

Abstract
Chronic inflammation is often associated with the development of tissue fibrosis, but how mesenchymal cell responses dictate pathological fibrosis versus resolution and healing remains unclear. Defining stromal heterogeneity and identifying molecular circuits driving extracellular matrix deposition and remodeling stands to illuminate the relationship between inflammation, fibrosis, and healing. We performed single-cell RNA-sequencing of colon-derived stromal cells and identified distinct classes of fibroblasts with gene signatures that are differentially regulated by chronic inflammation, including IL-11–producing inflammatory fibroblasts. We further identify a transcriptional program associated with trans-differentiation of mucosa-associated fibroblasts and define a functional gene signature associated with matrix deposition and remodeling in the inflamed colon. Our analysis supports a critical role for the metalloprotease Adamdec1 at the interface between tissue remodeling and healing during colitis, demonstrating its requirement for colon epithelial integrity. These findings provide mechanistic insight into how inflammation perturbs stromal cell behaviors to drive fibroblastic responses controlling mucosal matrix remodeling and healing.
Introduction
Fibroblasts are essential components of parenchymal tissues, providing the framework that is necessary for tissue structure. However, emerging evidence has revealed critical functions for fibroblast cells that extend beyond their traditional roles as structural scaffolds, including roles in regulating cell survival, differentiation, and migration [1–3].
This concept is exemplified in the gut, where fibroblasts have been shown to support mucosal crypt architecture, extracellular matrix (ECM) remodeling, and immune fitness [4–6]. By secreting factors like Wnt ligands and bone morphogenetic protein (BMP) antagonists, fibroblasts are critical in supporting colon crypt architecture, creating discrete anatomical zones that maintain the epithelial stem cell niche in defined areas, while supporting epithelial cell differentiation and inhibition of cell proliferation in others [2,6,7]. This functional compartmentalization is also reinforced through matrix-dependent signaling cues to neighboring cells that collectively contribute to crypt architecture [8,9].
The gastrointestinal tract represents a potential vantage point to study fibroblast-imposed immunoregulation, as it constitutes the largest reservoir of immune cells within the human body, ensuring protection from pathogenic infections while promoting mucosal tolerance against commensal microbes. Recent studies have provided foundational insights into how fibroblasts mediate immune activation and inflammation, thus expanding their roles in tissue homeostasis [10,11]. Intestinal stromal cells secrete the CCL19, CCL21, and CXCL13 chemokines to promote isolated lymphoid follicle formation and B cell recruitment [12,13]. Additionally, fibroblasts secrete proinflammatory cytokines within colon tissues from Crohn’s disease patients, establishing that these cells are important contributors of inflammation in the gut [14]. However, how these diverse functions are regulated by intestinal fibroblasts is incompletely understood. In particular, it remains unclear how mesenchymal cells in the gut may imprint on the ensuing inflammatory response, while also driving excessive production of ECM components that ultimately lead to fibrosis, a hallmark of chronic inflammation in mucosal tissue and a prominent cause of morbidity in diseases like inflammatory bowel disease (IBD) [15]. It is thus not known how inflammation in pathologies like IBD results in the progressive accumulation of ECM that compromises normal intestinal functions [15].
The coexistence of distinct subsets of fibroblasts is thought to account for their pleiotropic properties in supporting gut homeostasis and disease pathogenesis. Uncovering the heterogeneity of fibroblasts, however, has been hindered by a dearth of molecular tools available for experimental assessment, including molecular markers and transgenic mouse models. Recent advances in technologies such as single-cell RNA-sequencing (scRNA-seq) now permit the survey of stromal cell heterogeneity across organs [16–21].
Here, we employed orthogonal technologies, including next generation sequencing of mucosa-associated stroma to survey cellular heterogeneity in the colon. We identified phenotypic and functionally divergent fibroblast populations in mucosal tissues of the gastrointestinal tract and uncovered molecular circuitries governing inflammation and ECM remodeling during colitis, identifying a key role for Adamdec1 in mucosal matrix remodeling and healing.
Results
A cellular census of stromal cells in healthy and inflamed mucosal tissues
To better understand the cellular and molecular circuitries operating during inflammation, ECM remodeling, and wound healing in the intestine, we implemented a murine model of colonic inflammation based on oral administration of multiple cycles of low-dose dextran sulfate sodium (DSS) to induce epithelial injury and ECM deposition [22,23]. Mice subjected to 3 repetitive cycles of DSS displayed progressive accumulation of immune cell infiltrates associated with excessive deposition of collagen fibers (Fig 1A, S1A and S1B Fig). Strikingly, ECM deposition was also documented using high-resolution tissue scanning confocal microscopy (Fig 1B), showing alterations in the structural network of reticular fibers, as stained with ER-TR7. ECM protein deposition was similarly increased in mucosal and submucosal tissues of DSS-treated mice, together with muscularis thickening, therefore supporting the notion that chronic DSS treatment can be used to model fibroblast-mediated tissue remodeling in mice.
Fig 1. A cellular census of stromal cells in healthy and inflamed mucosal tissues.
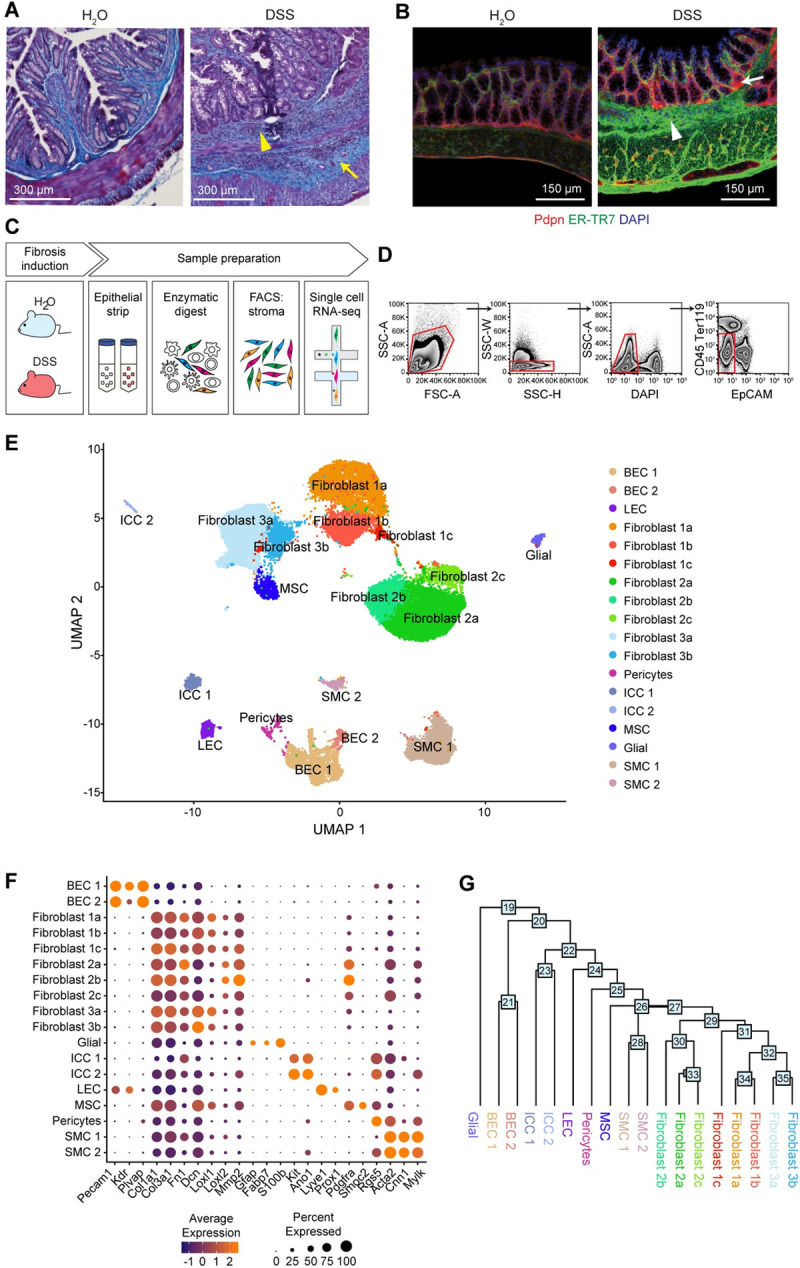
(A) Masson’s trichrome staining of colon from water- and chronic DSS (3 rounds)-fed mice. Collagen accumulation in blue, as demarcated by yellow arrows. Leukocyte infiltrates, as demarcated by yellow arrowheads. n = 2, Scale bar, 300 μm. representative of 2 experiments. (B) IF staining of colon from water- and chronic DSS-fed mice. DAPI (blue), Pdpn (red), ER-TR7 (green). ECM deposition, as demarcated by white arrowhead. Pdpn+ cell expansion, as demarcated by white arrow. Scale bar, 150 μm. n = 3. (C) Workflow depicts colon processing, epithelial strip, mechanical and enzymatic digestion dissociation, and sorting to enrich for stromal cells. (D) Gating strategy for FACS to enrich for colon stromal cells (DAPI− CD45− Ter119− EpCAM−) prior to performing single-cell transcriptomics. (E) Single-cell atlas of the murine colonic stroma. UMAP of approximately 34,000 single-cell (dots) profiles colored by cell type assignment. (F) Expression of common stromal marker genes across cell type subsets. Color represents average expression of marker gene within clusters; diameter represents percentage expression of marker gene within cluster. (G) A dendrogram of cell subset relationships based on the single-cell transcriptomic data. Numbers at nodes represent score of how closely related the clusters are, with the higher the number indicating transcriptional similarity. DSS, dextran sulfate sodium; ECM, extracellular matrix; IF, immunofluorescence; UMAP, uniform manifold approximation and projection.
We then employed scRNA-seq to survey stromal cell heterogeneity in response to chronic inflammation in the colon in this model (Fig 1C). We collected colons from water- and DSS-treated mice, prepared single-cell suspensions from the lamina propria by adapting a protocol that we have previously optimized to extract stromal cells from various organs (Fig 1C) [24,25], enriched for stromal cells by FACS using antibodies to exclude hematopoietic cells (CD45), epithelial cells (EpCAM), and erythrocytes (Ter119), and profiled the cells by droplet-based scRNA-seq (Fig 1D, Methods).
We identified 18 cell subsets by dimensionality reduction, alignment, and clustering of batch-corrected expression profiles of approximately 34,000 cells (22,949 from water-treated samples and 11,248 from DSS-treated samples) (Fig 1E, Methods). Following integrated analysis and clustering of cells from both water- and DSS-treated samples, we confirmed that all samples contributed to each cluster, suggesting that clustering was driven by subset-specific rather than treatment-related features (S1C Fig). We annotated the clusters post hoc by a combination of canonical lineage marker genes to identify blood endothelial cells (BECs), fibroblasts, lymphatic endothelial cells (LECs), smooth muscle cells (SMCs), pericytes, and interstitial cells of Cajal (ICCs) (Fig 1F, S1D Fig) [26–33].
Fibroblasts exhibited high expression of fibrillar collagen types I and III (Col1a1 and Col3a1) as well as the glycoprotein fibronectin (Fn1), and the proteoglycan decorin (Dcn) [6], but could be further distinguished into 3 major lineages (1, 2, and 3), each further partitioned into subsets (Fig 1E and 1G), which we labeled 1a-c, 2a-c, and 3a-b (Fig 1E). Additional stromal components, such as BECs, could be clearly identified based on expression of Pecam1 and Plvap, whereas LECs expressed Lyve1 and Prox1 and pericytes were distinguished by expression of Rgs5 (Fig 1E). We also identified a rare subset of cells resembling mesenchymal stem cells (MSCs) that expressed Ptgs2 and that were previously shown to establish a tumor-promoting niche through provision of PGE2 and induction of Yap [34]. Taken together, we note that the cellular composition of colonic stroma was remarkably heterogeneous before and after induction of the chronic DSS model. While the spectrum of stromal cell types was similar comparing chronic DSS to previous reports of acute DSS [16,35], the chronic model was associated with more extensive pathology related to aberrant ECM structure and accumulation. Thus, the chronic DSS model afforded an opportunity to identify cell type–specific transcriptional programs in the entire stromal compartment, including fibroblasts and endothelial cells.
Intestinal inflammation elicits a coordinated transcriptional response in the vascular endothelium
Among stromal responses in the setting of colitis, much emphasis has been placed on the activation of endothelial cells and the increased angiogenesis and lymphangiogenesis characteristic of dysregulated wound healing processes [36–39]. Supporting a critical role for endothelial cell dynamics during intestinal inflammation, therapeutic antibodies targeting endothelial-mediated intestinal T cell infiltration (natalizumab and vedolizumab) are efficacious in patients with Crohn’s disease and ulcerative colitis [40,41]. The clinical benefit of these therapies, however, is restricted to a subset of patients, highlighting the need to fully uncover the extent of endothelial cell activation and the implications for immune cell responses in the inflamed gut.
To further characterize the transcriptional diversity of endothelial cells in the colon with respect to their roles during colitis, we reanalyzed the subset of annotated endothelial cells (based on the expression of Pecam1, Plvap, and Lyve1), identifying 8 subclusters with distinct expression profiles (Fig 2A, S2A Fig). LEC and BEC subsets (artery, arteriole, capillary, venule, and vein cells) were identified by canonical markers (Fig 2B, S2B Fig) [42–45]. Arterial and venous endothelial cells were characterized by expression of Efnb2 and Ephb4 markers that are necessary for appropriate vessel development by regulating endothelial cell adhesion, migration, and sprouting angiogenesis (S2C Fig) [46]. Additionally, arterial endothelial cells were enriched for Notch4, the Notch ligand Jag1, and downstream Notch effector Hey1. Similarly, arteriole endothelial cells were enriched for Notch3, in concordance with the requirement for Notch signaling in vascular remodeling and development [47].
Fig 2. Intestinal inflammation elicits a coordinated transcriptional response in the vascular endothelium.

(A) Single-cell atlas of colon endothelial cells. UMAP of endothelial cell (dots) profiles colored by cell type assignment. (B) Expression of canonical markers across endothelial cell clusters. Color represents average expression of marker gene within clusters; diameter represents percentage expression of marker gene within cluster. (C) Volcano plots depicting DEGs between water- and DSS-treated samples for select endothelial cell clusters. Each dot represents an individual gene, and dot colors represent statistical significance. Gray, not differentially expressed; orange, differentially expressed but gene name is not annotated; blue, differentially expressed and gene name is annotated. Significant DEGs had FDR <0.05 using MAST (see Methods). (D-F) Violin plots of expression levels of select novel (D), chemokine (E), and adhesion (F) transcripts across endothelial cell clusters in water- and DSS-treated samples. Normalized gene expression levels are plotted on the y-axis. Significant DEGs had FDR <0.05, and respective adjusted p-values derived using MAST (see Methods); *p < 0.05, **p < 0.001, ***p < 1E-10. DEG, differentially expressed gene; DSS, dextran sulfate sodium; FDR, false discovery rate; LEC, lymphatic endothelial cell; UMAP, uniform manifold approximation and projection.
To determine how each subset may individually contribute to inflammation, we calculated cell frequencies and identified differentially expressed genes (DEGs) between baseline and DSS within the cells in each cluster (Fig 2C–2F, S2B Fig, S1 Table). In line with the role of endothelial cells as integral components of the intestinal architecture, we detected an enrichment for gene annotations related to “extracellular structure organization” concomitant with the up-regulation of many collagen genes in endothelial cells from inflamed colons (Fig 2C, S2D Fig, S1 Table), likely reflecting a function for endothelial cells in the remodeling of vessel basement membrane during inflammation [48]. Other ECM-related genes were also induced in endothelial cells after DSS treatment (Fig 2D, S1 Table), including Sparc, which encodes a matricellular binding protein that facilitates cell–matrix interactions and is expressed at high levels during tissue remodeling in many conditions [49–51]. Importantly, we also detected a striking up-regulation of Aqp1 in 2 of the endothelial cell subsets (Fig 2D, S1 Table), suggesting that these subsets may contribute to the regulation of fluid uptake during chronic inflammation.
In endothelial cells in DSS, enriched genes were associated with “response to interferon gamma” and “cytokine mediated signaling pathway” (S2D Fig), suggesting general activation of the endothelial compartment and their overall contribution to inflammation. Accordingly, DSS treatment impacted most endothelial clusters, displaying significant up-regulation of chemokines, such as CXCL9 and CXCL10 (Fig 2C and 2E, S1 Table), implicating these genes in recruitment of immune cells [52]. Genes encoding for adhesion molecules associated with immune cell recruitment and extravasation during inflammation were also selectively up-regulated in subsets of endothelial cells (Fig 2F). MadCAM1, in particular, was only expressed in venous endothelial cells, in line with its putative function in directing leukocyte migration through high endothelial venules (HEVs) by engaging integrin α4β7 on T cells homing to intestinal tissues [53]. As both natalizumab and vedolizumab target this interaction, these data support the concept that a deeper understanding of stromal heterogeneity can facilitate the development of targeted therapies.
Functional and spatial heterogeneity of colonic fibroblasts
Just as endothelial cells play key roles in intestinal homeostasis and inflammation, a diversity of specialized stromal cells of mesenchymal origin also execute important functions in maintaining organ integrity and function. In particular, intestinal fibroblasts regulate structural fitness and repair [2,6] but have also been associated with development of fibrosis in pathologies like IBD [15]. To better understand the molecular circuits driving wound healing versus fibrotic development in the colon, we reanalyzed the subset of intestinal fibroblast cells (Methods). As noted above (Fig 1E and 1G), we identified 3 main transcriptional fibroblast lineages (Fig 3A, S3A Fig), each with 2 to 3 subclusters (Figs 1G and 3A).
Fig 3. Single-cell profiling reveals functional and spatial heterogeneity of colonic fibroblasts.

(A) Single-cell atlas of colon fibroblasts. UMAP of fibroblast (dots) profiles colored by cell type assignment. (B) Expression of canonical and newly characterized markers across fibroblast clusters. Color represents average expression of marker gene within clusters; diameter represents percentage expression of marker gene within cluster. (C) Expression of genes involved in maintaining colon crypt architecture. Color represents average expression of marker gene within clusters; diameter represents percentage expression of marker gene within cluster. (D) GO enrichment of DEGs for each fibroblast cluster at baseline (water-fed mice). (E) IF staining of 3 fibroblasts classes from water-fed mouse colon. Boxes zoom onto fibroblast subsets: green box- fibroblast class 1, orange box- fibroblast class 2, and blue box- fibroblast class 3. Pcolce2 (red), CD55 (white), Procr (green), DAPI (blue). Scale bar, 25 μm. n = 3. (F) IF and FISH of various markers for fibroblast subsets. Fibroblast 1 (orange box): Grem1 (FISH). Fibroblast 2 (green box): Adamdec1 (IF), Agt (FISH), and Sox6 (IF). Fibroblast 3 (blue box): Pi16 (FISH), C3 (IF). Denoted stain (white), DAPI (blue). Scale bar, 50 μm. n = 3. (G) Proposed distribution of 3 fibroblast classes within the colon. BMP, bone morphogenetic protein; DEG, differentially expressed gene; FISH, fluorescence in situ hybridization; GO, gene ontology; IF, immunofluorescence; IstF, interstitial fibroblast; MAF, mucosa-associated fibroblast; UMAP, uniform manifold approximation and projection.
We characterized the fibroblast lineages and their subclusters by their DEGs, which included known markers of fibroblast-specific features (such as αSMA) and niche-associated markers (e.g., Grem1), as well as new genes predictive of functional specialization (including Pcolce2, Pi16, Has1, C3, CD81, CD55, Lpl, Agt, Sox6, Procr, Adamdec1, Ackr4) (Fig 3B, S3B Fig). We used these markers to define a gene signature for each of the 3 major fibroblast subsets: Fibroblast 1 was defined as CD55+Grem1+CD81−Procr−; Fibroblast 2 as αSMA+, together with high levels of Agt, Procr, and Adamdec1 and absence of CD55 and CD81; and Fibroblast 3 as CD55+CD81+Pcolce2+C3+Procr−. We also found the corresponding murine fibroblast subsets in human colon stroma of both male and female donors, suggesting sex-independent effects. (S4 Fig) [16,21].
Differential expression of genes associated with mucosal-specific functions pointed to divergent roles of the 3 lineages in intestinal homeostasis. A dominant role for the Fibroblast 1 subset in supporting the epithelial stem cell niche was suggested by elevated expression of Grem1, which is known to maintain Wnt/β-catenin signaling gradients by antagonizing BMPs [54,55] (Fig 3C, S3C Fig). These cells have also been referred to as crypt bottom fibroblasts (CBFs) [35,56] or trophocytes [57,58] that express Grem1, are Pdgfra low, and secrete Wnt ligands and BMP antagonists to support intestinal epithelial stem cell renewal [16,59].
The Fibroblast 2 subset-expressed genes associated with contractile features of myofibroblasts (aSMA), and genes that support the differentiative compartment such as Bmp2, Bmp5, Bmp7, and the atypical Wnt5a (Fig 3C, S3C Fig). Moreover, Fibroblast 2 subset genes were enriched in pathways associated with negative regulation of Wnt signaling (Fig 3D), in line with the notion that BMPs counteract β-catenin signals to prevent epithelial proliferation and promote cell differentiation in the differentiative crypt compartment [2,60–62]. This fibroblast 2 subset has also been referred to as crypt top fibroblasts (CTFs) [35,56] or telocytes [57,58] that express Foxl1, are Pdgfra high, Adamdec1 high, and secrete noncanonical Wnt ligands and BMP agonists to promote epithelial differentiation [16,59].
Together, Fibroblast 1s and 2s may maintain colon crypt architecture by establishing opposing Wnt and BMP growth factor gradients. Conversely, the Fibroblast 3 subset did not express genes related to regulation of growth factors necessary for crypt architecture. Rather, they expressed Ackr4, a receptor that functions as a chemokine sink, thus regulating chemokine gradients [63], and an array of ECM modifying genes, including Pi16, Has1, and Pcolce2 (Fig 3B), suggesting that Fibroblast 3 cells are mesenchymal cells that remodel the ECM in the intestine. These cells have also been referred to as crypt bottom fibroblast 2 (CBF2) [35,56] or interstitial stromal cells [57,58] that express Pi16, CD81, and are Pdgrfa1 low [16]. Importantly, these Pi16-expressing interstitial fibroblasts are a universal fibroblast subset found in all tissues [64].
To test if this functional heterogeneity is associated with discrete spatial niches, we defined the tissue localization of the 3 intestinal fibroblast populations by immunofluorescence (IF)- and in situ hybridization (ISH)-based analyses of subset-specific markers (Fig 3E and 3F). Consistent with a role in nurturing stem cells, Fibroblast 1 cells were marked by costaining of CD55 and Grem1 and predominantly localized at the base of crypts, associated with the epithelial stem cell niche. Fibroblast 2 cells (positive for Procr, Adamdec1, Agt, and Sox6) were found at the outer edge of the crypt, in line with their putative function in inhibiting Wnt signaling and sustaining the differentiative compartment along the crypt axis. Fibroblast 3 cells, coexpressing Pcolce2, CD55, Pi16, and C3, were localized within the muscularis mucosa. Taken together, these data suggest that each of the 3 major lineages of fibroblasts may have distinct phenotypic and functional attributes and different spatially restricted anatomical niches. Based on these functional features, we hereafter refer to these fibroblast lineages as mucosa-associated fibroblasts (MAFs: Fibroblast 1, CBF1, trophocytes), myofibroblasts (MyoFs: Fibroblast 2, CTF, telocytes), and interstitial fibroblasts (IstFs: Fibroblast 3, CBF2) (Fig 3G).
Intestinal inflammation elicits a dynamic fibroblast response
In order to delineate how the inflammatory response may impact fibroblast phenotypes, we compared the expression profiles of cells between water- and DSS-treated samples accounting for different levels of the lineage tree: (1) the broader lineage; and (2) each subcluster of a fibroblast lineage (Fig 4A, S5 Fig, S1 Table). We focused on DEGs and gene set enrichment analysis (Fig 4A, S5 Fig). Consistent with a general response to the inflammatory environment, we detected a significant induction of immunomodulatory factors, including complement genes (C3 and C4b), MHC-related molecules (B2m, Calr, H2-D1, H2-K1, H2-Q7, Psmb8), and chemokines (Ccl2, Ccl8, Cxcl5, Cxcl12, Cxcl14) (Fig 4B, S2 Table). DSS-induced genes in all fibroblast subsets were enriched for inflammatory responses (Fig 4A, S5 Fig). In all subsets, DSS induced redox regulators such as Gpx1, Gpx3, Prdx2, and Prdx5 (Fig 4A, S2 Table), suggesting that fibroblasts exhibit a metabolic adaptation to counterbalance oxidative stress associated with inflammation [65]. Notably, the antioxidant system appears to be a critical player in fibrosis development, which is prevalent across many pathological conditions [66–69], thereby suggesting that fibroblast activation in colitis may be linked, at least in part, to this process.
Fig 4. Intestinal inflammation elicits a dynamic fibroblast response.

(A) Volcano plots depicting DEGs between water- and DSS-treated samples for select fibroblast clusters. Each dot represents an individual gene, and dot colors represent the contributing fibroblast subset when that gene is differentially expressed. Significant DEGs had FDR <0.05 using MAST (see Methods). (B) GO enrichment of DEGs for each fibroblast cluster comparing water- and DSS-treated mice. Color represents adjusted p-value of GO enrichment annotation for each fibroblast cluster; diameter represents gene ratio for each fibroblast cluster. (C) Violin plots of expression levels of select IGFBP family members across all stromal clusters in water- and DSS-treated samples. Normalized gene expression levels are plotted on the y-axis. Significant DEGs had FDR <0.05, and respective adjusted p-values derived using MAST (see Methods); *p < 0.05, **p < 0.001. (D) Fibroblast frequency changes between water- and DSS-treated samples. Significant frequency changes derived using Dirichlet multinomial regression; *p < 0.05. (E) Feature plot for IL11 expression in fibroblasts from water- and DSS- treated samples; *p < 0.05 (F) Expression of select IL6 family receptors across fibroblast clusters in water- and DSS-treated samples. Color represents average expression of marker gene within clusters; diameter represents percentage expression of marker gene within clusters. DEG, differentially expressed gene; DSS, dextran sulfate sodium; FDR, false discovery rate; GO, gene ontology; IGFBP, insulin growth factor binding protein; IstF, interstitial fibroblast; MAF, mucosa-associated fibroblast; MyoF, myofibroblast.
Consistently, across fibroblast subsets and lineages, cells from DSS-treated mice up-regulated genes encoding ECM components and matrix modifiers (Fig 4A, S5 Fig, S2 Table). These included genes for fibrillar collagen type I and III, which are known to provide tensile strength, as well as for the glycoprotein fibronectin, reported to be deposited aberrantly in patients with IBD [70–74]. Myofibroblasts and interstitial fibroblasts showed a pronounced up-regulation of genes associated with the process of extracellular structure organization, as highlighted by up-regulation for genes such as Col1a1, Col3a1, and Fn1, and also the genes implicated in the propagation of the fibrotic response, such as Mmp3 and Mmp10 (Fig 4A and 4B, S5 Fig, S2 Table). We also observed a striking alteration in the expression of genes from the insulin growth factor binding protein (IGFBP) family that are known to regulate the local availability of insulin growth factor l [75] (Fig 4C, S5 Fig, S3 Table). Notably, the IGFBP1/IGFBP3 locus was recently associated by GWAS with poor prognosis in patients with Crohn’s disease, where IGFBPs have been suggested to play a role in both the inflammatory response and fibrosis [76–82]. The increased expression of IGFBP genes in mucosal fibroblasts and other stromal subsets may point to a pathological role for this pathway in colitis.
The frequency of the MAF 3 population, which expressed a potent inflammatory signature (S2 Table), increased dramatically in response to DSS treatment (P value = 0.04, Methods) (Fig 4E), suggesting that this subcluster may be analogous to the inflammation-associated fibroblasts (IAFs) we recently described in ulcerative colitis patients [21]. In fact, a gene expression correlation analysis corroborated this conclusion (S4 Fig). We also observed an increase in IL-11 expression in MAF subsets from DSS-treated mice (Fig 4E, S2 Table), an observation with potential clinical implications, as IL-11 is associated with fibrosis in other organs [83–85]. As most of the fibroblast subsets expressed both IL-11 receptor subunits IL11ra (even at baseline) and IL6st (DSS-induced) (Fig 4F), as well as Osmr (DSS-induced, Fig 4F), these data highlight a putative role for the IL-6 cytokine family in fibroblast intercellular communication during inflammation. In addition, receptor-ligand expression patterns suggest that IAFs communicate with endothelial cells through provision of Notch ligands, as previously described [86], and through IL-6 family cytokines such as IL-11 (S6 Fig). Altogether, these data identify changes in gene expression in mucosal fibroblasts that occur during chronic inflammation, which collectively provides a framework for functional dissection of the mechanisms driving fibrosis in the intestine.
The myofibroblast differentiation program confers matrix remodeling function
Next, we defined a shared transcriptional program associated with inflammation across fibroblast lineages to subsequently identify key effectors that coordinate matrix remodeling. To this end, we identified 10 genes that have high expression specificity within the gastrointestinal tract relative to other human tissues [87] and were also differentially expressed between water- and DSS-treated samples across many stromal lineages and that had an average log2 fold change >0.10 and showed differential expression in >2 cell types (Fig 5A, S7A–S7C Fig). This fibroblast gene signature included genes controlling ECM deposition and remodeling (Adamdec1, Ecm1), immune function (Cstb, Dpep1), and extracellular nucleotide processing (Gda, Gbp4, Enpp3). Among these, Adamdec1 was coordinately up-regulated in response to chronic inflammation in fibroblast subsets, both at the transcriptional and protein levels (Fig 5A–5C, S2 Table).
Fig 5. The myofibroblast differentiation program confers matrix remodeling function.

(A) Genes that are most often differentially expressed in all stromal clusters between water- and DSS-treated samples and whose expression is enriched in gastrointestinal tissues. The frequency of times they are a DEG is plotted on the y-axis. (B) Violin plot of Adamdec1 across fibroblast clusters in water- and DSS-treated samples. Normalized gene expression levels are plotted on the y-axis. Significant DEGs had FDR <0.05, and respective adjusted p-values derived using MAST (see Methods); *p < 0.05, **p < 0.001, ***p < 1E-10. (C) IF staining of colon from water- and chronic DSS-treated mice. Adamdec1 (green), DAPI (blue). Scale bar, 100 μm. n = 3. (D) Inferred differentiation trajectory for MAFs into myofibroblast subset populations. Each dot represents a cell, and color represents the estimated pseudotime for each cell. (E) Adamdec1 expression overlaid on top of inferred differentiation trajectory for MAF into myofibroblast subset populations. Each dot represents a cell, and color represents Adamdec1 expression. (F) Dynamics of Adamdec1 expression levels as a function of pseudotime. Each dot represents a cell, and color represents the annotated fibroblast subset. (G) Genes identified in association with MAF-to-myofibroblast inferred differentiation trajectory. Color indicates when gene expression peaks along differentiation trajectory along the x-axis (from left to right). DEG, differentially expressed gene; DSS, dextran sulfate sodium; FDR, false discovery rate; IF, immunofluorescence; MAF, mucosa-associated fibroblast; UMAP, uniform manifold approximation and projection.
Because myofibroblasts are thought to represent a terminally differentiated cell state [88], we reasoned that pseudotemporal ordering of cells based on their transcriptional profiles could provide insights into the dynamic transcriptional modules that may be associated with the fibroblast to myofibroblast transition. Using both tree-based and diffusion map-based inference methods, we learned the differentiation trajectory from the MAF populations to myofibroblasts and identified putative effector genes that may drive this transition (Fig 5D–5G, S8 Fig, S4 Table). Our model suggesting that myofibroblasts are derived from MAFs is supported by lineage tracing strategies, which showed that fibroblasts located at the base of the crypt differentiate into myofibroblasts along the outer edge of the crypt [55,89]. We sought to expand on these findings and define the gene signature associated with the MAF-to-myofibroblast trans-differentiation trajectory within the colon. The early stage of the trajectory is enriched for genes related to the ECM components Gsn, Fbln1, Eln, and Mfap5 and the immune mediators Ccl1, Cd34, Fos, and Jun. At later stages of transition, fibroblasts up-regulated genes associated with contractility such as Tagln, Myl9, and Acta2. Adamdec1 is up-regulated during the intermediate stages, at the boundary of the MAF to myofibroblast transition (Fig 5E and 5F), suggesting a role in myofibroblast function and matrix remodeling activity characteristic of the myofibroblast lineage.
Transcriptional modules associated with the pseudotime trajectory reveal distinct transition states that provide a higher resolution view of myofibroblast function (Fig 5G, S4 Table). Among these differentiation transition genes, Adamdec1 is unique in that it is a secreted metalloproteinase with no other paralogs [90,91], its expression is enriched in intestinal tissues [92], and a single nucleotide polymorphism (SNP) variant (8:24248756 T/C) in this gene locus is associated with rectal prolapse [93,94]). Taken together, these findings led us to hypothesize that Adamdec1 may play a key role in balancing homeostatic matrix remodeling versus development of pathological fibrosis.
Adamdec1 is required for matrix remodeling and healing in response to epithelial injury
To define the role of Adamdec1 in tissue homeostasis we generated Adamdec1 knockout (KO) mice (Fig 6A) and induced epithelial injury by administration of 2% DSS in drinking water for 7 days followed by 7 days of recovery. Adamdec1 KO mice were considerably more susceptible to epithelial injury compared to their wild-type (WT) littermate counterparts, as demonstrated by increased weight loss and reduced colon lengths (Fig 6B and 6C). Histological analysis indicated that Adamdec1 KO mice exhibited increased immune infiltration and mucosal erosion (Fig 6D, S9 Fig).
Fig 6. Adamdec1 is required for matrix remodeling and healing in response to epithelial injury.
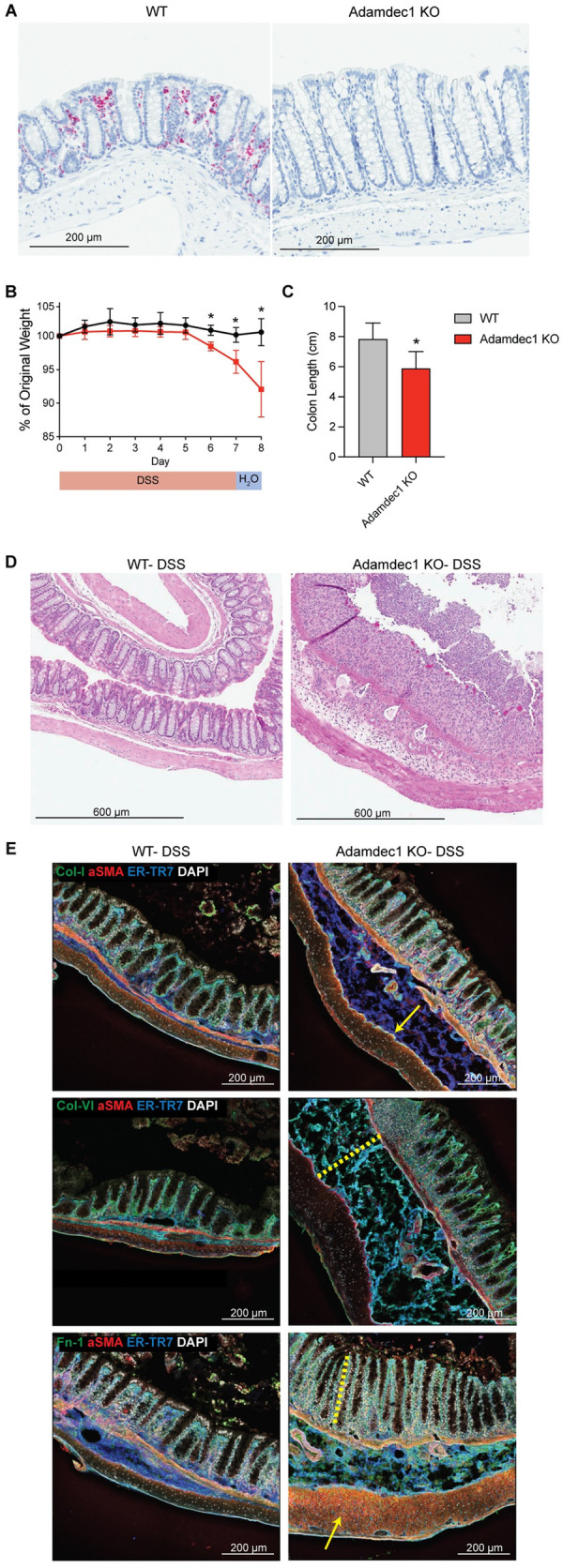
(A) ISH of Adamdec1 in WT or Adamdec1 KO colon at basal state (water-fed mice). Scale bar, 200 μm. n = 2 per cohort. (B) Weight loss in Adamdec1 KO versus WT mice administered 2% DSS for 7 days and followed by H2O for 7 days. n = 4 per cohort. Mann–Whitney test, *p < 0.05. (C) Colon length in Adamdec1 KO versus WT mice administered DSS as described above. n = 8 per cohort. Mann–Whitney test, *p < 0.05. For source data for panels B and C, see S1 Data. (D) HE staining of representative images of colon following DSS in WT and Adamdec1 KO mice. Mice were administered 2% DSS for 7 days, H2O for 1 day, and killed on day 8. Scale bar, 600 μm. n = 4 per cohort. (E) IF staining was performed on colons from Adamdec1 KO and WT mice with indicated markers. Mice were administered 2% DSS for 7 days and killed on day 7. αSMA (red), ER-TR7 (blue), DAPI (gray). Indicated ECM component (green). (Top row) Collagen type I (green). Yellow arrow denotes submucosal ECM accumulation. (Middle row) Collagen type VI (green). Yellow dotted line denotes submucosal thickening and edema. (Bottom row) Fibronectin (green). Yellow dotted line denotes hyperplastic response. Yellow arrow denotes muscle thickening. Scale bar, 200 μm. n = 3 per cohort, representative of 2 experiments. DSS, dextran sulfate sodium; ECM, extracellular matrix; HE, hematoxylin–eosin; IF, immunofluorescence; ISH, in situ hybridization; KO, knockout; WT, wild-type.
Because Adamdec1 is a metalloproteinase, and members of the ADAM family have been previously shown to modify the ECM [95,96], we analyzed ECM remodeling in colons from Adamdec1 KO and WT mice following 7 days of DSS treatment (Fig 6E, S10 Fig). We focused on collagen type I (Col I) and fibronectin (Fn1), due to their increased expression during fibrosis [73,74], as well as collagen type VI (Col VI), due to its role in promoting mesenchymal cell proliferation [97]. Although minor mucosal and submucosal ECM accumulation occurred in WT littermates following DSS, distinct and organized reticular fibers were observed by high-resolution tissue scanning and tiling by confocal microscopy. In striking contrast, the ECM was aberrantly remodeled in Adamdec1 KO mice following DSS treatment, as characterized by increased matrix deposition and disorganization of fibrillar structures, including fibrillar Col I, filamentous Col VI, and the glycoprotein Fn1. Strikingly, Adamdec1 KO mice exhibited aberrant submucosal Col VI and Fn1 deposition between the muscularis mucosa and the muscularis, where this deposition was nonlinear, fragmented, and enveloped within areas of edema (Fig 6E, S10 Fig). Adamdec1 KO mice also presented with hyperplasia, edema, increased immune infiltrates, and muscle thickening, altogether indicative of colitis disease pathology (Fig 6D, S9 Fig). These data suggest that Adamdec1 is necessary for ECM remodeling, and a deficiency in this gene results in an accelerated accumulation and disorganization of the ECM following inflammatory insult. Taken together, we have identified Adamdec1 as a pivotal effector regulating the balance between wound healing and ECM remodeling.
Discussion
In response to chronic inflammation, intestinal stromal cells adopt specialized functions to promote tissue repair and healing. We identified functional and locational attributes of fibroblast subsets that are regulated by chronic inflammation in the intestine. As the heterogeneity of stromal cell subsets is coming into focus from single-cell transcriptional profiling, classifying and naming these cell types remains challenging. In this context, a recent cross-tissue atlas of human and mouse fibroblasts was defined at baseline and during inflammation [64]. These datasets identified universal fibroblasts found in all tissues, tissue-specific cell states, and disease-associated activation programs [64]. Here, we identify 3 broad subtypes of fibroblasts that perform critical functions in the intestine to maintain mucosal homeostasis. Grem1+ MAFs and myofibroblasts establish the growth factor gradient along the crypt axis that promotes epithelial stem cell renewal in the base of the crypt and the differentiation compartment emanating upwards. Recent studies have described these Grem1+ MAFs as CBFs or trophocytes and myofibroblasts as CTFs or telocytes [35,56–58]. The third fibroblast subtype, interstitial fibroblasts, express genes encoding ECM components and have been found in multiple tissues and are variously referred to as CBF2 cells or Pi16 fibroblasts [35,56,64]. Although there remains a need for the field to define and adopt a unified nomenclature for stroma, these single-cell transcriptomic stroma atlases offer insights into mechanisms of fibroblast function in the context of inflammation and tissue homeostasis. Despite the well-documented role of fibrosis in many chronic inflammatory conditions, a lack of therapies to manage tissue fibrosis represents a significant unmet medical need [98]. A deeper understanding of the mechanisms operating in inflamed tissues and leading to pathological fibrosis may facilitate identification of novel targeting opportunities to bolster the currently limited therapeutic landscape.
Despite significant progress, it remains unclear how chronic inflammation activates fibroblasts, and, conversely, how activated fibroblasts amplify local inflammation in mucosal tissues. Recent studies highlighted fibroblasts as critical targets of IL-6 family cytokines such as OSM in the intestine [14]. In this context, myeloid cell–derived OSM was shown to activate fibroblasts to become inflammatory effectors producing chemokines such as CCL2, CXCL1, CXCL9, and CXCL11 to recruit neutrophils and monocytes, thereby acting as amplifiers of intestinal inflammation [14]. Similarly, in the context of rheumatoid arthritis, chronic inflammation was shown to drive differentiation of synovial fibroblasts (CD34− Thy1+) that share many features with IAFs [99]. Specifically, the presence of this fibroblast subset located within the sublining of the synovium correlated with increased infiltrating leukocytes and more severe clinical scores in rheumatoid arthritis patients [99]. Mechanistically, these expanded inflammatory Thy1+ fibroblasts were shown to drive inflammation in rheumatoid arthritis by secreting IL6 and chemokines such as CCL2, CX3CL1, CXCL9, and CXCL12 [100,101]. Taken together with our findings, these data demonstrate the utility of scRNA-seq for identifying unique subsets of inflammatory fibroblasts and elucidating pathogenic molecular mechanisms, such as IL-6–dependent inflammatory responses, driven by these expanded subsets.
Within the colonic mucosa, we identified a unique IL-11+ MAF subset arising in response to chronic inflammation and exhibiting up-regulation of inflammatory genes, including C4B, CXCL5, and SAA3. Notably, IL-11 is a member of the IL-6 family and has been previously implicated in an autocrine mechanism inducing ERK activation in fibroblasts to drive ECM deposition and inflammatory chemokine production [83–85]. Neutralization of IL-11 was shown to reduce pathological fibrosis in murine models of liver, cardiovascular, and lung fibrosis [83–85]. Genetic KO of Il11 or Il11ra in mice attenuated DSS-induced tumorigenesis [102]. Conversely, a transgenic mouse model driving Il11 expression in smooth muscle cells or fibroblasts was shown to spontaneously develop colitis, as well as inflammation in other organs [103]. In humans, expansion of IL-11+ IAFs was recently described in ulcerative colitis and Crohn’s disease patients [21,104]. Together, these results and our data suggest that IL-11 signaling by fibroblasts is a conserved molecular circuit that may be shared among diverse inflammatory fibrotic diseases and therefore represents a potential therapeutic target to prevent fibrosis.
Here, we expand on these findings showing both the fibroblast subset expressing Il11 and the subsets expressing its receptor subunits Il11ra and Il6st. While Il11 was strictly expressed in IAFs, the receptor was more broadly expressed among MAFs, myofibroblasts, interstitial fibroblasts, and endothelial cells. These findings are consistent with previous reports suggesting that local inflammatory cues drive Il11 expression in IAFs and engage an autocrine/paracrine mechanism of fibroblast matrix deposition and chemokine production [83–85]. Our findings also suggest that IL-11 derived from fibroblasts may act on endothelial cells to promote gut inflammation.
In concert with IAF-mediated amplification of inflammation, myofibroblasts promote fibrosis in chronic inflammatory diseases. Myofibroblast differentiation has been described in the context of wound healing in the dermis and shares several key features with wound healing in the mucosa. In the dermis, wound healing occurs in coordinated phases mediated by hemostasis, inflammation, reepithelialization, and remodeling [105,106]. During the reepithelialization phase, activated fibroblasts form granulation tissue, and differentiated myofibroblasts utilize contractile machinery to draw the wound margins together. During the remodeling phase, myofibroblasts remodel the ECM to replace granulation tissue with organized connective tissue. In mucosal tissues, the wound-associated epithelium (WAE) envelops over the wound to protect underlying tissues. Subsequently, mesenchymal cells form granulation tissue as proliferating epithelial cells regenerate mucosal crypts and myofibroblasts remodel the tissue site [107–109]. While the cellular and histological features of wound healing have been thoroughly characterized, insights into the molecular mechanisms driving myofibroblast differentiation are only starting to emerge. Using lineage tracing mice based on expression of the fibroblast markers Pdgfra, Dlk1, and/or En1, two studies demonstrated that murine dermal fibroblasts gave rise to early wound bed myofibroblasts that drove ECM deposition and fibrosis [110,111]. A similar mechanism was observed in human dermal fibroblasts identified using scRNA-seq, in which Sfrp2+Dpp4+ and Fmo1+Lsp1+ fibroblast subsets were implicated in matrix deposition and inflammatory cell retention, respectively [112]. Importantly, a subset of the matrix-depositing Sfrp2+Dpp4+ fibroblasts share expression of key lineage markers Pcolce2+ and CD55+, which we identified in MAFs and interstitial fibroblasts. Taken together, these findings suggest a concerted fibroblast differentiation program giving rise to myofibroblasts that promote wound healing in diverse tissues, including the intestinal mucosa.
In the context of tissue homeostasis, the stromal cellular compartment is dynamic. Specifically, we and others have demonstrated that intestinal myofibroblasts are derived from MAFs, reinforcing the idea that ECM-remodeling myofibroblasts are derived from lineage-restricted fibroblasts [55,89]. These studies leveraged lineage tracing strategies to show that fibroblasts located at the base of the crypt differentiate into myofibroblasts along the outer edge of the crypt. Here, we expand on these findings and define the gene signature associated with the MAF-to-myofibroblast trans-differentiation trajectory within the colon. While the early stage of differentiation is enriched for genes encoding ECM components and immune mediators, the later stages are enriched for genes involved in cytoskeletal contraction such as Acta2, Myl9, and Tagln. These genes were recently identified to be expressed also during the terminal phases of wound healing in injured skin [113], thereby highlighting a shared myofibroblast differentiation trajectory across organs and conditions.
We provide evidence that Adamdec1 is a key component of the MAF-to-myofibroblast trans-differentiation trajectory in the intestine, influencing the local microenvironment at the level of ECM remodeling. Our results indicate that Adamdec1 is a required component of the matrix remodeling program that helps orchestrate wound healing to maintain intestinal tissue homeostasis. Adamdec1 is a member of the ADAM metalloproteinase family, which includes members that have been shown to modulate inflammation, wound repair, and tissue development [95,114,115]. However, Adamdec1 is an unusual member of the ADAM metalloproteinase family in that its domain structure is unique, and there are no structurally similar paralogues in the human genome. Specifically, Adamdec1 is a secreted protein containing a prodomain, metalloproteinase domain, and a truncated disintegrin domain [90,91]. While the proteolytic substrates targeted by Adamdec1 are not well defined, our data suggest that Adamdec1 plays a key role in matrix remodeling. In addition, recent studies suggest that Adamdec1 may play a broader role in tissue homeostasis by solubilizing ECM-bound growth factors such as FGF2 [116,117].
Our genetic ablation studies provide additional insights into the function of Adamdec1 in tissue homeostasis. We demonstrated that genetic ablation of Adamdec1 in a mouse model impaired recovery from intestinal epithelial injury and aberrant ECM. Consistent with these findings, previous reports indicated that Adamdec1-deficient mice are more susceptible to Citrobacter rodentium and Salmonella typhimurium bacterial infections relative to WT controls [118]. However, these studies hypothesized a cell-intrinsic function for Adamdec1 in the myeloid lineage, whereas our analyses of stroma identify fibroblasts as the major source of Adamdec1 expression and identify a critical role for this gene in tissue remodeling and healing. In the human colon, the vast majority of Adamdec1-expressing cells are fibroblasts, whereas expression in myeloid cells was infrequent and quantitatively lower [21]. Thus, Adamdec1 performs key functions in fibroblast-mediated tissue homeostasis, and this may occur in cooperation with myeloid cells expressing Adamdec1. Together, these data implicate a critical and novel role for Adamdec1 in restoring mucosal integrity through matrix remodeling following tissue injury. Pinpointing molecular factors that dictate healing versus fibrosis will be instrumental in efforts to identify potential therapeutic targets that may prevent fibrosis pathologies that accompany chronic inflammatory diseases.
Methods
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Daniel B. Graham (dgraham@broadinstitute.org).
Experimental model and subject details
Mice
C57BL/6J mice were housed in specific pathogen free housing at Massachusetts General Hospital (MGH). For all experiments, 8- to 12-week-old mice were used. Adamdec1 KO and Balb/c W littermate mouse work was approved and performed at Novartis Institutes for BioMedical Research (NIBR). The mice were housed in specific pathogen free housing at NIBR. For all experiments, 8- to 12-week-old male mice were used. Adamdec1 KO mice were generated and derived from Balb/c mice using CRISPR technology. Two sgRNAs targeting exons 1 and 2 of Adamdec1 were combined with Cas9 and delivered to zygotes by microinjection. F1 offspring were backcrossed to the Balb/c background, and Adamdec1 genotype was confirmed by genomic DNA sequencing. The founder line selected for breeding contained a deletion in exons 1 and 2 spanning intron 1 and resulting in an out-of-frame KO allele. While Balb/c mice are less susceptible to DSS pathology compared to C57BL/6J, Adamdec1 deficiency in the Balb/c genetic background was still associated with morbidity, but tolerated well enough to achieve endpoints in this study.
DSS models
For chronic DSS, C57BL/6J mice were fed 2.5% (weight/volume) DSS salt (40,000 to 50,000 MW, Affymetrix #14489) dissolved in sterile water ad libitum for 7 days, and then returned to regular sterile water for 7 days; this cycle was repeated for a total of 3 times [22,23]. Mice were then killed, and colons were obtained. A total of 3 female mice (12 weeks old) were processed for each treatment group and analyzed by scRNA-seq. Female mice were utilized because they were able to withstand 3 cycles of DSS, whereas males exhibited morbidity.
For acute DSS, Adamdec1 KO and WT littermate control mice were fed 2.0% DSS dissolved in sterile water ad libitum for 7 days. Mice were then killed at noted time points, and colons were obtained. Male mice were utilized in the acute DSS model, because they exhibit more uniform disease penetrance relative to females.
Ethics statement
All animal procedures were conducted in accordance with protocols approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee (IACUC), and animals cared for according to the requirements of the National Research Council’s Guide for the Care and Use of Laboratory Animals. MGH IACUC protocol number, 2003N000158. NIBR IACUC protocol number, 17IMO018.
Method details
Immunofluorescence (IF) staining
Colons were obtained and flushed with ice-cold washing buffer (2% FBS in 1X PBS (Sigma-Aldrich, calcium and magnesium free)). Colons were cut open longitudinally, covered with Optimal Cutting Temperature medium (OCT, Sakura Finetek), and Swiss rolls were made and frozen at −80°C. Sections were cut on a cryostat with a thickness of 12 to 16 μm. Immunostaining was performed as follows: Sections were fixed with 4% PFA (Alfa Aesar) for 10 minutes at room temperature, permeabilized with 0.2% Triton X-100 (Sigma Aldrich) in 1X PBS for 2 minutes, and blocked with 2% BSA (Sera Care) in 1X PBS for 20 minutes. Tissues were immunostained with primary (1:100) and then secondary antibodies/DAPI (1:500 and 1:1,000, respectively), each for 1 hour at room temperature. Sections were mounted with Fluorescent Mounting Reagent (Dako), sealed, and imaged with a Leica SP5X laser-scanning confocal microscope.
Fluorescent in situ hybridization (FISH)
Flash frozen colon Swiss roll sections were prepared as described previously. FISH was performed using the RNAscope Multiplex Fluorescent Kit v2 (Advanced Cell Diagnostics) per the manufacturer’s recommendations with the following alterations. The protease treatment was adjusted and performed with Protease III for 20 minutes. Sections were mounted with Fluorescent Mounting Reagent (Dako), sealed, and imaged with a Leica SP5X laser-scanning confocal microscope.
Image analysis
Individual tiles were stitched together using the LAS X Life (Leica). Images were overlaid and cropped using Photoshop and Illustrator (Adobe).
HE/Trichrome
Colons were isolated and cleaned as previously described, fixed in 4% PFA, and paraffin embedded. Sections were stained for hematoxylin–eosin (HE) or Masson’s trichrome staining according to standard protocols.
Lamina propria single-cell isolation
To obtain a stromal single-cell suspension [24,25] for scRNA-seq, we collected colons from water- and chronic DSS-treated C57BL6J mice and flushed them with ice-cold washing buffer (2% FBS in 1X PBS (Sigma-Aldrich, calcium and magnesium free)). After removing fat and any connective tissue, colons were flipped inside out using forceps and cut into 2 cm chunks. Each colon was then incubated with 25 mL epithelial strip buffer (5 mM EDTA (Invitrogen), 1 mM DTT (Sigma Aldrich), 2.5 mM HEPES (Gibco), and 5% FBS in HBSS (GE Healthcare, calcium and magnesium free)) for 30 minutes at 37°C with stirring. Tissues were collected, rinsed with ice-cold washing buffer, minced using scissors and razor blades, and transferred into 50 mL conicals containing 5 mL enzyme digest buffer (0.2 mg/mL Collagenase P (Roche), 0.2 mg/mL Dispase II (Gibco), 0.1 mg/mL DNase I (Roche) in full media (CO2-independent media (Gibco), 2% FBS, 1X GlutaMAX (Gibco), 1X MEM NEAA (Gibco))) and placed into a bead bath at 40°C. The conicals were vortexed every 5 minutes for 10 minutes, tissue chunks were allowed to settle for 5 minutes, and the supernatants were collected into ice-cold collection media (full media with 10% FBS and 10 mM EDTA) that was kept on ice. Next, 5 mL enzyme digest media was added to the remaining colon fragments, the conicals were vortexed every 5 minutes for 5 minutes, tissue chunks were allowed to settle for 5 minutes, and the supernatants were added to previously collected fraction. Then, 3 mL enzyme digest media was added to the remaining tissue fragments, pipetted up and down for 2 minutes, allowed to settle for 3 minutes, and supernatants were collected and added to the previously collected fraction; this process was repeated for approximately 45 minutes until no colon fragments remained. The collection buffer and its contents were filtered using a 100-μm cell strainer (Falcon), centrifuged (10 minutes, 450g, 4°C), and resuspended in full media (2% FBS, 10 mM EDTA).
Antibodies and FISH probes
The following primary antibodies were used for FACS and IF staining with murine tissues: Ter-119 (TER-119, Biolegend), CD45 (30-F11, Biolegend), EpCAM (G8.8, Santa Cruz Biotechnology), Pdpn (8.1.1, Biolegend), CD31 (390, Biolegend), Procr (eBio1560, Invitrogen), CD90 (53–2.1, Biolegend), CD55 (RIKO-3, Biolegend), αSMA (1A4, Sigma Aldrich), Adamdec1 (Origine #TA323936), Pcolce2 (Proteintech #10607-I-AP), C3 (11H9, Abcam), Sox6 (Abcam #ab30455), ER-TR7 (Abcam #ab51824), Collagen I (Abcam #ab34710), Collagen VI (Abcam #ab6588), and Fibronectin (Abcam #ab2413). The following secondary antibodies were used: donkey anti-rabbit PE (Poly4064, Biolegend), Alexa Fluor 488-, 546-, 568-, and 647-conjugated secondary antibodies were obtained for goat anti-rabbit, goat anti-rat, goat anti-mouse, and goat anti-Syrian hamster from Life Technologies, and DyLight 488- and 649-conjugated secondary antibodies for goat anti-Syrian hamster were obtained from Biolegend. NucBlue viability dye (Invitrogen) was spiked into single-cell suspensions before flow analysis or FACS sorting.
The following probes from Advanced Cell Diagnostics were used for FISH in murine tissues: Pi16 (Mm-Pi16-C2), Grem1 (Mm-Grem1-C3), and Agt (Mm-Agt-C1).
Cell enrichment and sorting for single-cell RNA-seq
Lamina propria single-cell suspensions obtained as above were blocked with FcR blocking reagent (Miltenyi Biotec) for 10 minutes on ice, stained with primary antibodies previously listed for 20 minutes on ice, and cells were sorted using the Beckman Coulter MoFlo Astrios EQ (100 μm nozzle, 25 psi). For all steps, cells were kept in full media (2% FBS, 10 mM EDTA). Stromal cells that were sorted were defined as Ter119− CD45−EpCAM−, and dead cells were excluded using NucBlue (1 drop/500 μL cells). Cells were collected in full media, and purity was assessed by taking a fraction of sorted cells and reanalyzing them immediately on the MoFlo Astrios EQ. All samples were analyzed using FlowJo (Tree Star).
Droplet-based single-cell RNA-seq
Single-cell suspensions were processed using the Chromium Single-Cell 3′ Gene Expression kit (v2, 10x Genomics) per manufacturer’s instructions. Libraries were sequenced on the Illumina HiSeq 2500 per manufacturer’s instructions.
Quantification and statistical analysis
Analysis workflow—Gut stroma single-cell data
Data processing and QC. Digital gene expression (DGE) matrices for each individual cell were obtained by aligning the FASTQ sequence reads against the reference mm10 mouse transcriptome using CellRanger v2.2 software (10x Genomics). Cells that satisfied any one of the following criteria were removed: (1) <300 detected genes; (2) outlier number of unique molecular identifiers (UMIs), ranging from 7,500 to 15,000; (3) outlier proportion of mitochondrial gene expression were excluded ranging from 2.5% to 15%. Outlier cutoffs for each batch of samples were determined empirically based on the distribution of UMI and proportion of mitochondrial gene expression per cell; or (4) doublets identified by the python package Scrublet [119]. Overall, this led to removal of 4.7% of cells, retaining 35,072 cells for downstream analyses.
Normalization and batch correction. Normalized gene expression values were obtained by applying a regularized negative binomial (NB) regression model implemented in the SCTransform function in Seurat v3 [120]. Briefly, the function first applies an NB regression model to the raw UMI count of each gene using sequencing depth as a covariate. Next, by including a dispersion parameter that combines information across similar genes with similar abundances, the model uses a kernel regression to learn regularized parameters that are robust to sampling noise. Finally, a second round of NB regression is applied on the learned regularized parameters to derive residuals that are treated as normalized expression levels. To adjust for batch effects, we included the batch variable as an additional parameter in the NB regression model. DGE matrices generated for each mice colon were considered as a different batch. Batch-corrected normalized expression data were used for integration and clustering of datasets (below). Alternatively, log-normalized expression levels without batch correction were used for differential expression analysis.
Identification of shared cell types across colon sites and conditions. Samples collected from a different site in the colon (proximal and distal) or treated differently (water and DSS) were considered as a separate dataset for integration. To integrate the batch-normalized expression datasets for cell type identification across different sites and treatment conditions, we used the FindIntegrationAnchors and IntegrateData functions implemented in Seurat v3 to align the datasets [121]. Briefly, the method first performs joint dimensionality reduction using canonical correlation analysis (CCA) to identify latent gene level projections that are shared across datasets. This is achieved through a standard singular value decomposition (SVD) of the input matrices for the subset of highly variable genes to identify a set of canonical correlation projection vectors (dims = 1:50), which is then L2 normalized. Following this dimensionality reduction procedure, the algorithm next identifies K-nearest neighbors for each cell in one dataset with the cells in its paired dataset. This search is constrained on the mutual nearest neighbors (MNNs), i.e., to identify pairs of cells, also called “anchors,” each taken from the individual datasets being present mutually in each other’s nearest neighborhood (k.anchor = 5). Finally, the identified anchors are scored to ensure that low scoring correspondences are filtered out to prevent anchoring of cells that represent different biological states.
Clustering and visualization. We performed a principal component analysis (PCA) of the integrated anchor weights using the RunPCA function in Seurat v3 and selected the top 60 eigenvectors that explained a substantial proportion of the variance in the dataset. Subsequently, a k-NN graph was constructed with the top PCs and k = 200 using the FindNeighbors function, to which the louvain clustering algorithm was applied using FindClusters function. The resulting clusters were visualized using the RunUMAP function. Subclustering of annotated endothelial and fibroblast subsets was also performed by subsetting the integrated dataset. For endothelial cell subset analysis, clustering was parametrized at top 50 pcs and k = 50, whereas top 40 pcs and k = 200 was used for fibroblast subsets.
Cell lineage dendrogram. Hierarchical clustering was performed on the average gene expression levels for each cell type cluster using the BuildClusterTree function.
Differential expression analysis. We used MAST [122], which fits a hurdle model to log-normalized expression levels for each gene to identify DEGs between any 2 given conditions. For each lineage as well as subcluster separately, we compared gene expression levels between the 2 conditions (e.g., DSS-treated versus water-treated MSCs). GO term enrichment analysis was performed to identify biological processes enriched in differential expressed genes and visualized using ClusterProfiler [123].
Changes in cell proportions. To identify statistically significant changes in proportion of cell types in water- and DSS-treated samples, we used the Dirichlet multinomial regression model from the DirichletReg R package, and also Wilcoxon test as described previously [21]. Changes in cell proportions that were significant in both tests were considered to be relevant.
Selection of GI tract–specific genes. Data from [87] were used to identify genes elevated in the following annotated tissues: esophagus, stomach, small intestine, duodenum, appendix, colon, and rectum.
Pseudotime analysis. Trajectory inferences were performed using the R implementation of Monocle3 [124,125] and destiny. Log-normalized expression data for top 3,000 highly variable genes in the subset of MAF and myofibroblast populations in the water-treated dataset were used as input for both methods. In Monocle3, dimensionality reduction was performed to project the data into a lower dimensional space, and the top 20 PC components were corrected for batch effects and technical factors using the residual model: ~ number of UMIs + percentage of mitochondrial gene expression + percentage of ribosomal gene expression. Further dimensionality reduction into UMAP space was performed using the reduce_dimension function with the following parametrization: umap.min_dist = 0.1, umap.n_neighbors = 20. The trajectory graph was then learned on the louvain clusters, and pseudotime was estimated using a cell from MAF 1.1 population as the root index. To identify putative regulators associated with the differentiation trajectory, the graph_test function was applied on the principal_graph. Trajectory analysis was also performed using destiny to validate the observations using an alternative method. Diffusion components and diffusion pseudotime were estimated with the following parameters: n_pcs = 50 and sigma = local.
Supporting information
(A) Chronic DSS murine model: Mice were fed 3 iterative cycles of 2.5% DSS for 7 days followed by water for 7 days. (B) Masson’s trichrome staining of colons from water- and chronic DSS-fed mice after 1 round (day 14), 2 rounds (day 28), and 3 rounds (day 42) of DSS. Collagen accumulation in blue, as demarcated by yellow arrows. Leukocyte infiltrates, as demarcated by yellow arrowheads. Scale bar, 200 μm. n = 2, representative of 2 experiments. (C) Single-cell atlas of the murine colonic stroma. UMAP of stroma cells (dots) colored by cell type assignment from water (top) or DSS (middle) samples and combined embedding of both conditions (bottom panel). (D) Expression of lineage-specific marker genes across cell type subsets. Color represents average expression of marker gene within clusters; diameter represents percentage expression of marker gene within cluster. BEC, blood endothelial cell; DSS, dextran sulfate sodium; ICC, interstitial cell of Cajal; LEC, lymphatic endothelial cell; MSC, mesenchymal stem cell; SMC, smooth muscle cell; UMAP, uniform manifold approximation and projection.
(TIF)
(A) Single-cell atlas of colon fibroblasts UMAP of endothelial cell (dots) profiles (Methods) colored by cell type assignment from water (top) or DSS (bottom) samples. (B) Expression of canonical markers across endothelial cell clusters. Color represents average expression of marker gene within clusters; diameter represents percentage expression of marker gene within cluster. (C) Violin plots of Ephb4 and Efnb2 expression level across endothelial cell clusters. Normalized gene expression levels are plotted on the y-axis. (D) GO enrichment of DEGs for each endothelial cell cluster between water- and DSS-treated samples. Color represents adjusted p-value of GO enrichment annotation for each endothelial cell cluster; diameter represents gene ratio for each endothelial cell cluster. DEG, differentially expressed gene; DSS, dextran sulfate sodium; GO, gene ontology; LEC, lymphatic endothelial cell; UMAP, uniform manifold approximation and projection.
(TIF)
(A) Single-cell atlas of colon fibroblasts UMAP of fibroblast (dots) profiles (Methods) colored by cell type assignment from water (left) or DSS (right) samples. (B) Expression of canonical and newly characterized markers across fibroblast clusters. Color represents average expression of marker gene within clusters; diameter represents percentage expression of marker gene within cluster. (C) Expression of genes involved in maintaining colon crypt architecture. Color represents average expression of marker gene within clusters; diameter represents percentage expression of marker gene within cluster. DSS, dextran sulfate sodium; UMAP, uniform manifold approximation and projection.
(TIF)
(A) UMAP of combined analysis and clustering of stromal cell subsets identified in [21] and [16]. Cell clusters obtained in the combined analysis were annotated based on the cell annotations defined by Smillie colleagues, using the most frequently present broader cell type annotation to annotate each cluster. (B) Dotplot of gene set module scores of genes defined to be highly specific to broader level mouse fibroblast subsets (x-axis) computed for each redefined human fibroblast cell subsets from joint clustering analysis. Only specific marker gene lists with AUC >0.65 were considered for computing gene set module score. Mouse fibroblast cell subsets were redefined as broader level subsets: IstF, MAF, and MyoF. MAF3 cell subset from mouse colon data was renamed to MAF IL11-hi. (C) UMAP of combined analysis and clustering of stromal cell subsets identified in Smilie and colleagues and Kinchen and colleagues with redefined cell type annotations aligned with broader mouse fibroblast cell type annotations. (D) Spearman correlation estimates of pseudobulk gene expression levels for each fibroblast cell subset identified in our mouse colon stroma atlas compared to pseudobulk expression levels of orthologous genes in each fibroblast cell subsets identified in a combined analysis of stromal cell subsets from Smilie and colleagues and Kinchen and colleagues. Correlation estimates were scaled for each column. AUC, area under the curve; BEC, blood endothelial cell; IstF, interstitial fibroblast; LEC, lymphatic endothelial cell; MAF, mucosa-associated fibroblast; MyoF, myofibroblast; SMC, smooth muscle cell; UMAP, uniform manifold approximation and projection.
(TIF)
Heatmaps of select DEGs between water- and DSS-treated samples for all fibroblast clusters. Color represents normalized gene expression. Significant DEGs had FDR <0.05 using MAST (see Methods). DEG, differentially expressed gene; DSS, dextran sulfate sodium; FDR, false discovery rate; IstF, interstitial fibroblast; MAF, mucosa-associated fibroblast; MyoF, myofibroblast.
(TIF)
Heatmap of estimated interaction scores based on L–R pair expression between sender cell type MAF3 (IAFs) and receiver cell types: endothelial cell subsets (A) and fibroblast subsets (B). Interaction scores were computed separately for water-treated and DSS-treated mice. Heatmap displays L–R pairs that are differentially expressed between water-treated and DSS-treated mice; for example, ligands that are up-regulated in DSS in MAF3 and receptors that are up-regulated in receiving endothelial subsets (A) or other fibroblast subsets (B). DSS, dextran sulfate sodium; IAF, inflammation-associated fibroblast; L–R, ligand–receptor; MAF, mucosa-associated fibroblast; MyoF, myofibroblast.
(TIF)
(A) Average expression levels of top 10 genes enriched for expression in the gastrointestinal tissues and also differentially expressed in water- and DSS-treated conditions. Color represents average expression of marker genes across all stromal cell subsets; diameter represents percentage expression of marker genes. (B) Average expression of top 10 genes enriched for expression in the gastrointestinal tract and also differentially expressed in water- and DSS-treated conditions stratified by annotated stromal cell subsets. Color represents average expression of marker gene in each cluster; diameter represents percentage expression of marker gene within cluster. (C) Column scaled average transcript levels of GI tract–enriched genes in several human tissues profiled in the Human Protein Atlas study by Uhlen and colleagues. DEG, differentially expressed gene; DSS, dextran sulfate sodium; GI, gastrointestinal.
(TIF)
(A) UMAP projections of dimensionality reduction analysis on the subset of MAF and MyoF cells used for the construction of differentiation trajectory overlaid by their lineage subset labels. (B) Visualization of top 2 diffusion components (DC1) from diffusion map analysis of the subset of MAF and MyoF cells overlaid by their lineage subset labels. (C) Diffusion pseudotime reconstruction of the differentiation trajectory from MAFs to MyoFs. MAF, mucosa-associated fibroblast; MyoF, myofibroblast; UMAP, uniform manifold approximation and projection.
(TIF)
Mice were administered 2% DSS for 7 days, H2O subsequently, and killed on day 11, 13, or 24. HE staining (A) of representative images of colon following DSS in WT and Adamdec1 KO mice. Scale bar, 600 μm. Immunohistochemistry was performed for CD3 (B) and F4/80 (C) on day 11 or 13 after administration of DSS. Scale bars, 600 μm and 200 μm. Representative images, n = 2 per mice per genotype and time point. DSS, dextran sulfate sodium; HE, hematoxylin–eosin; KO, knockout; WT, wild-type.
(TIF)
IF imaging was performed on colons from Adamdec1 KO and WT mice with the indicated markers. Mice were administered 2% DSS for 7 days and killed on day 7. (A) Indicated ECM component (white). Scale bar, 200 μm. n = 3 per cohort, raw images representative of 2 experiments. (B) Quantification of indicated ECM components by % area. n = 3 mice per cohort. See S1 Data for source data. DSS, dextran sulfate sodium; ECM, extracellular matrix; IF, immunofluorescence; KO, knockout; WT, wild-type.
(TIF)
DEG, differentially expressed gene; DSS, dextran sulfate sodium.
(XLSX)
DEG, differentially expressed gene; DSS, dextran sulfate sodium.
(XLSX)
DEG, differentially expressed gene; DSS, dextran sulfate sodium.
(XLSX)
(XLSX)
(XLSX)
Acknowledgments
We thank the Broad Flow Cytometry Facility (Patricia Rogers, Chelsea Otis, Natan Pirete, and Stephanie Saldi), Elizabeth Creasey, Kathryn Devaney, Theresa Reimels, and Heather Kang for technical and editorial expertise.
Abbreviations
- BEC
blood endothelial cell
- BMP
bone morphogenetic protein
- CBF
crypt bottom fibroblast
- CCA
canonical correlation analysis
- CTF
crypt top fibroblast
- DEG
differentially expressed gene
- DGE
digital gene expression
- DSS
dextran sulfate sodium
- ECM
extracellular matrix
- FISH
fluorescent in situ hybridization
- HE
hematoxylin–eosin
- HEV
high endothelial venule
- IAF
inflammation-associated fibroblast
- IBD
inflammatory bowel disease
- ICC
interstitial cell of Cajal
- IF
immunofluorescence
- IGFBP
insulin growth factor binding protein
- ISH
in situ hybridization
- IstF
interstitial fibroblast
- KO
knockout
- LEC
lymphatic endothelial cell
- MAF
mucosa-associated fibroblast
- MNN
mutual nearest neighbor
- MSC
mesenchymal stem cell
- MyoF
myofibroblast
- NB
negative binomial
- PCA
principal component analysis
- scRNA-seq
single-cell RNA-sequencing
- SNP
single nucleotide polymorphism
- SVD
singular value decomposition
- UMAP
uniform manifold approximation and projection
- UMI
unique molecular identifier
- WAE
wound-associated epithelium
- WT
wild-type
Data Availability
Processed and raw data will be available on the Single Cell Portal: https://singlecell.broadinstitute.org/single_cell/study/SCP1711/mouse-colon-stroma-inflammation. Raw data is available at GEO (Accession Number: GSE172261). Source code for the analyses is deposited in GitHub (https://gitlab.com/xavier-lab-computation/public/stroma-dss).
Funding Statement
This study was supported by NIH grant (RC2 DK114784 to R.J.X., D.B.G., and A.R; DK043351 to R.J.X.) and the Klarman Cell Observatory. G.J.J. was supported by the Ford Foundation Fellowship and an NIH T32 grant (2T32DK007191-42). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505:327–34. doi: 10.1038/nature12984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gehart H, Clevers H. Tales from the crypt: new insights into intestinal stem cells. Nat Rev Gastroenterol Hepatol. 2019;16:19–34. doi: 10.1038/s41575-018-0081-y [DOI] [PubMed] [Google Scholar]
- 3.Chacón-Martínez CA, Koester J, Wickström SA. Signaling in the stem cell niche: regulating cell fate, function and plasticity. Development. 2018;145. doi: 10.1242/dev.165399 [DOI] [PubMed] [Google Scholar]
- 4.Nowarski R, Jackson R, Flavell RA. The Stromal Intervention: Regulation of Immunity and Inflammation at the Epithelial-Mesenchymal Barrier. Cell. 2017;168:362–75. doi: 10.1016/j.cell.2016.11.040 [DOI] [PubMed] [Google Scholar]
- 5.Owens BMJ. Inflammation, Innate Immunity, and the Intestinal Stromal Cell Niche: Opportunities and Challenges. Front Immunol. 2015;6:319. doi: 10.3389/fimmu.2015.00319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roulis M, Flavell RA. Fibroblasts and myofibroblasts of the intestinal lamina propria in physiology and disease. Differentiation. 2016;92:116–31. doi: 10.1016/j.diff.2016.05.002 [DOI] [PubMed] [Google Scholar]
- 7.Kosinski C, Li VSW, Chan ASY, Zhang J, Ho C, Tsui WY, et al. Gene expression patterns of human colon tops and basal crypts and BMP antagonists as intestinal stem cell niche factors. Proc Natl Acad Sci U S A. 2007;104:15418–23. doi: 10.1073/pnas.0707210104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gattazzo F, Urciuolo A, Bonaldo P. Extracellular matrix: a dynamic microenvironment for stem cell niche. Biochim Biophys Acta. 2014;1840:2506–19. doi: 10.1016/j.bbagen.2014.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meran L, Baulies A, Li VSW. Intestinal Stem Cell Niche: The Extracellular Matrix and Cellular Components. Stem Cells Int. 2017;2017:7970385. doi: 10.1155/2017/7970385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buechler MB, Turley SJ. A short field guide to fibroblast function in immunity. Semin Immunol. 2018;35:48–58. doi: 10.1016/j.smim.2017.11.001 [DOI] [PubMed] [Google Scholar]
- 11.Chang JE, Turley SJ. Stromal infrastructure of the lymph node and coordination of immunity. Trends Immunol. 2015;36:30–9. doi: 10.1016/j.it.2014.11.003 [DOI] [PubMed] [Google Scholar]
- 12.Comerford I, Harata-Lee Y, Bunting MD, Gregor C, Kara EE, McColl SR. A myriad of functions and complex regulation of the CCR7/CCL19/CCL21 chemokine axis in the adaptive immune system. Cytokine Growth Factor Rev. 2013;24:269–83. doi: 10.1016/j.cytogfr.2013.03.001 [DOI] [PubMed] [Google Scholar]
- 13.Luther SA, Bidgol A, Hargreaves DC, Schmidt A, Xu Y, Paniyadi J, et al. Differing activities of homeostatic chemokines CCL19, CCL21, and CXCL12 in lymphocyte and dendritic cell recruitment and lymphoid neogenesis. J Immunol. 2002;169:424–33. doi: 10.4049/jimmunol.169.1.424 [DOI] [PubMed] [Google Scholar]
- 14.West NR, Hegazy AN, Owens BMJ, Bullers SJ, Linggi B, Buonocore S, et al. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat Med. 2017;23:579–89. doi: 10.1038/nm.4307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rieder F, Fiocchi C, Rogler G. Mechanisms, Management, and Treatment of Fibrosis in Patients With Inflammatory Bowel Diseases. Gastroenterology. 2017;152:340–350.e6. doi: 10.1053/j.gastro.2016.09.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kinchen J, Chen HH, Parikh K, Antanaviciute A, Jagielowicz M, Fawkner-Corbett D, et al. Structural Remodeling of the Human Colonic Mesenchyme in Inflammatory Bowel Disease. Cell. 2018;175:372–386.e17. doi: 10.1016/j.cell.2018.08.067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Z, Wang L, Welch JD, Ma H, Zhou Y, Vaseghi HR, et al. Single-cell transcriptomics reconstructs fate conversion from fibroblast to cardiomyocyte. Nature. 2017;551:100–4. doi: 10.1038/nature24454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodda LB, Lu E, Bennett ML, Sokol CL, Wang X, Luther SA, et al. Single-Cell RNA Sequencing of Lymph Node Stromal Cells Reveals Niche-Associated Heterogeneity. Immunity. 2018;48:1014–1028.e6. doi: 10.1016/j.immuni.2018.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie T, Wang Y, Deng N, Huang G, Taghavifar F, Geng Y, et al. Single-Cell Deconvolution of Fibroblast Heterogeneity in Mouse Pulmonary Fibrosis. Cell Rep. 2018;22:3625–40. doi: 10.1016/j.celrep.2018.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8:14049. doi: 10.1038/ncomms14049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smillie CS, Biton M, Ordovas-Montanes J, Sullivan KM, Burgin G, Graham DB, et al. Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell. 2019;178:714–730.e22. doi: 10.1016/j.cell.2019.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chassaing B, Aitken JD, Malleshappa M, Vijay-Kumar M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr Protoc Immunol. 2014;104: 15.25.1–15.25.14. doi: 10.1002/0471142735.im1525s104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990;98:694–702. doi: 10.1016/0016-5085(90)90290-h [DOI] [PubMed] [Google Scholar]
- 24.Cremasco V, Astarita JL, Grauel AL, Keerthivasan S, MacIsaac K, Woodruff MC, et al. FAP Delineates Heterogeneous and Functionally Divergent Stromal Cells in Immune-Excluded Breast Tumors. Cancer Immunology. Research. 2018:1472–85. doi: 10.1158/2326-6066.cir-18-0098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fletcher AL, Malhotra D, Acton SE, Lukacs-Kornek V, Bellemare-Pelletier A, Curry M, et al. Reproducible isolation of lymph node stromal cells reveals site-dependent differences in fibroblastic reticular cells. Front Immunol Switzerland. 2011;2(35):2011. doi: 10.3389/fimmu.2011.00035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Banerji S, Ni J, Wang SX, Clasper S, Su J, Tammi R, et al. LYVE-1, a new homologue of the CD44 glycoprotein, is a lymph-specific receptor for hyaluronan. J Cell Biol. 1999;144:789–801. doi: 10.1083/jcb.144.4.789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Farr AG, Berry ML, Kim A, Nelson AJ, Welch MP, Aruffo A. Characterization and cloning of a novel glycoprotein expressed by stromal cells in T-dependent areas of peripheral lymphoid tissues. J Exp Med. 1992;176:1477–82. doi: 10.1084/jem.176.5.1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huizinga JD, Thuneberg L, Klüppel M, Malysz J, Mikkelsen HB, Bernstein A. W/kit gene required for interstitial cells of Cajal and for intestinal pacemaker activity. Nature. 1995;373:347–9. doi: 10.1038/373347a0 [DOI] [PubMed] [Google Scholar]
- 29.Malhotra D, Fletcher AL, Astarita J, Lukacs-Kornek V, Tayalia P, Gonzalez SF, et al. Immunological Genome Project Consortium Transcriptional profiling of stroma from inflamed and resting lymph nodes defines immunological hallmarks. Nat Immunol. 2012;13:499–510. doi: 10.1038/ni.2262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miano JM, Olson EN. Expression of the smooth muscle cell calponin gene marks the early cardiac and smooth muscle cell lineages during mouse embryogenesis. J Biol Chem. 1996;271:7095–103. doi: 10.1074/jbc.271.12.7095 [DOI] [PubMed] [Google Scholar]
- 31.Schlingemann RO, Dingjan GM, Blok J, Emeis JJ, Warnaar SO, Ruiter DJ. Monoclonal antibody PAL-E specific for endothelium. Lab Invest. 1985;52:71–6. [PubMed] [Google Scholar]
- 32.Stan RV, Ghitescu L, Jacobson BS, Palade GE. Isolation, cloning, and localization of rat PV-1, a novel endothelial caveolar protein. J Cell Biol. 1999;145:1189–98. doi: 10.1083/jcb.145.6.1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takahashi K, Hiwada K, Kokubu T. Vascular smooth muscle calponin. A novel troponin T-like protein. Hypertension. 1988:620–6. doi: 10.1161/01.hyp.11.6.620 [DOI] [PubMed] [Google Scholar]
- 34.Roulis M, Kaklamanos A, Schernthanner M, Bielecki P, Zhao J, Kaffe E, et al. Paracrine orchestration of intestinal tumorigenesis by a mesenchymal niche. Nature. 2020;580:524–9. doi: 10.1038/s41586-020-2166-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fazilaty H, Brügger MD, Valenta T, Szczerba BM, Berkova L, Doumpas N, et al. Tracing colonic embryonic transcriptional profiles and their reactivation upon intestinal damage. Cell Rep. 2021;36:109484. doi: 10.1016/j.celrep.2021.109484 [DOI] [PubMed] [Google Scholar]
- 36.Danese S, Sans M, de la Motte C, Graziani C, West G, Phillips MH, et al. Angiogenesis as a novel component of inflammatory bowel disease pathogenesis. Gastroenterology. 2006;130:2060–73. doi: 10.1053/j.gastro.2006.03.054 [DOI] [PubMed] [Google Scholar]
- 37.Alkim C, Alkim H, Koksal AR, Boga S, Sen I. Angiogenesis in Inflammatory Bowel Disease. Int J Inflamm. 2015:1–10. doi: 10.1155/2015/970890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rahier J-F, De Beauce S, Dubuquoy L, Erdual E, Colombel J-F, Jouret-Mourin A, et al. Increased lymphatic vessel density and lymphangiogenesis in inflammatory bowel disease. Aliment Pharmacol Ther. 2011;34:533–43. doi: 10.1111/j.1365-2036.2011.04759.x [DOI] [PubMed] [Google Scholar]
- 39.Pedica F, Ligorio C, Tonelli P, Bartolini S, Baccarini P. Lymphangiogenesis in Crohn’s disease: an immunohistochemical study using monoclonal antibody D2-40. Virchows Arch. 2008;452:57–63. doi: 10.1007/s00428-007-0540-2 [DOI] [PubMed] [Google Scholar]
- 40.Chudy-Onwugaje KO, Christian KE, Farraye FA, Cross RK. A State-of-the-Art Review of New and Emerging Therapies for the Treatment of IBD. Inflamm Bowel Dis. 2019;25:820–30. doi: 10.1093/ibd/izy327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neurath M. Current and emerging therapeutic targets for IBD. Nat Rev Gastroenterol Hepatol. 2017;14:688. doi: 10.1038/nrgastro.2017.138 [DOI] [PubMed] [Google Scholar]
- 42.Su T, Stanley G, Sinha R, D’Amato G, Das S, Rhee S, et al. Single-cell analysis of early progenitor cells that build coronary arteries. Nature. 2018;559:356–62. doi: 10.1038/s41586-018-0288-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Castillo M, Alvarez H. Artery or Vein: To Be or Not To Be? AJNR Am J Neuroradiol. 2011;32:791–3. doi: 10.3174/ajnr.A2266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adams RH. Molecular control of arterial—venous blood vessel identity. J Anat. 2003;202:105–12. doi: 10.1046/j.1469-7580.2003.00137.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.dela Paz NG, D’Amore PA. Arterial versus venous endothelial cells. Cell Tissue Res. 2009;335:5–16. doi: 10.1007/s00441-008-0706-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Füller T, Korff T, Kilian A, Dandekar G, Augustin HG. Forward EphB4 signaling in endothelial cells controls cellular repulsion and segregation from ephrinB2 positive cells. J Cell Sci. 2003;116:2461–70. doi: 10.1242/jcs.00426 [DOI] [PubMed] [Google Scholar]
- 47.Swift MR, Weinstein BM. Arterial–Venous Specification During Development. Circ Res. 2009;104:576–88. doi: 10.1161/CIRCRESAHA.108.188805 [DOI] [PubMed] [Google Scholar]
- 48.Zaferani A, Talsma DT, Yazdani S, Celie JWAM, Aikio M, Heljasvaara R, et al. Basement membrane zone collagens XV and XVIII/proteoglycans mediate leukocyte influx in renal ischemia/reperfusion. PLoS ONE. 2014;9:e106732. doi: 10.1371/journal.pone.0106732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trombetta-eSilva J. The Function of SPARC as a Mediator of Fibrosis. Open Rheumatol J. 2012:146–55. doi: 10.2174/1874312901206010146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang J-C, Lai S, Guo X, Zhang X, de Crombrugghe B, Sonnylal S, et al. Attenuation of fibrosis in vitro and in vivo with SPARC siRNA. Arthritis Res Ther. 2010;12:R60. doi: 10.1186/ar2973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ng Y-L, Klopcic B, Lloyd F, Forrest C, Greene W, Lawrance IC. Secreted Protein Acidic and Rich in Cysteine (SPARC) Exacerbates Colonic Inflammatory Symptoms in Dextran Sodium Sulphate-Induced Murine Colitis. PLoS ONE. 2013:e77575. doi: 10.1371/journal.pone.0077575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thomas S, Baumgart DC. Targeting leukocyte migration and adhesion in Crohn’s disease and ulcerative colitis. Inflammopharmacology. 2012:1–18. doi: 10.1007/s10787-011-0104-6 [DOI] [PubMed] [Google Scholar]
- 53.Ogawa H, Binion DG, Heidemann J, Theriot M, Fisher PJ, Johnson NA, et al. Mechanisms of MAdCAM-1 gene expression in human intestinal microvascular endothelial cells. Am J Physiol Cell Physiol. 2005;288:C272–81. doi: 10.1152/ajpcell.00406.2003 [DOI] [PubMed] [Google Scholar]
- 54.Hsu DR, Economides AN, Wang X, Eimon PM, Harland RM. The Xenopus dorsalizing factor Gremlin identifies a novel family of secreted proteins that antagonize BMP activities. Mol Cell. 1998;1:673–83. doi: 10.1016/s1097-2765(00)80067-2 [DOI] [PubMed] [Google Scholar]
- 55.Worthley DL, Churchill M, Compton JT, Tailor Y, Rao M, Si Y, et al. Gremlin 1 identifies a skeletal stem cell with bone, cartilage, and reticular stromal potential. Cell. 2015;160:269–84. doi: 10.1016/j.cell.2014.11.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brügger MD, Valenta T, Fazilaty H, Hausmann G, Basler K. Distinct populations of crypt-associated fibroblasts act as signaling hubs to control colon homeostasis. PLoS Biol. 2020;18:e3001032. doi: 10.1371/journal.pbio.3001032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McCarthy N, Manieri E, Storm EE, Saadatpour A, Luoma AM, Kapoor VN, et al. Distinct Mesenchymal Cell Populations Generate the Essential Intestinal BMP Signaling Gradient. Cell Stem Cell. 2020;26:391–402.e5. doi: 10.1016/j.stem.2020.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McCarthy N, Kraiczy J, Shivdasani RA. Cellular and molecular architecture of the intestinal stem cell niche. Nat Cell Biol. 2020;22:1033–41. doi: 10.1038/s41556-020-0567-z [DOI] [PubMed] [Google Scholar]
- 59.Loe AKH, Rao-Bhatia A, Kim J-E, Kim T-H. Mesenchymal Niches for Digestive Organ Development, Homeostasis, and Disease. Trends Cell Biol. 2021;31:152–65. doi: 10.1016/j.tcb.2020.11.010 [DOI] [PubMed] [Google Scholar]
- 60.Hardwick J, Van Den Brink G, Van Den Brande J, Van Deventer S. Bone morphogenetic protein 2 is expressed by, and acts upon, mature epithelial cells in the colon. Gastroenterology. 2003:A458. doi: 10.1016/s0016-5085(03)82315-6 [DOI] [PubMed] [Google Scholar]
- 61.He XC, Zhang J, Tong W-G, Tawfik O, Ross J, Scoville DH, et al. BMP signaling inhibits intestinal stem cell self-renewal through suppression of Wnt–β-catenin signaling. Nat Genet. 2004;36:1117–21. doi: 10.1038/ng1430 [DOI] [PubMed] [Google Scholar]
- 62.Qi Z, Li Y, Zhao B, Xu C, Liu Y, Li H, et al. BMP restricts stemness of intestinal Lgr5+ stem cells by directly suppressing their signature genes. Nat Commun. 2017;8:13824. doi: 10.1038/ncomms13824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ulvmar MH, Werth K, Braun A, Kelay P, Hub E, Eller K, et al. The atypical chemokine receptor CCRL1 shapes functional CCL21 gradients in lymph nodes. Nat Immunol. 2014;15:623–30. doi: 10.1038/ni.2889 [DOI] [PubMed] [Google Scholar]
- 64.Buechler MB, Pradhan RN, Krishnamurty AT, Cox C, Calviello AK, Wang AW, et al. Cross-tissue organization of the fibroblast lineage. Nature. 2021;593:575–9. doi: 10.1038/s41586-021-03549-5 [DOI] [PubMed] [Google Scholar]
- 65.Graham DB, Jasso GJ, Mok A, Goel G, Ng ACY, Kolde R, et al. Nitric Oxide Engages an Anti-inflammatory Feedback Loop Mediated by Peroxiredoxin 5 in Phagocytes. Cell Rep. 2018;24:838–50. doi: 10.1016/j.celrep.2018.06.081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hecker L, Logsdon NJ, Kurundkar D, Kurundkar A, Bernard K, Hock T, et al. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci Transl Med. 2014;6:231ra47. doi: 10.1126/scitranslmed.3008182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kavian N, Mehlal S, Jeljeli M, Saidu NEB, Nicco C, Cerles O, et al. The Nrf2-Antioxidant Response Element Signaling Pathway Controls Fibrosis and Autoimmunity in Scleroderma. Front Immunol. 2018;9:1896. doi: 10.3389/fimmu.2018.01896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim HJ, Vaziri ND. Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am J Physiol Renal Physiol. 2010;298:F662–71. doi: 10.1152/ajprenal.00421.2009 [DOI] [PubMed] [Google Scholar]
- 69.Kikuchi N, Ishii Y, Morishima Y, Yageta Y, Haraguchi N, Itoh K, et al. Nrf2 protects against pulmonary fibrosis by regulating the lung oxidant level and Th1/Th2 balance. Respir Res. 2010;11:31. doi: 10.1186/1465-9921-11-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mouw JK, Ou G, Weaver VM. Extracellular matrix assembly: a multiscale deconstruction. Nat Rev Mol Cell Biol. 2014;15:771–85. doi: 10.1038/nrm3902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Humphrey JD, Dufresne ER, Schwartz MA. Mechanotransduction and extracellular matrix homeostasis. Nat Rev Mol Cell Biol. 2014;15:802–12. doi: 10.1038/nrm3896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Petrey AC, de la Motte CA. The extracellular matrix in IBD: a dynamic mediator of inflammation. Curr Opin Gastroenterol. 2017;33:234–8. doi: 10.1097/MOG.0000000000000368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ippolito C, Colucci R, Segnani C, Errede M, Girolamo F, Virgintino D, et al. Fibrotic and Vascular Remodelling of Colonic Wall in Patients with Active Ulcerative Colitis. J Crohns Colitis. 2016;10:1194–204. doi: 10.1093/ecco-jcc/jjw076 [DOI] [PubMed] [Google Scholar]
- 74.Moriggi M, Pastorelli L, Torretta E, Tontini GE, Capitanio D, Bogetto SF, et al. Contribution of Extracellular Matrix and Signal Mechanotransduction to Epithelial Cell Damage in Inflammatory Bowel Disease Patients: A Proteomic Study. Proteomics. 2017;17. doi: 10.1002/pmic.201700164 [DOI] [PubMed] [Google Scholar]
- 75.Allard JB, Duan C. IGF-Binding Proteins: Why Do They Exist and Why Are There So Many? Front Endocrinol. 2018;9:117. doi: 10.3389/fendo.2018.00117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee JC, Biasci D, Roberts R, Gearry RB, Mansfield JC, Ahmad T, et al. Genome-wide association study identifies distinct genetic contributions to prognosis and susceptibility in Crohn’s disease. Nat Genet. 2017;49:262–8. doi: 10.1038/ng.3755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Eivindson M, Grønbæk H, Flyvbjerg A, Frystyk J, Zimmermann-Nielsen E, Dahlerup JF. The insulin-like growth factor (IGF)-system in active ulcerative colitis and Crohn’s disease: Relations to disease activity and corticosteroid treatment. Growth Horm IGF Res. 2007:33–40. doi: 10.1016/j.ghir.2006.10.003 [DOI] [PubMed] [Google Scholar]
- 78.Street ME, Miraki-Moud F, Sanderson IR, Savage MO, Giovannelli G, Bernasconi S, et al. Interleukin-1beta (IL-1beta) and IL-6 modulate insulin-like growth factor-binding protein (IGFBP) secretion in colon cancer epithelial (Caco-2) cells. J Endocrinol. 2003;179:405–15. doi: 10.1677/joe.0.1790405 [DOI] [PubMed] [Google Scholar]
- 79.Zimmermann EM, Li L, Hou YT, Mohapatra NK, Pucilowska JB. Insulin-like growth factor I and insulin-like growth factor binding protein 5 in Crohn’s disease. Am J Physiol Gastrointest Liver Physiol. 2001;280:G1022–9. doi: 10.1152/ajpgi.2001.280.5.G1022 [DOI] [PubMed] [Google Scholar]
- 80.Yasuoka H, Hsu E, Ruiz XD, Steinman RA, Choi AMK, Feghali-Bostwick CA. The fibrotic phenotype induced by IGFBP-5 is regulated by MAPK activation and egr-1-dependent and -independent mechanisms. Am J Pathol. 2009;175:605–15. doi: 10.2353/ajpath.2009.080991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nguyen X-X, Muhammad L, Nietert PJ, Feghali-Bostwick C. IGFBP-5 Promotes Fibrosis via Increasing Its Own Expression and That of Other Pro-fibrotic Mediators. Front Endocrinol. 2018. doi: 10.3389/fendo.2018.00601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Flynn RS, Mahavadi S, Murthy KS, Grider JR, Kellum JM, Akbari H, et al. Endogenous IGFBP-3 regulates excess collagen expression in intestinal smooth muscle cells of Crohnʼs disease strictures. Inflamm Bowel Dis. 2011:193–201. doi: 10.1002/ibd.21351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ng B, Dong J, Viswanathan S, D’Agostino GA, Widjaja AA, Lim W-W, et al. IL-11 Is a Therapeutic Target in Idiopathic Pulmonary Fibrosis. A29. Sci Transl Med. 2019. doi: 10.1126/scitranslmed.aaw1237 [DOI] [PubMed] [Google Scholar]
- 84.Schafer S, Viswanathan S, Widjaja AA, Lim W-W, Moreno-Moral A, DeLaughter DM, et al. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature. 2017;552:110–5. doi: 10.1038/nature24676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Widjaja AA, Singh BK, Adami E, Viswanathan S, Dong J, D’Agostino GA, et al. Inhibiting Interleukin 11 Signaling Reduces Hepatocyte Death and Liver Fibrosis, Inflammation, and Steatosis in Mouse Models of Nonalcoholic Steatohepatitis. Gastroenterology. 2019;157:777–792.e14. doi: 10.1053/j.gastro.2019.05.002 [DOI] [PubMed] [Google Scholar]
- 86.Wei K, Korsunsky I, Marshall JL, Gao A, Watts GFM, Major T, et al. Notch signalling drives synovial fibroblast identity and arthritis pathology. Nature. 2020;582:259–64. doi: 10.1038/s41586-020-2222-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
- 88.Evans RA, a Chung Tian Y, Steadman R, Phillips AO. TGF-β1-mediated fibroblast–myofibroblast terminal differentiation—the role of smad proteins. Exp Cell Res. 2003:90–100. doi: 10.1016/s0014-4827(02)00015-0 [DOI] [PubMed] [Google Scholar]
- 89.Pascal RR, Kaye GI, Lane N. Colonic Pericryptal Fibroblast Sheath: Replication, Migration, and Cytodifferentiation of a Mesenchymal Cell System in Adult Tissue. Gastroenterology. 1968:835–51. doi: 10.1016/s0016-5085(68)80155-6 [DOI] [PubMed] [Google Scholar]
- 90.Bates E, Fridman W, Mueller C. The ADAMDEC1 (decysin) gene structure: evolution by duplication in a metalloprotease gene cluster on Chromosome 8p12. Immunogenetics. 2002:96–105. doi: 10.1007/s00251-002-0430-3 [DOI] [PubMed] [Google Scholar]
- 91.Lund J, Olsen OH, Sørensen ES, Stennicke HR, Petersen HH, Overgaard MT. ADAMDEC1 Is a Metzincin Metalloprotease with Dampened Proteolytic Activity. J Biol Chem. 2013:21367–75. doi: 10.1074/jbc.M113.474536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Consortium GTEx. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45:580–5. doi: 10.1038/ng.2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.McInnes G, Tanigawa Y, DeBoever C, Lavertu A, Olivieri JE, Aguirre M, et al. Global Biobank Engine: enabling genotype-phenotype browsing for biobank summary statistics. Bioinformatics. 2019:2495–7. doi: 10.1093/bioinformatics/bty999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018:203–9. doi: 10.1038/s41586-018-0579-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jones JC, Rustagi S, Dempsey PJ. ADAM Proteases and Gastrointestinal Function. Annu Rev Physiol. 2016;78:243–76. doi: 10.1146/annurev-physiol-021014-071720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Seals DF. The ADAMs family of metalloproteases: multidomain proteins with multiple functions. Genes Dev. 2003:7–30. doi: 10.1101/gad.1039703 [DOI] [PubMed] [Google Scholar]
- 97.Atkinson JC, Rühl M, Becker J, Ackermann R, Schuppan D. Collagen VI Regulates Normal and Transformed Mesenchymal Cell Proliferationin Vitro. Exp Cell Res. 1996:283–91. doi: 10.1006/excr.1996.0328 [DOI] [PubMed] [Google Scholar]
- 98.Rockey DC, Darwin Bell P, Hill JA. Fibrosis—A Common Pathway to Organ Injury and Failure. N Engl J Med. 2015:1138–49. doi: 10.1056/NEJMra1300575 [DOI] [PubMed] [Google Scholar]
- 99.Mizoguchi F, Slowikowski K, Wei K, Marshall JL, Rao DA, Chang SK, et al. Functionally distinct disease-associated fibroblast subsets in rheumatoid arthritis. Nat Commun. 2018. doi: 10.1038/s41467-018-02892-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhang F, Accelerating Medicines Partnership Rheumatoid Arthritis and Systemic Lupus Erythematosus (AMP RA/SLE) Consortium, Wei K, Slowikowski K, Fonseka CY, Rao DA, et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat Immunol. 2019. pp. 928–42. doi: 10.1038/s41590-019-0378-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Croft AP, Campos J, Jansen K, Turner JD, Marshall J, Attar M, et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature. 2019;570:246–51. doi: 10.1038/s41586-019-1263-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nishina T, Deguchi Y, Ohshima D, Takeda W, Ohtsuka M, Shichino S, et al. Interleukin-11-expressing fibroblasts have a unique gene signature correlated with poor prognosis of colorectal cancer. Nat Commun. 2021;12:2281. doi: 10.1038/s41467-021-22450-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lim W-W, Ng B, Widjaja A, Xie C, Su L, Ko N, et al. Interleukin 11 expression causes murine inflammatory bowel disease. bioRxiv. 2019:756098. doi: 10.1101/756098 [DOI] [Google Scholar]
- 104.Martin JC, Chang C, Boschetti G, Ungaro R, Giri M, Grout JA, et al. Single-Cell Analysis of Crohn’s Disease Lesions Identifies a Pathogenic Cellular Module Associated with Resistance to Anti-TNF Therapy. Cell. 2019;178:1493–1508.e20. doi: 10.1016/j.cell.2019.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rodrigues M, Kosaric N, Bonham CA, Gurtner GC. Wound Healing: A Cellular Perspective. Physiol Rev. 2019:665–706. doi: 10.1152/physrev.00067.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rognoni E, Watt FM. Skin Cell Heterogeneity in Development, Wound Healing, and Cancer. Trends Cell Biol. 2018;28:709–22. doi: 10.1016/j.tcb.2018.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Miyoshi H. Wnt-expressing cells in the intestines: guides for tissue remodeling. J Biochem. 2017:19–25. doi: 10.1093/jb/mvw070 [DOI] [PubMed] [Google Scholar]
- 108.Miyoshi H, Ajima R, Luo CT, Yamaguchi TP, Stappenbeck TS. Wnt5a Potentiates TGF-β Signaling to Promote Colonic Crypt Regeneration After Tissue Injury. Science. 2012:108–13. doi: 10.1126/science.1223821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Leoni G, Neumann P-A, Sumagin R, Denning TL, Nusrat A. Wound repair: role of immune–epithelial interactions. Mucosal Immunol. 2015:959–68. doi: 10.1038/mi.2015.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Driskell RR, Lichtenberger BM, Hoste E, Kretzschmar K, Simons BD, Charalambous M, et al. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature. 2013;504:277–81. doi: 10.1038/nature12783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rinkevich Y, Walmsley GG, Hu MS, Maan ZN, Newman AM, Drukker M, et al. Identification and isolation of a dermal lineage with intrinsic fibrogenic potential. Science. 2015:aaa2151–1. doi: 10.1126/science.aaa2151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tabib T, Morse C, Wang T, Chen W, Lafyatis R. SFRP2/DPP4 and FMO1/LSP1 Define Major Fibroblast Populations in Human Skin. J Investig Dermatol. 2018:802–10. doi: 10.1016/j.jid.2017.09.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Guerrero-Juarez CF, Dedhia PH, Jin S, Ruiz-Vega R, Ma D, Liu Y, et al. Single-cell analysis reveals fibroblast heterogeneity and myeloid-derived adipocyte progenitors in murine skin wounds. Nat Commun. 2019. doi: 10.1038/s41467-018-08247-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lambrecht BN, Vanderkerken M, Hammad H. The emerging role of ADAM metalloproteinases in immunity. Nat Rev Immunol. 2018:745–58. doi: 10.1038/s41577-018-0068-5 [DOI] [PubMed] [Google Scholar]
- 115.Giebeler N, Zigrino P. A Disintegrin and Metalloprotease (ADAM): Historical Overview of Their Functions. Toxins. 2016:122. doi: 10.3390/toxins8040122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chen R, Jin G, McIntyre TM. The soluble protease ADAMDEC1 released from activated platelets hydrolyzes platelet membrane pro-epidermal growth factor (EGF) to active high-molecular-weight EGF. J Biol Chem. 2017:10112–22. doi: 10.1074/jbc.M116.771642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jimenez-Pascual A, Hale JS, Kordowski A, Pugh J, Silver DJ, Bayik D, et al. ADAMDEC1 Maintains a Growth Factor Signaling Loop in Cancer Stem Cells. Cancer. Discovery. 2019:1574–89. doi: 10.1158/2159-8290.CD-18-1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.O’Shea NR, Chew TS, Dunne J, Marnane R, Nedjat-Shokouhi B, Smith PJ, et al. Critical Role of the Disintegrin Metalloprotease ADAM-like Decysin-1 [ADAMDEC1] for Intestinal Immunity and Inflammation. J Crohns Colitis. 2016:1417–27. doi: 10.1093/ecco-jcc/jjw111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wolock SL, Lopez R, Klein AM. Scrublet: computational identification of cell doublets in single-cell transcriptomic data. doi: 10.1101/357368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hafemeister C, Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. doi: 10.1101/576827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, et al. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015. doi: 10.1186/s13059-015-0844-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R Package for Comparing Biological Themes Among Gene Clusters. OMICS, A Journal of Integrative Biology. 2012:284–7. doi: 10.1089/omi.2011.0118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature. 2019;566:496–502. doi: 10.1038/s41586-019-0969-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, et al. Reversed graph embedding resolves complex single-cell trajectories. Nat Methods. 2017:979–82. doi: 10.1038/nmeth.4402 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Chronic DSS murine model: Mice were fed 3 iterative cycles of 2.5% DSS for 7 days followed by water for 7 days. (B) Masson’s trichrome staining of colons from water- and chronic DSS-fed mice after 1 round (day 14), 2 rounds (day 28), and 3 rounds (day 42) of DSS. Collagen accumulation in blue, as demarcated by yellow arrows. Leukocyte infiltrates, as demarcated by yellow arrowheads. Scale bar, 200 μm. n = 2, representative of 2 experiments. (C) Single-cell atlas of the murine colonic stroma. UMAP of stroma cells (dots) colored by cell type assignment from water (top) or DSS (middle) samples and combined embedding of both conditions (bottom panel). (D) Expression of lineage-specific marker genes across cell type subsets. Color represents average expression of marker gene within clusters; diameter represents percentage expression of marker gene within cluster. BEC, blood endothelial cell; DSS, dextran sulfate sodium; ICC, interstitial cell of Cajal; LEC, lymphatic endothelial cell; MSC, mesenchymal stem cell; SMC, smooth muscle cell; UMAP, uniform manifold approximation and projection.
(TIF)
(A) Single-cell atlas of colon fibroblasts UMAP of endothelial cell (dots) profiles (Methods) colored by cell type assignment from water (top) or DSS (bottom) samples. (B) Expression of canonical markers across endothelial cell clusters. Color represents average expression of marker gene within clusters; diameter represents percentage expression of marker gene within cluster. (C) Violin plots of Ephb4 and Efnb2 expression level across endothelial cell clusters. Normalized gene expression levels are plotted on the y-axis. (D) GO enrichment of DEGs for each endothelial cell cluster between water- and DSS-treated samples. Color represents adjusted p-value of GO enrichment annotation for each endothelial cell cluster; diameter represents gene ratio for each endothelial cell cluster. DEG, differentially expressed gene; DSS, dextran sulfate sodium; GO, gene ontology; LEC, lymphatic endothelial cell; UMAP, uniform manifold approximation and projection.
(TIF)
(A) Single-cell atlas of colon fibroblasts UMAP of fibroblast (dots) profiles (Methods) colored by cell type assignment from water (left) or DSS (right) samples. (B) Expression of canonical and newly characterized markers across fibroblast clusters. Color represents average expression of marker gene within clusters; diameter represents percentage expression of marker gene within cluster. (C) Expression of genes involved in maintaining colon crypt architecture. Color represents average expression of marker gene within clusters; diameter represents percentage expression of marker gene within cluster. DSS, dextran sulfate sodium; UMAP, uniform manifold approximation and projection.
(TIF)
(A) UMAP of combined analysis and clustering of stromal cell subsets identified in [21] and [16]. Cell clusters obtained in the combined analysis were annotated based on the cell annotations defined by Smillie colleagues, using the most frequently present broader cell type annotation to annotate each cluster. (B) Dotplot of gene set module scores of genes defined to be highly specific to broader level mouse fibroblast subsets (x-axis) computed for each redefined human fibroblast cell subsets from joint clustering analysis. Only specific marker gene lists with AUC >0.65 were considered for computing gene set module score. Mouse fibroblast cell subsets were redefined as broader level subsets: IstF, MAF, and MyoF. MAF3 cell subset from mouse colon data was renamed to MAF IL11-hi. (C) UMAP of combined analysis and clustering of stromal cell subsets identified in Smilie and colleagues and Kinchen and colleagues with redefined cell type annotations aligned with broader mouse fibroblast cell type annotations. (D) Spearman correlation estimates of pseudobulk gene expression levels for each fibroblast cell subset identified in our mouse colon stroma atlas compared to pseudobulk expression levels of orthologous genes in each fibroblast cell subsets identified in a combined analysis of stromal cell subsets from Smilie and colleagues and Kinchen and colleagues. Correlation estimates were scaled for each column. AUC, area under the curve; BEC, blood endothelial cell; IstF, interstitial fibroblast; LEC, lymphatic endothelial cell; MAF, mucosa-associated fibroblast; MyoF, myofibroblast; SMC, smooth muscle cell; UMAP, uniform manifold approximation and projection.
(TIF)
Heatmaps of select DEGs between water- and DSS-treated samples for all fibroblast clusters. Color represents normalized gene expression. Significant DEGs had FDR <0.05 using MAST (see Methods). DEG, differentially expressed gene; DSS, dextran sulfate sodium; FDR, false discovery rate; IstF, interstitial fibroblast; MAF, mucosa-associated fibroblast; MyoF, myofibroblast.
(TIF)
Heatmap of estimated interaction scores based on L–R pair expression between sender cell type MAF3 (IAFs) and receiver cell types: endothelial cell subsets (A) and fibroblast subsets (B). Interaction scores were computed separately for water-treated and DSS-treated mice. Heatmap displays L–R pairs that are differentially expressed between water-treated and DSS-treated mice; for example, ligands that are up-regulated in DSS in MAF3 and receptors that are up-regulated in receiving endothelial subsets (A) or other fibroblast subsets (B). DSS, dextran sulfate sodium; IAF, inflammation-associated fibroblast; L–R, ligand–receptor; MAF, mucosa-associated fibroblast; MyoF, myofibroblast.
(TIF)
(A) Average expression levels of top 10 genes enriched for expression in the gastrointestinal tissues and also differentially expressed in water- and DSS-treated conditions. Color represents average expression of marker genes across all stromal cell subsets; diameter represents percentage expression of marker genes. (B) Average expression of top 10 genes enriched for expression in the gastrointestinal tract and also differentially expressed in water- and DSS-treated conditions stratified by annotated stromal cell subsets. Color represents average expression of marker gene in each cluster; diameter represents percentage expression of marker gene within cluster. (C) Column scaled average transcript levels of GI tract–enriched genes in several human tissues profiled in the Human Protein Atlas study by Uhlen and colleagues. DEG, differentially expressed gene; DSS, dextran sulfate sodium; GI, gastrointestinal.
(TIF)
(A) UMAP projections of dimensionality reduction analysis on the subset of MAF and MyoF cells used for the construction of differentiation trajectory overlaid by their lineage subset labels. (B) Visualization of top 2 diffusion components (DC1) from diffusion map analysis of the subset of MAF and MyoF cells overlaid by their lineage subset labels. (C) Diffusion pseudotime reconstruction of the differentiation trajectory from MAFs to MyoFs. MAF, mucosa-associated fibroblast; MyoF, myofibroblast; UMAP, uniform manifold approximation and projection.
(TIF)
Mice were administered 2% DSS for 7 days, H2O subsequently, and killed on day 11, 13, or 24. HE staining (A) of representative images of colon following DSS in WT and Adamdec1 KO mice. Scale bar, 600 μm. Immunohistochemistry was performed for CD3 (B) and F4/80 (C) on day 11 or 13 after administration of DSS. Scale bars, 600 μm and 200 μm. Representative images, n = 2 per mice per genotype and time point. DSS, dextran sulfate sodium; HE, hematoxylin–eosin; KO, knockout; WT, wild-type.
(TIF)
IF imaging was performed on colons from Adamdec1 KO and WT mice with the indicated markers. Mice were administered 2% DSS for 7 days and killed on day 7. (A) Indicated ECM component (white). Scale bar, 200 μm. n = 3 per cohort, raw images representative of 2 experiments. (B) Quantification of indicated ECM components by % area. n = 3 mice per cohort. See S1 Data for source data. DSS, dextran sulfate sodium; ECM, extracellular matrix; IF, immunofluorescence; KO, knockout; WT, wild-type.
(TIF)
DEG, differentially expressed gene; DSS, dextran sulfate sodium.
(XLSX)
DEG, differentially expressed gene; DSS, dextran sulfate sodium.
(XLSX)
DEG, differentially expressed gene; DSS, dextran sulfate sodium.
(XLSX)
(XLSX)
(XLSX)
Data Availability Statement
Processed and raw data will be available on the Single Cell Portal: https://singlecell.broadinstitute.org/single_cell/study/SCP1711/mouse-colon-stroma-inflammation. Raw data is available at GEO (Accession Number: GSE172261). Source code for the analyses is deposited in GitHub (https://gitlab.com/xavier-lab-computation/public/stroma-dss).