

Abstract
Objectives
OA is a complex genetic disease with different risk factors contributing to its development. One of the genes, TNFRSF11B, previously identified with gain-of-function mutation in a family with early-onset OA with chondrocalcinosis, is among the highest upregulated genes in lesioned OA cartilage (RAAK-study). Here, we determined the role of TNFRSF11B overexpression in development of OA.
Methods
Human primary articular chondrocytes (9 donors RAAK study) were transduced using lentiviral particles with or without TNFRSF11B. Cells were cultured for 1 week in a 3 D in-vitro chondrogenic model. TNFRSF11B overexpression was confirmed by RT-qPCR, immunohistochemistry and ELISA. Effects of TNFRSF11B overexpression on cartilage matrix deposition, matrix mineralization, and genes highly correlated to TNFRSF11B in RNA-sequencing dataset (r >0.75) were determined by RT-qPCR. Additionally, glycosaminoglycans and collagen deposition were visualized with Alcian blue staining and immunohistochemistry (COL1 and COL2).
Results
Overexpression of TNFRSF11B resulted in strong upregulation of MMP13, COL2A1 and COL1A1. Likewise, mineralization and osteoblast characteristic markers RUNX2, ASPN and OGN showed a consistent increase. Among 30 genes highly correlated to TNFRSF11B, expression of only eight changed significantly, with BMP6 showing the highest increase (9-fold) while expression of RANK and RANKL remained unchanged indicating previously unknown downstream pathways of TNFRSF11B in cartilage.
Conclusion
Results of our 3D in vitro chondrogenesis model indicate that upregulation of TNFRSF11B in lesioned OA cartilage may act as a direct driving factor for chondrocyte to osteoblast transition observed in OA pathophysiology. This transition does not appear to act via the OPG/RANK/RANKL triad common in bone remodeling.
Keywords: osteoarthritis, TNFRSF11B, matrix mineralization, cartilage hypertrophy, chondrogenesis, CCAL1
Rheumatology key messages
TNFRSF11B in cartilage is a direct driving factor for chondrocyte-to-osteoblast transition observed in OA pathophysiology.
Chondrocyte-to-osteoblast transition in cartilage does not act via the OPG/RANK/RANKL triad, common in bone remodeling.
Likely, the TNFRSF11B-induced cartilage mineralization is accomplished via BMP6 signalling.
Introduction
OA is a common degenerative disorder characterized by cartilage extracellular matrix degradation (ECM) and changes in subchondral bone. Being marked by pain and disability, no effective therapy is available, and current treatments are focussed on pain relief or joint surgery at end-stage disease, creating a great social and economic burden [1, 2]. To characterize the OA pathophysiological process, multiple studies [3, 4] have performed transcriptome-wide analyses of preserved and lesioned cartilage. Among the most consistent and highly upregulated genes in lesioned OA cartilage is the TNF receptor superfamily member 11 b (TNFRSF11B) at the CCAL1 (chondrocalcinosis) locus [4, 5], encoding osteoprotegerin (OPG). OPG is a decoy receptor for the binding of nuclear factor KB ligand (RANKL) to the receptor activator of the nuclear KB factor (RANK). Together, this triad is well known for tightly regulating osteoclastogenesis, hence playing a critical role in bone formation, endochondral ossification and bone remodelling [ 5, 6]. A gain of function mutation in OPG (c1205A=>T; p. Stop402Leu) was identified in multiple families with early onset OA (FOA) characterized by chondrocalcinosis [5, 7]. With this mutation, the underlying importance of OPG was further confirmed, not only in bone turnover but also in cartilage homeostasis and OA onset. Given the eminent cross-talk between bone and cartilage, it was suggested that aberrant OPG function can affect the delicate balance between subchondral bone formation and resorption [8], making OPG essential in joint homeostasis. A drug called strontium ranelate has been administered in the clinic in order to fight osteoporosis by increasing OPG expression and impairing bone resorption processes [9]. This drug is also used to treat OA, but controversial results have been shown in preclinical and clinical studies [10, 11]. To study whether the observed upregulation of TNFRSF11B in OA can trigger unbeneficial mineralization of cartilage, a tailored human 3 D in vitro OA tissue model was applied in which aberrant gene function was mimicked by lentiviral overexpression of TNFRSF11B in spherical cartilage pellets. Potential effects on anabolic, catabolic and mineralization markers involved in cartilage homeostasis were investigated. Moreover, to better comprehend the pathways in which TNFRSF11B acts in articular cartilage and during OA, a selected panel of genes that showed high co-expression with TNFRSF11B during OA pathophysiology was studied.
Materials and methods
Sample description
RNA sequencing data previously obtained of n = 57 preserved and n = 44 lesioned OA cartilage samples (RAAK study) and previously assessed [4], were taken for in silico TNFRSF11B correlation analyses. RNA sequencing was used to identify genes co-expressed with TNFRSF11B, where a Spearman correlation was performed. Genes were considered to be correlated if the P-value was lower than 0.05 and the absolute r-value was higher than 0.75. Quality control of the data was performed as previously described [4]. Human primary articular chondrocytes (hPACs) obtained from knee replacement surgeries of n = 9 participants (four females and five males with average age of 69.4 (±11.1)) of the RAAK study were isolated and cultured to perform lentiviral transduction.
The RAAK study was approved by the medical ethics committee of the Leiden University Medical Center (P08.239/P19.013). Written informed consent was obtained from all donors.
Cloning of TNFRSF11B in lentivirus plasmid
The Porf9-Htnfrsf11b V04 plasmid and the pLV.CMV.bc.eGFP lentivirus vector (kindly provided by Prof. Dr R. Hoeben) were digested with AgeI and NheI (New England Biolabs-BIOKÉ, Leiden, The Netherlands). The full gene TNFRSF11B was ligated into the AgeI and NheI sites of the K4_pLV.CMV.bc.eGFP plasmid by using the Takara Mighty Mix ligation kit (Takara Bio Europe AB, Göteborg, Sweden). DNA was obtained by Maxiprep Kit (ThermoFisher, Landsmeer, The Netherlands), and Sanger sequencing was performed to confirm successful cloning of the lentivirus plasmid.
Lentiviral production and cell culture
Lentiviral production was performed in HEK293T cells using the Lenti-vpak Lentiviral Packaging Kit (Origene Technologies, Rockville, MD 20850, USA). In short, HEK 293 T cells were expanded in DMEM high glucose (Life Technologies Europe BV, Bleiswijk, The Netherlands), supplemented with 10% foetal calf serum (FCS; Life Technologies Europe BV, Bleiswijk, The Netherlands) 100 U/ml penicillin, 100 μg/ml streptomycin (Life Technologies Europe BV, Bleiswijk, The Netherlands), and lentivirus particles were collected and titrated.
Following their isolation, hPACs were transduced at passage 1 with either control (pLV.CMV.bc.eGFP) or TNFRSF11B Lentivirus (MOI of 1). After 16 h, the medium was refreshed [DMEM high glucose (Life Technologies Europe BV, Bleiswijk, The Netherlands) supplemented with 10% FCS (Life Technologies Europe BV, Bleiswijk, The Netherlands), 100 U/ml penicillin and 100 μg/ml streptomycin (Life Technologies Europe BV, Bleiswijk, The Netherlands), and 0.5 ng/ml bFGF-2 (PeproTech, London, UK)] and hPACs were further cultured for another passage. Subsequently, neo-cartilage was generated from 250 000 transduced hPACs in 3 D pellets for seven days, as described before [12], and keeping the conditions between both groups equal. All data were analysed 7 days following the 3D chondrogenesis.
RNA isolation and RT-qPCR
RNA was isolated from four biological replicates for each patient and condition (control and TNFRSF11B-overexpressing chondrocytes) while pooling two pellets together to generate two independent samples for downstream analyses. Isolations were performed as described previously [12]. Total mRNA (150 ng) was processed with the first strand cDNA kit according to the manufacturer’s protocol (Roche Applied Science, Almere, The Netherlands). CDNA was further diluted five times, and preamplification with TaqMan preamp master mix (Thermo Fisher, Landsmeer, The Netherlands) was performed. Gene expression was measured (Supplementary Table S1, available at Rheumatology online) with RT-qPCR, and average of the two biological replicates was determined as relative levels (−ΔCt values) using expression levels of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and Acidic ribosomal phosphoprotein P0 (ARP) as housekeeping genes. Quality control of the results was performed as described before [12].
Selection criteria for gene expression analyses
TNFRSF11B expression was analysed to quantify overexpression. To determine matrix homeostasis, metabolic activity and mineralization status, a list of 16 relevant genes was selected from the literature (Supplementary Table S1, available at Rheumatology online). Additionally, to identify potential new downstream pathways, a two-step approach was taken for selection of genes. Firstly, a TNFRSF11B co-expression network was created based on correlations of genes significantly differentially expressed between preserved (n = 57) and lesioned (n = 44) OA cartilage from our previously published RNA sequencing dataset (n = 2387 genes) [4]. Genes with r ≥ 0.80 were selected for expression analysis (n = 21 genes). Secondly, genes with r > 0.75 were designated on the basis of their functionality in cartilage homeostasis and mineralization (six genes), or based on previously identified protein–protein interactions with TNFRSF11B within the online available webtool STRING [13] (three genes). This resulted in a total of 30 genes. Finally, GAPDH and ARP were used as housekeeping genes.
Histology and immunohistochemistry
Following chondrogenesis, pellets were fixed in 4% formaldehyde and embedded in paraffin. After sectioning, deparaffinization and rehydration sections were analysed by histology [1% Alcian Blue 8-GX (Sigma-Aldrich, Zwijndrecht, The Netherlands)] and immunohistochemistry [COL2 (MAB1330 1:100; Merck-Milipore, Zwijndrecht, The Netherlands), COL1 (ab34710 1:100; Abcam, Amsterdam, The Netherlands) and OPG (EPR3592 1:100; Epitomics-Abcam, Amsterdam, The Netherlands)], as described before [12]. Pixel intensity quantification was performed for Alcian Blue staining by ImageJ, and surface area of the pellets were measured with the CellSens Dimension software (Olympus, Leiderdorp, The Netherlands).
ELISA and DMMB assay
The osteoprotegerin human instant ELISATM Kit (Thermo Fisher, Landsmeer, The Netherlands) and the Dimethylmethylene Blue assay (DMMB; Sigma-Aldrich, Zwijndrecht, The Netherlands), respectively, were used following the manufacturer’s instructions for OPG and GAG quantification in the conditioned media of three independent pellets with or without TNFRSF11B overexpression for each patient.
Statistical analysis
To determine statistical differences between the controls and samples with TNFRSF11B overexpression, a paired sample t test was performed. P-value <0.05 was considered significant.
Results
No change in matrix deposition upon TNFRSF11B overexpression
Lentiviral transduction of primary chondrocytes resulted in consistent and significant upregulation of TNFRSF11B mRNA as well as OPG protein (Supplementary Fig. S1, available at Rheumatology online). Following TNFRSF11B overexpression, hPACs were subjected to a 3 D in vitro chondrogenesis model, and neo-cartilage formation was characterized at day seven.
Alcian Blue staining was performed to visualize pellets and the presence of glycosaminoglycans (GAGs). The relative pixel intensity of the GAG staining showed no significant differences in the presence of higher OPG levels (n = 18, P-value = 3.4× 10−1; Fig. 1). Likewise, GAG release in the medium was similar (n = 27, P-value = 5.3× 10−1). Furthermore, no significant difference in pellet size was observed between controls and TNFRSF11B (n = 72, P-value = 5.5× 10−1). Together, these data indicate that TNFRSF11B upregulation does not change capacity of chondrocytes to deposit matrix at day 7.
Fig. 1.

Alcian blue staining of neo-cartilage
(A) Representative images of 1-week neo-cartilage pellets as indicated (left: control chondrocytes; right: chondrocytes with TNFRSF11B overexpression). Scale bars: 50 μm. (B) Area of the pellets (n = 72), GAG-release in the medium (n = 27) and Alcian blue pixel intensity quantification (n = 18) for control and TNFRSF11B overexpressing chondrocytes.
Collagen type I and collagen type II become upregulated upon TNFRSF11B overexpression
To study the effect of TNFRSF11B overexpression on matrix characteristics, RT-qPCR was performed for anabolic and catabolic genes involved in cartilage homeostasis (Supplementary Table S2, available at Rheumatology online, Fig. 2). Of note was the high and significant upregulation of MMP13 (FD = 14.76, P-value = 2.0× 10−3) following overexpression of TNFRSF11B (Fig. 2). Furthermore, overexpression of TNFRSF11B resulted in significantly higher upregulation of COL2A1 (FD = 4.77, P-value = 4.8× 10−4) and COL1A1 (FD = 1.88, P-value = 1.3× 10−2) and a modest downregulation of COMP (FD = 0.69, P-value = 2.0× 10−2) during chondrogenesis. Hypertrophic marker COL10A1 showed no significant difference (FD = 4.24, P-value = 6.3× 10−1). Immunohistochemistry of collagen type 2 (COL2) and collagen type 1 (COL1) showed a visual higher expression for both collagens in the OPG overexpressing group concurrent with respective gene expression levels (Fig. 3). As such, COL1 staining showed a darker and wider layer of staining towards the edges of the pellet when compared with the control group. COL2 differences were less strong between both conditions; nevertheless, a more consistent staining was observed in the ECM and retained within the cells cytoplasm in the OPG overexpressing group.
Fig. 2.

Expression of matrix-related genes in neo-cartilage
-ΔCt values for anabolic and catabolic genes in 1-week neo-cartilage pellets [control chondrocytes vs chondrocytes with TNFRSF11B overexpression (n = 18; *P-value <0.05; **P-value <10−3; *** P-value <10−6)].
Fig. 3.
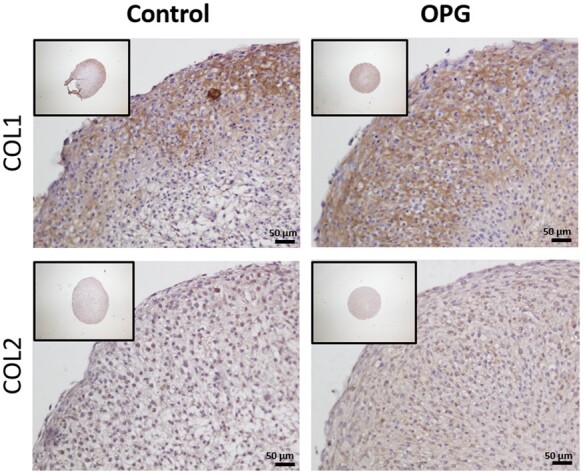
COL1 and COL2 immunohistochemistry of neo-cartilage
Representative images of 1-week neo-cartilage pellets as indicated (left: control chondrocytes; right: chondrocytes with TNFRSF11B overexpression). Scale bars: 50 µm.
High gene expression of osteogenic markers, yet no alteration in the TNFRSF11B triad upon TNFRSF11B overexpression
To investigate our hypothesis that upregulation of TNFRSF11B with OA pathophysiology directly induces cartilage mineralization, we next explored the expression of genes involved in matrix mineralization (Supplementary Table S2, available at Rheumatology online). First, we explored expression of TNFRSF11A encoding RANK and TNFSF11 encoding RANKL, which together with OPG are known to tightly regulate bone turnover. Remarkably (Fig. 4), neither TNFSF11 (FD = 1.06, P-value = 3.9× 10−1) nor TNFRSF11A (FD = 2.45, P-value = 7.8× 10−1) did significantly respond to the lentiviral-induced upregulation of TNFRSF11B. Nonetheless, the osteogenic markers RUNX2 (FD = 4.51, P-value = 4.0× 10−3), POSTN (FD = 1.75, P-value = 4.0× 10−2), OGN (FD = 1.68, P-value = 2.3× 10−2) and ASPN (FD = 2.61, P-value = 1.0× 10−2) were significantly higher upregulated in chondrocytes upon TNFRSF11B overexpression.
Fig. 4.

Expression of osteogenic-related genes in neo-cartilage
-ΔCt values for mineralization and bone formation genes in 1-week neo-cartilage pellets [control chondrocytes vs chondrocytes with TNFRSF11B overexpression (n = 18; *P-value <0.05; **P-value <10−3; ***P-value <10−6)].
A novel set of signalling pathways is discovered in highly correlated genes with TNFRSF11B upon TNFRSF11B overexpression
After assessing the effect of TNFRSF11B-induced overexpression on known related genes, we next performed an exploratory analysis to identify potential novel TNFRSF11B signalling pathways in cartilage. To do so, we generated a TNFRSF11B co-expression network with differentially expressed genes between preserved and lesioned OA cartilage as previously assessed (n = 2387 genes) [4]. We found 51 genes highly correlated with TNFRSF11B with absolute r-values ≥0.75 (Supplementary Table S3, available at Rheumatology online). Among the highest positively correlated genes, we found CDH19 (r = 0.88), ATP1A1 (r = 0.87) and DIXDC1 (r = 0.85), whereas the highest inverse correlation was observed for SLC15A3 (r =−0.81), MAPK11 (r = –0.81) and HLA-E (r =−0.8). Of these 51 genes, 30 were selected for expression analysis based on their correlation with TNFRSF11B and additional functional connection in STRING (Supplementary Fig. S2, available at Rheumatology online). As shown in Supplementary Table S4, available at Rheumatology online and Fig. 5, we found eight genes to be significantly differentially expressed upon lentiviral-induced TNFRSF11B overexpression. The strongest increased expression was found for BMP6 (FD = 9.34, P-value = 2.6× 10−2) while the SLC15A3 gene was 2.5-fold downregulated (FD = 0.4, P-value = 4.0× 10−3). Around 2-fold increase was observed for FITM2 (FD = 2.28, P-value = 1.4× 10−2), CDON (FD = 2.03, P-value = 5.0× 10−3), and SLC16A7 (FD = 1.97, P-value = 1.8× 10−2). Moderate effects were found for CDH19 (FD = 1.53, P-value = 4.5× 10−2), P3H2 (FD = 1.48, P-value = 4.7× 10−2) and WNT16 (FD = 0.81, P-value = 4.3× 10−2).
Fig. 5.

Expression of TNFRSF11B-correlated genes in neo-cartilage
-ΔCt values for TNFRSF11B-correlated genes in 1-week neo-cartilage pellets [control chondrocytes vs chondrocytes with TNFRSF11B overexpression (n = 18; *P-value <0.05; **P-value <10−3; ***P-value <10−6)].
Discussion
In the current study, we investigated the role of increased TNFRSF11B in OA pathophysiology. To this end, lentiviral upregulation of TNFRSF11B was established in a 3 D in vitro chondrogenic model (Supplementary Fig. S1, available at Rheumatology online). As reflected by the particularly high upregulation of MMP13 (FD = 14.76, P-value = 2.0× 10−3) in combination with the upregulation of characteristic osteogenic genes RUNX2, POSTN, BMP6, ASPN and OGN and in absence of differential expression of the mineralization markers COL10A1 and ALPL, we advocate that TNFRSF11B affects OA pathophysiology by advancing chondrocyte to osteoblast transition [14]. This finding is in line with the observed chondrocalcinosis phenotype observed in previously described members of the family with early-onset OA and carriers of read-through mutation in TNFRSF11B also known as the CCAL1 locus [7].
With TNFRSF11B encoding the decoy receptor OPG, which competes for binding of RANKL to the RANK receptor, we next examined expression of TNFRSF11A (encoding RANK) and TNFSF11 (encoding RANKL) upon TNFRSF11B upregulation. Even though this triad, and particularly the RANKL/OPG ratio, is known to be an important determinant of bone mass and skeletal integrity [5, 6], no significant changes in TNFRSF11A or TNFSF11 levels were observed (Fig. 4, Supplementary Table S2, available at Rheumatology online). This, together with the fact that we did not find a high correlation of expression between TNFRSF11B with TNFRSF11A or TNFSF11 in preserved and lesioned OA cartilage, would suggest that, in cartilage, the interaction among the triad may not play such an important role as in bone. This is in line with the findings of Komuro et al. and Tat et al. [15, 16], showing no alterations in RANK and OPG expression upon adding exogenous RANKL to chondrocytes.
OPG at high concentration is well known to decrease TNF-related apoptosis-inducing ligand (TRAIL) in chondrocytes, as such inhibiting apoptosis [17, 18]. Given that we observed high upregulation of MMP13 in combination with the upregulation of characteristic osteogenic genes RUNX2, POSTN, BMP6, ASPN and OGN (Fig. 4), which is an opposite response to that of OPG binding to TRAIL [19], we advocate that OPG rather affects OA pathophysiology in cartilage by advancing chondrocyte to osteoblast transition [14]. On the other hand, in our spherical neo-cartilage pellets model, we have studied the effect of OPG overexpression at an early timepoint in postmitotic chondrocytes that are stimulated to deposit matrix without further proliferation. As such, this model may not be optimal to provide insight into TRAIL-related signalling role of OPG.
In order to determine the co-expression network of OPG signalling in articular cartilage and with OA pathophysiology, we explored a previously assessed RNA sequencing dataset of preserved and lesioned OA cartilage for correlation with TNFRSF11B [4]. We found 51 genes that highly correlated with TNFRSF11B (r ≥ 0.75), such as CDH19 (r = 0.88) encoding for cadherins involved in calcium-dependent cell–cell adhesion, or SLC15A3 (r =−0.81) encoding histidine and osteoclast transporters [20]. From this network, expression of 30 genes were compared between control and TNFRSF11B overexpressing chondrocytes. Despite the high correlations with TNFRSF11B, notably only eight genes were found to be responsive to TNFRSF11B upregulation (26.6%; CDON, BMP6, CDH19, P3H2, WNT16, SLC16A7, SLC15A3 and FITM2). This may be explained partly by the fact that genes are upstream of OPG. Alternatively, genes may be correlated to TNFRSF11B as a general result of ongoing OA disease processes. Notable among the TNFRSF11B correlated and responsive genes were BMP6 and SLC15A3 (Fig. 5). BMP6 (r = 0.77), encoding bone morphogenic protein 6, is well known to be involved in bone formation [21], and SLC15A3 (r =−0.81) an osteoclast transporter of which lower expression would likely result in a reduction of the number of available osteoclasts. Additionally, we identified increased expression of FITM2 (r = 0.76) and SLC16A7 (r = 0.77), genes involved in lipid droplet formation and metabolite transport, respectively. Lipid droplets have been reported in OA cartilage [22] and during the osteogenesis process, where osteoprogenitors and osteoblasts synthesize them to use them as energy supplies for the differentiation process [23]. More importantly, it has recently been confirmed in mice that fat metabolism is a critical antagonist of cartilage health and integrity [24]. Notable as being co-expressed and highly responsive to TNFRSF11B was CDON (Cell Adhesion Associated, Oncogene Regulated; r = 0.83). Although little is known about its direct role in cartilage or bone homeostasis, cadherin signalling is known to be essential for successful cell differentiation, as it has been previously shown for osteogenesis [25, 26]. Lastly, expression of ANKH (r = 0.84), a gene previously associated with chondrocalcinosis and early OA [27, 28], was not affected by TNFRSF11B upregulation. This would confirm the work performed in porcine chondrocytes by Williams et al. [5] and translate it to primary human chondrocytes where ANKH would affect chondrocalcinosis by a TNFRSF11B-independent mechanism.
Remarkably, the study by Zhu et al. [29] showed a different signalling outcome upon overexpression of OPG (CCAL1) in primary human chondrocytes from OA patients. In contrast to the results shown here, they observed a fibrotic effect, dominated by reduced expression of COL2A1 and SOX9 and a higher expression of COL1A1. Several factors may have contributed to this disparity in results. Likely, the most important difference is the use of a 2 D model that was previously demonstrated to rather result in a hypertrophic phenotype [30].
Additionally, a previously published trial claimed minimal but debatable effects in OA joints upon treatment with strontium ranelate, a drug licensed for osteoporosis [31, 32].
Strontium ranelate increased bone formation while decreasing bone resorption via stimulation of OPG and was thought to target unbeneficial changes in subchondral bone with OA. Considering our current results showing the effect of OPG on cartilage, we advocate that the risk of such an oral treatment to OA patients is seriously underestimated and bound to considerably increase the burden of OA.
A potential limitation of our study is that we have mainly focussed on gene expression responses of hPACs by RT-qPCR at day 7 of matrix deposition. Henceforth, due to the early timepoint taken for these analyses and the inherently lower sensitivity and more challenging quantification methods that regular protein analyses such as immunoblotting offer, we have not extensively quantified our changes at a protein level. To further confirm, for example, whether the high upregulation of MMP13 results in significant changes in protein expression or, for that matter matrix degeneration, later harvesting timepoints (day 14 or day 21) and increasing sample sizes may be required.
In conclusion, we here highlighted the role of TNFRSF11B upregulation in OA pathophysiology. Results of our 3 D in vitro chondrogenesis model indicate that the observed consistent upregulation of TNFRSF11B in lesioned OA cartilage may act as a direct driving factor for chondrocyte to osteoblast transition occurring in OA pathophysiology. Moreover, we showed that this transition does not act via the OPG/RANK/RANKL triad, known for that matter in bone remodeling. Together, our results merit further exploration of TNFRSF11B as a promising disease OA modifying factor.
Supplementary Material
Acknowledgements
We thank all the participants of the RAAK study. The LUMC has and is supporting the RAAK study. We thank all the members of our group for valuable discussion and feedback, especially Evelyn Houtman and Ritchie Timmermans. We also thank Enrike van der Linden, Demiën Broekhuis, Peter van Schie, Shaho Hasan, Maartje Meijer, Daisy Latijnhouwers and Geert Spierenburg for collecting the RAAK material. We thank the Sequence Analysis Support Core (SASC) of the Leiden University Medical Center for their support.
The Medical Ethics Committee of the LUMC gave approval for the RAAK study (P08.239). Written informed consent was obtained from all donors .
Funding: Research leading to these results has received funding from the Dutch Arthritis Society (DAF-16–1-406), and the Dutch Scientific Research council NWO/ZonMW VICI scheme (nr 91816631/528). Data is generated within the scope of the Medical Delta programs Regenerative Medicine 4D (‘Generating complex tissues with stem cells and printing technology and improving mobility with technology’).
Disclosure statement The authors have declared no conflicts of interest.
Data availability statement:
The data that support the findings of this study are available from the corresponding author upon request.
Contributor Information
Alejandro Rodríguez Ruiz, Department of Biomedical Data Sciences, Section Molecular Epidemiology.
Margo Tuerlings, Department of Biomedical Data Sciences, Section Molecular Epidemiology.
Ankita Das, Department of Biomedical Data Sciences, Section Molecular Epidemiology.
Rodrigo Coutinho de Almeida, Department of Biomedical Data Sciences, Section Molecular Epidemiology.
H Eka D Suchiman, Department of Biomedical Data Sciences, Section Molecular Epidemiology.
Rob G H H Nelissen, Department of Orthopaedics, Leiden University Medical Center, Leiden, The Netherlands.
Yolande F M Ramos, Department of Biomedical Data Sciences, Section Molecular Epidemiology.
Ingrid Meulenbelt, Department of Biomedical Data Sciences, Section Molecular Epidemiology.
Supplementary data
Supplementary data are available at Rheumatology online.
References
- 1. Chen D, Shen J, Zhao W et al. Osteoarthritis: toward a comprehensive understanding of pathological mechanism. Bone Res 2017;5:16044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cucchiarini M, de Girolamo L, Filardo G et al. Basic science of osteoarthritis. J Exp Orthop 2016;3:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dunn SL, Soul J, Anand S et al. Gene expression changes in damaged osteoarthritic cartilage identify a signature of non-chondrogenic and mechanical responses. Osteoarthritis Cartilage 2016;24:1431–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Coutinho de Almeida R, Ramos YFM, Mahfouz A et al. RNA sequencing data integration reveals an miRNA interactome of osteoarthritis cartilage. Ann Rheum Dis 2019;78:270–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Williams CJ, Qazi U, Bernstein M et al. Mutations in osteoprotegerin account for the CCAL1 locus in calcium pyrophosphate deposition disease. Osteoarthritis Cartilage 2018;26:797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Theoleyre S, , WittrantY, , Tat SK et al. . The molecular triad OPG/RANK/RANKL: involvement in the orchestration of pathophysiological bone remodeling. Cytokine Growth Factor Rev 2004;15:457–75. [DOI] [PubMed] [Google Scholar]
- 7. Ramos YF, Bos SD, van der Breggen R et al. A gain of function mutation in TNFRSF11B encoding osteoprotegerin causes osteoarthritis with chondrocalcinosis. Ann Rheum Dis 2015;74:1756–62. [DOI] [PubMed] [Google Scholar]
- 8. Pelletier JP, Boileau C, Brunet J et al. The inhibition of subchondral bone resorption in the early phase of experimental dog osteoarthritis by licofelone is associated with a reduction in the synthesis of MMP-13 and cathepsin K. Bone 2004;34:527–38. [DOI] [PubMed] [Google Scholar]
- 9. Cianferotti L, D'Asta F, Brandi ML. A review on strontium ranelate long-term antifracture efficacy in the treatment of postmenopausal osteoporosis. Ther Adv Musculoskelet Dis 2013;5:127–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rodrigues TA, Freire AO, Bonfim BF, Cartagenes MSS, Garcia JBS. Strontium ranelate as a possible disease-modifying osteoarthritis drug: a systematic review. Braz J Med Biol Res 2018;51:e7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chu JG, Dai MW, Wang Y et al. Strontium ranelate causes osteophytes overgrowth in a model of early phase osteoarthritis. BMC Musculoskelet Disord 2017;18:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bomer N, den Hollander W, Ramos YF et al. Underlying molecular mechanisms of DIO2 susceptibility in symptomatic osteoarthritis. Ann Rheum Dis 2015;74:1571–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Szklarczyk D, Gable AL, Lyon D et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 2019;47:D607–D13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aghajanian P, Mohan S. The art of building bone: emerging role of chondrocyte-to-osteoblast transdiffe rentiation in endochondral ossification. Bone Res 2018;6:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Komuro H, , OleeT, , Kühn K. et al. The osteoprotegerin/receptor activator of nuclear factor?B/receptor activator of nuclear factor KappaB ligand system in cartilage. Arthritis & Rheumatism 2001;44:2768–76. [DOI] [PubMed] [Google Scholar]
- 16. Tat SK, , PelletierJ-P, , VelascoCR, , PadrinesM, , Martel-Pelletier J. New perspective in osteoarthritis: the OPG and RANKL system as a potential therapeutic target? The Keio journal of medicine 2009;58:29–40. 10.2302/kjm.58.29 19398882 [DOI] [PubMed] [Google Scholar]
- 17. Park DR, Kim J, Kim GM et al. Osteoclast-associated receptor blockade prevents articular cartilage destruction via chondrocyte apoptosis regulation. Nat Commun 2020;11:4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shimizu S, Asou Y, Itoh S et al. Prevention of cartilage destruction with intraarticular osteoclastogenesis inhibitory factor/osteoprotegerin in a murine model of osteoarthritis. Arthritis Rheum 2007;56:3358–65. [DOI] [PubMed] [Google Scholar]
- 19. Qin X, Jiang Q, Nagano K et al. Runx2 is essential for the transdifferentiation of chondrocytes into osteoblasts. PLoS Genet 2020;16:e1009169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Song F, Yi Y, Li C et al. Regulation and biological role of the peptide/histidine transporter SLC15A3 in Toll-like receptor-mediated inflammatory responses in macrophage. Cell Death Dis 2018;9:770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu M, Chen G, Li YP. TGF-beta and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res 2016;4:16009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mansfield JC, Winlove CP. Lipid distribution, composition and uptake in bovine articular cartilage studied using Raman micro-spectrometry and confocal microscopy. J Anat 2017;231:156–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rendina-Ruedy E, Guntur AR, Rosen CJ. Intracellular lipid droplets support osteoblast function. Adipocyte 2017;6:250–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Collins KH, Lenz KL, Pollitt EN et al. Adipose tissue is a critical regulator of osteoarthritis. Proc Natl Acad Sci USA 2021;118:e2021096118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee JK, Hu JC, Yamada S, Athanasiou KA. Initiation of chondrocyte self-assembly requires an intact cytoskeletal network. Tissue Eng Part A 2016;22:318–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kawaguchi J, Kii I, Sugiyama Y, Takeshita S, Kudo A. The transition of cadherin expression in osteoblast differentiation from mesenchymal cells: consistent expression of cadherin-11 in osteoblast lineage. J Bone Miner Res 2001;16:260–9. [DOI] [PubMed] [Google Scholar]
- 27. Gruber BL, Couto AR, Armas JB, Brown MA et al. Novel ANKH amino terminus mutation (Pro5Ser) associated with early-onset calcium pyrophosphate disease with associated phosphaturia. J Clin Rheumatol 2012;18:192–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Abhishek A, Doherty M. Pathophysiology of articular chondrocalcinosis–role of ANKH. Nat Rev Rheumatol 2011;7:96–104. [DOI] [PubMed] [Google Scholar]
- 29. Zhu H, Yan H, Ma J et al. CCAL1 enhances osteoarthritis through the NF-kappaB/AMPK signaling pathway. FEBS Open Bio 2020;10:2553–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Caron MM, Emans PJ, Coolsen MM et al. Redifferentiation of dedifferentiated human articular chondrocytes: comparison of 2D and 3D cultures. Osteoarthritis Cartilage 2012;20:1170–8. [DOI] [PubMed] [Google Scholar]
- 31. Reginster JY. Efficacy and safety of strontium ranelate in the treatment of knee osteoarthritis: results of a double-blind randomised, placebo-controlled trial. Ann Rheum Dis 2014;73:e8. [DOI] [PubMed] [Google Scholar]
- 32. Lafeber FP, van Laar JM. Strontium ranelate: ready for clinical use as disease-modifying osteoarthritis drug? Ann Rheum Dis 2013;72:157–61. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request.