurrence. Radiother Oncol. 2020;150:211–216. doi: 10.1016/j.radonc.2020.06.045. [DOI] [PubMed] [Google Scholar]
- 18.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 19.McShane LM, Altman DG, Sauerbrei W, Taube SE, Gion M, Clark GM. REporting recommendations for tumour MARKer prognostic studies (REMARK) Eur J Cancer. 2005;41:1690–1696. doi: 10.1038/sj.bjc.6602678. Statistics Subcommittee of the NCI-EORTC Working Group on Cancer Diagnostics. [DOI] [PubMed] [Google Scholar]
- 20.Nygaard AD, Holdgaard PC, Spindler KL, Pallisgaard N, Jakobsen A. The correlation between cell-free DNA and tumour burden was estimated by PET/CT in patients with advanced NSCLC. Br J Cancer. 2014;110:363–368. doi: 10.1038/bjc.2013.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thomsen CB, Hansen TF, Andersen RF, Lindebjerg J, Jensen LH, Jakobsen A. Early identification of treatment benefit by methylated circulating tumor DNA in metastatic colorectal cancer. Ther Adv Med Oncol. 2020;12(1758835920918472) doi: 10.1177/1758835920918472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spindler KL, Pallisgaard N, Appelt AL, Andersen RF, Schou JV, Nielsen D, Pfeiffer P, Yilmaz M, Johansen JS, Hoegdall EV, et al. Clinical utility of KRAS status in circulating plasma DNA compared to archival tumour tissue from patients with metastatic colorectal cancer treated with anti-epidermal growth factor receptor therapy. Eur J Cancer. 2015;51:2678–2685. doi: 10.1016/j.ejca.2015.06.118. [DOI] [PubMed] [Google Scholar]
- 23.Hamfjord J, Guren TK, Dajani O, Johansen JS, Glimelius B, Sorbye H, Pfeiffer P, Lingjærde OC, Tveit KM, Kure EH, et al. Total circulating cell-free DNA as a prognostic biomarker in metastatic colorectal cancer before first-line oxaliplatin-based chemotherapy. Ann Oncol. 2019;30:1088–1095. doi: 10.1093/annonc/mdz139. [DOI] [PubMed] [Google Scholar]
- 24.Spindler KG, Demuth C, Sorensen BS, Johansen JS, Nielsen D, Pallisgaard N, Hoegdall E, Pfeiffer P, Vittrup Jensen B. Total cell-free DNA, carcinoembryonic antigen, and C-reactive protein for assessment of prognosis in patients with metastatic colorectal cancer. Tumour Biol. 2018;40(1010428318811207) doi: 10.1177/1010428318811207. [DOI] [PubMed] [Google Scholar]
- 25.Pallisgaard N, Spindler KL, Andersen RF, Brandslund I, Jakobsen A. Controls to validate plasma samples for cell free DNA quantification. Clin Chim Acta. 2015;446:141–146. doi: 10.1016/j.cca.2015.04.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.