

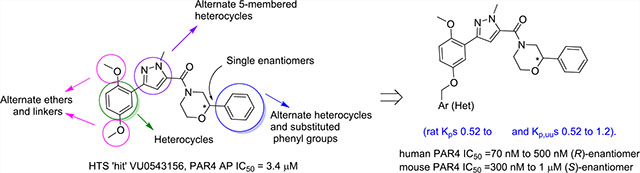
Abstract
The detailed pharmacology and therapeutic potential of the central PAR4 receptors are poorly understood due to a lack of potent, selective, and brain-penetrant tool compounds. Despite this, robust data with biochemical and genetic tools show the therapeutic potential of PAR4 antagonists in traumatic brain injury, Alzheimer’s disease, Parkinson’s disease, and other neurodegenerative disorders with a neuroinflammatory component. Thus, we performed a functional HTS campaign, identified a fundamentally new PAR4 competitive inhibitor chemotype, optimized this new series (increased potency >45-fold), discovered enantiospecific activity (though opposing preference for human versus mouse PAR4), and engendered high central nervous system penetration (rat Kp’s of 0.52 to 4.2 and Kp,uu’s of 0.52 to 1.2).
Keywords: PAR4, protease-activated receptor, CNS penetrant, SAR, selectivity
Graphical Abstract
INTRODUCTION
Protease activated receptors (PARs) are family A G protein-coupled receptors (GPCRs) that signal in response to the serine protease cleavage of their N-terminal domains to reveal an encrypted tethered ligand (TL) that interacts with and activates the receptor.1–6 Protease-activated receptor 4 (PAR4) is essential for the thrombin-induced procoagulant effect on platelets, and a recent noncompetitive antagonist (BMS-986120, 1) demonstrated clinical efficacy for antiplatelet therapy to treat thrombosis without bleeding.7–9 However, the therapeutic potential of inhibiting PAR4 in the central nervous system (CNS) has only recently been recognized and appreciated. In the CNS, PAR4 is expressed on astrocytes, dendrites in the hippocampus, neurons, and glial cells; moreover, activation of PAR4 leads to a breakdown of the blood–brain barrier (BBB), suggesting that inhibition of PAR4 could be of benefit across a broad range and neurodegenerative and neuroinflammatory disorders, such as Alzheimer’s disease and Parkinson’s disease.10,11 To support this, cerebral ischemia/reperfusion injury was attenuated in PAR4 KO mice, demonstrating a greater than 80% reduction of infarct volume with improved motor and neurologic functions as compared to wild-type mice. Moreover, BBB disruption was attenuated in PAR4 KO mice, relative to wild-type mice, as was the severity of cerebral edema.12 This year, Qu and co-workers showed that 1 protected against traumatic brain injury (TBI) in mice by suppression of neuroinflammation in astrocytes.13 These data, despite in our hands exhibiting low brain penetration (Kp’s ≤ 0.1) for the BMS series of PAR4 ligands, clearly garner a need for highly CNS-penetrant PAR4 inhibitors. Not unlike the BMS series, other reported PAR4 inhibitors from our lab and others, 1–8 (Figure 1), display low brain penetration (Kp’s ≤ 0.2) in rodents.14–20 Therefore, we needed a new PAR4 chemotype and elected to initiate a high-throughput screening (HTS) campaign to identify fundamentally new chemical matter as starting points for the development of highly CNS-penetrant PAR4 inhibitors.
Figure 1.
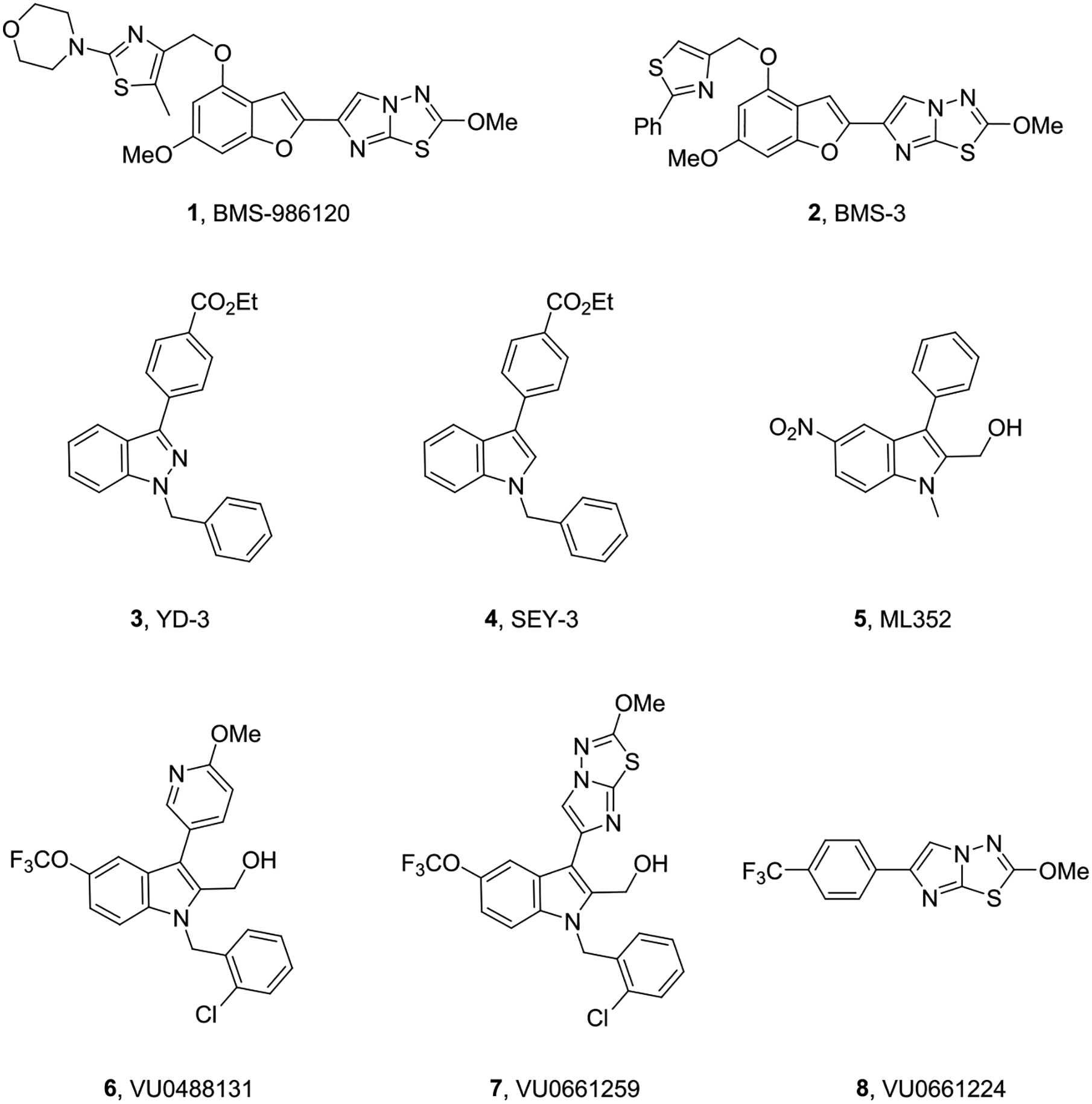
Structures of reported PAR4 competitive and noncompetitive inhibitors 1–8. All of the known ligands show poor CNS penetration and/or, in the case of 3–8, insufficient potency at γ-thrombin and unacceptable DMPK profiles.
RESULTS AND DISCUSSION
Toward the Next Generation of PAR4 Antagonists.
To identify fundamentally new PAR4 chemotypes, we performed a high-throughput screening (HTS) of a 25,000-membered general screening deck employing a platelet flow cytometry assay and the PAR4-activating peptide mimic (PAR4-AP, AYPGKF) to determine potency and efficacy.19,21 From this exercise, we discovered a weak, racemic “hit”, VU0543156 (9), with a PAR4-AP IC50 value of 3.4 μM, yet 9 was found to inhibit PAR4-AP-induced aggregation by ~95% and show selectivity versus PAR1. While the latent quinone of 9 was a concern, overall, the 2,5-dialkoxy-(1-methyl-1H-pyrazol-5-yl)(2-arylmorpholino) methanone chemotype possessed many avenues for chemical optimization (Figure 2) and was distinct from previously reported PAR4 chemotypes 1–8.
Figure 2.
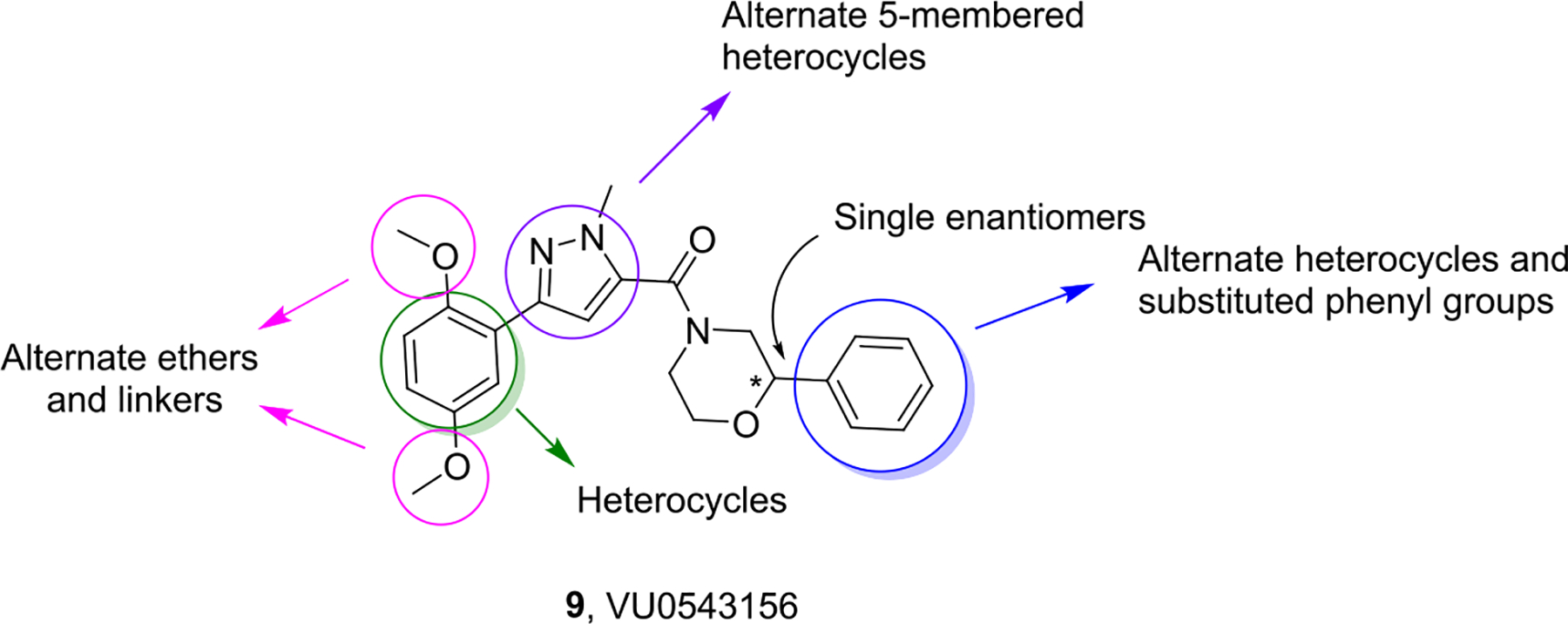
Structures of 9 (VU0543156) and the initial optimization plan to improve potency, assess enantiopreference, and engender CNS penetration.
Chemistry and Initial Structure–Activity Relationships (SARs).
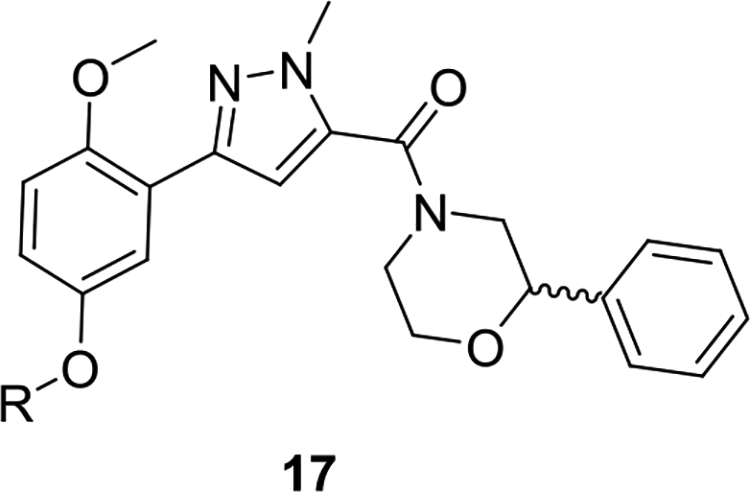
The chemistry to access this new series of PAR4 antagonists was straightforward (Scheme 1), and an early focus was to bulk up either of the methoxy ethers to minimize the potential for quinone formation while screening the racemic variant of the 2-phenyl morpholine.21 Commercial 10 smoothly coupled with racemic 11 under HATU conditions to provide 12 in 80–88% yield. A Suzuki coupling between 12 and either regioisomeric phenol 13 or 14 delivers 15 and 16, respectively, in good isolated yields. Finally, alkylation of the regioisomeric phenols 15 and 16 could be accomplished through either SN2 chemistry with suitably substituted bromide or via a Mitsunobu or Ullman coupling with the alcohol to provide analogs 17 and 18, respectively. Interestingly, substitution of the 2-position led to inactive congeners 18, whereas the 5-position was tolerant, and a simple benzyl derivative 17a afforded an ~10-fold increase in potency (PAR4-AP IC50 = 0.36 μM). Thus, SAR exploration focused on this region, and Table 1 highlights the impact of substituted benzyl congeners, incorporating heterocycles and the first glimpse of enantioselective inhibition. Incorporation of a racemic α-methyl moiety on the benzylic carbon as in 17b was detrimental to potency as were lipophilic substituents on the aromatic ring (e.g., 17f–17h). A pyridine survey proved interesting, with 2-pyridyl losing activity (17c, IC50 = 1.6 μM), whereas the 3- and 4-congeners, 17d (IC50 = 0.35 μM) and 17e (IC50 = 0.47 μM), respectively, were equipotent to 17a but offered opportunities to improve solubility. A racemic 2-cyano derivative (17i) showed improved potency (IC50 = 0.14 μM) and thus warranted synthesis of the discrete enantiomers. While the (S)-enantiomer (S)-17i lost ~3-fold (IC50 = 0.48 μM), the (R)-enantiomer (R)-17i was as potent as the racemate and indicated enantioselective human PAR4 inhibition. The same enantiopreference was noted for the 2-OCF3 congener 17l, with the (R)-enantiomer (R)-17l (IC50 = 0.39 μM) ~3-fold more potent than (S)-17l (IC50 = 0.93 μM). Lipophilic groups, such as regioisomeric CF3 congeners 17f–17h, showed “flat” SAR with weak activity. All analogs 17 were inactive on PAR1.21
Scheme 1.
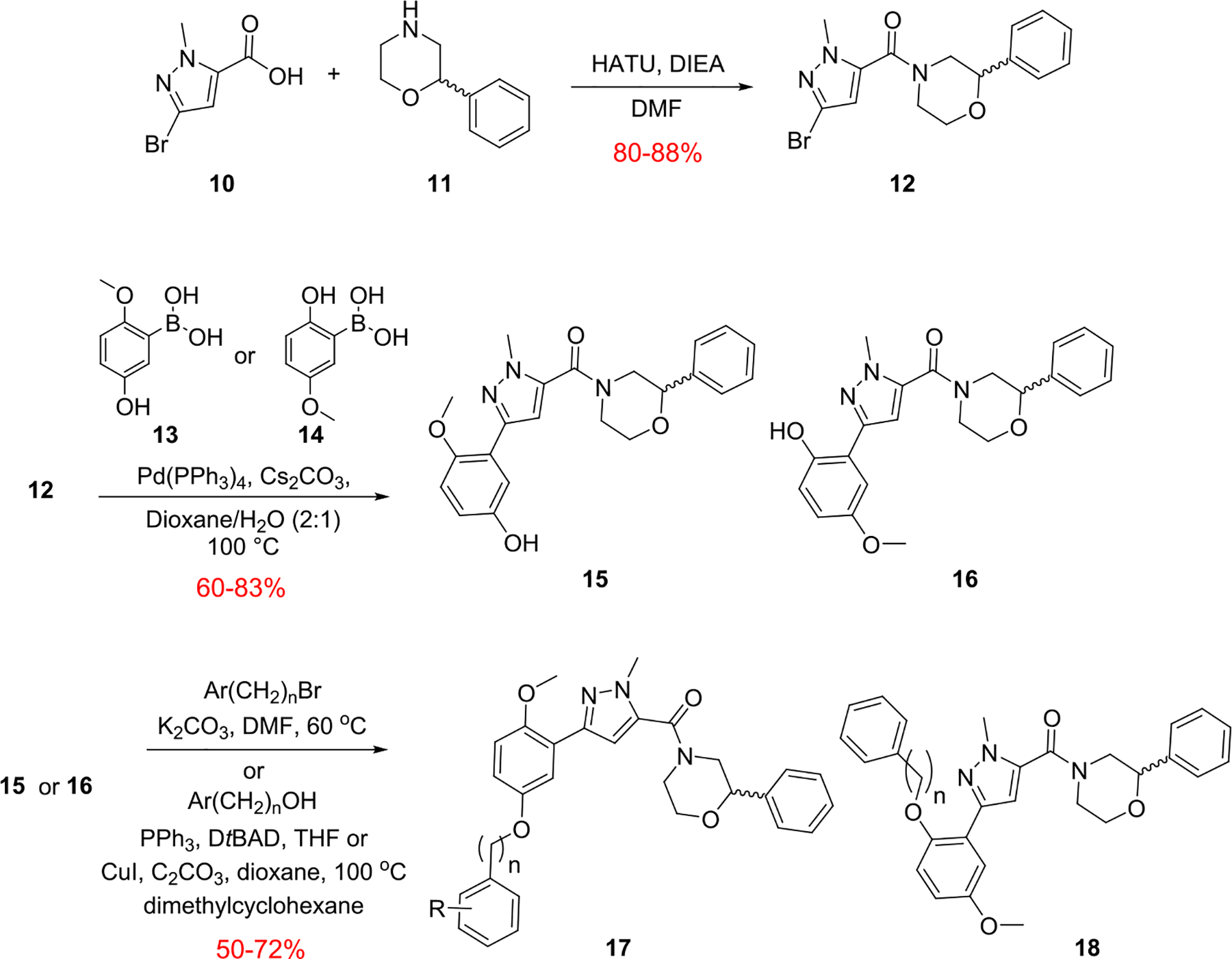
Synthesis of Regioisomeric Ether Analogs 17 and 18
Table 1.
Structure and Activities of Analogs 17a
| Compound | R | PAR4-AP PAC-1 IC50 (μM)b (pIC50±SEM) | PAR4-AP % Max PAC-1a | PAR1 %Maxc |
|---|---|---|---|---|
| 7 | 0.18 (6.74±0.06) | 0.32 | 98 | |
| 17a |
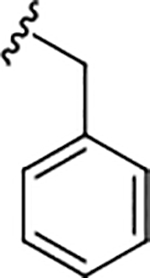
|
0.36 (6.44±0.08) | 0.72 | 99 |
| 17b |
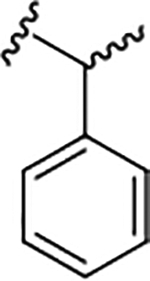
|
1.7 (5.76±0.13) | 6.8 | 83 |
| 17c |
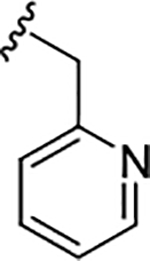
|
1.6 (5.79±0.09) | 3.9 | 99 |
| 17d |
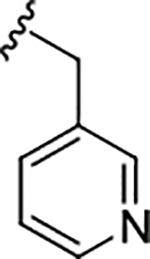
|
0.35 (6.45±0.10) | 0.75 | 88 |
| 17e |
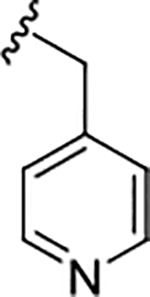
|
0.47 (6.32±0.11) | 0.89 | 98 |
| 17f |
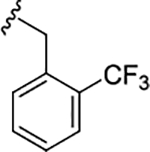
|
1.2 (5.92±0.16) | 4.7 | 84 |
| 17g |
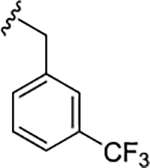
|
1.2 (5.92±0.12) | 5.7 | 84 |
| 17h |
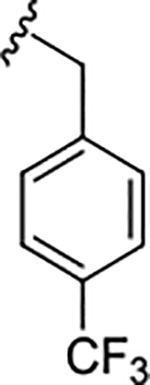
|
1.3 (5.88±0.14) | 8.7 | 83 |
| 17i |
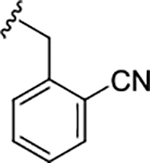
|
0.14 (6.85±0.06) | 0.47 | 92 |
| (S)-17i |
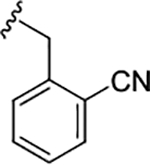
|
0.48 (6.31±0.12) | 5.9 | 94 |
| (R)-17i |
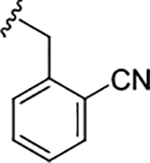
|
0.14 (6.85±0.02) | 1.3 | 95 |
| 17j |
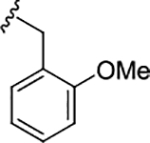
|
0.83 (6.08±0.13) | 3.4 | 82 |
| 17k |
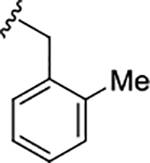
|
0.17 (6.76±0.12) | 1.2 | 77 |
| 17l |
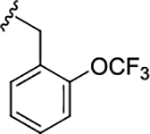
|
1.50 (5.82±0.09) | 5.9 | 75 |
| (S)-17l |
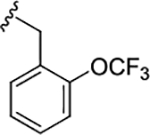
|
0.93 (6.03±0.06) | 0.93 | 80 |
| (R)-17l |
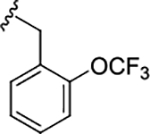
|
0.39 (6.40±0.03) | 0.39 | 76 |
Values indicate the percentage of max PAC-1 binding after PAR4-AP stimulation of human platelets.
Average of three independent determinations.
Values indicate the percentage PAR1 activity in the presence of 10 μM compound.
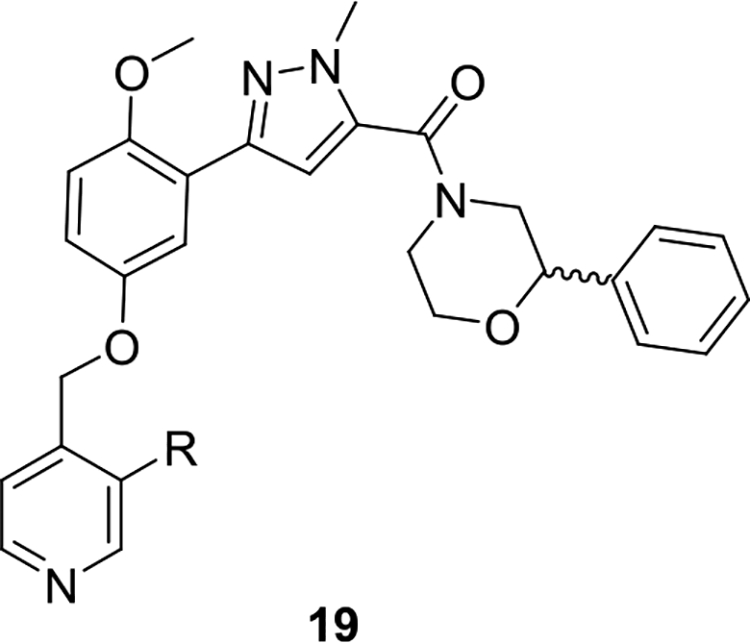
Based on these SAR findings, we combined the favorable pyridyl moiety with various 2-substituents (Table 2) in analogs 19. With a 2-cyano moiety, 19 was a potent human PAR4 inhibitor (IC50 = 0.09 μM) and exhibited ~37-fold improvement over the HTS hit 9. Preparation of the discrete enantiomers of 19 once again showed a preference for the (R)-enantiomer (R)-19a (IC50 = 0.09 μM) and (S)-19a (IC50 = 0.66 μM). This trend is consistent whether the 2-substituent is CN (19a), Me (19b), or Cl (19d). All analogs 19 were inactive on PAR1.
Table 2.
Structure and Activities of Analogs 19a
| ||||
|---|---|---|---|---|
| compound | R | PAR4-AP PAC-1 IC50 (μM)b (pIC50 ± SEM) | PAR4-AP % max PAC-1a | PAR1% maxc |
| 7 | 0.18 (6.74 ± 0.06) | 0.32 | 98 | |
| 19a | CN | 0.09 (7.04 ± 0.06) | 0.27 | 79 |
| (S)-19a | CN | 0.66 (6.18 ± 0.11) | 3.3 | 83 |
| (R)-19a | CN | 0.09 (7.04 ± 0.05) | 0.33 | 95 |
| 19b | Me | 0.09 (7.04 ± 0.10) | 0.74 | 103 |
| (S)-19b | Me | 0.44 (6.33 ± 0.10) | 2.3 | 95 |
| (R)-19b | Me | 0.07 (7.15 ± 0.03) | 0.37 | 101 |
| 19c | F | 0.30 (5.92 ± 0.12) | 9.8 | 85 |
| 19d | Cl | 0.08 (7.09 ± 0.14) | 11 | 79 |
| (S)-19d | Cl | 0.07 (6.85 ± 0.06) | 0.68 | 80 |
| (R)-19d | Cl | 0.45 (6.31 ± 0.12) | 2.6 | 94 |
Values indicate the percentage of max PAC-1 binding after PAR4-AP stimulation of human platelets.
Average of three independent determinations.
Values indicate the percentage PAR1 activity in the presence of 10 μM compound.
Computational docking studies of (R)-19b in our PAR4 homology model indicated that the methylpyridine moiety was situated near the surface of the receptor, extending into the extracellular space (Figure 3).21 Homologation of the benzyl linkage in (R)-19b afforded equipotent analogs and overall “flat” SAR, supporting the prediction that this region extends into the extracellular space. Thus, further optimization in this region of the compound stopped. The model also suggested a possible π-stacking interaction between the phenyl substituent off the morpholine and tyrosine residue 93; thus, we would later explore this region to build further confidence in the homology model.
Figure 3.
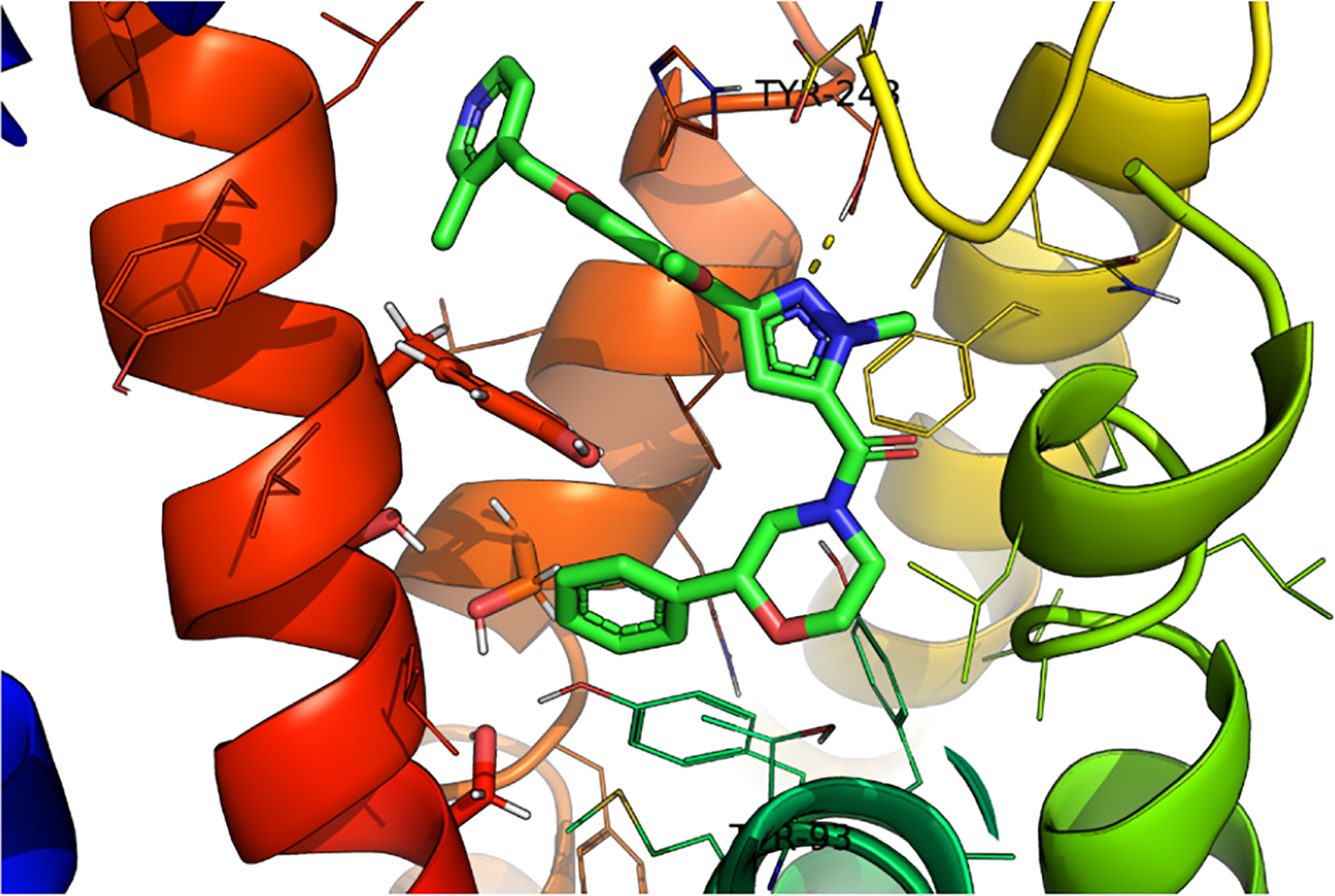
Structure of (R)-19b (VU6019097) docked in our PAR4 homology model.
Fully saturated alternatives (cyclohexyl, cylopentyl, pyranyl, and furyl) for the benzyl/pyridyl moiety in 19a led to flat SAR, with all analogs displaying PAR4-AP IC50’s in the ~1 μM potency range. Similarly, all replacements for the OMe group, analogs 20, proved inactive, and the OMe, while a potential liability, proved to be an essential pharmacophore. As shown in Figure 4, attempts to replace the 3,5-disubstituted pyrazole in (R)-19a were ineffective, leading to inactive congeners 20a–20d. Here, regioisomeric pyrazoles lost >100-fold in PAR4 activity, as did a regioisomerically related pyrrole, as did ring expansion to a pyridine analog. An attempt to cyclize the pyrazole moiety into the amide to provide a 5,6-fused heteroaryl ring system, [1,2,4]-triazolo[4,3-b]pyridazine, led to the discovery of racemic 21 (VU6016706) and a “switch” from a PAR4 antagonist to a PAR1-preferring antagonist (Figure 5). Upon preparation of the single enantiomers, we found that (R)-21 was a weak inhibitor of both PAR1 and PAR4, while (S)-21 was clearly PAR1-preferring (PAR1 PAC-1 % max @ 10 μM = 6). Thus, further modifications to the central 3,5-pyrazole core were stopped in favor of exploring other regions of the lead scaffold.
Figure 4.
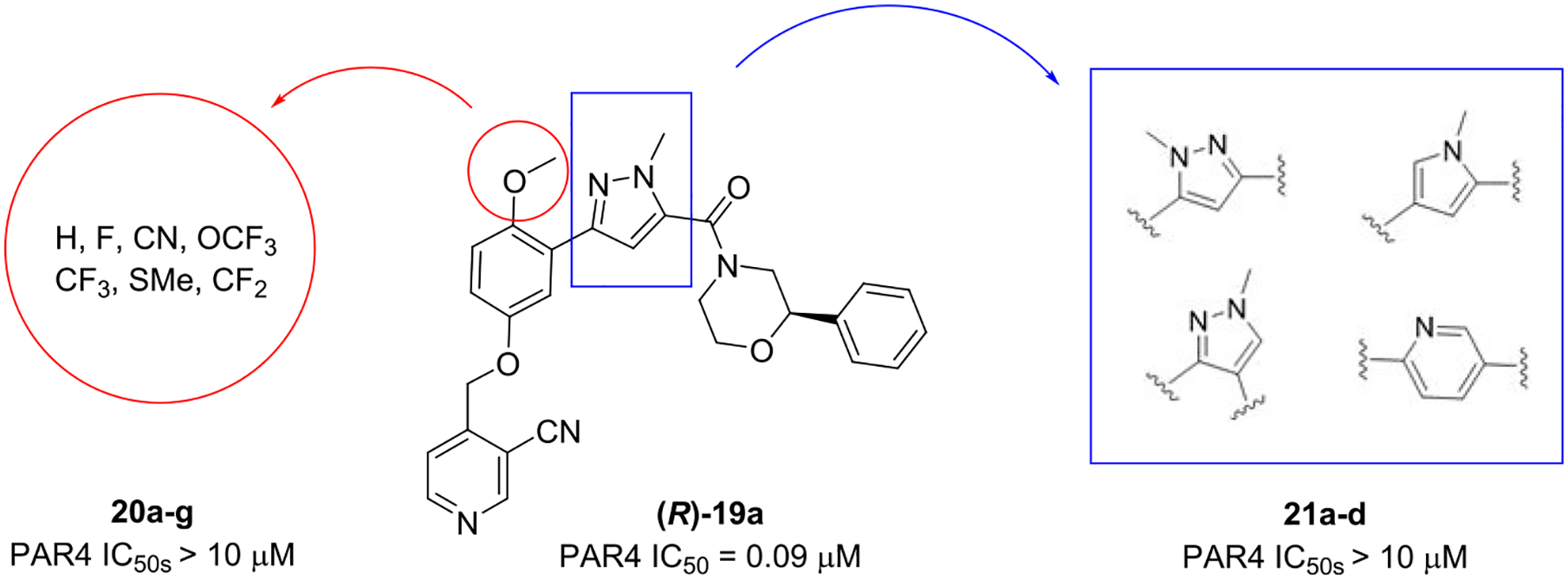
Inactive congeners of 19 with alternative moieties 20 for the OMe and heterocyclic replacements for the core pyrazole 21.
Figure 5.
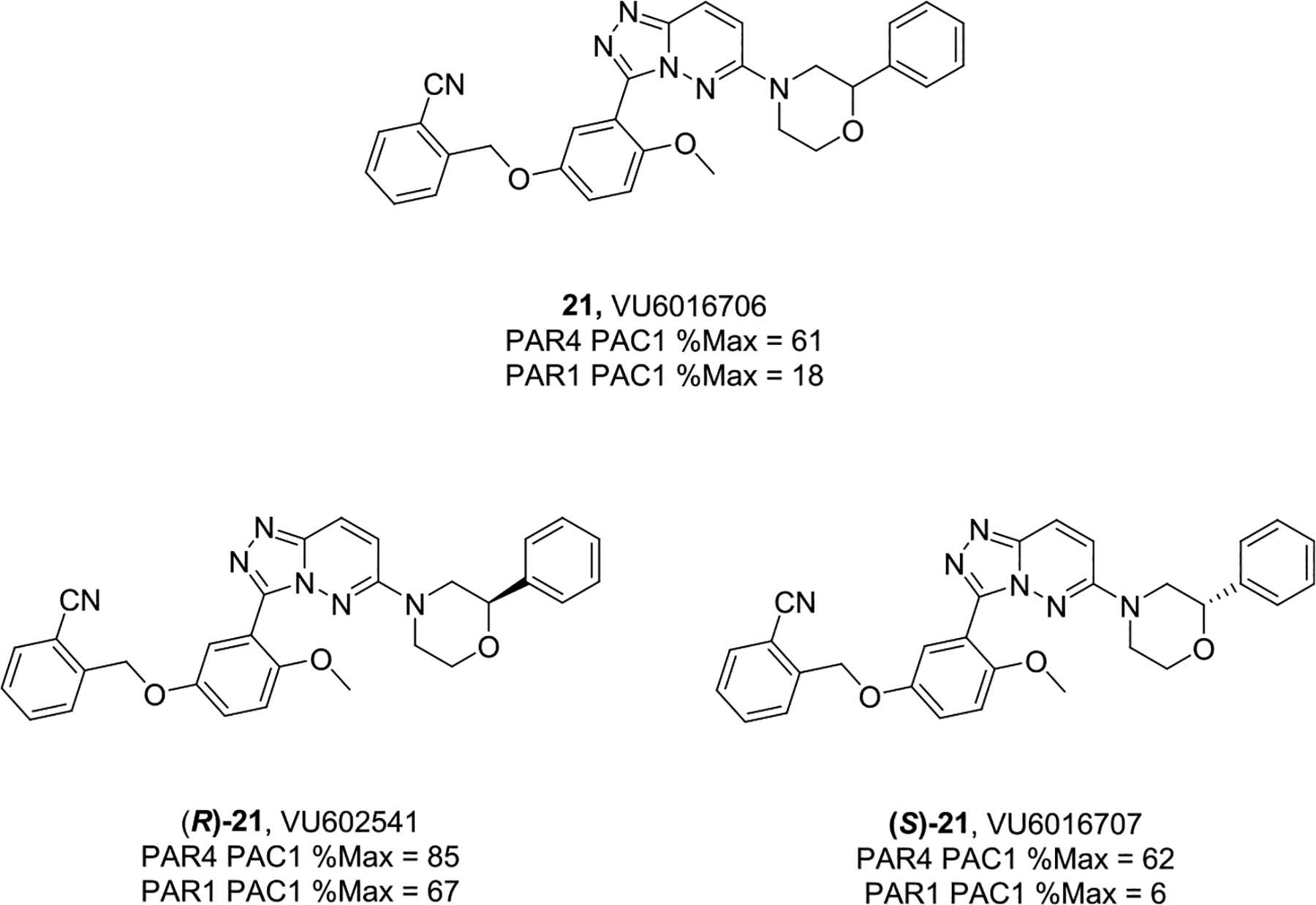
Cyclized, constrained congeners 21, of 19, led to a PAR1-preferring inhibitor chemotype.
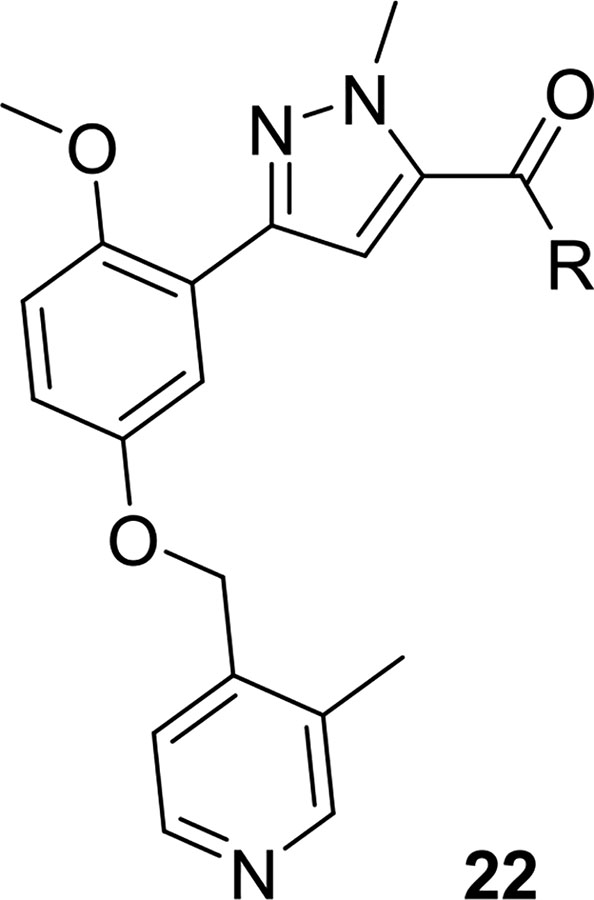
As the last region of the HTS hit to explore, our attention focused on the chiral 3-position of the morpholine and we explored the potential π-stacking interaction between the phenyl substituent off the morpholine and tyrosine residue 93. Alternate moieties were prepared and screened initially in a racemic form, and if there was interesting activity, they would be either resolved by chiral SFC or synthesized enantioselectively.21 Interestingly, replacement of the phenyl ring with a saturated cyclohexyl group 22a only lost ~3.5-fold (PAR4-AP IC50 = 0.32 μM), relative to racemic 19b (PAR4-AP IC50 = 0.09 μM). Truncation to a methyl substituent, as in 22b, did lose significant potency (PAR4-AP IC50 = 2.4 μM). Replacement of the morpholine with either 2-phenyl pyrrolidine (22c) or 3-phenyl piperidine (22d) retained PAR4 activity, PAR4-AP IC50 = 0.16 μM and PAR4-AP IC50 = 0.63 μM, respectively; however, an isosteric piperazine congener (22e) lost activity (PAR4-AP IC50 = 2.4 μM). As a classical GPCR “privileged structure”,22 the 4-cyano-4-phenyl-piperidine derivative 22f also retained PAR4 activity (PAR4-AP IC50 = 0.24 μM), whereas linear amide congers 22g and 22h lost activity. Although not shown, heterocycles (2-, 3-, and 4-pyridinyl analogs) replacing the phenyl moiety of 19 proved inactive as well. Overall, this region of the hit displayed some flexibility but did not fully support the potential π-stacking interaction between the phenyl substituent off the morpholine and tyrosine residue 93 noted in the homology model. Based on the predicted pose, this region was buried within a hydrophobic pocket and limited the potential for greater potency optimization (Table 3).
Table 3.
Structure and Activities of Analogs 22a
| Compound | R | PAR4-AP PAC-1 IC50 (μM)b (pIC50±SEM) | PAR4-AP % Max PAC-1a | PARI %Maxc |
|---|---|---|---|---|
| 7 | 0.18 (6.74±0.06) | 0.32 | 98 | |
| 22a |
|
0.32 (6.49±0.03) | 0.66 | 84 |
| 22b |
|
2.4 (5.61±0.13) | 5.1 | 80 |
| 22c |
|
0.16 (6.79±0.04) | −1.2 | 92 |
| 22d |
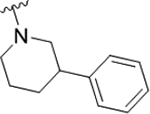
|
0.63 (6.2±0.09) | 1.3 | 81 |
| 22e |
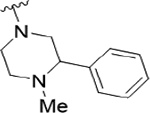
|
2.1 (5.67±0.10) | 11 | 90 |
| (5.67±0.10) | ||||
| 22f |
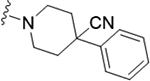
|
0.24 (6.61±0.05) | 0.32 | 92 |
| 22g |
|
1.3 (5.88±0.13) | 4.2 | 85 |
| 22h |
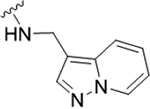
|
1.2 (5.92±0.11) | 2.84 | 76 |
Values indicate the percentage of max PAC-1 binding after PAR4-AP stimulation of human platelets.
Average of three independent determinations.
Values indicate the percentage PAR1 activity in the presence of 10 μM compound.
Mode of PAR4 Inhibition.
With a subhundred nanomolar PAR4 antagonist in hand, we were able to evaluate the mode of action for the PAR4 antagonist (R)-19a in a progressive fold-shift assay/Schild analysis (Figure 6).23 The Schild analysis results are consistent with a competitive binding mode of inhibition, denoted by the parallel leftward shift of the curve with increasing concentrations of PAR4-AP. This is in stark contrast to the noncompetitive mode of inhibition of the BMS series 1 and 2 but comparable to 3–8.23 As such, it was clear from our work, and that of others, that competitive inhibition would lead to weak activity at the native γ-thrombin, as opposed to the soluble PAR4 peptide mimic (AYPGKF; PAR4-AP).14–20 Thus, at 10 μM, all of our most potent ligands afford only ~65–43% of the PAC-1% max of γ-thrombin. Of these, 19 showed the greatest activity against γ-thrombin (γ-thrombin PAC-1 43% max @ 10 μM, PAR4 IC50 = 6.4 μM). However, once we achieve these ~10 μM potencies, SAR can initiate employing γ-thrombin, which is currently underway.
Figure 6.
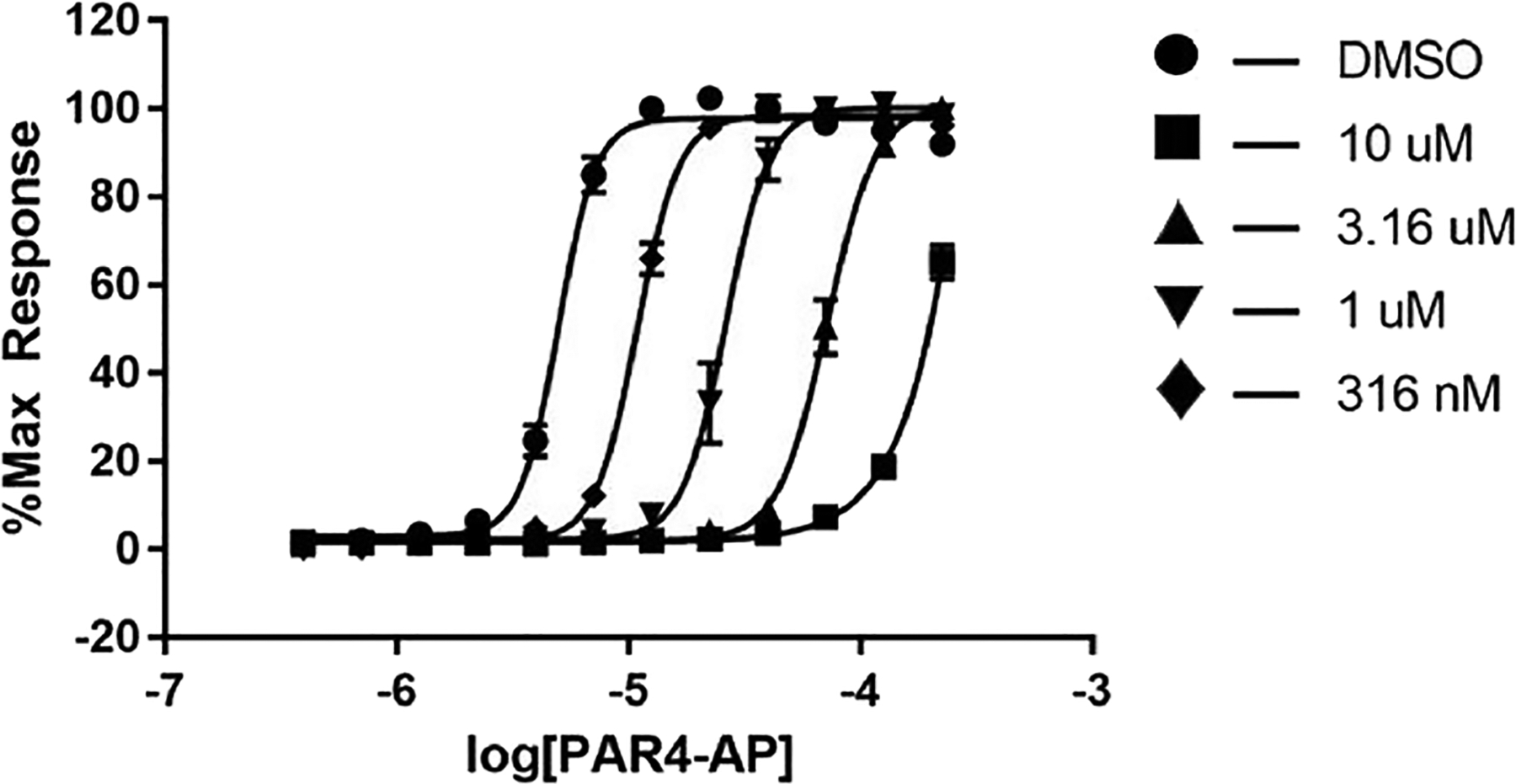
Progressive fold-shift inhibition assay with (R)-19a also showing a competitive mode of PAR4 inhibition.
Activity at Mouse PAR4.
At this stage, with our PAR4 antagonists achieving low nanomolar potency against PAR4-AP, and gaining traction against γ-thrombin inhibition, we assessed our library of inhibitors in mouse platelets (first in single point @ 10 μM and then in full CRC). Interestingly, the mouse PAR4 (mPAR4) receptor has an enantiopreference for the (S)-enantiomer, whereas the hPAR4 receptor displayed clear enantiopreference for the (R)-enantiomer (Table 4). The analog 19 remains the most active chemotype within this series, with the S-enantiomer (S)-19a being our most potent compound in mPAR4 and the (R)-enantiomer (R)-19a being the most potent against hPAR4. This was a truly unanticipated and unprecedented finding among known PAR4 ligands. We believe that the divergence in stereochemical activity between mouse and hPAR4 receptors could provide insight into the structural differences among the two active sites. After analyzing our human PAR4 homology model, we believe that 2-phenylmorpholine in the (S)-conformation could possibly be clashing with HIS165, which causes it to lose its critical interaction with TYR243, in addition to losing the π-stacking interaction with the (R)-conformation (Figure 7). Based on our working hypothesis that Tyr243 is making a key hydrogen-bonding interaction with the nitrogen of the pyrazole, combined with mPAR4 and hPAR4 receptors having the opposite enantiopreference, we looked at in silico mutations in our hPAR4 homology model. With the congener (R)-17l docked in our hPAR4 homology model (magenta), the native hPAR4 Ala172 residue was replaced with native mPAR4 Glu172 (cyan) (Figure 7). Computational modeling studies suggest that the larger sidechain, glutamate, would likely sterically hinder the crucial hydrogen-bonding interaction between Tyr243 and the central pyrazole, which could potentially explain the loss of activity in mouse platelets. To test this, mutation studies are planned to mutate mouse PAR4 Glu172 to alanine to observe whether affinity is rescued in the mouse platelet assays using the R-analog (R)-17l.
Table 4.
Structure and Activities of Analogs 17 and 19 at Mouse and Human PAR4a
| Compound | R | Mouse PAR4-AP PAC-1 IC50 (μM)b (pIC50±SEM) | Human PAR4-AP PAC-1 IC50 (μM)b (pIC50±SEM) | Human PAR4-AP % Max PAC-1a |
|---|---|---|---|---|
| 7 | 0.18 (6.74±0.06) | 0.32 | 98 | |
| 17i |
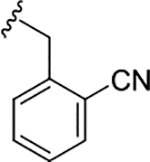
|
0.37 (6.43±0.07) | 0.14 (6.85±0.06) | 0.47 |
| (S)-17i |
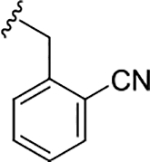
|
0.35 (6.45±0.04) | 0.48 (6.31±0.12) | 5.8 |
| (R)-17i |
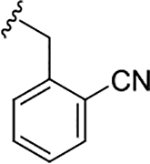
|
2.1 (5.67±0.03) | 0.14 (6.85±0.02) | 1.9 |
| 19a |
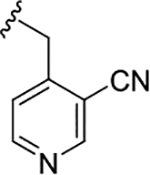
|
0.42 (6.37±0.11) | 0.09 (6.85±0.06) | 0.27 |
| (S)-19a |
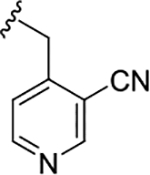
|
0.30 (6.52±0.14) | 0.66 (6.18±0.11) | 3.3 |
| (R)-19a |
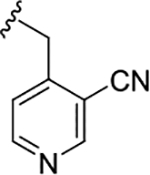
|
2.1 (5.67±0.15) | 0.09 (7.04±0.05) | 0.33 |
| 17l |
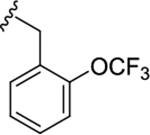
|
0.70 (6.15±0.08) | 1.50 (5.82±0.09) | 5.9 |
| (S)-17l |
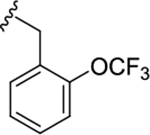
|
0.45 (6.34±0.04) | 0.93 (6.03±0.06) | 0.93 |
| (R)-17l |
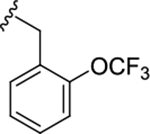
|
1.1 (5.95±0.03) | 0.39 (6.40±0.03) | 0.39 |
| 19d |
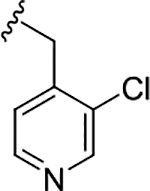
|
1.0 (6.00±0.14) | 0.08 (7.09±0.14) | 11 |
| (S)-19d |
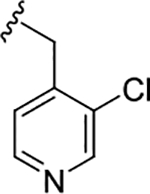
|
0.53 (6.27±0.06) | 0.07 (6.85±0.06) | 0.68 |
| (R)-19d |
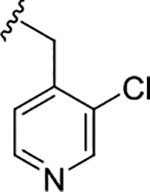
|
1.0 (6.00±0.12) | 0.45 (6.31±0.12) | 2.6 |
Values indicate the percentage of max PAC-1 binding after PAR4-AP stimulation of human or mouse platelets.
Average of three independent determinations.
Figure 7.
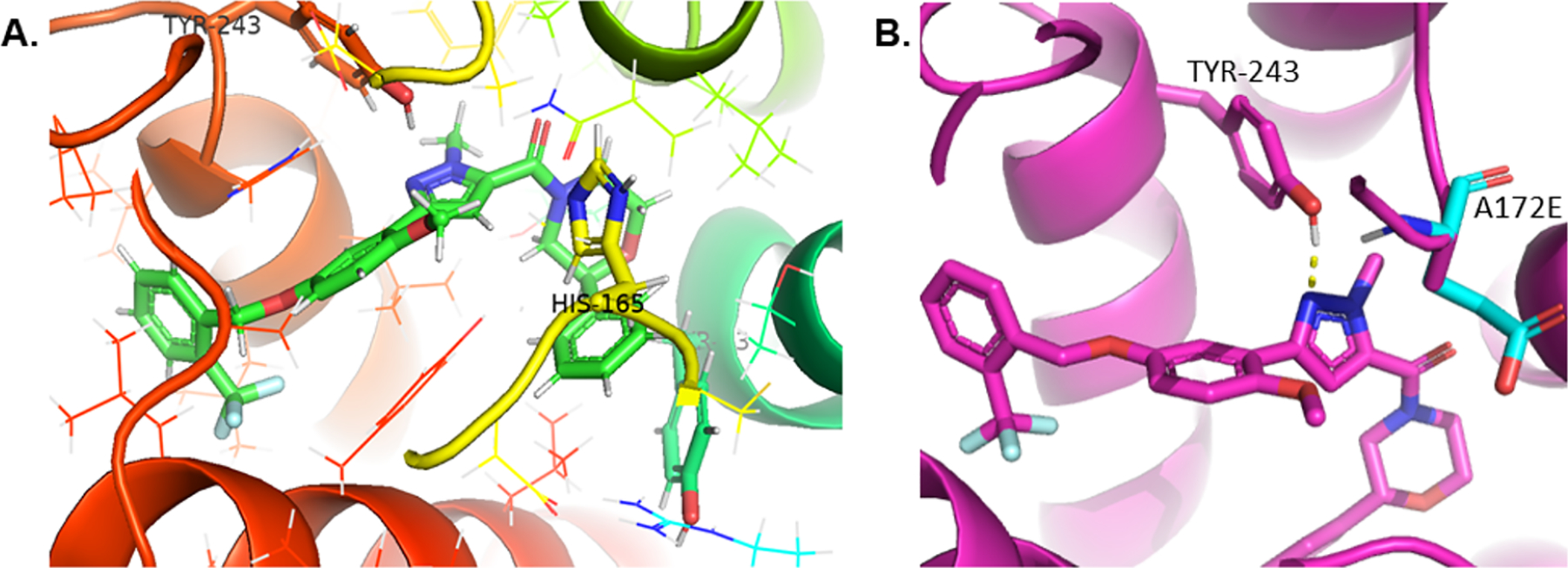
Congener (R)-17l docked in the hPAR4 homology model (A) highlighting the histidine residue situated directly above 2-phenylmorpholine and (B) depicting in silico mutation A172E and its potential clashing interaction with TYR243.
DMPK Profiles of Lead Mouse PAR4 Antagonists.
To rapidly assess the ability of our top four rodent PAR4 inhibitors to achieve meaningful brain exposure, compounds were evaluated in our standard rat IV plasma:brain level (PBL) cassette paradigm, as well as rat plasma protein binding and rat brain homogenate binding (Table 5). Excitingly, and unlike all previously reported PAR4 inhibitors 1–8,14–20 this series of PAR4 antagonists ((S)-17i, (S)-17l, (S)-19a, and (S)-19d) was found to be highly CNS-penetrant, with Kp’s of 0.52 to 4.2, and Kp,uu’s of 0.52 to 1.2. However, plasma protein binding and brain homogenate binding were high, and this parameter will need to be addressed in future analogs in the γ-thrombin optimization campaign. Of these, (S)-17l showed the highest plasma concentrations (30.9 ng/mL) and good Kp (0.52) and Kp,uu (0.52) and was selected for further PK profiling in mice.
Table 5.
Rat Plasma Protein and Brain Homogenate Binding and Rat IV PBL Cassette Data
| rat in vitro PPB/BHB and rat IV cassette PBL | ||||||
|---|---|---|---|---|---|---|
| compound number | rat PPB (fu) | rat BHB (fu) | plasma (ng/mL) | brain (ng/g) | K p | K p,uu |
| (S)-17l | 0.001 | 0.001 | 30.9 | 16 | 0.52 | 0.52 |
| (S)-17i | 0.004 | 0.002 | 15.7 | 37.8 | 2.41 | 1.2 |
| (S)-19a | 0.022 | 0.005 | 9.21 | 20.4 | 2.22 | 0.5 |
| (S)-19d | 0.017 | 0.004 | 10.9 | 46.1 | 4.23 | 1.0 |
As shown in Table 6, all four analogs displayed high predicted hepatic clearance (CLhep > 79 mL/min/kg) in mouse liver microsomes (at mouse hepatic clearance); however, (S)-17l showed moderate clearance in a mouse IV cassette study (CLp = 58 mL/min/kg), whereas the other analogs displayed superhepatic in vivo clearance in mouse. Based on these data, we performed a mouse IP PK study dosing (S)-17l at 10 mg/kg to avoid first pass metabolism and found favorable parameters (t1/2 = 8.6 h, tmax = 0.5 h, and Cmax = 225 ng/mL). When pretreated with 1-aminobenzotriazole (ABT) to inhibit the CYP450 machinery in the rat,24 the Cmax from the 10 mg/kg dose could be improved by another 3.75-fold (Cmax = 844 ng/mL). Thus, in the γ-thrombin optimization campaign, metabolite identification/soft spot analysis will be essential to develop PAR4 inhibitors with balanced properties to serve as first-generation, CNS-penetrant in vivo tool compounds.
Table 6.
Mouse In Vitro and In Vivo DMPK Profiles
| mouse in vitro CLhep and IV cassette PK | |||||
|---|---|---|---|---|---|
| compound number | predicted CLhep | CLp (mL/min/kg) | t1/2 (h) | Vss (L/kg) | Cmax (ng/mL) |
| (S)-17l | 79.7 | 58 | 0.70 | 2.2 | 153 |
| (S)-17i | 87.1 | 111 | 0.71 | 5.3 | 45 |
| (S)-19a | 87.6 | 230 | 0.29 | 4.9 | 43 |
| (S)-19d | 88.1 | 268 | 0.18 | 3.8 | 58 |
CONCLUSIONS
As tools to study the therapeutic potentials of PAR4 inhibition the CNS were lacking, we performed a high-throughput screening (HTS) of a 25,000-membered library employing a platelet flow cytometry assay and the PAR4-activating peptide mimic (PAR4-AP, AYPGKF) to determine potency and efficacy. Here, we discovered a weak (IC50 of 3.4 μM), racemic “hit”, VU0543156 (9), based on a novel 2,5-dialkoxy-(1-methyl-1H-pyrazol-5-yl)(2-arylmorpholino)methanone scaffold that was selective versus PAR1. Multidimensional optimization of this new series increased potency >45-fold, discovered enantiospecific activity (though opposing preference for human versus mouse PAR4 receptors), and engendered high CNS penetration (rat Kp’s of 0.52 to 4.2 and Kp,uu’s of 0.52 to 1.2). Further optimization against γ-thrombin and activity in CNS rodent models will be reported in due course.
METHODS
Chemical Synthesis and Purification.25
All reactions were carried out employing standard chemical techniques under an inert atmosphere. The solvents used for extraction, washing, and chromatography were of HPLC grade. All reagents were purchased from commercial sources and were used without further purification. Analytical HPLC was performed on an Agilent 1200 LCMS with UV detection at 215 and 254 nm along with ELSD detection and electrospray ionization, with all final compounds showing ≥95% purity and a parent mass ion consistent with the desired structure. All NMR spectra were recorded on a 400 MHz Brüker AV-400 instrument. 1H chemical shifts were reported as δ values in ppm relative to the residual solvent peak (MeOD = 3.31, CDCl3 = 7.26). Data are reported as follows: chemical shift, multiplicity (br = broad, s = singlet, d = doublet, t = triplet, q = quartet, and m = multiplet), coupling constant (Hz), and integration. 13C chemical shifts are reported as δ values in ppm relative to the residual solvent peak (MeOD = 49.0 and CDCl3 = 77.16). When visible, minor rotamer peaks are denoted with an asterisk (*) in the 1H NMR spectra. The low-resolution mass spectra were obtained on an Agilent 1200 LCMS with electrospray ionization, with a gradient of 5–95% MeCN in 0.1% TFA water solution over 1.5 min. The high-resolution mass spectra were obtained on an Agilent 6540 UHD Q-TOF with an ESI source. Automated flash column chromatography was performed on an Isolera One by Biotage. Preparative purification of library compounds was performed on a Gilson 215 preparative LC system. Optical rotations were acquired on a Jasco P-2000 polarimeter at 23 °C and 589 nm. The specific rotations were calculated according to the equation [α]23/D = (100∝)/(l × c), where l is the path length in decimeters and c is the concentration in g/100 mL.25 For full experimental procedures, please see the Supporting Information.
Blood Collection and Platelet Isolation.
Human platelets were obtained from healthy volunteers. The studies were approved by the Vanderbilt University Internal Review Board. Informed consent was S12 obtained from all individuals prior to the blood draw. Blood was collected into sodium citrate anticoagulant (final concentration, 0.32%) through a 19-gauge needle. Blood was centrifuged at 1100 rpm for 15 min at room temperature (Thermo Forma 400 ML GP, Aerocarrier rotor 236). The platelet-rich plasma layer was isolated and treated with acid citrate dextrose (0.25% citrate, 0.15% citric acid, and 0.2% glucose final concentrations) for 10 min. The platelets were then pelleted by centrifugation at 2400 rpm and washed with Tyrode’s buffer (15 mM HEPES, 0.33 mM NaH2PO4 (pH 7.4), 138 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 5.5 mM dextrose, and 0.1% BSA). The platelets were counted on a Coulter Counter and diluted with Tyrode’s buffer to the indicated concentration.
Flow Cytometry.
Washed platelets were diluted to 1.5 × 107 platelets/mL with Tyrode’s buffer. The platelets were pre-incubated with the compound or vehicle control and a combination of PE-conjugated CD62p and FITC-conjugated PAC-1 (Becton Dickinson, Franklin Lakes, NJ) for 20 min. The reaction was stimulated with agonist for 10 min and then fixed with 1% paraformaldehyde (final concentration) for 20 min. PAR4-AP (AYPGKF) was from GL Biochem (Shanghai, China). γ-Thrombin was from Enzyme Research Laboratories (South bend, IN). γ-Thrombin was treated for 20 min with 1 unit/mL hirudin (Sigma-Aldrich, St. Louis, MO) to sequester residual α-thrombin within the preparation. The samples were analyzed on a BD LSR II. Compensation controls for dual color labeling (PE and FITC) were run each day of data collection. Data were analyzed using FlowJo (Ashland, OR). Mean fluorescence intensity (MFI) was determined by taking the geometric mean of the 20,000 fluorescence recordings within the platelet gate (forward versus side-scatter plot). MFI was normalized to internal vehicle-treated control samples. Concentration–response curves were generated by nonlinear regression analysis in Prism (La Jolla, CA). IC50 values with SEM were also calculated with Prism.
Schild Analysis Competition Binding Assay.
Schild analysis and identification of competitive PAR4 antagonists were performed. Progressive fold-shift experiments and accompanying Schild analysis with VU were also performed. Platelet activation was monitored by PAC-1 binding. The platelets were pretreated with increasing concentrations of each antagonist for 20 min prior to activation with increasing concentrations of PAR4-AP. Each curve was constructed from at least three independent experiments. DRs were calculated from the EC50’s of each individual experiment (vehicleEC50/VU#EC50) and plotted against the administered concentration of antagonist. (A) Shown on the right are the means 6 S.E.M. of log DR-1 (n = 3). In the graph insert, m is the slope from linear regression. DMSO, dimethylsulfoxide.
Drug Metabolism Methods: In Vitro.
For plasma protein binding, the protein binding of each compound was determined in plasma via equilibrium dialysis employing RED Plates (Thermo Fisher Scientific, Rochester, NY). Plasma was added to the 96-well plate containing test compound and mixed thoroughly for a final concentration of 5 μM. Subsequently, an aliquot of the plasma–compound mixture was transferred to the cis chamber (red) of the RED plate, with a phosphate buffer (25 mM, pH 7.4) in the trans chamber. The RED plate was sealed and incubated for 4 h at 37 °C with shaking. At completion, aliquots from each chamber were diluted 1:1 with either plasma (cis) or buffer (trans) and transferred to a new 96-well plate, at which time ice-cold acetonitrile containing an internal standard (50 ng/mL carbamazepine) (2 volumes) was added to extract the matrices. The plate was centrifuged (3000 rcf, 10 min) and supernatants were transferred and diluted 1:1 (supernatant:water) into a new 96-well plate, which was then sealed in preparation for LC/MS/MS analysis. Each compound was assayed in triplicate within the same 96-well plate. Fraction unbound was determined using the following equation. For intrinsic clearance, human or rat hepatic microsomes (0.5 mg/mL) and 1 μM test compound were incubated in 100 mM potassium phosphate pH 7.4 buffer with 3 mM MgCl2 at 37 °C with constant shaking. After a 5 min pre-incubation, the reaction was initiated by addition of NADPH (1 mM). At S13 selected time intervals (0, 3, 7, 15, 25, and 45 min), aliquots were taken and subsequently placed into a 96-well plate containing cold acetonitrile with an internal standard (50 ng/mL carbamazepine). The plates were then centrifuged at 3000 rcf (4 °C) for 10 min, and the supernatant was transferred to a separate 96-well plate and diluted 1:1 with water for LC/MS/MS analysis. The in vitro half-life (T1/2, min, eq 1), intrinsic clearance (CLint, mL/min/kg, eq 2), and subsequent predicted hepatic clearance (CLhep, mL/min/kg, eq 3) were determined employing the following equations:
| (1) |
where k represents the slope from the linear regression analysis of the natural log percent remaining of test compound as a function of the incubation time.
| (2) |
where the value with superscript a is a scale-up factor that is species-specific.
| (3) |
where Qh (hepatic blood flow) is species-specific.
LC/MS/MS Bioanalysis of Samples from Plasma Protein Binding and Intrinsic Clearance Assays.
Samples were analyzed on a Thermo Electron TSQ Quantum Ultra triple quad mass spectrometer (San Jose, CA) via electrospray ionization (ESI) with two Thermo Electron Accela pumps (San Jose, CA), and a Leap Technologies CTC PAL autosampler (Carrboro, NC). The analytes were separated by gradient elution on a dual column system with two Thermo Hypersil Gold (2.1 × 30 mm, 1.9 μm) columns (San Jose, CA) thermostated at 40 °C. HPLC mobile phase A was 0.1% formic acid in water, and mobile phase B was 0.1% formic acid in acetonitrile. The gradient started at 10% B after a 0.2 min hold and was linearly increased to 95% B over 0.8 min, held at 95% B for 0.2 min, and returned to 10% B in 0.1 min. The total run time was 1.3 min, and the HPLC flow rate was 0.8 mL/min. While pump 1 ran the gradient method, pump 2 equilibrated the alternate column isocratically at 10% B. Compound optimization, data collection, and processing were performed using Thermo Electron’s QuickQuan software (v2.3) and Xcalibur (v2.0.7 SP1).
Rat Plasma:Brain Level Studies.
Male Sprague–Dawley rats (n = 2) weighing around 300 g were purchased from Harlan Laboratories (Indianapolis, IN) and implanted with catheters in the carotid artery and jugular vein. The cannulated animals were acclimated to their surroundings for approximately 1 week before dosing and provided food and water ad libitum. IV cassette PK experiments in rats were carried out according to methods described previously (Bridges et al. Pharmacol. Res. Perspect. 2014; reference 49). Briefly, a cassette of compounds (n = 4–5/cassette) was formulated from 10 mM solutions of compounds in DMSO. To reduce the absolute volume of DMSO that was administered, the compounds were combined and diluted with ethanol and PEG 400 to achieve a final concentration of 0.4–0.5 mg/mL for each compound (2 mg/mL total) administered in each cassette. The final dosing solutions consisted of approximately 10% ethanol, 40% PEG400, and 50% DMSO (v/v). Each cassette dose was administered IV via the jugular vein to two dual-cannulated (carotid artery and jugular vein) adult male Sprague–Dawley rats, each weighing between 250 and 350 g (Harlan, Indianapolis, IN) for a final dose of 0.2 mg/kg per compound. Whole blood collections via the carotid artery were performed at 0.033, 0.117, 0.25, 0.5, 1, 2, 4, 7, and 24 h post dose and plasma samples were prepared for bioanalysis. For tissue distribution studies in cassette format, brain dissection and blood collections via the carotid artery were performed at 0.25 h post dose. Blood samples were collected into chilled, EDTA-fortified tubes, and centrifuged for 10 min at 3000 rpm (4 °C), and the resulting plasma was aliquoted into 96-well plates for LC/MS/MS analysis. The brain samples were rinsed with PBS, snap-frozen, and stored at −80 °C. Prior to LC/MS/MS analysis, brain samples were thawed to room temperature and subjected to mechanical homogenation employing a Mini-Beadbeater and 1.0 mm zirconia/silica Beads (BioSpec Products).
Liquid Chromatography/Mass Spectrometry Analysis.
PAR4 antagonists were analyzed via electrospray ionization (ESI) on an AB Sciex API-4000 (Foster City, CA) triple-quadrupole instrument that was coupled with Shimadzu LC-10 AD pumps (Columbia, MD) and a Leap Technologies CTC PAL autosampler (Carrboro, NC). Analytes were separated by gradient elution using a Fortis C18 2.1 × 50 mm, 3.5 μm column (Fortis Technologies Ltd., Cheshire, UK) thermostated at 40 °C. HPLC mobile phase A was 0.1% formic acid in water (pH unadjusted), and mobile phase B was 0.1% formic acid in acetonitrile (pH unadjusted). The gradient started at 30% B after a 0.2 min hold and was linearly increased to 90% B over 0.8 min, held at 90% B for 0.5 min, and returned to 30% B in 0.1 min followed by a re-equilibration (0.9 min). The total run time was 2.5 min, and the HPLC flow rate was 0.5 mL/min. The source temperature was set at 500 °C, and mass spectral analyses were performed using multiple reaction monitoring (MRM) utilizing a Turbo Ion Spray source in positive ionization mode (5.0 kV spray voltage). All data were analyzed using AB Sciex Analyst 1.4.2 software. For in vivo studies, the final PK parameters were calculated by noncompartmental analysis using Phoenix (version 6.2) (Pharsight Inc., Mountain View, CA).
Animal Care and Use.
All animal study procedures were approved by the Institutional Animal Care and Use Committee and were conducted in accordance with the National Institutes of Health regulations of animal care covered in Principles of Laboratory Animal Care (National Institutes of Health). All animals were group-housed under a 12 h light/dark cycle with food and water available ad libitum. For behavioral studies, 8–10-week-old male C57Bl/6 mice were used.
Molecular Modeling.
See the Supporting Information for full details.
Supplementary Material
ACKNOWLEDGMENTS
We thank William K. Warren, Jr., and the William K. Warren Foundation who funded the William K. Warren, Jr. Chair in Medicine (to C.W.L.) and endowed the Warren Center for Neuroscience Drug Discovery.
Funding
We warmly thank the NIH for support of our programs (1R01AG068623, 1R01HL133923, and 1R21AG073891).
ABBREVIATIONS
- PAR4
protease-activated receptor 4
- CRC
concentration–response curve
- PBL
plasma:brain level
- DMPK
drug metabolism and pharmacokinetics
- AP
activated peptide
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.1c00557.
Compound characterization and computational/modeling methods (PDF)
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/acschemneuro.1c00557
Contributor Information
Jeanette L. Bertron, Department of Chemistry and Warren Center for Neuroscience Drug Discovery, Vanderbilt University, Nashville, Tennessee 37232, United States.
Matthew T. Duvernay, Department of Pharmacology, Vanderbilt University, Nashville, Tennessee 37232, United States; Warren Center for Neuroscience Drug Discovery, Vanderbilt University, Nashville, Tennessee 37232, United States.
Sidnee G. Mitchell, Department of Pharmacology, Vanderbilt University, Nashville, Tennessee 37232, United States
Shannon T. Smith, Chemical and Physical Biology Program, Center for Structural Biology, Vanderbilt University, Nashville, Tennessee 37232, United States
Jae G. Maeng, Department of Pharmacology, Vanderbilt University, Nashville, Tennessee 37232, United States
Anna L. Blobaum, Department of Pharmacology, Vanderbilt University, Nashville, Tennessee 37232, United States; Warren Center for Neuroscience Drug Discovery, Vanderbilt University, Nashville, Tennessee 37232, United States
Dexter C. Davis, Department of Pharmacology, Vanderbilt University, Nashville, Tennessee 37232, United States; Warren Center for Neuroscience Drug Discovery, Vanderbilt University, Nashville, Tennessee 37232, United States
Jens Meiler, Department of Chemistry, Vanderbilt University, Nashville, Tennessee 37232, United States; Institute for Drug Discovery, Leipzig University, Saxony 04109, Germany.
Heidi E. Hamm, Department of Pharmacology, Vanderbilt University, Nashville, Tennessee 37232, United States;
Craig W. Lindsley, Department of Pharmacology, Vanderbilt University, Nashville, Tennessee 37232, United States; Department of Chemistry, Department of Biochemistry, and Warren Center for Neuroscience Drug Discovery, Vanderbilt University, Nashville, Tennessee 37232, United States;.
REFERENCES
- (1).Kahn ML; Nakanishi-Matsui M; Shapiro MJ; Ishihara H; Coughlin SR Protease-activated receptors 1 and 4 mediate activation of human platelets by thrombin. J. Clin. Invest 1999, 103, 879–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Vu TKH; Wheaton VI; Hung DT; Charo I; Coughlin SR Domains specifying thrombin-receptor interaction. Nature 1991, 353, 674–677. [DOI] [PubMed] [Google Scholar]
- (3).Lindahl TL; Macwan AS; Ramstrom S PAR4 is more important than PAR1 for the thrombin-induced procoagulant effect on platelets. J. Thromb. Haemostasis 2016, 14, 1639–1641. [DOI] [PubMed] [Google Scholar]
- (4).Duvernay M; Young S; Gailani D; Schoenecker J; Hamm HE Protease-activated receptor (PAR) 1 and PAR4 differentially regulate factor V expression from human platelets. Mol. Pharmacol 2013, 83, 781–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Coughlin SR Thrombin signalling and protease-activated receptors. Nature 2000, 407, 258–264. [DOI] [PubMed] [Google Scholar]
- (6).Faruqi TR; Weiss EJ; Shapiro MJ; Huang W; Coughlin SR Structure-function analysis of protease-activated receptor 4 tethered ligand peptides. Determinants of specificity and utility in assays of receptor function. J. Biol. Chem 2000, 275, 19728–19734. [DOI] [PubMed] [Google Scholar]
- (7).Wong PC; Seiffert D; Bird JE; Watson CA; Bostwick JS; Giancarli M; Allegretto N; Hua J; Harden D; Guay J; Callejo M; Miller MM; Lawrence RM; Banville J; Guy J; Maxwell BD; Priestley ES; Marinier A; Wexler RR; Bouvier M; Gordon DA; Schumacher WA; Yang J Blockade of protease-activated receptor-4 (PAR4) provides robust antithrombotic activity with low bleeding. Sci. Transl. Med 2017, 9, aaf5294. [DOI] [PubMed] [Google Scholar]
- (8).Miller MJ; Banville J; Friends TJ; Gagnon M; Hangeland JJ; Lavallee J-F; Martel A; O’Grady H; Remillard R; Ruediger E; Tremblay F; Posy SL; Allegretto NJ; Guarino VR; Harden DG; Harper TW; Hartl K; Josephs J; Malmstrom S; Watson C; Yang Y; Zhang G; Wong P; Yang J; Bouvier M; Seiffert DA; Wexler RR; Lawrence RM; Priestly ES; Marinier A Discovery of Potent Protease-Activated Receptor 4 Antagonists with in Vivo Antithrombotic Efficacy. J. Med. Chem 2019, 62, 7400–7416. [DOI] [PubMed] [Google Scholar]
- (9).Wilson SJ; Ismat FA; Wang Z; Cerra M; Narayan H; Raftis J; Gray TJ; Connell S; Garonzik S; Ma X; Yang J; Newby DE PAR4 (Protease-Activated Receptor 4) Antagonism With BMS-986120 Inhibits Human Ex Vivo Thrombus Formation. Arterioscler., Thromb., Vasc. Biol 2018, 38, 448–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Suo Z; Wu M; Citron BA; Gao C; Festoff BW Persistent protease-activated receptor 4 signaling mediates thrombin-induced microglial activation. J. Biol. Chem 2003, 278, 31177–31183. [DOI] [PubMed] [Google Scholar]
- (11).Sokolova E; Reiser G Prothrombin/thrombin and the thrombin receptors PAR-1 and PAR-4 in the brain: localization, expression and participation in neurodegenerative diseases. Thromb. Haemostasis 2008, 100, 576–581. [PubMed] [Google Scholar]
- (12).Mao Y; Zhnag M; Tuma RF; Kunapuli SP Deficiency of PAR4 attenuates cerebral ischemia/reperfusion injury in mice. J. Cereb. Blood Flow Metab 2010, 30, 1044–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Luo J; Wu X; Liu H; Cui W; Guo W; Guo K; Guo H; Tao K; Shi Y; Li F; Shi Y; Feng D; Yan H; Gao G; Qu Y Antagonism of Protease-Activated Receptor 4 Protects Against Traumatic Brain Injury by Suppressing Neuroinflammation via Inhibition of Tab2/NF-jB Signaling. Neurosci. Bull 2021, 37, 242–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Lee F-Y; Lien J-C; Huang L-J; Huang T-M; Tsai S-C; Teng C-M; Wu C-C; Cheng F-C; Kuo S-C Synthesis of 1-benzyl-3-(5′-hydroxymethyl-2′-furyl)indazole analogues as novel antiplatelet agents. J. Med. Chem 2001, 44, 3746–3749. [DOI] [PubMed] [Google Scholar]
- (15).Wu CC; Hwang TL; Liao CH; Kuo SC; Lee FY; Lee CY; Teng CM Selective inhibition of protease-activated receptor 4-dependent platelet activation by YD-3. Thromb. Haemostasis 2002, 87, 1026–1033. [PubMed] [Google Scholar]
- (16).Chen HS; Kuo SC; Teng CM; Lee FY; Wang JP; Lee YC; Kuo CW; Huang CC; Wu CC; Huang LJ Synthesis and antiplatelet activity of ethyl 4-(1-benzyl-1H-indazol-3-yl)benzoate (YD-3) derivatives. Bioorg. Med. Chem 2008, 16, 1262–1278. [DOI] [PubMed] [Google Scholar]
- (17).Young SE; Duvernay MT; Lindsley CW; Hamm HE Synthesis and characterization of indole-derived protease activated receptor-4 (PAR4) antagonists. PLoS One 2013, 8, No. e65528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Wen W; Young SE; Duvernay MT; Schulte ML; Nance KD; Melancon BJ; Engers J; Locuson CW 2nd; Wood MR; Daniels JS; Wu W; Lindsley CW; Hamm HE; Stauffer SR Substituted indoles as selective protease activated receptor 4 (PAR-4) antagonists: Discovery and SAR of ML354. Bioorg. Med. Chem. Lett 2014, 24, 4708–4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Temple KJ; Duvernay MT; Young SE; Wen W; Wu W; Maeng JG; Blobaum AL; Stauffer SR; Hamm HE; Lindsley CW Development of a series of (1-benzyl-3-(6-ethoxypyrimidin-3-yl)-5-(trifluoromethoxy)-1H-indol-2-yl)-methanol as selective protease activated receptor 4 (PAR4) antagonists with in vivo utility and activity against γ-thrombin. J. Med. Chem 2016, 59, 7690–7695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Temple KJ; Duvernay MT; Maeng JG; Blobaum AL; Stauffer SR; Hamm HE; Lindsley CW Identification of the minimum PAR4 inhibitor pharmacophore and optimization of a series of 2-methoxy-6-arylimidazo[2,1-b][1,3,4]thiadiazoles. Bioorg. Med. Chem. Lett 2016, 26, 5481–5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).See the Supporting Information for full details. ACS Publications website at DOI:
- (22).Evans BE; Rittle KE; Bock MG; DiPardo RM; Freidinger RM; Whitter WL; Lundell GF; Veber DF; Anderson PS; Chang RSL; Lotti VJ; Cerino DJ; Chen TB; Kling PJ; Kunkel KA; Springer JP; Hirshfield J Methods for drug discovery: Development of potent, selective orally effective cholecystokin antagonists. J. Med. Chem 1988, 31, 2235–2246. [DOI] [PubMed] [Google Scholar]
- (23).Duvernay MT; Temple KJ; Maeng JG; Blobaum AL; Stauffer SR; Lindsley CW; Hamm HE Contributions of PAR1 and PAR4 to thrombin induced GPIIbIIIa activation in human platelets. Mol. Pharmaceutics 2017, 91, 39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Bridges TM; Rook JM; Noetzel MJ; Morrison RD; Zhou Y; Gogliotti RD; Vinson PN; Jones CK; Niswender CM; Lindsley CW; Stauffer SR; Conn PJ; Daniels JS Biotransformation of a novel positive allosteric modulator of metabotropic glutamate receptor subtype 5 contributes to seizures in rats involving receptor agonism-dependent mechanism. Drug Metab. Dispos 2013, 41, 1703–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Bender AM; Cho HP; Nance KD; Lingenfelter KS; Luscombe VB; Gentry PR; Voigtritter K; Berizzi AE; Sexton PM; Langmead CJ; Christopoulos A; Locuson CW; Bridges TM; Niswender CM; Jones CK; Conn PJ; Lindsley CW Discovery and Optimization of Potent and CNS Penetrant M5-Preferring PAMs Derived From a Novel, Chiral N-(Indanyl)-piperidine Amide Scaffold. ACS Chem. Neurosci 2018, 9, 1572–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.