

Abstract
Steroid metabolism in humans originates from cholesterol and involves several enzyme reactions including dehydrogenation, hydroxylation, and carbon–carbon bond cleavage that occur at regio- and stereo-specific points in the four-membered ring structure. Cytochrome P450s occur at critical junctions that control the production of the male sex hormones (androgens), the female hormones (estrogens) as well as the mineralocorticoids and glucocorticoids. An important branch point in human androgen production is catalyzed by cytochrome P450 CYP17A1 and involves an initial Compound I-mediated hydroxylation at the 17-position of either progesterone (PROG) or pregnenolone (PREG) to form 17-hydroxy derivatives, 17OH-PROG and 17OH-PREG, with approximately similar efficiencies. Subsequent processing of the 17-hydroxy substrates involves a C17–C20 bond scission (lyase) activity that is heavily favored for 17OH-PREG in humans. The mechanism for this lyase reaction has been debated for several decades, some workers favoring a Compound I-mediated process, with others arguing that a ferric peroxo- is the active oxidant. Mutations in CYP17A1 can have profound clinical manifestations. For example, the replacement of the glutamic acid side with a glycine chain at position 305 in the CYP17A1 structure causes a clinically relevant steroidopathy; E305G CYP17A1 displays a dramatic decrease in the production of dehydroepiandrosterone from pregnenolone but surprisingly increases the activity of the enzyme toward the formation of androstenedione from progesterone. To better understand the functional consequences of this mutation, we self-assembled wild-type and the E305G mutant of CYP17A1 into nanodiscs and examined the detailed catalytic mechanism. We measured substrate binding, spin state conversion, and solvent isotope effects in the hydroxylation and lyase pathways for these substrates. Given that, following electron transfer, the ferric peroxo- species is the common intermediate for both mechanisms, we used resonance Raman spectroscopy to monitor the positioning of important hydrogen-bonding interactions of the 17-OH group with the heme-bound peroxide. We discovered that the E305G mutation changes the orientation of the lyase substrate in the active site, which alters a critical hydrogen bonding of the 17-alcohol to the iron-bound peroxide. The observed switch in substrate specificity of the enzyme is consistent with this result if the hydrogen bonding to the proximal peroxo oxygen is necessary for a proposed nucleophilic peroxoanion-mediated mechanism for CYP17A1 in carbon–carbon bond scission.
Graphical Abstract

INTRODUCTION
Cytochrome P450 CYP17A1 is a heme-containing monooxygenase involved in the production of an array of steroid products, including the male and female sex hormones as well as glucocorticoids that regulate many biological processes.1–4 It is a critical enzyme and drug target responsible for conversion of the steroids progesterone (PROG) and pregnenolone (PREG) to androgens androstenedione (AD) and dehydro-epiandrosterone (DHEA), respectively, also referred to hereafter as the Δ4 and Δ5 pathways.5,6 Specifically, CYP17A1 performs standard hydroxylase chemistry, presumably utilizing a “Compound I” intermediate to initiate hydrogen abstraction at the C17 position, and radical recombination in the classic “oxygen-rebound” mechanism,7,8 generating 17α-OH pregnenolone (17OH-PREG) or 17α-OH progesterone (17OH-PROG). In forming the androgens, these hydroxylated products then undergo a second oxidative “lyase” cycle to cleave the C17–C20 bond to form DHEA and AD. Interestingly, different organisms have different preferences for the pregnenolone (Δ5) pathway as compared to that using the Δ4 (progesterone) route.6 Human CYP17A1 dramatically favors the Δ5 pathway to produce DHEA with little androstenedione (AD), the efficiency for 17OH-PREG being 50-fold greater than that for 17OH-PROG.9 CYP17A1 is also at a critical branch point between androgen production and the formation of glucocorticoids (e.g., cortisol) and mineralocorticoids (e.g., aldosterone). The latter two steroid classes require hydroxylation at the 17-position, but not side-chain cleavage, and are then oxygenated by a different cytochrome P450 at the 21-position.6 The control of the carbon–carbon bond scission reaction of CYP17A1 is critical. If this lyase reaction is inhibited or slowed, a lack of androgen and estrogen production can lead to the various signs of disordered sexual development.10,11
Thus, a detailed understanding of the molecular mechanisms of CYP17A1 is important (Scheme 1). While the classical mechanisms of aliphatic carbon hydroxylation through the Compound I mechanism are well understood, there has been an ongoing debate as to the chemical process of C–C bond cleavage in the CYP17A1 lyase step. While atmospheric dioxygen and two reducing equivalents are required, debate centers whether Compound I is involved or the precursor heme-peroxoanion is the “active oxygen” intermediate. Much work by Guengerich and co-workers, as well as others,12 supports the compound I-mediated C–C bond cleavage pathway for several cytochromes P450, including CYP17A1 (green arrows in Scheme 1). On the other hand, in the case of CYP17A1, strong evidence for the major pathway involving the ferric peroxoanion (red arrows in Scheme l) has accumulated from measurements of an inverse solvent isotope effect,13 active site proton delivery mutations,14 trapping of the key heme-peroxo-hemiketal intermediate at low temperature,15–19 and detailed QM/MM calculations.20 As shown in Scheme 1, a key feature enabling this pathway is the potential formation of a hydrogen bond between the 17-OH group and the proximal oxygen atom of the bound peroxo fragment, as observed by Raman spectroscopy.16,17,19 The distal oxygen atom of the heme-bound peroxo is free for a nucleophilic attack on the 20-keto group forming a peroxohemiketal intermediate. The α-hydroxyketone sets up an ideal precursor to a six-membered transition state, which can break, either homolytically or heterolytically20 to yield the lyase products. Positioning of the Δ4 and Δ5 substrates, together with their 17-OH precursors has been shown to contribute to the prevalence of the PREG/17OH-PREG verses PROG/17OH-PROG in the human enzyme.9,21 The high-resolution X-ray structures provide an explanation, pointing to a more favorable orientation of the Δ5 substrate.21 Using resonance Raman (rR) spectroscopy on cryo-stabilized peroxo states with bound lyase substrates, the origin of this preference for the Δ5 pathway was explained as a critical positioning of the 17-OH hydrogen bond donor between the distal and proximal peroxo oxygen atoms.15–19 This shift is due to the difference in A-ring interactions with Asn202 and either the Δ4 3-keto or Δ5 3-hydroxyl group. Mutation of asparagine to serine, which switches the interactions at the A-ring, was shown to reverse the preference to favor the Δ4 pathway.22
Scheme 1. Reactions of the Ferric Peroxo Anion of CYP17A1 with Bound Lyase Substratesa.

aShown are the Compound I (green arrows) and the nucleophilic (red arrows) mechanisms of carbon–carbon bond scission. Proton-dependent uncoupling pathways that lead to hydrogen peroxide or reduction of Compound I to water are shown with blue arrows.28–31 In a separate set of experiments, gas chromatography mass spectrometry (GCMS) analyses of annealed samples of the cryoreduced oxy complexes of nanodics incorporated CYP17A1 with each of the lyase substrates was shown to contain significant amounts of the expected products, DHEA or AD.
It is important to consider the implications of these two mechanisms for interpreting experimental data acquired for the lyase reaction. The Compound I-driven catalysis (green arrows) is common for most P450-catalyzed reactions.7,8,12 According to this mechanism, two protons from water are required to make Compound I and for product formation. Therefore, P450 catalysis is slower in D2O, with kinetic solvent isotope effect (KSIE) usually observed in the range 1.3–3.5 for CYP10123–25 and for CYP19A1.13 The alternative pathway (green arrows) does not involve protonation of the peroxo-ferric intermediate and a step, at least partially rate limiting, involving cleavage of the O–O bond and Compound I formation. If the product is predominantly generated via the peroxoanion that avoids the proton-dependent pathway, and if the protonation leads to mostly unproductive uncoupling, then the overall product formation rate is expected to be faster in D2O than in H2O. This is indeed the case for the CYP17A1-catalyzed lyase reaction, as an inverse KSIE from 0.4 to 0.83 was repeatedly observed by our laboratory, and others, for both substrates 17OH-PREG and 17OH-PROG and with the wild-type protein. In addition, mutations of the add/alcohol pair that are thought to be involved in P450 proton transfer events also led to an inverse isotope effect for the lyase reaction.11,14,19,26,27 These observations strongly indicate that the peroxo anion pathway is the main mechanism for the C–C bond scission in CYP17A1.
Variations in residues at the active site of P450s are known to alter the proton delivery pathways that are necessary for generating the Compound I intermediate.32 Others perturb the binding of redox partners, generate covalent inhibition of both hydroxylase and lyase activities, or alter substrate binding.11 Various mutations in CYP17A1 have profound clinical relevance. An interesting set are those that cause “isolated 17,20-lyase deficiency (ILD)”,33,34 with multiple clinical manifestations. A particularly relevant mutation is E305G. This position is part of a largely conserved “acid–alcohol” pair in the cytochrome P450s that has been extensively studied in several proteins.23,35,36 This residue, in the classic P450cam (CYP101), is involved in a mechanistically important structural change in the I-helix that alters the proton delivery pathway necessary for efficient hydroxylase activity.37,38 However, in other P450 systems, there does not appear to be this major rearrangement39 and the glutamate side chain appears to play a less critical role. Auchus and co-workers described a E305G variant in a clinical presentation of gynecomastia.10 In this pioneering work and subsequent investigations,40,41 the E305G mutation was also found to alter the preference for the Δ4 and Δ5 substrates. Given our understanding of a potential peroxo reactivity in this enzyme, we undertook this study of the E305G mutation in human CYP17A1 to explore the mechanistic role of this residue and its impact on Δ4/Δ5 lyase specificity.
RESULTS
Functional Studies.
The effect of the mutation of glutamic acid to glycine at position 305 (E305G) in the CYP17A1 sequence is reminiscent of the results when residues impinging on the A-ring of the steroid nucleus are altered. In recent publications, we showed that mutation of asparagine 202 to serine (N202S) had a dramatic effect on the lyase activities of CYP17A1.22 Noteworthy is the shift from a preference of the Δ5 steroid 17-hydroxypregnenolone (17OH-PREG) to favor the Δ4 17-hydroxyprogesterone (17OH-PROG). Not only were the binding preferences of the lyase substrates switched with the mutation, but the turnover rate of the mutant was faster for the Δ4 lyase precursor, as reflected in a 775-fold change in kcat/Km for the Δ5/Δ4 ratio in the N202S mutant. To examine the role of E305 in the metabolism of 17OH-PREG and 17OH-PROG by CYP17A1, we generated the E305G mutation and self-assembled the protein in nanodiscs to provide a nativelike membrane environment.
We first measured hydroxylase activities. With progesterone (PROG) as substrate, the wild-type enzyme generated the 17-hydroxy product with rates of 4.77 ± 0.2 and 3.76 ± 0.06 nmol/(min nmol) in H2O and D2O, respectively. This yields a KSIE of 1.27 ± 0.074. The E305G mutant, under the same conditions, has corresponding rates of 0.57 ± 0.02 and 0.30 ± 0.01 nmol/(min nmol) for a KSIE of 1.90 ± 0.13. For PREG hydroxylations, the rates are also slower in the E305G mutant (1.1 ± 0.36 in H2O and 0.74 ± 0.21 in D2O, KSIE = 1.48) than in the wild-type CYP17A1 (3.2 ± 0.2 in H2O and 2.5 ± 0.13 in D2O, KSIE = 1.28); all rates are in units of nmol/(min nmol) P450. The significant decrease of hydroxylation rates observed in the E305G mutant with both substrates suggests a perturbation of the proton delivery required for Compound I formation and the oxygen-rebound mechanism for P450 hydroxylations.6 In addition, the slower rates of hydroxylation can be due, in part, to the rearrangement of substrates in the active site of the mutant, resulting in the less optimal positioning of C17 for the attack by the Compound I ferryl oxygen. Note that the KSIE for the hydroxylation reactions are both larger in the “normal” direction, consistent with the need for efficient proton delivery required for the generation of Compound I. A similar, although more dramatic, effect was noted in the D251N mutant of CYP101A1 when an alternate water pathway of proton delivery was opened.24
The maximal rates of product formation for both lyase substrates were then quantitated. The lyase activities in both H2O and D2O were measured (Figure 1), and the kinetic solvent isotope effect (KSIE) calculated for both wild-type and mutant protein, Figure 2. Interestingly, the mutation decreases the activity with 17OH-PREG (Δ5) as substrate but increases the activity of 17OH-PROG (Δ4) carbon–carbon bond scission. This dramatic reversal of the Δ5/Δ4 specificity is remarkably similar to that seen with the N202S mutation20 and in the clinical presentation of the E305 mutation. Clearly, two different enzymatic mechanisms are operating in hydroxylation and lyase activities. While the hydroxylation reaction is accepted to proceed through a Compound I-mediated pathway7,8,12 there has been a long-standing debate regarding the nature of the key intermediate involved in the lyase reaction, some arguing for a Compound I intermediate,12,26 while others have suggested a peroxo-mediated pathway.5,6,15,19,22,26
Figure 1.
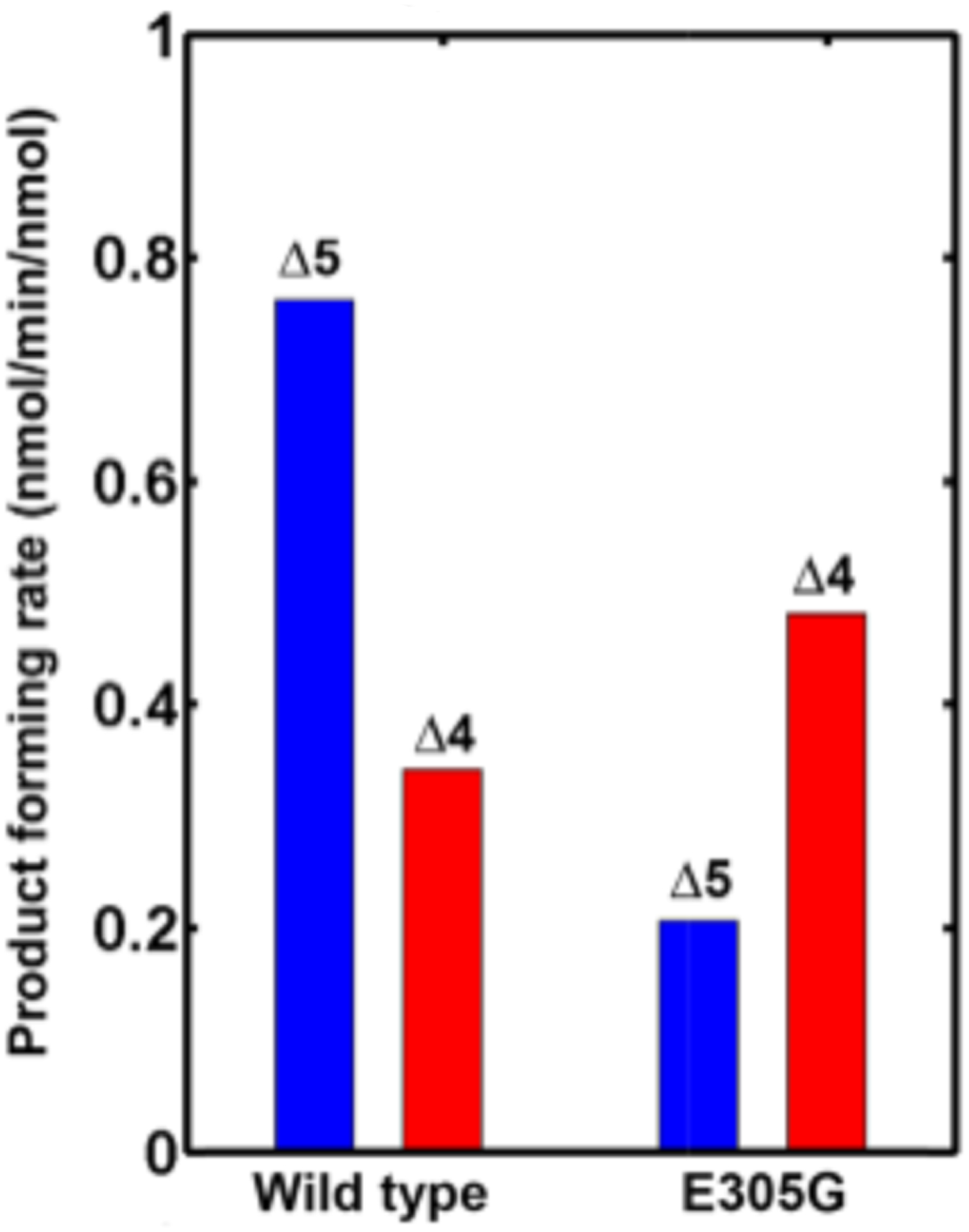
Product forming rates for wild-type CYP17A1 and the E305G mutant for the Δ5 lyase substrate (17OH-pregnenolone) and the Δ4 substrate (17OH-progesterone).
Figure 2.

Comparison of the wild-type and mutant kinetic solvent isotope effects (KSIE) for the Δ5 (17OH-PREG) and Δ4 (17OH-PROG) lyase substrates.
The observation of an unusual inverse kinetic solvent isotope effect (KSIE), wherein the lyase reaction actually runs faster in a deuterated solvent,14 is consistent with the nucleophilic peroxoanion mechanism proposed by us and others.5,6,15–18 As the KSIE reports on the proton-dependent uncoupling pathway and is dependent on the positioning of the 17-hydroxyl hydrogen bonding with the iron-peroxo oxygen, we measured the rate of lyase product formation for the E305G mutant in both D2O and H2O and compared them to that of the wild-type protein, Figure 2 Strikingly, the E305G mutation also reverses the Δ4/Δ5 pattern in the measured solvent isotope. The KSIE still remains less than unity, an inverse isotope effect.
Spectroscopic Studies.
Given that the KSIE is directly probing the peroxoanion reactivity, we elected to conduct the same series of cryoradiolytic trapping and resonance Raman spectroscopic characterizations previously reported for the wild-type protein.16–18 This approach provides a direct structural picture of the hydrogen-bonding interactions of the Fe–O–O fragment with active site proton donors. Based on the crystallographic studies of CYP17 by Scott and co-workers,21 the most reasonable candidates for such donors are the C17–OH groups of the two lyase substrates, as depicted in Scheme 1. Figure 3 shows the 16O2/18O2 difference resonance Raman (rR) spectra of the (A) 17OH-PREG and (B) 17OH-PROG ferrous dioxygen complexes of the CYP17A1 E305G mutant in nanodiscs. The ν(16O–16O) modes appear near 1132 cm−1, while the ν(18O–18O) stretching modes occur near 1065 cm−1, with the observed isotope shift being close to that expected for a harmonic oscillator.42 These ν(O–O) stretching frequencies are indicative of an O–O fragment influenced by a hydrogen bond interaction, in this case provided by the C17–O–H fragment of the substrates as was described and characterized in our previous work.15–18 The Fe–O–O fragment of the OH-PREG-bound sample exhibits a ν-(Fe–16O) mode at 540 cm−1, with the corresponding ν(Fe–18O) mode appearing at 513 cm−1, the expected isotopic shift for an Fe–O–O fragment, and a three-body oscillator (Fe–O–O). The corresponding spectral data for the 17OH-PROG-bound E305G protein show a ν(Fe–16O) mode at 528 cm−1, with the corresponding ν(Fe–18O) mode occurring at 501 cm−1. Again, this isotopic shift is in good agreement with expectations. These observed rR spectral patterns are of crucial importance as our earlier works have shown that such Fe–O–O fragments of P450 dioxygen adducts exhibit a ν(Fe–16O) near 535 cm−1 for a nonhydrogen bound Fe–O–O fragment, near 525 cm−1 with an H-bond directed to the proximal oxygen (Op) and nearer to 545 cm−1 for a fragment involved in an H-bonding interaction with the terminal oxygen (Ot) of the Fe–Op–Ot fragment.15–18,43
Figure 3.

16O2–18O2 Difference rR spectra of (A) 17OH-PREG-bound and (B) 17OH-PROG-bound dioxygen adducts of the E305G CYP17A1 mutant. Spectra were measured using 413.1 nm excitation.
Comparison of these results for the E305G variant with those previously obtained for the wild-type enzyme supports the conclusion that the hydrogen-bonding patterns observed for these two lyase substrates are shifted. That is, with wild type, the bound 17OH-PREG provides an H-bond to the proximal oxygen, exhibiting a ν(Fe–16O) at 526 cm−1, whereas for the E305G variant, this same substrate is oriented to generate an H-bonding interaction primarily with the terminal oxygen, exhibiting a ν(Fe–16O) at 540 cm−1. Conversely, for the 17OH-PROG complex in the E305G mutant, an H-bonding interaction with the proximal oxygen is documented, with a ν(Fe–16O) appearing at 528 cm−1, whereas the corresponding complex with the wild-type enzyme generates an H-bonding interaction with the terminal oxygen; i.e., ν(Fe–16O) at 542 cm−1. Thus, the rR data for E305G and wild-type enzyme show that the H-bond interactions with the Fe–O–O fragment are reversed.
To ascertain if the mutation also alters hydrogen bonding in the case of the ferric peroxo-intermediate, as in our earlier works,16–18 we generated the peroxo states in CYP17A1 by cryoreduction of the ferrous dioxygen adduct of E305G CYP17A1 at 77 K using cobalt-60 radiation. The rR difference spectrum obtained with the lyase substrate 17OH-PREG is shown in Figure 4A. Clearly seen are two positive bands at 797 and 779 cm−1 in the mid-frequency region, both shifting as expected (38 and 40 cm−1) upon 18O2 substitution. Following our earlier work,16–18,43 these features are most reasonably assigned to the ν(O–O) modes of the peroxo- and hydroperoxo-intermediates, respectively. The corresponding ν(Fe–O) modes are seen at 560 cm−1 (peroxo) and 570 cm−1 (hydroperoxo). Interestingly, in the corresponding difference trace seen for the 17OH-PROG-bound E305G sample, Figure 4B, only the ferric peroxo-intermediate forms, as confirmed by the observation of its ν(Fe–16O) mode at 553 cm−1 (Δ16/18 = 28 cm−1) and its ν(16O–16O) mode at 811 cm−1 (Δ16/18 = 37 cm−1). It is noted that this latter mode occurs about 10–15 cm−1 higher than is typically seen for P450 peroxo-intermediates,16–18,43,44 i.e., 790–795 cm−1. It is also important to emphasize that the 17OH-PREG bound E305G mutant generates not only the peroxo-intermediate but also some hydroperoxo, even at 77 K. Importantly, the relative ease of formation of hydroperoxo intermediates for a given substrate/enzyme complex can provide useful insight regarding the branch point for uncoupling and Compound I formation versus peroxoanion reaction with a susceptible carbon.
Figure 4.

rR Spectra data for the peroxo/hydroperoxo states of E305G CYP17A1 with (A) 17OH-PREG or (B) 17OH-PROG bound. These are formed at 77 K from irradiation of the dioxygen adducts. Spectra were measured with 442 nm excitation.
Previous work in our laboratory discovered a key intermediate in the nucleophilic mechanism of carbon–carbon bond scission by gently raising the temperature of the trapped peroxo states. In wild-type CYP17A1, this allowed the nucleophilic attack of the peroxo anion on the 20-carbonyl of the steroid substrate to form a stabilized peroxohemiketal intermediate (central structure in Scheme l) characterized by unique optical and Raman signatures, the ν/(16O–16O) and ν(Fe–16O) modes being observed at 791 and 579 cm−1 (Table 1).16,18,22 The rR spectra for E305G mutant bound with 17OH-PREG was obtained after annealing of the cryoreduced sample at 165 K for 1 min then returning to 77 K for data collection (Figure 5A). Excitation of the Raman bands with 442 nm radiation, close to the 440 nm Soret peak of the peroxo states,16,18,22 enhances Fe–O–O modes and reveals that the peroxo anion intermediate has disappeared, leaving only weak difference features observed at 779/740 cm−1 which are associated with the ν(O–O) mode of the hydroperoxo-species, whose ν(Fe–O) mode is seen at 570/545 cm−1. These data are entirely consistent with those observed for the hydroperoxo intermediates of cryoradiolytically trapped states of other P450s;16–18,44 noting however that this intermediate was not observed for wild-type CYP17A1 bound with OH-PREG, the peroxo-intermediate efficiently converting to the hemiketal (Table 1).16 It is also noted that after annealing to 190 K and employing the 406.7 nm excitation line (Figure 5B), no evidence was obtained for the accumulation and persistence of the hemiketal intermediate of the OH-PREG substrate, i.e., apparently, its accumulation was minimal, though the possibility it may have formed and disappeared at some temperatures between 77 and 190 K cannot be ruled out.
Table 1.
Summary of the ν(Fe–O) and ν(O–O) Modes in the Peroxo, Hydroperoxo, and Hemiketal Forms for Wild-Type and E305G CYP17A1a
Figure 5.

rR Spectral data for irradiated dioxygen adducts of the E305G mutant of CYP17A1 with bound 17OH-PREG.(A) 165 K and measured with the excitation at 442 nm. and (B) after annealing to 190 K and measured with excitation at 406.7 nm, which closely matches the absorption maximum of the hemiketal intermediate. Trace C shows the 16O2/18O2 difference spectrum for 17-OHPROG after annealing to 16S K for 1 min and exciting with 406 nm light. (Methods section) (D) The rR spectrum measured with this 406 nm excitation after further annealing of the 17OH-PROG to 190 K where enhancement is highest for the hemiketal intermediate.16
In contrast to this behavior observed for 17OH-PREG, samples containing the Δ4 lyase substrate, 17OH-PROG, when annealed at 165 K and measured with the 442 nm excitation line (spectrum not shown), show no evidence for remaining peroxo intermediate seen in Figure 4B. When the sample was further measured with 406 nm excitation the rR spectrum showed a ν(O–O) mode occurring at 786 cm−1, with a corresponding ν(Fe–O) mode at 571 cm−1 (Figure 5C). These are quite close to those seen before, employing 406.7 nm excitation, for the hemiketal intermediate of the wild-type enzyme arising from peroxo-attack on 17OH-PROG, i.e., those were seen at 785 and 573 cm−1 (Table 1).17 This behavior reveals that the peroxo intermediate formed at 77K efficiently converts to the hemiketal intermediate at temperatures between 77K and 165K. Further resonance Raman studies were performed after carefully annealing the 17OH-PROG sample to a higher temperature (190 K), using the 406.7 nm excitation line, which more effectively enhances the modes of the hemiketal intermediate.16,17 As seen in Figure 5D, the intensity of the 585 cm−1 mode has increased relative to the intensity of the 1133 cm−1 mode observed for the residual oxy precursor, which is a reliable standard. It is important to note that earlier studies have shown that peroxo- and hydroperoxo intermediates are typically not stable above ~170 K and are not observable using the 406 nm excitation line.16,17 Consequently, we attribute the observed spectral difference pattern to the peroxohemiketal intermediate, consistent with that reported previously for other P450 hemiketal intermediates.16–18 As is discussed further below, these observations are consistent with the functional studies, which document much more favorable lyase efficiency for the OH-PROG substrate compared to the OHPREG substrate with the E305G mutant.
DISCUSSION
Cytochrome P450 CYP17A1 plays a critical role in human steroid metabolism, being the committed step in the production of androgens via a carbon–carbon bond cleavage between carbon-17 at the apex of the steroid D ring and the 20-carbonyl of the methylketone side chain. The enzyme functions via a 17-hydroxylation followed by the C17–20 lyase chemistry, which also uses atmospheric dioxygen and two reducing equivalents. Isomerization of the A-ring, with an alcohol (Δ5) or ketone (Δ4) at the 3-position, is connected through a steroid dehydrogenase. The precursor substrates pregnenolone (Δ5) and progesterone (Δ4) are also feedstocks for the production of the mineralocorticoids and glucocorticoids, and hence, the activity of CYP17A1 serves as a regulatory branch point. A clinical presentation10 provided for the isolation of a point mutation, E305G, with the patient displaying significant steroidopathy. Detailed in vitro investigations, summarized in several references,10,40,45 revealed a shift in the human preference for the Δ5 substrate over Δ4 in metabolism with this amino acid change. This shift was reminiscent of our observations and conclusions for the effects of a mutation at position-202, wherein the normal asparagine was converted to serine.22 Given this mechanistic similarity, we hypothesize that the clinical effect of these two mutations could share a mechanistic origin. With this in mind, we undertook a functional characterization of the E305G mutation in CYP17A1 self-assembled into nanodiscs and used resonance Raman spectroscopy to determine the vibrational frequencies of iron-peroxo fragments as altered by the E305 mutation.
Table 1 summarizes the vibrational data for cryoradiolytically generated intermediates for the Δ4 and Δ5 hydroxylation substrates (PREG and PROG) together with those for the lyase substrates (17OH-PREG and 17OH-PROG). Both wild-type and the E305G mutant are shown. The peroxo level states are generated at 77 K by radiolysis. Interestingly, as noted before,9,10 with the preferred Δ5 substrate bound, only the peroxoanion is generated, yet with 17OH-PROG bound, the hydroperoxo is also generated at low temperature, consistent with proton delivery inhibiting turnover. However, replacing the glutamate at position-305 with a glycine reverses this preference. Both peroxoanion and hydroperoxo are found at 77 K for 17OH-PREG but only the peroxoanion with 17OH-PROG. Similarly, the stronger inhibiting effect of E305G mutation on the Compound I-driven hydroxylation of the PROG, as compared to the PREG, suggests the less favorable protonation of peroxo-intermediate with the PROG substrate in the mutant. This provides a rationalization of the increased turnover of the Δ4 substrates in the E305G mutant, exactly as noted in previous work with the N202S mutant.22
Figure 6 shows the structure of CYP17A1 from Scott and co-workers21 focusing on the I-helix and the interactions of E305 and N202 with themselves and the steroid A-ring. Molecular dynamic simulations40 have indicated that the I-helix is fairly immobile in CYP17A1, and hydrogen-bonding interactions between E305, N202, and the A-ring alcohol or keto group can be a dominant player in the repositioning of the substrate in the active site and subsequent alteration in the lyase activity preference for Δ5 or Δ4 substrates. This reorientation is crucial because a critical point in the lyase cycle, shown in Scheme 1, is the formation of the ferric peroxo-intermediate. If the proton shuttle assembly is highly functional, proton transfer is facile and formation of the hydroperoxo-intermediate readily occurs, followed by peroxide release and second protonation leading to cleavage of the Fe–O–O linkage to form the highly reactive Compound I intermediate. On the other hand, if a susceptible (electrophilic) carbon, such as the C20 carbonyl of the two lyase substrates, is present in the active site, attack of the nucleophilic terminal oxygen of the peroxo-intermediate can compete with proton delivery, thereby diverting the cycle away from the Compound I pathway. Clearly, the efficiency of this diversion depends on the relative nucleophilicity of the terminal oxygen, which is modulated by the directionality of H-bond donors to either the proximal or terminal oxygen atoms of the Fe–Op–Ot fragment. In addition, the relative efficiency of the lyase reactions depends critically on the presence and directionality of the hydrogen bond from the C17-alcohol to the heme-bound peroxo oxygens. The rR data obtained for the E305G variant reveal differential H-bonding interactions that adequately explain the observed functional differences.
Figure 6.

Structure of the CYP17A1 active site (A) with the Δ5 substrate 17-hydroxypregnenolone or (B) the Δ4 substrate 17-hydroxyprogesterone bound. The positions and interactions between N202, E305, and the substrates are indicated. This figure is comparable to one published by Scott and co-workers,21 which was made from the PDB files 4NKZ (PREG) and 4NKY (PROG) from Petrunak et al.21
Thus, in addition to providing a mechanistic origin for the clinical presentation of an impaired steroidogenic pathway, we have additional experimental evidence for the detailed chemistry of carbon–carbon bond scission in CYP17A1. An important factor is the placement of the lyase substrates in the active site such that a hydrogen bond formed between the 17-alcohol of the steroid and the proximal oxygen atom of the heme-bound peroxoanion. This facilitates generation of the key hemiketal intermediate, following the nucleophilic attack of the distal oxygen of the peroxo anion on the 20-carbonyl, that can subsequently decompose to the observed products via a homolytic or heterolytic process. The reaction coordinates for both were documented using QM/MM calculations,20 which identified the transition state as the main pathway for lyase activity with equal barriers to either homolysis or heterolysis. While cytochrome P450s are involved in hydroxylation and carbon–carbon bond cleavage reactions at multiple places in steroid metabolism, the case of CYP17A1 seems to be unique in being able to take advantage of an α-hydroxyketone structure, wherein the alcohol can hydrogen bond with the peroxo oxygen, to set up facile nucleophilic mechanism.
METHODS
The protocols for the experiments described in this manuscript duplicate those reported in earlier publications. Briefly, the E305G mutant of full-length human CYP17A1 was generated using a codon-optimized full-length synthetic gene (DNA 2.0) cloned into the pCWori plasmid.47 Mutagenesis was performed using iProof polymerase with primers obtained from Integrated DNA Technologies. Sequencing was performed by ACGT Inc. Expression, purification, and nanodisc incorporation of CYP17A1, for both wild-type and the E305G mutant were performed as previously described.48
Spectral dissociation constants were determined by the sequential addition of the substrate dissolved in methanol to a cuvette containing 1.5 μM CYP17A1 in 100 mM potassium phosphate (pH 7.4) and 50 mM NaCl. Absorption spectra were recorded from 350 to 800 nm in a Cary 300 spectrophotometer at 25 °C. Spectral dissociation constants were estimated by plotting the change in the peak to trough A390 – A420 difference spectra versus substrate concentration and fitting the resultant binding isotherm to a quadratic tight-binding model. For each substrate/enzyme pair, titrations were performed in triplicate, and the percent high spin was calculated at each substrate concentration using a peak to trough extinction coefficient in the difference spectra (ε390 – ε420) of 100 mM−1 cm−1.
Rat cytochrome P450 oxidoreductase and cytochrome b5 were expressed and purified as described previously15,17 and self-assembled into nanodiscs containing either wild-type or E305G CYP17A1 as described.48 Product formation rates were determined under saturating concentrations (50 μM) of PREG, PROG, OHPREG, or OHPROG in 100 mM potassium phosphate buffer (pH 7.4) containing 50 mM NaCl. One milliliter of reaction mixtures contained 0.2 nmol of wild-type or E305G CYP17A1 incorporated in nanodiscs, 0.8 nmol of cytochrome P450 oxidoreductase, and, for lyase reaction, 0.8 nmol of cytochrome b5. The samples were incubated at 37 °C for 30 min before the reactions were initiated by the addition of 400 nmol of NADPH. After 10 min, reactions were quenched by the addition of 50 μL of 8.9 N sulfuric acid, and the mixtures were flash-frozen in liquid nitrogen and stored at −80 °C.
Methods for extraction and quantitation of the products have been described in detail elsewhere.13,17 Briefly, products were extracted with 1.5 mL of dichloromethane after the addition of internal standards and dried under a stream of nitrogen. Products of the reactions with progesterone and 17OH-PROG were analyzed by reversed-phase high-performance liquid chromatography after reconstitution in 100 μL 50% methanol with cortexolone as an internal standard. Separation was achieved using an ACE3 C18 column and a linear gradient beginning with a 5:5:90 methanol/acetonitrile/water ratio and ending with a 45:45:10 methanol/acetonitrile/water mixture. Mobile phases were supplemented with 0.2% formic acid, and the product was observed optically at 240 nm. Products of the reactions with pregnenolone and 17OH-PREG were derivatized and analyzed by GCMS using deuterated hydroxypregnenolone and DHEA as an internal standards. For derivatization, the dried dichloromethane extracts were reconstituted in 50 uL of pyridine followed by addition of 50 uL of MSTFA + 1% TMCS derivatization reagent (Thermo Scientific), and incubated at 70 °C for 90 min.
Sample Preparation for Resonance Raman (rR) Spectroscopy.
Ferric rR samples contained 150 μM E305G CYP17A1 nanodiscs in 100 mM potassium phosphate buffer (pH 7.4) with 15% (v/v) glycerol and 450 μM 17OH-PREG or 17OH-PROG. Oxy–ferrous samples contained 150 μM E305G CYP17A1 nanodiscs in 100 mM potassium phosphate (pH 7.4), 250 mM NaCl, and 30% (v/v) distilled glycerol. 17OH-PREG, 17-OHPROG, PREG, or PROG was added to a final concentration of 450 μM. Samples were deoxygenated under argon and reduced with a 1.5-fold molar excess of sodium dithionite in the presence of 3.8 μM methyl viologen. Each sample was then chilled to −15 °C in a dry ice/ethanol bath and oxygenated by being bubbled with 16O2 or 18O2 for 5 s. Samples were immediately flash-frozen in liquid N2 and stored at 77 K. Cryoradiolytic reduction was performed as described.16,17,39
Resonance Raman spectra of the oxy complexes and cryo-irradiated samples were recorded using a Spex 1269 spectrometer attached to a Spec-10 liquid nitrogen-cooled detector with a 2048-pixel resolution (Princeton Instruments, Princeton, NJ). The slit width was set at 150 μm, and a 1200 g/mm grating was used. Spectra were calibrated with fenchone (Sigma-Aldrich, Milwaukee, WI) and processed with Grams/32 AI software (Galactic Industries, Salem, NH). Spectra were collected using back scattering (180°) geometry with the laser beam being focused by a cylindrical lens to form a line image on the frozen sample contained in 5 mm outside diameter NMR tubes (WG-5 ECONOMY, Wilmad). The NMR tubes were positioned in a homemade double-walled quartz low-temperature cell filled with liquid nitrogen, i.e., all rR measurements were performed at 77 K. The sample tubes were spun to avoid local heating. The rR data for the dioxygen adducts were measured using the 413.1 nm line from a Kr+ laser (Coherent Innova Sabre Ion Laser). The laser power was adjusted to ≤1 mW and the total collection time was ~4 h. Samples of the cryo-irradiated oxy ND:CYP17A1 were excited using 441.6 nm line provided by a He–Cd laser (IK Series He–Cd laser, Kimmon Koha Co., Ltd.) or the 406.7 nm excitation line from the Kr+ laser. The total acquisition time for these cryo-irradiated samples was 6 h for the samples excited with the 441.6 nm laser line and 8–9 h when employing the 406.7 nm excitation line.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health MIRA award to SGS (2R35 GM118145) and by NIGMS RO1 GM125303 to J.R.K. The authors thank Dr. Jay A. LaVerne, Notre Dame Radiation Laboratory (Notre Dame University, IN), a facility of the US Department of Energy, Office of Basic Energy Science for his assistance with the cryo-irradiation procedures.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.biochem.1c00282
The authors declare no competing financial interest.
Contributor Information
Yilin Liu, Department of Chemistry, Marquette University, Milwaukee, Wisconsin 53233, United States.
Yelena Grinkova, Department of Biochemistry, University of Illinois Urbana-Champaign, Urbana, Illinois 61801, United States.
Michael C. Gregory, Department of Biochemistry, University of Illinois Urbana-Champaign, Urbana, Illinois 61801, United States; Present Address: Merck Pharmaceutical, 2000 Galloping Hill Road, Kenilworth, New Jersey 07033, United States
Ilia G. Denisov, Department of Biochemistry, University of Illinois Urbana-Champaign, Urbana, Illinois 61801, United States
James R. Kincaid, Department of Chemistry, Marquette University, Milwaukee, Wisconsin 53233, United States;.
Stephen G. Sligar, Department of Biochemistry and Department of Chemistry, University of Illinois Urbana-Champaign, Urbana, Illinois 61801, United States;.
REFERENCES
- (1).Nebert DW; Russell DW Clinical importance of the cytochromes P450. Lancet 2002, 360, 1155–1162. [DOI] [PubMed] [Google Scholar]
- (2).Gomez L; Kovac JR; Lamb DJ CYP17A1 inhibitors in castration-resistant prostate cancer. Steroids 2015, 95, 80–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Porubek D CYP17A1: A Biochemistry, Chemistry, and Clinical Review. Curr. Top. Med. Chem 2013, 13, 1364–1384. [DOI] [PubMed] [Google Scholar]
- (4).Yoshimoto FK; Auchus RJ The diverse chemistry of cytochrome P450 17A1 (P450c17, CYP17A1). J. Steroid Biochem. Mol. Biol 2015, 151, 52–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Akhtar M; Wright JN; Lee-Robichaud P A review of mechanistic studies on aromatase (CYP19) and 17α-hydroxylase-17,20-lyase (CYP17). J. Steroid Biochem. Mol. Biol 2011, 125, 2–12. [DOI] [PubMed] [Google Scholar]
- (6).Gilep AA; Sushko TA; Usanov SA At the crossroads of steroid hormone biosynthesis: The role, substrate specificity and evolutionary development of CYP17. Biochim. Biophys. Acta, Proteins Proteomics 2011, 1814, 200–209. [DOI] [PubMed] [Google Scholar]
- (7).Groves JT; McClusky GA; White RE; Coon MJ Aliphatic hydroxylation by highly purified liver microsomal cytochrome P-450. Evidence for a carbon radical intermediate. Biochem. Biophys. Res. Commun 1978, 81, 154–160. [DOI] [PubMed] [Google Scholar]
- (8).Ortiz de Montellano PR Substrate Oxidation by Cytochrome P450 Enzymes. In Cytochrome P450: Structure, Mechanism, and Biochemistry, 4th ed.; Ortiz de Montellano PR (Ed.); Springer International Publishing: New York, 2015; pp 111–176. [Google Scholar]
- (9).Mathieu AP; LeHoux JG; Auchus RJ Molecular dynamics of substrate complexes with hamster cytochrome P450c17 (CYP17): mechanistic approach to understanding substrate binding and activities. Biochim. Biophys. Acta, Gen. Subj 2003, 1619, 291–300. [DOI] [PubMed] [Google Scholar]
- (10).Sherbet DP; Tiosano D; Kwist KM; Hochberg Z; Auchus RJ CYP17 mutation E305G causes isolated 17,20-lyase deficiency by selectively altering substrate binding. J. Biol. Chem 2003, 278, 48563–48569. [DOI] [PubMed] [Google Scholar]
- (11).Auchus RJ Steroid 17-hydroxylase and 17,20-lyase deficiencies, genetic and pharmacologic. J. Steroid Biochem. Mol. Biol 2017, 165, 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Guengerich FP; Yoshimoto FK Formation and Cleavage of C-C Bonds by Enzymatic Oxidation Reduction Reactions. Chem. Rev 2018, 118, 6573–6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Khatri Y; Gregory MC; Grinkova YV; Denisov IG; Sligar SG Active site proton delivery and the lyase activity of human CYP17A1. Biochem. Biophys. Res. Commun 2014, 443, 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Gregory MC; Denisov IG; Grinkova YV; Khatri Y; Sligar SG Kinetic solvent isotope effect in human P450 CYP17A1-mediated androgen formation: Evidence for a reactive peroxoanion intermediate. J. Am. Chem. Soc 2013, 135, 16245–16247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Gregory M; Mak PJ; Sligar SG; Kincaid JR Differential Hydrogen Bonding in Human CYP17 Dictates Hydroxylation versus Lyase Chemistry. Angew. Chem., Int. Ed 2013, 52, 5342–5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Mak PJ; Gregory MC; Denisov IG; Sligar SG; Kincaid JR Unveiling the crucial intermediates in androgen production. Proc. Natl. Acad. Sci. U.S.A 2015, 112, 15856–15861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Mak PJ; Duggal R; Denisov IG; Gregory MC; Sligar SG; Kincaid JR Human Cytochrome CYP17A1: The Structural Basis for Compromised Lyase Activity with 17-Hydroxyprogesterone. J. Am. Chem. Soc 2018, 140, 7324–7331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Liu Y; Denisov I; Grinkova Y; Sligar S; Kincaid JR P450 CYP17A1 variant with a disordered proton shuttle assembly retains peroxo-mediated lyase efficiency. Chem. - Eur. J 2020, 26, 16846–16852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Liu YL; Denisov IG; Sligar SG; Kincaid JR Substrate-Specific Allosteric Effects on the Enhancement of CYP17A1 Lyase Efficiency by Cytochrome b(5). J. Am. Chem. Soc 2021, 143, 3729–3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Bonomo S; Jorgensen FS; Olsen L Mechanism of Cytochrome P450 17A1-Catalyzed Hydroxylase and Lyase Reactions. J. Chem. Inf. Model 2017, 57, 1123–1133. [DOI] [PubMed] [Google Scholar]
- (21).Petrunak EM; DeVore NM; Porubsky PR; Scott EE Structures of Human Steroidogenic Cytochrome P450 17A1 with Substrates. J. Biol. Chem 2014, 289, 32952–32964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Gregory MC; Mak PJ; Khatri Y; Kincaid JR; Sligar SG Human P450 CYP17A1: Control of Substrate Preference by Asparagine 202. Biochemistry 2018, 57, 764–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kim SH; Yang TC; Perera R; Jin SX; Bryson TA; Sono M; Davydov R; Dawson JH; Hoffman BM Cryoreduction EPR and C-13, F-19 ENDOR study of substrate-bound substates and solvent kinetic isotope effects in the catalytic cycle of cytochrome P450cam and its T252A mutant. Dalton Trans 2005, 3464–3469. [DOI] [PubMed] [Google Scholar]
- (24).Vidakovic M; Sligar SG; Li HY; Poulos TL Understanding the role of the essential Asp251 in cytochrome P450cam using site-directed mutagenesis, crystallography, and kinetic solvent isotope effect. Biochemistry 1998, 37, 9211–9219. [DOI] [PubMed] [Google Scholar]
- (25).Makris TM; von Koenig K; Schlichting I; Sligar SG Alteration of P450 distal pocket solvent leads to impaired proton delivery and changes in heme geometry. Biochemistry 2007, 46, 14129–14140. [DOI] [PubMed] [Google Scholar]
- (26).Yoshimoto FK; Gonzalez E; Auchus RJ; Guengerich FP Mechanism of 17,20-Lyase and New Hydroxylation Reactions of Human Cytochrome P450 17A1: O-18 Labeling and Oxygen Surrogate Evidence for a Role of a Perferryl Oxygen. J. Biol. Chem 2016, 291, 17143–17164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Bhatt MR; Khatri Y; Rodgers RJ; Martin LL Role of cytochrome b5 in the modulation of the enzymatic activities of cytochrome P450 17 alpha-hydroxylase/17,20-lyase (P450 17A1). J. Steroid Biochem. Mol. Biol 2017, 170, 2–18. [DOI] [PubMed] [Google Scholar]
- (28).Loida PJ; Sligar SG Molecular recognition in cytochrome P-450: mechanism for the control of uncoupling reactions. Biochemistry 1993, 32, 11530–11538. [DOI] [PubMed] [Google Scholar]
- (29).Gorsky LD; Koop DR; Coon MJ On the stoichiometry of the oxidase and monooxygenase reactions catalyzed by liver microsomal cytochrome P-450. Products of oxygen reduction. J. Biol. Chem 1984, 259, 6812–6817. [PubMed] [Google Scholar]
- (30).Atkins WM; Sligar SG Metabolic switching in cyctochrome P-450cam: deuterium isotope effects on regiospecificity and the monooxygenase/oxidase ratio. J. Am. Chem. Soc 1987, 109, 3754–3760. [Google Scholar]
- (31).Atkins WM; Sligar SG Deuterium isotope effects in norcamphor metabolism by cytochrome P-450cam: kinetic evidence for the two-electron reduction of a high-valent iron-oxo intermediate. Biochemistry 1988, 27, 1610–1616. [DOI] [PubMed] [Google Scholar]
- (32).Martinis SA; Atkins WM; Stayton PS; Sligar SG A conserved residue of cytochrome-P-450 is involved in heme- oxygen stability and activation. J. Am. Chem. Soc 1989, 111, 9252–9253. [Google Scholar]
- (33).Flück CE; Miller WL; Auchus RJ The 17, 20-lyase activity of cytochrome p450c17 from human fetal testis favors the Delta 5 steroidogenic pathway. J. Clin. Endocrinol. Metab 2003, 88, 3762–3766. [DOI] [PubMed] [Google Scholar]
- (34).Petrunak EM; Rogers SA; Aube J; Scott EE Structural and functional evaluation of clinically relevant inhibitors of steroidogenic cytochrome P450 17A1. Drug Metab. Dispos 2017, 45, 635–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Nagano S; Poulos TL Crystallographic study on the dioxygen complex of wild-type and mutant cytochrome P450cam - Implications for the dioxygen activation mechanism. J. Biol. Chem 2005, 280, 31659–31663. [DOI] [PubMed] [Google Scholar]
- (36).Sjodin T; Christian JF; Macdonald IDG; Davydov R; Unno M; Sligar SC; Hoffman BM; Champion PM Resonance Raman and EPR investigations of the D251N oxycytochrome p450(cam)/putidaredoxin complex. Biochemistry 2001, 40, 6852–6859. [DOI] [PubMed] [Google Scholar]
- (37).Martinis SA; Atkins WM; Stayton PS; Sligar SG A Conserved Residue of Cytochrome-P-450 Is Involved in Heme-Oxygen Stability and Activation. J. Am. Chem. Soc 1989, 111, 9252–9253. [Google Scholar]
- (38).Gerber NC; Sligar SG Catalytic Mechanism of Cytochrome-P-450 - Evidence for a Distal Charge Relay. J. Am. Chem. Soc 1992, 114, 8742–8743. [Google Scholar]
- (39).Murarka VC; Batabyal D; Amaya JA; Sevrioukova IF; Poulos TL Unexpected Differences between Two Closely Related Bacterial P450 Camphor Monooxygenases. Biochemistry 2020, 59, 2743–2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Tiosano D; Knopf C; Koren I; Levanon N; Hartmann MF; Hochberg Z; Wudy SA Metabolic evidence for impaired 17 alpha-hydroxylase activity in a kindred bearing the E305G mutation for isolate 17,20-lyase activity. Eur. J. Endocrinol 2008, 158, 385–392. [DOI] [PubMed] [Google Scholar]
- (41).Miller WL The Syndrome of 17,20 Lyase Deficiency. J. Clin. Endocrinol. Metab 2012, 97, 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Nakamoto K Infrared and Raman Spectra of Inorganic and Coordination Compounds, Part A: Theory and Applications in Inorganic Chemistr, 6th ed.; Wiley: Hoboken, NJ, 2009. [Google Scholar]
- (43).Spiro TG; Soldatova AV; Balakrishnan G CO, NO and O-2 as vibrational probes of heme protein interactions. Coord. Chem. Rev 2013, 257, 511–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Denisov IG; Mak PJ; Makris TM; Sligar SG; Kincaid JR Resonance Raman Characterization of the Peroxo and Hydroperoxo Intermediates in Cytochrome P450. J. Phys. Chem. A 2008, 112, 13172–13179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Lee-Robichaud P; Akhtar ME; Akhtar M An analysis of the role of active site protic residues of cytochrome P-450s: mechanistic and mutational studies on 17 alpha-hydroxylase-1 7,20-lyase (P-450(17 alpha) also CYP17). Biochem. J 1998, 330, 967–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Cui Y-L; Zheng Q-C; Zhang J-L; Xue Q; Wang Y; Zhang H-X Molecular Dynamic Investigations of the Mutational Effects on Structural Characteristics and Tunnel Geometry in CYP17A1. J. Chem. Inf. Model 2013, 53, 3308–3317. [DOI] [PubMed] [Google Scholar]
- (47).Imai T; Globerman H; Gertner JM; Kagawa N; Waterman MR Expression and Purification of Functional Human 17-Alpha-Hydroxylase/17,20-Lyase (P450c17) in Escherichia-Coli - Use of This System for Study of a Novel Form of Combined 17-Alpha-Hydroxylase/17,20-Lyase Deficiency. J. Biol. Chem 1993, 268, 19681–19689. [PubMed] [Google Scholar]
- (48).Luthra A; Gregory M; Grinkova YV; Denisov IG; Sligar SG Nanodiscs in the Studies of Membrane-Bound Cytochrome P450 Enzymes. In Methods in Molecular Biology; Springer, 2013; Vol. 987, pp 115–127. [DOI] [PMC free article] [PubMed] [Google Scholar]