

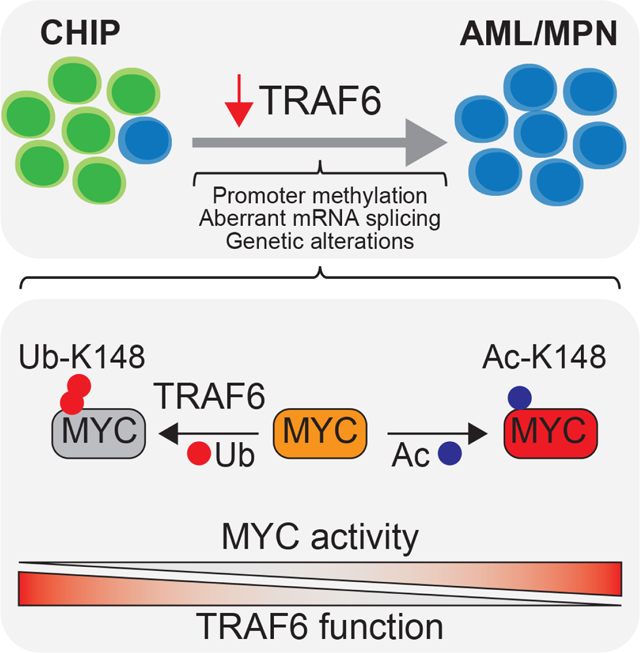
Summary
Clonal hematopoiesis (CH) is an aging-associated condition characterized by the clonal outgrowth of pre-leukemic cells that acquire specific mutations. Although individuals with CH are healthy, they are at an increased risk of developing myeloid malignancies, suggesting that additional alterations are needed for the transition from a pre-leukemia stage to frank leukemia. To identify signaling states that cooperate with pre-leukemic cells we used an in vivo RNAi screening approach. One of the most prominent genes identified was the ubiquitin ligase TRAF6. Loss of TRAF6 in pre-leukemic cells results in overt myeloid leukemia and is associated with MYC-dependent stem cell signatures. TRAF6 is repressed in a subset of patients with myeloid malignancies, suggesting that subversion of TRAF6 signaling can lead to acute leukemia. Mechanistically, TRAF6 ubiquitinates MYC, an event that does not affect its protein stability, but rather represses its functional activity by antagonizing an acetylation modification.
eTOC Blurb
Individuals with clonal hematopoiesis have an increased risk of developing hematopoietic malignancies. Starczynowski and colleagues demonstrate that loss of TRAF6 signaling contributes to the development of AML in the context of clonal hematopoiesis by activating MYC. TRAF6 functions as a tumor-suppressor by ubiquitinating MYC and repressing its transcriptional activity.
Graphical Abstract
Introduction
Hematopoietic stem cells (HSC) accumulate somatic mutations with age in otherwise healthy individuals (Alexandrov et al., 2013). Although most of these mutations are inconsequential, HSC can acquire specific mutations providing a competitive advantage leading to clonal hematopoiesis of indeterminant potential (CHIP)(Mardis et al., 2009; Welch et al., 2012). The most common mutations identified in CHIP occur in DNMT3A and TET2(Challen et al., 2012; Wu et al., 2015), which are also prevalent in myelodysplastic syndrome (MDS), acute myeloid leukemia (AML), and myeloproliferative neoplasms (MPN). Moreover, CHIP mutations are a strong risk factor for subsequent hematologic cancer(Busque et al., 2012; Genovese et al., 2014; Jaiswal et al., 2014; Xie et al., 2014). In line with these clinical observations, inactivation of Tet2 or Dnmt3a in mice confers a competitive advantage of hematopoietic stem and progenitor cells (HSPCs), and susceptibility to hematologic malignancies(Challen et al., 2011; Mayle et al., 2015; Moran-Crusio et al., 2011; Quivoron et al., 2011); yet, overt leukemia occurs with low frequency(Moran-Crusio et al., 2011; Muto et al., 2015; Muto et al., 2013b; Quivoron et al., 2011). These findings indicate that additional alterations are needed for malignant transformation of pre-leukemic HSPCs. However, the factors - beyond mutated genes - that cooperate to induce myeloid malignancies with CHIP-associated pre-leukemic mutations, such as TET2 and DNMT3A, remain largely uncharacterized.
To identify disease-modifying signaling states that cooperate with pre-leukemic HSPCs, we used in vivo RNAi screening and identified that downregulation of certain mediators of innate immune signaling promote development of myeloid malignancies. The innate immune system recognizes pathogens and host cellular by-products by pattern recognition receptors, such as the toll-like receptor (TLR) family. More recently, TLR signaling has been implicated in HSC development, homeostasis, and differentiation(Capitano, 2019). Dysregulation of innate immune pathways is also widely implicated in myeloid malignancies(Barreyro et al., 2018; Sallman and List, 2019; Trowbridge and Starczynowski, 2021). Innate immune pathway activation in HSPCs can lead to hematopoietic defects associated with MDS, and in some cases to leukemic transformation, in the absence of infection(Fang et al., 2017; Muto et al., 2020; Rhyasen et al., 2013; Varney et al., 2015). Moreover, inhibition of innate immune signaling can suppress defined subsets of MDS- and AML-propagating cells, indicating that dysregulated innate immune signaling in HSPCs is associated with certain myeloid malignancies(Ågerstam et al., 2015; Askmyr et al., 2013; Bennett and Starczynowski, 2021; Fang et al., 2012; Rhyasen et al., 2013; Smith et al., 2019). Paradoxically, TLR activation has also been demonstrated to impede AML development by inducing differentiation of leukemic blasts through unknown mechanisms(Eriksson et al., 2017). Herein we describe an unexpected tumor suppressor-like function of innate immune signaling via TRAF6 in myeloid malignancies. These studies implicate subversion of TLR-TRAF6 signaling in the transformation of pre-leukemic HSPCs to overt AML/MPN, and uncover the complex dependencies of dysregulated innate immune pathways in myeloid malignancies.
Results
In vivo RNAi screen identifies Traf6 as putative disease-modifying gene in myeloid leukemia.
Tet2 is a common mutation in pre-leukemic HSCs, however, secondary events are required for malignant transformation(Bowman et al., 2018; Steensma et al., 2015). To identify such cooperating events, we performed an in vivo RNAi screen using Tet2−/− HSPCs (Fig. 1A). We utilized a focused shRNA library targeting regulators of ubiquitination and proteasomal function as dysregulation of these processes and somatic mutations at ubiquitin sites are frequently associated with blood cancer (Narayan et al., 2016)(Table S1). Tet2−/− lineage−cKit+ HSPCs were transduced with four pools containing 5–6 shRNA per gene and a dsRed fluorescence marker. Transduced cells were transplanted into lethally-irradiated recipient mice, and then monitored for disease (Fig. 1A). We used sh-p53 and sh-Ncst as positive controls due to their role in myeloid neoplasms(Klinakis et al., 2011; Loizou et al., 2019). Indeed, both of these clones expanded significantly in peripheral blood (PB) over time, and developed a myeloid disease, while the control group (Tet2−/− HSPCs expressing shRNA-Renilla) did not develop disease (Fig. 1B). Likewise, mice transplanted with Tet2−/− HSPCs transduced with the shRNA pools showed expansion of dsRed+ cells and signs of myeloid disease (Fig. 1B,C). We used deep sequencing to readout the relative representation of shRNA in the Tet2−/− HSPCs pre-transplantation (T0) and in the primary leukemic cells (L1-L4) isolated from the spleen of sick mice (Fig. 1D). Similar to the representation of shRNAs in the original pool of shRNA plasmids (P1-P4), pre-transplanted cells retained more than 98% of the shRNA in an equivalent representation (Fig. 1D and Fig. S1A,B). In contrast, a small fraction of the shRNAs (3–28%) was represented in the primary leukemic cells (Fig. 1D and Fig. S1B) collected from the sick mice and even less (53 out of 1739 shRNAs) showed >5-fold average enrichment. As a result, the distribution of reads was dominated by 1–6 shRNAs in the leukemic cells (Fig. 1E and Fig. S1C), as is evident in the strong shifts in their respective cumulative fractions (Fig. S1A). We didn’t observe much overlap between the dominant shRNAs from primary samples transduced with the same pool (Fig.1E and Fig. S1C), likely due to the strong competition between the malignant clones expanding in the same mice. We ranked the shRNAs based on: a) the average fold-enrichment of individual shRNA per primary malignant sample, b) the number of primary malignant samples showing >1.5 enrichment of an individual shRNA and, c) the number of shRNA >5-fold increased per gene. Genes targeted by the top ranked shRNAs were involved in cellular process such as post-replication repair (Rad18), gene silencing (Trim28), or innate immune/inflammatory signaling (Traf6). PANTHER pathway analysis of the genes targeted by shRNAs with >5-fold increase revealed enrichment of the TLR pathway (Fig. S1D), mainly due to the presence of the E3 ubiquitin ligase Traf6 (one of the top ranked genes in our screen) and its conjugating enzymes (Ube2v1, Ube2n)(Fig.1E). Thus, we hypothesized that TRAF6 loss-of-function may cooperate with TET2 mutations in the development of myeloid malignancies.
Figure 1. In vivo shRNA screening identifies Traf6 in the transformation of pre-leukemic HSPC.
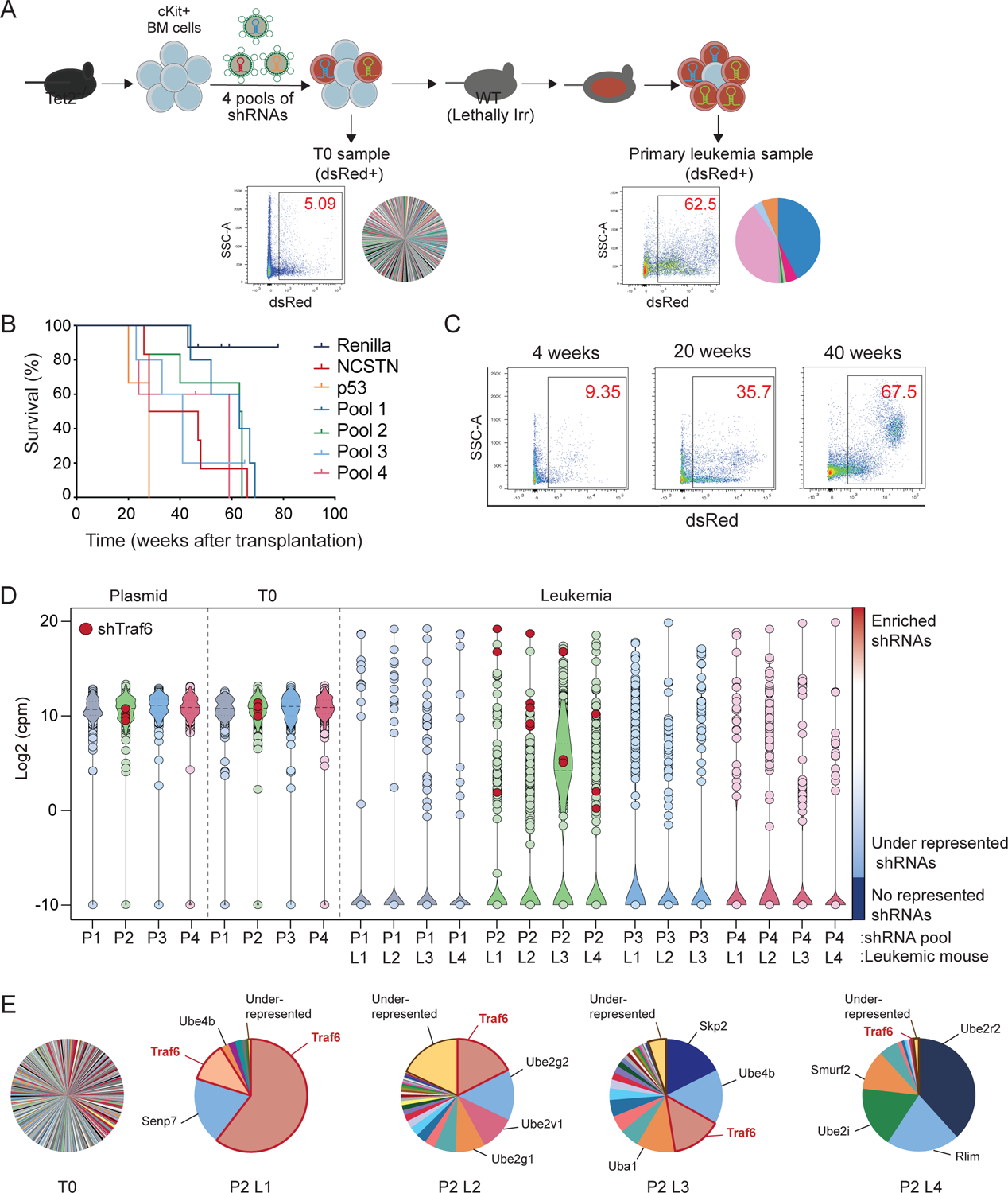
(A) Outline of in vivo shRNA screen to identify cooperating genes/pathways in the transformation of Tet2−/− c-Kit+ BM cells. (B) Kaplan Meier survival analysis of mice transplanted with Tet2−/− cells transduced with the shRNA library (n = 4 mice per pool), shp53 (n = 3), shNCSTN (n = 3), or shRenilla (n = 4). (C) Flow cytometry analysis of dsRed-positive PB cells isolated from mice transplanted with Tet2−/− cells transduced with the shRNA library. (D) Violin plot of the normalized counts for the shRNAs in the plasmid pools (P1-P4), progenitor cells before transplantation (T0) and leukemic cells (L1-L4) collected from moribund mice. Enrichment of shRNAs targeting Traf6 are indicated. (E) Pie charts of the cells transduced with Pool 2 before transplantation and leukemia collected from 4 mice. The area of each shRNA corresponds to the fraction of total reads.
Traf6 loss in pre-leukemic HSPC results in a myeloid malignancy.
To determine the consequences of TRAF6 deletion on pre-leukemic cells, we crossed Traf6f/f and Tet2f/f mice to hematopoietic-specific (VavCre) and inducible (Mx1Cre) recombinase strains (Fig. S2A)(Han et al., 2013; Moran-Crusio et al., 2011). We first analyzed the progenitor phenotype of Traf6−/−;Tet2−/−;VavCre+ (Traf6−/−;Tet2−/−) lineage−Sca1+cKit+ (LSK) compared with control, Traf6−/−;VavCre+ (Traf6−/−) and Tet2−/−;VavCre+ (Tet2−/−) LSK cells using methylcellulose assays. Initially, all LSK populations formed equivalent number of colonies. After the 3rd replating, wild-type (WT) and Traf6−/− LSK cells were unable to form colonies, while Tet2−/− LSK cells formed colonies beyond the 5th replating (Fig. 2A). However, Traf6−/−;Tet2−/− LSK cells exhibited an increased colony replating potential as compared to Tet2−/− cells (P < 0.001)(Fig. 2A). We next performed in vivo competitive repopulation assays using BM cells from Mx1Cre-expressing mice transplanted with equal numbers of WT competitor cells into lethally-irradiate recipient mice. Following engraftment, mice were administered with poly(IC) to induce Cre-mediated excision of the floxed alleles and then analyzed every 4 weeks for hematopoietic reconstitution. As expected, donor-derived Tet2−/− cells out-competed WT cells in primary and secondary BM transplants (Fig. 2B,C). Traf6−/− cells exhibited a significant reduction in donor PB chimerism as previously observed (P < 0.0001)(Fig. 2B,C)(Fang et al., 2018). However, deletion of Tet2 restored the repopulating potential defect of Traf6-deficient cells (Fig. 2B) and contributed to a significant expansion of donor-derived myeloid cells in primary (P < 0.05) and secondary BM transplants (P < 0.0001)(Fig. 2C). Since combined loss of Traf6 and Tet2 resulted in expansion of phenotypically-defined myeloid cells in the PB, we next examined the HSPC proportions in the BM of these mice. In the primary and secondary competitive BM transplantation, the proportions of all donor-derived Traf6−/− HSPCs were significantly reduced as compared to WT donor-derived populations (Fig. 2D,E and Fig. S2B). In contrast, deletion of Tet2 restored the defect of Traf6-deficient HSPCs in the primary competitive BM transplantation (Fig. 2D and Fig. S2B). Moreover, combined loss of Traf6 and Tet2 resulted in an expansion of HSPCs in secondary competitive BM transplantations as compared to deletion of Tet2 alone (Fig. 2E). These observations suggest that loss of TET2 is necessary to prevent attrition of TRAF6-deficient HSPCs, while loss of TRAF6 promotes myeloid-biased hematopoiesis of pre-leukemic TET2-deficient HSPCs.
Figure 2. Deletion of Traf6 in pre-leukemic HSPCs results in myeloid progenitor expansion.
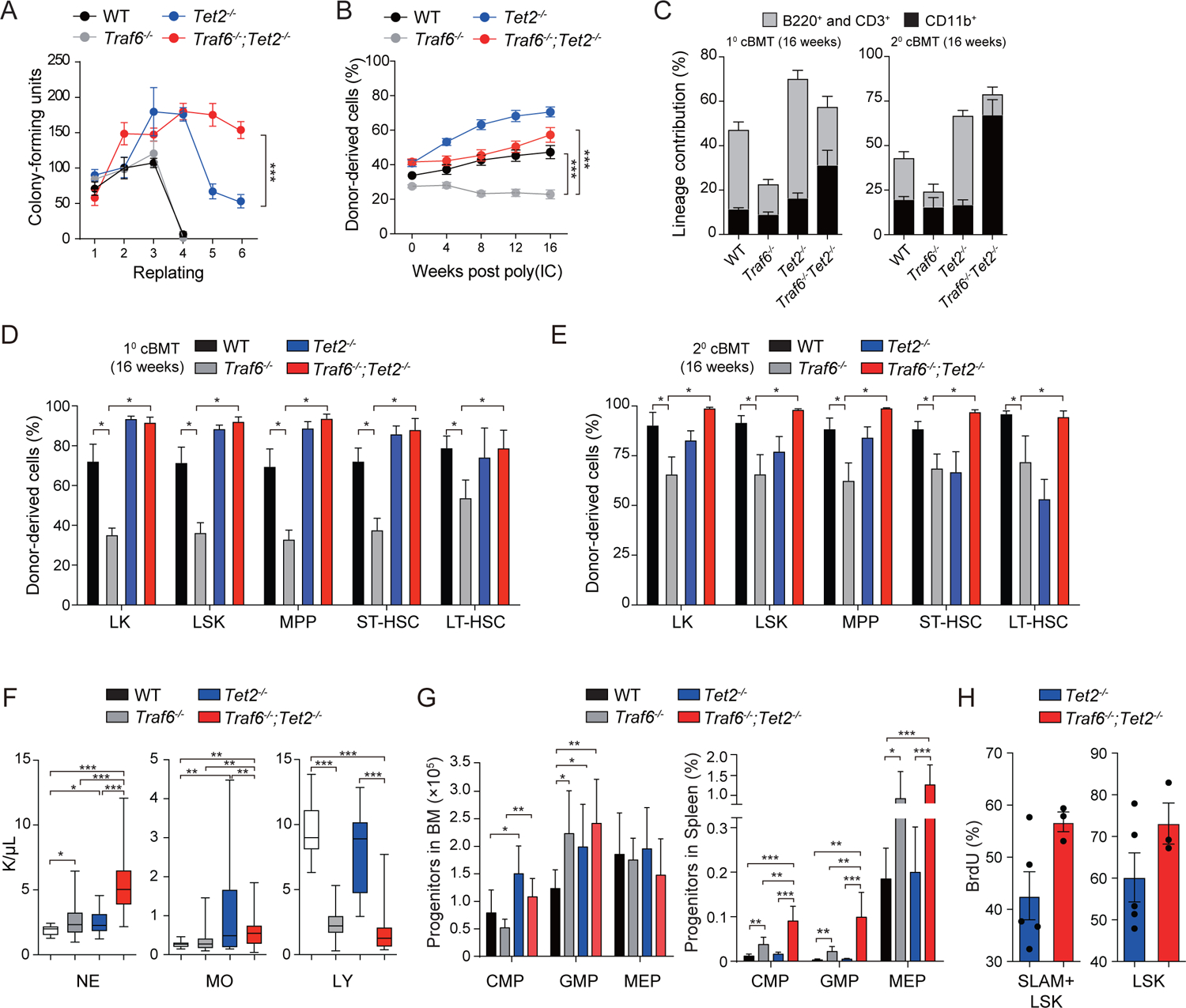
(A) Colony replating potential in methylcellulose using BM cells. Error bars represent the S.D.; n = 3 independent biological replicates. (B) Summary of donor-derived PB proportions at the indicated time points (n = 6 per group). Error bars represent the S.E.M. (C) Proportion of donor-derived PB cells from primary (left) or secondary (right) recipient mice transplanted with WT (n = 6), Traf6−/− (n = 5–6), Tet2 −/− (n = 5–6), and Traf6−/−;Tet2−/− (n = 4–6) BM cells. Statistical analysis was performed between WT and Traf6−/−;Tet2−/− mice. Error bars represent the S.E.M. (D) Proportions of donor-derived BM cells from primary recipient mice is reported 4 months after pIpC injection. Error bars represent the S.E.M. (n = 5–6 per group). (E) Proportions of donor-derived BM cells from secondary recipient mice 4 months post-transplantation. Error bars represent the S.E.M. (n = 5–6 per group). (F) PB counts of WT (n = 18), Traf6−/− (n = 34), Tet2−/− (n = 28) and Traf6−/−;Tet2−/− (n = 31) mice at 2 months after transplantation. (G) Absolute number of cells in the BM (left) and spleen (right) 2 months after transplantation (n = 6–9 per group). Error bars represent the S.E.M. (H) Proportion of BrdU-positive cells within the indicated BM cell populations from Tet2−/− (n = 5) and Traf6−/−;Tet2−/− (n = 3) mice treated with BrdU. Error bars represent the S.E.M. Significance was determined with a Student’s T test for two groups or ANOVA for multiple groups (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
We next evaluated the effect of concurrent deletion of Traf6 and Tet2 on hematopoiesis in a non-competitive BM transplantation model using BM cells from VavCre-expressing mice. Mice transplanted with Traf6−/− BM cells exhibited increased number of neutrophils (P < 0.05), and reduced lymphocytes (P < 0.001) and platelets (P < 0.001) 4 months post transplantation as compared to mice transplanted with WT BM cells (Fig. 2F and Fig. S2D,E). Mice transplanted with Tet2−/− BM cells had moderately increased neutrophils, otherwise the PB cell counts were unremarkable (Fig. 2F). In contrast, mice transplanted with Traf6−/−;Tet2−/− BM cells developed neutrophilia (P < 0.001), lymphopenia (P < 0.001), and mild anemia (P < 0.05) as compared to mice transplanted with WT or Tet2−/− BM cells (Fig. 2F and Fig. S2D,E). Mice transplanted with Traf6−/−;Tet2−/− BM cells also resulted in increased myeloid progenitors in the spleen and disruption of spleen architecture as compared to the other groups (Fig. 2G and Fig. S2E). Moreover, Traf6−/−;Tet2−/− SLAM+LSK and LSK BM cells exhibited increased proliferation as compared to Tet2−/− cells (Fig. 2H), but without changes in cell survival (Figure S2F). Although both Traf6−/− and Traf6−/−;Tet2−/− exhibited overlapping hematopoietic defects, the phenotype was more severe in Traf6−/−;Tet2−/− mice.
We next evaluated the effect of concurrent deletion of Traf6 and Tet2 on the development of hematological malignancies. BM cells from VavCre-expressing were transplanted into lethally-irradiated WT mice and then monitored for hematological disease. A subset of experimental animals reconstituted with Tet2−/− BM cells developed a fatal myeloid disease after a long latency consistent with previous reports (Fig. 3A)(Moran-Crusio et al., 2011). All mice reconstituted with Traf6−/−;Tet2−/− BM cells developed a fully penetrant, fatal, and transplantable disease resembling MPN/AML (Fig. 3A,B). MPN/AML-like Traf6−/−;Tet2−/− mice manifested an expansion of myeloid cells (Fig. 3C), which coincided with neutrophilia and elevated hemoglobin (Fig. 3D). MPN/AML-like Traf6−/−;Tet2−/− mice presented with a hyper-cellular BM containing myeloid blasts, disruption of spleen architecture, and infiltration of leukemic cells into the lungs (Fig. 3E). Based on the Bethesda classification, the Traf6−/−;Tet2−/− mice developed a myeloid leukemia(Kogan et al., 2002). Myeloid progenitor cell expansion in the BM and spleen of MPN/AML-like Traf6−/−;Tet2−/− mice is likely the reason for the myeloid-biased disease manifestation (Fig. 3F). Mice reconstituted with Traf6fl/fl;VavCre+ BM cells also developed a lethal phenotype including sustained myelopoiesis, lymphopenia, and increased hemoglobin levels, which coincided with loss of BM HSPCs (Fig. 3A and Fig. S3). However, the disease manifest by Traf6 deletion was not transplantable into secondary recipient mice (Fig. 3B,C), suggesting that TRAF6 loss alone is insufficient to sustain a hematologic disease. Since deletion of TRAF6 in immature hematopoietic cells elicits a diseased phenotype, we next deleted Traf6 in mature myeloid cells by crossing Traf6fl/fl mice to the myeloid-specific recombinase strain (LysM-Cre). Mice reconstituted with Traf6−/−;LysMCre+ BM cells did not develop overt hematologic disease (Fig. S4), indicating that the initial myeloid expansion and eventual hematologic disease associated with TRAF6 deletion initiates within HSPCs. These findings indicate that loss of TRAF6 contributes to leukemic transformation of pre-leukemic HSPCs, a phenotype resembling human MPN/AML. Moreover, loss of TRAF6 mediates a diseased phenotype only in the context of a CHIP-associated mutation, such as TET2, which is necessary to prevent attrition of TRAF6-deficient HSPCs.
Figure 3. Loss of TRAF6 results in myeloid leukemia.
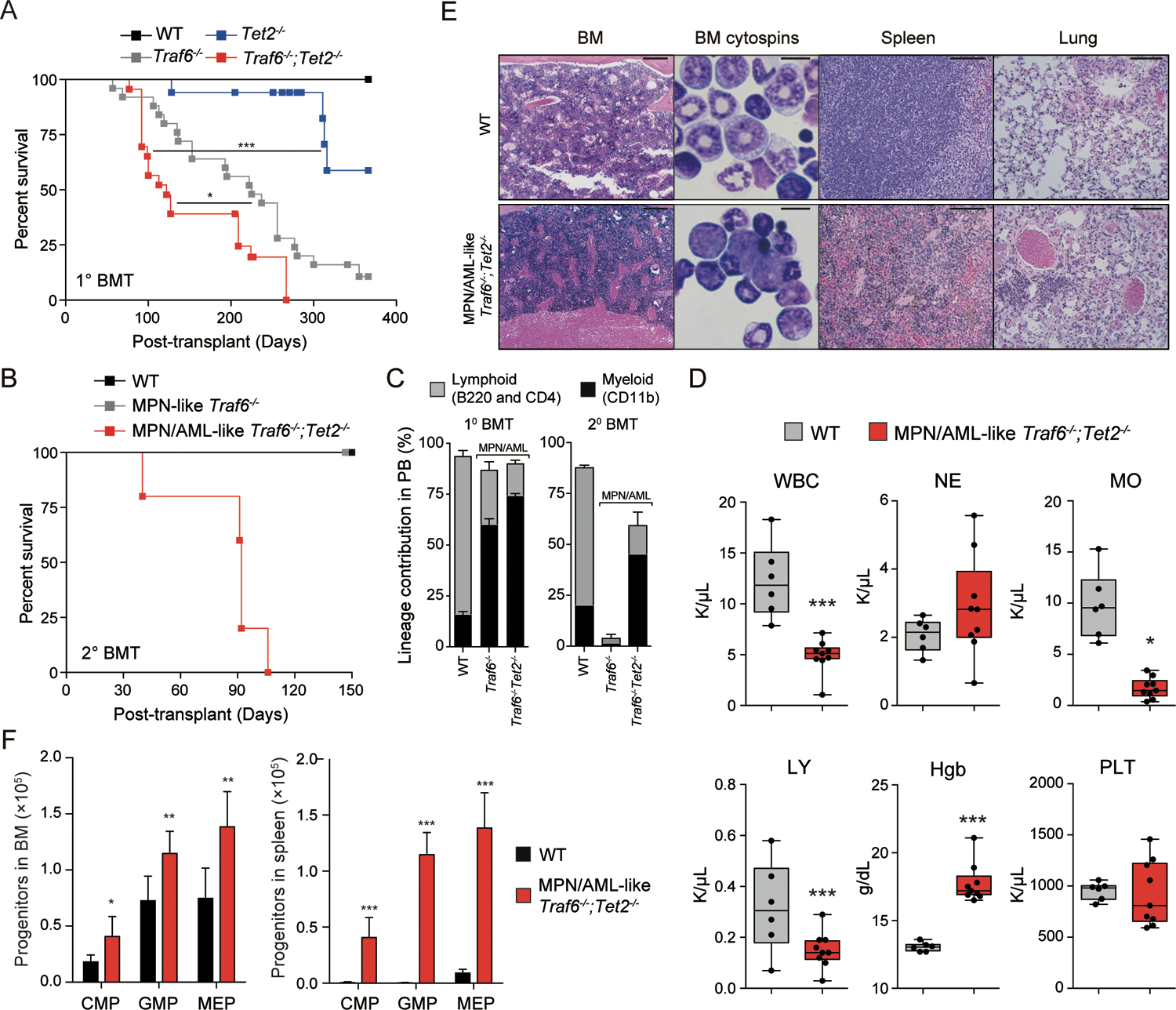
(A) Overall survival of mice transplanted with WT (n = 12), Traf6−/− (n = 25), Tet2 −/− (n = 17), and Traf6−/−;Tet2−/− (n = 23) BM cells. (B) Overall survival of secondary recipient mice transplanted with BM cells from WT primary recipient mice (n = 6), or diseased (MPN-like) Traf6 −/− (n = 4) and (MPN/AML-like) Traf6−/−;Tet2−/− (n = 5) mice. (C) Proportion of donor-derived PB cells from WT primary (10 BMT) recipient mice (n = 4) or diseased (20 BMT) MPN-like Traf6 −/− (n = 4), and MPN/AML-like Traf6−/−;Tet2−/− (n = 6) mice. Error bars represent the S.D. (D) PB counts from mice transplanted with WT (n = 6) or diseased (MPN/AML-like) Traf6−/−;Tet2−/− (n = 9) mice ~100 days post transplantation. (E) Hematoxylin and eosin staining of BM, spleen, and lung and Wright-Giemsa staining of BM from WT and diseased (MPN/AML-like) Traf6−/−;Tet2−/− mice. For BM, spleen and lung, scale bars is 100 μm; for BM cytospins, the scale bar is 10 μm. (F) Proportions of donor-derived cells within the BM (left) or spleen (right) of recipient mice transplanted with BM cells from WT or diseased (MPN/AML-like) Traf6−/−;Tet2−/− mice. Error bars represent the S.E.M. Significance was determined with a Student’s T test for two groups (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
Multiple mechanisms lead to reduced TRAF6 expression in human MPN/AML.
Since reduced expression of TRAF6 can promote a myeloid leukemia in mouse models, we wanted to determine whether genetic alterations targeting TRAF6 are observed in human myeloid malignancies. Upon inspection of publicly available data, TRAF6 copy number deletions (11p13) occur in AML at a frequency of ~3 percent (Fig. 4A). Moreover, mutations in TRAF6 are observed in ~1 percent of AML patients(Edberg et al., 1990; Tate et al., 2019; Tyner et al., 2018). Based on a case report, TRAF6 mutations that are predicted to disrupt its signaling function are secondary events that co-occur with CHIP mutations, suggesting that loss of TRAF6 occurs later in the evolution of AML(Engen et al., 2020). Genetic alterations affecting TRAF6 are observed in AML, albeit at a relatively low frequency, therefore we evaluated TRAF6 mRNA expression in AML and MPN(Nischal et al., 2013; Rampal et al., 2014; Tyner et al., 2018). Reduced TRAF6 expression occurs in ~30% of AML patients as compared to healthy controls (Fig. 4B). TRAF6 expression was lowest in more differentiated AML subtypes while highest in the less differentiated AML subtypes (Fig. 4C). We also examined expression of TRAF6 in de novo AML and AML following antecedent MDS. De novo AML patients exhibit reduced TRAF6 expression as compared to patients with an antecedent MDS (Fig 4D), suggesting that TRAF6Low AML may emerge from a distinct pre-leukemic subclone and not in a step-wise progression from MDS(Chen et al., 2019a). Stratification of AML patients revealed that TRAF6Low corresponds with shorter overall survival (P = 0.02)(Fig. 4E), and significantly elevated blasts and WBC counts at diagnosis (Fig. 4F) as compared to TRAF6High AML patients.
Figure 4. Reduced TRAF6 expression in human myeloid leukemia.
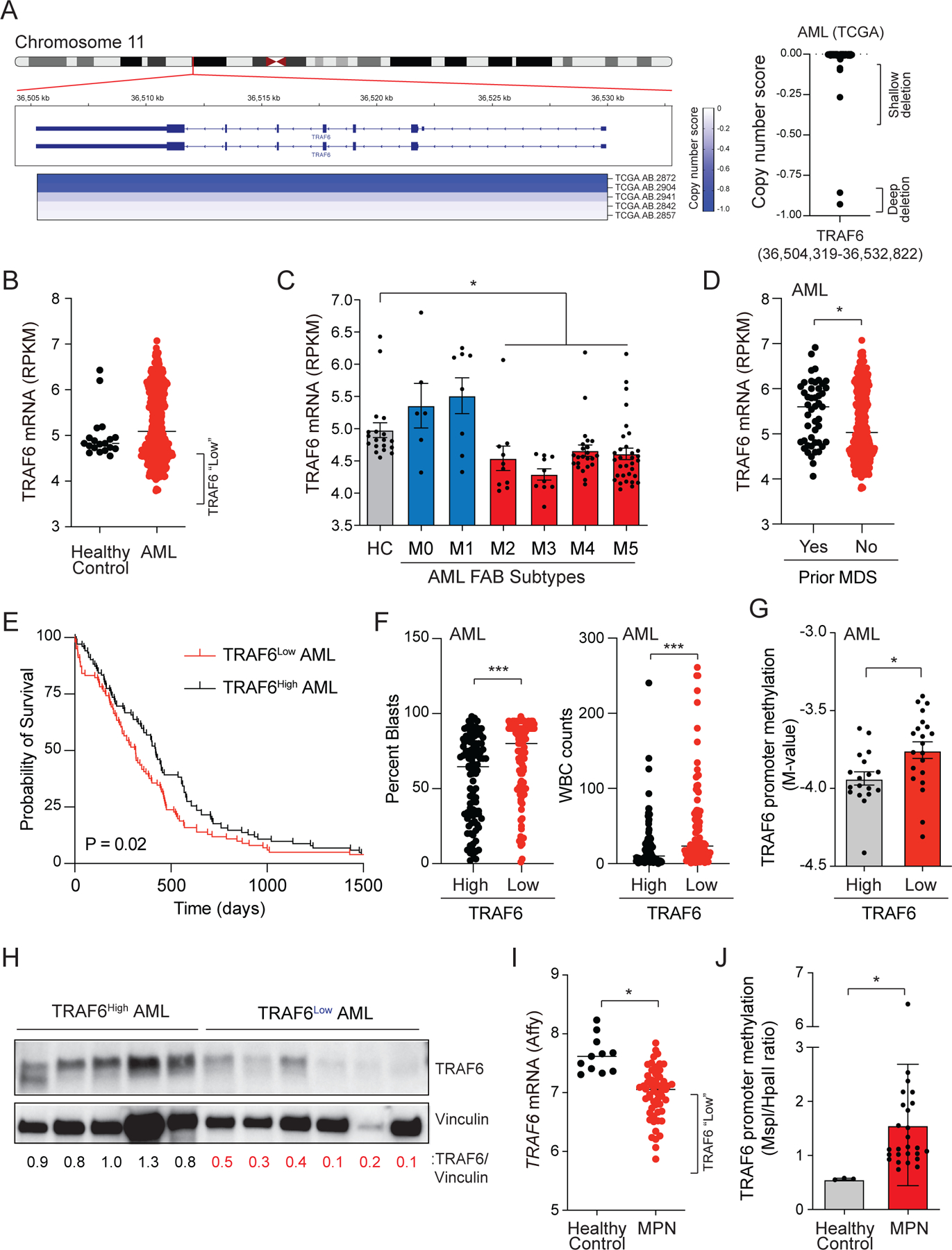
(A) Human chromosome 11p13 is shown for AML. The TRAF6 copy number score is depicted in the heatmap (TCGA sample numbers are indicated below). Negative scores indicate chromosome deletions. The copy number scores (<0) for TRAF6 are summarized for all AML patients within the TCGA data set (right panel). (B) TRAF6 mRNA expression in healthy age-matched BM CD34+ cells (n = 12) and AML (n = 451). (C) TRAF6 mRNA expression in AML stratified on FAB classification. (D) TRAF6 mRNA expression in de novo AML (“no”) and in AML patients with prior MDS (“yes”). (E) Overall survival of AML patients stratified on TRAF6 expression (highest/lowest 25%). (F) Percent blasts and WBC counts in AML patients stratified on TRAF6 expression (highest/lowest 25%). (G) TRAF6 promoter methylation in AML patients stratified on TRAF6 expression. (H) TRAF6 protein expression in primary AML. Relative TRAF6 expression was calculated by determining the densitometric values of TRAF6 as compared to Vinculin. (I) TRAF6 mRNA expression in healthy age-matched controls (n = 11) and MPN patients (n = 93). (J) TRAF6 promoter methylation in MPN patients and healthy controls. Error bars represent the S.E.M. Significance was determined with a Student’s T test for two groups or ANOVA for multiple groups (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
To establish the basis for reduced TRAF6 expression in AML, we first evaluated the epigenetic state of the TRAF6 locus in AML patient samples and found significant hypermethylation of CpG sites proximal to the TRAF6 promoter as compared to age-matched controls (Fig. 4G). Moreover, the methylation status of the TRAF6 promoter inversely correlated with TRAF6 mRNA in individual AML patient samples (Fig. S4A). Inspection of RNA-sequencing reads showed that the majority of AML samples with low levels of TRAF6 displayed aberrant processing of the 5’-end of the TRAF6 transcript (Fig. S4B–E). TRAF6 protein was significantly reduced in approximately half of the AML patients examined, indicating that reduced TRAF6 mRNA corresponds with lower TRAF6 protein expression (Fig. 4H). Consistent with AML data, TRAF6 mRNA levels were reduced in MPN patients (Fig. 4I), which correlated with increased methylation of the TRAF6 promoter (Fig. 4J and Fig S4E). Collectively, suppression of TRAF6 can occur through various molecular and genetic mechanisms in a significant proportion of AML and MPN.
A broad meta-analysis was performed on AML patients stratified on TRAF6 expression using the BEAT-AML data set(Tyner et al., 2018). TET2 mutations are observed in AML patients with low expression of TRAF6, particularly in the differentiated subtypes, indicating that the two alterations could cooperate in AML (Fig. S4F). A similar correlation was observed in AML patients with DMNT3a mutations. To determine whether TET2 mutations directly impact TRAF6 expression, we examined TRAF6 mRNA levels in mouse Tet2−/− LSK and human CD34+ BM cells with deletion of TET2. We observed that deletion of TET2 in mouse (Fig. S4G) or human (Fig. S4H,I) HSPCs did not affect TRAF6 expression, indicating that TRAF6 is not directly regulated by TET2. Although lower expression of TRAF6 does co-occur with TET2 and DNMT3A mutations in certain cohorts of AML patients, we propose that mechanisms that suppress TRAF6 are likely secondary genetic events that are independent of the initiating mutation. This analysis also revealed that FLT3-ITD mutations coincide with low TRAF6 expression (Hypergeometric test, P < 0.04)(Fig. S4J). To test whether TRAF6 overexpression could prevent malignant transformation of FLT3-mutant disease, FLT3-ITD transgenic mice were crossed to mice that overexpress TRAF6 in hematopoietic cells (Vav-TRAF6)(Fang et al., 2017). Biallelic FLT3-ITD mice develop a fatal MPN associated with elevated neutrophil counts (Fig. S4K)(Chu et al., 2012). In contrast, overexpression of TRAF6 in FLT3-ITD mice restored myeloid cell counts back to normal levels and rescued the survival of the animals (Fig. S4K,L). These findings demonstrate that TRAF6 can act as a context-specific tumor suppressor in human myeloid malignancies.
Traf6 loss results in MYC activation.
To identify the signaling pathways and gene expression changes following deletion of Traf6 in pre-leukemic HSPCs, we performed RNA-sequencing on LSK cells from Vav-Cre-expressing BM cells. Deletion of Traf6 and Tet2 resulted in significant differentially expressed genes as compared to Traf6−/− (6645 vs 1238) or Tet2−/− (6645 vs 1698) LSK (Fig. 5A and Table S2–5). Pathway analysis revealed that Traf6−/−;Tet2−/− LSK exhibit an enrichment of genes associated with mitochondrial function and cell cycle as compared to WT LSK. The enrichment of these pathways was not observed in Tet2−/− LSK but was present in Traf6−/− LSK, suggesting that loss of TRAF6 establishes gene expression networks associated with HSPC expansion as shown in Fig. 2H. Consistent with the finding that deletion of Traf6 and Tet2 leads to expansion of myeloid progenitors, Traf6−/−;Tet2−/− LSK have an enrichment of myeloid progenitor and leukemic stem cell gene programs at the expense of HSC gene programs as determined by gene set enrichment analysis (GSEA)(Fig. 5B). These gene expression changes were observed in Traf6−/− and Tet2−/− LSK but were much less pronounced. These results suggest that pre-leukemic HSPCs lacking TRAF6 have acquired gene programs congruent with a myeloid malignancy. Among the top enriched pathways, Traf6−/−;Tet2−/− LSK exhibited MYC gene signatures (Fig. 5C). The activation of MYC target genes is driven by loss of TRAF6 in normal and pre-leukemic HSPCs as elevated MYC regulated genes was observed in Traf6−/− and Traf6−/−;Tet2−/− LSK (Fig. 5D and Fig. S4M). In contrast, Tet2−/− LSK did not exhibit activation of MYC-regulated genes (Fig. 5D). We next determined whether low TRAF6 expression correlates with increased MYC activity in AML and MPN patients. MYC signatures were significantly and positively enriched in TRAF6Low AML and MPN relative to TRAF6High patient samples (Fig. 5E). We observed a similar enrichment of MYC signatures in additional TRAF6High human cancers (Fig. S4N), suggesting that the anti-correlation between TRAF6 expression and MYC activation extends beyond myeloid leukemia. To test this hypothesis at the single cell level in human BM, we performed single cell CITE-Seq(Stoeckius et al., 2017) from five healthy donors (Fig. S5). These samples were selected as they do not harbor somatic mutations that would confound the interpretation of TRAF6 expression and MYC activation. Consistent with previous findings(King et al., 2013), the highest levels of a MYC gene signature were detected in HSPC populations. On the other hand, these cell types had the lowest levels of TRAF6 expression, a gene mainly expressed in monocytes. To further validate these findings, we separated the BM in TRAF6Low and TRAF6High clusters. In agreement with our bulk RNA analysis, TRAF6Low total BM cells and HSPCs had the highest level of expression of a MYC gene signature. Interestingly, TRAF6 levels do not predict MYC expression (Fig. S5G). These data demonstrate that low levels of TRAF6 expression tightly correlate with enhanced MYC activity in healthy and leukemic BM cells.
Figure 5. Loss of TRAF6 results in a MYC-dependent AML.
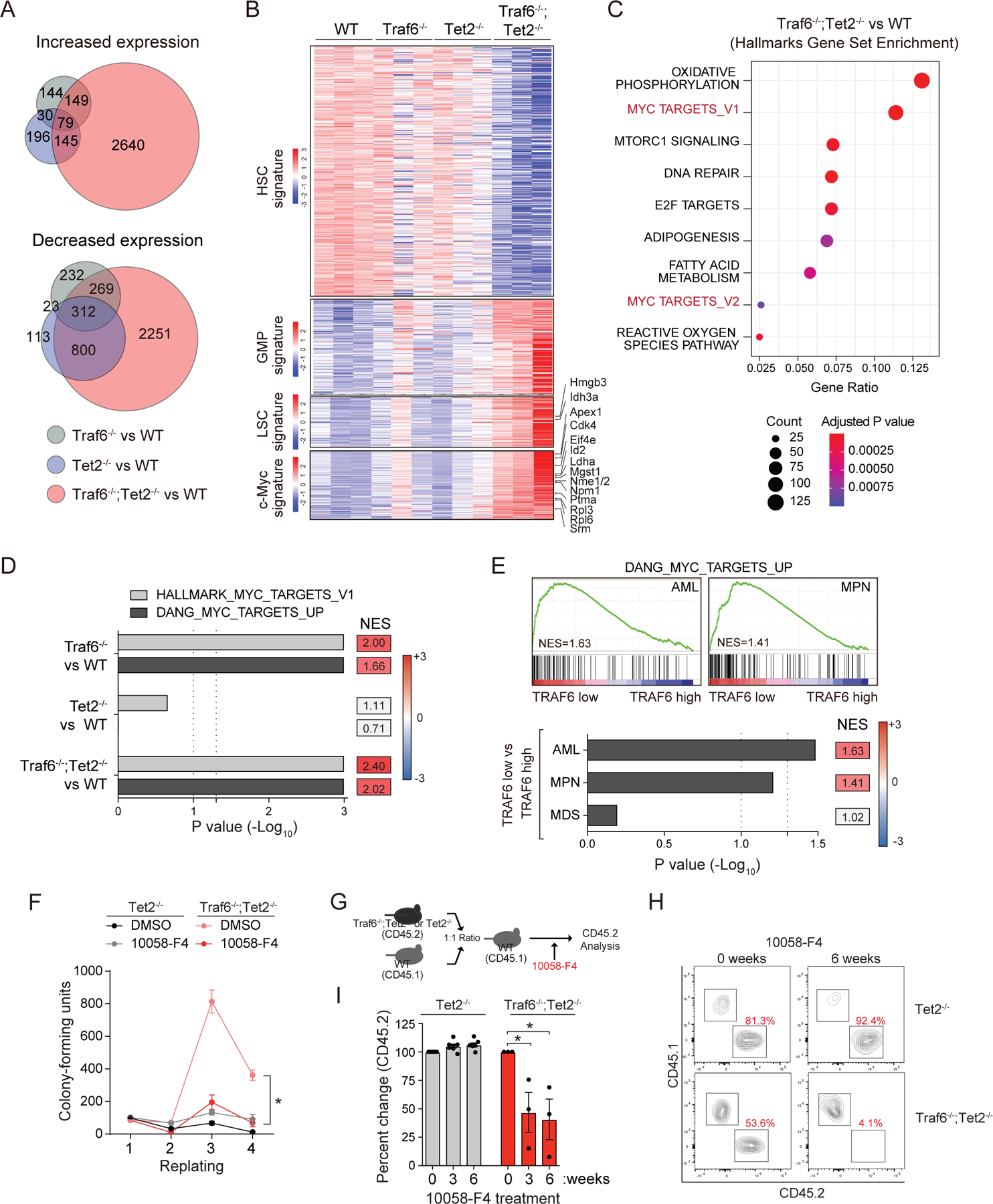
(A) Genes up/down-regulated in Traf6−/−, Tet2 −/−, and Traf6−/−;Tet2−/− BM LSK cells relative to WT LSK cells. (B) Differentially expressed genes in BM LSK cells (1.5-fold; P < 0.05; n = 3 per group). Genes were selected from HSC signature (Pietras et al. Cell Stem Cell 2015), granulocyte-monocyte progenitor (GMP) signature (Pietras et al. Cell Stem Cell 2015), and Leukemic stem cell (LSC) signature (Somervaille et al. Cell Stem Cell 2009). (C) The top Hallmark gene sets significantly enriched in Traf6−/−;Tet2−/− BM LSK cells relative to WT LSK cells. (D) Enrichment analysis of MYC signatures in LSK cells from the indicated genotypes. NES, normalized enrichment score. (E) GSEA plots of MYC signatures of MPN and AML patients stratified based on low/high TRAF6 expression (top). Enrichment analysis of MYC signatures in MDS, MPN, and AML patients stratified based on low/high TRAF6 expression (bottom). (F) Colony replating potential of BM HSPC treated with 10058-F4 (50 μM). *, P < 0.05. Error bars represent the S.E.M.; n = 3 independent biological replicates. (G) Outline of competitive BM transplantations. One month post-transplantation, mice were treated with 10058-F4 (20 mg/kg IP) 3 times/week, and analyzed for PB chimerism. (H) FACS plot at 0 and 6 weeks of 10058-F4 treatment. (I) Summary of PB chimerism in mice determined before and after treatment with 10058-F4 for the indicated number of weeks. Error bars represent the S.E.M. Significance was determined with a Student’s T test for two groups or ANOVA for multiple groups (*, P < 0.05).
To determine whether the leukemic phenotype driven by TRAF6 loss is mediated by MYC, we first treated Traf6-deficient Tet2−/− BM HSPCs (and Tet2−/− as a control) with a MYC-Max dimerization inhibitor (10058-F4) and performed hematopoietic colony replating assays(Huang et al., 2006; Sayyadi et al., 2020; Sheikh-Zeineddini et al., 2019). Treatment with 10058-F4 resulted in a significant reduction in colony-replating of Traf6−/−;Tet2−/− HSPCs as compared to vehicle-treated Traf6−/−;Tet2−/− HSPCs, while 10058-F4 had negligible effects on Tet2−/− HSPCs (Fig. 5F). To validate this observation, we also knocked down MYC in Traf6−/−;Tet2−/− BM HSPCs by expressing shRNAs (shMYC) (Fig. S6A). Knockdown of MYC in Traf6−/−;Tet2−/− HSPCs reduced colony replating as compared to Traf6−/−;Tet2−/− HSPCs expressing a non-targeting shRNA (P < 0.001)(Fig. S6B). Next, we generated chimeric mice engrafted with CD45.1+ WT and CD45.2+ Traf6−/−;Tet2−/− or Tet2−/− BM cells, followed by treatment with 10058-F4 every other day for 3 weeks (Fig 5G). Mice engrafted with Traf6−/−;Tet2−/− BM cells had a reduction in hematopoietic chimerism following treatment with 10058-F4 (Fig. 5H,I). In contrast, treatment of mice engrafted with Tet2−/− BM cells with 10058-F4 did not affect PB chimerism (Fig. 5H,I). These findings indicate that MYC function is essential for the malignant phenotype of TRAF6-deficient leukemic cells.
MYC is ubiquitinated and functionally repressed by TRAF6.
Since loss of TRAF6 represents a secondary disease-modifying alteration that contributes to the transformation of pre-leukemic HSPCs, we sought to focus on the mechanistic consequences resulting from loss of TRAF6 on MYC function. TRAF6, an E3 ligase that catalyzes the formation of Lysine (K) 63-linked ubiquitin (Ub) chains, is a signal transducer for TLRs. As such, we performed a global quantitative Ub capture proteomic screen using human leukemia cell line (TF-1) expressing a doxycycline (DOX)-inducible shRNA targeting TRAF6. Ubiquitinated peptides immunoprecipitated from DOX-treated TF-1 cells (shTRAF6) or from vehicle-treated cells as controls were analyzed by mass spectrometry (Fig. S6C). The proteomic analysis identified 51 proteins that were differentially ubiquitinated in shTRAF6 cells as compared to vehicle-treated cells (P < 0.05). Of the top ranked candidates, TRAF6 knockdown resulted in reduced ubiquitination of MYC at K148 (Fig. 6A and Table S6), a post-translational modification implicated in MYC function(Faiola et al., 2005; Gonzalez-Prieto et al., 2015). Substrates related to canonical NF-κB signaling did not reach significance in the screen, suggesting the NF-κB pathway is not a major effector of TRAF6 in AML. To validate the screen, we first transfected WT or ligase-defective TRAF6 (TRAF6-C70A) in HEK293 cells along with MYC. WT TRAF6, but not TRAF6-C70A, resulted in increased MYC ubiquitination (Fig. S6D), indicating that the E3 ligase domain of TRAF6 is required MYC ubiquitination. We did not detect direct interaction between TRAF6 and MYC in cells (data not shown), which is not unexpected as E3-substrate complexes commonly exhibit weak physical interaction and rapid dissociation rates(Pierce et al., 2009). Next, we transfected TRAF6 with either WT MYC or a mutant MYC, in which K148 was substituted for arginine (K148R). Unlike WT MYC, TRAF6 expression did not result in ubiquitination of MYC(K148R) in HEK293 cells (Fig. 6B), suggesting that K148 on MYC is the major ubiquitination site targeted by TRAF6. In addition to its central role in TLR signaling within the cytoplasm, TRAF6 has also been shown to function in the nucleus(Pham et al., 2008). As such, we sought to determine the cellular compartment in which TRAF6 ubiquitinates MYC. We found that TRAF6 mediates ubiquitination of MYC primarily in the nucleus (Fig. S6E). Although the proteomic screen does not discern between distinct ubiquitin linkages, TRAF6 preferentially forms K63-linked ubiquitin chains on its substrates. Expression of TRAF6 resulted in increased MYC ubiquitination in the presence of WT Ub; in contrast, expression of TRAF6 with Ub that is unable to form K63-linkages (Ub-K63R) did not result in MYC ubiquitination (Fig. 6C). As expected, TRAF6 expression did not affect the half-life of MYC (Fig. S6F). To test the ability of TRAF6 to directly ubiquitinate MYC, we utilized recombinant proteins in a cell-free biochemical assay. In the presence of TRAF6, ubiquitination of MYC was readily observed (Fig. S6G). In contrast, MYC(K148R) was not ubiquitinated when incubated with TRAF6 (Fig. S6G), indicating that MYC is a direct substrate of TRAF6-mediated ubiquitination. Immunoblotting c-Kit+ BM from Traf6−/−;Tet2−/− or Tet2−/− mice confirmed that MYC is less ubiquitinated following deletion of Traf6 (Fig. 6D). Finally, we next investigated whether ubiquitination of MYC at K148 by TRAF6 affects MYC transcription factor activity using reporter assays. Overexpression of TRAF6 repressed MYC-dependent reporter activity, but the inhibitory effect of TRAF6 was attenuated in the presence of MYC(K148R) (Fig. 6E), suggesting that TRAF6 can repress MYC transcriptional activity via ubiquitination of K148.
Figure 6. MYC activation is regulated by TRAF6-dependent post-translational modifications.
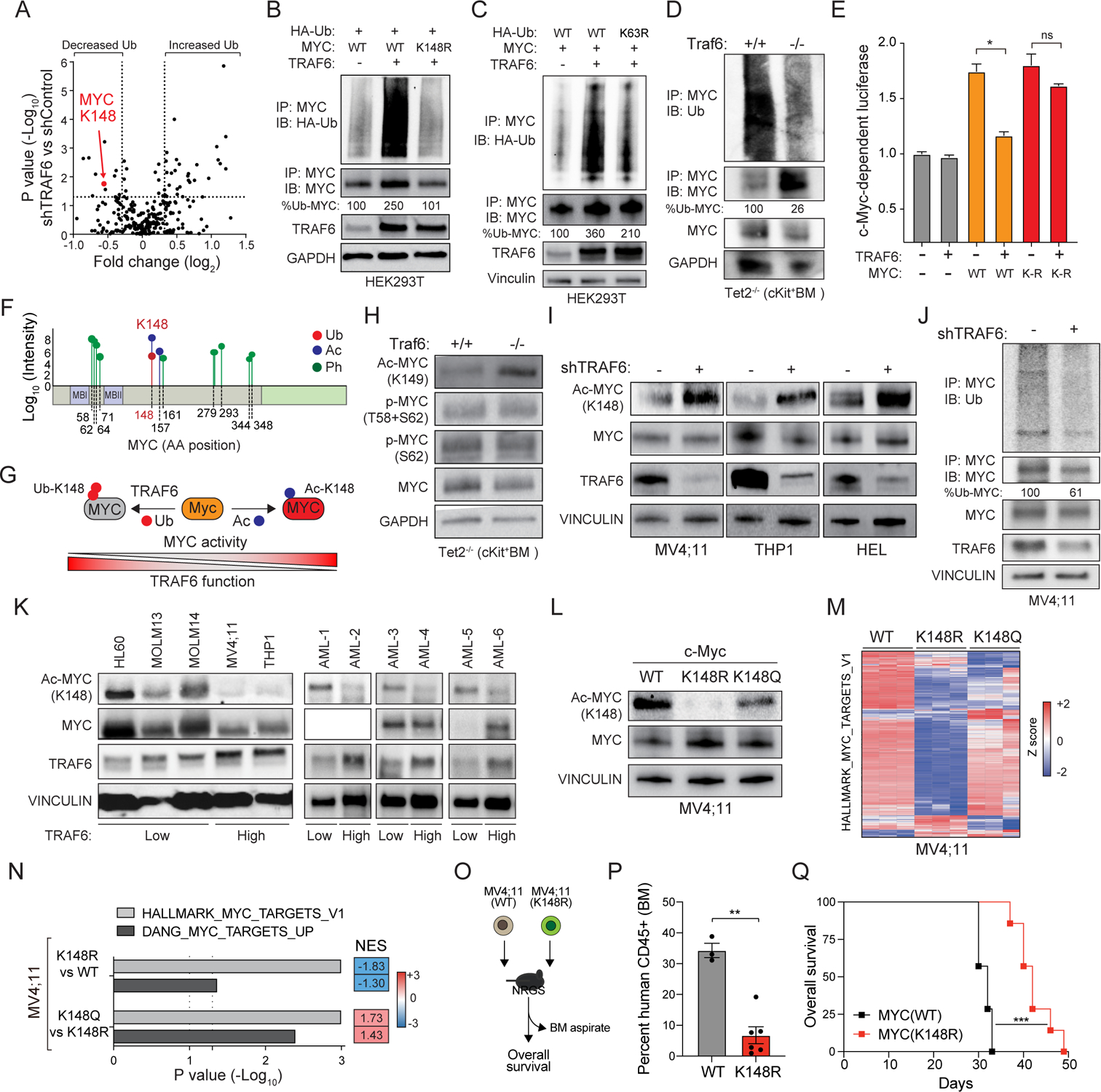
(A) Differentially ubiquitinated substrates in TF-1 cells expressing a doxycycline (DOX) inducible shTRAF6 (+DOX) relative to control cells (−DOX)(n = 3 independent experiments). (B) MYC ubiquitination was determined in HEK293 cells transfected with WT or K148R MYC and TRAF6. Shown is a representative image from 3 independent replicates. (C) MYC ubiquitination was determined in HEK293 cells transfected with WT or K63R ubiquitin (HA-Ub), MYC and TRAF6. Percent of ubiquitinated MYC was determined using densitometric values by calculating ubiquitinated MYC relative to total immunoprecipitated MYC. Show is a representative image from 3 independent replicates. (D) Endogenous MYC ubiquitination was determined in Tet2 −/− and Traf6−/−;Tet2−/− cKit+ BM cells. Percent of ubiquitinated MYC was calculated by determining the densitometric values of ubiquitinated MYC relative to total immunoprecipitated MYC. (E) MYC activation was measured by MYC-site containing reporter assays in HEK293 cells. Values are normalized to Renilla-luciferase and empty vector from the average of 3 independent experiments. Error bars represent the S.E.M. (F) Mass spectrometry analysis of MYC post-translational modifications in AML. (G) Schematic of MYC oncogenic activation regulated by opposing TRAF6-dependent ubiquitination and acetylation at K148. (H) Immunoblot analysis of acetylated (K149) and phosphorylated (T58 and S62) MYC in c-Kit+ BM cells. Show is a representative image from 3 independent replicates. (I) Immunoblot analysis of acetylated (K148) and total MYC in the indicated cell lines expressing shTRAF6 or scrambled shRNA control. (J) Endogenous MYC ubiquitination was determined in MV4;11 cells expressing shTRAF6 or scrambled shRNA control. Percent of ubiquitinated MYC was calculated by determining the densitometric values of ubiquitinated MYC relative to total immunoprecipitated MYC. (K) Immunoblotting of human AML cell lines and patient samples. (L) Immunoblot analysis of acetylated (K148) and total MYC in MV4;11 cells edited at K148 to arginine (R) or glutamine (Q). (M) Heat map of differentially expressed MYC signature genes from parental (WT) and gene edited K148R or K148Q MV4;11 cells. (N) Enrichment analysis of MYC signatures in parental (WT) and K148R or K148Q MV4;11 cells. NES scores in blue indicate gene enrichment in WT relative to K148R MV4;11 cells; NES scores in red indicate gene enrichment in K148Q relative to K148R MV4;11 cells. (O) Outline of xenograft experiment. (P) Percent of human CD45+ populations were evaluated in the BM at 3 weeks post engraftment. Error bars represent the S.E.M. (Q) Overall survival of mice transplanted with WT MV4;11 cells (n = 5 recipient mice) and MV4;11(K148R) (n = 6 recipient mice). Significance was determined with a Student’s T test for two groups (*, P < 0.05; **, P < 0.01).
MYC is regulated by competing ubiquitin and acetylation modifications on K148.
To map the post-translational modifications on MYC relevant to human AML, we immunoprecipitated MYC from MV4;11 AML cells and performed mass spectroscopy. We identified several of the previously described phosphorylated residues (Fig. 6F). Interestingly, we found that both acetylation and ubiquitination modifications are found on K148 in AML (Fig 6F). To determine the impact of TRAF6-mediated ubiquitination on MYC post-translational modifications, we first examined the phosphorylation status and total levels of MYC. Total and phosphorylated MYC levels were unchanged in Traf6-deficient HSPCs (Fig. 6H), suggesting that K148 (K149 in mouse) modifications do not affect phosphorylation or protein abundance of MYC. Although the functional relevance of K148 acetylation on K148 is unclear, such a post-translational modification of MYC could have pleotropic effects. We posited that TRAF6 ubiquitination of MYC at K148 prevents its acetylation, which results in diminished MYC activity (Fig. 6G). Immunoblotting of Traf6−/−;Tet2−/− or Tet2−/− c-Kit+ BM progenitors with an Ac-K148-specific MYC antibody (which recognizes K149 in murine MYC) revealed that MYC exhibits reduced Ub-K149 and a corresponding increase in Ac-K149 upon deletion of Traf6 (Fig. 6H). Knockdown of TRAF6 in human AML cells resulted in increased MYCK148 acetylation (Fig. 6I), which correlated with loss of MYC ubiquitination (Fig. 6J). We also examined human AML cell lines and primary AML samples to correlate TRAF6 protein expression and MYCK148 acetylation. As a general trend, TRAF6Low AML cell lines and patient samples exhibited increased MYCK148 acetylation (Fig 6K). These findings revealed that TRAF6 ubiquitinates MYCK148, which prevents acetylation on the same residues. In contrast, reduction in TRAF6-mediated ubiquitination of MYCK148 permits acetylation on the free lysine residue.
To investigate the effects of acetylated K148 on MYC activity, we generated MV4;11 cells gene-edited at K148 to glutamine (K148Q), which is predicted to mimic MYC lysine acetylation (Fig. S6H). We also generated MV4;11 cells with K148 to arginine (K148R), which results in the expression of MYC that is unable to undergo acetylation at K148 (Fig. 6L) or ubiquitination (Fig. S6I). MV4;11 cells were selected as they exhibited intermediate levels of MYCK148 acetylation. As predicted, the Ac-K148-specific antibody did not detect Ac-MYC in MV4;11(K148R) cells (Fig. 6L). In contrast, the structural change of K148Q mutation in MYC was recognized by the Ac-K148-specific antibody (Fig. 6L). RNA-sequencing revealed that MYC-related gene signatures were negatively enriched in MV4;11(K148R) cells as compared to parental MV4;11, suggesting that lack of MYCK148 acetylation results in diminished activity (Fig. 6M,N). The consequences of K148 modifications on MYC-induced gene expression are not due to differences in MYC protein stability (Fig. S6J). MV4;11(K148Q) cells exhibit a positive enrichment of MYC-regulated gene signatures as compared to MV4;11(K148R) cells, confirming that loss of MYCK148 acetylation results in diminished transcriptional activity in AML (Fig. 6M,N). We next investigated the consequences of MYCK148 acetylation on AML development. MV4;11(K148R) and parental MV4;11 cells were engrafted into NOD-scid IL2Rγ−/−3/GM/SF (NSGS) mice (Fig 6O). Mice engrafted with MV4;11(K148R) cells had reduced leukemic burden as compared to mice engrafted with parental MV4;11 cells (Fig. 6P). Moreover, the development of leukemia was delayed in mice with MV4;11(K148R) cells as compared to parental MV4;11 cells (P < 0.001)(Fig. 6Q). To determine the consequences of K148 acetylation on MYC function on AML, we also expressed MYC(K148Q) or WT MYC (MSCV-IRES-GFP) in Tet2−/− c-Kit+ BM cells, which do not have steady state MYCK148 acetylation (Fig. S6K). Transduced cells were transplanted into lethally-irradiated recipient mice, which were then monitored for leukemic progression. Overexpression of MYC(K148Q) in Tet2−/− HSPCs resulted in an increase in myeloid blasts (Fig. S6L) and splenomegaly (Fig. S6M) after 8 weeks post transplantation as compared to Tet2−/− HSPCs expressing WT MYC. As such, loss of TRAF6 and a corresponding increase in MYCK148 acetylation results in enhanced MYC-induced oncogenic function and development of AML.
TLR-induced TRAF6 activation suppresses MYC in AML.
Finally, we determined whether TLR-mediated activation of TRAF6 alters MYC transcriptional function. Treatment of MV4;11 cells with CU-T12–9 increased ubiquitination of MYC and correspondingly resulted in reduced MYCK148 acetylation (Fig. 7A). The reduction of acetylated MYCK148 by CU-T12–9 was abrogated following knockdown of TRAF6 (Fig. 7B), indicating that TRAF6-mediated K63-linked ubiquitination is required for interfering with acetylation of MYCK148. Since TLR-mediated activation of TRAF6 results in increased ubiquitination and a corresponding reduction in acetylation of MYCK148, we therefore investigated whether the ubiquitination of MYCK148 affects its functional activity by mapping promoter and enhancer occupancy of MYC in AML. Chromatin immunoprecipitation coupled to high-throughput sequencing revealed a significant loss of MYC occupancy in the genome following TLR1/2 activation with CU-T12–9 (Fig. 7C and Fig. S7A–D). Diminished MYC binding resulted in gene downregulation of MYC targets (Fig. 7D,E). In addition, we observed TLR1/2-mediated depletion of MYC binding in the promoter of a number of cancer- and AML-related genes (Fig. S7E). To validate whether TLR-TRAF6 stimulation affects MYC target gene expression via modification of K148 in AML cells, we treated MV4;11 or MV4;11(K148Q) cells with CU-T12–9 and assessed MYC target genes. Treatment with CU-T12–9 coincided with reduced expression of MYC target genes CDK4 and NPM1 (Fig. 7F,G). In contrast, MV4;11(K148Q) cells treated with CU-T12–9 maintained CDK4 and NPM1 expression at levels comparable to unstimulated MV4;11 cells (Fig. 7G). Lastly, we examined the effects of TLR-TRAF6 signaling on cell growth of AML lines. CU-T12–9 treatment resulted in a dose-dependent reduction of AML cell viability (Fig. S7F). Although CU-T12–9 impaired the viability of MV4;11 cells, MV4;11(K148Q), which maintain an acetylated state at K148, are resistant to the anti-leukemic effects of the TLR agonist (Fig 7H,I). These findings suggest that TRAF6-mediated ubiquitination of MYC at K148 inhibits activation of MYC by interfering with its acetylation at the same residue. These findings suggest that MYC acquires oncogenic activity via K148 acetylation.
Figure 7. TLR activation via TRAF6 suppresses MYC function in AML.
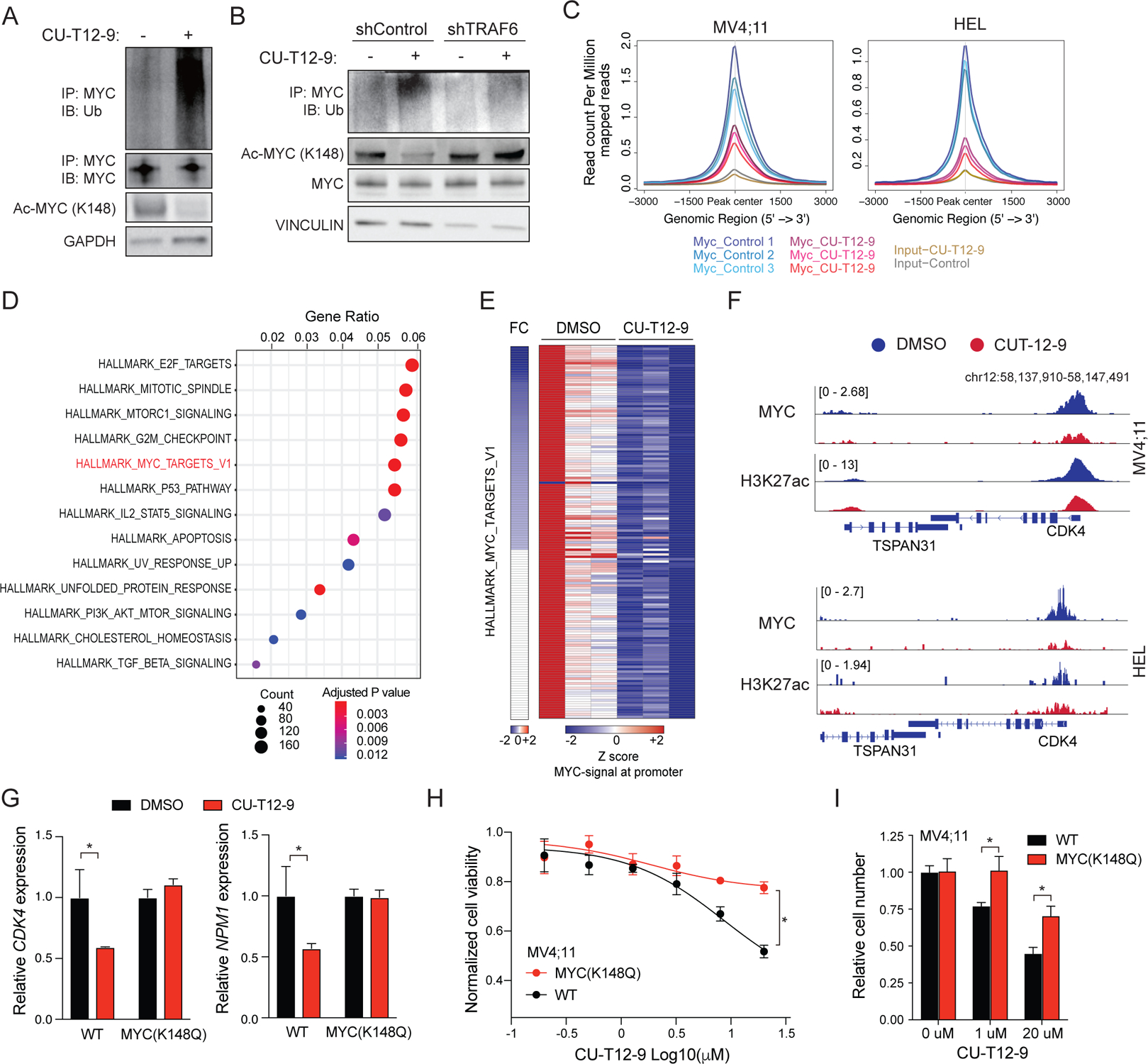
(A) MYC ubiquitination was determined in MV4;11 cells treated with TLR1/2 agonist (CU-T12–9; 20 uM) for 24 hours. Shown is a representative image from 3 independent replicates. (B) Immunoblot analysis of acetylated (K148) and total MYC in MV4;11 cells transduced with the indicated shRNAs and treated with TLR1/2 agonist (CU-T12–9; 20 μM). (C) Density plot of MYC ChIP reads within +/− 3 kb of all the MYC binding sites in MV4;11 or HEL cells treated with the TLR1/2 agonist CU-T12–9 and DMSO (control). (D) Dot plot graphs showing Hallmark gene sets significantly enriched in genes that have lost MYC binding at their promoter following CU-T12–9 treatment (MV4;11 cell line). (E) Heatmap of MYC signal at the promoter of the MYC target genes in MV4;11 cells treated with the TLR1/2 agonist CU-T12–9. (F) MYC ChIP tracks showing reduced binding to target genes (CDK4) with CU-T12–9 in MV4;11 (top) and HEL (bottom). (G) Expression levels of MYC target genes examined by q-PCR in MV4;11 cells 24 hours after DMSO or CU-T12–9 (10 μM) from the average of 3 independent experiments. Error bars represent the S.E.M. (H) Viability of WT (sg-CTL) and gene edited K148Q MV4;11 cells following treatment with TLR1/2 agonist (CU-T12–9) for 72 hours at the indicated concentrations. Error bars represent the S.E.M.; n = 3 independent biological replicates. (I) The number of WT (sg-CTL) and gene edited K148Q MV4;11 cells following treatment with TLR1/2 agonist (CU-T12–9) was determined at the indicated concentrations by Trypan blue exclusion. Error bars represent the S.E.M.; n = 3 independent biological replicates. Significance was determined with a Student’s T test for two groups (*, P < 0.05; **, P < 0.01).
Discussion
Here we identify TRAF6 as a context-dependent disease modifying gene in myeloid malignancies. TRAF6 function and expression are diminished in MPN/AML patients and correlate with TET2 mutations. CHIP-associated mutations, such as in TET2 and DNMT3a, are common founding mutations with the ability to modify the epigenome of pre-leukemic cells, and enable gradual accumulation of additional events essential for transformation, such as observed with TRAF6. TRAF6 is critical for the function of immune effector cells(Han et al., 2013; King et al., 2006; Kobayashi et al., 2009; Muto et al., 2013a), and is a regulator of HSC homeostasis(Fang et al., 2018). Previous findings reported that TRAF6 expression is broadly increased in MDS HSPCs (Boldin et al., 2011; Fang et al., 2014; Starczynowski et al., 2010; Varney et al., 2017; Varney et al., 2015; Zhao et al., 2011; Zhao et al., 2013). In this context TRAF6 overexpression contributes to MDS-associated HSPC defects(Fang et al., 2017). It was thus unexpected that the in vivo RNAi screen identified TRAF6 as a cooperating gene in Tet2 deficient pre-leukemic cells and that a subset of MPN/AML patients exhibit decreased TRAF6 expression. TRAF6 mutations and copy number deletions occur in AML but at a relative low frequency. In a case report, discrete AML clones harbor co-existing CHIP and TRAF6 mutations(Engen et al., 2020), supporting a tumor suppressor-like role of TRAF6 in AML. The paradoxical observation that TRAF6 is overexpressed in MDS, but its expression and function is reduced in a subset of MPN/AML patients perhaps suggest that TRAF6Low AML emerges from a distinct subclone and not in a step-wise progression from MDS(Chen et al., 2019a). This is supported by the observation that de novo AML patients exhibit reduced TRAF6 expression as compared to patients with an antecedent MDS. Collectively, these findings reveal that cellular states are sensitive to TRAF6 dosage and signaling thresholds and that TRAF6 exhibits both oncogenic- and tumor suppressive-like qualities in hematopoietic cells.
Inflammation and innate immune signaling dysregulation is implicated in the pathogenesis of myeloid malignancies; therefore, why would loss of TRAF6 signaling contribute to myeloid progenitor cell expansion and MPN/AML? One potential explanation is that microenvironmental factors in patients with myeloid malignancies are not conducive for the function of normal HSPCs. In this scenario, the altered microenvironment would confer a relative competitive advantage to pre-leukemic HSPCs that have overcome the suppressive effects of such microenvironmental factors. Age- and disease-related microenvironment changes occur in patients with MPN/AML(Oishi and Manabe, 2016). TLR-TRAF6 activation in pre-leukemic cells can result in HSPCs that persist during chronic inflammation, however these TLR-TRAF6-primed HSPCs do not readily transform to overt AML(Muto et al., 2020). Alternatively, loss of TRAF6 could subvert the inflammatory state and unleash MYC activation facilitating MPN/AML development. A related bi-phasic phenomenon has been described in AML cells that can shift from highly proliferative cells to ones that acquire a diapaus-like molecular adaptation following chemotherapy(Duy et al., 2021). These treatment-persistent residual leukemia cells exhibit suppression of MYC and an accompanied activation of inflammatory transcriptional programs.
Altered MYC function is implicated in many human cancers, including AML (Huang et al., 2006; Zuber et al., 2011). The most prominent mechanism ensuring proper regulation of MYC involves degradation by the ubiquitin-proteasome system following K48-linked ubiquitination(Chen et al., 2019b). MYC is also known to interact with histone acetyltransferases, which results in acetylation of MYC at specific lysine residues(Farrell and Sears, 2014), resulting in increased MYC protein stability(Vervoorts et al., 2003). In separate studies, MYC ubiquitination has been associated with increased transcriptional activity(Zhang et al., 2013). Despite evidence of interplay between acetylation and ubiquitination of MYC, the consequences of these modifications and specifically the role of non-degradative ubiquitination on MYC function and relevance to cancer are not well understood(Farrell and Sears, 2014). We reveal that TRAF6 targets MYC by K63-linked ubiquitination of K148, and that this modification antagonizes K148 acetylation leading to a decrease of MYC activity without affecting its protein abundance. However, in the absence of TRAF6, acetylation of K148 results in enhanced MYC function and oncogenic potential. In summary, TLR triggering in certain contexts could suppress myeloid leukemia due to its ability to target the TRAF6/MYC signaling axis.
Limitations of the studies
We report that combined loss of TRAF6 and TET2 leads to AML in murine models. Studies on human MPN and AML will be required to confirm if reduced TRAF6 expression combined with TET2 mutation is sufficient to cause leukemia in humans. Mutations in TET2 do not directly result in suppression of TRAF6 expression, thus repression of TRAF6 is an independent event and not simply a downstream consequences of a CHIP mutation. Although our study focused on the cooperation between TRAF6 and TET2, our study does not address whether loss of TRAF6 can cooperate with additional CHIP mutations. We have demonstrated that loss of TRAF6 results in increased MYC function via acetylation of K148, however the mechanistic basis for increased MYC function upon acetylation of K148 remains unknown. Since TRAF6 exhibits both proto-oncogenic and tumor suppressor functions, a better understanding of TRAF6 function in other human cancers is warranted.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Daniel Starczynowski (Daniel.Starczynowski@cchmc.org).
Materials Availability
Plasmids and mouse lines generated for this study are available upon request and will be fulfilled by the lead contacts, Iannis Aifantis (Ioannis.Aifantis@nyulangone.org) and Daniel Starczynowski (Daniel.Starczynowski@cchmc.org).
Data and Code Availability
Data
Next Generation Sequencing data, including data for mouse RNA-Seq, have been deposited at GEO and are publicly available as of the date of publication.
Next Generation Sequencing data, including data for mouse and human RNA-Seq have been deposited at GEO: GSE19429, GSE54646, GSE184937, GSE185295, and GSE185299.
Published microarray data of patients with MDS(Pellagatti et al., 2010), MPN(Rampal et al., 2014), and respective age-matched controls were downloaded from GEO series GSE19429 and GSE54646. RNA-seq data of patients with AML(Cancer Genome Atlas Research et al., 2013) were downloaded from the GDC Data Portal (https://portal.gdc.cancer.gov/). AML RNA-seq data was also obtained from the BEAT AML data viewer (Vizome, http://www.vizome.org/aml/)(Tyner et al., 2018). DNA methylation data of AML and MPN was obtained from the GDC Data Portal(Cancer Genome Atlas Research et al., 2013) and GSE4272(Nischal et al., 2013), respectively. Mutation data of AML, MDS and MPN were downloaded from GDC Data Portal(Cancer Genome Atlas Research et al., 2013) and the study by Gerstung M et al.(Gerstung et al., 2015).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Code
This paper does not report original code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Traf6fl/fl mice (C57Bl/6) were a kind gift from Dr. Yongwon Choi (University of Pennsylvania)(Han et al., 2013). Traf6fl/fl mice and Tet2fl/fl mice (Jackson Laboratory, 017573) were crossed with Mx1-Cre (Jackson Laboratory, 003556) and Vav-Cre mice (Jackson Laboratory, 008610) for inducible or conditional deletion, respectively. Vav-TRAF6 mice(Fang et al., 2017) were crossed with FLT3ITD mice(Lee et al., 2007). All mice were bred, housed and handled in the Association for Assessment and Accreditation of Laboratory Animal Care-accredited animal facility of Cincinnati Children’s Hospital Medical.
Cell lines
MV4;11, THP1 and HEL cell lines were purchased from American Type Culture Collection (ATCC). The cells were cultured in RPMI 1640 medium with 10% fetal bovine serum (FBS) and 1% penicillin and streptomycin (P/S). HEK 293T cells was purchased from ATCC, and were cultured in DMEM with 10% FBS and 1% P/S. PLAT-E cells were cultured in DMEM with 10% FBS, 1 μg/mL puromycin, 10 μg/mL blasticidin, and 1% P/S.
METHOD DETAILS
Reagents
10058-F4 (F3680) were purchased from Sigma. CU-T12–9 (5414) and Poly(I:C) (4287) were purchased from Tocris Bioscience. Cycloheximide (sc-3508) was purchased from Santa Cruz.
Hematological and histological analysis
Blood counts were measured with a hemacytometer (HEMAVET). BM, spleens and lung were fixed with 10% formalin, sectioned and stained with hematoxylin and eosin stain. Blood smear and BM cytospins were stained with Wright-Giemsa.
BM transplantation
For non-competitive BM transplantations, 1 × 106 CD45.2+ BM cells were transplanted into lethally-irradiated recipient mice (CD45.1+ B6.SJLPtprca Pep3b/Boy; 6–10 weeks of age) as described(Fang et al., 2018; Smith et al., 2020). For competitive repopulation, BM cells from 8-week-old Mx1Cre+, Traf6fl/fl;Mx1Cre+, Tet2fl/fl;Mx1Cre+ and Traf6fl/fl;Tet2fl/fl;Mx1Cre+ mice were transplanted into lethally irradiated recipient mice with CD45.1+ B6.SJLPtprca Pep3b/BoyJ BM cells. At 4 weeks after transplantation, recipient mice were injected with 200μL of polyinosinic-polycytidylic acid (pIpC) (4287, Tocris Bioscience) dissolved in phosphate-buffered saline (PBS) at a concentration of 1.5 mg/mL intraperitoneally every other day 5 times to delete Traf6 and Tet2. For serial transplantation, BM cells were collected from all recipient mice 20 weeks after transplantations and pooled together. Then, 2 × 106 BM cells were transplanted into lethally-irradiated recipient mice (CD45.1+ B6.SJLPtprca Pep3b/Boy; 6–10 weeks of age) without competitor cells. For in vivo MYC inhibitor experiment, 5 × 105 BM cells from Tet2fl/fl;Vav1Cre+ mice and Traf6fl/fl;Tet2fl/fl;Vav1Cre+ mice were transplanted were transplanted into lethally irradiated recipient mice with 5 × 105 CD45.1+ B6.SJLPtprca Pep3b/BoyJ BM cells. Six weeks post-transplant, the recipient mice were treated with 10058-F4 dissolved in corn oil with 5 % DMSO at a concentration of 2.5 mg/mL intraperitoneally every other day for 3 weeks. For overexpression of MYC(K148Q), BM cells were transduced with retroviruses as previously described(Yokota et al., 2019). Plat-E packaging cells were transfected with retrovirus vectors using TransIT®-LT1 Transfection Reagent (MIR2305, Mirus) according to the manufactures’ recommendation. BM cells were harvested from Tet2fl/fl;Vav1Cre+ mice (6 to 8 weeks of age) treated with 5-fluorouracil (5-FU) (150 mg/kg, intraperitoneally). BM cells were cultured at 37°C for 48 h in IMDM containing 15% FBS, 50 μM 2-mercaptoethanol, 50 ng/ml mouse stem cell factor, 50 ng/ml human thrombopoietin), 50 ng/ml mouse FMS-like tyrosine kinase 3 ligand, and 10 ng/ml mouse interleukin-6. First-round retroviral infection was carried out using RetroNectin (Takara Bio, Otsu, Japan) in the same medium. For the second round infection, polybrene was added with the retroviral supernatant. GFP+ MYC (K148Q)–transduced mouse BM cells were transplanted (1 × 105 cells/recipient) into lethally irradiated recipient mice (CD45.1+ B6.SJLPtprca Pep3b/Boy; 6–10 weeks of age) with 2 × 105 helper cells.
Xenograft experiments
MV4;11 and MV4;11(K148R) cells (5 × 105 cells per recipient) were tail vein transplanted into sublethally irradiated NOD-scid IL2Ry−/− (NSG) mice (250 rads whole body irradiation).Bone marrow aspirates were taken 3 weeks after transplantation to assess engraftment by flow cytometry using a BD FACS Canto System (BD Biosciences, San Jose, CA). Time of death was recorded, and Kaplan Meier survival analysis was performed using GraphPad Prism version 7.00 for Mac (GraphPad Software, La Jolla California USA, www.graphpad.com).
Flow cytometry
For immunophenotypic analysis of lineage positive cells, PB, BM and spleen samples were processed with 1 x RBC lysis buffer, and then incubated with CD11b-PE-cy7 (25–0112-81, eBiosciences), Gr1-eFluor450 (48–5931-82, eBiosciences), CD3-PE (12–0031-83, eBiosciences), and B220-APC (17–0452-82, eBiosciences). For the analysis of erythroid differentiation, BM cells and splenocytes were stained with Ter119-APC (17–5921-82, eBiosciences), CD71-PE (12–0711-81, eBiosciences), CD45-APC-Cy7 (557659, BD) and CD44-BV421 (563970, BD)(Liu et al., 2013). To distinguish donor from recipient hematopoietic cells, PB were stained with CD45.1-Brilliant Violet 510 (110741, BioLegend) and CD45.2-APC-eFluor780 (47–0454-82, eBiosciences) or CD45.2- eFluor450 (48–0454-82, eBiosciences). For SLAM-HSC analysis, BM cells and splenocytes were washed and incubated for 30 minutes with biotin conjugated lineage markers (CD11b, Gr1, Ter119, CD3, B220, mouse hematopoietic lineage biotin panel, [88–7774-75 eBiosciences]), followed by staining with streptavidin eFluor780 (47–4317-82, eBiosciences), Sca-1-PE (12–5981-82, eBiosciences), c-Kit-APC (17–1171-81, eBiosciences), CD48-FITC (11–0481-85, affymetrix), CD150-PE-cy7 (115914, BioLegend). SLAM-HSC were identified based on expression of Lin−Sca−1+c-Kit+CD150+CD48−. For progenitor analysis, BM cells and splenocytes were washed and incubated for 30 minutes with biotin conjugated lineage markers, followed by staining with streptavidin eFluor780 (47–4317-82, eBiosciences), Sca-1-PE-cy7 (25–5981-82, eBiosciences), c-Kit-APC (17–1171-81, eBiosciences), CD34-FITC (11–0341-82, eBiosciences), CD16/32-PE (12–0161-82, eBioscience). Common myeloid progenitor cells (CMP) were identified base on expression of Lin−Sca−1−c-Kit+CD34+CD16/32−; megakaryocyte-erythroid progenitor cells (MEP) were identified base on expression of Lin−Sca−1−c-Kit+CD34−CD16/32−; granulocyte-macrophage progenitor cells (GMP) were identified base on expression of Lin−Sca−1−c-Kit+CD34+CD16/32+.
Cell Cycle Analysis
BrdU (Sigma-Aldrich) was administered continuously to mice via drinking water (0.5 mg/ml). After 1 week, BrdU incorporation was analyzed using a BrdU Flow Kit (BD Biosciences) according to the manufacture’s recommendation.
Colony forming assay
Twenty thousand BM cells per replicate were plated in methylcellulose (3434; Stemcell Technologies). Colonies propagated in culture were scored at day 12–14, pooled and replated at 7.5 × 103 cells per replicate into the same medium.
In vivo RNAi screening
A customized shRNA library (Table S1) was cloned as previously described into the LMS-vector (Scouppo C., et al Nat, 2012). Retroviruses were produced by transient transfection of PlatE packaging cells. The virus-containing medium was collected after 72 hrs post-transfection and optimized dilution was used to infect <8% of ckit+ cell collected from Tet2−/− mice. Cells from Tet2 −/− mice were transduced with four different pools containing 5–6 shRNA per gene and a dsRed fluorescence marker. Two days later, an aliquot of transduced and dsRed sorted Tet2−/− cells (T0) were sequenced for shRNA library representation. In parallel, 8 × 106 total cKit+ cells (including 500.000 dsRED+ cells) were transplanted into lethally irradiated mice. Sorted DsRed+ cells before transplantation (T0) and sorted dsRED+ cells collected from the spleen of sick mice were used for genomic extraction and deep sequencing as described previously (Zuber et al., Nature 2011).
RNA sequencing
Total RNA was extracted from bone marrow sorted samples (Lineage negative, CD117+, SCA-1+) using RNeasy Plus Micro Kit (Qiagen #74034). Libraries were generated with SMART-Seq v4 Ultra™ Low Input RNA Kit for Sequencing and Low Input Library Prep Kit v2 (Takara Bio # 634899). Libraries were sequenced on an Illumina HiSeq 4000 sequencer. Sequencing results were demultiplexed and converted to FASTQ format using Illumina bcl2fastq software. The sequencing reads were aligned to the mouse genome (mm10/GRCm38) using the splice-aware STAR aligner. The feature Counts program (Liao, Y., Bioinformatics 2014) was utilized to generate counts for each gene based on how many aligned reads overlap its exons. These counts were then normalized and used to test for differential expression using negative binomial generalized linear models implemented by the DESeq2 R package. For gene-edited MV4;11 cells, RNA was isolated using Quick-RNA MiniPrep (Zymo Research, Cat#R1055) from MV4;11 cells treated with DMSO or CU-T12–9 (10μM) for 24 hours in biological triplicates. RNA libraries were prepared according to the Illumina TruSeq Stranded mRNA (polyA capture) library protocol by the DNA Sequencing and Genotyping Core at CCHMC.
Gene set enrichment analysis
We performed GSEA as described(Subramanian et al., 2005). Patients with the indicated cancers were stratified based on low and high TRAF6 expression as defined by: TRAF6 high, Z score > 1; TRAF6 low, Z score < 1.
Single cell RNA-seq analysis
Raw Illumina sequencing data was converted to FASTQ format using Illumina bcl2fastq software. We used Cell Ranger Single Cell Gene Expression Software (version 3.1.0, 10x Genomics) to demultiplex and align raw reads to GRCh38 (version 3.0.0). All following downstream analysis was performed using the Seurat R package (version 3.2.2)(Butler et al., 2018). To exclude low-quality cells and multiple cells captured in droplets, we excluded cells with less than 400 or more than 6000 unique feature counts, as well as cells with more than 15% transcripts originating from mitochondrial genes. After quality filtering, RNA expression data was normalized by total expression, multiplied by a scaling factor of 10,000 and log-transformed. To account for batch and patient differences, we integrated the 5 Control samples using Harmony (Korsunsky et al., 2019). The first 20 harmony embeddings were used as an input to uniform manifold approximation and projection (UMAP)(Becht et al., 2018), shared nearest neighbor graph (SNN) construction using 20 nearest neighbors and clustering using a resolution of 0.5. To identify clusters containing the AML relevant hematopoietic stem, progenitor and myeloid populations, we performed reference-based cell type annotation in individual cells with SingleR based on the Human Primary Cell Atlas reference (Aran et al., 2019). Clusters with >90% stem, progenitor or myeloid cell content were selected for downstream analysis. To refine the annotations for AML relevant clusters, we repeated the integration, UMAP, SNN graph construction and clustering for the relevant clusters as described above. To identify cluster markers, we performed differential expression analysis for cells within a cluster against the remaining cells using Wilcoxon rank sum test with Bonferroni multiple-comparison correction (gene expressed in at least 10% of cells, fold-change difference >10%, adjusted p-value <0.05). Cluster markers were used for manual cell type annotation. We used single-cell analysis via expression recovery (SAVER) to improve the visualization and coverage of genes with low or moderate expression (Huang et al., 2018). The MYC target score was calculated based on expression of CDK4, NPM1, HMGB3, APEX1, EIF4E, ID2, LDHA, MGST1, NME1, PTMA, RPL3, RPL6, SRM, CCNB1, HMGA1, RPL26, XIAP, MCM4, EIF4H and NME1. Gene expression counts were normalized to counts per million, filtered for genes expressed in any cell and normalized relative to the maximum expression level across cells. The rescaled values for the MYC target genes were averaged and used as the MYC target score. All analysis was performed using R, version 3.6.1.
Quantitative RT–PCR
Total RNA was extracted and purified using Quick-RNA MiniPrep (Zymo research, R1055) and reverse transcription was carried out using SuperScript cDNA Synthesis Kit (Invitrogen) or High-Capacity cDNA Reverse Transcription Kit (Thermo-Fisher). Quantitative RT–PCR was performed with the Taqman Master Mix.
Subtractive proteomics for TRAF6 ubiquitin substrate identification
Identification and quantification of ubiquitinated peptides was accomplished by coupling ubiquitin enrichment with a label-free, data independent acquisition (DIA) mass spectrometry workflow. Specifically, TF1 cells transduced with a doxycycline-inducible TRAF6 shRNA were grown in DMEM supplemented with FBS. Cells were treated with or without doxycycline for 24 hours. After treatment, cells were lysed, the proteins were digested with trypsin and the peptide desalted on a C18 SepPak (Waters). Ubiquitinated peptides were immuno-enriched with anti-diGlycine agarose beads using the PTMScan Ubiquitin Proteomics System (Cell Signaling Technology) using the vendor protocol. Eluted peptides were analyzed by nanoLC-MS/MS on a Sciex 5600+ TripleTOF mass spectrometer coupled to an Eksigent nanoLC ultra nanoflow HPLC system as reported previously (Heaven, 2016) but with the following instrument parameters. Data independent acquisition was set with a collection range from 480–760 m/z using a 6 m/z isolation window and 1 m/z overlap. Each scan cycle included a 250msec TofMS spectrum follow by 56 × 50msec overlapping 6 m/z region for a total duty cycle of 3089 msec. 1892 cycles were collected over the course of the 100 min gradient collection run. Comparative quantification of peptides from a 3 v 3 TRAF6-expressing (without doxycycline) v TRAF6-depleted (with doxycycline) TF1 cells were generated using PROTALIZER DIA software (Vulcan Analytical, Birmingham) as described in detail previously with the output data including the fold-change and p value for each identified ubiquitinated peptide(Heaven et al., 2016).
Proteins alkylation and digestion
Immunoprecipitated MYC eluted from beads in 100 mM NH4HCO3 and 1% SDS (typically 50–100 μl) were incubated at 56°C for 10 min followed by 10-min cooling at room temperature. Iodoacetamide was then added to 15 mM final concentration and samples were incubated at room temperature in dark for 30 min. To remove SDS and iodoacetamide, proteins were precipitated with 4 vol. of cold acetone at −20°C for 1 hour. Acetone precipitates were collected by 10-min centrifugation at 14000g. Pellets were rinsed with 80% acetone, dried on air and re-dissolved in 5 μl 50 mM NH4HCO3, 8 M urea. To digest proteins, urea-dissolved samples were mixed with 50 μl 50 mM NH4HCO3 containing 20 ng/μl trypsin (Sigma) followed by overnight incubation at 25°C. Digestion reactions were then acidified by mixing with 50 μl 2% heptafluorobutyric acid and peptides were desalted on C18 spin tips (Pierce) following manufacturer instructions. Desalted peptides were dried under vacuum and re-dissolved in 0.1% formic acid. Peptides concentration was measured on Nanodrop One (Thermo Scientific) at 205 nm.
LC-MS analysis of peptides
LC-MS/MS analysis of peptides was done on Orbitrap Fusion Lumos mass spectrometer coupled with Dionex Ultimate 3000 UHPLC (Thermo Scientific). During each run, 1–2 μg of peptides were injected and resolved on a 50-cm long EASY-Spray C18 column (Thermo Scientific) over 90-min long gradient 2–32% acetonitrile in 0.1% formic acid, followed by 5-min steep increase to 96% acetonitrile and 5-min step at 96% acetonitrile. Data-dependent acquisition was based on published protocol (Davis et al., 2017) except each cycle was set to last for 2 s instead of 3 s.
Mass spectrometry data analysis
Raw mass spectrometry data were processed using Proteome Discoverer 2.1 (Thermo Scientific). Sequest HT search engine was supplied with protein data base consisting of human (www.uniprot.org/proteomes/UP000005640) or mouse (www.uniprot.org/proteomes/UP000000589) proteome and list of known protein contaminants, parameters for mass tolerance were left at their default values. Peptides termini were set to fully trypsin specific with at most two missed cleavages allowed. Variable post-translational modifications were set to: cysteine carbamidomethylation; methionine oxidation; serine, threonine and tyrosine phosphorylation; lysine acetylation or ubiquitination (GlyGly dipeptide remained after trypsin digestion); and acetylation of protein N-terminus. MS1-based peptide quantitation was done using Precursor Ions Area Detector module in Proteome Discoverer. Visualization was done using ggplot2 (Wickham, 2016) within R environment for statistical computing (www.r-project.org).
In Vitro ubiquitin conjugation reaction
In Vitro Ubiquitin Conjugation Reaction was performed with ubiquitinylation kit (Enzo Life Science, #BML-UW9920) according to manufacturer’s protocol. Recombinant MYC (ab169901) and TRAF6 (UB312) were purchased from Abcam and Life Sensors, respectively. Recombinant MYC (K148R) was generated by GeneScript.
Plasmids
FLAG-tagged TRAF6 vectors have been described previously(Rhyasen et al., 2013). pRK5-HA-Ubiquitin-WT (17608) and pcDNA3-MYC (16011) was purchased from Addgene. Flag tagged TRAF6-C70A, MYC(K148R) and Ub-K63R cDNA was created by custom gene synthesis (IDT). TRAF6-C70A and Ub-K63R was cloned into pcDNA3 with EcoRI and XhoI. MYC(K148R) was cloned into pcDNA3 with EcoRI and XbaI. MYC and MYC(K148Q) were cloned into MSCV-IRES-GFP with EcoRI and XbaI.
Transfection
Transfection was performed with HEK293T cells with TransIT®-LT1 Transfection Reagent (MIR2305, Mirus) according to the manufactures’ recommendation.
Lentivirus production and infection
An inducible knockdown of human TRAF6 was performed in pTRIPZ system (RHS4696–101315918; OpenBiosystems). The leukemic cell lines transduced with pTRIPZ were generated by infection with the supernatants from transfected HEK293T cells in the presence of 4μg/mL of polybrene (TR-1003-G, Millipore). After transduction of pTRIPZ, 3μg of puromycin was used for selection. Doxycycline was used to induce knockdown of TRAF6. For overexpression of TRAF6 to THP1 and MV4;11 cells, LeGO-iG2 vector and MIEG3 (MSCV-IRES-GFP) was used, respectively.
Luciferase assay
HEK293T cells were transfected for 48 h with MYC-luciferase (pBV-Luc MBS1–4) (16564, Addgene) and pGL4.70[hRluc] Vector (E688A, Promega) plasmids along with TRAF6, TRAF6-C70A, MYC or MYC(K148R). Lysates were analyzed for MYC reporter activity using the dual luciferase reporter assay system (E1910, Promega).
Immunoblotting and immunoprecipitation
Cell extract was prepared by lysing cells in sodium dodecyl sulfate (SDS) sample buffer followed by incubation with benzonase (70746, Millipore) on ice for 10 minutes. Nuclear and cytoplasmic fractionation was performed with Nuclear Extract Kit (40010, Active Motif) according to the manufacture’s protocol. Samples were boiled at 95 °C for 5 minutes and loaded to SDS-polyacrylamide gel electrophoresis (PAGE) and electrotransferred to nitrocellulose membranes (162–0112, Bio-Rad). For immunoprecipitation, cell extract was prepared by lysing cells in cold RIPA buffer in the presence of PMSF, sodium orthovanadate, protease and phosphatase inhibitors. Protein concentration was evaluated by a BCA assay (32106, Pierce). MYC antibody (9402, Cell Signaling) or normal rabbit IgG antibody (2729, Cell Signaling) was added to cell lysates (>2 mg) for 1h at 4°C following by the addition of Protein A/G Plus beads (sc-2003; Santa Cruz) at 4°C overnight. Western blot analysis was performed with the antibodies listed in the Key Resource Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-TRAF6 antibody (H-274) | Santa Cruz | Cat# sc-2003 |
| Rabbit monoclonal anti-Vinculin antibody (E1E9V) | Cell Signaling Technology | Cat# 13901 |
| Rabbit monoclonal anti-TET2 Antibody (D6B9Y) | Cell Signaling Technology | Cat# 18950 |
| Mouse monoclonal anti- Ubiquitin antibody (P4D1) | Santa Cruz | Cat# sc-8017 |
| Monoclonal anti- FLAG® M2 antibody in mouse | Sigma | Cat# F1804 |
| Rabbit polyclonal anti-c-Myc Antibody | Cell Signaling Technology | Cat# 9402 |
| Rabbit monoclonal anti-phospho-c-Myc (Ser62) antibody (E1J4K) | Cell Signaling Technology | Cat# 13748 |
| Rabbit monoclonal anti-phospho-c-Myc (Thr58/Ser62) antibody (E203) | Millipore | Cat# 04-217 |
| Rabbit polyclonal anti-acetyl-c-Myc (Lys148) antibody | Millipore | Cat# ABE25 |
| Rabbit monoclonal anti-GAPDH antibody (D16H11) | Cell Signaling Technology | Cat# 5174 |
| Rabbit polyclonal anti-α/β-Tubulin antibody | Cell Signaling Technology | Cat# 2148 |
| Rabbit monoclonal anti-Lamin B1 antibody (D4Q4Z) | Cell Signaling Technology | Cat# 12586 |
| Normal Rabbit IgG | Cell Signaling Technology | Cat# 2729 |
| Anti-CD11b PE-Cy7 | eBioscience | Cat# 25-0112-81 |
| Anti-Gr1 eFluor450 | eBioscience | Cat# 48-5931-82 |
| Anti-CD3 PE | eBioscience | Cat# 12-0031-83 |
| Anti-B220 APC | eBioscience | Cat# 17-0452-82 |
| Anti-Ter119 APC | eBiosceince | Cat# 17-5921-82 |
| Anti-CD71 PE | eBioscience | Cat# 12-0711-81 |
| Anti-CD45-APC-Cy7 | BD Biosciences | Cat# 557659 |
| Anti-CD44-BV421 | BD Biosciences | Cat# 563970 |
| Anti-CD45.1-Brilliant Violet 510 | BioLegend | Cat# 110741 |
| Anti-CD45.2-APC-eFluor780 | eBiosciences | Cat# 47-0454-82, |
| Anti-CD45.2- eFluor450 | eBiosciences | Cat# 48-0454-82 |
| Mouse hematopoietic lineage biotin panel | eBiosciences | Cat# 88-7774-75 |
| Anti-streptavidin eFluor780 | eBiosciences | Cat# 47-4317-82 |
| Anti-Sca-1-PE | eBiosciences | Cat# 12-5981-82 |
| Anti-c-Kit-APC | eBiosciences | Cat# 17-1171-81 |
| Anti-CD48-FITC | Affymetrix | Cat# 11-0481-85 |
| Anti-CD150-PE-cy7 | BioLegend | Cat# 115914 |
| Anti-Sca-1-PE-cy7 | eBiosciences | Cat# 25-5981-82 |
| Anti-CD34-FITC | eBiosciences | Cat# 11-0341-82 |
| Anti-CD16/32-PE | eBiosciences | Cat# 12-0161-82 |
| Anti-cMyc | Cell Signaling Technology | Cat # 18583S |
| Bacterial and virus strains | ||
| OneShot TOP10 Competent Cells | ThermoFisher | Cat# C404010 |
| Chemicals, peptides, and recombinant proteins | ||
| 5-Bromo-2-deoxyuridine (BrdU) | Sigma | Cat# B5002 |
| 10058-F4 | Sigma | Cat# F3680 |
| CU-T12-9 | Tocris Bioscience | Cat# 5414 |
| Cycloheximide | Santa Cruz | Cat# sc-3508 |
| Methocult GF M3434 | Stem Cell Technologies | Cat# 03434 |
| Mouse Flt3-ligand cytokine | PeproTech | Cat# 250-31 |
| Human IL-6 cytokine | PeproTech | Cat# 200-06 |
| Mouse SCF cytokine | PeproTech | Cat# 250-03 |
| Human TPO cytokine | PeproTech | Cat# 300-18 |
| RPMI | Fisher | Cat# SH30027.01 |
| DMEM | Fisher | Cat# SH30022FS |
| Fetal Bovine Serum | Atlanta Biologicals | Cat# S11550 |
| Penicillin/Steptomycin | ThermoFisher | Cat# SV30010 |
| TransIT-LT1 transfection reagent | Mirus | Cat# MIR2306 |
| IMDM | Corning | Cat# 10-016-CV |
| Benzonase | Millipore | Cat# 70746 |
| Nitrocellulose membrane | Bio-Rad | Cat# 162-0112 |
| RIPA Lysis Buffer | Santa Cruz | Cat# sc-24948 |
| Protein A/G Plus beads-Agarose | Santa Cruz | Cat# sc-2003 |
| TrueCut Cas9 Protein v2 | Thermo Fisher | Cat# A36498 |
| SuperScript™ II Reverse Transcriptase | Invitrogen | Cat# 18064014 |
| TaqMan™ Universal PCR Master Mix | Life Technologies | Cat# 4324018 |
| Recombinant c-Myc (WT) | Abcam | Cat# ab169901 |
| Recombinant c-Myc (K148R) | GeneScript | N/A |
| Recombinant TRAF6 | Life Sensors | Cat# UB312 |
| Polybrene | Millipore | Cat# TR-1003-G |
| EcoRI-HF | New England Biolabs | Cat# R3101L |
| XhoI | New England Biolabs | Cat# R0146S |
| XbaI | New England Biolabs | Cat# R0145S |
| Quick Ligation kit | New England Biolabs | Cat# M2200S |
| Puromycin | Corning | Cat# 61-385-RA |
| Doxycycline | Fisher Scientific | Cat# BP26535 |
| Poly(I:C) | Tocris Bioscience | Cat# 4287 |
| High-Capacity cDNA Reverse Transcription Kit | ThermoFisher | Cat# 4368814 |
| 5-fluorouracil (5-FU) | CCHMC pharmacy | NDC#63323-117-10 |
| 2-mercaptoethanol | Fisher | Cat#03446I |
| Blasticidin | Gibco | Cat# R21001 |
| RetroNectin | Takara | Cat# T100B |
| AMPURE XP 60ML | Thermo Fisher Scientific | Cat#NC9933872 |
| FORMALDEHYDE 20% 10X10ML | Thermo Fisher Scientific | Cat# NC9611804 |
| Critical commercial assays | ||
| FITC BrdU Flow Kit | BD Biosciences | Cat# 559619 |
| CellTiterGlo kit | Promega | Cat# G7570. |
| Zymo Quick-RNA Miniprep Kit | Zymo Research | Cat# R1050 |
| Mouse hematopoietic progenitor cell enrichment kit | Stem Cell Technologies | Cat# 19856 |
| Nuclear Extract Kit | Active Motif | Cat# 40010 |
| BCA assay | Pierce | Cat# 32106 |
| Neon™ Transfection System 10 μL Kit | Invitrogen | Cat# MPK1025 |
| RNeasy Plus Micro Kit | Qiagen | Cat# 74034 |
| SMART-Seq® v4 Ultra® Low Input RNA Kit for Sequencing | Takara Bio | Cat# 634899 |
| Taqman Assay human CDK4 probe | Thermo Fisher | Cat# 4453320 Assay ID: Hs00364847_m1 |
| Taqman Assay Human NPM1 probe | Thermo Fisher | Cat# 4448892 Assay ID: Hs02339479_g1 |
| Taqman Assay Human GAPDH probe | Thermo Fisher | Cat# 4351370 Assay ID: Hs02758991_g1 |
| Taqman Assay Mouse Cdk4 | Thermo Fisher | Cat# 4331182 Assay ID: Mm00726334_s1 |
| Taqman Assay Mouse Npm1 | Thermo Fisher | Cat# 4331182 Assay ID: Mm02391781_g1 |
| Taqman Assay Mouse Eif4e | Thermo Fisher | Cat# 4331182 Assay ID: Mm05786298_g1 |
| Taqman Assay Mouse Rpl6 | Thermo Fisher | Cat# 4351372 Assay ID: Mm05899694_g1 |
| Taqman Assay Mouse Ccna2 | Thermo Fisher | Cat# 4331182 Assay ID: Mm01292244_m1 |
| Taqman Assay Mouse Ldha | Thermo Fisher | Cat# 4331182 Assay ID: Mm01612132_g1 |
| Taqman Assay Mouse Gapdh | Thermo Fisher | Cat# 4331182 Assay ID: Mm99999915_g1 |
| PTMScan® Ubiquitin Remnant Motif (K-ε-GG) Kit | Cell Signaling Technology | Cat# 5562 |
| Ubiquitinylation Kit | Enzo Life Science | Cat# BML-UW9920 |
| Dual luciferase reporter assay system | Promega | Cat# E1910 |
| ThruPLEX® DNA-Seq Kit | Takara | Cat# R400675 |
| Dynabeads Protein G for Immunoprecipitation | Life Technologies | Cat# 10004D |
| Deposited data | ||
| RNA-Seq with human leukemia cells | This study |
GSE185295 GSE185299 |
| RNA-Seq with mouse HSPC | This study | GSE184937 |
| Microarray with MDS BM cells | Pellagatti et al. Leukemia 2010 | GSE19429 |
| Experimental models: Cell lines | ||
| MV4;11 | ATCC | Cat# CRL-9591 |
| HEL | ATCC | Cat# TIB-180 |
| THP-1 | ATCC | Cat# TIB-202 |
| HEK293T | ATCC | Cat# CRL-3216 |
| Plat-E cells | Cell biolabs inc. | RV-101 |
| Experimental models: Organisms/strains | ||
| Mouse: Traf6fl/fl mice (C57Bl/6) | Gift from Yongwon Choi (Han et al. Immunity 2013) | N/A |
| Mouse: Tet2fl/fl mice (C57Bl/6) | Jackson | Stock No: 017573 |
| Mouse: Mx1-Cre (C57Bl/6) | Jackson | Stock No: 003556 |
| Mouse: Vav-Cre (C57Bl/6) | Jackson | Stock No: 008610 |
| Mouse: Vav-TRAF6 (C57Bl/6) | Starczynowski Lab (Fang et al. Nat Immunol 2017) | N/A |
| Mouse: FLT3-ITD (C57Bl/6) | Jackson | Stock No: 011112 |
| Oligonucleotides | ||
| sgRNA and ssODN listed in Supplemental Table 7 | This study | N/A |
| Recombinant DNA | ||
| pCDLN packaging vector | Gift from Gang Huang laboratory | N/A |
| pMD.2 VSV-G envelope vector | Gift from Gang Huang laboratory | N/A |
| M57 retroviral gag/pol plasmid | Gift from Gang Huang laboratory | N/A |
| pcDNA3- FLAG-tagged TRAF6 | Starczynowski Lab (Rhyasen et al. Cancer Cell 2013) | N/A |
| pRK5-HA-Ubiquitin-WT | Addgene | Cat# 17608 |
| pcDNA3-MYC (WT) | Addgene | Cat# 16011 |
| pcDNA3- FLAG-tagged TRAF6-C70A | This study | N/A |
| pRK5-HA-Ubiquitin-K63R | This study | N/A |
| pcDNA3-MYC(K148R) | This study | N/A |
| pBV-Luc wt MBS1-4 | Addgene | Cat# 16564 |
| pGL4.70[hRluc] | Promega | Cat# E688A |
| MSCV-IRES-GFP | Gift from Jose Cancelas Lab (Chang et al. Cell Rep 2014) | N/A |
| MSCV-MYC (WT)-IRES-GFP | This study | Cloned from Cat# 16011 |
| MSCV- MYC (K148Q)-IRES-GFP | This study | N/A |
| pTRIPZ shTRAF6 | Open Biosystems | Cat# RHS4696-101315918 Clone Id: V3THS_372743 |
| LeGO-iG2-IRES-GFP | Addgene | Cat# 27341 |
| LeGO-iG2-FLAG-tagged TRAF6-IRES-GFP | Starczynowski Lab | N/A |
| Software and algorithms | ||
| iGeak | Starczynowski Lab (Choi et al. BMC Genomics. 2019) | N/A |
CellTiter Glo assay
CellTiter Glo Luminescent Viability Assay (Promega, Cat#G7572) was performed according to manufacturer protocol. Analysis was performed using GloMax 96 microplate Luminometer (Promega) with GloMax Software.
Chromatin immunoprecipitation with sequencing (ChIP-seq)
For MYC Chip-seq 2 million cells were fixed in PBS containing 1% formaldehyde for 15 min at RT in rotation. The reaction was quenched with 0.125 M glycine for 5 min at room temperature. After washing with PBS, cells were collected and the pellet was lysed in 50 mM Hepes pH 8.0 at 4C, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% Igepal CA-630, and 0.25% Triton X-100. After centrifugation, isolated nuclei were washed once in 10 mM Tris pH 8.0 at 4C, 200 mM NaCl, 1 mM EDTA and 0.5 mM EGTA, then resuspended in 130 ul of 10 mM Tris pH 8.0 at 4C, 1 mM EDTA, 0.5 mM EGTA and 0.5% N-Lauryl Sarcosine, and sonicated using COVARIS E220. Chromatin was diluted 1:1 with IP buffer to a final concentration of 1% Igepal CA-630, 0.25% N-LaurylSarcosine, 150 mM NaCl, 10 mM Tris pH 8.0 at 4C. Antibodies were at a final concentration of 1:50 and incubated at 4°C overnight. Protein G Dynabeads (Invitrogen) were blocked with BSA for 1h at 4C, and were added to each IP. After 2 hr of incubation at 4°C, the beads were washed 2X with low salt buffer (0.1% SDS, 1% TRITON X-100, 2mM EDTA, 20mM Tris pH8 150mM NaCl), 2x High Salt Buffer (0.1% SDS, 1% TRITON X-100, 2mM EDTA, 20mM Tris pH8 500mM NaCl), 2X Li Buffer (10mM Tris pH8, 250 mM LiCl, 1% NP-40, 1% deoxycholic acid, 1mM EDTA). The beads were then washed with 750 μl of 10 mM Tris pH 8.0 at 4C, 1 mM EDTA and 50 mM NaCl. Then each sample and 10% of the input was resuspended in 100 mM sodium bicarbonate, 200 mM NaCl, 1% SDS and 10 μ g of RNase A and incubated for 30 min at 37C. Finally, 10 μg of proteinase K was added to each sample and cross-linking was reversed at 65°C overnight. DNA was extracted by Phenol/Chloroform. Library were generated by amplification with ThruPLEX DNA-seq Kit following manufacture’s indications. For K27Acetylation we followed the Chipmentation protocol (Schmidl C Mat Protocols 2015). For Chip-seq analysis, reads were mapped to the human reference genome (GRCh37/hg19) using the Bowtie2 (v2.2.4)(Langmead and Salzberg, 2012) and duplicate reads were removed using Picard tools (v.1.126) (http://broadinstitute.github.io/picard/). Low quality mapped reads (MQ<20) were removed from the analysis. The read per million (RPM) normalized BigWig files were generated using BEDTools (v.2.17.0)(Quinlan and Hall, 2010) and the bedGraphToBigWig tool (v.4). Peak calling was performed using MACS (v1.4.2)(Zhang et al., 2008) and peak count tables were created using BEDTools. Differential peak analysis was performed using DESeq2(Love et al., 2014). ChIPseeker (v1.8.0)(Yu et al., 2015) R package was used for peak annotations and motif discovery was performed using HOMER (v4.10)(Heinz et al., 2010). ngs.plot (v2.47)(Heinz et al., 2010) and ChIPseeker were used for TSS site visualizations and quality controls. KEGG pathway analysis and Hallamark geneset analysis was performed using the clusterProfiler R package (v3.0.0)(Yu et al., 2012). To compare the level of similarity among the samples and their replicates, we used two methods: principal-component analysis and Euclidean distance-based sample clustering. The downstream statistical analyses and generating plots were performed in R environment (v3.1.1) (https://www.r-project.org/).
sgRNA Design and Synthesis
Protospacer sequence for human MYC gene and human TET2 was identified using the web tool, CRISPOR (http://crispor.tefor.net/crispor.py)(Haeussler et al., 2016). The targeted sequence of the gRNAs (sg-MYC and sgTET2) is shown in Table S7. The gRNA targeting MYC as well as non-targeting gRNA (sg-CTL) was directly purchased from Synthego. Two single-stranded oligodeoxynucleotides (ssODNs), ssODN-MYCK148R and ssODN-MYCK148Q, were designed with 3 silent mutations within the recognition sequence of sg-MYC to avoid repetitive digestions (Table S3). Alexa Fluor-488 NHS Ester was added to 5’-end the ssODNs. The synthesized single stranded oligonucleotides were purchased from Integrated DNA Technologies (IDT).
Cas9-sgRNA Pre-complexing and Transfection
To obtain Cas9-sgRNA RNPs, 200 ng of synthetic sgRNA (sg-CTL, sg-MYC or sg-TET2) was incubated with 1 μg Cas9 (TrueCut, ThermoFisher) for 10 min at room temperature. MV4;11 cells (3 × 105 cells) were electroporated in Buffer R (ThermoFisher) with either ssODN-MYC(K148R) or ssODN-MYC(K148Q). Electroporation was performed using the Neon Transfection System. Electroporation conditions were 1350 V, 35 ms, 1 pulse. Electroporation conditions for human CD34+BM cells were 1600V, 10ms, 3 pulses.
Cloning of MV4;11-sg-CTL, -sg-K148R and -sg-K148Q
Alexa Fluor-488 positive single MV4;11 cells were sorted into 96-well plates using the BD FACSAria™ II (BD Biosciences) flow cytometer (cell sorter) 24 h post-electroporation. After expansion of each clone, MYC mutation status was confirmed by PCR, followed by Sanger sequence.
QUANTIFICATION AND STATISTICAL ANALYSIS
The number of animals, cells, and experimental/biological replicates can be found in the figure legends. Differences among multiple groups were assessed by one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison posttest for all possible combinations. Comparison of two group was performed using a Student’s t-test (unpaired, two tailed). Unless otherwise specified, results are depicted as the mean ± standard deviation. For correlation analysis, Pearson correlation coefficient (r) was calculated. D’Agostino and Pearson and Shapiro-Wilk tests were performed to assess data distributions. For Kaplan-Meier analysis, Mantel-Cox test was used. GraphPad Prism (v5, GraphPad) was used for statistical analysis. For correlative analyses, Spearman rank test was used.
Supplementary Material
Table S1. Genes targeted in shRNA screen, Related to Figure 1.
Table S2. Differentially expressed genes in Tet2−/− vs WT LSK, Related to Figure 5.
Table S3. Differentially expressed genes in Traf6−/− vs WT LSK, Related to Figure 5.
Table S4. Differentially expressed genes in Traf6−/−;Tet2−/− vs WT LSK, Related to Figure 5.
Table S5. Differentially expressed genes in Tet2−/− vs Traf6−/−;Tet2−/− LSK, Related to Figure 5.
Table S6. Differential ubiquitin profile following knockdown of TRAF6 in TF1 cells, Related to Figure 6A.
Table S7. sgRNA sequences used in this study, Related to STAR Methods.
Highlights.
Loss of TRAF6 result in progression of clonal hematopoiesis to AML
Loss of TRAF6 results in increased MYC activation
TRAF6 ubiquitinates MYC and represses its transcriptional activity
TLR-induced TRAF6 activation suppresses MYC function in AML
Acknowledgements
This work was supported by the NIH (R01CA216421, R01CA173636, R01CA228135, R01CA242020, 1RO1CA266212, 1RO1HL159175 to IA; R35HL135787, R01DK102759, R01DK113639 to DTS; L40HL143713 to LHL; R01CA190261 to SWL), The Edward P. Evans MDS Foundation (IA), The Leukemia and Lymphoma Society (TM, DTS and IA), The Uehara Memorial Foundation (TM), The NY State Department of Health IDEA and IIRP programs (IA), The Waksman Foundation of Japan (TM), The Mochida Memorial Foundation for Medical and Pharmaceutical Research (TM), Cincinnati Children’s Hospital Research Foundation (DTS, LHL), Japan Society for the Promotion of Science (TM), Cancer Free Kids (DTS, LHL), and Pelotonia Fellowship (TM). MG is supported by the American Society of Hematology and Gilead Sciences Inc. SWL is a Howard Hughes Medical Institute investigator.
Footnotes
Declaration of Interests
DTS serves on the scientific advisory board at Kurome Therapeutics, and is a consultant for Kymera Therapeutics, Kurome Therapeutics, Captor Therapeutics, and Tolero Therapeutics. DTS has equity in Kurome Therapeutics. The other authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ågerstam H, Karlsson C, Hansen N, Sandén C, Askmyr M, von Palffy S, Högberg C, Rissler M, Wunderlich M, Juliusson G, et al. (2015). Antibodies targeting human IL1RAP (IL1R3) show therapeutic effects in xenograft models of acute myeloid leukemia. Proceedings of the National Academy of Sciences 112, 10786–10791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. (2013). Signatures of mutational processes in human cancer. Nature 500, 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, et al. (2019). Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol 20, 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askmyr M, Ågerstam H, Hansen N, Gordon S, Arvanitakis A, Rissler M, Juliusson G, Richter J, Järås M, and Fioretos T (2013). Selective killing of candidate AML stem cells by antibody targeting of IL1RAP. Blood 121, 3709–3713. [DOI] [PubMed] [Google Scholar]
- Barreyro L, Chlon TM, and Starczynowski DT (2018). Chronic immune response dysregulation in MDS pathogenesis. Blood 132, 1553–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, Ginhoux F, and Newell EW (2018). Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol. [DOI] [PubMed] [Google Scholar]
- Bennett J, and Starczynowski DT (2021). IRAK1 and IRAK4 as emerging therapeutic targets in hematologic malignancies. Curr Opin Hematol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldin MP, Taganov KD, Rao DS, Yang L, Zhao JL, Kalwani M, Garcia-Flores Y, Luong M, Devrekanli A, Xu J, et al. (2011). miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J Exp Med 208, 1189–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman RL, Busque L, and Levine RL (2018). Clonal Hematopoiesis and Evolution to Hematopoietic Malignancies. Cell Stem Cell 22, 157–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busque L, Patel JP, Figueroa ME, Vasanthakumar A, Provost S, Hamilou Z, Mollica L, Li J, Viale A, Heguy A, et al. (2012). Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet 44, 1179–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research N, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, Hoadley K, Triche TJ Jr., Laird PW, et al. (2013). Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 368, 2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capitano ML (2019). Toll-like receptor signaling in hematopoietic stem and progenitor cells. Curr Opin Hematol 26, 207–213. [DOI] [PubMed] [Google Scholar]
- Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, Bock C, Vasanthakumar A, Gu H, Xi Y, et al. (2011). Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet 44, 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, Bock C, Vasanthakumar A, Gu H, Xi Y, et al. (2012). Dnmt3a is essential for hematopoietic stem cell differentiation. In Nat Genet (United States), pp. 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Kao YR, Sun D, Todorova TI, Reynolds D, Narayanagari SR, Montagna C, Will B, Verma A, and Steidl U (2019a). Myelodysplastic syndrome progression to acute myeloid leukemia at the stem cell level. Nat Med 25, 103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Sun XX, Sears RC, and Dai MS (2019b). Writing and erasing MYC ubiquitination and SUMOylation. Genes Dis 6, 359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu SH, Heiser D, Li L, Kaplan I, Collector M, Huso D, Sharkis SJ, Civin C, and Small D (2012). FLT3-ITD knockin impairs hematopoietic stem cell quiescence/homeostasis, leading to myeloproliferative neoplasm. Cell Stem Cell 11, 346–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duy C, Li M, Teater M, Meydan C, Garrett-Bakelman FE, Lee TC, Chin CR, Durmaz C, Kawabata KC, Dhimolea E, et al. (2021). Chemotherapy Induces Senescence-Like Resilient Cells Capable of Initiating AML Recurrence. Cancer Discov 11, 1542–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edberg SC, Allen MJ, Smith DB, and Kriz NJ (1990). Enumeration of total coliforms and Escherichia coli from source water by the defined substrate technology. Appl Environ Microbiol 56, 366–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engen C, Hellesøy M, Dowling TH, Eldfors S, Ferrell B, Gullaksen S-E, Popa M, Brendehaug A, Karjalainen R, Mejlænder-Andersen E, et al. (2020). Converging molecular evolution in acute myeloid leukaemia. medRxiv, 2020.2011.2003.20222885. [Google Scholar]
- Eriksson M, Pena-Martinez P, Ramakrishnan R, Chapellier M, Hogberg C, Glowacki G, Orsmark-Pietras C, Velasco-Hernandez T, Lazarevic VL, Juliusson G, et al. (2017). Agonistic targeting of TLR1/TLR2 induces p38 MAPK-dependent apoptosis and NFkappaB-dependent differentiation of AML cells. Blood Adv 1, 2046–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faiola F, Liu X, Lo S, Pan S, Zhang K, Lymar E, Farina A, and Martinez E (2005). Dual regulation of c-Myc by p300 via acetylation-dependent control of Myc protein turnover and coactivation of Myc-induced transcription. Mol Cell Biol 25, 10220–10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J, Barker B, Bolanos L, Liu X, Jerez A, Makishima H, Christie S, Chen X, Rao DS, Grimes HL, et al. (2014). Myeloid malignancies with chromosome 5q deletions acquire a dependency on an intrachromosomal NF-kappaB gene network. Cell Rep 8, 1328–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J, Bolanos LC, Choi K, Liu X, Christie S, Akunuru S, Kumar R, Wang D, Chen X, Greis KD, et al. (2017). Ubiquitination of hnRNPA1 by TRAF6 links chronic innate immune signaling with myelodysplasia. Nat Immunol 18, 236–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J, Muto T, Kleppe M, Bolanos LC, Hueneman KM, Walker CS, Sampson L, Wellendorf AM, Chetal K, Choi K, et al. (2018). TRAF6 Mediates Basal Activation of NF-kappaB Necessary for Hematopoietic Stem Cell Homeostasis. Cell Rep 22, 1250–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J, Rhyasen G, Bolanos L, Rasch C, Varney M, Wunderlich M, Goyama S, Jansen G, Cloos J, Rigolino C, et al. (2012). Cytotoxic effects of bortezomib in myelodysplastic syndrome/acute myeloid leukemia depend on autophagy-mediated lysosomal degradation of TRAF6 and repression of PSMA1. Blood 120, 858–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell AS, and Sears RC (2014). MYC degradation. Cold Spring Harb Perspect Med 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, et al. (2014). Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 371, 2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstung M, Pellagatti A, Malcovati L, Giagounidis A, Porta MG, Jadersten M, Dolatshad H, Verma A, Cross NC, Vyas P, et al. (2015). Combining gene mutation with gene expression data improves outcome prediction in myelodysplastic syndromes. Nat Commun 6, 5901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Prieto R, Cuijpers SA, Kumar R, Hendriks IA, and Vertegaal AC (2015). c-Myc is targeted to the proteasome for degradation in a SUMOylation-dependent manner, regulated by PIAS1, SENP7 and RNF4. Cell Cycle 14, 1859–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeussler M, Schonig K, Eckert H, Eschstruth A, Mianne J, Renaud JB, Schneider-Maunoury S, Shkumatava A, Teboul L, Kent J, et al. (2016). Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol 17, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D, Walsh MC, Cejas PJ, Dang NN, Kim YF, Kim J, Charrier-Hisamuddin L, Chau L, Zhang Q, Bittinger K, et al. (2013). Dendritic cell expression of the signaling molecule TRAF6 is critical for gut microbiota-dependent immune tolerance. Immunity 38, 1211–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaven MR, Funk AJ, Cobbs AL, Haffey WD, Norris JL, McCullumsmith RE, and Greis KD (2016). Systematic evaluation of data-independent acquisition for sensitive and reproducible proteomics-a prototype design for a single injection assay. J Mass Spectrom 51, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M, Wang J, Torre E, Dueck H, Shaffer S, Bonasio R, Murray JI, Raj A, Li M, and Zhang NR (2018). SAVER: gene expression recovery for single-cell RNA sequencing. Nat Methods 15, 539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang MJ, Cheng YC, Liu CR, Lin S, and Liu HE (2006). A small-molecule c-Myc inhibitor, 10058-F4, induces cell-cycle arrest, apoptosis, and myeloid differentiation of human acute myeloid leukemia. Exp Hematol 34, 1480–1489. [DOI] [PubMed] [Google Scholar]
- Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, et al. (2014). Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 371, 2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King B, Trimarchi T, Reavie L, Xu L, Mullenders J, Ntziachristos P, Aranda-Orgilles B, Perez-Garcia A, Shi J, Vakoc C, et al. (2013). The ubiquitin ligase FBXW7 modulates leukemia-initiating cell activity by regulating MYC stability. Cell 153, 1552–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King CG, Kobayashi T, Cejas PJ, Kim T, Yoon K, Kim GK, Chiffoleau E, Hickman SP, Walsh PT, Turka LA, et al. (2006). TRAF6 is a T cell-intrinsic negative regulator required for the maintenance of immune homeostasis. Nat Med 12, 1088–1092. [DOI] [PubMed] [Google Scholar]
- Klinakis A, Lobry C, Abdel-Wahab O, Oh P, Haeno H, Buonamici S, van De Walle I, Cathelin S, Trimarchi T, Araldi E, et al. (2011). A novel tumour-suppressor function for the Notch pathway in myeloid leukaemia. Nature 473, 230–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Kim TS, Jacob A, Walsh MC, Kadono Y, Fuentes-Panana E, Yoshioka T, Yoshimura A, Yamamoto M, Kaisho T, et al. (2009). TRAF6 is required for generation of the B-1a B cell compartment as well as T cell-dependent and -independent humoral immune responses. PLoS One 4, e4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogan SC, Ward JM, Anver MR, Berman JJ, Brayton C, Cardiff RD, Carter JS, de Coronado S, Downing JR, Fredrickson TN, et al. (2002). Bethesda proposals for classification of nonlymphoid hematopoietic neoplasms in mice. Blood 100, 238–245. [DOI] [PubMed] [Google Scholar]
- Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh PR, and Raychaudhuri S (2019). Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 16, 1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BH, Tothova Z, Levine RL, Anderson K, Buza-Vidas N, Cullen DE, McDowell EP, Adelsperger J, Frohling S, Huntly BJ, et al. (2007). FLT3 mutations confer enhanced proliferation and survival properties to multipotent progenitors in a murine model of chronic myelomonocytic leukemia. Cancer Cell 12, 367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Zhang JH, Ginzburg Y, Li HH, Xue FM, De Franceschi L, Chasis JA, Mohandas N, and An XL (2013). Quantitative analysis of murine terminal erythroid differentiation in vivo: novel method to study normal and disordered erythropoiesis. Blood 121, E43–E49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loizou E, Banito A, Livshits G, Ho YJ, Koche RP, Sanchez-Rivera FJ, Mayle A, Chen CC, Kinalis S, Bagger FO, et al. (2019). A Gain-of-Function p53-Mutant Oncogene Promotes Cell Fate Plasticity and Myeloid Leukemia through the Pluripotency Factor FOXH1. Cancer Discov. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardis E, Ding L, Dooling D, Larson D, McLellan M, Chen K, Koboldt D, Fulton R, Delehaunty K, McGrath S, et al. (2009). Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 361, 1058–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayle A, Yang L, Rodriguez B, Zhou T, Chang E, Curry CV, Challen GA, Li W, Wheeler D, Rebel VI, et al. (2015). Dnmt3a loss predisposes murine hematopoietic stem cells to malignant transformation. Blood 125, 629–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, Figueroa ME, Vasanthakumar A, Patel J, Zhao X, et al. (2011). Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 20, 11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muto G, Kotani H, Kondo T, Morita R, Tsuruta S, Kobayashi T, Luche H, Fehling HJ, Walsh M, Choi Y, et al. (2013a). TRAF6 is essential for maintenance of regulatory T cells that suppress Th2 type autoimmunity. PLoS One 8, e74639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muto T, Sashida G, Hasegawa N, Nakaseko C, Yokote K, Shimoda K, and Iwama A (2015). Myelodysplastic syndrome with extramedullary erythroid hyperplasia induced by loss of Tet2 in mice. Leuk Lymphoma 56, 520–523. [DOI] [PubMed] [Google Scholar]
- Muto T, Sashida G, Oshima M, Wendt GR, Mochizuki-Kashio M, Nagata Y, Sanada M, Miyagi S, Saraya A, Kamio A, et al. (2013b). Concurrent loss of Ezh2 and Tet2 cooperates in the pathogenesis of myelodysplastic disorders. J Exp Med 210, 2627–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muto T, Walker CS, Choi K, Hueneman K, Smith MA, Gul Z, Garcia-Manero G, Ma A, Zheng Y, and Starczynowski DT (2020). Adaptive response to inflammation contributes to sustained myelopoiesis and confers a competitive advantage in myelodysplastic syndrome HSCs. Nat Immunol 21, 535–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan S, Bader GD, and Reimand J (2016). Frequent mutations in acetylation and ubiquitination sites suggest novel driver mechanisms of cancer. Genome Med 8, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nischal S, Bhattacharyya S, Christopeit M, Yu Y, Zhou L, Bhagat TD, Sohal D, Will B, Mo Y, Suzuki M, et al. (2013). Methylome profiling reveals distinct alterations in phenotypic and mutational subgroups of myeloproliferative neoplasms. Cancer Res 73, 1076–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oishi Y, and Manabe I (2016). Macrophages in age-related chronic inflammatory diseases. Npj Aging And Mechanisms Of Disease 2, 16018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellagatti A, Cazzola M, Giagounidis A, Perry J, Malcovati L, Della Porta MG, Jadersten M, Killick S, Verma A, Norbury CJ, et al. (2010). Deregulated gene expression pathways in myelodysplastic syndrome hematopoietic stem cells. Leukemia 24, 756–764. [DOI] [PubMed] [Google Scholar]
- Pham LV, Zhou HJ, Lin-Lee YC, Tamayo AT, Yoshimura LC, Fu L, Darnay BG, and Ford RJ (2008). Nuclear tumor necrosis factor receptor-associated factor 6 in lymphoid cells negatively regulates c-Myb-mediated transactivation through small ubiquitin-related modifier-1 modification. J Biol Chem 283, 5081–5089. [DOI] [PubMed] [Google Scholar]
- Pierce NW, Kleiger G, Shan SO, and Deshaies RJ (2009). Detection of sequential polyubiquitylation on a millisecond timescale. Nature 462, 615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quivoron C, Couronne L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, Do Cruzeiro M, Delhommeau F, Arnulf B, Stern MH, et al. (2011). TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 20, 25–38. [DOI] [PubMed] [Google Scholar]
- Rampal R, Al-Shahrour F, Abdel-Wahab O, Patel JP, Brunel JP, Mermel CH, Bass AJ, Pretz J, Ahn J, Hricik T, et al. (2014). Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood 123, e123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhyasen GW, Bolanos L, Fang J, Jerez A, Wunderlich M, Rigolino C, Mathews L, Ferrer M, Southall N, Guha R, et al. (2013). Targeting IRAK1 as a therapeutic approach for myelodysplastic syndrome. Cancer Cell 24, 90–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallman DA, and List A (2019). The central role of inflammatory signaling in the pathogenesis of myelodysplastic syndromes. Blood 133, 1039–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayyadi M, Safaroghli-Azar A, Pourbagheri-Sigaroodi A, Abolghasemi H, Anoushirvani AA, and Bashash D (2020). c-Myc Inhibition Using 10058-F4 Increased the Sensitivity of Acute Promyelocytic Leukemia Cells to Arsenic Trioxide Via Blunting PI3K/NF-kappaB Axis. Arch Med Res. [DOI] [PubMed] [Google Scholar]
- Sheikh-Zeineddini N, Bashash D, Safaroghli-Azar A, Riyahi N, Shabestari RM, Janzamin E, and Safa M (2019). Suppression of c-Myc using 10058-F4 exerts caspase-3-dependent apoptosis and intensifies the antileukemic effect of vincristine in pre-B acute lymphoblastic leukemia cells. J Cell Biochem 120, 14004–14016. [DOI] [PubMed] [Google Scholar]
- Smith MA, Choudhary GS, Pellagatti A, Choi K, Bolanos LC, Bhagat TD, Gordon-Mitchell S, Von Ahrens D, Pradhan K, Steeples V, et al. (2019). U2AF1 mutations induce oncogenic IRAK4 isoforms and activate innate immune pathways in myeloid malignancies. Nat Cell Biol 21, 640–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Culver-Cochran AE, Adelman ER, Rhyasen GW, Ma A, Figueroa ME, and Starczynowski DT (2020). TNFAIP3 Plays a Role in Aging of the Hematopoietic System. Front Immunol 11, 536442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starczynowski DT, Kuchenbauer F, Argiropoulos B, Sung S, Morin R, Muranyi A, Hirst M, Hogge D, Marra M, Wells RA, et al. (2010). Identification of miR-145 and miR-146a as mediators of the 5q- syndrome phenotype. Nat Med 16, 49–58. [DOI] [PubMed] [Google Scholar]
- Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, and Ebert BL (2015). Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 126, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, Satija R, and Smibert P (2017). Simultaneous epitope and transcriptome measurement in single cells. Nat Methods 14, 865–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E, et al. (2019). COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res 47, D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trowbridge JJ, and Starczynowski DT (2021). Innate immune pathways and inflammation in hematopoietic aging, clonal hematopoiesis, and MDS. J Exp Med 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SL, Long N, Schultz AR, Traer E, Abel M, et al. (2018). Functional genomic landscape of acute myeloid leukaemia. Nature 562, 526–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varney ME, Choi K, Bolanos L, Christie S, Fang J, Grimes HL, Maciejewski JP, Inoue JI, and Starczynowski DT (2017). Epistasis between TIFAB and miR-146a: neighboring genes in del(5q) myelodysplastic syndrome. Leukemia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varney ME, Niederkorn M, Konno H, Matsumura T, Gohda J, Yoshida N, Akiyama T, Christie S, Fang J, Miller D, et al. (2015). Loss of Tifab, a del(5q) MDS gene, alters hematopoiesis through derepression of Toll-like receptor-TRAF6 signaling. J Exp Med 212, 1967–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vervoorts J, Luscher-Firzlaff JM, Rottmann S, Lilischkis R, Walsemann G, Dohmann K, Austen M, and Luscher B (2003). Stimulation of c-MYC transcriptional activity and acetylation by recruitment of the cofactor CBP. EMBO Rep 4, 484–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, Wartman LD, Lamprecht TL, Liu F, Xia J, et al. (2012). The origin and evolution of mutations in acute myeloid leukemia. Cell 150, 264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Bekker-Jensen IH, Christensen J, Rasmussen KD, Sidoli S, Qi Y, Kong Y, Wang X, Cui Y, Xiao Z, et al. (2015). Tumor suppressor ASXL1 is essential for the activation of INK4B expression in response to oncogene activity and anti-proliferative signals. Cell research 25, 1205–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, McMichael JF, Schmidt HK, Yellapantula V, Miller CA, et al. (2014). Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 20, 1472–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota A, Hirai H, Sato R, Adachi H, Sato F, Hayashi Y, Sato A, Kamio N, Miura Y, Nakano M, et al. (2019). C/EBPbeta is a critical mediator of IFN-alpha-induced exhaustion of chronic myeloid leukemia stem cells. Blood Adv 3, 476–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, and He QY (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, and He QY (2015). ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Spears E, Boone DN, Li Z, Gregory MA, and Hann SR (2013). Domain-specific c-Myc ubiquitylation controls c-Myc transcriptional and apoptotic activity. Proc Natl Acad Sci U S A 110, 978–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao JL, Rao DS, Boldin MP, Taganov KD, O’Connell RM, and Baltimore D (2011). NF-kappaB dysregulation in microRNA-146a-deficient mice drives the development of myeloid malignancies. Proc Natl Acad Sci U S A 108, 9184–9189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao JL, Rao DS, O’Connell RM, Garcia-Flores Y, and Baltimore D (2013). MicroRNA-146a acts as a guardian of the quality and longevity of hematopoietic stem cells in mice. Elife 2, e00537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, et al. (2011). RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 478, 524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Genes targeted in shRNA screen, Related to Figure 1.
Table S2. Differentially expressed genes in Tet2−/− vs WT LSK, Related to Figure 5.
Table S3. Differentially expressed genes in Traf6−/− vs WT LSK, Related to Figure 5.
Table S4. Differentially expressed genes in Traf6−/−;Tet2−/− vs WT LSK, Related to Figure 5.
Table S5. Differentially expressed genes in Tet2−/− vs Traf6−/−;Tet2−/− LSK, Related to Figure 5.
Table S6. Differential ubiquitin profile following knockdown of TRAF6 in TF1 cells, Related to Figure 6A.
Table S7. sgRNA sequences used in this study, Related to STAR Methods.
Data Availability Statement
Data
Next Generation Sequencing data, including data for mouse RNA-Seq, have been deposited at GEO and are publicly available as of the date of publication.
Next Generation Sequencing data, including data for mouse and human RNA-Seq have been deposited at GEO: GSE19429, GSE54646, GSE184937, GSE185295, and GSE185299.
Published microarray data of patients with MDS(Pellagatti et al., 2010), MPN(Rampal et al., 2014), and respective age-matched controls were downloaded from GEO series GSE19429 and GSE54646. RNA-seq data of patients with AML(Cancer Genome Atlas Research et al., 2013) were downloaded from the GDC Data Portal (https://portal.gdc.cancer.gov/). AML RNA-seq data was also obtained from the BEAT AML data viewer (Vizome, http://www.vizome.org/aml/)(Tyner et al., 2018). DNA methylation data of AML and MPN was obtained from the GDC Data Portal(Cancer Genome Atlas Research et al., 2013) and GSE4272(Nischal et al., 2013), respectively. Mutation data of AML, MDS and MPN were downloaded from GDC Data Portal(Cancer Genome Atlas Research et al., 2013) and the study by Gerstung M et al.(Gerstung et al., 2015).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Code
This paper does not report original code.