

Abstract
This study aims to provide a detailed analysis of allogeneic transplant (allo-SCT) outcomes in a large T-ALL cohort with a specific emphasis on the effects of pre-transplant minimal residual disease (MRD) and disease subtype, including the aggressive early-thymic precursor (ETP) subtype. Data for 102 allo-SCT patients with a diagnosis of T-ALL from three centers were retrospectively analyzed. Patients were grouped into four T-ALL subtypes: ETP, early, cortical, and mature. At three years, overall survival (OS), progression-free survival (PFS), non-relapse mortality (NRM), and cumulative incidence progression (CIP) were 35%, 33%, 11%, and 55%. Patients transplanted in first complete remission (CR1) had 3-year OS (62%), versus those transplanted in CR2 or greater (24%, HR 1.6, p=0.2). Patients with MRD positivity at the time of transplant had significantly higher rates of progression compared to those with MRD negativity (76% vs 34%, HR 2.8, p=0.006). There was no difference in OS, PFS, or CIP between disease subtypes, including ETP (n=16). ETP patients transplanted in CR1 (n=10) had OS of 47%, comparable to other disease subtypes, suggesting allo-SCT can overcome the poor prognosis associated with ETP. MRD status at transplant was highly predictive of disease relapse, suggesting novel therapies are necessary to improve transplant outcomes.
INTRODUCTION
T-cell acute lymphoblastic leukemia (T-ALL) represents approximately 20–25% of all cases of acute lymphoblastic leukemia (ALL) diagnoses per year.1 Given the rarity of T-ALL, patients are typically treated in a similar fashion to B-cell acute lymphoblastic leukemia (B-ALL) with dose-intense, multi-agent chemotherapy regimens.2–4 While the MRC UKALL XII/ECOG E2993 trial suggested there may be an overall survival (OS) benefit to allo-SCT from a sibling donor in first remission (61% vs 46%, p=0.02); increasingly, allo-SCT in first remission is utilized primarily for high-risk disease subtypes such as early-thymic precursor T-ALL (ETP-ALL) ALL, or minimal residual disease (MRD) positivity after induction.5
There are few transplant-specific studies, with an in-depth evaluation of conditioning and impact of prognostic factors, to guide the transplant management of T-ALL. The largest published report on transplant-specific outcomes in T-ALL is from Saudi Arabia, in which 53 patients received allo-SCT. OS was 43.5%, and patients in first remission (CR1) had improved OS versus those in second remission (CR2) (53.5% vs 31.9%).6 A large study from the EBMT in abstract form suggested patients transplanted in CR1 with TBI-based conditioning had improved outcomes, though data from this study are limited.7 An abstract by Hoelzer et al. suggested a benefit of allo-SCT for patients in CR1 with specific disease subtypes including early (CD1aneg, sCD3−), and mature (CD1aneg,sCD3+) T-ALL, though these results were not statistically validated.8 Additionally, MRD and complete immune-phenotyping data, particularly as it relates to ETP-ALL are not available in these studies. Recently, a report by Dhedin et al. demonstrated that allo-SCT improved relapse-free survival versus chemotherapy alone in patients with ALL who had MRD >10−3 after induction, suggesting MRD positivity should be the primary determining factor for proceeding with allo-SCT in first remission.9
Minimal residual disease (MRD) after therapy is well established as a risk factor for relapse in B-ALL,10, 11 and subsequent studies have demonstrated that the presence of MRD resulted in impaired progression-free survival (PFS) in T-ALL.12, 13 Few studies outside of the pediatric literature have addressed the potential of MRD detection at the time of transplant as a predictor of impaired outcomes in T-ALL. One study with primarily B-ALL, demonstrated a trend towards worse PFS in patients with MRD by flow cytometry at time of transplant, though this was not conclusive due to low patient numbers.14 A second study with 160 patients (only 24 T-ALL) demonstrated increased risk of relapse in patients with MRD positivity prior to myeloablative (MAC) SCT with 3-year relapse-free survival of 61% in MRD-negative versus 34% in MRD positive patients.15
T-ALL subtype has also been associated with chemotherapy outcomes. In the MRC study, CD1a negative T-ALL was associated with higher rates of relapse and death compared to CD1a+ (cortical-type) disease (OS 64% vs 39%), though the effect of other subtypes is unknown. In other studies, the highest-risk subtype identified is the ETP-ALL, which is a precursor T-ALL that expresses myeloid and/or immature markers. This subtype of T-ALL was found to have a 72% risk of relapse when first identified by St. Jude investigators, and similar findings were subsequently confirmed in the UK.16, 17 While recent evidence in children and young adults suggests that ETP may be a more intermediate-risk subtype, particularly when treated with an intensive consolidation/maintenance strategy with nelarabine, a recent analysis from MD Anderson by Jain et al reported poor outcomes for adult patients with ETP, with median survival of only 20 months with chemotherapy alone (Jain et al., under review).13, 18 Whether allo-SCT can overcome the poor risk associated with ETP is unknown. For patients who do relapse with T-ALL, results are poor, with long-term survival of 7% seen in the MRC study, and 11% with the novel agent nelarabine without transplantation.19, 20
Here, we present the results of a multi-center analysis of 102 patients who received allo- SCT for T-ALL. This study aims to provide detailed transplant outcomes in a large T-ALL cohort of patients with a specific emphasis on the effects of pre-transplant MRD, subtype, including ETP, and other prognostic markers on outcomes for patients receiving allo-SCT.
METHODS
Patients
Data was collected from three institutions including: University of Texas MD Anderson Cancer Center (UTMDACC), Oregon Health and Science University (OHSU), and National University Cancer Institute of Singapore (NUH). All patients with a diagnosis of T-ALL who received a first allo-SCT after January 1, 2000 through January 1, 2015 were included in the analysis. Patients with T-cell lymphoblastic lymphoma, defined as primarily nodal involvement with <25% bone marrow involvement, mixed-lineage ALL, bi-phenotypic leukemia, or human T-cell lymphotrophic virus (HTLV) positive adult T-cell leukemia/lymphoma were excluded. Patients who underwent ex vivo T-cell depleted allo-SCT were excluded. This study was approved by the institutional review board at each institution.
T-ALL Subtypes
Flow cytometry immunophenotyping (FCI) data was collected from pathology reports, FCI reports, and primary flow cytometry data and evaluated centrally by JEB and JLJ. Pathology data were reviewed upon diagnosis when available, and upon relapse if no reports were available upon diagnosis. All patients had a diagnosis of T-ALL as outlined by WHO criteria.21 T-ALL subtypes were determined utilizing a modified WHO/EGIL scheme initially developed by Hoelzer et al, into Pre-T, Pro-T, Mature (Medullary), and Cortical (Thymic) subtypes (Table 1).8, 21, 22 For these cases, attempting to apply criteria based on expression patterns of CD4 and CD8 left many cases unclassifiable, and therefore these markers were not used in this classification scheme. Since initial analysis showed that the outcomes of Pro-T and Pre-T were the same, this group was then combined into an ‘Early’ T-ALL subtype for the purpose of further analysis. ETP was classified as: CD1aneg, cytoplasmic CD3+, CD8−, T-ALL, with <75% expression of CD5, and the presence of 1 or more myeloid markers on at least 25% of lymphoblasts including: CD117, CD34, HLA-DR, CD13, CD33, CD11b, and/or CD65.16
Table 1:
Immunologic Classification of T-ALL
| Subtype | CD1a | CD2 | cCD3 | sCD3 | CD5 | CD8 | Myeloid/Immature | CD34 | 3-year OS |
|---|---|---|---|---|---|---|---|---|---|
| ETP | − | +/− | + | − | <75% | − | 25%* | +/− | 29% |
| Pro-T± | − | − | + | − | +/− | +/− | +/− | +/− | 47% |
| Pre-T± | − | + | + | − | +/− | +/− | +/− | +/− | 31% |
| Early | − | +/− | + | − | +/− | +/− | +/− | +/− | 41% |
| Cortical | + | +/− | + | − | +/− | +/− | +/− | +/− | 27% |
| Mature# | − | +/− | + | + | +/− | +/− | +/− | +/− | 31% |
ETP-Early-Thymic Precursor. cCD3: cytoplasmic CD3, sCD3: surface CD3 Note: all subtypes express Variable CD4/CD8 positivity. CD8 positivity was only considered in patients with ETP- ALL.
CD4/CD8 expression was not used for this classification scheme, given attempting to apply this criteria left many cases unclassifiable
Patients with ETP-ALL have 1 or more myeloid markers on at least 25% of lymphoblasts including: CD117, CD34, HLA-DR, CD13, CD33, CD11b, and/or CD65
There was no statistical difference between Pro-T and Pre-T ALL(p=0.57); given these are both similar variants of T-ALL, they were considered as one ‘Early’ group for the purpose of analysis.
Minimal Residual Disease
Minimal residual disease (MRD) was defined as the presence of T-ALL on FCI on bone marrow biopsy within one month prior to allo-SCT. Patients entering transplant in aplasia or with active disease were not considered to have MRD. Given detection of MRD by flow cytometry became routine by 2004, for the purpose of MRD analysis, only patients who received transplantation after January 1, 2004 were considered.
Minimal residual disease was assessed utilizing multi-parameter FCI with a sensitivity of 0.01% at UTMDACC and OHSU, and sensitivity of 0.04% at NUH. Bone marrow aspirate samples of 200,000 nucleated bone marrow cells were analyzed using a panel of markers including: CD1a, CD2, cytoplasmic CD3, surface CD3, CD4, CD5, CD7, CD8, CD10, CD13, CD33, CD34, CD45, CD56, HLA-DR, and terminal deoxynucleotidyl transferase. For most cases, cytoplasmic CD3 staining was used for gating, and surface CD3 was used to identify immature (CD3 negative) cells. Starting in 2012, cases at UTMDACC were analyzed with initial gating on CD7+CD45 dim+ cells. MRD was considered positive if a cluster of at least 20 cells was present on bivariate dot plots, with a significant difference in expression (>/= 3–fold) of 2 or greater antigens, compared to the known phenotype of mature marrow T and natural killer cells was present. MRD was reported as a fraction of total nucleated bone marrow cells. Each center certified their process and level of detection of MRD, with a level detection on the order of 10−3 (0.01%). All centers were academic transplant centers, with frequent utilization of MRD assessments in the transplant and non-transplant setting. When evaluating MRD as a prognostic marker, patients not in CR were omitted from MRD analysis.
Statistical Analysis
The primary objective of the analysis was to compare outcomes according to T-ALL subtype. OS and PFS were estimated using the Kaplan-Meier method. Overall survival was estimated from the date of transplant until death from any cause, and PFS was estimated from the date of transplant until disease progression or death from any cause. The rate of disease progression, non-relapse mortality (NRM), acute GVHD (aGVHD), and chronic GVHD (cGVHD) was estimated using the cumulative incidence (CI) method to account for competing risks. NRM was defined as death before disease progression and in the absence of persistent disease. NRM was considered a competing risk for disease progression; disease progression or death with persistent disease were considered competing risks for NRM, and disease progression or death of any cause before the development of GVHD were considered competing risks for GVHD. Acute GVHD was graded on the Glucksberg scale from grade I-IV, whereas cGVHD was graded as extensive/limited.23, 24 Cox Proportional Hazards regression was used on univariate and multivariate analysis to assess predictors of disease progression. Reference for regression analysis was determined either based upon known prognostic factors, or if unknown, by convention the group with the highest number was used as the reference group. Patients with missing data for a particular risk factor were excluded from the risk factors analysis. Because MRD data became systematically available after 2003, the impact of MRD on disease progression was evaluated in a subset analysis including patients transplanted starting January 2004 (N=84). Statistical significance was defined at the 0.05 level. Statistical analyses were primarily performed using STATA 11.0 (StataCorp. 2009. Stata Statistical Software: Release 11. College Station, TX: StataCorp LP).
RESULTS
Baseline Characteristics
102 patients received first allo-SCT for T-ALL between January 1, 2000 and January 1, 2015. The median age at transplant was 31 years (range 2–72 years), and median follow-up time in alive patients was 2.9 years (range 0.3–11 years). Only 37% of patients were in CR1 at the time of allo-SCT and 14% had high-risk disease/complex karyotype. The majority of patients received myeloablative conditioning (MAC) (77%). The most common MAC regimen was etoposide or cyclophosphamide/12Gy total body irradiation (52%), followed by busulfan /clofarabine +/− Thiotepa (24%), busulfan/melphalan (11%), busulfan/fludarabine +/− clofarabine (6%), busulfan/cyclophosphamide +/− Thiotepa (3%), carmustine/etoposide/cytarabine/melphalan (BEAM)/alemtuzumab (3%), and fludarabine/9.9Gy total body irradiation (1%). There was no statistically significant difference between OS or PFS between patients who received MAC versus RIC/NMA conditioning (p=0.8, 0.9, respectively). Patient characteristics are provided in Table 2, and transplant characteristics are presented in Table 3.
Table 2:
Patient Characteristics (n=102)
| Characteristic | n (%) |
|---|---|
| Institution | |
| MDACC | 72 (71) |
| OHSU | 21 (21) |
| NUH | 9 (9) |
|
| |
| Age | |
| <18 | 11 (11) |
| 18–39 | 63 (62) |
| >40 | 28 (27) |
|
| |
| Sex | |
| Male | 77 (75) |
| Female | 25 (25) |
|
| |
| Subtype | |
| Early* | 28 (27) |
| Mature | 17 (17) |
| Cortical | 27 (26) |
| ETP | 16 (16) |
| Unknown | 14 (14) |
|
| |
| WBC × 10 9 /L (Diagnosis) | |
| >100 | 63 (62) |
| </=100 | 18 (18) |
| Unknown | 21 (21) |
|
| |
| Extra-Medullary CNS Disease at Diagnosis | |
| Yes | 11 (11) |
| No | 90 (88) |
| Unknown | 1 (1) |
|
| |
| Extra-Medullary Disease (Any) at Diagnosis | |
| Yes | 46 (45) |
| No | 56 (55) |
|
| |
| SWOG Cytogenetic Risk at Diagnosis | |
| High/Very High | 14 (14) |
| Intermediate | 66 (65) |
| Unknown | 22 (22) |
|
| |
| Disease Status at Transplant | |
| CR1 | 38 (37) |
| CR2+ | 40 (39) |
| PIF/CR1 | 6 (6) |
| No CR | 18 (18) |
|
| |
| MRD at Transplant (After 2004, n=84) | |
| Yes | 18 (21) |
| No | 47 (55) |
| Unknown | 7 (7) |
| No CR | 12 (14) |
|
| |
| HCT-CI | |
| 0–2 | 60 (59) |
| 3+ | 30 (29) |
| Unknown | 12 (12) |
Abbreviations: ETP: Early-Thymic Precursor; WBC: White Blood Cell; CR: Complete Remission; PIF: Primary Induction Failure
12 patients had pre-T and 14 patients had pro-T, though there was no statistical difference between the two groups (p=0.571), they were classified together as Early T-ALL.
Table 3:
Transplant Characteristics (n=102)
| Characteristic | n (%) |
|---|---|
|
| |
| Stem Cell Source | |
| Peripheral Blood | 60 (59) |
| Bone Marrow | 27 (26) |
| Cord Blood | 15 (15) |
|
| |
| Donor Source | |
| Matched Related | 43 (42) |
| Matched Unrelated | 36 (35) |
| Other Mismatch | 23 (23) |
| Cord Blood | 15 |
| Haploidentical* | 2 |
| 1 Ag Mismatch Sibling | 4 |
| 1 Ag Mismatch Parent | 1 |
| 1 Ag Mismatch MUD | 1 |
|
| |
| Conditioning Regimens | |
| Myeloablative | 79 (77) |
| BEAM/Campath | 2 (2) |
| Bu-Based | 35 (34) |
| TBI-Based | 42 (41) |
| Reduced-Intensity | 16 (16) |
| FluMel +/− Tt | 15 (15) |
| TreoFlu | 1 (1) |
| Non-Myeloablative | 7 (7) |
| FluCyTBI# | 5 (5) |
| BuFluTBI | 2 (2) |
|
| |
| Total Body Irradiation (>2 Gy) | |
| Yes | 42 (41) |
| No | 60 (59) |
|
| |
| GVHD Prophylaxis ± | |
| CNI/Methotrexate | 76 (74) |
| Triple Prophylaxis | 12 (12) |
| Other | 14 (14) |
|
| |
| Anti-Thymocyte Globulin (ATG) | |
| Yes | 24 (24) |
| No | 78 (76) |
Abbreviations; BEAM: Carmustine, etoposide, ARA-C; Bu: Busulfan; TBI:Total Body Irradiation; Flu: Fludarabine; Mel:Melphalan; Tt: Thiotepa; Treo; Treosulfan; Cy: Cytoxan; GVHD: Graft-Versus-Host Disease; CNI: Calcineurin Inhibitor; HCT-CI: Hematopoietic Cell Transplant Co-Morbidity Index
CNI/Methotrexate: Tacrolimus or Cyclosporine; Triple Prophylaxis: CNI, Methotrexate, Prednisone
Haploidentical donors: 1 received Post-Cy GVHD prophylaxis, 1 Received standard MTX/Tacrolimus GVHD prophylaxis
All patients who received FluCyTBI received umibilical cord blood transplant
Survival Outcomes
There was no statistically significant difference in survival outcomes between the three centers, with 3-year PFS of 33% at UTMDACC (used as reference for PFS), 38% at OHSU (p=0.6), and 26% at NUH (p=0.6). Among the entire cohort of 102 patients, the 3-year OS and PFS was 35% and 33%, respectively. The cumulative incidence of NRM was 5% at 100 days, 10% at 1-year, and 11% at three years. For patients transplanted in CR1, the actuarial OS and PFS were 60% and 58%, respectively. Progression was the primary cause of treatment failure, and the CI progression was 44% at 1-year and 55% at 3-years. (Figure 1). The CI grade II-IV and III-IV aGVHD was 22% and 18%, respectively at day 100 and the CI cGVHD was 32% at three years.
Figure 1:
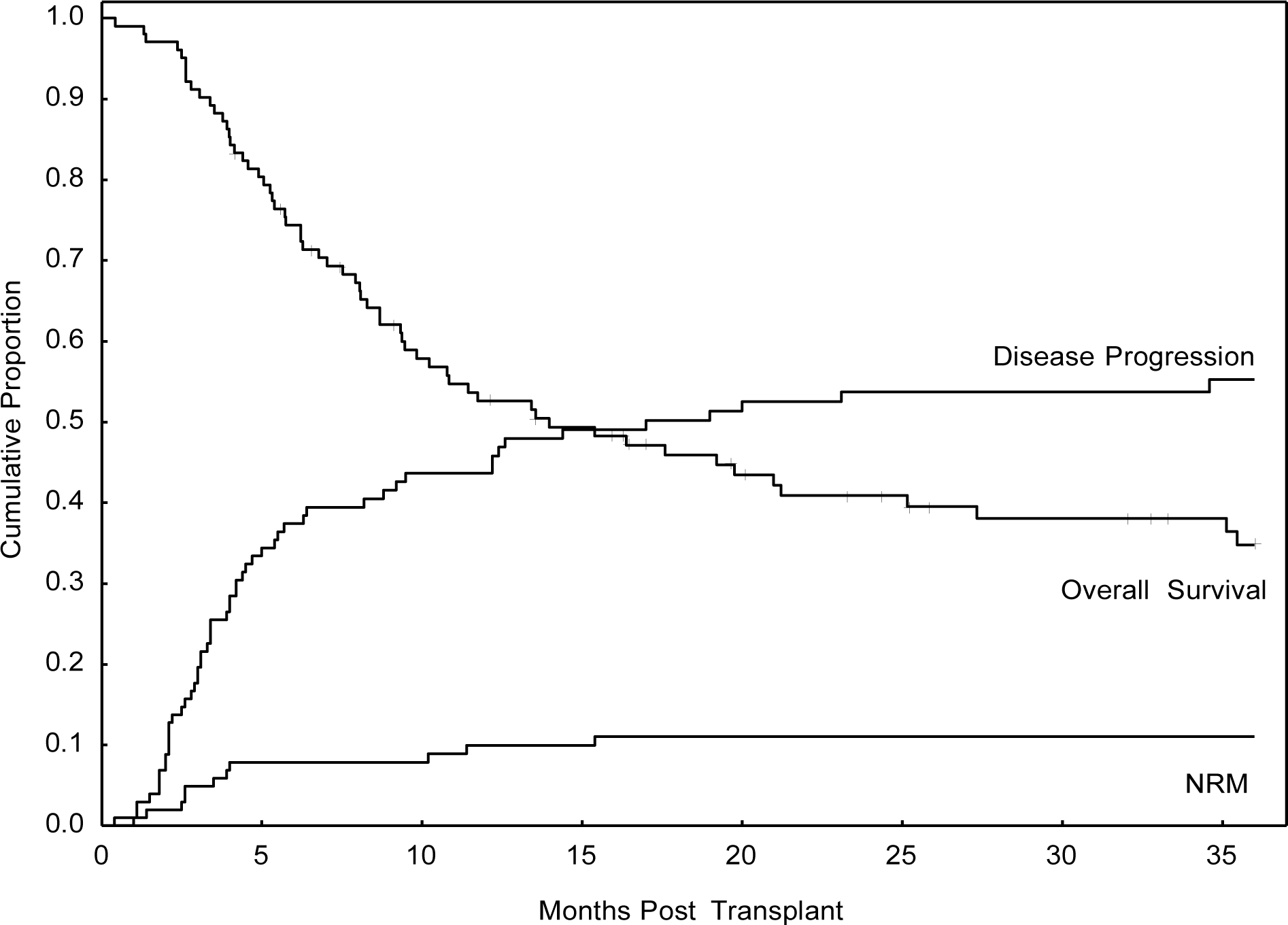
Overall Transplant Outcomes for Entire Cohort (n=102)
T-ALL Subtype Analysis
T-ALL subtype was determined in 88 patients (86%) who had available FCI data. There was no difference in OS between the Pro-T and Pre-T ALL groups (47% vs 31%, p=0.57) so they were considered together as ‘Early T-ALL’. At three years, there was no difference in OS, PFS, or CI Progression according to T-ALL subtypes, including ETP. (Figure 2A, Supplementary Table 1) OS for all patients with ETP was 29%, with a 63% rate of progression at 3 years. However, when patients with ETP underwent allo-SCT in CR1, OS was 47% at three years, which was not statistically significant from all other disease subtypes (63%), p=0.6, (Figure 2B, Supplementary Table 1)
Figure 2A:
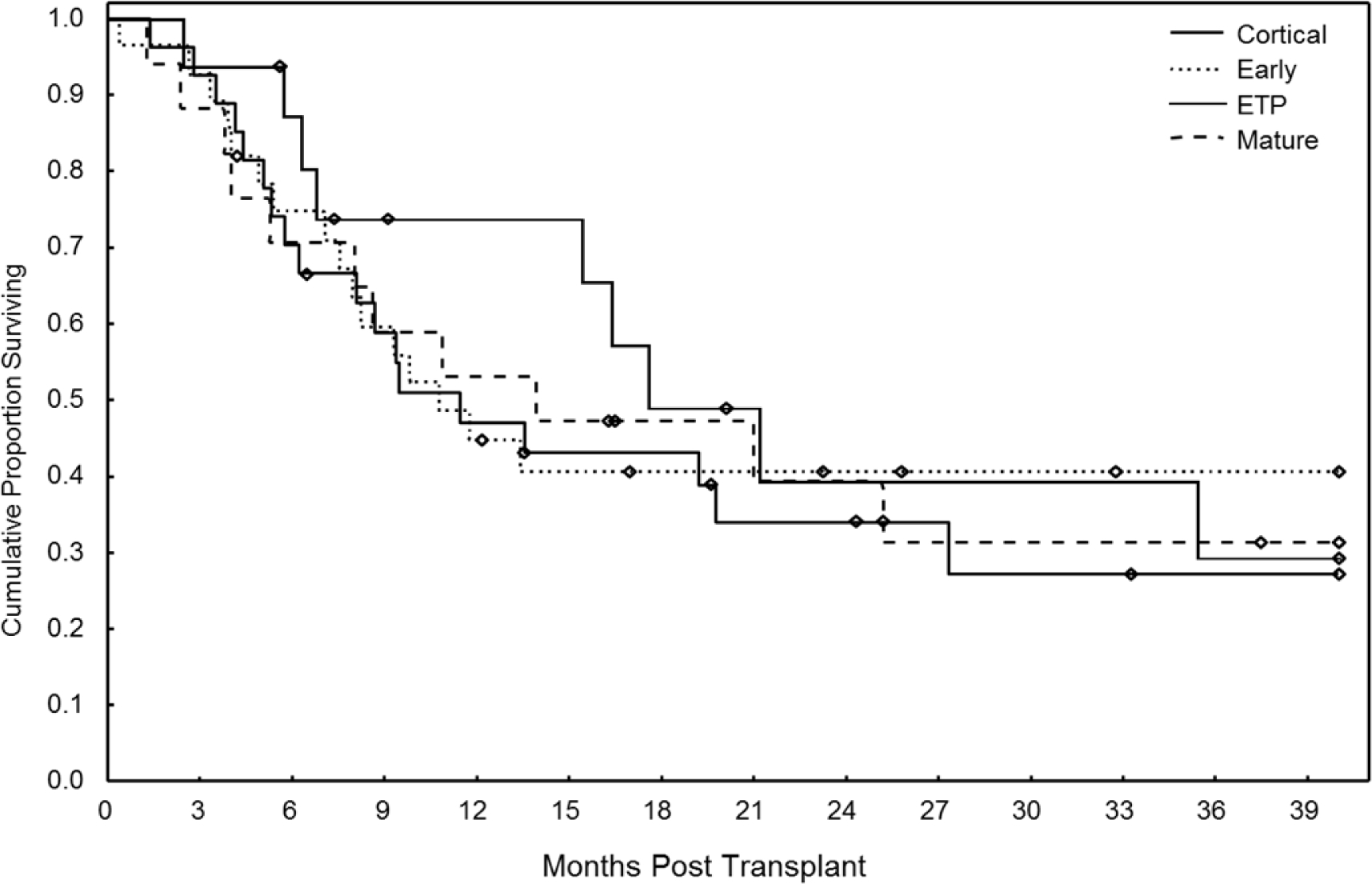
Overall Survival after Transplant According to T-ALL Subtype (n=102)
Figure 2B:
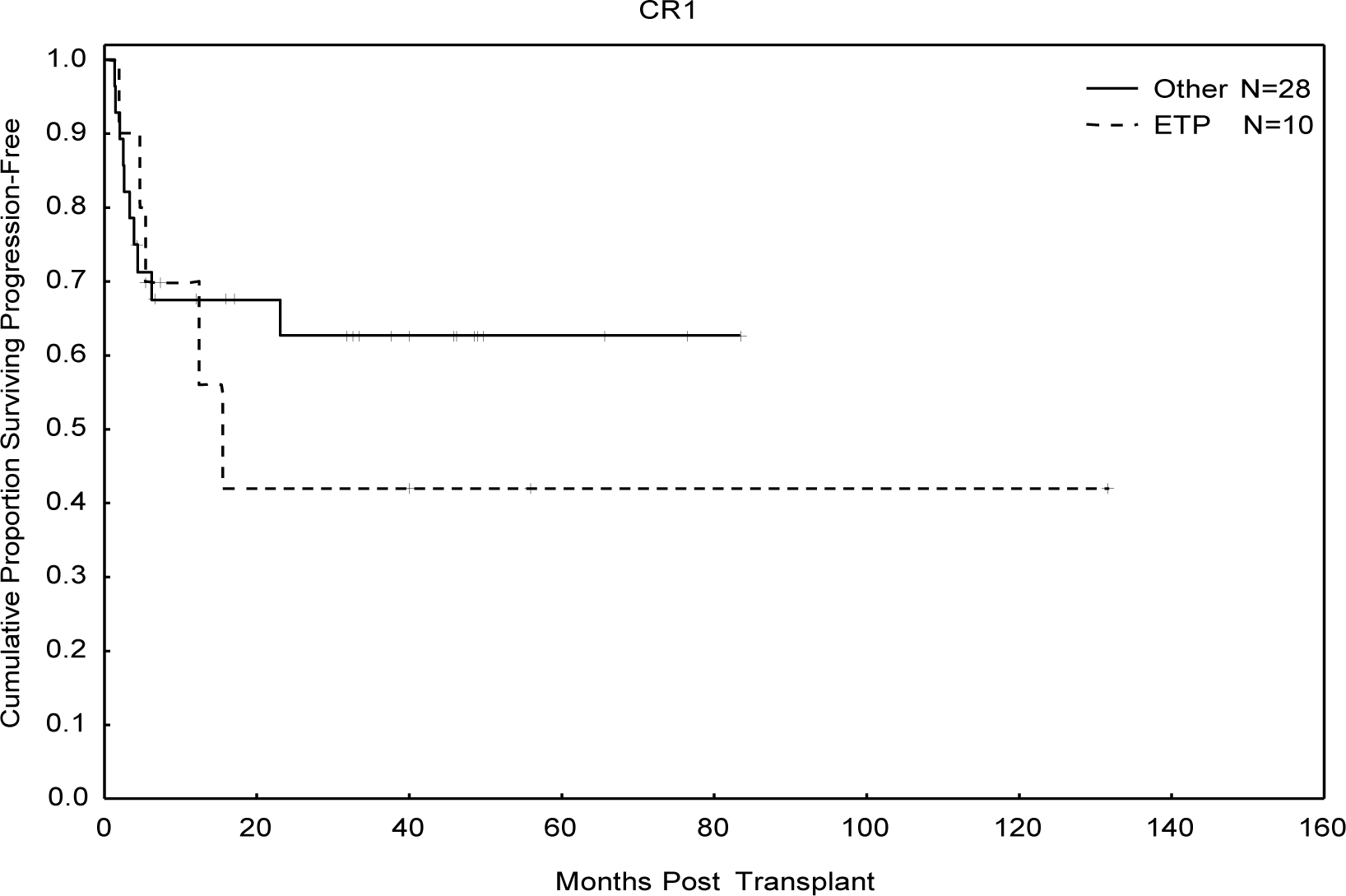
Overall Survival in ETP Patients Receiving Allogeneic Transplant in First Complete Remission
MRD and Univariate Risk Factor Analysis
Given NRM was low, and progression was the primary cause of treatment failure, risk factors for disease progression were evaluated. On univariate analysis for progression, there was no difference in progression rates according to institution, age, HCT-CI, white blood count, presence of extra-medullary disease at diagnosis, presence of CNS involvement at diagnosis, stem cell source, donor type, TBI >2 Gy, conditioning intensity, high risk/complex cytogenetics (SWOG classification scheme), or use of ATG.25 (Table 4) T-ALL subtype, including ETP, was not prognostic for progression, though the mature subtype was borderline significant with a wide confidence interval (HR 2.3 1.01–5.2, p=0.05), even though there was no difference in OS for the mature subset (Table 4, Supplementary Table 1). Patients in CR2+ at transplant trended towards increased risk for progression, while patients with PIF/CR1 and no CR had increased risk of progression. (Table 4). With a median follow-up time of 2 years, the actuarial OS for patients who were in CR1, CR2+, PIF/CRI, and no CR were 60%, 24%, 33%, and 0% respectively. Additionally, MRD positivity at transplant was associated with increased risk of progression. CI progression was 45% at 1-year and 76% at 3-years in patients who were MRD positive at the time of transplant (Figure 3). Given the potential heterogeneity across institutions in terms of monitoring MRD, we did a univariate evaluation of MRD assessment only among MDACC patients. In this analysis, MRD positivity at transplant remained a negative prognostic factor for disease progression (HR 2.3 1–5.3, p=0.049).
Table 4:
Univariate Outcomes for Progression (n=102)
| Risk Factor | n | HR | 95% CI | P |
|---|---|---|---|---|
| Institution | ||||
| MDACC | 72 | Ref. | ||
| OHSU | 21 | 0.8 | 0.4–1.6 | 0.5 |
| Singapore | 9 | 0.6 | 0.3–1.4 | 0.3 |
| MDACC vs other | 1.4 | 0.8–2.5 | 0.3 | |
| ALL type | ||||
| Early | 28 | Ref. | ||
| Mature | 17 | 2.3 | 1.01–5.2 | 0.05 |
| Cortical | 27 | 1.5 | 0.7–3.3 | 0.3 |
| ETP | 16 | 1.2 | 0.5–2.6 | 0.7 |
| Unknown | 14 | Excluded | ||
| Age | ||||
| <18 | 11 | Ref. | ||
| 18–39 | 63 | 1 | 0.4–2.4 | 0.9 |
| >=40 | 28 | 0.9 | 0.3–2.4 | 0.8 |
| Sex | ||||
| Male | 77 | Ref. | ||
| Female | 25 | 0.65 | 0.3–1.2 | 0.2 |
| Disease Status at Transplant | ||||
| CR1 | 38 | Ref. | ||
| CR2 | 40 | 1.8 | 0.8–3.7 | 0.09 |
| PIF/CR1 | 6 | 3.7 | 1.4–9.6 | 0.008 |
| Not CR | 18 | 4.9 | 2.1–11 | <0.001 |
| WBC | ||||
| >100K | 63 | Ref. | ||
| <=100K | 18 | 1.2 | 0.6–2.3 | 0.6 |
| Unknown | 21 | Excluded | ||
| Extra-medullary disease | ||||
| No | 46 | Ref. | ||
| Yes | 56 | 1.7 | 0.98–2.9 | 0.06 |
| CNS disease | ||||
| Yes | 11 | Ref. | ||
| No | 90 | 2.15 | 0.97–4.8 | 0.06 |
| Unknown | 1 | Excluded | ||
| MRD at Transplant after 2004 (N=84) | ||||
| Yes | 18 | 2.6 | 1.3–5.4 | 0.007 |
| No | 46 | Ref | ||
| Unknown | 6 | Excluded | ||
| Stem Cell source | ||||
| PB | 60 | Ref. | ||
| BM | 27 | 0.99 | 0.5–1.9 | 0.97 |
| CBT | 15 | 0.7 | 0.3–1.5 | 0.3 |
| Donor Type | ||||
| MRD | 43 | Ref. | ||
| MUD | 36 | 0.9 | 0.5–1.6 | 0.7 |
| Other | 23 | 0.5 | 0.2–1.1 | 0.08 |
| Prep | ||||
| Ablative | 79 | Ref. | ||
| RIC | 16 | 1.5 | 0.7–3.0 | 0.3 |
| NMA | 7 | 0.8 | 0.2–2.5 | 0.6 |
| ATG | ||||
| Yes | 24 | 0.68 | 0.34–1.36 | 0.28 |
| No | 78 | Ref | ||
| TBI >2 Gy | ||||
| Yes | 42 | 1.02 | 0.6–1.7 | 0.9 |
| No | 60 | Ref. | ||
| HCT-CI | ||||
| 0–2 | 60 | Ref. | ||
| >=3 | 30 | 1.2 | 0.7–2.2 | 0.5 |
| Unknown | 12 | Excluded | ||
| Cytogenetics | ||||
| High / v High | 14 | Ref. | ||
| Intermediate | 66 | 1 | 0.5–2.1 | 0.9 |
| Unknown | 22 | Excluded |
Given MRD measurement for MRD in ALL became standard in 2004, only patients transplanted after 2004 were analyzed in the multivariate analysis for progression (n=84). Disease status and MRD were prognostic factors for progression.
Abbreviations: CR:complete remission; PIF/CR1: patients with primary induction failure who eventually achieved first CR; WBC: White Blood Count; CNS: Central nervous system; MRD: minimal residual disease; PB: peripheral blood; BM: bone marrow; CBT: cord blood transplant; ATG: anti-thymocyte globulin, TBI: total body irradiation, HCT-CI: Hematopoietic cell transplantation-comorbidity index
Figure 3:
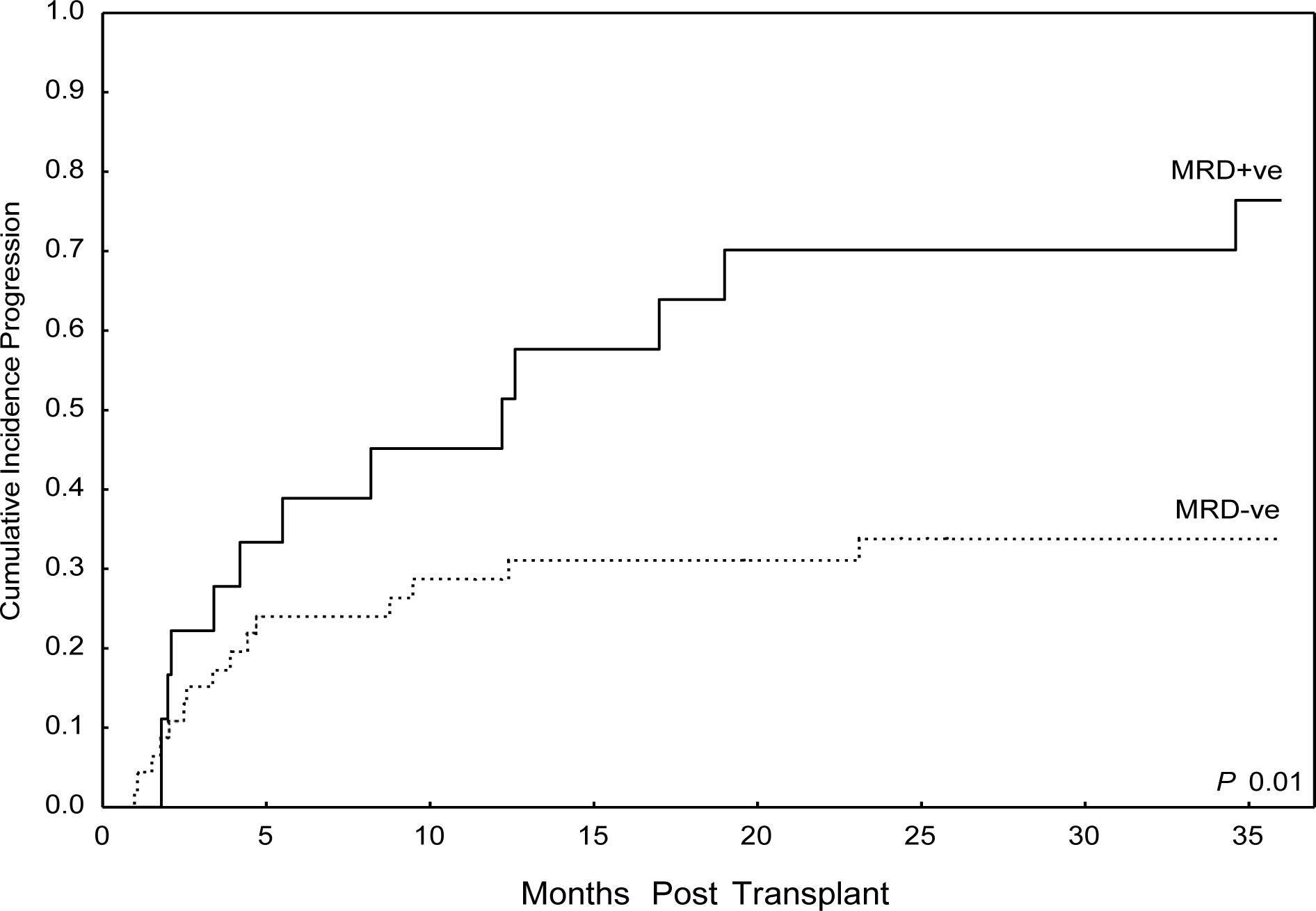
Cumulative Incidence Progression by MRD Status at Time of Transplant
Multivariate Analysis
CR status and MRD status were highly correlated variables, so each variable was evaluated independently. On multivariate analysis only MRD status and disease status (CR1/PIF and No CR), were predictive of increased risk of progression. (Table 5) Even when CR status and MRD status were taken into account, ETP was not a statistically significant predictor for progression in either of two models. (Table 5). CR status and MRD status were highly correlated variables, so each variable was evaluated independently.
Table 5:
Multivariate Analysis for Progression
| Model 1: Disease Status (n=88) | HR | 95% CI | P |
| ETP | 0.98 | 0.6–1.7 | 0.9 |
| CR2+ | 1.6 | 0.8–3.3 | 0.2 |
| PIF/CR1 | 2.9 | 1.1–7.5 | 0.03 |
| No CR | 3.5 | 1.5–7.9 | 0.003 |
| Model 2: MRD Status (n=64) |
HR | 95% CI | P |
| ETP | 0.7 | 0.3–1.6 | 0.4 |
| MRD Positive | 2.8 | 1.3–5.9 | 0.006 |
CR status and MRD status were highly correlated variables, so each variable was evaluated independently on multivariate analysis. 88 patients had known T-ALL subtype data, and were considered in model 1. 64 patients had known MRD data by flow cytometry after 2004, when flow cytometry assessment prior to transplant became routine.
DISCUSSION
Herein we present one of the largest series of T-ALL patients undergoing allo-SCT with detailed analyses of disease status, MRD, histology/immune phenotyping, including ETP-ALL, and transplant preparative regimens.
Our analysis demonstrated no difference in allo-SCT outcomes between the disease subtypes, including ETP. ETP is associated with increased risk of relapse and death, particularly in the adult population when treated with chemotherapy alone, with a median OS of 20 months in the UTMDACC analysis (Jain et al, Under Review). Given many adults may have difficulty tolerating an intensive course of nelarabine, which was demonstrated to overcome the negative prognostic implication of ETP in children and young adults, consolidation with allo-SCT is indicated for this patient population and was the conclusion of that study. In the present analysis, patients who received allo-SCT for ETP in CR1 had OS and PFS of 47% and 42%, respectively, suggesting that allo-SCT can abrogate the negative prognostic impact of ETP. While the numbers of patients with ETP-ALL in CR1 (n=10) are low, given the rarity of ETP-ALL, it is unlikely that large studies in adult patients will be undertaken looking into this question. Therefore, when taken in context with the results reported by Jain et al, our data suggest that strong consideration should be given for allo-SCT in patients with ETP-ALL after attainment of first remission. For patients with non-ETP T-ALL, patients should proceed to allo-SCT based upon other prognostic factors such as MRD at the end of induction therapy, as disease subtype does not appear to effect transplantation outcomes (Figure 2).
The presence of MRD at the end of induction therapy is a clear prognostic marker for increased disease relapse, and is considered a standard indication for allo-SCT, even for patients in CR1.12, 13 Increasing data in adult ALL corroborates the pediatric literature that MRD status immediately prior to allo-SCT predicts for poor transplant outcomes. However, the majority of studies are in patients with B-ALL.14, 15 Here we report a strong correlation between the presence of MRD at the time of transplant, and increased risk of disease progression (78% MRD+ vs 31% MRD−, HR 2.6 (1.3–5.4), p=0.01), in patients with T-ALL. Indeed, patients in CR1 with MRD at the time of transplant had similar outcomes to those transplanted in CR2+ (PFS 33% at 3 years, Supplementary Table 3). This suggests that MRD positivity, regardless of remission status, is predictive of relapse post allo-SCT in T-ALL. However, despite the strong correlation of MRD with risk of disease progression, due to low numbers of MRD-positive patients, we could not definitively determine the effect of remission status on MRD. While some patients will be cured with allo-SCT even with MRD positivity immediately prior to transplant, these data suggest a possible benefit for MRD-positive patients from additional therapy or novel therapies either pre- transplant to induce MRD negativity, or post-transplant in order to prevent disease relapse.
Given the large number of patients available for analysis, we also evaluated the impact of other prognostic factors on transplant outcomes in T-ALL. Conditioning chemotherapy in ALL has been historically with MAC TBI-based regimens (etoposide or Cytoxan), and this remains the standard of care in many institutions. Indeed, a registry analysis from EBMT presented in abstract form suggested TBI may have a protective effect in T-ALL; however, details of this study are not yet available.7 Alternatively, we found no difference in progression rates between TBI-based, or busulfan-based conditioning, either among all patients, or patients with extra- medullary disease, who had a trend toward higher progression on univariate analysis (Table 4). Therefore, we conclude busulfan-based conditioning regimens are an acceptable alternative to TBI-based conditioning in T-ALL. Although the majority of patients (79%) received MAC conditioning, we did not see any difference in outcomes in patients receiving RIC/NMA conditioning, including cord blood transplants. While MAC conditioning is preferred given the aggressive nature of T-ALL, RIC (particularly Flu/Mel based) may be an acceptable alternative for those who cannot tolerate MAC conditioning. A report from the EBMT has also suggested acceptable results utilizing RIC versus MAC in T-ALL.26
Remission status was a clear prognostic indicator for progression and treatment failure. Patients with delayed remission (PIF/CR1), and particularly patients transplanted with active disease, had poor outcomes. All patients in the PIF/CR1 group had MRD at the time of transplantation, which was likely the primary driver of poor outcomes, though this analysis is limited to low numbers. For this group of patients, post-transplant therapy, such as maintenance therapy may help to improve outcomes. Novel agents and approaches are necessary for those patients not in remission prior to allo-SCT given poor long-term outcomes in this group of patients. There was a trend (HR 1.6, p=0.2) for increased risk of progression for those transplanted in CR2+ vs CR1, though this did not reach statistical significance. Our data suggest that patients in CR1 with MRD or ETP-ALL should proceed with allo-SCT. In keeping with this recommendation, improved outcomes were noted in a recent paper by Dhedin et al for patients transplanted in CR1 who were MRD positive.9
Our study has several limitations. It is a retrospective analysis, and although FCI data was reviewed centrally, primary flow cytometry data was not available for all patients. Additionally, the relative numbers of patients in each disease subtype, with and without MRD, or other risk factors is relatively small, thus broad conclusions cannot be made. Despite similar procedures of detecting MRD across institutions, MRD detection is not standardized across institutions, which could confound the data. Data on recently described genetic mutations, such as NOTCH1/FBXW7 which has been associated with favorable outcomes in T-ALL, is unknown as these data was not routinely collected during the study period.27, 28 Furthermore, data on pre- transplant chemotherapy regimens, information regarding time from diagnosis to transplant, and the number of patients with similar diagnoses treated across the institutions were unavailable. Furthermore, there was significant heterogeneity of conditioning regimens. Nevertheless, the correlation of MRD with progression was strong, and is in keeping with prior studies in the non- transplant setting. Additionally, the lack of impact of disease subtype on transplant outcomes is consistent with the data by Jain et al, with the exception that ETP outcomes are better with allo- SCT. Finally, despite the limitations posed by the retrospective nature of this analysis, we present the largest, most detailed analysis of T-ALL allo-SCT outcomes to date which may inform future T-ALL studies in allo-SCT, and help guide treatment decisions for the rare ETP subtype. Ultimately, a large registry analysis will be needed to corroborate these findings.
Conclusions:
Here we present the largest and most detailed analysis of allo-SCT outcomes in T-ALL to date. Patients with ETP had long-term survival equivalent to other disease subtypes, suggesting allo-SCT can overcome the poor prognosis associated with ETP, particularly when performed in first remission. There was no difference in outcomes between patients treated with TBI-based versus busulfan-based conditioning, suggesting TBI may not be necessary to achieve cure in T- ALL. MRD status at transplant was highly predictive of disease relapse, suggesting novel therapies before or post-SCT are necessary to improve transplant outcomes in this subset of patients.
Supplementary Material
Acknowledgements:
The authors would like to acknowledge William Dibb for his contributions to data collection and Guang Fan for her contributions regarding pathology data from OHSU.
Sources of Funding:
The authors have no source of income for this study to declare
Footnotes
Conflict of Interest:
The authors declare no conflict of interest with this report.
Supplementary Information:
Supplementary information is available at the Leukemia journal website.
References
- 1.Han X, Kilfoy B, Zheng T, Holford TR, Zhu C, Zhu Y et al. Lymphoma survival patterns by WHO subtype in the United States, 1973–2003. Cancer causes & control : CCC 2008; 19(8): 841–858. doi: 10.1007/s10552-008-9147-4 [DOI] [PubMed] [Google Scholar]
- 2.Kantarjian HM, O’Brien S, Smith TL, Cortes J, Giles FJ, Beran M et al. Results of treatment with hyper-CVAD, a dose-intensive regimen, in adult acute lymphocytic leukemia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2000; 18(3): 547–561. [DOI] [PubMed] [Google Scholar]
- 3.Rytting ME, Thomas DA, O’Brien SM, Ravandi-Kashani F, Jabbour EJ, Franklin AR et al. Augmented Berlin-Frankfurt-Munster therapy in adolescents and young adults (AYAs) with acute lymphoblastic leukemia (ALL). Cancer 2014; 120(23): 3660–3668. doi: 10.1002/cncr.28930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang JE, Medlin SC, Kahl BS, Longo WL, Williams EC, Lionberger J et al. Augmented and standard Berlin-Frankfurt-Munster chemotherapy for treatment of adult acute lymphoblastic leukemia. Leukemia & lymphoma 2008; 49(12): 2298–2307. doi: 10.1080/10428190802517732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marks DI, Paietta EM, Moorman AV, Richards SM, Buck G, DeWald G et al. T-cell acute lymphoblastic leukemia in adults: clinical features, immunophenotype, cytogenetics, and outcome from the large randomized prospective trial (UKALL XII/ECOG 2993). Blood 2009; 114(25): 5136–5145. doi: 10.1182/blood-2009-08-231217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bakr M, Rasheed W, Mohamed SY, Al-Mohareb F, Chaudhri N, Al-Sharif F et al. Allogeneic hematopoietic stem cell transplantation in adolescent and adult patients with high-risk T cell acute lymphoblastic leukemia. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 2012; 18(12): 1897–1904. doi: 10.1016/j.bbmt.2012.07.011 [DOI] [PubMed] [Google Scholar]
- 7.Cahu X LM, Giebel S, Socie G, et al. Myeloablative Allogeneic Hematopoietic Stem Cell Transplantation for Adult Patients with T-Cell Acute Lymphoblastic Leukemia: A Survey From the Acute Leukemia Working Party of the EUropean Group for Blood and Marrow Transplantation (EBMT). Blood 2012: Abstract 356 2012. [Google Scholar]
- 8.Hoelzer D TE, Arnold R, Beck Joachim, Beelen D, et al. Successful Subtype Oriented Treatment Strategies in Adult T-ALL; Results of 744 Patients Treated in Three Consecutive GMALL studies. Blood 2009: Abstract 324 2009. [Google Scholar]
- 9.Dhedin N, Huynh A, Maury S, Tabrizi R, Beldjord K, Asnafi V et al. Role of allogeneic stem cell transplantation in adult patients with Ph-negative acute lymphoblastic leukemia. Blood 2015; 125(16): 2486–2496; quiz 2586. doi: 10.1182/blood-2014-09-599894 [DOI] [PubMed] [Google Scholar]
- 10.Bruggemann M, Raff T, Flohr T, Gokbuget N, Nakao M, Droese J et al. Clinical significance of minimal residual disease quantification in adult patients with standard-risk acute lymphoblastic leukemia. Blood 2006; 107(3): 1116–1123. doi: 10.1182/blood-2005-07-2708 [DOI] [PubMed] [Google Scholar]
- 11.Raff T, Gokbuget N, Luschen S, Reutzel R, Ritgen M, Irmer S et al. Molecular relapse in adult standard-risk ALL patients detected by prospective MRD monitoring during and after maintenance treatment: data from the GMALL 06/99 and 07/03 trials. Blood 2007; 109(3): 910–915. doi: 10.1182/blood-2006-07-037093 [DOI] [PubMed] [Google Scholar]
- 12.Willemse MJ, Seriu T, Hettinger K, d’Aniello E, Hop WC, Panzer-Grumayer ER et al. Detection of minimal residual disease identifies differences in treatment response between T-ALL and precursor B-ALL. Blood 2002; 99(12): 4386–4393. [DOI] [PubMed] [Google Scholar]
- 13.Wood BL WSS, Dunsmore KP, Devidas M, et al. T-Lymphoblastic Leukemia (T-ALL) Shows Excellent Outcome, Lack of Significance of Early Thymic Precursor (ETP) Immunophenotype, and Validation of the Prognostic Value of End-Induction Minimal Residual Disease (MRD) in Children’s Oncology Group (COG) Study AALL0434. Blood 2014;124(21) Abstract 1 2014. [Google Scholar]
- 14.Zhou Y, Slack R, Jorgensen JL, Wang SA, Rondon G, de Lima M et al. The effect of peritransplant minimal residual disease in adults with acute lymphoblastic leukemia undergoing allogeneic hematopoietic stem cell transplantation. Clinical lymphoma, myeloma & leukemia 2014; 14(4): 319–326. doi: 10.1016/j.clml.2014.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bar M, Wood BL, Radich JP, Doney KC, Woolfrey AE, Delaney C et al. Impact of minimal residual disease, detected by flow cytometry, on outcome of myeloablative hematopoietic cell transplantation for acute lymphoblastic leukemia. Leukemia research and treatment 2014; 2014: 421723. doi: 10.1155/2014/421723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coustan-Smith E, Mullighan CG, Onciu M, Behm FG, Raimondi SC, Pei D et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. The Lancet. Oncology 2009; 10(2): 147–156. doi: 10.1016/S1470-2045(08)70314-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Allen A, Sireci A, Colovai A, Pinkney K, Sulis M, Bhagat G et al. Early T-cell precursor leukemia/lymphoma in adults and children. Leukemia research 2013; 37(9): 1027–1034. doi: 10.1016/j.leukres.2013.06.010 [DOI] [PubMed] [Google Scholar]
- 18.Patrick K, Wade R, Goulden N, Mitchell C, Moorman AV, Rowntree C et al. Outcome for children and young people with Early T-cell precursor acute lymphoblastic leukaemia treated on a contemporary protocol, UKALL 2003. British journal of haematology 2014; 166(3): 421–424. doi: 10.1111/bjh.12882 [DOI] [PubMed] [Google Scholar]
- 19.Gokbuget N, Basara N, Baurmann H, Beck J, Bruggemann M, Diedrich H et al. High single-drug activity of nelarabine in relapsed T-lymphoblastic leukemia/lymphoma offers curative option with subsequent stem cell transplantation. Blood 2011; 118(13): 3504–3511. doi: 10.1182/blood-2011-01-329441 [DOI] [PubMed] [Google Scholar]
- 20.Fielding AK, Richards SM, Chopra R, Lazarus HM, Litzow MR, Buck G et al. Outcome of 609 adults after relapse of acute lymphoblastic leukemia (ALL); an MRC UKALL12/ECOG 2993 study. Blood 2007; 109(3): 944–950. doi: 10.1182/blood-2006-05-018192 [DOI] [PubMed] [Google Scholar]
- 21.Borowitz MJ CJ. T-Lymphoblastic Leukaemia/Lymphoma. In: Swerdlow SH, Campo E, Harris NL, et al. , eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues: World Health Organization Classification of Tumors., International Agency for Research on Cancer (IARC): Lyon, 2008. [Google Scholar]
- 22.Bene MC, Castoldi G, Knapp W, Ludwig WD, Matutes E, Orfao A et al. Proposals for the immunological classification of acute leukemias. European Group for the Immunological Characterization of Leukemias (EGIL). Leukemia 1995; 9(10): 1783–1786. [PubMed] [Google Scholar]
- 23.Glucksberg H, Storb R, Fefer A, Buckner CD, Neiman PE, Clift RA et al. Clinical manifestations of graft-versus-host disease in human recipients of marrow from HL-A-matched sibling donors. Transplantation 1974; 18(4): 295–304. [DOI] [PubMed] [Google Scholar]
- 24.Shulman HM, Sullivan KM, Weiden PL, McDonald GB, Striker GE, Sale GE et al. Chronic graft-versus-host syndrome in man. A long-term clinicopathologic study of 20 Seattle patients. Am J Med 1980; 69(2): 204–217. [DOI] [PubMed] [Google Scholar]
- 25.Pullarkat V, Slovak ML, Kopecky KJ, Forman SJ, Appelbaum FR. Impact of cytogenetics on the outcome of adult acute lymphoblastic leukemia: results of Southwest Oncology Group 9400 study. Blood 2008; 111(5): 2563–2572. doi: 10.1182/blood-2007-10-116186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mohty M, Labopin M, Volin L, Gratwohl A, Socie G, Esteve J et al. Reduced-intensity versus conventional myeloablative conditioning allogeneic stem cell transplantation for patients with acute lymphoblastic leukemia: a retrospective study from the European Group for Blood and Marrow Transplantation. Blood 2010; 116(22): 4439–4443. doi: 10.1182/blood-2010-02-266551 [DOI] [PubMed] [Google Scholar]
- 27.Mansour MR, Sulis ML, Duke V, Foroni L, Jenkinson S, Koo K et al. Prognostic implications of NOTCH1 and FBXW7 mutations in adults with T-cell acute lymphoblastic leukemia treated on the MRC UKALLXII/ECOG E2993 protocol. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2009; 27(26): 4352–4356. doi: 10.1200/JCO.2009.22.0996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Asnafi V, Buzyn A, Le Noir S, Baleydier F, Simon A, Beldjord K et al. NOTCH1/FBXW7 mutation identifies a large subgroup with favorable outcome in adult T-cell acute lymphoblastic leukemia (T-ALL): a Group for Research on Adult Acute Lymphoblastic Leukemia (GRAALL) study. Blood 2009; 113(17): 3918–3924. doi: 10.1182/blood-2008-10-184069 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.