

Abstract
Rationale:
PM2.5-induced adverse effects on respiratory health may be driven by epigenetic modifications in airway cells. The potential impact of exposure duration on epigenetic alterations in the airways is not yet known.
Objectives:
We aimed to study associations of fine particulate matter PM2.5 exposure with DNA methylation in nasal cells.
Methods:
We conducted nasal epigenome-wide association analyses within 503 children from Project Viva (mean age 12.9 y), and examined various exposure durations (1-day, 1-week, 1-month, 3-months and 1-year) prior to nasal sampling. We used residential addresses to estimate average daily PM2.5 at 1 km resolution. We collected nasal swabs from the anterior nares and measured DNA methylation (DNAm) using the Illumina MethylationEPIC BeadChip. We tested 719,075 high quality autosomal CpGs using CpG-by-CpG and regional DNAm analyses controlling for multiple comparisons, and adjusted for maternal education, household smokers, child sex, race/ethnicity, BMI z-score, age, season at sample collection and cell-type heterogeneity. We further corrected for bias and genomic inflation. We tested for replication in a cohort from the Netherlands (PIAMA).
Results:
In adjusted analyses, we found 362 CpGs associated with 1-year PM2.5 (FDR < 0.05), 20 CpGs passing Bonferroni correction (P < 7.0x10−8) and 10 Differentially Methylated Regions (DMRs). In 445 PIAMA participants (mean age 16.3 years) 11 of 203 available CpGs replicated at P < 0.05. We observed differential DNAm at/near genes implicated in cell cycle, immune and inflammatory responses. There were no CpGs or regions associated with PM2.5 levels at 1-day, 1-week, or 1-month prior to sample collection, although 2 CpGs were associated with past 3-month PM2.5.
Conclusion:
We observed wide-spread DNAm variability associated with average past year PM2.5 exposure but we did not detect associations with shorter-term exposure. Our results suggest that nasal DNAm marks reflect chronic air pollution exposure.
1. Introduction
Air pollution exposures are known to affect incidence and severity of multiple chronic health conditions, (Garcia et al., 2019; Schraufnagel et al., 2019) and in particular may exacerbate respiratory symptoms in asthma (Orellano et al., 2017) and chronic obstructive pulmonary disease (Hansel et al., 2016). Respirable air pollution particles of 2.5 μm diameter or less (PM2.5) have been shown to disrupt the airway epithelial barrier (Zhao et al., 2018), enhance responses to inhaled allergens, (He et al., 2017) and promote oxidative stress response pathways (Feng et al., 2016). The specific mechanisms of action for PM2.5-induced adverse effects are not entirely elucidated, (Rider and Carlsten, 2019) and may potentially be driven by epigenetic modifications, which in turn alter gene expression levels, potentially shifting both local immune responses and epithelial barrier function in the airways.
The majority of studies that have examined air pollution exposures and their associations with epigenetic modifications have focused on DNA methylation (DNAm) patterns in cord blood (Gruzieva et al., 2019) or blood leukocytes (Bind et al., 2014, 2015; Prunicki et al., 2018). These studies (Bind et al., 2014; Gruzieva et al., 2019; Prunicki et al., 2018) have shown epigenetic modification of immune signaling and inflammatory genes, specifically differential DNAm in or near FOXP3, IL-10, IL-6, IFN-γ, ICAM, and NOTCH4 which may be distinct from localized responses in the airways. Epigenetic analyses in airway cells may identify pathways relevant to air pollution-induced damage and repair responses in the tissue that is first to encounter these exposures, and may also shed light on the underlying mechanisms in air pollution induced exacerbation of existing respiratory disease. Nasal epithelial cells are the most relevant tissue type for research on allergic rhinitis phenotypes and upper airway irritant responses. The nasal epithelium can also serve as a useful surrogate for cells in the lower airway, given the similarities with respect to airway epithelial cell markers (cytokeratin 19, CD44), and the positive correlation between epithelial cell cytokine production (including IL-6, RANTES, vascular endothelial growth factor, monocyte chemoattractant protein-1,and MMP-9) in response to stimulation with IL-1Beta, TNF-alpha, and IL-13 for these two tissue types (McDougall et al., 2008; Thavagnanam et al., 2014). Thus far, studies of air pollution exposures and epigenetic alterations in airway epithelial cells have been very limited. One cross-sectional study among 24 participants identified nasal cell DNAm changes in TET1 (a gene for a dioxygenase that plays an active role in demethylation of DNA) associated with traffic-related air pollution (TRAP) (Somineni et al., 2016).
In this work our aim was to determine the association of residence-specific air pollutant exposures (PM2.5) with genome-wide DNA methylation in the nasal epithelium of children. We conducted our study in 503 participants in Project Viva, (Oken et al., 2015) a pre-birth cohort study, and examined the potential relationship between PM2.5 and nasal epithelium DNAm at over 700,000 CpG sites. We replicated our findings in an independent cohort from the Netherlands, the PIAMA birth cohort (N = 445) (Wijga et al., 2014). We hypothesized that prior PM2.5 exposure would be associated with altered nasal epithelium DNAm in oxidative stress and epithelial barrier function genes, and we studied various exposure time windows to determine the potential relevance of exposure duration on epigenetic modifications. We also determined whether DNAm patterns associated with PM2.5, an exposure known to exacerbate asthma, overlap with the altered nasal DNAm patterns we previously observed with asthma and airway inflammation phenotypes (Cardenas et al., 2019).
2. Methods
2.1. See supplement for additional details on all methods.
Study Populations.
Study participants were enrolled in Project Viva, a prospective pre-birth cohort study (Oken et al., 2015). Of the total 2,128 live births, 1,038 children were re-contacted at mean 12.9y (11.9 to 15.4y) and attended an early-teen in-person visit, of whom 547 provided consent for nasal swab sample collection. Of these, 503 also had residential specific PM2.5 exposure assessment. All study protocols were approved by the Institutional Review Board of Harvard Pilgrim Health Care.
PM2.5 Exposure Assessment.
We estimated ambient PM2.5 exposure at each participant’s residential address using a spatio-temporal model for the Northeastern USA, as previously described (Kloog et al., 2014; Rice et al., 2019). The PM2.5 model utilized satellite-based Aerosol Optical Depth data, retrieved using the Multi- Angle Implementation of Atmospheric Correction algorithm at 1 × 1 km resolution, and ground-level daily PM2.5 mass measurements, land use terms and meteorological covariates to estimate daily PM2.5 at 200 × 200 m resolution. Predictions from this model had an excellent mean out-of-sample R2 (0.88) and excellent fit of predictions when compared with withheld measurements (slope = 0.99).
DNAm Measurements.
Trained research assistants collected nasal swabs from the anterior nares. Sterile cotton swabs used for sampling were placed in DNA lysis buffer (Promega, Madison, WI, USA) after collection. DNA was isolated using the Maxwell 16 Buccal Swab LEV DNA Purification Kit following the manufacturer’s instructions (Promega, Madison, WI, USA). Swab samples from the anterior nares have been previously demonstrated to yield respiratory epithelial cells (Lai et al., 2015). We analyzed extracted DNA using the Infinium MethylationEPIC BeadChip (Illumina, San Diego, CA) to obtain epigenome-wide DNAm measurements.
Statistical Analyses.
Among 503 participants with high quality DNAm data eligible for analyses, we performed epigenome-wide association analyses (EWAS) by fitting linear regression models using limma with moderated test-statistics using an empirical Bayes estimation (pollution levels were modeled as predictors of DNAm) (Smyth, 2004). We adjusted for variables selected a priori and based on PCA plots in EWAS models: child race/ethnicity, sex, age at sample collection in days, age and sex-specific BMI z-score using US national reference data; smokers currently living in the house, sine and cosine of season of sample collection, and maternal education in pregnancy. To account for cell type heterogeneity, we used a bioinformatic method (ReFACTor), adjusting for the first 10 ReFACTor PCs in our analyses (Rahmani et al., 2017). To remove residual inflation and reduce the influence of potential unmeasured confounders, we used the R package bacon, which constructs an empirical null distribution using a Gibbs sampling algorithm, to adjust our EWAS results (van Iterson et al., 2017). We modeled DNA methylation as M values (methylation intensity) but include the beta value (proportion methylated) coefficients here for interpretability. Coefficients are expressed as % change in DNAm for a 1 μg/m3 increase in PM2.5 exposure level. We performed 5 independent EWAS to analyze nasal DNAm in relation to: 1) PM2.5 levels during 24 h prior to nasal sample collection 2) 7-day average PM2.5 levels prior to nasal sample collection 3) prior 30-day average PM2.5 levels 4) prior 90-day average PM2.5 levels 5) prior 365-day (past year) average PM2.5 levels. The EWAS was controlled for the false discovery rate (FDR) at 5% and for the familywise error rate using a Bonferroni correction (P < 6.95x10−8). In addition to the EWAS on all available participants (as described above), we also performed a sensitivity analysis removing individuals with current asthma.
After conducting individual CpG by CpG analyses, we performed regional DNAm analyses using DMRff (Suderman et al., 2018) to identify differentially methylated regions (DMRs) associated with PM2.5 exposure.
We also conducted pathway analyses of differentially methylated individual CpGs using the R package MissMethyl (Phipson et al., 2015). For pathway analyses, CpGs with FDR < 0.05 were examined for KEGG pathway enrichment.
External Replication Analysis.
We sought to replicate our findings in PIAMA (Wijga et al., 2014)(Prevention and Incidence of Asthma and Mite Allergy), a birth cohort study of children born in 1996–1997 in the Netherlands. In PIAMA participants (age 16 years), nasal epithelial cells were collected by nasal brushings in two study centers (Groningen and Utrecht) (Xu et al., 2018). The lateral area underneath the inferior turbinate was sampled using a Cytosoft brush (CP-5B, Cyto-Pak). Further details on nasal sampling and sample preparation have been previously described (Xu et al., 2018). DNA extracted from nasal brushes was analyzed using the Infinium HumanMethylation450 BeadChip array (Illumina, San Diego, CA, USA). DNA methylation data were pre-processed in R with the Bioconductor package minfi (Aryee et al., 2014) using the original IDAT files, and quality control procedures were performed as previously described (Qi et al., 2020). Exposure to PM2.5 was assessed as the annual average at the home address at the time of the age 16 medical examination using land-use regression models that have been described elsewhere (Eeftens et al., 2012). The replication analyses included 445 participants. Analyses were performed with stratification by PIAMA study center, and inverse variance–weighted fixed-effects meta-analyses on results from the two PIAMA centers were performed with METAL. Covariate adjustment in replication analyses included age, sex, batch, secondhand smoking, maternal education, season, and 3 surrogate variables. The 3 surrogate variables were added to adjust for unknown confounders, including cell type.
3. Results
Participant Characteristics.
We assessed the nasal methylome in Project Viva participants who were in early adolescence (mean age 12.9 years, SD 0.65) (Table 1). Half of the participants were female (50.1%), and the majority were of white race/ethnicity (68.6%) with representation from other racial/ethnic groups (15.7% Black, 3.6% Hispanic, 3.0% Asian, 9.1% more than one race). Nasal swab sampling was conducted in all seasons (about one third of samples were collected in summer, while 19–26% were collected in fall, winter and spring). On average, participants were exposed to PM2.5 below the EPA air quality standard levels. Average past week PM2.5 exposure was 7.42 μg/m3 (SD 2.00), past month average PM2.5 was 7.44 μg/m3 (SD 1.53), past 90 day average was 7.50 μg/m3 (SD 1.09) and past year average PM2.5 exposure was 7.76 μg/m3 (SD 0.55). Past year PM2.5 showed very low correlations with past day, past 7 day and past month exposures at the same address (Pearson r = 0.03, 0.18 and 0.19, respectively). Past year PM2.5 showed a moderate correlation with past 90 day exposure (0.53). Correlations for all exposures are shown in supplemental table 1.
Table 1.
Characteristics of Project Viva Adolescents with PM2.5 Exposure and Nasal DNA Methylation Data.
| Characteristic (N = 503) | N (%) or mean (SD) |
|---|---|
| Sex | |
| Female | 252 (50.1%) |
| Male | 251 (49.9%) |
| Age at sample collection (years) | 12.9 (0.65) |
| BMI z-score | 0.39 (1.06) |
| Maternal Education | |
| College graduate | 357 (71.0%) |
| Non-college graduate | 146 (29.0%) |
| Smoker Living in Household | |
| Yes | 59 (11.8%) |
| No | 444 (88.2%) |
| Current Asthma * | |
| Cases | 59 (13.6%) |
| Controls | 375 (86.4%) |
| Race/Ethnicity | |
| White | 345 (68.6%) |
| Black | 79 (15.7%) |
| Hispanic | 18 (3.6%) |
| Asian | 15 (3.0%) |
| More than one race | 46 (9.1%) |
| Season of Sample Collection | |
| Summer | 166 (33.0%) |
| Fall | 108 (21.5%) |
| Winter | 96 (19.0%) |
| Spring | 133 (26.4%) |
| PM2.5 (μg/m3) | |
| Day of sample collection | 7.44 (3.05) |
| 7-day average | 7.42 (2.00) |
| 30-day average | 7.44 (1.53) |
| 90-day average | 7.50 (1.09) |
| 365-day average | 7.76 (0.55) |
Participants with past asthma and no current wheeze/asthma medication use or current wheeze without asthma not included in N; percentages are out of N = 434
Global DNA DNAm Variability.
Univariate predictors of global DNAm PCs are shown in Fig. 1. We examined associations of participant characteristics and surrogate variables for cell type (ReFACTor PCs) with the top 30 global DNAm PCs explaining 59% of DNAm variability. As expected, the cell type surrogate variables (PCs on the y-axis of Fig. 1) showed the strongest associations with global DNAm. Season was associated with the first global DNAm PC at p < 0.01. Age, race and sex were also associated with some of the top 10 global DNAm PCs. PM2.5 exposure (past year and past 90 day levels) was also associated with global DNAm.
Fig. 1.
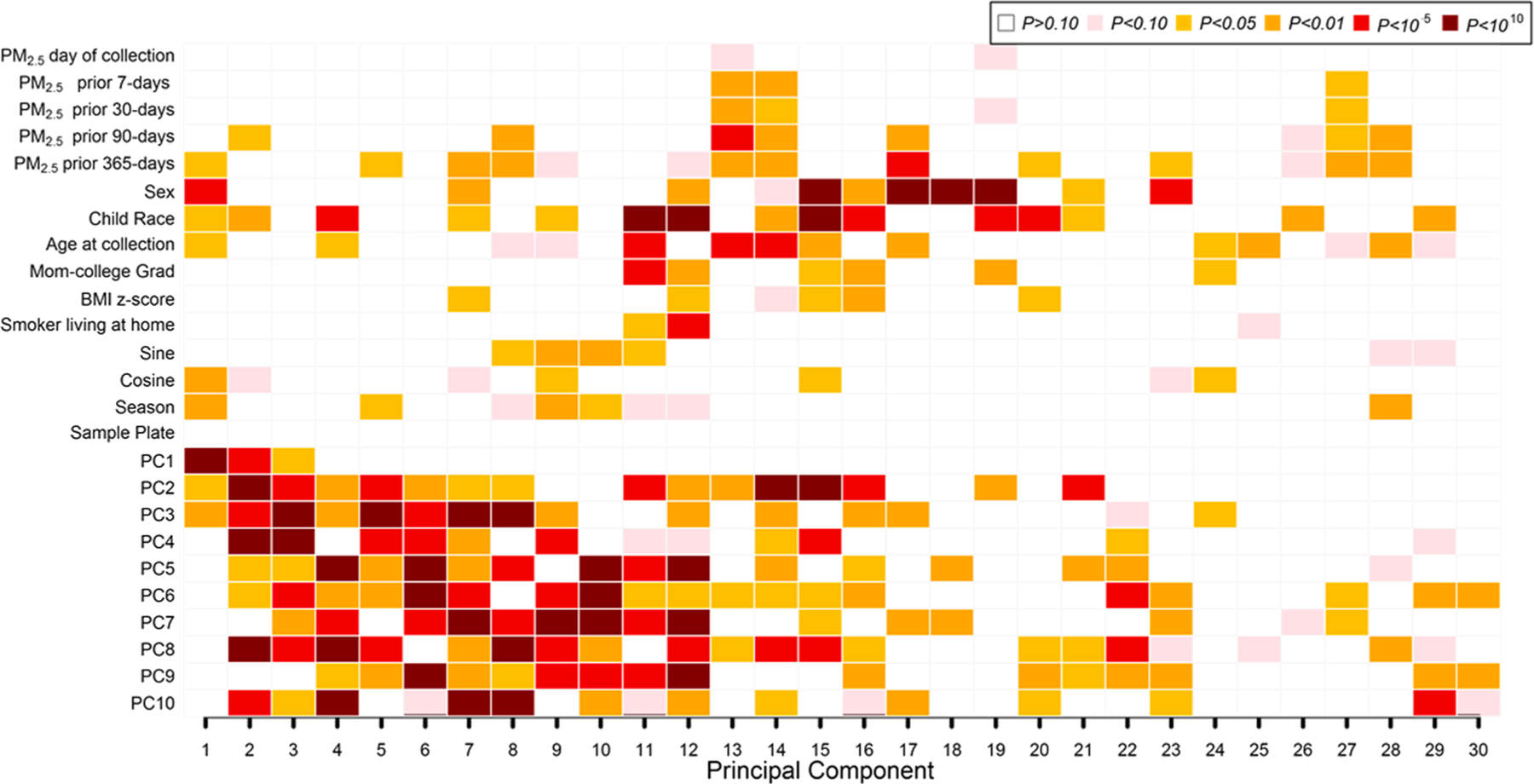
Associations of PM2.5 exposures, participant characteristics, and cell type heterogeneity with Global Nasal DNA methylation (DNAm) variability. PCs 1–10 on the vertical axis reflect DNAm differences in cell types estimated using the bio-informatic method ReFACTor. Univariate regression analysis (with global DNA methylation principal components as outcomes) was performed. P values for univariate associations between all covariates of interest and the top 30 global DNA methylation PCs (shown on the horizontal axis) are color-coded by smallest P value (dark red; P < 10–10) to largest (blank; P > 0.10). In all, global DNA methylation principal components explained 59% of the variance of the nasal DNA methylation data in the x-axis.
PM2.5 and DNAm Analyses.
In our EWAS of PM2.5 exposure 1-year prior to sample collection, we identified 362 differentially methylated individual CpGs at FDR < 0.05 and 20 CpGs that were statistically significant at the Bonferroni adjusted threshold (P < 6.95x10−8) (Table 2). We used the R package bacon to adjust for inflation in our EWAS results. A Manhattan plot of the past year PM2.5 exposure EWAS is shown in Fig. 2. Cell type adjustment reduced our FDR significant findings in the 90 day and past year PM2.5 exposure models (Supplemental Table 2). After applying bacon, genomic inflation was similar for cell type adjusted models vs. models that did not include cell type (λ = 1.15 vs. 1.10 for past year PM2.5 exposure).
Table 2.
Summary of differentially methylated CpGs and DMRs of Nasal EWAS for PM2.5*
| λ | FDR < 0.05 | Bonferroni | DMRs | |
|---|---|---|---|---|
| Day of sample collection | 0.93 | 0 | 0 | 0 |
| Prior 7-days | 0.93 | 0 | 0 | 0 |
| Prior 30-days | 1.03 | 0 | 0 | 0 |
| Prior 90-days | 1.03 | 9 | 2 | 0 |
| Prior 365-days | 1.15 | 362 | 20 | 10 |
Bacon-adjusted EWAS results, adjusted for cell type (ReFACTor PCs), child race/ethnicity, sex, BMI z-score in early teen, age at nasal swab collection (days), maternal education, smoker living at home, sine and cosine for season at sample collection.
λ = Genomic inflation
FDR: False Discovery Rate (5%)
Bonferroni: P < 6.953x10−8
DMR: Differentially Methylated Region (≥2-CpGs and Stouffer P < 0.05)
Fig. 2.
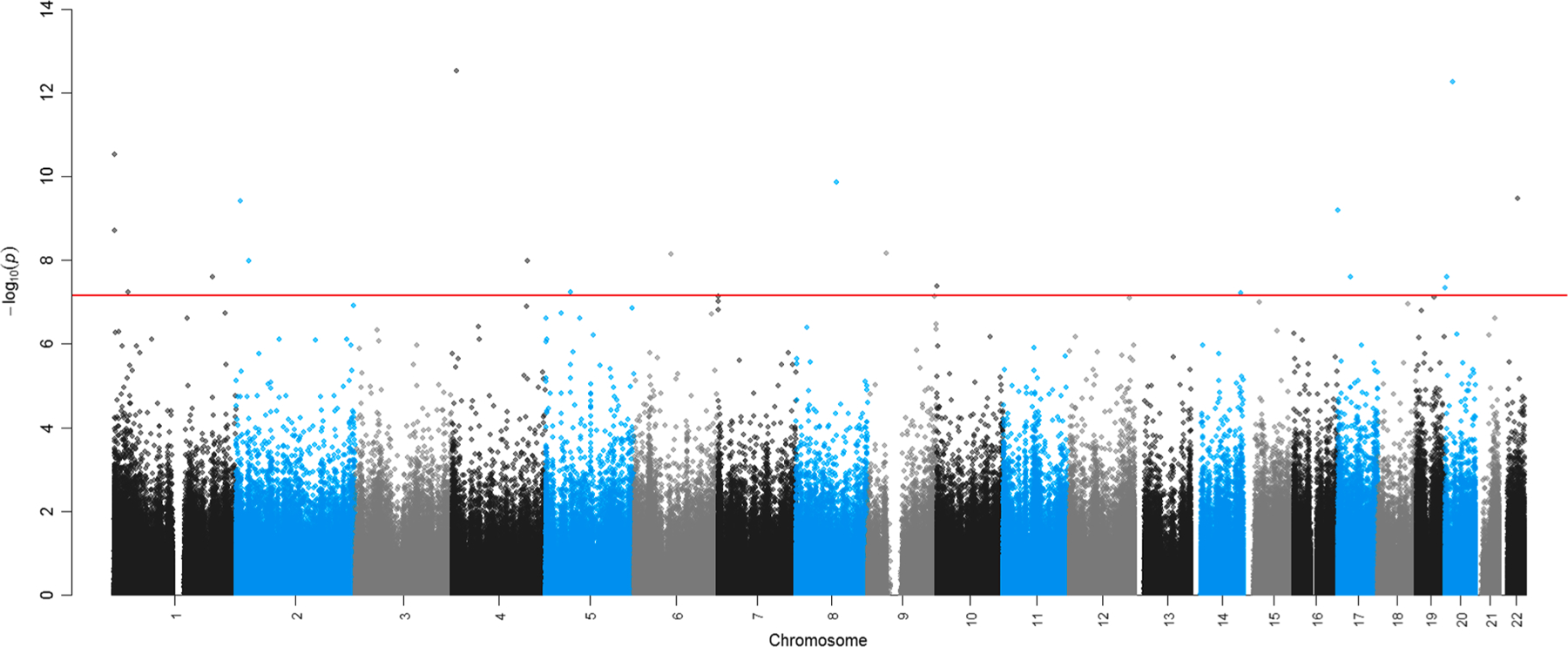
Nasal epigenome-wide associations for residence specific past year (365-day) PM2.5 exposure. Y axis shows uncorrected – log10(P values) plotted for each CpG site sorted by chromosomal and genomic position (as shown on the x-axis). Adjustment for multiple testing was accounted for in the epigenome-wide association analyses. Bonferroni threshold for statistical significance (P < 6.95 × 10–8) shown in the solid red horizontal line.
We did not find strong evidence for enrichment based on positional location of CpGs (i.e. CpG position relative to islands) (Supplemental table 3).
The top 20 CpG sites ranked on p-value associated with past year PM2.5 exposure and reaching statistical significance after Bonferroni correction are shown in Table 3. Findings included differential methylation of CpGs in solute transport protein genes (SLC2A9), epithelial membrane sodium ion transport genes (SCNN1D), fibronectin binding domains (ELFN2), a phosphoprotein gene associated with T-cell responses (PAG1) and a zinc finger protein/ innate immune response gene (RBCK1). Other top differentially methylated CpGs were located within genes promoting cell division (PPP2R5C), apoptosis (TMEM214), DNA repair/carcinogenesis (LGR6) and cellular responses to DNA damage (MACROD2). Higher previous year PM2.5 exposure was associated with lower DNAm of CpG sites in some of the cancer pathway and cell cycle genes in our EWAS results (TMEM214, LGR6), while in others DNAm levels were higher with higher PM2.5 exposure (PPP2R5C). FDR adjusted results (N = 362 sites at FDR < 0.05) are shown in supplemental table 4. These results included additional genes associated with DNA repair/carcinogenesis (RAD52, UIMC1, WNT7A, GSK3B), notch signaling (NOTCH4), immune response (VDR, NFKB2, PSTPIP1, TICAM1), the mTOR pathway (VDR, ULK1, FGF3) and MAPK signaling (TAOK1, CACNA1D, DUSP4, FGF3).
Table 3.
Bonferroni Significant Differentially Methylated CpGs associated with a 1 μg/m3 increase in past year PM2.5 Exposure.
| CpG | chr | Average % Methylation | Regression Coefficient (M value) | % Diff in DNAm (Beta) | P value | Gene/Closest gene | Relation_to_Island |
|---|---|---|---|---|---|---|---|
| cg26210521 | chr4 | 88.6 | −0.256 | −1.70 | 3.08E-13 | SLC2A9 | OpenSea |
| cg20396870 | chr20 | 66.5 | −0.182 | −2.74 | 5.53E-13 | MACROD2 | OpenSea |
| cg12120973 | chr1 | 87.3 | −0.203 | −1.44 | 3.03E-11 | SCNN1D | OpenSea |
| cg22118655 | chr22 | 11.1 | 0.226 | 1.47 | 3.29E-10 | ELFN2 | Island |
| cg01108434 | chr8 | 86.9 | −0.254 | −1.96 | 1.42E-10 | PAG1 | OpenSea |
| cg07769732 | chr2 | 81.8 | − 0.169 | −1.69 | 3.92E-10 | KIDINS220 | N_Shore |
| cg09969776 | chr17 | 93.8 | −0.186 | −0.72 | 6.32E-10 | VPS53 | OpenSea |
| cg13388025 | chr1 | 84.1 | −0.162 | −1.40 | 1.98E-09 | ATAD3C | N_Shelf |
| cg02180798 | chr9 | 83.6 | −0.141 | −1.28 | 6.91E-09 | SHB | OpenSea |
| cg07582070 | chr6 | 85.7 | −0.140 | −1.13 | 7.33E-09 | IMPG1 | OpenSea |
| cg18168844 | chr5 | 13.4 | 0.253 | 2.60 | 5.76E-08 | EMB | N_Shelf |
| cg08163906 | chr14 | 9.4 | 0.288 | 1.76 | 6.17E-08 | PPP2R5C | Island |
| cg13315471 | chr2 | 74.6 | −0.121 | −1.55 | 1.02E-08 | TMEM214 | S_Shelf |
| cg23369179 | chr4 | 74.0 | −0.104 | −1.34 | 1.03E-08 | DCHS2 | OpenSea |
| cg05103574 | chr1 | 90.4 | −0.116 | −0.67 | 5.84E-08 | MECR | N_Shore |
| cg04351903 | chr1 | 14.4 | −0.116 | −0.96 | 2.46E-08 | LGR6 | OpenSea |
| cg06118847 | chr17 | 78.7 | −0.108 | −1.21 | 2.48E-08 | WSB1 | N_Shore |
| cg01219087 | chr20 | 87.2 | −0.152 | −1.11 | 2.53E-08 | PNRP | Island |
| cg17608529 | chr10 | 96.9 | −0.084 | −0.16 | 4.12E-08 | DIP2C | Island |
| cg10495669 | chr20 | 9.0 | 0.319 | 1.74 | 4.69E-08 | RBCK1 |
We also performed a sensitivity analysis with individuals with current asthma removed (N = 375 controls without current asthma were analyzed). For the top differentially methylated CpG sites, comparisons between the original EWAS coefficients versus coefficients with current asthmatics removed showed little change (supplemental table 5).
Our Bonferroni and FDR adjusted findings for past year PM2.5 exposure were entirely distinct from the CpG sites identified as associated with asthma in our previous work in Project Viva (Cardenas et al., 2019), however we did identify overlap in the genes annotated to FDR significant CpGs in both the asthma and past year PM2.5 exposure EWAS. Although the CpGs themselves were different, both analyses identified sites associated with the following 11 genes: EPS15L1, NARF, NCOR2, PTPRS, RNF40, TPCN1, LGR6, PTPRE, RIN3, SLC45A4 and TBC1D22A. Our previously identified differentially methylated CpG sites for asthma are shown in supplemental table 6 for reference. A sex-stratified EWAS of past year PM2.5 exposure and the nasal methlyome did not show sex-specific differences (supplemental table 7).
While the majority of differentially methylated CpGs were detected in our analysis of past year PM2.5 exposure, we also detected a minor signal from our cell-type adjusted EWAS of past 90-day PM2.5 exposure (2 CpGs were significant at the Bonferroni threshold level, and 9 CpGs that were significant at FDR < 0.05) (Supplemental table 8). One of the CpGs significant at FDR < 0.05 (cg02548780) overlapped with the differentially methylated CpGs associated with past year PM2.5 exposure, with consistent direction of association. We did not identify any differentially methylated CpG sites associated with past month, past week or past day PM2.5 exposure.
Replication Analysis Results.
A comparison of baseline demographic characteristics of participants in PIAMA and Project Viva is shown in supplemental table 9, and a methods comparison table is shown in supplemental table 10. PIAMA participants (N = 445) were on average 3.4 year older than Project Viva participants, and experienced approximately double the PM2.5 exposure levels (mean 16.1 μg/m3 (SD 0.7)) as compared to Project Viva (mean 7.76 μg/m3 (SD 0.55)). Given that the epigenome was interrogated using a 450 K array in PIAMA (vs. the 850 K array in Project Viva), not all sites were available for replication analysis (10 out of 20 Bonferroni adjusted sites and 203 out of 362 FDR adjusted sites were available for replication analysis in PIAMA). In the replication analysis in PIAMA, we did not observe replication of the 10 available differentially methylated CpGs that met Bonferroni correction in our analysis. However, we did observe replication of 11 out of the 203 CpGs identified as FDR significant (FDR adjusted p value (q) < 0.05) in our analysis. These EWAS associations were replicated in PIAMA at nominal significance level (p < 0.05), but were not significant at the Bonferroni adjusted p value threshold (p < 2.46x10−4). Replicated associations for past year PM2.5 exposure and differential CpG DNAm are shown in Table 4. The majority of the differentially methylated CpGs that replicated are involved in carcinogenesis pathways (LASS4, IRX2, AGAP1, MEIS1/2, TNFRSF21, GRIK3, UIMC1), and several have been specifically implicated in lung cancer (LASS4, IRX2, MEIS1/2, GRIK3). TNFRSF21, one of the carcinogenesis genes with a replicated CpG site with differential DNAm, is also associated with T cell activation. Overall, average PM2.5 exposure in PIAMA was significantly higher as compared to Project Viva. See supplemental Fig. 1 for plot of PIAMA PM2.5 exposure levels.
Table 4.
Replication of Project Viva past year PM2.5 exposure FDR adjusted results in the PIAMA Cohort.
| Project Viva |
PIAMA |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| CpG | Chr | Average % Methylation | Beta coefficient (Mval) | % Diff DNAm (Beta) | q value | Unadjusted P value | % Diff DNAm (Beta) | Unadjusted P value | Gene/Closest Gene |
| cg15187788 | 19 | 1.9 | 0.109 | 0.13% | 0.035 | 1.290E-05 | (Beta) | 0.003 | LASS4 |
| cg24105287 | 2 | 12.4 | −0.295 | −2.31% | 0.032 | 9.000E-06 | 0.44% | 0.004 | MEIS1 |
| cg22986569 | 5 | 83.4 | −0.258 | −3.11% | 0.008 | 7.710E-07 | −2.34% | 0.005 | IRX2 |
| cg08805497 | 2 | 97.2 | −0.082 | −0.15% | 0.021 | 4.250E-06 | −2.90% | 0.006 | AGAP1 |
| cg17171920 | 15 | 93.9 | −0.158 | −1.04% | 0.045 | 2.350E-05 | −0.32% | 0.011 | MEIS2 |
| cg11524642 | 7 | 85.9 | −0.188 | −1.59% | 0.002 | 9.490E-08 | −0.92% | 0.022 | C7orf50 |
| cg02643778 | 6 | 44.6 | 0.146 | 2.56% | 0.015 | 2.140E-06 | −0.79% | 0.025 | TNFRSF21 |
| cg04817870 | 17 | 85.8 | −0.139 | −1.08% | 0.041 | 2.020E-05 | 1.52% | 0.033 | ASPSCR1 |
| cg07680195 | 19 | 43.6 | 0.114 | 2.00% | 0.012 | 1.710E-06 | −0.65% | 0.035 | PGLS |
| cg05589454 | 1 | 89.5 | −0.150 | −1.12% | 0.021 | 4.240E-06 | 0.76% | 0.045 | GRIK3 |
| cg19755108 | 5 | 49.2 | 0.150 | 2.66% | 0.003 | 1.390E-07 | −0.51% | 0.050 | UIMC1 |
Regional and Pathway Level DNAm Analyses.
Results from differentially methylated regions are shown in Table 5. Although the specific genes identified in the regional analysis were different than those differentially methylated at the individual CpG level (Table 3), the pathways and cellular processes represented were quite similar. We identified differential regional methylation of cell cycle genes (CDK2AP1, ZC3HC1, MAD1L1) and genes involved in carcinogenesis (CDK2AP1, ZC3HC1, MAD1L1, PHLPP1, SFRP2, PLCH1). Regional analyses also identified differential methylation of NXN (nucleoredoxin), which is associated with responses to oxidative stress and PHLPP1, a gene involved in modulation of innate immune responses.
Table 5.
Differentially Methylated Regions (DMRs) associated with past year PM2.5 Exposure.
| region | chr | UCSC RefGene | CpGs (N) | Start (bp) | End (bp) | Z statistic | Adjusted P value |
|---|---|---|---|---|---|---|---|
| 1 | 10 | WDFY4 | 6 | 49,892,943 | 49,893,406 | −5.573 | 0.020 |
| 2 | 12 | CDK2AP1 | 3 | 123,752,805 | 123,752,916 | −6.317 | <0.001 |
| 3 | 17 | NXN | 4 | 800,100 | 800,717 | −5.804 | 0.005 |
| 4 | 7 | ZC3HC1 | 3 | 129,691,325 | 129,691,449 | 5.659 | 0.012 |
| 5 | 18 | PHLPP1 | 4 | 60,381,593 | 60,382,247 | 6.064 | 0.001 |
| 6 | 7 | MAD1L1 | 4 | 1,991,345 | 1,991,534 | −5.831 | 0.004 |
| 7 | 11 | 4 | 12,088,146 | 12,088,476 | −6.306 | <0.001 | |
| 8 | 3 | PLCH1 | 3 | 155,422,754 | 155,423,168 | 5.510 | 0.029 |
| 9 | 2 | SPEG | 3 | 220,355,154 | 220,355,252 | −5.515 | 0.028 |
| 10 | 4 | SFRP2 | 5 | 154,711,620 | 154,711,906 | −5.470 | 0.036 |
Pathway analysis based on differentially methylated CpGs showed pathway enrichment for ABC (ATP binding cassette) transporters, glycosaminoglycan degradation, and mineral absorption pathways (Table 6).
Table 6.
Biological pathway enrichment analysis of differentially methylated CpGs.
| KEGG Biological Pathway | Pathway Name | Genes on Pathway | Differentially methylated genes | Unadjusted P value |
|---|---|---|---|---|
| path:hsa04978 | Mineral absorption | 58 | 4 | 0.0056 |
| path:hsa00531 | Glycosaminoglycan degradation | 19 | 2 | 0.036 |
| path:hsa02010 | ABC (ATP Binding Cassette) Transporters | 45 | 3 | 0.045 |
4. Discussion
In this work, we identified associations of exposure to PM2.5 air pollution in the previous year with the nasal cell epigenome in over 500 children from Project Viva in eastern Massachusetts. Our study showed three major findings. First, even at the relatively low air pollution exposure levels experienced by our Project Viva participants, we observed differences in nasal cell DNAm with exposure to PM2.5. Second, a consistent signal for differential DNAm of cell cycle and innate immune response genes emerged across our site specific CpG and regional DNAm analyses. Third, it was average past year exposure to PM2.5, but not the shorter-term exposures (day of, past week, or past month) that was correlated with altered nasal DNAm profiles. This last finding suggests that average exposure levels must be elevated for an extended period of time in order to alter epigenetic profiles in the upper airways.
Our top results included differential DNAm of RBCK1, a cell cycle and innate immune signaling gene. RBCK1, is a zinc finger protein gene involved in immune dysfunction (Krenn et al., 2018), inflammatory pathways (Tian et al., 2007) and carcinogenesis (Liu et al., 2019). RBCK1 is a negative regulator of TNF and IL-1 driven inflammation (Taminiau et al., 2016). Higher PM2.5 exposure was associated with higher DNAm of a CpG site in the promoter of RBCK1, suggesting that the inflammatory dampening effects of this gene may be reduced with higher exposure to air pollution. In addition to its capacity to modulate inflammation, RBCK1 may also play a role in carcinogenesis (Yu et al., 2019). Similarly, other differentially methylated CpG sites were located within pathways broadly associated with both inflammatory processes and the cell cycle, including two genes from the mTOR signaling pathway in FDR adjusted analyses (ULK1, RRN3P2). The potential relevance of mTOR in human airway response to PM2.5, identified here in our EWAS study, is recapitulated in data from experimental models. In vitro studies of human bronchial epithelial cells show that inhibition of mTOR following PM2.5 exposure promotes production of inflammatory cytokines IL-6 and IL-8 (thereby increasing epithelial inflammation), and enhances autophagy/cellular degradation (thereby increasing epithelial cell damage) (Wu et al., 2019).
CpG site specific and regional analysis also identified differential methylation of genes that have been linked specifically to cancers of the airways. Previous epidemiological studies indicate that PM2.5 exposure is carcinogenic (Harrison et al., 2004), and in vitro studies suggest that PM2.5 may promote carcinogenesis through epigenetic regulation of the tumor suppressor gene p53 (Zhou et al., 2016). Our results did not show alteration of p53 DNA methylation, although we did identify PM2.5 associated changes in methylation of of CDK2AP1 (associated with lung and nasopharyngeal carcinomas), (Sun et al., 2013; Wu et al., 2019) SFRP2 (modulator of lung cancer cell apoptosis) (Li et al., 2019), and PLCH1 (associated with non-small cell lung cancer risk) (Zhang et al., 2013). Furthermore, the majority of the CpGs replicated in an external cohort were found in cancer/cell cycle pathways (MEIS1, IRX2, GRIK3, AGAP1, U1MC1) reinforcing the global pattern we observed for epigenetic modifications associated with exposure to PM2.5 air pollution. Differential DNAm of MEIS1, IRX2, and GRIK3 are known epigenetic biomarkers of lung cancer (however, it is worth noting that MEIS1 and IRX2 were hyper rather than hypo-methylated as is observed in cancer studies; GRIK3 is hypomethylated in our study as well as in carcinogenesis) (Pradhan et al., 2013; Rauch et al., 2012). AGAP1 controls cancer cell invasion, (Tsutsumi et al., 2020) and U1MC1 is involved in recognition and repair of DNA lesions (Hamdi et al., 2019). However, it is important to note that, given the overlaps between inflammatory pathways and those in carcinogenesis (Greten and Grivennikov, 2019), the pollution-related differential methylation of genes associated with cancer in the airways in our study may simply reflect alterations in inflammatory processes, including pathways involved in tissue homeostasis and repair that do not necessarily give rise to carcinogenesis.
Findings from our pathway analysis did not highlight either immune or cell cycle/cancer pathways, but instead identified enrichment of ABC (ATP binding cassette) transporters. In vitro studies of human bronchial epithelial cells exposed to air pollutant particles show altered ABC transporter gene expression, suggesting that the detoxifying action of these proteins may be compromised with exposure to air pollution, with potential implications for airway epithelial barrier integrity (Le Vée et al., 2019).
As far as we are aware, our study is the first large-scale analysis of air pollution exposures and the nasal cell DNA methylation. One previous study by Somineini et al, (Somineni et al., 2016) focused on the association between TRAP (traffic associated air pollutant) exposure and nasal DNAm (assessed using a 450 K array) in a very small group of 12 African American children with asthma and their non-asthmatic siblings. That report identified lower DNAm of TET1 in the nasal brushings of participants exposed to higher levels of TRAP; however the investigators did not have the statistical power to look at epigenome-wide associations. In contrast, we did not identify differential DNAm of TET1 or any other genes associated with global DNAm (i.e. DNA methyl-transferases) in our analyses.
Other studies of air pollution exposure and DNAm have examined circulating blood leukocytes. A comparison of our nasal EWAS findings to studies of PM2.5 and blood cell DNAm reveal some genes and pathways related to those identified in our nasal analysis. For example, a previous study in whole blood identified differential methylation of CpGs within the NXN gene (Panni et al., 2016). Our analysis also demonstrated differenial DNA methylation of NXN in our regional (but not CpG site specific) findings. NXN encodes for nucleoredoxin, a redox dependent regulator which, when activated, may promote Wnt-mediated cell growth and differentiation under conditions of oxidative stress. It is conceivable that air pollutant exposures trigger a cellular turnover process mediated through oxidative stress response genes such as NXN. Other reports on PM2.5 exposure and blood DNA methylation show differences in immune signaling genes. For example, in candidate gene studies, PM2.5 exposure was associated with hypermethylation of the IL-6 gene in circulating leukocytes (Bind et al., 2014). In our nasal EWAS we observed differential DNAm of genes within the mTOR pathway, which is known to modulate IL-6 levels, but we did not observe differential DNAm of the IL-6 gene itself. (This could be because nasal swab samples are enriched for epithelial cells (upstream effector cells) whereas the blood tissue compartment has a higher proportion of cells (i.e. monocytes) that are major producers of IL-6). Other candidate gene epigenetic studies have found associations with differential DNAm of Foxp3 and IL-10, which suggest alterations in adaptive immune response (specifically T-cell responses) with PM2.5 exposure (Prunicki et al., 2018). In our nasal EWAS, altered DNAm patterns with higher PM2.5 exposure levels suggests shifts in innate, rather than adaptive, immune responses in the local environment of the airway. This is perhaps unsurprising, given that the target tissue sampled in our nasal swabs (the respiratory epithelium) plays a major role in modulating innate immune response to inhaled agents (Diamond et al., 2000).
Greater PM2.5 exposure was associated with a far greater number of differentially methylated CpGs in our nasal samples as compared to other EWAS studies that examined differentially methylated CpGs in blood. The largest EWAS of PM2.5 exposure to date (>8,000 participants) (Gondalia et al., 2019) quantified site-specific DNAm of blood leukocytes on the 450 K array, and identified only one differentially methylated CpG in the CFTR gene (which did not overlap with our nasal EWAS). A meta-analysis of PM2.5 exposure and the blood DNAm in 9 birth cohort studies identified 14 differentially methylated CpGs (none of which replicated in our cohort) (Gruzieva et al., 2019). Overall, our results suggest that nasal cells are more sensitive to PM2.5 induced modifications in DNAm as compared to blood leukocytes, with some overlap observed for the two tissue types. The larger number of differentially methylated sites in our nasal EWAS as compared to blood EWAS is perhaps expected, given the direct contact of nose with airborne particulates, including PM2.5.
Epigenetic changes associated with PM2.5 exposure in our nasal study and in blood EWAS tend to be relatively small, and additional studies are necessary to understand the functional impact of these changes. However, it is important to note that persistence and reproducibility of similar small scale changes have been observed in response to other types of exposures (particularly maternal smoking in pregnancy), (Breton et al., 2017) suggesting that effect changes of this magnitude may indeed have biological relevance.
Given that PM2.5 exposure is associated with both asthma incidence and severity (Guarnieri and Balmes, 2014) it is reasonable to hypothesize that some nasal DNAm signatures in response to PM2.5 may overlap with differential DNAm patterns associated with asthma. Interestingly, when we compared differentially methylated CpGs associated with PM2.5 exposure to those we previously found to be associated with current asthma in our Project Viva participants, (Cardenas et al., 2019) we found no overlap for Bonferroni adjusted EWAS hits. While PM2.5 associated differential DNAm patterns were contained mainly in pathways associated with the cell cycle and DNA damage/repair mechanisms, asthma associated differential DNAm included multiple genes involved in Th2 response, T cell activation, and mucin production. Examination of beta coefficients in PM2.5 nasal EWAS after removing individuals with current asthma from the analysis showed minimal change, suggesting that current asthma status is not a meaningful effect modifier of epigenetic responses to PM2.5 exposure.
Our study has several strengths. First, we had a large sample size. Second, we examined differential DNAm in the nasal cells, which is a first point of contact for air pollution exposures, and may also serve as a surrogate for responses in the lower airways. Lastly, we were able to assess exposure to PM2.5 with a spatial resolution of 1 1 km with time specific resolution. We should also acknowledge some limitations. We may have had more exposure misclassification of short term PM2.5 relative to long-term average exposures, which could potentially explain why most differential DNAm was observed with past year, rather than past week or past day exposures. While outdoor PM2.5 exposures are known to correlate with indoor PM2.5 (both in terms of composition and concentration), (Liu and Zhang, 2019) we did not have direct assessment of indoor PM2.5 exposure levels. We did not have complementary gene expression data, in order to determine whether methylation changes were associated with changes in gene expression levels. Our replication analysis may also have been hindered by differences in DNA methylation assays (450 K vs. 850 K array), differences in PM2.5 exposure levels (which were higher in the Netherlands), and differences in nasal sampling procedures and extracted cells (anterior nares vs. inferior turbinate cells). (Cells collected from the anterior nares have very similar methylation patterns and gene expression profiles as those collected from the inferior turbinate, although the proportion of respiratory epithelial cells in anterior nares samples tend to be lower (65% vs. 99%)) (Lai et al). Even given these differences across cohorts, we were still able to detect replication of multiple sites.
Future studies focused on the consistency of CpG assessment between the 450 K and 850 K platforms, specifically utilizing nasal tissue samples, will help inform replication efforts that compare findings across arrays. Specifically, findings from EPIC and 450 K arrays might differ due enrichment of regulatory elements in the EPIC array. Development of an ideal cell type reference panel for epigenetic studies of the upper airways would also reduce bias in nasal epigenomic studies. Lastly, new studies ought to consider potential effect modifiers of air pollutant exposures and nasal epigenome associations. As highlighted in a recent review, physical activity and diet (particularly B vitamins) may modulate the effects PM2.5 on DNA methylation (Rider and Carlsten, 2019). In conclusion, we report multiple associations of long term (past year) PM2.5 exposure and the nasal methylome, that were not observed for shorter term exposure windows. Site specific and regional DNA methylation changes were mainly observed in cell cycle, cancer and immune/inflammatory pathways. Our results suggest that the nasal methylome is sensitive to long-term ambient PM2.5 exposure.
Supplementary Material
Funding
This research was supported by the National Institutes of Health grants R01 AI102960, R01 034568, UH3 OD023286, P30ES000002, R01HL111108, P01 HL132825, HL P01114501, R01 ES031259; and by the Environmental Protection Agency grants US EPA (R832416, RD834798). The PIAMA study was supported by The Netherlands Organization for Health Research and Development; The Netherlands Organization for Scientific Research; the Lung Foundation of the Netherlands (with methylation studies supported by AF 4.1.14.001); The Netherlands Ministry of Spatial Planning, Housing, and the Environment; and The Netherlands Ministry of Health, Welfare, and Sport. C. Q. was supported by a grant from the China Scholarship Council.
This publication’s contents are solely the responsibility of the grantee and do not necessarily represent the official views of the US EPA. Further, US EPA does not endorse the purchase of any commercial products or services mentioned in the publication.
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.envint.2021.106505.
References
- Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA, 2014. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369. 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bind M-A, Lepeule J, Zanobetti A, Gasparrini A, Baccarelli A, Coull BA, Tarantini L, Vokonas PS, Koutrakis P, Schwartz J, 2014. Air pollution and gene-specific methylation in the Normative Aging Study: association, effect modification, and mediation analysis. Epigenetics 9, 448–458. 10.4161/epi.27584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bind M-AC, Coull BA, Peters A, Baccarelli AA, Tarantini L, Cantone L, Vokonas PS, Koutrakis P, Schwartz JD, 2015. Beyond the Mean: Quantile Regression to Explore the Association of Air Pollution with Gene-Specific Methylation in the Normative Aging Study. Environ. Health Perspect 123, 759–765. 10.1289/ehp.1307824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breton CV, Marsit CJ, Faustman E, Nadeau K, Goodrich JM, Dolinoy DC, Herbstman J, Holland N, LaSalle JM, Schmidt R, Yousefi P, Perera F, Joubert BR, Wiemels J, Taylor M, Yang IV, Chen R, Hew KM, Freeland DMH, Miller R, Murphy SK, 2017. Small-Magnitude Effect Sizes in Epigenetic End Points are Important in Children’s Environmental Health Studies: The Children’s Environmental Health and Disease Prevention Research Center’s Epigenetics Working Group. Environ Health Perspect 125, 511–526. 10.1289/EHP595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas A, Sordillo JE, Rifas-Shiman SL, Chung W, Liang L, Coull BA, Hivert M-F, Lai PS, Forno E, Celedón JC, Litonjua AA, Brennan KJ, DeMeo DL, Baccarelli AA, Oken E, Gold DR, 2019. The nasal methylome as a biomarker of asthma and airway inflammation in children. Nat Commun 10, 3095. 10.1038/s41467-019-11058-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond G, Legarda D, Ryan LK, 2000. The innate immune response of the respiratory epithelium. Immunol. Rev 173, 27–38. 10.1034/j.1600-065x.2000.917304.x. [DOI] [PubMed] [Google Scholar]
- Eeftens M, Beelen R, de Hoogh K, Bellander T, Cesaroni G, Cirach M, Declercq C, Dėdelė A, Dons E, de Nazelle A, Dimakopoulou K, Eriksen K, Falq G, Fischer P, Galassi C, Gražulevičienė R, Heinrich J, Hoffmann B, Jerrett M, Keidel D, Korek M, Lanki T, Lindley S, Madsen C, Mölter A, Nádor G, Nieuwenhuijsen M, Nonnemacher M, Pedeli X, Raaschou-Nielsen O, Patelarou E, Quass U, Ranzi A, Schindler C, Stempfelet M, Stephanou E, Sugiri D, Tsai M-Y, Yli-Tuomi T, Varró MJ, Vienneau D, von Klot S, Wolf K, Brunekreef B, Hoek G, 2012. Development of Land Use Regression models for PM(2.5), PM(2.5) absorbance, PM(10) and PM(coarse) in 20 European study areas; results of the ESCAPE project. Environ. Sci. Technol 46, 11195–11205. 10.1021/es301948k. [DOI] [PubMed] [Google Scholar]
- Feng S, Gao D, Liao F, Zhou F, Wang X, 2016. The health effects of ambient PM2.5 and potential mechanisms. Ecotoxicol. Environ. Saf 128, 67–74. 10.1016/j.ecoenv.2016.01.030. [DOI] [PubMed] [Google Scholar]
- Garcia E, Berhane KT, Islam T, McConnell R, Urman R, Chen Z, Gilliland FD, 2019. Association of Changes in Air Quality With Incident Asthma in Children in California, 1993–2014. JAMA 321, 1906–1915. 10.1001/jama.2019.5357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gondalia R, Baldassari A, Holliday KM, Justice AE, Méndez-Giráldez R, Stewart JD, Liao D, Yanosky JD, Brennan KJM, Engel SM, Jordahl KM, Kennedy E, Ward-Caviness CK, Wolf K, Waldenberger M, Cyrys J, Peters A, Bhatti P, Horvath S, Assimes TL, Pankow JS, Demerath EW, Guan W, Fornage M, Bressler J, North KE, Conneely KN, Li Y, Hou L, Baccarelli AA, Whitsel EA, 2019. Methylome-wide association study provides evidence of particulate matter air pollution-associated DNA methylation. Environ Int 132, 104723. 10.1016/j.envint.2019.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greten FR, Grivennikov SI, 2019. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 51, 27–41. 10.1016/j.immuni.2019.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruzieva O, Xu C-J, Yousefi P, Relton C, Merid SK, Breton CV, Gao L, Volk HE, Feinberg JI, Ladd-Acosta C, Bakulski K, Auffray C, Lemonnier N, Plusquin M, Ghantous A, Herceg Z, Nawrot TS, Pizzi C, Richiardi L, Rusconi F, Vineis P, Kogevinas M, Felix JF, Duijts L, den Dekker HT, Jaddoe VWV, Ruiz JL, Bustamante M, Antó JM, Sunyer J, Vrijheid M, Gutzkow KB, Grazuleviciene R, Hernandez-Ferrer C, Annesi-Maesano I, Lepeule J, Bousquet J, Bergström A, Kull I, Söderhäll C, Kere J, Gehring U, Brunekreef B, Just AC, Wright RJ, Peng C, Gold DR, Kloog I, DeMeo DL, Pershagen G, Koppelman GH, London SJ, Baccarelli AA, Melén E, 2019.Prenatal Particulate Air Pollution and DNA Methylation in Newborns: An Epigenome-Wide Meta-Analysis. Environ. Health Perspect 127, 57012. 10.1289/EHP4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarnieri M, Balmes JR, 2014. Outdoor air pollution and asthma. Lancet 383, 1581–1592. 10.1016/S0140-6736(14)60617-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdi Y, Leclerc M, Dumont M, Dubois S, Tranchant M, Reimnitz G, Soucy P, Cassart P, Ouimet M, Sinnett D, Chaieb MLL, Simard J, 2019. Functional Analysis of Promoter Variants in Genes Involved in Sex Steroid Action, DNA Repair and Cell Cycle Control. Genes (Basel) 10. 10.3390/genes10030186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansel NN, McCormack MC, Kim V, 2016. The Effects of Air Pollution and Temperature on COPD. COPD 13, 372–379. 10.3109/15412555.2015.1089846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison RM, Smith DJT, Kibble AJ, 2004. What is responsible for the carcinogenicity of PM2.5? Occup Environ Med 61, 799–805. 10.1136/oem.2003.010504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M, Ichinose T, Yoshida Y, Arashidani K, Yoshida S, Takano H, Sun G, Shibamoto T, 2017. Urban PM2.5 exacerbates allergic inflammation in the murine lung via a TLR2/TLR4/MyD88-signaling pathway. Sci Rep 7, 11027. 10.1038/s41598-017-11471-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloog I, Chudnovsky AA, Just AC, Nordio F, Koutrakis P, Coull BA, Lyapustin A, Wang Y, Schwartz J, 2014. A New Hybrid Spatio-Temporal Model For Estimating Daily Multi-Year PM2.5 Concentrations Across Northeastern USA Using High Resolution Aerosol Optical Depth Data. Atmos Environ (1994) 95, 581–590. 10.1016/j.atmosenv.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenn M, Salzer E, Simonitsch-Klupp I, Rath J, Wagner M, Haack TB, Strom TM, Schänzer A, Kilimann MW, Schmidt RLJ, Schmetterer KG, Zimprich A, Boztug K, Hahn A, Zimprich F, 2018. Mutations outside the N-terminal part of RBCK1 may cause polyglucosan body myopathy with immunological dysfunction: expanding the genotype-phenotype spectrum. J. Neurol 265, 394–401. 10.1007/s00415-017-8710-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai PS, Liang L, Cibas ES, Liu AH, Gold DR, Baccarelli A, Phipatanakul W, 2015. Alternate methods of nasal epithelial cell sampling for airway genomic studies. J. Allergy Clin. Immunol 136, 1120–1123.e4. 10.1016/j.jaci.2015.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Vée M, Bacle A, Jouan E, Lecureur V, Potin S, Fardel O, 2019. Induction of multidrug resistance-associated protein 3 expression by diesel exhaust particle extract in human bronchial epithelial BEAS-2B cells. Toxicol In Vitro 58, 60–68. 10.1016/j.tiv.2019.03.021. [DOI] [PubMed] [Google Scholar]
- Li P, Zhao S, Hu Y, 2019. SFRP2 modulates non-small cell lung cancer A549 cell apoptosis and metastasis by regulating mitochondrial fission via Wnt pathways. Mol Med Rep 20, 1925–1932. 10.3892/mmr.2019.10393. [DOI] [PubMed] [Google Scholar]
- Liu C, Zhang Y, 2019. Relations between indoor and outdoor PM2.5 and constituent concentrations. Front. Environ. Sci. Eng 13, 5. 10.1007/s11783-019-1089-4. [DOI] [Google Scholar]
- Liu M-L, Zang F, Zhang S-J, 2019. RBCK1 contributes to chemoresistance and stemness in colorectal cancer (CRC). Biomed. Pharmacother 118, 109250 10.1016/j.biopha.2019.109250. [DOI] [PubMed] [Google Scholar]
- McDougall CM, Blaylock MG, Douglas JG, Brooker RJ, Helms PJ, Walsh GM, 2008. Nasal epithelial cells as surrogates for bronchial epithelial cells in airway inflammation studies. Am J Respir Cell Mol Biol 39, 560–568. 10.1165/rcmb.2007-0325OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oken E, Baccarelli AA, Gold DR, Kleinman KP, Litonjua AA, Meo DD, Rich-Edwards JW, Rifas-Shiman SL, Sagiv S, Taveras EM, Weiss ST, Belfort MB, Burris HH, A, J.C.C., Huh SY, Mantzoros C, Parker MG, Gillman MW, 2015. Cohort profile: project viva. International journal of epidemiology 44, 37–48. 10.1093/ije/dyu008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orellano P, Quaranta N, Reynoso J, Balbi B, Vasquez J, 2017. Effect of outdoor air pollution on asthma exacerbations in children and adults: Systematic review and multilevel meta-analysis. PLoS ONE 12, e0174050. 10.1371/journal.pone.0174050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panni T, Mehta AJ, Schwartz JD, Baccarelli AA, Just AC, Wolf K, Wahl S, Cyrys J, Kunze S, Strauch K, Waldenberger M, Peters A, 2016. Genome-Wide Analysis of DNA Methylation and Fine Particulate Matter Air Pollution in Three Study Populations: KORA F3, KORA F4, and the Normative Aging Study. Environ. Health Perspect 124, 983–990. 10.1289/ehp.1509966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phipson B, Maksimovic J, Oshlack A, et al. , 2015. missMethyl: an R package for analysing methylation data from Illuminas HumanMethylation450 platform. Bioinformatics btv560. [DOI] [PubMed] [Google Scholar]
- Pradhan MP, Desai A, Palakal MJ, 2013. Systems biology approach to stage-wise characterization of epigenetic genes in lung adenocarcinoma. BMC Syst Biol 7, 141. 10.1186/1752-0509-7-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prunicki M, Stell L, Dinakarpandian D, de Planell-Saguer M, Lucas RW, Hammond SK, Balmes JR, Zhou X, Paglino T, Sabatti C, Miller RL, Nadeau KC, 2018. Exposure to NO2, CO, and PM2.5 is linked to regional DNA methylation differences in asthma. Clin. Epigenetics 10, 2. 10.1186/s13148-017-0433-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi C, Jiang Y, Yang IV, Forno E, Wang T, Vonk JM, Gehring U, Smit HA, Milanzi EB, Carpaij OA, Berg M, Hesse L, Brouwer S, Cardwell J, Vermeulen CJ, Acosta-Pérez E, Canino G, Boutaoui N, van den Berge M, Teichmann SA, Nawijn MC, Chen W, Celedón JC, Xu C-J, Koppelman GH, 2020. Nasal DNA methylation profiling of asthma and rhinitis. J. Allergy Clin. Immunol 145, 1655–1663. 10.1016/j.jaci.2019.12.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmani E, Zaitlen N, Baran Y, Eng C, Hu D, Galanter J, Oh S, Burchard EG, Eskin E, Zou J, Halperin E, 2017. Correcting for cell-type heterogeneity in DNA methylation: a comprehensive evaluation. Nat. Methods 14, 218–219. 10.1038/nmeth.4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch TA, Wang Z, Wu X, Kernstine KH, Riggs AD, Pfeifer GP, 2012. DNA methylation biomarkers for lung cancer. Tumour Biol 33, 287–296. 10.1007/s13277-011-0282-2. [DOI] [PubMed] [Google Scholar]
- Rice MB, Li W, Schwartz J, Di Q, Kloog I, Koutrakis P, Gold DR, Hallowell RW, Zhang C, O’Connor G, Washko GR, Hunninghake GM, Mittleman MA, 2019. Ambient air pollution exposure and risk and progression of interstitial lung abnormalities: the Framingham Heart Study. Thorax 74, 1063–1069. 10.1136/thoraxjnl-2018-212877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rider CF, Carlsten C, 2019. Air pollution and DNA methylation: effects of exposure in humans. Clin Epigenetics 11, 131. 10.1186/s13148-019-0713-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schraufnagel DE, Balmes JR, Cowl CT, De Matteis S, Jung S-H, Mortimer K, Perez-Padilla R, Rice MB, Riojas-Rodriguez H, Sood A, Thurston GD, To T, Vanker A, Wuebbles DJ, 2019. Air Pollution and Noncommunicable Diseases: A Review by the Forum of International Respiratory Societies’ Environmental Committee, Part 1: The Damaging Effects of Air Pollution. Chest 155, 409–416. 10.1016/j.chest.2018.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK, 2004. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3, Article3. 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- Somineni HK, Zhang X, Biagini Myers JM, Kovacic MB, Ulm A, Jurcak N, Ryan PH, Khurana Hershey GK, Ji H, 2016. Ten-eleven translocation 1 (TET1) methylation is associated with childhood asthma and traffic-related air pollution. J. Allergy Clin. Immunol 137, 797–805.e5. 10.1016/j.jaci.2015.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suderman M, Stanley J, French R, et al. , 2018. dmrff: identifying differentially methylated regions efficiently with power and control 10.1101/508556v1. [DOI] [Google Scholar]
- Sun M, Jiang R, Wang G, Zhang C, Li J, Jin C, Zhang X, 2013. Cyclin-dependent kinase 2-associated protein 1 suppresses growth and tumorigenesis of lung cancer. Int. J. Oncol 42, 1376–1382. 10.3892/ijo.2013.1813. [DOI] [PubMed] [Google Scholar]
- Taminiau A, Draime A, Tys J, Lambert B, Vandeputte J, Nguyen N, Renard P, Geerts D, Rezsöhazy R, 2016. HOXA1 binds RBCK1/HOIL-1 and TRAF2 and modulates the TNF/NF-κB pathway in a transcription-independent manner. Nucleic Acids Res 44, 7331–7349. 10.1093/nar/gkw606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thavagnanam S, Parker JC, McBrien ME, Skibinski G, Shields MD, Heaney LG, 2014. Nasal epithelial cells can act as a physiological surrogate for paediatric asthma studies. PLoS One 9, e85802. 10.1371/journal.pone.0085802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Zhang Y, Zhong B, Wang Y-Y, Diao F-C, Wang R-P, Zhang M, Chen DY, Zhai Z-H, Shu H-B, 2007. RBCK1 negatively regulates tumor necrosis factor- and interleukin-1-triggered NF-kappaB activation by targeting TAB2/3 for degradation. J. Biol. Chem 282, 16776–16782. 10.1074/jbc.M701913200. [DOI] [PubMed] [Google Scholar]
- Tsutsumi K, Nakamura Y, Kitagawa Y, Suzuki Y, Shibagaki Y, Hattori S, Ohta Y, 2020. AGAP1 regulates subcellular localization of FilGAP and control cancer cell invasion. Biochem. Biophys. Res. Commun 522, 676–683. 10.1016/j.bbrc.2019.11.147. [DOI] [PubMed] [Google Scholar]
- van Iterson M, van Zwet EW, BIOS Consortium, Heijmans BT, 2017. Controlling bias and inflation in epigenome- and transcriptome-wide association studies using the empirical null distribution. Genome Biol 18, 19. 10.1186/s13059-016-1131-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijga AH, Kerkhof M, Gehring U, de Jongste JC, Postma DS, Aalberse RC, Wolse APH, Koppelman GH, van Rossem L, Oldenwening M, Brunekreef B, Smit HA, 2014. Cohort profile: the prevention and incidence of asthma and mite allergy (PIAMA) birth cohort. Int J Epidemiol 43, 527–535. 10.1093/ije/dys231. [DOI] [PubMed] [Google Scholar]
- Wu Y-F, Li Z-Y, Dong L-L, Li W-J, Wu Y-P, Wang J, Chen H-P, Liu H-W, Li M, Jin C-L, Huang H-Q, Ying S-M, Li W, Shen H-H, Chen Z-H, 2019. Inactivation of MTOR promotes autophagy-mediated epithelial injury in particulate matter-induced airway inflammation. Autophagy 1–16. 10.1080/15548627.2019.1628536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C-J, Söderhäll C, Bustamante M, Baïz N, Gruzieva O, Gehring U, Mason D, Chatzi L, Basterrechea M, Llop S, Torrent M, Forastiere F, Fantini MP, Carlsen KCL, Haahtela T, Morin A, Kerkhof M, Merid SK, van Rijkom B, Jankipersadsing SA, Bonder MJ, Ballereau S, Vermeulen CJ, Aguirre-Gamboa R, de Jongste JC, Smit HA, Kumar A, Pershagen G, Guerra S, Garcia-Aymerich J, Greco D, Reinius L, McEachan RRC, Azad R, Hovland V, Mowinckel P, Alenius H, Fyhrquist N, Lemonnier N, Pellet J, Auffray C, BIOS Consortium, van der Vlies P, van Diemen CC, Li Y, Wijmenga C, Netea MG, Moffatt MF, Cookson WOCM, Anto JM, Bousquet J, Laatikainen T, Laprise C, Carlsen K-H, Gori D, Porta D, Iñiguez C, Bilbao JR, Kogevinas M, Wright J, Brunekreef B, Kere J, Nawijn MC, Annesi-Maesano I, Sunyer J, Melén E, Koppelman GH, 2018. DNA methylation in childhood asthma: an epigenome-wide meta-analysis. Lancet Respir Med 6, 379–388. 10.1016/S2213-2600(18)30052-3. [DOI] [PubMed] [Google Scholar]
- Yu S, Dai J, Ma M, Xu T, Kong Y, Cui C, Chi Z, Si L, Tang H, Yang L, Sheng X, Guo J, 2019. RBCK1 promotes p53 degradation via ubiquitination in renal cell carcinoma. Cell Death Dis 10, 254. 10.1038/s41419-019-1488-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Hua S, Zhang A, Kong X, Jiang C, Deng D, Wenlong B, 2013. Association between polymorphisms in COMT, PLCH1, and CYP17A1, and non-small-cell lung cancer risk in Chinese nonsmokers. Clin Lung Cancer 14, 45–49. 10.1016/j.cllc.2012.04.004. [DOI] [PubMed] [Google Scholar]
- Zhao R., Guo Z., Zhang R., Deng C., Xu J., Dong W., Hong Z., Yu H., Situ H., Liu C., Zhuang G., 2018. Nasal epithelial barrier disruption by particulate matter 2.5 μm via tight junction protein degradation. J Appl Toxicol 38, 678–687. 10.1002/jat.3573. [DOI] [PubMed] [Google Scholar]
- Zhou W, Tian D, He J, Wang Y, Zhang Lijun, Cui L, Jia L, Zhang Li, Li L, Shu Y, Yu S, Zhao J, Yuan X, Peng S, 2016. Repeated PM2.5 exposure inhibits BEAS-2B cell P53 expression through ROS-Akt-DNMT3B pathway-mediated promoter hypermethylation. Oncotarget 7, 20691–20703. 10.18632/oncotarget.7842. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.