

Abstract
This Review focuses on the establishment and development of self-assemblies governed by the supramolecular interactions between cyclic peptides. The Review first describes the type of cyclic peptides able to assemble into tubular structures to form supramolecular cyclic peptide nanotubes. A range of cyclic peptides have been identified to have such properties, including α-peptides, β-peptides, α,γ-peptides, and peptides based on δ- and ε-amino acids. The Review covers the design and functionalization of these cyclic peptides and expands to a recent advance in the design and application of these materials through their conjugation to polymer chains to generate cyclic peptide–polymer conjugates nanostructures. The Review, then, concentrates on the challenges in characterizing these systems and presents an overview of the various analytical and characterization techniques used to date. This overview concludes with a critical survey of the various applications of the nanomaterials obtained from supramolecular cyclic peptide nanotubes, with a focus on biological and medical applications, ranging from ion channels and membrane insertion to antibacterial materials, anticancer drug delivery, gene delivery, and antiviral applications.
1. Introduction
Supramolecular chemistry represents the area of chemistry focusing on chemical systems with spatially organized molecules via noncovalent interactions.1 These interactions include hydrogen bonding, metal coordination, hydrophobic interactions, van der Waals interactions, π–π interactions, and electrostatic interactions. Hydrogen bonding, arising from the dipole–dipole attraction between an electronegative atom with a lone pair of electrons and a hydrogen atom, which is covalently bound to another electronegative atom, such as nitrogen, oxygen, or fluorine, was first suggested by Moore and Winmill in 1912.2 Nowadays, it has become one of the most widely used noncovalent interactions in the field of supramolecular chemistry. While a single hydrogen bond is normally too weak to drive the formation of ordered supramolecular structures, hydrogen bonding arrays built by aligning multiple hydrogen bonds show increased strength and directionality. Different multiple-hydrogen-bonding arrays have been designed with binding constants ranging from 102 M–1 to as high as 109 M–1 (Figure 1a–c), which are subsequently used to fabricate supramolecular assemblies.3−10 In the meantime, because hydrogen bonding is drastically weakened in the presence of hydrogen bonding competitors or in very polar media, it is challenging to transfer these hydrogen bonding motifs in aqueous environment. Delicate molecular design by means of hydrophobic shielding is typically required for the construction of supramolecular structures in water.11
Figure 1.

Examples of multiple-hydrogen-bonding arrays: (a) triple-hydrogen-bonding, (b) quadruple-hydrogen-bonding, (c) sextuple-hydrogen-bonding, (d) β-Sheet structure of a protein, and (e) double helix structure of DNA (the dashed lines in orange represent intermolecular hydrogen bonds, while the dashed lines in blue represent intramolecular hydrogen bonds).
Similar hydrogen bonding arrays have already been widely employed by nature (Figure 1d and e). For example, the secondary structures of proteins, such as α-helix and β-sheet formation, are held in shape by hydrogen bonds between the carbonyl and amide groups of the corresponding amino acid. The double helix structure of DNA is mainly attributed to the complementary hydrogen bonds between the nucleotides. These biobased building blocks have been adopted and further adapted by supramolecular chemists to facilitate the construction of supramolecular assemblies.12,13 Because of the ease of synthesis, derivation, and functionalization, along with the biocompatibility, these building blocks have witnessed a tremendous increase of focus over the past few decades, in the fields of both fundamental studies and practical applications.
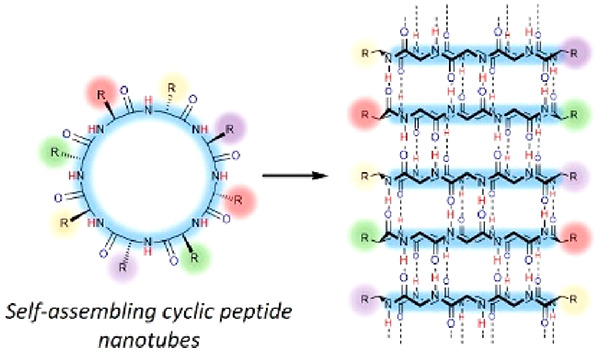
Self-assembling cyclic peptides (CPs) are bioinspired supramolecular building blocks, which stack into supramolecular cyclic peptide nanotubes (SCPNs), driven by β-sheet-like hydrogen bonding. Considering the rare supramolecular strategies of fabricating nanotubes, they offer a versatile route toward the construction of a diverse range of nanotubes.14−16 More interestingly, the hydrophobic side chains of the cyclic peptides can effectively shield the hydrogen bonds from the access of water molecules when forming nanotubular structures, which guarantee the extraordinary stability of SCNPs in water. This allows a whole range of applications from the construction of various supramolecular assemblies in aqueous media to the exploration of biological functions.
In this Review, we first provide a detailed description of different types of self-assembling cyclic peptides, covering their structures, physical properties, self-assembling characteristics, and synthetic approaches. We then introduce some general strategies toward the design and functionalization of these cyclic peptides, including both within the peptide ring and external modifications to the peptide cycle. In the latter case, we put specific emphasis on the conjugation of polymers. The conjugation of synthetic polymers to biomolecules provides easy access to biohybrid materials, which combines advantages from both the synthetic world and nature. Since the model of synthetic polymer–drug conjugates proposed by Ringsdorf in 1975,17 synthetic polymers have been successfully conjugated to proteins/peptides, nucleic acids, carbohydrates, lipids, and even live cells.18−23 With no exception, cyclic peptide–polymer conjugates have also been widely explored, and polymer conjugation has dramatically enhanced the application of the peptide assemblies. We cover the synthetic procedure to obtain cyclic peptide–polymer conjugates and the characterization of the resulting supramolecular polymers. The last section presents the most recent applications of these systems, ranging from functional materials to bioapplications, such as ion transfer channels and drug delivery. It is hoped that this review will provide a comprehensive overview of the rapidly expanding field that is supramolecular polymers obtained from cyclic peptide self-assembly.
2. Tubular Ensembles of Cyclic Peptides
Self-assembling cyclic peptides are important building blocks to construct synthetic nanotubes. To date, there are several types of cyclic peptides that are able to stack into SCPNs, which can be mainly categorized as cyclic α-alt(d,l)-peptides, cyclic β-peptides, cyclic α,γ-peptides, and cyclic peptides containing δ- or ε-amino acids on the basis of their composition (Figure 2). Although different in composition, they all adopt flat ring-like conformations with all of the amino acid side chains pointing outward in a pseudoequatorial position. At the same time, the carbonyl and amide protons of the peptide backbone are oriented perpendicular to the plane of the ring. The ability to form β-sheet-type hydrogen bonds between the adjacent peptide subunits on both sides of the ring structures leads to the formation of SCPNs.
Figure 2.

Classes of cyclic peptides that assemble into SCPNs through hydrogen bonds: (a) cyclic α-alt(d,l)-peptide 1, (b) cyclic β-peptide 2, (c) cyclic α,γ-peptide 3, and (d) cyclic peptide containing ε-amino acids 4.
2.1. Tubular Ensembles of Cyclic α-alt(d,l)-Peptides
2.1.1. Preliminary Studies
In 1974, within the context of theoretical analysis, De Santis et al. concluded that cyclic peptides comprised of an even number of alternating d- and l-amino acid residues could adopt β-type conformations with CO and NH bonds pointing in opposite directions and nearly parallel to the symmetry axis.24 Therefore, cylindrical structures were expected to form via the stacking of cyclic peptide rings through a parallel or antiparallel configuration, stabilized by both van der Waals and hydrogen-bond interactions. Initially, attempts to demonstrate these predictions experimentally were carried out by Lorenzi and co-workers in the late 1980s.25,26 Cyclic oligopeptides, including cyclo-[(d-Val-l-Val)n-, n = 2, 3, 4], cyclo-[(d-Leu-l-Leu)3-], and cyclo-[(d-Phe-l-Phe)3-], were found to be insoluble in most of the organic solvents, while IR results showed the presence of strong hydrogen bonds (amide band at 3270–3290 cm–1). However, single-crystal X-ray analysis of cyclo-[(d-Val-l-Val)3-] and cyclo-[(d-Phe-l-Phe)3-] showed that the cyclic peptides were more strongly hydrogen-bonded to the solvent molecules (trifluoroacetic acid) than to themselves. This may have been because the single crystals were grown by adding chloroform into the trifluoroacetic acid solutions of cyclic peptides.
2.1.2. Solid-State Studies
In 1993, Ghadiri and co-workers reported for the first time a well-characterized peptide nanotube formed by a cyclic α-alt(d,l)-peptide.27 An octapeptide 5 with the sequence of cyclo-[(l-Gln-d-Ala-l-Glu-d-Ala)2-] was designed and synthesized (Figure 3). Two glutamic acids were introduced to prevent subunit association through Coulombic repulsion in a basic aqueous solution. The formation of rod-shaped crystals was triggered by the controlled acidification of the alkaline cyclic peptide solution. The resulting peptide assembly was fully characterized by transmission electron microscopy (TEM), electron diffraction (ED), Fourier-transform infrared (FT-IR) spectroscopy, and molecular modeling. These results clearly indicated the formation of ordered tubular structures with internal diameters of 7.5 Å and distances between ring-shaped subunits of 4.73 Å with an antiparallel orientation through the backbone–backbone intermolecular hydrogen bonding interactions.
Figure 3.

(a) Chemical structure of the cyclic α-alt(d,l)-peptide 5. (b) Schematic representation of the SCPN emphasizing the antiparallel stacking and the extensive network of intermolecular hydrogen-bonding interactions.
A follow-up study was carried out using molecular dynamics simulation on the same cyclic peptide.28 During the simulation, 10 cyclic peptide subunits in water were shown to form a stable tubular structure, with average intersubunit distances between 4.75 and 4.80 Å, in good agreement with the experimental results. More interestingly, the behavior of water molecules was studied. The average number of water molecules inside the 10-peptide tube was found to be 32.8, clearly showing the hydrophilic nature of the internal cavity. However, it is concluded that water within the tubes does not compete significantly with the peptide–peptide hydrogen-bonding system after studying the distribution functions relating to the water interactions with hydrophilic components of the peptide tube. Additionally, the access of water outside the tubes to the backbone amides is blocked by the peptide side chains, which might explain the significant stability of the nanotubes even in aqueous environments. Analysis of the motion of water molecules inside the peptide channel structure gives a diffusion constant of 4.4 × 10–6 cm2 s–1. This is approximately one-sixth of the self-diffusion constant of bulk water and much faster than either molecular dynamics or experimental measurements of the diffusion constant of water in the structurally related gramicidin A transmembrane channel, highlighting its potential high efficiency as transmembrane channels. Indeed, an artificial membrane ion channel built based on a more hydrophobic octapeptide with the sequence of cyclo-[(l-Trp-d-Leu)3-l-Gln-d-Leu-] shows ion transport rates of 2.2 × 107 ions s–1 for K+ and 1.8 × 107 ions s–1 for Na+, which are almost three times faster than those of gramicidin A under similar conditions.29
It should be noted cyclic α-alt(d,l)-peptides containing 10 or 12 residues are also capable of self-assembling into nanotubes, even though their N-alkylated counterparts fail to dimerize in solutions. In this way, the internal diameter of the nanotube ensemble can be tailored by adjusting the ring size of the peptide subunit. As shown in Figure 4, an 8-residue cyclic peptide 6 can form nanotubes with internal diameters of 7.5 Å, while 10- and 12-residue cyclic peptides 7 and 8 form nanotubes with internal diameters of 10 and 13 Å, respectively.30 As a result, the desired transport of different ions and small hydrophilic molecules through the cylindrical cavity of the tubular structures could be realized by using cyclic peptides with variable internal diameters.
Figure 4.

Representative chemical structures of cyclic α-alt(d,l)-peptides containing 8, 10, and 12 amino acids 6–8.
Given the feasibility of designing and synthesizing cyclic α-alt(d,l)-peptides with various sequences and functionalities, it is vital to investigate the influence of peptide sequences on the formation of SCNPs. Similar to natural proteins, different amino acids are expected to play different roles in noncovalent interactions such as hydrogen bonding, electrostatic interactions, van der Waals interactions, and hydrophobic effect in directing the arrangement of the cyclic peptide subunits. Four cyclic peptides were studied and compared by Ghadiri and co-workers to address this issue; these included cyclo-[(l-Gln-d-Ala)4-], cyclo-[(l-Gln-d-Val)4-], cyclo-[(l-Gln-d-Leu)4-], and cyclo-[(l-Gln-d-Phe)4-].31 All four cyclic peptides formed needle-shaped microcrystals under appropriate conditions, which were subsequently studied by low-dose cryogenic electron microscopy, FT-IR, ED, and molecular modeling. Without exception, all four ensembles displayed highly ordered axial periodicities (along the tube axis a) in the range of 4.75–4.85 Å, suggesting they all stacked in an ideal antiparallel β-sheet arrangement. However, the lateral aggregation of the nanotubes differed from one to another. Cyclo-[(l-Gln-d-Leu)4-] and cyclo-[(l-Gln-d-Phe)4-] showed tighter packing than cyclo-[(l-Gln-d-Ala)4-], while cyclo-[(l-Gln-d-Val)4-] exhibited less ordered lateral packing. The results indicate that peptide sequences are more likely to affect the lateral packing of the nanotubes rather than the stacking of the cyclic peptide subunits. Therefore, it is anticipated that more ordered self-assembled structures might be realized by the rational design of the corresponding cyclic peptide sequences. For example, Montenegro et al. reported a cyclic peptide capable of undergoing sequential 1D-to-2D self-assembly in an aqueous medium.32 As indicated in Figure 5, an asymmetric cyclic peptide 9 with the sequence of cyclo-[d-Leu-l-Trp-d-Leu-l-His-d-Glu-l-Gln-d-His-l-Glu-] was designed to have four distinct domains: (i) a hydrophobic tripeptide domain of Leu-Trp-Leu, (ii) two hydrophilic ionizable domains on either side of the peptide ring comprising pH-sensitive Glu and His, and (iii) a neutral hydrophilic domain of Gln opposite to the hydrophobic domain. In solution, an amphiphilic nanotube is expected to form based on the multiple hydrogen bonding interactions between the peptide rings, which could subsequently assemble into a nanotubular bilayer to bury its hydrophobic face from the aqueous environment. Meanwhile, at physiological pH, the surface of the tubular ensemble will have a highly anionic character due to the deprotonated glutamate residues (pKa ≈ 4.2) and the neutral histidine residues (pKa ≈ 6.0). The aggregation of the individual layers is prevented by the strongly anionic surface, leading to the formation of large and flat nanosheets with lateral dimensions in the high micrometer range (e.g., 260 μm × 50 μm). At the same time, the thickness is only 3.2 nm. The formed nanosheets are highly dynamic and can reversibly transform into 1D nanotubes under the stimuli of pH or temperature. A follow-up study emphasized the necessity of the aforementioned four domains.33 A systematic structural modification at key positions of the original 2D sheet-forming cyclic peptide was carried out to assess their effect on 2D nanosheet assembly. It was found that the mutation of the central hydrophobic residue Trp in the original peptide was well tolerated; 2D nanosheets could be observed when replacing Trp with Ala, Leu, or Phe. Similarly, the two Leu moieties could also be substituted with Phe moieties. However, variations in the two hydrophilic ionizable domains of the parent peptide, originally consisting of alternating glutamic acid and histidine residues, revealed the importance of balancing electrostatic repulsion and net charge. In other words, ionizable glutamic acid and histidine residues must be placed together in the peptide sequence regardless of their order but never segregated on different sides of the cyclic peptide. Cyclic peptides with no electrostatic repulsion or with a very strong one could not form nanosheets. It is, therefore, believed that the large abundance of cyclic peptide compositions will result in a large variety of ordered supramolecular structures.
Figure 5.

Proposed model for the sequential 1D-to-2D self-assembly of cyclic peptide 9. (a) Chemical structure of cyclic peptide 9.(b) Formation of amphiphilic nanotubes based on the multiple hydrogen-bonding interactions between the peptide rings.(c) Formation of 2D nanosheet assembly by the amphiphilic nanotubes. Reproduced with permission from ref (32). Copyright 2020 American Chemical Society.
An antiparallel arrangement of the nanostructures formed by cyclic α-alt(d,l)-peptides is widely recognized in literature; however, in principle, similar SCPNs can be formed by either parallel or antiparallel stacking. The evidence that a particular nanotube is formed from antiparallel stacked cyclic peptides is indirect as crystal structures of continuous nanotubes have not been reported until very recently. In 2017, the first two crystal structures of cyclic α-alt(d,l)-peptides were reported by Chalmers and co-workers.34 Two water-soluble cyclic peptides with the sequences of cyclo[-(l-Asp-d-Leu-l-Lys-d-Leu)2-] (10) and cyclo[-(l-Asp-d-Ala-l-Lys-d-Ala)2-] (11) were used to grow crystals in aqueous media (Figure 6). The crystal structure of 10 shows that the peptide rings arrange in continuous β-sheet-like nanotubes with extensive hydrogen-bonding interactions with two adjacent peptides. Within each nanotube, the peptides are assembled in an antiparallel fashion with like residues aligned along the nanotubes. Two conformations of 10 are present within the unit cell, differing slightly in backbone conformation and side-chain rotamers. Some disorder is observed in the amide carbonyls of the Asp residues as well as the side chains of Asp and Lys. The positively charged Lys side chains of each nanotube form a continuous network through ionic interactions with the negatively charged Asp side chains of an adjacent tube. Adjacent nanotubes run antiparallel to one another. Unlike peptide 10, the crystal structure of peptide 11 shows the peptides are stacked in parallel and separated by layers of hexafluoroisopropanol (HFIP). Along the nanotube, each peptide is in alignment with its neighboring peptides. Although the CO and NH groups of the peptides interact through hydrogen bonds that are slightly offset, the CO···HN distances are similar to those found in the antiparallel crystal structure. The short Ala side chains allow the SCPNs to pack closely together, and each peptide forms two ionic interactions with its neighboring peptides. In the meantime, the less bucky Ala residues in 11 increase the flexibility of the peptide backbone, which is evident in the disorder of all backbone amide carbonyl groups, and the more ovular pore shape than for 10. These findings of both antiparallel and parallel stacking in cyclic peptides with similar sequences show that, contrary to the assumptions that have been made to-date, cyclic α-alt(d,l)-peptides do not necessarily adopt an antiparallel arrangement in the solid state. This implies that the steric hindrance caused by the side chains of amino acids also plays an important role in directing the stacking behaviors of the cyclic peptides.
Figure 6.

Chemical structures of cyclic peptides 10 and 11 and crystal structures of their SCNPs. Reproduced with permission from ref (34). Copyright 2017 The Royal Society of Chemistry.
Utilizing steric hindrance, Undén and co-workers developed a new strategy for a stereochemical control of peptide nanotubes.35 As shown in Figure 7, a new cyclic peptide 12 with a sequence of cyclo-[(l-Gln-d-Tle-l-Glu-d-Tle)2-] was designed by replacing the d-Leu amino acids of the nanotube-forming cyclic peptide cyclo-[(l-Gln-d-Leu-l-Glu-d-Leu)2-] with a bulkier amino acid tert-leucine (Tle). It fails to assemble into nanotubes either by the formation of parallel or antiparallel hydrogen bonds. This is attributed to the steric clashes between near-neighboring homochiral residues of Tle. However, nanotubes are found to form when mixing 12 with its enantiomer cyclo-[(d-Gln-l-Tle-d-Glu-l-Tle)2-] 13 through close antiparallel hydrogen bonding. The bulky tert-butyl side chains are positioned above the homochiral Gln and Glu residues of the enantiomeric peptide to overcome the steric hindrance, leading to the formation of a repetitive layered pattern of enantiomers. It is anticipated that this will provide a powerful strategy of precisely aligning different functional groups of the side chains of a given residue Xaa (amino acid X) and Yaa (amino acid Y) along the SCPNs by coassembling cyclic peptides with the general structures cyclo-[(l-Xaa-d-Tle-l-Xaa-d-Tle)2-] and cyclo-[(l-Xaa-d-Tle-l-Xaa-d-Tle)2-].
Figure 7.

Chemical structures of cyclic peptides 12 and 13 and the proposed layered structure of enantiomers. Reproduced with permission from ref (35). Copyright 2004 American Chemical Society.
2.1.3. Solution-Phase Studies Using N-Alkylated Cyclic α-Alt(d,l)-peptides
To better understand the stacking interactions between the cyclic peptides, simplified dimeric motifs of cyclic peptides have been studied in detail. These dimers were afforded by selective N-alkylation of the backbone amino acids with the same chirality. All the N-alkyl groups will point toward the same face of the peptide ring, thus preventing it from unlimited stacking and only allowing the formation of dimeric assemblies.
The first example of N-alkylated cyclic α-alt(d,l)-peptide was reported by the Lorenzi group.26,36 An N-methylated hexapeptide with the sequence of cyclo-[(d-Leu-l-MeN-Leu)3-] was synthesized and studied. It was found that unlike cyclo-[(d-Leu-l-Leu)3-], which is soluble only in the presence of strong acids, the N-methylated hexapeptide showed good solubility in solvents such as CHCl3 and CCl4. X-ray crystallographic studies showed the anticipated dimeric antiparallel β-sheet structures, which were made up of pairs of cyclic peptide rings facing each other with their nonmethylated face, connected by six intermolecular hydrogen bonds. Its association behavior in the solution-phase was studied by 1H NMR spectroscopy. In polar solvents, such as CDCl3 or CD2Cl2, no dimerization was observed. However, in apolar solvents, including CCl4 and CCl4/C6D12 mixture, a strong downfield shift of the signal attributed to the three NH protons was observed upon increasing the concentration. The association constant (Ka) in CCl4/C6D12 mixture was determined to be 80 M–1 at 25 °C.
The first investigation of the N-methylated octapeptide 14 with a sequence of cyclo-[(l-Phe-d-MeN-Ala)4-] was reported by Ghadiri’s group in 1995 (Figure 8).37 Single-crystal X-ray structural analysis gave a cylindrical dimeric ensemble, showing two peptide rings closely stacked in an antiparallel orientation driven by eight intersubunit hydrogen bonds with an intersubunit N–O distance of 2.90 Å. The cylindrical dimer has an approximate van der Waals internal diameter of 7.5 Å, which is filled with partially disordered water molecules, confirming the hydrophilic internal nature of the peptide nanotubes. NMR investigations gave a much higher Ka value (1.4 × 104 M–1) in apolar solvent CCl4. Moreover, a Ka value of 2.54 × 103 M–1 was reported in CDCl3, emphasizing the stronger binding affinity between octapeptides than hexapeptides.
Figure 8.

(a) Schematic representation of the chemical structure of cyclic peptide 14 and the cylindrical dimeric ensemble. (b) Crystal structure of the cylindrical dimeric ensemble. Reproduced with permission from ref (37). Copyright 1995 John Wiley and Sons.
A direct evaluation of the thermodynamic preference for the antiparallel versus parallel β-sheet formation was carried out using two enantiomeric forms of the same peptide sequence, cyclo-[(l-Phe-d-MeN-Ala)4-] (15) and cyclo-[(d-Phe-l-MeN-Ala)4-] (16) (Figure 9).38 A racemic solution containing an equal number of peptides 15 and 16 can produce an equilibrium mixture of the parallel ensemble (15·16) and the enantiomeric antiparallel ensembles (15·15 and 16·16), which could easily be differentiated by 1H NMR spectroscopy. By analyzing the relative intensity of different species, the free energy of stabilization of the two β-sheet arrangement could be obtained, showing that the antiparallel β-sheet structure (ΔG = 4.56 kcal mol–1, Ka(CDCl3) = 2540 M–1) is 0.8 kcal mol–1 more stable than the parallel arrangement (ΔG = 3.76 kcal mol–1, Ka(CDCl3) = 640 M–1).
Figure 9.

Schematic representation showing the two N-methylated cyclic peptides 15 and 16 and the corresponding parallel and antiparallel ensembles. Reproduced with permission from ref (38). Copyright 1995 John Wiley and Sons.
A systematic study was carried out by Ghadiri and co-workers, later on, aiming to explore the scope and limitations of this mode of self-assembly using 20 cyclic peptides with variations in ring size, backbone alkylation, and amino acid composition.39 The effect of cyclic peptide ring size on solution dimerization affinity was examined using a series of N-methylated cyclic peptides consisting of 6, 8, 10, 12 amino acids with the same alternating sequence of cyclo[-(l-Phe-d-MeN-Ala)n-]. It was found that the octapeptide was the only one that could self-associate in CDCl3. Consistent with reports by Lorenzi et al., the hexapeptide showed no evidence of dimerization in CDCl3, most likely because of the low binding affinity of the hydrogen bonds in polar solvents. Both decapeptide and dodecapeptide failed to self-associate in CDCl3, which was attributed to greater conformational flexibility and diminished preorganization for self-assembly through stronger hydrogen bonding interactions were anticipated. The effect of identity and location of backbone alkyl substituents was also studied. The dimerization process was found to tolerate various amide N-alkyl substituents, including methyl, allyl, and n-propyl groups. Replacing the four methyl groups with allyl groups results in similar dimerization affinity, while n-propyl substituents lead to a 5-fold decrease of the association constant, implying the self-association might be largely affected by the steric hindrance between the two cyclic peptides. An MeN-Phe-containing cyclic peptide with the sequence of cyclo-[(MeN-l-Phe-d-Ala)4-] showed no evidence of dimer formation, which may arise from unfavorable steric interactions between backbone N-methyl substituents and side-chain β-phenyl groups of Phe. For the same reason, α,α-disubstituted peptides exhibited no dimer formation in CDCl3. Finally, the effect of amino acid composition was explored. A decreasing trend of Ka was witnessed when replacing the two Phe amino acids with Leu, Hag, Ile, and Val amino acids because of the increased steric hindrance.
2.1.4. Fibrillation of Cyclic α-Alt(d,l)-peptides
The hierarchical self-assembly of the cyclic α-alt(d,l)-peptides leads to materials mimicking natural nano- and microfibers. In this way, cyclic α-alt(d,l)-peptides share certain similarities with amyloid fibrils, including self-assembly, dense intermolecular hydrogen bonding along the peptide backbone, as well as chemical and mechanical stability. While amyloids represent some of the stiffest known protein-based materials, the mechanical properties of SCPNs formed by cyclic peptides remained unexplored until 2015. As shown in Figure 10, Joshi and co-workers utilized a combination of electron microscopy, nanomechanical characterization, and molecular modeling methods to evaluate the mechanical properties of the nanotube assemblies formed by cyclo-[(d-Leu-l-Gln)4-] 17.40 The average elastic modulus and hardness were determined to be 11.3 ± 3.3 GPa and 387 ± 136 MPa. In contrast, the bending modulus and strength were 10.5 ± 0.9 GPa and 97.8 ± 18.8 MPa, respectively. By comparing these values with other natural and synthetic materials, the cyclic peptide fibers are most directly comparable to bone and outperform several biological materials, including collagen and enamel. The outstanding mechanical performance of cyclic peptides and their potential use as structural components of composite materials is largely unexplored at this time.41
Figure 10.

(a) Chemical structure of the cyclic peptide 17 and schematic of the cyclic peptide fiber. (b) Molecular dynamics simulation. (c) Nanoindentation and bending experiments. Reproduced with permission from ref (40). Copyright 2015 American Chemical Society.
The fibrillation of cyclic α-alt(d,l)-peptides can generate physically cross-linked networks in solutions, which subsequently causes gelation in either organic or aqueous solutions. Peltier, Perrier, and co-workers developed a range of cyclic peptide-based gelators.42 For example, pH-responsive hydrogels were built from cyclic peptides containing two charged residues, either Lys or Glu residues, while pH-independent hydrogels were constructed using quaternized residues. Further, the hydrogelation of a cyclic peptide containing two His and two Lys residues was studied by Granja, Montenegro, and co-workers in confined water microdroplets, which could be controlled spatially by microfluidics.43,44
2.1.5. Summary
The stacking of cyclic peptides to assemble into SCNPs is proved to be compatible with a wide range of peptide sequences, provided they are comprised of an even number of alternating d- and l-amino acid residues. The strong multiple hydrogen bonds between the cyclic peptides with flat ring-like conformation are the main driving force. In the meantime, the lateral aggregation of the nanotubes is mainly guided by the interactions between the side chains. While assemblies of 1D fibers and 2D sheets have been reported so far, it is believed that a large variety of other ordered self-assembled structures are accessible by the rational design of the cyclic peptides. Furthermore, the diverse self-assembled structures of cyclic peptides would provide valuable building blocks in the field of supramolecular systems chemistry.45−47 While an initial attempt has been carried out on the light-fuelled self-assembly of cyclic peptides into SCPNs, more studies focusing on the transient assembly of cyclic peptides are anticipated in the next years.48
There is no doubt that the study of dimers formed by N-alkylated cyclic α-alt(d,l)-peptides in solution offers a great tool in understanding the physical properties of cyclic α-alt(d,l)-peptides. Subsequently, these findings will help guide researchers to rationally design cyclic peptides with desired properties. However, the studies until now are not adequate to conclude precise design principles. Moreover, studies in different solvents, especially in water, are necessary considering the widely explored applications in biology.
The unique nanotubular structures of the cyclic peptides and their capability to interact with lipid bilayers enable a wide range of applications, including antibacterial agents and artificial transmembrane ion channels. Moreover, the stacking of cyclic peptides is unlikely to be affected by the side chains. This provides powerful scaffolds to fabricate supramolecular structures with molecularly aligned functional moieties, as well as polymers, allowing further applications in the fields of biosensing, pharmacology, catalysis, and electronics.
2.2. Tubular Ensembles of Cyclic β-Peptides
Cyclic β-peptides, consisting of a certain number of β-amino acids, are proved to be capable of forming SCPNs similar to cyclic α-alt(d,l)-peptides. Despite the possible difficulty related to the synthesis of unnatural amino acids, the ability of β-peptides to resist enzymatic degradation and their favorable conformational properties emphasize their potential advantages over cyclic α-alt(d,l)-peptides.
Seebach and co-workers pioneered the synthesis and characterization of cyclic β-peptides in 1997.49 As shown in Figure 11, three stereoisomeric cyclic β-tetrapeptides made of 3-amino butanoic acids, cyclo[-(β3-HAla)4-], were designed and synthesized. Using powder diffraction, the crystal structures of the three isomers could be determined. All three isomers were found to adopt tubular structures via four intermolecular hydrogen bonds in a parallel manner. However, they adopt totally different conformations. The (all-S)-derivative 18 exhibits a flat parallelogram conformation in a C2-symmetrical arrangement. The amide planes are within the sides, and the two tetrahedral C atoms form the bridges between these sides. Interestingly, the four C=O and the four N–H bonds (mutually antiperiplanar) point, respectively, in the same direction, while all four Me groups are in pseudoequatorial positions. As a result, the peptide rings stack in a parallel arrangement, which leads to a macrodipole along the nanotube. The (R,S,R,S)-isomer 19 has a flat square shape with a S4 symmetry. Opposite pairs of C=O bonds point in the same direction, as do the N–H bonds, which is very similar to the conformation of cyclic α-alt(d,l)-peptides. In this case, the peptide rings also stack in a parallel way. However, no macrodipole is expected. As for the (R,R,S,S)-isomer 20, a Ci-symmetrical rectangular appearance is witnessed with the parallel C=O and N–H bonds on neighboring sides. In contrast to the stacking of 18 and 19, the peptide rings are tilted with respect to the stacking direction. All three isomers exhibit tubular stacking through nonlinear hydrogen bonds. It is, therefore, clear that, in terms of their stacking ability, the cyclic β-peptides are much more versatile. It is believed that the inclusion of the additional CH2 units allows the rings more freedom to adopt a conformation suitable for stacking, and hence, the cyclic β-peptides do not suffer from the restrictions imposed on their α-counterparts.
Figure 11.
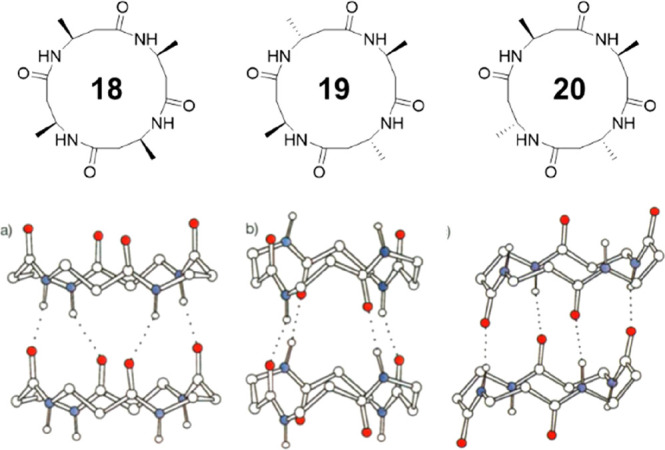
Chemical and crystal structures of the three stereoisomeric cyclic β-tetrapeptides 18–20. Reproduced with permission from ref (49). Copyright 1997 John Wiley and Sons.
The macrodipole caused by the stacking of homochiral cyclic β-peptides is expected to influence conductance through effects such as voltage gating and current rectification, which might exhibit better performance as artificial transmembrane ion channels. As shown in Figure 12, Ghadiri and co-workers explored the ion transport activities of the channel-forming cyclic β-peptides using three homochiral cyclic β-tetrapeptides, cyclo[-(β3-HTrp)4-] (21), cyclo[-(β3-HTrp-β-HLeu)2-] (22), and cyclo[-(β-HLeu)4-] (23).50 Different from the C2 symmetrical conformation in which the central hole of the peptide ring is collapsed reported by Seebach,49 they proposed that the cyclic β-tetrapeptides will adopt an alternative C4 symmetrical conformation which gives an internal diameter of 2.6–2.7 Å in a lipid membrane environment. As a result, cyclic peptide 21 exhibited an ion transport rate of 1.9 × 107 ions s–1 for K+ ion, which is comparable with the K+ transport rate of the previously reported cyclic α-alt(d,l)-peptide, cyclo-[(l-Trp-d-Leu)3-l-Gln-d-Leu-]. Considering the significantly smaller internal diameter of the nanotube formed by 21 compared to its cyclic α-alt(d,l)-peptide counterpart (7–8 Å), the result might emphasize the importance of the existing macrodipole. However, a direct comparison using cyclic β-peptide and cyclic α-alt(d,l)-peptide with similar chemical structure and the internal diameter is necessary to draw a firm conclusion.
Figure 12.

(a) Chemical structures of the three cyclic β-tetrapeptides 21–23. (b) Predicted tubular structure with a parallel arrangement, showing a macrodipole along the nanotube. Reproduced with permission from ref (50). Copyright 1998 American Chemical Society.
More recently, a novel class of cyclic β-peptides constituted by sugar-derived amino acids (SAAs) were developed by Kimura and co-workers. β-SAA with a pyranose ring as the side chain can improve the solubility of the corresponding cyclic β-peptides due to the multiple hydroxyl groups. Further, the pyranose ring is expected to promote the planar structure of the cyclic peptide, thus favoring molecular stacking and the formation of tubular structures. Cyclic β-peptides containing 3-, 4-, and 6-acetylated β-glycosamino acid units were synthesized and investigated (Figure 13).51,52 They are all capable of forming nanotubular structures, while showing good solubility in organic solvents, such as DMSO, DMF, TFE, HFIP, and pyridine. Water-soluble cyclic β-peptides could be, subsequently, obtained by the deprotection of acetyl groups, forming nanotubes endowed with affinity sites for proteins (particularly lectins).53 Interestingly, the cyclic hexa-β-peptide is found to form a polypseudorotaxane with a polyethylene glycol (PEG) chain in water.54
Figure 13.

Chemical structures of cyclic β-peptides 24-26 containing 3-, 4-, and 6-acetylated β-glycosamino acid units.
Similar to cyclic α-alt(d,l)-peptides, more ordered self-assembling structures could be realized by using noncovalent interactions between the side chains. For example, an amphiphilic cyclic peptide composed of two hydrophilic β-glucosamino acids (GA) and one hydrophobic trans-2-aminocyclohexylcarboxylic acid (ACHC) was designed, aiming to control the lateral aggregation of peptide nanotubes.55 In aqueous media, the cyclic peptides are expected to stack together to form amphiphilic nanotubes, which could further associate to form nanotube bundles with the hydrophobic ACHC residue facing inward and the two hydrophilic GA residues pointing outward. It was found that as the increase of the peptide concentrations in the assembly process, the nanotube bundles became thicker (from ∼5 to ∼100 nm in diameter), driven by the hydrogen bonds between amides of the cyclic peptides and interbundle hydrogen bonds through hydroxyl groups.
Apart from cyclic β-peptides, a cyclic hexapeptide composed of four β-amino acids and two α-amino acids with a sequence of cyclo[-l-naphthylalanine-β3-HAla-β3-HAla-d-anthrylalanine-β3-HAla-β3-HAla-] was also shown to assemble into SCPNs, offering a new strategy of designing self-assembling cyclic peptides.
Cyclic β-peptides are far less explored compared to cyclic α-alt(d,l)-peptides. Because all the N–H bonds point toward the same direction, it is impossible to achieve simplified dimeric motifs by selective N-alkylation of the backbone amino acids, making it difficult to reveal the physicochemical properties of the self-assemblies. The unique macrodipole within the nanotubes formed by cyclic β-peptides suggests that they might exhibit better performance as artificial transmembrane ion channels. However, their applications as artificial transmembrane ion channels are still rarely explored at the moment.
2.3. Tubular Ensembles of Cyclic α,γ-Peptides
A new type of self-assembling cyclic peptides with novel structural and internal cavity properties was proposed and developed by Granja’s group.56 These peptides are composed of an even number of alternating α-amino acids and cyclic γ-amino acids. Planar ring conformations similar to cyclic α-alt(d,l)-peptides and cyclic β-peptides lead to the formation of nanotubes driven by intermolecular hydrogen bonding interactions. As mentioned earlier, almost all the cyclic peptide nanotubes that have been developed, including cyclic α-alt(d,l)-peptides and cyclic β-peptides, possess unmodifiable hydrophilic internal cavities and can only be permeated by polar molecules. In the case of cyclic α,γ-peptides, the incorporation of cyclic γ-residues results in the projection of one of the cycloalkane methylenes into the lumen, thus generating a partially hydrophobic cavity. The cavity can be further modulated by simple chemical modification, allowing, in principle, finer control of the transport properties of the nanotube.
Within the flat ring-like conformation, the α and γ alternation makes all the NH and CO groups of the γ-amino acid on one face (γ-face) and the amide groups of the α-amino acid on the other (α-face). Because of the different CO···HN spacings on the α- and γ-faces, the orientations of the cyclic peptide rings are expected to alternate between making sure the selective α–α and γ–γ face stacking, leading to an antiparallel arrangement through two types of hydrogen bonding patterns. However, the parallel arrangement through interactions between α-face and γ-face is also possible, which is proved to be more stable, according to a very recent study.57
2.3.1. Solution-Phase Studies Using N-alkylated Cyclic α,γ-Peptides
Because of the anticipated antiparallel arrangement between the cyclic α,γ-peptide rings, dimers featuring each of the two hydrogen bonding patterns could be obtained by blocking the NH groups of either α- or γ-residues via N-methylation. Extensive studies have been conducted on the dimeric motifs formed by N-alkylated cyclic α,γ-peptides, providing useful physicochemical information including conformation, stacking manner, binding affinity, etc.
In 2003, Granja and co-workers reported the first examples of N-methylated cyclic hexapeptides using 3-aminocyclohexanecarboxylic acid (γ-Ach-OH) and d-α-amino acids.56 By selective methylation of γ-Ach-OH or d-α-amino acids, dimers formed based on α–α or γ–γ interactions could be obtained, as shown in Figure 14. Cyclic peptide 28 with the sequence of cyclo-[(1R,3S)-γ-Ach-d-MeN-Ala-]3 was synthesized to study the γ–γ hydrogen bonding interactions. An association constant of 230 M–1 was reported in CDCl3, while a higher value of 2.5 × 104 M–1 was obtained in a less polar solvent (2:3 CDCl3/CCl4). The α–α interactions were studied using cyclic peptide 29 with the sequence of cyclo-[d-Phe-(1R,3S)-MeN-γ-Ach-]3 which methylated γ-Ach-OH, displaying a much larger Ka in chloroform (>105 M–1). Single-crystal X-ray structural analysis shows an unsymmetrical dimeric ensemble that corroborates the nanotube structure. The two peptide subunits are closely stacked in an antiparallel orientation stabilized by six intersubunit hydrogen bonds. Moreover, the cylindrical dimer cavity can be filled with either one molecule of chloroform or two disordered water molecules (depending on how crystallization is performed), highlighting the amphiphilic character of the internal cavity, which is distinct from the hydrophilic cavities of either cyclic α-alt(d,l)-peptides or cyclic β-peptides. The mechanism behind these two types of dimers was revealed by computational studies.58 It was concluded that the preference for α–α interactions is governed by differences between the deformation energies in the α and γ monomers, rather than by differences between the relative strengths of the α–α and γ–γ hydrogen bonding patterns.
Figure 14.

Schematic representation showing the formation of a SCNP by a cyclic α,γ-peptide 27 and the dimeric ensembles by two N-methylated cyclic peptides 28 and 29 via two different hydrogen bonding patterns. Reproduced with permission from ref (56). Copyright 2003 American Chemical Society.
To investigate whether the dimerization via α–α interactions could be affected by interactions between the side chains of one ring and those of the other, the dimers of cyclic peptides composed of alternating methylated γ-Ach-OH and different d-α-amino acids (Phe, Ser, benzyl ether of Ser) were studied.59 Surprisingly, all cyclic peptides display high dimerization association strength in CDCl3 (Ka > 105 M–1), showing negligible cross-strand side-chain interactions.
Similar studies were carried out with other cyclic peptides that differ in the number of amino acids, resulting in peptide dimers with different diameters.60,61 Cyclic octapeptides showed similar dimerization behavior as cyclic hexapeptides, with a Ka = 340 M–1 for the γ–γ interactions and a Ka greater than 105 M–1 for the α–α interactions in CDCl3. However, cyclic tetrapeptides, the smallest cyclic α,γ-peptides, were found to dimerize by forming hydrogen bonds only between their α-faces but not between their γ-faces. Specifically, although cyclo-[l-MeN-Ala-γ-Ach-]2 was found to adopt the flat ring conformation in solutions, no evidence of dimer formation was observed in either CDCl3 or CCl4/CDCl3, suggesting the γ–γ quadruple hydrogen bonding interactions are too weak to drive the dimer formation. On the other hand, cyclo-[d-Phe-(1R,3S)-MeN-γ-Ach-]2 was shown to dimerize in CDCl3 through α–α interactions with a small Ka (15 M–1).
Similar to the studies of N-alkylated cyclic α-alt(d,l)-peptides, a direct comparison between the stability of the antiparallel and parallel arrangement of the N-alkylated cyclic α,γ-peptides was carried out.57 Two N-methylated forms of the same hexapeptide sequence, cyclo-[(1R,3S)-γ-Ach-d-MeN-Ala-]3(30) and cyclo-[(1R,3S)-MeN-γ-Ach-d-Ala-]3(31) were studied (Figure 15). The former one could form homodimer through γ–γ interactions in an antiparallel manner, while the latter one could homodimerize through α–α interactions in the same antiparallel manner. The combination of the two will lead to a heterodimer representing the parallel β-sheet through α–γ interactions. Surprisingly, mainly heterodimer was observed when mixing the two peptides in CDCl3, as probed by 1H NMR spectroscopy, indicating that the parallel dimer is more stable than the antiparallel structure. This is in accordance with computational calculations, which shows that the parallel β-sheet leads to shorter H-bond distances compared to the antiparallel dimers, and also to more favorable O···H–N angles for the H-bonds, resulting in stronger hydrogen bonding interactions. This line of research suggests that instead of the previously expected antiparallel arrangement through two types of hydrogen bonding patterns, a parallel stacking through α–γ hydrogen bonding interactions is more likely to happen during the formation of SCNPs by cyclic α,γ-peptides. Nevertheless, direct evidence is required to draw a firm conclusion.
Figure 15.

Structures of the two N-methylated cyclic α,γ-peptides 30 and 31, and the corresponding dimers via parallel or antiparallel interactions. Reproduced with permission from ref (57). Copyright 1995 John Wiley and Sons.
Granja’s group extended their studies by replacing γ-Ach with other γ-Aca residues, including cis-3-aminocyclopentanecarboxylic acid (γ-Acp-OH) and cis-4-aminocyclopent-2-enecarboxylic acid (γ-Ace-OH).62 Compared to γ-Ach, the angle defined in the plane of the cyclic peptide ring by the C–N and C–C(O) bonds radiating from the cycloalkane ring is wider (140° compared to 120°), making it more suitable for the construction of larger cyclic α,γ-peptides. For this reason, hexa-, octa-, deca-, and dodecapeptides were synthesized using N-methylated γ-Acp-OH alternated with α-amino acids, with internal diameters ranging from 7 to 17 Å (Figure 16). All of these cyclic peptides are able to form dimers through α–α interactions and have large Ka values (>105 M–1) in CDCl3. On the contrary, the cyclic decapeptide composed of alternating methylated γ-Ach-OH and α-amino acids fail to dimerize, further emphasizing that γ-Acp is more suitable than γ-Ach for the design of cyclic peptides capable of forming nanotubes with large pore diameters.
Figure 16.

Representative structures of cyclic α,γ-peptides 32–35 composed of 4, 6, 8, 10, and 12 amino acids. Reproduced with permission from ref (62). Copyright 2007 The Royal Society of Chemistry.
Similar to the aforementioned strategy, which uses the steric hindrance of side-chains to fabricate heterodimers in the case of cyclic α-alt(d,l)-peptides, heterodimers formed by cyclic α,γ-peptides have also been reported (Figure 17).63 It was found that heterodimers could be formed when mixing Ach-based hexapeptides with Acp-based hexapeptides, which is about 30 times more stable than the corresponding homodimers. The cause of the higher stability of the heterodimers is mainly due to more favorable packing between cyclopentyl and cyclohexyl than cyclopentyl-cyclopentyl or cyclohexyl-cyclohexyl ring packing, which leads to an improved alignment of hydrogen-bond donors and acceptors.
Figure 17.

(a) Schematic representation of homo- and heterodimerization of the two Ach- and Acp-based cyclic α,γ-peptides 37 and 38. (b) Crystal structures of the two homodimers and the heterodimer. Reproduced with permission from ref (63). Copyright 2005 John Wiley and Sons.
The dimers of N-alkylated cyclic α,γ-peptides process cavities with tunable properties, which could be used as molecular capsules to entrap guest molecules. For example, a hydrogen-bonded dimer formed by a cyclic hexapeptide 39 with three pyridyl moieties connected to its backbone was reported to encapsulate a single xenon atom (Figure 18).64 Single-crystal X-ray diffraction shows the drum-shaped dimer has an approximate van der Waals internal equatorial diameter of 5.4 Å and a height appropriate for hosting a sphere with a maximum diameter of 4.5 Å, which fits well with the size of a single xenon atom (4.3 Å). It is highly anticipated that by rational designing the N-alkylated cyclic α,γ-peptides, the resulting dimeric motifs with desired cavity properties could be used as supramolecular hosts, which bind with guest molecules with high affinity and selectivity.
Figure 18.

(a) Chemical structures of the cyclic hexapeptide 39 and the corresponding dimer. (b) Crystal structures of the dimeric ensemble containing a Xe atom in the intradimer cavity. Reproduced with permission from ref (64). Copyright 2019 John Wiley and Sons.
Apart from constructing dimeric motifs from N-alkylated cyclic α,γ-peptides, an alternative strategy was also reported whereby α-amino acids were replaced with α,α-dialkylated amino acids.65 A cyclic hexapeptide with a sequence of alternating 2-aminoisobutyric acid (Aib) and 4-aminocyclopent-2-enecarboxylic acid (γ-Ace) was studied. The steric hindrance caused by the two methyl substituents at axial α-positions of every second residue can block one face of the peptide ring, resulting in the formation of dimeric structures instead of nanotubes. 1H NMR dilution experiment proves the formation of dimers with a binding constant of 51 M–1 in CDCl3. However, a similar cyclic hexapeptide with a sequence of cyclo[-(α-Aib-γ-Acp)3-] failed to assemble into dimers, suggesting that this strategy is sensitive to the choice of amino acids.
2.3.2. Solid-State Studies
While extensive studies have been carried out on the dimeric assemblies of N-methylated cyclic α,γ-peptides because of their relative ease of characterization, the formation of nanotubes by cyclic α,γ-peptides has been far less explored. With the preacquired information on the X-ray crystal structure of a dimer-forming methylated cyclic peptide, cyclo-[d-Phe-(1R,3S)-MeN-γ-Ach-]3, the self-assembling nanotube of a cyclic hexapeptide consisting of alternating γ-Ach and d-α-amino acid was studied by theoretical analysis, allowing a full characterization of the structural, energetic, dynamic, and transport properties.66 The nanotube structure could be well preserved in solvents, including water, methanol, and chloroform. Still, the structure in water is more flexible than in the other two solvents because of its hydrogen-bond capability, which competes with the intermolecular hydrogen bonds between cyclic peptides. As predicted, hydrogen bonds between α-faces are more stable than those between γ-faces, especially in water. The association free energy is calculated to be between 57 and 61 kJ mol–1 in chloroform, significantly higher than that of the α-alt(d,l)-cyclic octapeptides (29.3 ± 4.2 kJ mol–1). However, as the parallel arrangement through interactions between α-face and γ-face is also possible, a model consisting of parallelly stacked cyclic α,γ-peptides should be comparatively evaluated with the established antiparallel model.
The nanotube structure of cyclic α,γ-peptides was experimental confirmed by Granja’s group in 2009.67 As shown in Figure 19, a cyclic octapeptide 40 with the sequence of cyclo-[d-γ-Acp-l-Cys-d-γ-Acp-l-Arg-d-γ-Acp-l-Lys-d-γ-Acp-l-Glu-] was designed and synthesized. Hydrophilic residues were introduced, aiming to increase the water solubility of the cyclic peptide and, at the same time, induce the self-assembly process under appropriate and controlled conditions. Moreover, the positively charged Arg residue and negatively charged Glu residue were expected to form complementary interstrand salt-bridge interactions, which might induce the predominant formation of only one β-sheet pattern in the nanotube and also stabilize the nanotube. As a result, long fibrous structures were visualized by atomic force microscopy (AFM), which consisting of several nanotubes packed in a parallel manner. In the meantime, shorter needle-shaped structures with 2.5–3.0 nm in height were also observed, corresponding to the expected single-nanotubes. Furthermore, a fullerene moiety was then attached to the cyclic peptide to aid in the characterization. TEM revealed the presence of long filaments (∼200 nm) that were ∼2.5 nm in width, consistent with the width of the nanotubes. Combining the results obtained by TEM and scanning tunneling microscope (STM), it is concluded that the cyclic peptides are able to align the C60 moieties to form 1D fullerene arrangements in which the fullerenes point outward from the nanotube on both sides of the nanotubes. In other words, guided by the cyclic peptides, the fullerenes form two parallel wires separated by the peptide nanotube, which opens up opportunities for 1D alignment of functional materials.
Figure 19.

(a) Chemical structure of the cyclic octapeptide 40. (b) Two possible structures of the SCNP. Reproduced with permission from ref (67). Copyright 2009 American Chemical Society.
The ability of the nanotubes to act as selective transporters or containers for small molecules were also evaluated.68 Computational analysis shows a tremendous difference in the transport properties of the nanotubes in different solvents. Chloroform molecules show slow diffusion, probably because of the polar nature of the external border of the nanotube (multi-nanosecond scale). The chloroform molecules inside the nanotube remain stable, and no transport of chloroform across the internal pore is observed, suggesting that the hydrophobic environment of the nanotube interior can act as a suitable container for nonpolar molecules, such as chloroform but not a good transporter. On the other hand, a significant amount of water molecules is found inside the nanotube within a very short simulation time (<600 ps), confirming its ability to capture water. The interior of the nanotube can dynamically form transient hydrogen bonds with water, leading to a slow diffusion of water molecules along the channel (0.0025–0.13 × 10–5 cm2 s–1) compared to the pure solvent (3.5 × 10–5 cm2 s–1). However, bigger polar molecules such as methanol refuse to spontaneously enter the nanotubular cavity during the simulation period (20 ns). As theoretical studies show that in aqueous media, the nanotubes are filled with water molecules, the nanotubes are expected to be capable of acting as ion channels. The transmembrane ion transport in lipid bilayers by self-assembling cyclic α,γ-peptide nanotubes was therefore experimentally studied. Two cyclic α,γ-peptides with different internal diameter were designed and synthesized, a cyclic hexapeptide with the sequence of cyclo-[(l-Trp-d-γ-Ach)2-l-Gln-d-γ-Ach-] with an internal diameter of ∼5 Å, and a cyclic octapeptide of cyclo-[(l-Trp-d-γ-Ach)3-l-Gln-d-γ-Ach-] with an internal diameter of ∼7.5 Å. The hexapeptide was found to support only proton transport but not allow the passage of metal ions, which was attributed to the combination of its narrow diameter and its hydrophobic inner wall. The octapeptide, however, showed ion transport selectivity toward alkaline metal ions (Na+, K+, Cs+), while no transport of Ca2+ or Cl– was observed. The rates of transport of the alkaline ions exhibited the same order as their mobilities in solution, but Na+ flux was ∼25% greater than expected on this basis. Such a preference is tentatively attributed to the hydrophobicity of the methylene groups projected toward the center of the nanotube by its γ-amino acids, which must limit the mobility of larger cations.
2.3.3. Other Cyclic Peptides Containing γ-Amino Acids
The conformational rigidity of γ-Aca segments implies that the cycloalkane ring of a γ-Aca can be regarded as a superatom, and (1S,3R)- and (1R,3S)-γ-Aca residues as equivalent to d- and l-α-amino acid residues, respectively. Thus, considering the correspondence between α- and γ-amino acids, cyclic peptides with other α- and γ-amino acid combinations are also likely to form nanotubes, which offers more precise tuning of the parameters and properties of the self-assembling structures.
The above-mentioned cyclic α,γ-peptides (42) can be regarded as replacing all the α-amino acid residues of the same chirality in the parent cyclic α-alt(d,l)-peptides (41) with the corresponding γ-Aca residues. Alternatively, by only substituting half of the α-amino acid residues of the same chirality, a new class of cyclic peptides (43, namely cyclic 3α,γ- peptides) in which the α-amino acids and γ-Acas are combined in a 3:1 ratio could be obtained (Figure 20). Studies carried out by Amorin et al. with N-methylated cyclic 3α,γ-peptides showed the formation of dimeric assemblies via antiparallel β-sheet-type hydrogen bonding through either α, γ–α, γ, or α–α interaction in most of the cases, while N-methylation of certain residues led to twisted, double reverse turn structures.69
Figure 20.

Schematic representation showing the design of cyclic α,γ-peptide 42 and cyclic 3α,γ-peptide 43 from cyclic α-alt(d,l)-peptide 41 and the α- and γ-amino acid equivalence.
Similarly, cyclic peptides that have one α-amino acid residue substituted with the corresponding γ-amino acid are also capable of stacking into SCPNs. For example, Helms, Xu, and co-workers reported a single γ-amino acid-substituted cyclic peptide.70,71 This was achieved by replacing one of the l-Leu in a sequence of cyclo-[(d-Ala-l-Lys-d-Ala-l-Leu)2-] of a cyclic α-alt(d,l)-octapeptide with a γ-amino acid, 3-amino-2-methyl- benzoic acid (γ-Mba-OH) (Figure 21a). The cyclic peptide 44 readily assembles into nanotubes in acetonitrile with a methyl group pointing toward the interior. Finally, cyclic peptides composed of only γ-amino acids could be designed by replacing any α-amino acid residue with its chiral counterpart of γ-Aca (Figure 21b).72−74 The self-assembling behavior of the γ-cyclic peptides were shown to vary depending on the ring size. Tetrapeptides (45) or hexapeptides (46) self-assemble into nanotubes through parallel-type hydrogen bonding interactions, while octapeptides (47) interact with each other via antiparallel β-sheet-type hydrogen bonding.
Figure 21.

(a) Chemical structure of a single γ-amino acid-substituted cyclic peptide 44. (b) Chemical structures of cyclic peptides 45–47 composed by only γ-amino acids.
2.3.4. Summary
The introduction of γ-amino acids greatly enriches the diversity of cyclic peptides and allows the fabrication of nanotubes with larger internal diameters. More importantly, the modifiable internal properties of nanotubes built by cyclic α,γ-peptides open up the potential for a whole range of new applications in areas such as artificial ion channels, molecular recognition, drug delivery, and smart materials.75,76
2.4. Tubular Ensembles of Cyclic Peptides Containing δ- or ε-Amino Acids
The introduction of δ- or ε-amino acids will further allow the design of new cyclic peptides with various self-assembling structures and properties. However, to retain the necessary rigid shape of the cyclic peptide rings to facilitate intermolecular hydrogen bonding, δ- or ε-amino acids with more rigid conformation need to be used.
δ-Amino acid-containing cyclic peptides were initially explored by Dory and co-workers.77 A cyclic tripeptide composed of α,β-unsaturated δ-amino acid residues with a trans geometry for the vinyl group was found to crystallize as bundles of nanotubes. The detailed nanotubular structure was obtained by X-ray single crystal analysis. The cyclic peptide ring adopts a rigid triangular structure with the three CO, and the three NH bonds pointing, respectively, in the same direction. Thus, the peptide rings stack in a parallel manner, which leads to a macrodipole along the nanotube, similar to cyclic β-peptides. In the meantime, the cyclic tetrapeptide and tripeptide composed of β,γ-unsaturated δ-amino acid residues showed the formation of undesired β-turns because of their flexibility. As a result, the cyclic tetrapeptide fails to assemble into nanotubes; the crystal structure of the cyclic tripeptide shows the stacking of macrocycles, which have a rectangular shape due to one β-turn, and only two out of three amides are involved in the stacking. Later on, groups of Zhao and Dory developed simple strategies to restrict the structural flexibility of aforementioned cyclic tetrapeptide composed of β,γ-unsaturated δ-amino acid residues using either liquid crystal or hydrogen-bonded polymer, inducing the formation of hexagonal hollow nanotubes.78,79
Similar to γ-Ach used to construct cyclic peptides containing γ-amino acid residues, trans-4-aminocyclohexanecarboxylic acid (δ-Ach) is a useful building block to fabricate cyclic peptide containing δ-amino acid residues. While δ-Ach is rigid enough for the planar ring-like conformation, the 180° angle described by the bonds between the carboxylic acid–Cα and the amino group–Cδ suggests that cyclic peptides made only by δ-Ach residues cannot form a cyclic structure. Instead, they must be combined with α-residues to create nanotube forming cyclic peptides. Thus, the δ-residue mainly functions as a rigid extender for the design of nanotubes with large diameter and hydrophobic internal properties and could be inserted after each α-amino acid of a cyclic α-alt(d,l)-peptide to obtain a new cyclic peptide, namely cyclic d,l-α,δ-peptide. N-Methylated cyclic peptides were first studied due to their simplicity.57 To ensure that all the N-methyl groups are pointing in the same direction, the N-methylated δ-Ach residues should be coupled to the N-methylated α-residues, while the nonmethylated δ-Ach residues should be attached to the α-residues of opposite chirality. Consequently, derived from dimer-forming cyclic α-alt(d,l)-peptides (tetramers, hexamers, and octamers), three cyclic d,l-α,δ-peptides were designed, including octa-, dodeca-, and hexadecapeptides. The octapeptide remains as unimers in nonpolar solvents, but the other two are able to self-assemble into dimers with a large association constant (Ka > 105 M–1). Because of the incorporation of rigid δ-Ach residues, the internal diameters of the nanotubular structures could be drastically extended, resulting in diameters as big as 21 and 30 Å, respectively. The stability differences between the parallel and antiparallel interactions of these dimer-forming cyclic d,l-α,δ-peptides were thoroughly studied. A pseudoenantiomeric form of the aforementioned dodecapeptide was, therefore, designed, as shown in Figure 22. The two pseudoenantiomers are able to form homodimers individually in an antiparallel manner, while heterodimers could be formed through a parallel arrangement between the two pseudoenantiomers. Experimental studies indicate that the heterodimers are ∼1.80 kcal mol–1 more stable than the homodimers. Computational calculations suggest that the difference in stability is most likely due to the relative disposition of the axial protons of the cyclohexyl moieties. In other words, the steric hindrance between the cyclohexyl moieties allows the parallel heterodimer to establish shorter hydrogen bonds, therefore exhibiting higher stability. Moreover, the nonstacking octapeptide and its pseudoenantiomer could form parallel heterodimers, further highlighting the higher stability of the parallel β-sheet structure. But possibilities exist for the control of the parallel and antiparallel arrangements in cyclic d,l-α,δ-peptides by peptide sequence, and the resulting cross-strand interactions established between the residues.
Figure 22.

Structures of the two N-methylated cyclic d,l-α,δ-peptides 48 and 49, and the corresponding dimers via either parallel or antiparallel interactions. Reproduced with permission from ref (57). Copyright 2020 John Wiley and Sons.
After confirming the assembling properties of the cyclic d,l-α,δ-peptides with dimeric models, the studies were extended to the formation of nanotubes. To this end, a cyclic dodecapeptide 50 was designed, with the incorporation of Glu, Lys, and His residues to improve solubility in aqueous media and provide control over the assembling process (Figure 23).80 With the help of Arg residue to facilitate the deposition of the nanotube on an anionic mica surface, fibrous structures with lengths of a few μm and average heights of 3.3–3.9 nm were observed using AFM by drop-casting the aqueous solution on mica, proving the formation of nanotubes. Moreover, the nanotubes with hydrophobic, large internal cavities were further used for the efficient encapsulation of fullerene C60 to form peptide nano-peapod-type structures. It is, thus, anticipated that these new nanotubes with large internal diameters and hydrophobic properties will function as efficient platforms such as nanocarriers and nanochannels.
Figure 23.

(a) Chemical structure of the cyclic d,l-α,δ-peptide 50. (b) Side and top views of a computer-assisted model of C60 encapsulated in the inner cavity of SCPN. (c) AFM topography micrographs from aqueous solutions of cyclic peptide 50 and C60 deposited over mica and AFM height profiles along the transects. (d) STEM image of an aqueous solution of cyclic peptide 50 and C60 after deposition on a holey carbon grid (scale bar 100 nm). Reproduced with permission from ref (80). Copyright 2018 The Royal Society of Chemistry.
Cyclic d,l-α,ε-peptides could be designed following the same principle. A novel 1,4-disubstituted-1,2,3-triazole ε-amino acid synthesized via copper-catalyzed azide–alkyne cycloaddition (CuAAC) was initially used to obtain a cyclic d,l-α,ε-tetrapeptide 51 (Figure 24).81,82 The anticipated hydrogen-bond-driven intermolecular aggregation behavior in CDCl3 was confirmed by 1H NMR spectroscopy, which gave relatively high association constants (>104 M–1). Direct evidence of the nanotubular structure was given by X-ray crystallography. The crystal structure indicates that the peptide subunits adopt the expected flat ring-shaped conformation and stack in a parallel manner into tubular assemblies. In the meantime, only three of the four amide bonds in the ring backbone form a network of intermolecular hydrogen bonds, while the remaining one is involved in an ethanol-mediated bridging hydrogen bond. Furthermore, the triazole ring can also function as an amide group to participate in noncovalent interactions during the nanotube formation, which was mainly investigated by Chattopadhyay’s group.83 A hybrid triazole/amide macrocycle 52 was designed by replacing two of the amide bonds in a cyclic β-tetrapeptide constructed by cis-β-furanoid sugar amino acids. Detailed studies show that it forms a well-defined tubular nanostructure through both an antiparallel backbone to backbone intermolecular hydrogen bonding involving amide NH and triazole N2/N3, and parallel hydrogen-bonded stacking between amide NH and carbonyl oxygen. However, a triazole-substituted cyclic peptide 53 derived from a cyclic β-tetrapeptide containing cis-β-furanoid sugar and β-alanine residues undergoes only parallel stacking via amide NH and amide carbonyl oxygen–hydrogen bonding.84 Last but not least, a larger triazole-containing cyclic peptide 54 with modified macrodipole was also reported by inserting extra α-amino acids into the peptide backbone.85
Figure 24.

Examples of cyclic peptides 51–54 containing ε-amino acids.
2.5. Synthetic Approaches
Linear peptides with desired sequences could be readily synthesized using standard peptide chemistry techniques (especially solid-phase peptide synthesis, SPPS). The availability of a large range of natural and novel amino acid building blocks and the ability to readily incorporate these into linear peptide sequences enable the fine-tuning of cyclic peptide structures and functionalities. Although peptide cyclization can be generally categorized into four types: head-to-tail, head-to-side chain, side chain-to-tail, and side chain-to-side chain cyclization within a peptide, most above-mentioned self-assembling cyclic peptides can only be synthesized via head-to-tail cyclization, which involves the two terminal amino and carboxyl groups through amidation.86 Two main strategies are developed for the synthesis of cyclic peptides: cyclization on resin and cyclization in solution.
Cyclization on resin is a powerful method to obtain cyclic peptides with various sequences.87 Typically, a specific amino acid is first anchored on the resin via the side-chain carboxyl group (such as Asp and Glu) or amino group (such as Lys) through a cleavable linker. Using standard coupling and deprotection procedures (either Fmoc- or Boc-), the linear peptide precursor is then assembled on the resin. After selectively removing the carboxyl and amino-terminal protecting groups, the peptide is cyclized on resin with the addition of coupling reagents. The targeted cyclic peptide is eventually obtained by cleaving the linker between the peptide and the resin and the protecting groups of certain amino acids. An extra purification by preparative HPLC might be needed depending on the cyclization efficiency. By immobilizing the linear peptide precursor on a solid support, a pseudodilution is achieved to minimize the oligomerization of linear peptides. Thus, cyclization on resin is proved to be efficient and could be used to synthesize a wide range of cyclic peptides. Moreover, it is particularly useful when synthesizing an extensive library of cyclic peptides. There are two main limitations: (i) an amino acid with either carboxyl or amino side chain need to be used to be anchored on the resin, which makes it impossible to synthesize a cyclic peptide without these specific amino acids in its sequence and (ii) several orthogonal protecting chemistries are required to sequentially cleave the carboxy and amino-terminal protecting groups, other protecting groups of the cyclic peptide, as well as the linker between cyclic peptide and resin. Consequently, amino acids with different protecting groups are needed.
Cyclization in solution is also widely used for preparing cyclic peptides.88 A linear peptide with free carboxyl and amino-terminal groups is first synthesized using standard peptide chemistry. The cyclization is then carried out by dissolving the linear peptide at a low concentration (typical 1 mM in DMF), with the addition of coupling reagents. The final cyclic peptide is obtained by separating the peptide from the reaction mixture (by precipitation, for example) and removing the other protecting groups, if there are any. Compared to cyclization on resin, this strategy is, in theory, suitable for the synthesis of any cyclic peptides, as no anchoring group is needed. Although the tendency to form oligomers is a big concern for general cyclic peptide synthesis, the pseudocyclic conformation of the linear peptide precursor in the case of self-assembling cyclic peptide greatly inhibits intermolecular reactions. Fewer orthogonal protecting groups are required, which offers opportunities to modify the cyclic peptides with different functional moieties or polymeric chains. In the meantime, it should be noted that cyclization in solution is typically more time-consuming because of the slower cyclization carried out in dilute solutions and the isolation/purification steps required by solution synthesis. In addition, the solubility of some fully protected linear peptide precursors in organic solvents are poor, which can reduce the cyclization efficiency.
Figure 25.

Schematic representation showing the synthesis of cyclic peptides via cyclization on resin and cyclization in solution methods.
To conclude, although these two synthetic approaches are general enough for most of the cyclic peptides, considerations need to be made on a case-by-case basis. In addition, some other approaches have also been reported targeting some specific cyclic peptides, allowing more choices when designing and synthesizing cyclic peptides.25,82,89,90
3. Design and Functionalization of Cyclic Peptides
The internal diameter of the SCPN is determined by the number and type of amino acids that constitute the cyclic peptide. As shown in Table 1, within the range of the cyclic peptides described above, the size of the internal cavity could be precisely tuned from 1 to 30 Å, allowing their applications as nanocarriers or nanochannels for different targeting molecules. The external properties of the SCPNs are dependent on the side chains of the amino acids. Thus, SCPNs with desired properties could be easily obtained by the rational design of the peptide sequences. Moreover, the internal and external properties of the SCPNs could be further tuned by covalent modification of the corresponding cyclic peptides.
Table 1. Summary of Internal Diameters of Different Cyclic Peptides.
| type | no. of amino acids | internal diameter (Å) | examples |
|---|---|---|---|
| α-alt(d,l)-CP | 8 | 7–8 | 5, 6, 10, 11, 14, 15, 16, 17 |
| 10 | 10 | 7 | |
| 12 | 13 | 8 | |
| β-CP | 3 | 24 | |
| 4 | 2.6–2.7/2.0 | 18, 21, 22, 23, 25 | |
| 6 | 5.1 | 26 | |
| α,γ-CP | 4 | 1 | 32 |
| 6 | 7 | 27, 28, 29, 30, 31, 33, 37, 38, 39 | |
| 8 | 10 | 34, 40, 42 | |
| 10 | 13 | 35 | |
| 12 | 17 | 36 | |
| d,l-α,δ-CP | 12 | 21 | 48, 49, 50 |
| 16 | 30 |
3.1. Internal Functionalization
The introduction of γ-, δ-, and ε-amino acids into the peptide sequences allows the projection of functional groups into the lumen of the SCPNs. It, therefore, enables the modification of the internal properties of the nanotubes. An initial attempt was carried out using 4-amino-3-hydroxytetrahydrofuran-2-carboxylic acid (γ-Ahf-OH).91 In this case, a cyclic tetrapeptide with the sequence of cyclo-[d-Leu-l-MeN-γ-Acp-d-Tyr-l-γ-Ahf-] was synthesized, with the hydroxyl group projected into the cavity in a pseudoequatorial orientation. Moreover, compared to cyclo-[d-Phe-(1R,3S)-MeN-γ-Ach-]2, which showed a very small Ka (15 M–1), a significantly higher association constant of 1.2 × 104 M–1 in CDCl3 was determined, which is most likely because of the extra hydrogen bonding interactions contributed by the equatorially oriented hydroxy group. Later on, the same strategy was employed to construct a cyclic octapeptide 55 with a larger cavity (Figure 26).92 Briefly, a hydrophilic cavity with two hydroxyl groups was obtained, allowing the encapsulation of various polar molecules, including methanol and water via hydrogen bonds between the solvent molecules and the hydroxyl groups. Moreover, another attractive feature of these ensembles is their simple chemical modification, which allows the cavity properties to be tuned. For example, a pyridine moiety was incorporated through a simple ester linkage into the cyclic octapeptide (56), allowing different metals and other guests to be trapped inside the dimeric structure, such as silver ion and oxalic acid. In addition, a cyclic decapeptide 57 that contains a carboxylic acid group projected toward the internal cavity of the dimeric structure was also reported, which is capable of encapsulating platinum complexes of cis-coordination.93 It is highly anticipated that by projecting certain functional groups into the internal cavity of the cyclic peptide, stronger and more specific encapsulation of guest molecules could be realized, offering new tools in the field of host–guest chemistry as well as drug delivery.
Figure 26.

(a) Chemical structure of the cyclic octapeptide 55 with two hydroxyl groups in its cavity. (b) Chemical structures of the pyridine-modified cyclic octapeptide 56 and the dimeric ensemble. (c) Computational structure of the dimeric ensemble showing the incorporation of the silver ion (gray color) within the dimer cavity coordinating the two picolinates (orange). (d) Chemical structure of the cyclic decapeptide 57. Reproduced with permission from ref (92). Copyright 2016 The Royal Society of Chemistry.
Modulating the cavity properties by a simple chemical modification of β-carbon of the cyclic γ-Acas allows, in principle, finer control of the transport properties of a very wide range of molecules in the nanotube. Apart from dimers formed by N-methylated cyclic α,γ-peptides, nanotubes by cyclic α,γ-peptides with modified internal cavities were also reported. For example, cyclic tetra- and hexapeptides consisting of fluorinated sugar amino acids were synthesized, forming nanotubular structures with a parallel orientation of cyclic rings.94 The fluorine atom could act as a reverse turn inducer, which leads to flat oval- and triangular-ring shaped conformations of cyclic tetra- and hexapeptides, respectively. Moreover, the projection of fluorine atoms into the nanotube cavity is expected to impact its physicochemical properties, which could be reflected by the transmembrane ion transport activity. The cyclic hexapeptide with a larger internal diameter of 9.7 Å showed better activity compared to the tetrapeptide with a smaller internal diameter of 5.7 Å. Moreover, the nanotubes formed by the hexapeptide showed anion-selective transport, with NO3– showing the highest ion transport rate.
The use of ε-amino acids also makes it possible to modify the properties of the internal cavity of the SCPNs. Recently, a novel ε-amino acid-containing cyclic peptide was reported by Helms, Xu, and co-workers, which could form organic nanotubes with a pH-responsive lumen.95 To this regard, a benzimidazole-containing ε-amino acid was used to replace two α-amino acids of an α-alt(d,l)-cyclic octapeptide, which forms a nanotubular structure where each subunit projects one of two ionizable nitrogen groups to the interior. Hence, the nanotube lumen is endowed with pH-responsive properties, bringing new opportunities toward constructing smart transmembrane channels.
Alternative strategies to achieve internal functionalization of the nanotubes have also been reported using different types of cyclic peptides other than cyclic α,γ-peptides. One of the first approaches was the use of cyclic α-alt(l,l,l,d) peptides.96 A cyclic dodecylpeptide was designed with the sequence of cyclo[-l-Trp-l-Dap-l-Leu-d-Ala-l-Trp-l-Ser-l-Val-d-Ala-l-Trp-l-Ser-l-Ile-l-Gly-], which could adopt a planar ring conformation with the side chain of the second l-amino acid residue among every four amino acids pointing toward the cavity. As a result, the hydroxyl groups (from l-Ser) and the β-amino group (from l-Dap, l-diaminopropionic acid) projected into the nanotubular channel were able to create a more polar environment, which enabled designable ion selectivity, as well as pH-responsiveness as artificial ion channels.
The ability to modify the internal properties of nanotubes opens new opportunities in synthetic ion channels, molecular motors, separation technologies, catalysts, and so on. Up to now, while carbon nanotubes with functionalized inner cavities are still far from being achieved experimentally, SCPNs may offer easier ways of preparing nanotubular structures with desired internal properties.
3.2. External Functionalization
The amino acid side chains which point outward not only decide the external properties of the SCNPs, but also offer reactive sites for further chemical modification. As mentioned above, the nanotubular structures formed by the cyclic peptides are mainly attributed to the backbone-to-backbone interactions but not the cross-strand interactions between the side chains. This allows the use of side chains as anchor points to achieve an efficient alignment of functional moieties without interfering with the self-assembly process. Therefore, the conjugation of different molecules to cyclic peptides expands the properties and applications of SCPNs.
As many organic semiconducting and conducting materials are based on one-dimensional stacking of aromatic molecules through π–π interactions and partial charge transfer, initial works have aimed to control and direct the stacking of aromatic moieties with the aid of cyclic peptide ensembles, greatly expanding the properties of cyclic peptides. Ghadiri’s group investigated whether the arrangement of naphthalenediimide (NDI) moieties could be directed or promoted by the display of such species as side chains of cyclic peptides.97 To this regards, one or two NDIs were attached to N-methylated α-alt(d,l)-cyclic peptides via the side chains of lysine residues. The role of cyclic peptide backbone self-assembly in enhancing NDI-NDI interactions was probed through fluorescence measurements. As a consequence of enhanced aromatic ring stacking between NDI moieties, all the NDI-modified peptides showed the formation of excimers indicated by the presence of a broad peak at a longer wavelength. Later on, a nanotube-forming α-alt(d,l)-cyclic peptide bearing four NDI units was shown to self-assemble into electronically delocalized nanotubes in aqueous media because of the alignment of the NDI moieties promoted by the directed backbone hydrogen bonding interactions between the cyclic peptides.98 Moreover, the alignment of other π-conjugated moieties such as fullerene, pyrene, and tetrathiafulvalene was also realized by the use of either cyclic α,γ-peptides or cyclic β-peptides as scaffolds.67,99−107 More interestingly, as shown in Figure 27, the pyrene-modified SCPNs were further applied to noncovalently interact with single-walled carbon nanotubes (SWCNTs) or subnanometric silver metal clusters, resulting in functional hybrid nanomaterials.108,109 Thus, intermolecular peptide self-assembly proved to be an effective template for the construction of one-dimensional nanomaterials with potential applications, such as optical and electronic devices.
Figure 27.

(a) Chemical structure of the pyrene-modified cyclic peptide 58 and its assembly. (b) Plausible model showing the formation of SWCNT/SCPN hybrids. (c) Plausible model showing the formation of silver cluster/SCPN hybrids. Reproduced with permission from refs (108 and 109). Copyright 2014 and 2015 American Chemical Society.
The self-assembly process of the cyclic peptide monomers with Cn symmetry should in theory, produce a mixture of n nonequivalent regioisomeric dimers. Each dimer features a different cross-strand pairwise relationship between the amino acid components of the monomers. This subsequently leads to different relative arrangements of the attached aromatic moieties, including the staggered arrangement and the eclipsed arrangement. Introducing specific cross-strand interactions between the side chains is expected to control the selective formation of one of the possible regioisomeric dimers. Strategies of using an external component capable of forming a ditopic interaction with efficient binding sites covalently attached to the side chains have proven successful in controlling the relative arrangement of the monomers within the dimeric assemblies. For example, Zn-porphyrins coordinated with diamines such as 1,4-diazabicyclo[2.2.2]octane (DABCO) were shown to form molecular tweezers that could achieve regioisomeric control of the self-assembled dimeric mixtures.110 Thus, using DABCO as an external trigger, the exclusive formation of self-assembled dimers with a staggered arrangement with Zn-porphyrin moieties stacking on top of each other was realized. Similarly, complementary interactions, such as metal–ligand coordination, hydrogen bonding between nucleobases, were also used to precisely control the arrangement of homo- or hetero- dimers.111−113
As discussed earlier, heterodimers could be formed when using cyclic peptides with specific sequences. Up to now, three strategies have been reported regarding the construction of heterodimers (Figure 28). In those cases, the formation of heterodimers is mainly due to the interactions established at the level of the peptide backbone and not by the side-chain/side-chain interactions. This allows the anchoring of diverse functional moieties at the same time to obtain more ordered alignment, generating novel functional materials including electron or energy transfer active systems. In the meantime, it should be noted that strategy I is only suitable for SCNPs, strategy II is only suitable for dimeric ensembles, while strategy III can be applied to both dimeric ensembles and SCNPs.
Figure 28.

Design strategies to construct heterodimers.
For example, an Acp-based cyclic hexapeptide 65 and an Ach-based cyclic hexapeptide 66 were decorated with a fullerene moiety (C60) as an electron acceptor and a π-extended tetrathiafulvalene moiety (exTTF) as an electron donor, respectively (Figure 29).114 The formation of the heterodimers brings C60 in close proximity to exTTF, which subsequently facilitates the occurrence of photoinduced electron transfer process from exTTF to C60. Later on, a heterodimeric assembly featuring a more efficient photoinduced electron transfer process was established by replacing exTTF with a stronger electron-donating moiety, Zn-porphyrin.115 The charge transfer interaction between C60 and Zn-porphyrin was found to further stabilize the heterodimer, resulting in a binding constant as high as 1.8 × 108 M–1 in dichloromethane. In return, an eclipsed conformation was favored over the staggered conformation. Therefore, an increased electron transfer (∼75%) was observed compared to the former heterodimeric system (∼33%). Besides electron transfer active systems, the fabrication of energy transfer systems was also demonstrated by Granja’s group. Efficient energy transfer could be realized via the formation of heterodimers between Acp- and Ach-based cyclic α,γ-peptides, which bring together in space the attached energy donors and acceptors, including Förster resonance energy transfer (FRET) pairs, such as pyrene/dapoxyl, pyrene/perylene.116,117 The heterodimeric ensembles with a distinctive organization of energy donor and acceptor units show great potential as efficient artificial light-harvesting/light-converting photosystems.
Figure 29.

(a) Chemical structures of the C60- and exTTF-containing cyclic peptides 65 and 66. (b) Formation of the heterodimer which brings C60 in close proximity to exTTF. Reproduced with permission from ref (114). Copyright 2007 National Academy of Sciences.
3.3. Summary
In conclusion, the functionalization of the internal channel of SCNPs is still in its infancy. As synthetic procedures have now been established, there is an exciting opportunity to design nanomaterials with functional internal channels. On the other hand, the functionalization of the outer ring has been exploited widely, and in particular, the conjugation of polymers has revolutionized the applications of the resulting SCNPs, which will be extensively reviewed in the next section.
4. Cyclic Peptide–Polymer Conjugates
One of the major drawbacks of self-assembling cyclic peptide nanotubes is their poor solubility in solvents and the propensity to laterally aggregate in large bundles. Inspired by the concept of synthetic polymer–biomolecule conjugates initially proposed by Ringsdorf, polymers have been attached to cyclic peptides, resulting in cyclic peptide–polymer conjugates. The introduction of polymers onto the periphery of cyclic peptide cores not only improve the solubility of these cyclic peptide nanotubes but provides the steric hindrance to avoid their lateral aggregation. Consequently, the cyclic peptide–polymer conjugate is expected to self-assemble into a supramolecular cyclic peptide–polymer nanotube (SCPPN) in solutions with a well-defined core–shell structure (Figure 30). The cyclic peptide constitutes the core, with polymer chains wrapped as the shell. The conjugation of polymers to cyclic peptides allows for a detailed study of the self-assembly mechanism in solutions and enables more control over the self-assembling behaviors of the cyclic peptides. Moreover, the combination of properties of both cyclic peptides and polymers will expand the functionality and application of the system.
Figure 30.

(a) Chemical structure of a cyclic peptide–polymer conjugate 67. (b) Schematic representation of the self-assembled structure of the cyclic peptide–polymer conjugate. Reproduced with permission from ref (123). Copyright 2011 The Royal Society of Chemistry.
4.1. Synthetic Approaches of Cyclic Peptide–Polymer Conjugates
Cyclic peptide–polymer conjugates can be synthesized either by divergent growth of a polymer from the peptide scaffold or by convergent coupling of a polymer to the cyclic peptide (Figure 31).
Figure 31.

Schematic illustration of two synthetic methods for cyclic peptide–polymer conjugates.
In the divergent method, polymer chains are grown from the peptide subunits or peptide nanotubes using diverse polymerization techniques. Biesalski and co-workers first reported the synthesis of a series of cyclic peptide–polymer conjugates using the divergent method.118−120 The cyclic peptide was first modified with atom-transfer radical polymerization (ATRP) initiators. The assembled SCNP thus provided a structurally defined template from which N-isopropylacrylamide (NIPAm) could polymerize to obtain the desired cyclic peptide–polymer conjugate. Similarly, the cyclic peptides could also be modified with chain transfer agents (CTAs) for reversible addition–fragmentation chain transfer (RAFT) polymerization.121
In the convergent method, polymers are first synthesized and then attached to the cyclic peptide. This approach permits more flexibility in the choice of cyclic peptides and the coupling strategies, as compared to the demanding step of premodified cyclic peptides in the divergent approach. Perrier’s group has explored this strategy to construct a wide range of conjugates. RAFT polymerization was used to synthesize polymers with various functionalities, as it not only enables the preparation of homopolymers or copolymers with a low dispersity but also provides functional handles for conjugation at the same time. Subsequently, these polymers were attached to the cyclic peptide using a great range of highly efficient conjugation methodologies, including strained-promoted or copper-catalyzed alkyne–azide cycloaddition (SPAAC or CuAAc),122−132 isocyanate-amine addition,133,134 activated ester-mediated chemistry,132,135−137 thiol–ene click chemistry,129 and pyridyl disulfide chemistry.137
In general, both convergent and divergent approaches are effective in the fabrication of cyclic peptide–polymer conjugates using a great range of functional polymers. Nevertheless, there are some factors that cannot be ignored when making a choice of appropriate synthetic methods. Larnaudie et al. carried out a systematic comparison between these two synthetic routes.121 RAFT was used to control the polymerizations and the couplings between cyclic peptide and RAFT agent or polymer were realized using N-hydroxysuccinimide (NHS) active ester ligation. A wide range of monomers were chosen to evaluate the efficiency of both approaches. Both the convergent and divergent approaches were found to be efficient for most monomers, including N-acryloylmorpholine (NAM), butyl acrylate (BA), NIPAm, hydroxyethyl acrylate (HEA), and methyl methacrylate (MMA). However, for the monomer with strong basicity like dimethylaminoethyl methacrylate (DMAEMA), the divergent approach showed a higher synthetic efficiency than the convergent one, due to the fact that the hydrolysis of the NHS end groups reduced the conjugation efficiency remarkably. On the other hand, for bulky monomers like (polyethylene glycol) acrylate (PEGA), the convergent approach could promise a better controlled polymerization with lower dispersity than the divergent one. Furthermore, the types of polymers for the divergent method are limited by the polymerization techniques. In the convergent method, any polymer can be attached to the cyclic peptide provided it has a suitable functional group for the coupling chemistry.
Solvents also play a key role on the choice of the two synthetic routes. Generally, the cyclic peptide is only soluble in such strong hydrogen bonding competitive solvents as DMSO and DMF. However, some polymers like poly(2-(diisopropylamino)ethyl methacrylate) (pDPAEMA) had limited solubility in them, making it very challenging for the convergent approach.138 To ensure the initial solubilization and prevent the precipitation during the polymerization, these conjugates were synthesized using the divergent approach. However, it is possible to optimize the convergent method by using cosolvents.
Given the two different synthetic pathways, the characterization and purification are obviously different. For the divergent method, although CTA- or initiator-modified cyclic peptides can be easily characterized by mass spectroscopy (MS) and NMR, the final conjugate is sometimes difficult to be analyzed by size exclusion chromatography (SEC), as the stacking nature of the cyclic peptide often leads to the aggregation of the conjugate. Moreover, the kinetics of the polymerization is challenging to be monitored on a small scale. Conversely, the convergent pathway allows full characterization of both the cyclic peptide and the polymer before the coupling reaction. As for the purification, the divergent method could permit a pure synthesis without free polymer chains in the system. However, to achieve a complete reaction, excess polymers are added in the convergent approach, making the removal of excess unreacted polymers a compulsory step. In theory, both solution sedimentation and dialysis are effective for the removal of unreacted polymers, which are based on the solubility and size difference between polymers and their conjugates, respectively. Despite this, it is still challenging to choose an appropriate solvent mixture for different systems, making it more time-consuming compared with the divergent method. The dialysis method is only efficient for the purification of conjugates attached with water-soluble homopolymers.
4.2. Self-Assembling Characteristics of Cyclic Peptide–Polymer Conjugates
With the successful synthesis of cyclic peptide–polymer conjugates, the investigation of the influence of polymers on their self-assembling behaviors has attracted increasing attention. By understanding these, researchers will be able to rationally construct SCPPNs with desired structures and properties. Moreover, the versatility of polymers will greatly endow these SCPPNs with diverse functions, thus opening up much potential for applications including antibacterial treatments, transmembrane ion channels, and anticancer drug delivery. In this section, we will introduce the self-assembling characteristics of conjugates attached with homopolymers, copolymers, and two different polymers, respectively. We will, then, discuss strategies toward the design and construction of stimuli-responsive cyclic peptide–polymer conjugates.
4.2.1. Cyclic Peptide–Homopolymer Conjugates
Cyclic peptide–homopolymer conjugates are the most widely studied cyclic peptide–polymer conjugates because of the ease of synthesis and the simplicity in structures. Up to now, a large range of polymers have been used to construct cyclic peptide–polymer conjugates, resulting SCPPNs with distinct structures and properties.
4.2.1.1. Factors Affecting the Self-assembly of Conjugates
The study of the self-assembling properties of cyclic peptide–polymer conjugates was initiated with the ones grafted with homopolymers. The dimensions (i.e., length and diameter) of these nanotubes are expected to be strongly affected by the size of the attached polymers. This was investigated by Biesalski and co-workers using conjugates of different polymers, including poly(styrene) (PS), pBA, and pNIPAM, with different molar mass synthesized using a divergent route.120 It was found that, regardless of the chemical nature of polymers, with increasing molar mass of the grafted polymer chains, the diameter increased while the length decreased. The former was due to the increase of thickness of the polymeric corona with the increase of polymer chain length. The latter was ascribed to the strong segment–segment repulsions between the bulky polymer chains. Similar results were reported by Chapman et al. for SCPPNs synthesized via a convergent method. The length of CP-pBA nanotubes reduced dramatically from 100 to 15 nm with the increasing molecular weight of pBA from 2300 to 14 100 g mol–1, in agreement with the expectation that longer polymer chains should sterically hinder the ability of conjugates to stack together.123 The influence of the steric effect of polymer chains on the resulting nanotube structures was further emphasized by attaching PEG chains with different architectures (linear vs brush).139 The introduction of two linear PEG chains (Mn = 11080 g mol–1) helped to form a cylindrical nanotube with the number of aggregation (Nagg) of 22 by small-angle neutron scattering (SANS), while the conjugate grafted with two pPEGA brushes (Mn = 5080 g mol–1) assembled in very small aggregates (Nagg = 3) because of the steric hindrance of bulky polymers.
Apart from the effect of polymer properties, solvents also play a vital role in the self-assembly of SCPPNs. A study later on showed that in solvents with strong hydrogen bonding competitivity, the length of polymers had a remarkably different effect on the length of nanotubes. This is most likely due to the fact that longer polymer chains may provide better shielding from the competitive solvent to facilitate the self-assembling process.127 Given this, Mansfield et al. studied the self-assembling behavior of a series of one-, two-, and three-arm CP–PEG conjugates (68, 69, 70) in different solvents with different polarity and hydrogen bonding capacity (Figure 32).139 In toluene, all conjugates formed long nanotubes due to the low hydrogen bonding capacity of the solvent. On the contrary, in strong hydrogen bonding competitive solvents, such as DMF and DMSO, all the conjugates remained as unimers or formed oligomers regardless of the number of polymer arms. Although water has a high hydrogen-bonding capacity, the high polarity greatly lowers the solubility of cyclic peptides, thus preventing water from penetrating the cyclic peptide core. As a result, long nanotubes were found to exist for 68, while the length decreased dramatically for 69 and 70. In solvents of intermediate hydrogen bonding basicity like THF, two PEG arms were believed to provide a better shielding than one arm, resulting in longer nanotubes, while the three-arm conjugate 70 assembled into shorter nanotubes because of the increased steric hindrance. The results confirm that the self-assembly of cyclic peptide–polymer conjugates in solutions is determined by a coordination of the competition of hydrogen bonding between conjugates and solvent molecules, as well as solvent polarity. Thus, one needs to take both the polymer architectures and solvents into consideration when designing cyclic peptide–polymer conjugates with desired self-assembling structures.
Figure 32.

(a) Chemical structures of the one- (68), two- (69), and three-arm (70) CP–PEG conjugates. (b) Aggregation number of the three conjugates in several solvents. (c) Contour plots of the degree of aggregation as a function of the hydrogen bond capacity and solvent polarity. Reproduced with permission from ref (139). Copyright 2018 The Royal Society of Chemistry.
4.2.1.2. Dynamic Nature
Supramolecular polymers are brought together by various noncovalent interactions, making them dynamic and reversible in nature. Meanwhile, these desirable attributes make them challenging to characterize. In contrast to conventional covalent polymers, any change in temperature, solvent, or external stimuli from the surrounding environment may remarkably influence the self-assembly they adopt.140 Therefore, a better understanding of the dynamic nature can help tune their structure and widen their applications. To investigate the dynamic nature of self-assembling cyclic peptide–polymer conjugates, Rho et al. designed a series of cyclic peptide–polymer conjugates modified with fluorescent dyes (71, 73) to study the dynamic behavior via FRET.136 In this case, fluorescent dyes Cy3 (donor) and Cy5 (acceptor) were chosen as the FRET pair to study the proximity-dependent energy transfer process from Cy3 to Cy5. The synthetic route is shown in Figure 33. When two dye-attached conjugates are mixed, the decrease in the donor emission and increase in the acceptor emission due to the energy transfer between the pair showed that the assemblies were highly dynamic and exchanging rapidly in solution. This dynamic exchange was shown to be solvent dependent. In highly hydrogen-bonding competitive solvents like DMF, the FRET ratio remained constant at a low value because of their existence as mainly unimers. In water and toluene, the FRET ratio increased over time, while the rate of exchange in water was faster than that in toluene resulting from the stronger hydrogen bonding competitive ability of water than toluene. Furthermore, in vitro studies demonstrated that this self-assembling system was robust enough to maintain its self-assembly in cells. The dynamic exchange was also shown to take part inside the cells using a study that looked at the FRET emission upon sequential injection of the two different dye conjugates. This system provides a general strategy to study the dynamics of other cyclic peptide-based systems, allowing further investigation on manipulating these dynamics.
Figure 33.

(a) Scheme of the synthesis of dye-modified cyclic peptide–polymer conjugates 71 and 73 and control conjugate 72. (b) Schematic description of the FRET occurring between the donor (Cy3) and acceptor (Cy5) dyes in SCPPNs. (c) Evolution of normalized FRET ratio of the mixed system in DMF, water, and toluene as a function of time. Reproduced with permission from ref (136). Copyright 2018 John Wiley and Sons.
4.2.2. Cyclic Peptide–Copolymer Conjugates
Given the diversity of polymers, efforts have been made to extend the library of cyclic peptide–polymer conjugates by the attachment of copolymers. Among those, conjugates bearing amphiphilic diblock copolymers are attracting the most attention. These conjugates could self-assemble into polymer-wrapped peptide nanotubes. The polymer corona is expected to phase-separate into two distinct layers in aqueous media: a hydrophobic inner layer in close contact with the cyclic peptides and a hydrophilic outer layer solvated by water. The hydrophobic inner layer is believed to prevent the peptide core from the hydrogen bonding competition of water molecules and significantly improve the stability of the SCPPNs in water. The hydrophilic outer layer is applied to increase the solubility in water. A better control over the size and dynamic property of the SCPPNs might be realized. In this regard, a model cyclic peptide–diblock copolymer conjugate system was investigated by the Perrier group using pBA as the hydrophobic block and poly(N,N-dimethylacrylamide) (pDMA) as the hydrophilic block.141 Two amphiphilic diblock copolymer chains (pBA-b-pDMA) were attached to a cyclic peptide. The self-assembling structure of the diblock conjugate in water was first investigated. It was found that compared to the control cyclic peptide–homopolymer conjugate, CP-(pDMA)2, the diblock conjugate formed much longer nanotubes (273 vs 12 nm), emphasizing the effect of the hydrophobic inner layer on promoting the stacking of the cyclic peptides. The dynamic nature of these nanotubes was probed via FRET using conjugates attached with Cy3 and Cy5 dyes (74–77). The change in FRET ratio over time was observed to be extremely slow, while in the control conjugate system, the increase in FRET ratio was very fast, and the plateau was reached within 60 min. Moreover, the mixing degree of the diblock conjugates was only 41% even after 7 days, which was far lower than that of the control conjugate (89%). The results confirm that the introduction of hydrophobic segments effectively stabilizes the hydrogen-bonded cyclic peptide ensemble, affording a much less dynamic system (Figure 34).
Figure 34.

(a) Chemical structures of dye-functionalized cyclic peptide–diblock polymer conjugates 74 and 75. (b) Structure of the SCPPN formed by the cyclic peptide–diblock polymer conjugate. (c) Scheme showing no FRET occurring in a stable nondynamic system. (d) Scheme showing FRET occurring in a dynamic system. Reproduced with permission from ref (141) and available under a CC BY 4.0 License (http://creativecommons.org/licenses/by/4.0/).
Up to now, examples of cyclic peptide–diblock copolymer conjugates using pBA, pDPAEMA, and pDEAEMA as hydrophobic blocks and pDMA and poly(2-hydroxypropyl methacrylamide) (pHPMA) as hydrophilic blocks have been reported. By introducing a secondary hydrophobic driving force, the self-assembling behavior of the conjugates could be rationally controlled. Furthermore, the hydrophobic segments might act as nanocarriers for applications, such as drug delivery and controlled release.
4.2.3. Asymmetric Cyclic Peptide–Polymer Conjugates
Block copolymers have attracted considerable attention for several decades because of their ability to generate diverse self-assembling morphologies, including spheres, cylinders, vesicles, and other complex or hierarchical assemblies.142 Among these, amphiphilic block copolymers have been studied most extensively. Their thermodynamically stable aggregation in aqueous solutions provides a wide range of potential or practical applications. Inspired by block copolymers, Danial et al. first reported a class of asymmetric cyclic peptide–polymer conjugates bearing two different polymers.129 Depending on the compatibility of the two polymers, the asymmetric conjugates could self-assemble into either Janus nanotubes with “demixed” polymeric corona or hybrid nanotubes with “mixed” polymeric corona. The polymeric corona properties could be distinguished by two-dimensional (2D) 1H–1H nuclear Overhauser effect spectroscopy (NOESY) NMR in solution and differential scanning calorimetry (DSC) in bulk. Specifically, the conjugate bearing a pBA chain and a PS chain formed Janus nanotubes because of the phase separation of the two polymer chains, while that with miscible poly(cyclohexyl acrylate) (pCHA) and PS formed nanotubes wrapped with mixed polymeric corona.
Similar to amphiphilic block copolymers, asymmetric cyclic peptide–polymer conjugates with amphiphilic properties were also studied. Brendel et al. designed an asymmetric cyclic peptide–polymer conjugate, pBA-CP-pPEGA (78), bearing one hydrophobic polymer pBA and one hydrophilic polymer pPEGA (Figure 35).133 The conjugate assembles into amphiphilic Janus nanotubes, which is expected to self-assemble further into a more complex core–shell cylindrical assembly in aqueous solution, named tubisome. The core of the tubisome consists of the hydrophobic pBA, while the hydrophilic pPEGA forms the shell. The tubisome structure is proved by SANS. The core diameter of these cylindrical structures was around 15 nm, much larger than the internal diameter of a single nanotube (0.75 nm). Such a large diameter confirmed these structures were composed of several nanotubes. The tubisome structure is driven by both the intermolecular hydrogen bonding interaction between peptide rings and the hydrophobic effect of amphiphilic polymers in water.
Figure 35.

Chemical structure of the asymmetric cyclic peptide–polymer conjugate pBA-CP-pPEGA 78 and its self-assembled tubisome structure in water. Reproduced with permission from ref (133). Copyright 2018 John Wiley and Sons.
To further understand the effect of polymers on their self-assembly, a family of pBA-CP-pPEGA conjugates with different block ratios were synthesized and the corresponding block copolymers were used as a control.134 With the increase of hydrophobic mass fraction, the block copolymers witnessed a pronounced structural change from spherical micelles (15 wt %) to more elliptical micelles (22 wt %), and finally to cylindrical/wormlike micelle vesicles (35 wt %). In sharp contrast, the asymmetric conjugates remained as tubisomes with ratios of the hydrophobic content varying from 14 to 35 wt %. SANS showed the diameter of tubisomes was dependent on the molar mass of hydrophobic segments, which varied from 7.8 to 9.7 nm, while the hydrophilic moieties had no significant effect on the self-assembly, except for the control of the corona size. In summary, the assembly process of these tubisomes is first triggered by the hydrogen bonding between the cyclic peptides to form single nanotubes, followed by a phase separation due to the hydrophobic effect between the polymers.
4.3. Stimuli-Responsive Cyclic Peptide–Polymer Conjugates
Stimuli-responsiveness is one of the key features of supramolecular materials, which permits control over the structures and properties of the supramolecular assemblies using physicochemical stimuli such as pH, temperature, redox, light, etc. Stimuli-responsive materials have shown applications in fields, such as sensing, controlled drug delivery, smart coating, artificial muscles, and actuators.143−147 Stimuli-responsive cyclic peptide–polymer conjugates have drawn increasing attention recently, as they will offer opportunities not only for the precise control over self-assembling structures but also for biorelated applications. In general, a cyclic peptide–polymer conjugate is divided into three segments: a cyclic peptide, a polymer, and a linker. Thus, one can introduce stimuli-responsiveness into any of these three segments.
4.3.1. Introduction of Stimuli-Responsive Polymers
A wide range of pH-sensitive polymers have been used to construct pH-responsive materials, including poly(acrylic acid) (pAA), pDMAEMA, poly(ethylene imine), and so on. These polymers can be reversibly protonated or deprotonated upon the change of pH, thus resulting in a change of hydrophobicity and charge state. pH-responsive cyclic peptide–polymer conjugates can be easily constructed by attaching these polymers onto cyclic peptides. In most cases, the electrostatic repulsion between the charged polymer chains will lead to the disassembly of the formed SCPPNs when changing the pH.
Chapman et al. first reported a water-soluble cyclic peptide–polymer conjugate bearing two pAA polymers, which showed pH-responsiveness in solutions.128 At pH = 3.0, the carboxylic acid groups, were in an uncharged state, and the conjugates could stack into nanotubes by intermolecular hydrogen bonding. At higher pH, the electrostatic repulsion between the charged pAA polymers led to the disassembly of the nanotubes. Likewise, Catrouillet et al. introduced the pH-responsive pDMAEMA onto the cyclic peptide, realizing the control over self-assembly and disassembly of the conjugates using pH in aqueous solutions.148 These conjugates self-assembled into supramolecular nanotubes at pH = 9 with a Nagg of 15. By subsequently adding acid into the solution, Nagg was reduced significantly to 5 at pH = 7, and finally to 1 at pH = 2. A linear inverse proportionality was observed between the Nagg and the degree of ionization, confirming the increased charge density of polymers promoted the disassembly. The pH-responsiveness could also be introduced to cyclic peptide–block copolymer conjugates. For example, Larnaudie et al. designed a pH-responsive cyclic peptide–polymer conjugate bearing two amphiphilic block copolymer chains: hydrophobic segment pDPAEMA and hydrophilic segment pHPMA.138 These conjugates could form a stable core–shell cylindrical nanotubes at physiological pH, which disassemble in a more acidic environment because of the protonation of the pDPAEMA core. The aggregation number of hybrid compounds at pH 5.0 (Nagg = 3) was far smaller than that in pH 7.4 (Nagg = 22), confirming the electrostatic interactions of protonated pDPAEMA chains hindered the self-assembly and shortened the aggregation length.
Thermoresponsive polymers, such as pDMAEMA, pNIPAm, poly(2-oxazoline), enable the reversible switching between hydrophobicity and hydrophilicity using temperature. Conjugating these polymers onto cyclic peptides will endow nanotubes with thermoresponsive properties. As no charge repulsion is involved, a change between two assembled states is therefore expected, other than between assembled and disassembled states. Chapman et al. reported a class of thermoresponsive cyclic peptide–polymer conjugates bearing two poly(ethyl oxazoline) (pEtOx) chains. The CP-(pEtOx40)2 conjugate showed a clear transition at 70 °C in water, representing its cloud point temperature (TCP).125 These conjugates could reversibly form short nanotubes with an average hydrodynamic diameter of 20–100 nm below TCP, whereas large spherical particles with a diameter of 2.5–5.0 μm would occur when reaching TCP. Furthermore, these cyclic peptide−polymer conjugates have been utilized to construct thermoresponsive artificial transmembrane channels. For example, CP-pNIPAm conjugates were water-soluble below the lower critical solution temperature (LCST) (i.e., 22 °C) and formed cylindrical nanotubes with the length of 30–70 nm in aqueous media. They became lipophilic above the LCST (i.e., 35 °C), making them easier to interact with lipid bilayers to form transmembrane channels.135
Apart from the above-mentioned responsive factors, photoresponsiveness can also be used. Yang et al. reported the fabrication of hierarchical self-assembled photoresponsive tubisomes in water using a photosensitive polymer, poly(2-nitrobenzyl methacrylate (pNBMA).132 Upon UV irradiation, the hydrophobic segment pNBMA was transformed into a hydrophilic block poly(methacrylic acid) (pMAA) by the cleavage of the o-nitrobenzyl ester groups. The combination of the lack of hydrophobic effect, the electrostatic repulsion between the deprotonated pMAA in neutral aqueous solutions, and the steric hindrance of bulky pPEGA caused the disassembly of tubisomes.
To conclude, with the well-established knowledge of stimuli-responsive polymers, a wide range of stimuli-responsive conjugates could be designed.149,150 The capability of switching between hydrophobicity and hydrophilicity using extra stimuli is proved to be extremely attractive for drug delivery systems and artificial transmembrane channels. In the future, systems which could respond to other stimuli, such as near-infrared (NIR), enzyme, redox, will demonstrate more potential in biological applications. Furthermore, the possibilities of tuning the dynamic property of the nanotubes using stimuli will enable better control over the self-assembling process but remain largely unexplored.
4.3.2. Introduction of Cleavable Linkers
A cleavable linker between the cyclic peptide and polymer is also able to endow the cyclic peptide–polymer conjugates with stimuli-responsive properties.151,152 For example, Hartlieb et al. reported an example of redox-responsive cyclic peptide–polymer conjugates using a cleavable disulfide bond as the linker.153 As shown in Figure 36, two water-soluble pEtOx chains were attached to the cyclic peptide with a cleavable linker in between, enabling the redox-responsive detachment of the polymer corona. As a result, the addition of 1,4-dithiothreitol (DTT) led to a structural transition from self-assembled polymeric nanotubes to larger agglomerates due to the lateral aggregation between nanotubes. This material showed redox-responsive membrane activity. More recently, pyridyl disulfide reaction chemistry was developed as a straightforward strategy toward redox-responsive cyclic peptide–polymer conjugates.137 The chemistry involves the reaction between a pyridyl disulfide (PDS) moiety and a free thiol group, which is proved to be highly efficient under mild conditions.154,155 Thus, a polymer bearing a PDS moiety made by RAFT polymerization could be efficiently attached onto a cyclic peptide bearing a thiol group, resulting in a redox-responsive conjugate with a disulfide linker. Besides, the orthogonality between pyridyl disulfide reaction chemistry and other conjugation chemistries allows the facile synthesis of asymmetric conjugates with redox-responsiveness.
Figure 36.

(a) Schematic description of cyclic peptide–polymer conjugates bearing cleavable linkers and reduction-induced membrane interaction. (b) Synthesis of pEtOx bearing either cleavable or noncleavable linkers. (c) Synthesis of the stimuli-responsive and nonresponsive conjugates. Reproduced with permission from ref (153). Copyright 2019 The Royal Society of Chemistry.
The introduction of cleavable linkers permits more freedom when choosing either polymers or cyclic peptides, which enables the integration of novel functionalities while maintaining the stimuli-responsiveness. The dramatic difference between polymer-wrapped nanotubes and “naked” SCPNs when interacting with biomembranes opens new opportunities for the design of smart antimicrobial materials.
4.3.3. Introduction of Stimuli-Responsive Cyclic Peptides
Apart from the utilization of stimuli-responsive polymers and cleavable linkers, it is also possible to directly use stimuli-responsive cyclic peptides. However, given the relatively strict requirements on their chemical structures, only limited examples of stimuli-responsive cyclic peptides have been reported so far. Most cases involved pH-responsive cyclic peptides, where nanotube disassembly was triggered by the electrostatic repulsion of charged side chains when changing the pH. Therefore, pH-responsive cyclic peptide–polymer conjugates could be obtained by attaching polymers onto these cyclic peptides. Alternatively, steric hindrance could be used to control the self-assembly of cyclic peptides. Recently, Perrier and co-workers showed an example of switching the self-assembly of cyclic peptide–polymer conjugates via host–guest chemistry (Figure 37).130 Cucurbit[7]uril (CB[7]) was used as a macrocyclic host because its diameter is greater than the distance between cyclic peptide rings. A conjugate 81 with two phenylalanine moieties coupled onto the cyclic peptide, Phe2-CP-PEG, was designed, allowing the incorporation of CB[7] by host–guest interactions. Thus, the steric hindrance caused by the two CB[7]s will lead to the disassembly of the polymeric nanotubes. Before introducing CB[7], conjugate 81 formed a core–shell cylinder in water with a length of 16.8 ± 0.5 nm from SANS. The complexation with CB[7] caused the disassembly of the polymeric nanotubes, which was evidenced by SANS indicating the presence of merely polymer chains. Moreover, this process is reversible. When a competitive guest molecule (1-adamantanamine, ADA) was incorporated into this system, CB[7] could be released from conjugates, which allowed the polymeric nanotubes reform in aqueous solutions. Compared to the other two strategies, the introduction of stimuli-responsive cyclic peptides is believed to be the most efficient because it interferes directly with the interactions between the cyclic peptides.
Figure 37.

(a) Schematic representation of the reversible self-assembly of a cyclic peptide–polymer conjugate via host–guest chemistry. (b) Chemical structures of the cyclic peptide–polymer conjugate Phe2-CP-PEG 81, CB[7], and ADA. Reproduced with permission from ref (130). Copyright 2019 The Royal Society of Chemistry.
4.4. Summary
The development of cyclic peptide–polymer conjugates has greatly advanced both the fundamental studies and practical applications of self-assembling cyclic peptides in solutions. The attachment of polymers offers more control over the self-assembling process of the cyclic peptides. By the rational design of the cyclic peptide–polymer conjugates, supramolecular assemblies with targeted structures and dynamics could be obtained. Furthermore, the capability of introducing stimuli-responsiveness emphasizes the advantage of a supramolecular system, which will greatly expand its application as “smart” supramolecular materials. While continuous efforts are to be made to fabricate self-assemblies with more architectures using cyclic peptide–polymer conjugates, the application of these materials is still in its infancy and is expected to witness enormous growth in the future.
5. Characterization of Supramolecular Structures Formed by Cyclic Peptides
The development of any class of self-assembled materials comes a requirement to understand the fundamental mechanism driving their behavior. In the case of supramolecular structures formed by self-assembling cyclic peptides, much of our understanding of the mechanism of their self-assembly has been derived from seminal studies performed in the 1990s. As discussed in detail in previous sections, these initial design principles have been substantially expanded upon, yielding a wealth of materials with diverse chemistries imparting distinct characteristics. As the breadth of supramolecular assemblies based on cyclic peptides has increased, so too has the means by which the resultant assemblies have been characterized.
In this section, we will discuss the initial characterization of the mechanism of cyclic peptide self-assembly, summarizing the methodologies which have been employed and how the information gained from each technique has synergistically provided the understanding we have today. While it is beyond the scope of this Review to cover all characterization methodologies applicable to supramolecular cyclic peptide assemblies, we will subsequently discuss the most frequently utilized structural characterization methodologies.
5.1. Determination of the Mechanism of Self-Assembly
As discussed in the previous section, the first indication of the mechanism of CP nanotube self-assembly came from pioneering studies in the 1990s, using a combination of ED and FT-IR. Both cyclo-[(l-Gln-d-Ala-l-Glu-d-Ala)2-] (5) and cyclo-[(l-Gln-d-Ala-l-Glu-d-Ala)3-] (8) were found to form rod-shaped crystals composed of CP nanotubes with CPs adopting similar conformations by cryo-ED, where the peptide backbone adopts a planar conformation with side-chains extending out parallel to the plane of the ring. Diffraction patterns indicate spacing between the CP units was 4.78 Å, with molecular models suggesting CPs stack forming extensive intrananotube hydrogen bonding contacts between the backbone amide groups and side chains participating in internanotube hydrogen bonding. This suggested that CPs within a nanotube stack in an extensive hydrogen-bonded antiparallel β-sheet structure. The ability of FT-IR to identify specific vibrational modes characteristic of bonds in different environments provided further evidence of this stacked β-sheet structure. Specifically, in both cases, C=O stretching amide-I and N–H bending amide-II bands indicated an extended, planar backbone conformation with N–H stretching bands identified at frequencies indicative of tightly packed hydrogen bonding interactions.
To further confirm the self-assembly mechanism of CP nanotubes is mediated via hydrogen bonding interactions between backbone amide residues, Ghadiri and co-workers synthesized cyclo-[(l-Phe-d-MeN-Ala)4-] (14), containing N-methylated d-alanine residues (Figure 8).37 This results in one face of the CP ring being devoid of hydrogen bond donors, restricting CP self-assembly to dimers. The inclusion of aromatic phenylalanine residues facilitated crystallization. Analysis of X-ray diffraction data indicated peptide dimers associate via hydrogen bonds between the amide backbones, consistent with previous ED studies. Notably, while π–π stacking between phenyl rings could be thought to contribute to the self-assembly of CP-dimers, the aromatic rings sit out of plane, contributing instead to lateral interdimer interactions. To investigate the thermodynamics of CP nanotube formation, 1H NMR experiments were performed, allowing identification of N–H resonances corresponding to unimeric CPs or dimeric CP species, which are forming hydrogen bonding interactions. This allowed the determination of the association constant of CP dimers in CDCl3 and CCl4 solvents. Interestingly, the association constant was an order of magnitude larger in less polar CCl4 (Ka = 1.4 × 104 M–1) compared to CDCl3 (Ka = 1.26 × 103 M–1), indicating the equilibrium position is shifted more toward dimers in less polar solvent with reduced capacity for competing for hydrogen bonding interactions with backbone amide groups. Performing this analysis at multiple temperatures allowed the construction of Van’t Hoff plots, demonstrating dimer self-assembly is driven by a large enthalpic contribution (−11 kcal mol–1) with only a minor entropic penalty (−23.7 cal K–1 mol–1). The large negative enthalpy change associated with dimer self-assembly further supported hydrogen bond formation as the primary driving force, with the sum of individual enthalpy changes for each expected hydrogen bonding interaction accounting for the majority of the experimentally determined value. It is worth noting that the entropic penalty for self-assembly would be expected to be larger for each additional CP unimer binding to a CP nanotube, while the enthalpy change would be constant. This provides a thermodynamic framework to govern the ultimate length of a given SCPN in a given solvent.
Because of the N-methylated d-alanine residues utilized in the above study, the orientation of CPs within the dimer was restricted to an antiparallel β-strand arrangement. For CPs with hydrogen bond donor sites on both faces of the peptide, it is feasible that CPs could interact via a parallel or antiparallel arrangements, potentially with both orientations being present within a single nanotube. To investigate this further, an enantiomeric N-methylated peptide was synthesized, cyclo-[(d-Phe-l-MeN-Ala)4-] (16, Figure 9).38 Similarly to cyclo-[(l-Phe-d-MeN-Ala)4-], enantiomerically pure 16 dimers are only capable of forming antiparallel β-strands, while the formation of a racemic heterodimer would be restricted to the parallel arrangement. 1H NMR is able to distinguish between dimeric species through the difference in amide proton J coupling constant between the two arrangements. Upon preparing a racemic solution of these enantiomeric CPs in CDCl3, 1H NMR was used to determine the relative concentrations of each species; monomeric CPs, enantiomeric pure homodimers (antiparallel) and racemic heterodimers (parallel). This allowed the determination of the association constants for dimers with antiparallel (Ka = 2540 M–1) and parallel (Ka = 640 M–1) β-strand arrangements, indicating that the antiparallel β-strand stacking is the more thermodynamically favorable mode of interaction.
While the studies discussed in this section thus far have focused on the interaction between the CP backbone as the predominant factor determining the mechanism of self-assembly, the choice of side chains has also been found to have a substantial impact on both the resultant structural and thermodynamic properties of CP nanotubes. Ghadiri and co-workers designed a series of cyclic octapeptides, containing alternating l-glutamine and hydrophobic d-residues, either Ala, Val, Leu or Phe.31 Cryo-TEM and FT-IR analysis indicated all CPs formed nanotubular structures stabilized by a tight β-sheet hydrogen bonding network. Analysis of cryo-ED data indicated that cyclo-[(l-Gln-d-Val)4-] was the only compound found not to form extended nanotube crystals, though short individual nanotubes, approximately 10 nm long, could be identified. Broadened diffraction peaks suggest that these nanotubes exhibited poor lateral packing, preventing single crystal formation. As evidenced from crystal structures, glutamine side chains contribute to both intra- and internanotube hydrogen bonding, demonstrating that the choice of side chains have important implications in the thermodynamics of CP nanotube self-assembly. The authors hypothesize that the cumulative sum of hydrogen bonds still enthalpically drives self-assembly, though hydrophobic effect imparted by hydrophobic side chains help protect the intrananotube hydrogen bonding from competing solvent interactions.
The mechanism of self-assembly could be revealed by fluorescence spectroscopy with the aid of fluorescent probes. Thioflavin T (ThT) is a fluorescent probe that enhances its fluorescence emission in its planar state when trapped within β-sheets.156 Hence, the formation of SCPNs with β-sheet structures could be confirmed by fluorescence spectroscopy using ThT as an external probe. For example, the effect of temperature on preassembled nanosheets of CP 9 could be investigated by measuring ThT emission during heating and cooling steps.32 Results showed the quenching of ThT emission with the increase of temperature, indicating a progressive disassembling route of the nanosheet assembly. The conjugation of fluorophores onto the cyclic peptides can also provide direct and valuable information on the self-assembly of cyclic peptides. The alignment of the attached fluorophores during the self-assembling process leads to the change of their relative spatial distance, which may be reflected by fluorescence spectroscopy. Among those, pyrene has been widely used as an internal probe to study the self-assembling behavior of cyclic peptides. The stacking of the cyclic peptides brings the pyrene fluorophores in close proximity, thus resulting in a significant bathochromic shift of the fluorescence emission due to excimer formation.43,44,108,109 Last but not least, as mentioned earlier, by attaching a FRET pair (i.e., Cy3, Cy5) onto cyclic peptides, the dynamic behavior could be obtained by fluorescence spectroscopy.136,141
5.2. Structural Characterization
5.2.1. Imaging Techniques
It is well-known that the self-assembled structure of supramolecular aggregates greatly impacts their potential application. Within the context of CP assemblies, multiple studies have demonstrated a structural dependence on their biological applications.131,157,158 Structural investigation is, therefore, a crucial aspect in their design process, and this is frequently done by employing high-resolution microscopy techniques, such as TEM and AFM.
Both techniques have the potential to provide subnanometer spatial resolution,159−161 which has been exploited to great effect, as discussed above, in determining the fundamental mechanism of CP nanotube self-assembly. Although the information provided by TEM and AFM are similar, TEM relies on the absorption and scattering of an incident high energy beam of electrons by electron density within a sample, whereas AFM provides a surface topography map of a sample adsorbed to a solid substrate through deflection of a nanoscale cantilever tip. A requirement for successful TEM imaging is that there is sufficient electron density contrast between the sample and substrate to resolve particles. AFM, while not dependent on electron density, requires similar differentiation between sample and substrate through differences in both the height of the adsorbed sample and its physicochemical properties.
While the application of TEM in structural and mechanistic studies of CP self-assembly was touched on in previous sections, TEM has also been comprehensively employed for the structural investigation of CP–polymer conjugate self-assembly,126,127,131 where the focus is on the large-scale structure of individual nanotubes, rather than the crystallization of CP nanotubes in the solid-state. A recent reference is the work of Rho et al. featured in section 4.2.1.2.136 Here, TEM was applied to confirm the morphology of the CP-PEG conjugates in an aqueous environment (71–73), as shown in Figure 38. The collected TEM images were also statistically analyzed to obtain distribution information on the SCPPNs’ measured lengths and widths. For example, an average width ranging from 7 to 8 nm was noted for the dye conjugates, which was consistent with the lack of lateral aggregation as also seen in the images.
Figure 38.

(a) TEM image of the PEG-CP-Cy3 conjugate 71 stained with UOAc against nanotubular length (b) and diameter (c) distributions extracted from the TEM images. Reproduced with permission from ref (136). Copyright 2017 John Wiley and Sons.
Comparable structural studies of CP–polymer conjugate assemblies have also been performed using AFM.70,120,157,162,163 Hourani et al. investigated the thermoreversible assembly process and nanotube growth of a CP–PEG conjugate 82 through AFM imaging.70 Collected images showed that consecutive annealing and quenching of the conjugate disrupts its assembly process resulting in the dissociation of its nanotubes (Figure 39). However, if allowed to slowly cooled following heating, high aspect ratio nanotubes could be reformed. Size dimensions of the conjugate’s nanotubes could also be derived from the images whereby they averaged at a width of ∼5.5 nm, the height of ∼0.5 nm, and a length range of 50–150 nm. These dimensions corresponded to those of individual nanotubes stacks, highlighting the role of polymers in decreasing the lateral aggregation earlier noted with the unconjugated γ-Mba-containing CP.
Figure 39.

(a) Chemical structure of conjugate 82. AFM images of (b) a spin-casted THF solution of conjugate 82, (c) the conjugate solution after solvent-annealing and quenching, and (d) the conjugate solution following thermal annealing at 80 °C for 1 h and slow cooling. Reproduced with permission from ref (70). Copyright 2011 American Chemical Society.
The presence of either external or internal fluorescent probes makes it possible to utilize fluorescence microscopy techniques to characterize the cyclic peptide assemblies. While traditional fluorescence microscopy is only suitable for large assemblies,32,33 super-resolved fluorescence microscopy, such as Stochastic Optical Reconstruction Microscopy (STORM), is more powerful for the acquisition of finer structural information on the cyclic peptide assemblies. STORM is a super-resolution (up to 20–30 nm) method that manipulates fluorophores in a system to emit light at distinct times, thereby making them resolvable.164,165 This is achieved by either repeatedly activating and deactivating photoswitchable fluorophores (e.g., Cy5) or by incorporating dye pairs with distinctive wavelength emissions (e.g., Cy5, Cy3).166,167 Not only is this useful for the structure elucidation of complex systems, but information on self-assembly pathways can also be deduced. An illustration of this is detailed in the work of Rho et al., who used STORM to assess the mechanism of mixing between two types of dye-modified conjugates (74 and 75).141 Upon being premixed, colocalization was observed, suggesting that units from both conjugates integrated with each other (Figure 40a) in contrast to when the conjugates were coinjected. In the latter case, the conjugates aggregated into separate nanotube populations, as seen in Figure 40b.
Figure 40.

STORM images of conjugates 74 and 75 showing their assembly mechanism following (a) premixing and (b) coinjection. Reproduced with permission from ref (141) and available under a CC BY 4.0 License (http://creativecommons.org/licenses/by/4.0/).
5.2.2. Scattering Techniques
While imaging techniques discussed to this point are all able to provide detailed information on the structure of individual self-assembled cyclic peptide-based aggregates, they all are limited by requiring immobilization, either by surface adsorption or by vitrification, which can potentially affect the structural behavior and may result in analyses that are not characteristic of the solution structures.168,169 Furthermore, as a result of the practicality of imaging a large number of particles at sufficient magnification to resolve structural information, the low sampling of particles can limit the statistical significance of any structural conclusions drawn solely from imaging techniques.159,170 Scattering techniques, however, are able to provide high resolution structural and compositional information on a population of particles in bulk solution, providing a wealth of complementary structural information. However, these techniques rely on modeling and data fitting and often require a good knowledge of scattering theory. This section reviews the various uses of scattering techniques for the characterization of SCNPs, by first providing the basis of typical scattering techniques.
The most widely available scattering technique with application in the structural investigation of cyclic peptide self-assembly is static light scattering (SLS). SLS measures the angular dependence of scattered light, usually a visible laser source, by a sample in solution. Supramolecular CP aggregates in solution are of a size where, provided there are no interparticle interactions, samples should produce isotropic scattering over the angular range typically measured.171 Provided both the refractive index of the solvent and the refractive index increment of the sample are known, measuring a range of sample concentrations allows the construction of the Zimm plot.172 This allows extrapolation to the “zero-angle” intensity of a sample at infinite dilution, providing both the radius of gyration (Rg) and molecular weight (Mw) of the aggregates in solution. Given the known molecular weight of each CP-based unimer, this has been extensively applied to determine Nagg of resultant supramolecular CP assemblies.136,141,153,158,173
Although not classified as scattering techniques, diffusion ordered spectroscopy (DOSY) is included here due to the great similarity regarding the size information it reveals. DOSY is a well-established NMR technique that reports diffusion coefficients for individual resonances in NMR spectra. It has been widely used to analyze polymers and supramolecular assemblies to measure the diffusion coefficient of a species in solutions.174−177 Hydrodynamic radius (Rh) of the species could then be calculated using the Stokes–Einstein equation by assuming it is hydrodynamically spherical. Despite being a semiquantitive method, it is proved to be beneficial as a complementary technique for the characterization of stimuli-responsive CP–polymer conjugates.130
Other than an indication of size in the form of Rg, Mw, or Rh, SLS and DOSY provide limited information on the structure of the CP-assemblies themselves. In order to provide high resolution information on the structure of self-assembled CP aggregates, incident radiation is required with a wavelength suitable for resolving structural components within the self-assembled aggregates. Hard X-rays (those with a wavelength of around 0.07–0.2 nm) are ideal for probing the length scales of self-assembled aggregates of cyclic peptides. Small-angle X-ray scattering (SAXS) measures the intensity of monochromatic X-rays scattered at small angles by a sample as a function of the momentum transfer Q (Q = 4π sin θ/λ, where 2θ is the scattering angle, and λ is the X-ray wavelength) providing information on the size, shape and to a limited extent, composition of CP aggregates, covering length scales from ∼1–100 nm. The “scattering power” of a sample is defined by the scattering length density (SLD). As X-rays scatter from the electron density within a sample the SLD is defined as the sum over all atoms within a particle (N) of the product of the atomic number of each atom (Z) and the scattering length of the electron (be, the propensity of a single electron to interact with an X-ray of a given energy) divided by the total volume of the particle (V)
For a successful scattering experiment to be performed, a requirement for SAXS is that there needs to be a substantial difference between the SLD of the particle and the solvent. While not a problem for electron-rich particles, this can prove problematic for CP-based materials which are of a similar atomic composition to the solvents frequently employed. To overcome this limitation, SANS, a similar technique utilizing a collimated beam of neutrons as the incident radiation, has been frequently employed. As opposed to X-rays, which scatter from electron density, neutrons scatter from nuclear density, where the nuclear scattering length density (nSLD) is given by
where bc describes the coherent atomic nuclear scattering length. The atomic nuclear scattering length shows no trend with increasing atomic number, with large differences between different isotopes of the same element. Notably, the difference in nuclear scattering length between the two most abundant hydrogen isotopes, protium and deuterium is particularly pronounced. Through deuteration of either the sample or the solvent, the scattering contrast can be substantially enhanced.
While a detailed description of small-angle scattering (SAS) theory, instrumentation, and analysis are beyond the scope of this Review (for more information see the following references178−182), a brief description will be presented in the following section. For all SAS measurements, the angular dependence of scattered intensity (I(Q)) is given by
where ΔSLDi is the difference in contrast between the particle and the solvent (which is dependent not only on the composition of the particle/solvent but also the radiation used), Vi is the volume of the particle, Pi(Q) represents the form factor (describing the structure of the particle), and S(Q) represents the structure factor (describing the interaction between discrete particles).
Because of the inherent property of CP nanotubes to laterally aggregate in solution, reducing their solubility, SAS in the solution state has seen limited application in their characterization to date. There are, however, applications of CP nanotubes to which SAS has provided unique information not accessible by other structural techniques. One such example is the utilization of CPs as low molecular weight gelators.42 Here, both the tendency of individual CP building blocks to form elongated tubular structures and the tendency of individual nanotubes to laterally aggregate was exploited. The authors systematically investigated the influence of side-chain chemistry on the gelation performance of CP nanotubes, carefully balancing hydrophobic aromatic tryptophan residues with hydrophilic residues, either basic lysine or acidic glutamic acid. Initially, the influence of CP composition was investigated on their gelation performance. It was found that two symmetric cyclic peptides, cyclo-[(d-Leu-l-Trp-d-Leu-l-Lys)2-] (83) and cyclo-[(d-Leu-l-Trp-d-Leu-l-Glu)2-] (84), formed hydrogel networks of entangled nanotube fibers at pH 3 and 10, respectively. Interestingly, increasing the number of Trp residues resulted in insolubility while increasing the number of charged residues resulted in the formation of individual soluble nanotubes, where electrostatic repulsion restricted the internanotube contacts required to form extended networks of cyclic peptide fibers. Unsurprisingly, a clear pH dependence was identified where partial deprotonation (of 83) or protonation (of 84) resulted in hindered solubility, restricting their gelation. SAXS measurements were performed to provide further structural insight into the nanostructure of hydrogels formed by 83 and 84 (Figure 41). While substantial differences in the scattering pattern were observed for each sample, neither gel showed a clear plateau at low values of Q, indicating the formation of entangled networks of elongated particles in both cases. Data were fit to models containing two components: a cylindrical form factor describing the structure of individual CP nanotubes, and a mass fractal model describing the extent of entanglement between CP nanotubes. Both gels contained CP-nanotube fibers over 200 nm in length, though a greater entanglement was observed for the cationic CP 83. It was also found that the individual CP 84 nanotubes within the fibers were substantially shorter than CP 83 nanotubes, explaining differences in entanglement between the two gels. Combined, these observations highlight the importance of the surface properties of resultant CP-nanotubes in controlling internanotube interactions in solution.
Figure 41.

(a) Chemical structures of cyclic peptide 83 and 84 found to function as pH-responsive low-molecular weight gelators; (b) SAXS data and fits of supramolecular gels formed by 83 and 84. Reproduced with permission from ref (42). Copyright 2018 John Wiley and Sons.
While the inherent insolubility of “naked”, uncharged cyclic peptide nanotubes limits their characterization by SAXS in the solution state, their self-assembly at the air–water interface has been characterized by grazing-incidence SAXS (GISAXS), providing information on the interaction between CP nanotubes.183 The first peptide investigated, cyclo[(l-Phe-d-N-Me-Ala-)4] (CP 85, Figure 42 a), containing N-methylated d-alanine residues, restricting self-assembly to CP dimers as discussed in Sections 2.1.3 and 5.1. Indexing of Bragg rods from GISAXS patterns indicate the CP-dimers order within a square lattice with a structure similar to that determined for the 3-dimensional crystal (Figure 42b).37 Interestingly, the CP-dimers orientate in a thin film approximately three dimers thick with the plane of the peptide ring oriented parallel to the interface. Conversely, cyclo[(l-Trp-d-Leu)3-l-Ser-d-Leu-] (86) was found to form long nanotubes over 100 nm in length oriented with the long axis of the nanotube parallel to the interface. These nanotubes displayed lateral 1-dimensional aggregation with individual CPs within adjacent nanotubes offset relative to each other (Figure 42c). This orientates amino acid side chains such that internanotube interactions are mediated by hydrogen-bonding between serine hydroxyl groups and π–π stacking of tryptophan indole rings from adjacent nanotubes. Importantly, no ordered clustering of cyclo[(l-Trp-d-Leu-)4] (87) nanotubes could be identified, highlighting the importance of peptide side-chain sterics in controlling the interaction between CP nanotubes.
Figure 42.

(a) Chemical structures of 85, 86, and 87 investigated using GISAXS. (b) Unit cell structure of 85, self-assembled at the air–water interface. (c) Self-assembled structure of 86 at the air–water interface. Overlay shows the unit cell as determined by GISAXS. Reproduced with permission from ref (183). Copyright 1999 American Chemical Society.
For structural determination of CP–polymer conjugate assemblies in solution by SAS, it is primarily the form factor which is of interest in describing the structure of individual nanotubes. It is therefore desirable to adjust sample conditions such that particles in solution can be considered dilute and noninteracting such that S(Q) approximates to unity. Through model-dependent analysis, structural parameters comprising the form factor can be obtained to describe the size and shape of the CP–polymer conjugate assemblies in solution. CP–polymer conjugates can be considered core–shell cylinders with a sufficient difference in scattering contrast (especially in the neutron case) to differentiate between the cyclic peptide core and the solvated polymer corona. Exploiting the enhanced contrast available using SANS, multiple studies have confirmed the core–shell cylinder structure formed by CP–polymer conjugates in both organic and aqueous media.127,128,136,141,148,153,158,173
Further confirming the presence of nanotubes, SANS has also been applied to great effect in gaining a fundamental understanding of the influence of polymer grafting density and solvent polarity on the structure of CP–PEG conjugate nanotubes.139 SANS patterns of the conjugates (68–70) discussed in Section 4.2.1 show a Q–1 dependency at low values of Q in all solvents investigated, characteristic of cylindrical structures (Figure 43). However, differences in the onset of the Guinier region, describing the length of the nanotubes, and in the higher-Q regions describing the radius of gyration of the polymer chains can identify structural features beyond large-scale morphology. Interpretation of structural parameters derived from SANS data found that solvents with a high capacity for accepting hydrogen bonds will compete with hydrogen bonding between CP backbones, restricting nanotube self-assembly to unimeric or oligomeric assemblies. As the hydrogen bonding capacity of the solvent decreases, a higher grafting density will shield the CP core from competing hydrogen bonds to solvent molecules, resulting in longer nanotubes. Finally, in nonpolar solvents, the lack of solvent competition for hydrogen bonding to CP backbones promotes the formation of long nanotubes, where increasing grafting density can improve the solubility of the CP–PEG conjugates.
Figure 43.
Example SANS data demonstrating the influence of solvent polarity on the self-assembly of conjugate 68. Reproduced with permission from ref (139). Copyright 2018 The Royal Society of Chemistry.
Because of the starkly different form factors describing individual polymer chains and SCPPNs, SANS is an ideal tool for investigating the structural basis of stimuli-responsive cyclic peptide–polymer conjugates. A unimeric cyclic peptide–polymer conjugate (in a good solvent) can be approximated as a Gaussian coil, whereas, as discussed above, a tubular assembly of conjugates are best described as core–shell cylinders. The difference in appearance between these form factors can be exploited to understand the stimuli-responsive reversible assembly and disassembly of cyclic peptide–polymer conjugates. Larnaudie et al. conjugated diblock copolymers to a cyclic peptide, where a short pH-responsive pDPAEMA was introduced between the cyclic peptide and the hydrophilic but nonresponsive pHPMA block.138 At pH 7.4, where pDPAEMA side chains are deprotonated, SANS data indicated the formation of core–shell cylindrical micelles. Upon decreasing the pH to 5.0, where pDPAEMA side chains are protonated and cationic, SANS data showed disassembly of the nanotubes, driven by electrostatic repulsion between charged pDPAEMA blocks. Similarly, SANS has been able to demonstrate redox-responsive self-assembly of CP–polymer conjugates.137 Here, an asymmetric conjugate was synthesized containing a linear PEG and a pPEGA brush, where the latter is conjugated by a redox-responsive disulfide linker (88, Figure 44a). As with the previous observations,139 SANS demonstrated the intact conjugate did not undergo self-assembly into long nanotubes (Figure 44b). Upon cleavage of the disulfide linker with the addition of reductant, the loss of pPEGA chains removes the steric constraints which restrict nanotube self-assembly. Similarly to the examples discussed above, SANS data confirmed redox-responsive behavior, with the scattering pattern being described to a core–shell cylindrical form factor, characteristic of nanotubes.
Figure 44.

(a) Chemical structure of PEG-CP–S-S-pPEGA (88) and a schematic representation of the redox-responsive self-assembly. (b) SANS data and fits corresponding to 88 before and after exposure to reducing agent demonstrating redox-responsive self-assembly. Reproduced with permission from ref (137). Copyright 2019 American Chemical Society.
As discussed previously, the synthesis of asymmetric cyclic peptide–polymer conjugates containing hydrophilic and hydrophobic polymers has dramatic effects on their self-assembly, resulting in the formation of so-called “tubisomes”. SANS is uniquely able to differentiate between the formation of hierarchical tubisomes and individual nanotubes. As the hydrophobic effect leads to phase separation of hydrogen-rich hydrophobic polymer chains into the tubisome core, this leads to sufficient contrast to distinguish between the hydrophobic core and the hydrophilic, solvated corona. SANS data collected for the asymmetric pBA23-CP-pPEGA45 conjugate (Figure 45a) demonstrates the formation of a core–shell cylindrical micelle, but in contrast to conventional SCPPNs, the core radius increases to 76 Å, far larger than the diameter of the cyclic peptide ring alone, confirming the formation of tubisomes.133 Further investigation into the ratio of hydrophobic to hydrophilic polymer lengths conjugated to the cyclic peptide on tubisome self-assembly also used SANS to demonstrate the formation of tubisomes, irrespective of the ratios employed (Figure 45b and c).134 Similar core radii were determined to that found previously, with an increase in the length of the hydrophobic chain intuitively leading to an increase in the radius of the hydrophobic core.
Figure 45.

SANS data and fits of tubisomes formed by pBA-CP-pPEGA with differing ratios of hydrophobic and hydrophilic polymer chain lengths. Reproduced with permission from ref (134). Copyright 2019 The Royal Society of Chemistry.
These examples demonstrate the power of SAS techniques in not only defining the global morphology of the supramolecular assemblies formed by CPs but allow detailed structural characterization of the internal and surface structure of the particles and interactions between particles. Through detailed structural characterization by SAS, it has been possible to determine the structural effects of a broad range of chemical modifications, expanding on the range of accessible morphologies and applications.
5.2.3. Chromatographic Techniques
While a common goal in the synthesis of supramolecular materials is to produce monodisperse assemblies, in reality, a size/molar mass distribution will always be observed. In the case of CP self-assembly, this dispersity occurs as a result of not only the stochastic self-assembly of CP unimers resulting in a population of nanotubes of varying Nagg, but also, in the case of CP–polymer conjugates, the dispersity associated with polymer chains as a result of synthesis conditions.136,184−186 Both distributions are expected to have a significant impact on the self-assembly behavior and functionality of cyclic peptide systems, as in the case of polymers,185,187,188 therefore their measurement is important and multiple methods can be implemented to do so. These include the imaging and scattering techniques discussed thus far; however, their use for this purpose is limited by sampling and modeling restrictions, respectively. A more preferred way to estimate dispersity, is the coupling of chromatographic techniques with the appropriate detector setup (e.g., light scattering–refractive index–UV–vis). Doing so enables the determination of representative molar mass or size values for every sample fraction as opposed to the biased averaged values obtained from the batch analysis.
Historically, the most established separation method for determining molar mass distributions is SEC because of its sensitivity, robustness, and reproducibility. Separation is functioned on the hydrodynamic volume of analytes in solution and their ability to permeate through a heterogeneous porous column media. As it stands, however, its use for supramolecular CP assemblies has been confined to their analysis in unimeric forms and an explanation for this will be provided later on. SEC in this case is employed to confirm the complete coupling of polymers to CPs as well to determine the dispersity of the grafted polymers as reported by various authors.121,122,141,153 An example SEC chromatogram of the conjugates (68–70) studied by Mansfield et al. is shown below (Figure 46).139
Figure 46.
SEC traces of an unconjugated linear PEG alongside unimeric CP–PEG conjugates having one (68), two (69), or three (70) linear polymer arms. Reproduced with permission from ref (139). Copyright 2018 The Royal Society of Chemistry.
As previously stated, analysis of SCPPNs by SEC is confined due to limitations associated with its use of a column. These include the risk of nonspecific adsorption in addition to degradation effects induced by shear forces in the column packing which would compromise the structural integrity of large assemblies with high molecular weights.189−191 An innovative approach that circumvents these issues is the use of asymmetric flow field-flow fractionation (AF4), which conducts separation within an open channel. Sample constituents are fractionated by a gentle hydrodynamic force field that drives them into different parabolic laminar flow velocities according to their diffusion coefficients.192,193 Its successful use for the characterization of cyclic peptide-based assemblies has been achieved by our group whereby well-resolved fractograms of assembled CP–polymer conjugates were collected for the determination of their distribution. This approach, therefore, provides a route to assessing the mechanism of assembly which consequently promotes a better understanding of SCPPN structures. An illustrative AF4 result of a cyclic peptide conjugate (89) is shown in Figure 47.
Figure 47.

(a) Chemical structure of conjugate 89. (b) AF4 fractogram of 89 with signals from MALS90°– RI detectors, solvent: 0.1 M NaCl + 0.02% NaN3.
5.3. Summary
The growth of supramolecular cyclic peptide materials and their targeted applications has created the need for the continuous progression of multiple characterization techniques in order to thoroughly assess the functionality, behavior, and design efficiency of the cyclic peptide structures. An array of techniques are available today, with the choice of their application influenced by factors such as the properties of the assemblies, the characterization environment, and the sensitivity range of the applied instrumentation. While a lot of information can be derived from the techniques individually, the complementary use of different methods is the most effective way in acquiring cross-validated and accurate data. This has been demonstrated throughout the section whereby the pairing of scattering and chromatographic techniques enables the collection of representative unbiased data. A second example is the validation of size information derived from imaging techniques via scattering experiments or in the reciprocal case, confirming shape estimates. The dynamic nature of the supramolecular assemblies, particularly in solution, is however a complicated aspect of their characterization. This is in addition to their self-assembly mechanisms, and therefore further advancements of analysis or sample preparation techniques are required to gain more comprehensive insight. An interesting future research area would be in modulating the cyclic peptides self-assembly kinetics such that it is confined in a time frame that is compatible with the processing window of numerous techniques. The development of standardized characterization protocols would also be a welcomed future goal so as to ensure comparability of results between different materials and laboratories.
6. Biological Applications of Self-Assembling Cyclic Peptides
6.1. Ion Channels, Lipid Interactions, and Membrane Insertion
Protein channels, peptides and metabolites embedded in cell membranes play a fundamental role in the transport of small molecules.194 These features are necessary for the regulation of intracellular processes and signaling cascades and, therefore, investigation of ways to artificially manage the exchange of cargo is of paramount importance for biomedical and disease research initiatives. The propensity of cyclic peptides to stack into elongated structures has naturally led to the investigation of how they might form “channels” that either (a) allow transport of small molecules through the confined space of the internal core, (b) interact and insert into lipid membranes on account of potential binding with certain amino acid combinations, leading to structural disruption, or (c) interact uniquely with native peptide or lipid species in a biological environment to alter another specific process. The occurrence of these behaviors has been investigated by various methods, starting with Ghadiri and co-workers who administered peptide nanotube assemblies to the phosphatidylcholine liposome model (with differing internal/external pH) and used fluorescence dye spectroscopy to track the collapse of the pH gradient, rationalizing formation of transmembrane channels to be the cause.29 Notably, control peptides without both the requisite properties for lipid partitioning and extended self-assembly did not manifest the same behaviors. Subsequently, various possible orientations of nanotube insertion into lipid membranes were accounted for using polarized attenuated total reflectance (ATR) FT-IR; nanotubes laying perpendicular to the bilayer normal in a regular environment but parallel in the absence of lipid matrix support, suggesting a possibility to manipulate functionality in different biological environments, for example in biosensor applications.195 Furthermore, the synthesis of cyclic peptides with diverse internal core radii, self-assembly properties (length), and external functional chemistries permits a wide range of unique transport behaviors to be unearthed for ions (Na+, K+, or Ca2+) and small molecules, such as water, ammonia, or glutamic acid.68,196−201 Moreover, Danial et al. have shown that the formation of transmembrane channels can be tuned according to peptide composition; the presence of charged side chains affording large pores, and neutral side chains forming unimeric pores, with a nonsymmetrical or amphiphilic amino acid sequence translating to higher membrane activity in calcein entrapped LUV assays (Escherichia coli lipid extract).202 Decoration of peptides with mixed polymer arms to form “Janus” nanotubes have similarly exhibited artificial pore formation in phospholipid bilayer models. The versatility of polymers synthesized by RAFT provides a degree of tunability in artificial channel formation, for instance, by mixing hydrophilic, hydrophobic, or thermoresponsive polymers to control bilayer interactions.129,135 More recently, Zhao et al. have exploited interactions of peptide nanotubes with lipidic membranes as an antiatherosclerotic agent to regulate the formation of HDL’s (high-density lipoprotein complexes which reduce cholesterol levels).203 Incubation of peptides in human plasma was analyzed via enzyme-linked immunosorbent assay (ELISA) to generate a structure–activity relationship validated library of sequences that highlighted the importance of specific amino acid topologies for effective promotion of pre-β HDL formation. They found that in contrast to apolipoprotein mimetic peptides which actively package lipids into “HDL-like” nanoparticles, Fast Protein Liquid Chromatography (FPLC) and liposome clearance assays indicate the nanotubes displace apoA-I from HDL’s to generate a lipid-poor environment capable of binding to cholesterol and are instead “modulators”. These systems are an exciting new avenue for tackling vascular diseases. A further novel application was discovered by Richman and Chemerovski who determined that self-assembled cyclic α-alt(d,l)-peptides that formed fibrils were able to bind to and stabilize the nonamyloid-β region of α-synucleins, thereby preventing aggregation and the downstream pathways which lead to amyloidogenic diseases, for instance, Parkinson’s or Alzheimer’s.204−206 Hence, the advantageous capability for producing diverse types of self-assemblies from such basic building blocks was clearly evident in the early years of research. Although most of the early literature concerns fundamental studies on the ion transport or membrane insertion behaviors with classical techniques and simplified bilayer models, it nonetheless strengthened the knowledge database of peptide nanotubes and laid the important groundwork for more advanced biological applications, which will next be evaluated.
6.2. Antibacterial Treatments
One of the greatest challenges facing the healthcare system in recent years has been the emergence of “superbacteria” which have developed resistance to traditional antibiotic medications due to overuse by the general public.207 A major concern is that gradually, medications will lose their effectiveness and thus research is now driving toward finding alternatives to replace clinical treatments that will soon become obsolete. Cyclic peptide nanotubes have emerged as an attractive solution to these challenges as they exhibit strong antibacterial properties on both Gram-negative and Gram-positive bacteria. In addition, the rigidity of the ring affords superior proteolytic stability. The relative ease of synthesizing these materials and the specificity for different membrane types which can be achieved from manipulating the amino acid sequences have allowed structure–activity relationships to be investigated, as was first demonstrated by Lopez et al., who presented the first report defining SCPNs as a class of novel antibacterial agents.208 They established that amphiphilic cyclic α-alt(d,l)-peptides composed from an alternating l-Trp and d-Leu segment and three consecutive hydrophilic amino acids were acting as ion transporters in lipid bilayer models and potentially also embed into bacterial membranes. A typical cyclic peptide 93 (Table 2) from this study, cyclo-[d-Lys-l-Glu-d-Arg-l-Trp-d-Leu-l-Trp-d-Leu-l-Trp-] exhibited potency against multiple Gram-positive and negative bacterial strains. Lopez et al. hypothesized that this is caused by insertion into bacterial membranes, disrupting transmembrane ion channels and increasing permeability, which induces eventual cell death by a “carpet-like” mechanism. Modifications of cyclic peptides to generate a library of compounds elucidated the influence of hydrophobicity and charge upon activity where replacement of lysine with histidine hampered any antibacterial effects. The introduction of glutamic acid also hindered the activity due to the electrostatic repulsion of negative charges with the membrane. Hence, it is clear that the rational design of these materials can facilitate advantageous behaviors upon a range of antibacterial models.
Table 2. Library of Biological Data for Compounds 90–151 with Antibacterial Activitya.
| MIC
(μM) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| compound number | peptide sequence | S. aureus 29213 | MRSA | MRSA in FBS | Bacillus cereus | VRE | MRSA | E. coli | Ulva linza (LD 66) | Navicula perminuta (LD 66) | HD50 (μM) | ref |
| 90 | d-Ser-l-Lys-d-Ser-l-Trp-d-Leu-l-Trp-d-Leu-l-Trp | ND | 8 | ND | ND | ND | ND | 8 | ND | ND | 24 | (208) |
| 91 | d-Thr-l-His-d-Ser-l-Trp-d-Leu-l-Trp-d-Leu-l-Trp | ND | 80 | ND | ND | ND | ND | 80 | ND | ND | 80 | |
| 92 | d-Arg-l-Gly-d-Asp-l-Trp-d-Leu-l-Trp-d-Leu-l-Trp | ND | 80 | ND | ND | ND | ND | 80 | ND | ND | 80 | |
| 93 | d-Lys-l-Glu-d-Arg-l-Trp-d-Leu-l-Trp-d-Leu-l-Trp | ND | 4 | ND | ND | ND | ND | 60 | ND | ND | 34 | |
| 94 | l-Lys-d-Glu-l-Arg-d-Trp-l-Leu-d-Trp-l-Leu-d-Trp | ND | 7 | ND | ND | ND | ND | 66 | ND | ND | 33 | |
| 95 | d-Arg-l-Glu-d-Arg-l-Trp-d-Leu-l-Trp-d-Leu-l-Trp | ND | 15 | ND | ND | ND | ND | 73 | ND | ND | 20 | |
| 96 | d-Lys-l-Glu-d-Lys-l-Trp-d-Leu-l-Trp-d-Leu-l-Trp | ND | 38 | ND | ND | ND | ND | 84 | ND | ND | 42 | |
| 97 | d-Lys-l-Ser-d-Lys-l-Trp-d-Leu-l-Trp-d-Leu-l-Trp | ND | 4 | ND | ND | ND | ND | 31 | ND | ND | 79 | |
| 98 | d-Ser-l-His-d-Lys-l-Trp-d-Leu-l-Trp-d-Leu-l-Trp | ND | 8 | ND | ND | ND | ND | 78 | ND | ND | 27 | |
| 99 | l-Ser-l-Lys-l-His-l-Trp-l-Leu-l-Trp-l-Leu-l-Trp | ND | 13 | ND | ND | ND | ND | 86 | ND | ND | 17 | |
| 100 | d-Ser-l-His-d-His-l-Trp-d-Leu-l-Trp-d-Leu-l-Trp | ND | 16 | ND | ND | ND | ND | 78 | ND | ND | 16 | |
| 101 | d-Glu-l-Lys-d-His-l-Trp-d-Leu-l-Trp-d-Leu-l-Trp | ND | 76 | ND | ND | ND | ND | 76 | ND | ND | 76 | |
| 102 | d-Lys-l-Lys-d-Lys-l-Trp-d-Leu-l-Trp-d-Leu-l-Trp | ND | 6 | ND | ND | ND | ND | 53 | ND | ND | 38 | |
| 103 | d-Arg-l-Arg-d-Lys-l-Trp-d-Leu-l-Trp-d-Leu-l-Trp | ND | 4 | ND | ND | ND | ND | 11 | ND | ND | 37 | |
| 104 | d-Lys-l-Arg-d-Lys-l-Trp-d-Leu-l-Trp-d-Leu-l-Trp | ND | 7 | ND | ND | ND | ND | 30 | ND | ND | 37 | |
| 105 | d-Arg-l-Arg-d-Arg-l-Trp-d-Leu-l-Trp-d-Leu-l-Trp | ND | 7 | ND | ND | ND | ND | 36 | ND | ND | 25 | |
| 106 | d-His-l-Lys-d-His-l-Trp-d-Leu-l-Trp-d-Leu-l-Trp | ND | 9 | ND | ND | ND | ND | 11 | ND | ND | 19 | |
| 107 | d-Lys-l-His-d-Lys-l-Trp-d-Leu-l-Trp-d-Leu-l-Trp | ND | 8 | ND | ND | ND | ND | 61 | ND | ND | 23 | |
| 108 | H2N-l-Trp-d-Lys-l-Lys-d-Lys-l-Trp-d-Leu-l-Trp-d-Leu-COOH | ND | 48 | ND | ND | ND | ND | ND | ND | ND | 80 | |
| 109 | H2N-d-Lys-l-Lys-d-Lys-l-Trp-d-Leu-l-Trp-d-Leu-l-Trp-CONH2 | ND | 40 | ND | ND | ND | ND | ND | ND | ND | 80 | |
| 110 | Ac-d-Lys-l-Lys-d-Lys-l-Trp-d-Leu-l-Trp-d-Leu-l-Trp-CONH2 | ND | 62 | ND | ND | ND | ND | ND | ND | ND | 80 | |
| 111 | d-Lys-l-Lys-d-Leu-l-Trp-d-Leu-l-Trp | ND | 10 | ND | ND | ND | ND | 18 | ND | ND | 80 | |
| 112 | d-Lys-l-His-d-Leu-l-Trp-d-Leu-l-Trp | ND | 10 | ND | ND | ND | ND | 103 | ND | ND | 26 | |
| 113 | d-Lys-l-Ser-d-Leu-l-Trp-d-Leu-l-Trp | ND | 81 | ND | ND | ND | ND | 109 | ND | ND | 98 | |
| 114 | d-Arg-l-Arg-d-Leu-l-Trp-d-Leu-l-Trp | ND | 34 | ND | ND | ND | ND | 5 | ND | ND | 88 | |
| 115 | l-Ser-d-Trp-l-Phe-d-Lys-l-Thr-d-Lys-l-Ser-d-Lys | 7 | ND | ND | ND | ND | ND | ND | ND | ND | >352 | (209) |
| 116 | d-Ser-l-Trp-d-Phe-l-Lys-d-Thr-l-Lys-d-Ser-l-Lys | 7 | ND | ND | ND | ND | ND | ND | ND | ND | >352 | |
| 117 | l-Ser-d-Trp-l-Phe-d-Lys-l-His-d-Lys-l-Ser-d-Lys | 10 | ND | ND | ND | ND | ND | ND | ND | ND | >341 | |
| 118 | l-Ser-d-Trp-O-benzyltyrosine-d-Lys-l-Asn-d-Lys-l-Ser-d-Lys | 2 | ND | ND | ND | ND | ND | ND | ND | ND | 161 | |
| 119 | l-Ile-l-Leu-d-Trp-l-His-d-Ornithine-l-Lys | 2 | ND | ND | ND | ND | ND | ND | ND | ND | 211 | |
| 120 | d-Lys-l-Lys-d-His-l-Lys-d-Trp-l-Leu-d-Trp-l-Lys | 2 | ND | ND | ND | ND | ND | ND | ND | ND | 79 | |
| 121 | d-Lys-l-Trp-d-Phe-l-His-d-Trp-l-Lys | ND | 30 | ND | ND | ND | 30 | 15 | 10 | 20 | >100 | (210) |
| 122 | l-Lys-d-Trp-l-Phe-d-His-l-Trp-d-Lys | ND | >100 | ND | ND | ND | >100 | 7.5 | 12.5 | ND | ND | |
| 123 | l-Lys-d-Trp-l-Phe-d-Phe-l-Leu-d-His | ND | 10 | ND | ND | ND | 10 | >100 | 10 | >100 | >100 | |
| 124 | l-Lys-d-Trp-l-Leu-d-Phe-l-Phe-d-Lys | ND | 7.5 | ND | ND | ND | 7.5 | 40 | 40 | 20 | 50 | |
| 125 | l-Lys-d-Trp-l-Trp-d-Phe-l-Trp-d-Lys | ND | 30 | ND | ND | ND | 30 | 100 | 10 | 40 | >100 | |
| 126 | l-Lys-d-Trp-l-Phe-d-Trp-l-Trp-d-Lys | ND | 100 | ND | ND | ND | 100 | >100 | 30 | 20 | >100 | |
| 127 | l-Lys-d-Trp-l-Phe-d-Lys-l-Trp-l-Ser | ND | >100 | ND | ND | ND | >100 | >100 | 50 | 30 | >100 | |
| 128 | l-Lys-d-Trp-l-Phe-d-Lys-l-Lys-d-Leu | ND | 50 | ND | ND | ND | 50 | 20 | >100 | 30 | >100 | |
| 129 | l-Lys-d-Trp-l-Phe-d-Leu-l-Trp-d-His | ND | 100 | ND | ND | ND | 100 | >100 | >100 | 40 | >100 | |
| 130 | d-His-l-Trp-d-Phe-l-His-d-Trp-l-Lys | ND | 15 | ND | ND | ND | 15 | 20 | 50 | 20 | >100 | |
| 131 | d-Arg-l-Trp-d-Phe-l-His-d-Trp-l-Lys | ND | 30 | ND | ND | ND | 30 | 20 | 10 | 30 | >100 | |
| 132 | d-Ser-l-Trp-d-Phe-l-His-d-Trp-l-Lys | ND | 10 | ND | ND | ND | 10 | 40 | 5 | 8 | >100 | |
| 133 | d-Leu-l-Trp-d-Phe-l-His-d-Trp-l-Lys | ND | 10 | ND | ND | ND | 10 | >100 | >100 | >100 | >100 | |
| 134 | d-Trp-l-Trp-d-Phe-l-His-d-Trp-l-Lys | ND | >100 | ND | ND | ND | >100 | >100 | >100 | >100 | >100 | |
| 135 | d-Tyr-l-Trp-d-Phe-l-His-d-Trp-l-Lys | ND | >100 | ND | ND | ND | >100 | >100 | >100 | >100 | >100 | |
| 136 | d-Phe-l-Trp-d-Phe-l-His-d-Trp-l-Lys | ND | >100 | ND | ND | ND | >100 | >100 | >100 | >100 | >100 | |
| 137 | l-Lys-d-Trp-l-Phe-l-Phe-d-Phe-d-His | ND | 100 | ND | ND | ND | 100 | >100 | 1 | >100 | >100 | |
| 138 | l-Lys-d-Trp-l-Phe-d-Phe-l-Trp-d-His | ND | 50 | ND | ND | ND | 50 | >100 | 30 | >100 | >100 | |
| 139 | l-Lys-d-Trp-l-Phe-d-Phe-l-Lys-d-His | ND | 15 | ND | ND | ND | 15 | 10 | 10 | >100 | >100 | |
| 140 | l-Lys-d-Trp-l-Phe-d-Phe-l-His-d-His | ND | 7.5 | ND | ND | ND | 7.5 | 50 | 8 | 90 | 90 | |
| 141 | l-Lys-d-Trp-l-Phe-d-Phe-l-Ser-d-His | ND | 40 | ND | ND | ND | 40 | 50 | 5 | >100 | >100 | |
| 142 | d-Arg-l-Arg-d-Lys-l-Trp-d-Leu-l-Trp-d-Leu-l-Trp | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | (211) |
| 143 | l-Glu-d-Lys-l-Ser-d-Lys-l-Ser-d-Trp-l-propylargylglycine-d-Lys | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | |
| 144 | l-Trp-d-Leu-l-Trp-d-Lys-l-Ser-d-Lys-l-Ser-l-Ser(βGlcNH2) | ND | 5 | 15 | 10 | 10 | ND | ND | ND | ND | 85 | (212) |
| 145 | l-Trp-d-Leu-l-Trp-d-Lys-l-Ser-d-(βGlcNH2)-l-Ser-d-Lys | ND | 5 | 10 | 10 | 10 | ND | ND | ND | ND | 140 | |
| 146 | l-Trp-d-Leu-l-Trp-d-(βGlcNH2)-l-Ser-d-Lys-l-Ser-d-Lys | ND | 5 | 5 | 15 | 10 | ND | ND | ND | ND | 150 | |
| 147 | l-Trp-d-Leu-l-Trp-d-Lys-l-Ser-d-Lys-l-Ser(βGal)-d-Lys | ND | 5 | 10 | 10 | 15 | ND | ND | ND | ND | 260 | |
| 148 | l-Trp-d-Leu-l-Trp-d-Lys-l-Ser(βGal)-d-Lys-l-Ser-d-Lys | ND | 5 | 5 | 5 | 10 | ND | ND | ND | ND | 190 | |
| 149 | l-Trp-d-Leu-l-Trp-d-Lys-l-Ser-d-Lys-l-Ser(αMan)-d-Lys | ND | 2.5 | 5 | 10 | 10 | ND | ND | ND | ND | 120 | |
| 150 | l-Trp-d-Leu-l-Trp-d-Lys-l-Ser(αMan)-d-Lys-l-Ser-d-Lys | ND | 2.5 | 10 | 5 | 5 | ND | ND | ND | ND | 105 | |
| 151 | l-Trp-d-Leu-l-Trp-d-Lys-l-Ser-d-Lys-l-Ser-d-Lys | ND | 5 | 5 | 5 | 10 | ND | ND | ND | ND | 120 | |
LD50 values have been attained via the MTT or XTT assay protocol. (ND: Not detected. MIC: Minimum inhibitory concentration. HD50: Dosage required for 50% hemolysis detection. MRSA: Methicillin-resistant Staphylococcus aureus bacterial strain. VRE: Vancomycin-resistant Enterococcus faecalis bacterial strain. FBS: Fetal bovine serum.)
It was also shown that nanotube activity was not affected by the presence of up to 50% FBS (fetal bovine serum), and the balance between bacterial inhibition and hemolysis of red blood cells was favorable, attesting to their biocompatibility. To verify this, toxicology studies on the peptides 102, 103, and 111 were performed via intraperitoneal administration in mice and healthy responses without signs of toxicity were reported at concentrations up to 12, 17.5, and 50 mg kg–1, respectively. Next, to assess an infected model in vivo, mice were injected with a lethal dose of MRSA and peptides 102, 103, and 111 administered 60 min later. Remarkably, 67% and 50% (intraperitoneal and subcutaneous injection respectively) in the study survived (compared to 100% death in a control group) and recovered after being given a single dose, which equated to PD50 of 6 ± 2, 5 ± 2, and 10 ± 2 mg kg–1, respectively.
Following this proof of concept, Dartois et al. later expanded the library to 140 000 compounds via a comprehensive screening process on hexa or octapeptides of similar composition with discrete hydrophobic and hydrophilic regions.209 Hit compounds that produced an 80 + % inhibition effect against in vitro S.aureus strains advanced to the modification stage with point substitutions affording 6 optimized second generation cationic compounds 115–120. Evaluation of the in vitro behavior revealed a notable difference in the therapeutic index window of the peptides 115, 116, 117 and 118, 119, 120, which was confirmed by differing in vivo tolerance dosages. While peptides 118, 119, 120 strongly disrupted antimicrobial longevity, their toxicity against mammalian cells was significant. Interestingly, the enantiomeric pair, 115 and 116 exhibited very similar results, which supports a hypothesis that small antimicrobial peptides might act via a nonchiral lipid membrane interaction. However, upon advancing to in vivo infection models, the peptides exhibited a conflicting behavior where the first three aforementioned compounds impede growth far more than the latter three, which have minimal effect despite their strong potency in vitro. Examination of the pharmacokinetic properties shed light on this result where the first three are retained in blood plasma in vivo for significantly longer, with a greater chance to inhibit the bacteria. Peptides 119 and 120 had low peak concentrations and AUC (area under the concentration–time curve) values and an extremely high volume of distributions, which may have resulted in the weak efficacy and limited tolerability of the rat/mice due to off-target accumulations. They suggest that the poor tolerability of certain compounds is caused by vascular leakage and diffusion into tissues upon injection, which would correlate with their observation of a rapid decrease in plasma concentrations. Therefore, these results demonstrate two important points: (1) cationically charged nanotubes could match current treatments such as vancomycin and accordingly be considered a strong alternative that does not incur antimicrobial resistance and (2) in vivo activity cannot necessarily be predicted from in vitro results and, therefore, the relationship between structure and pharmacokinetic observations should be the next focus to bring these novel materials to the stipulations of clinical application.
An overall review of the literature in this area shows there are some key structure–activity relationships that are emerging for the design of cyclic peptide antibacterials. For instance, Hsieh et al. have identified a negligible difference between enantiomeric sequences, alongside the need for amphiphilic contrast, and the necessity of charged hydrophilic residues such as lysine, arginine and histidine for broad spectrum treatment.213 Having also recognized this, Fletcher et al. combined targeted screening methods with synthetic optimization to design a cationic, amphiphilic hexapeptide 137, which displayed a 100-fold selectivity toward eukaryotic algae, Ulva linza, which may be used as a biofouling agent for marine ecology applications.210 This demonstrates the potential of utilizing screening methods traditionally used for small molecule medicines on macromolecular species to reveal diverse relevancy and biocompatibility outside of the clinical setting. Claro et al. have also used logic based prediction methods like computational simulation to model the interactions of various cyclic α-alt(d,l)-peptides with antimicrobial membrane targets, arguing that a combined analysis of biophysical characterization and coarse-grained molecular–dynamics (CG-MD) can support understanding of membrane/antimicrobial agent interactions. The two peptides studied, 142 (known antimicrobial activity with amphipathic sequence and three basic residues) and 143 (improved solubility from three Lys residues) were tested against DMPE and DMPG liposomal models in mixed ratios to elucidate the role of negatively charged membrane density. DSC, ATR-FTIR, and CG-MD were rigorously used to corroborate the previous speculation that hydrophobic peptide residues are critical for a significant effect against Gram-positive bacteria and the results suggest that the assembled peptides lay in parallel to membrane surfaces in most cases (Figure 48).211
Figure 48.

Chemical structures of two cyclic peptides 142 and 143 modeled by Claro et al. and illustration of the parallel conformation in which they are thought to lay in regard to membrane surfaces. Reproduced with permission from ref (211). Copyright 2020 Elsevier.
Cyclic peptide nanotubes have also taken inspiration from existing active compounds. Motiei et al. have observed the similarity in the design of cyclic peptides and mannopeptimycins, a class of cationic glycopeptide antibiotic materials with activity against Gram-positive bacteria.212 Accounting for the known targeting properties of sugars toward bacteria, a membrane-active cyclic α-alt(d,l)-peptide 151 was modified by replacement of lysine with sugar-based serine residues (d-glucosamine, d-galactose, and d-mannose) (compounds 144–150). Each of the modified peptides formed the expected β-sheet nanotubes and were found to inhibit the growth of both MRSA and vancomycin-resistant Enterococcus faecalis. This demonstrates a remarkable breakthrough of the nanotubes, which can outperform vancomycin and potentially become a replacement once it inevitably becomes redundant in the clinic. Furthermore, cyclic peptide nanotubes might again resolve another issue in the antimicrobial arena; where mannopeptimycins face significant synthetic challenges, cyclic peptides are noncomplex, easy to modify, and therefore, capable of undergoing high-throughput screening protocols for optimizations.
6.3. Anti-Cancer Drug Delivery
Given the aforementioned ability of cyclic peptides to reversibly stack into tubular structures, to date, they have chiefly been explored as a possible tool for anticancer drug delivery since their synthetic pliability, high aspect ratio, and supramolecular composition make them attractive materials for this type of application. The task to define a nanoparticle that can overcome biological hurdles including circulation, retention, distribution, cellular uptake, and phagocytotic or renal clearance is a significant challenge facing scientists wanting to advance synthetic materials for in vivo use; with the most crucial features of nanoparticle design being size, morphology, and surface chemistry.214,215 In addition, the high aspect ratio has also attracted attention as a property which may improve the time of circulation and ensure better cellular uptake.216,217 Hence, the elongated structures formed by cyclic peptides are of increasing interest for improving the bioavailability and activity of hydrophobic, poorly soluble anticancer actives, which are toxic but nonselective and can produce undesirable side effects. A central principle of optimal drug delivery has been to transport through the cell membrane and release cargo at intracellular compartments for processing. Given the former evidence that nanotubes can bury themselves into membranes and facilitate the transport of ions through channels, Chen et al. attempted the selective transport of 5-fluorouracil (5-FU, diameter < 1.0 nm) through 152 cyclo-[l-Gln-(d-Leu-l-Trp)4-d-Leu-] (internal diameter = 1.0 nm) for in vitro liposomal models.218 Efficacy of the delivery vehicle was evident by the significantly reduced IC50 values for 5-FU in the BEL7402, HeLa, and S180 cell lines and further analysis confirmed the synergistic or additive effect imparted by the combination of drug and carrier (Table 3). A promising feature of these materials was made apparent by 125I radiolabeling imaging in vivo, which revealed that the nanotubes were localized in tumor tissue and distribution to other organs was negligible. The effect of this direct transport to tumor tissues resulted in a positive effect upon administration of 5-FU with SCPNs, where a clear inhibition of tumor growth was observed.
Table 3. Library of In Vitro Biological Data for Compounds 152–157 with Antiproliferative Effects on Cancer Cell Lines.
| in
vitro |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (μM) |
||||||||||||
| compound number | peptide sequence | conjugate polymer | drug type | BEL7402 | HeLa | S180 | A2780 | A2780cis | MCF-7 | MCF-7/ADR | MDA-MB-231 | ref |
| 152 | none (drug only) | none | 5-fluorouracil | 596 | 1460 | 1790 | ND | ND | ND | ND | ND | (218) |
| Gln-(d-Leu-l-Trp)4-d-Leu | none | 5-fluorouracil | 241 | 819 | 389 | ND | ND | ND | ND | ND | ||
| 153 | none (drug only) | none | doxorubicin | ND | ND | ND | ND | ND | 0.53 ± 0.05 | 4.21 ± 0.65 | ND | (219) |
| l-Gln-d-Ala-l-Glu-d-Ala-l-Gln-d-Ala-l-Cys-d-Ala | PEG | doxorubicin | ND | ND | ND | ND | ND | 0.16 ± 0.03 | 0.84 ± 0.04 | ND | ||
| 154 | none (drug only) | none | ruthenium RAPTA-C | ND | ND | ND | 271 | 266 | ND | ND | ND | (131) |
| (l-Lys(N3)-d-Leu-l-Trp-d-Leu)2 | pHEA-co-pCEMA | ruthenium RAPTA-C | ND | ND | ND | 15 | 22 | ND | ND | ND | ||
| none (drug only) | none | iridium | ND | ND | ND | 0.95 ± 0.03 | ND | ND | ND | ND | (173) | |
| 155 | none (drug + polymer only) | pHPMA-co-pPUEMA | iridium | ND | ND | ND | 1.80 ± 0.09 | ND | ND | ND | ND | |
| 156 | (d-Leu-l-Lys-d-Leu-l-Trp)2 | pHPMA-co-pPUEMA | Iridium | ND | ND | ND | 0.61 ± 0.02 | ND | ND | ND | ND | |
| 157 | none (drug only) | none | doxorubicin | ND | ND | ND | ND | ND | ND | ND | 17.85 | (132) |
| l-Lys-d-Leu-l-Trp-d-Leu-l-Lys(N3)-d-Leu-l-Trp-d-Leu | pNBMA-CP-pPEGA | doxorubicin | ND | ND | ND | ND | ND | ND | ND | 13.61 | ||
However, one setback of SCPNs is that depending on the sequence, they can aggregate uncontrollably into arrays that are undefined and may precipitate in an aqueous solution, negating their suitability for delivering hydrophobic drugs. Furthermore, the large size can cause recognition by the phagocytic system and result in transport to the liver and reduced bioavailability. Therefore, most attention has shifted toward using cyclic peptide–polymer conjugates since the polymer can be used to control the nanotube size better, elicit a large increase in solubility, and often act as a biological shield via the stealth effect. Some of the first evidence for this shift is found in the work of Wang et al., who modified their SCPN aggregates with PEG.219 By neutralizing the negatively charged glutamic acid residue in 153 with a sequence of cyclo-[l-Gln-d-Ala-l-Glu-d-Ala-l-Gln-d-Ala-l-Cys-d-Ala-] via ion-complexation with doxorubicin; the assembly could proceed to make microscaled aggregates. After orthogonal conjugation of a maleimide PEG onto the thiol-containing residue, these bundles could undergo reduction to a nanoscale material as a consequence of the steric and solubility effects from the hydrophilic polymer. These entities, which scaled 50 nm in diameter and 200–300 nm in length, achieved a high drug loading ratio (54.2% at pH 7.0) and displayed greater cytotoxicity toward MCF-7 breast cancer cells than the free doxorubicin administration. This suggested that the nanotube bundles were able to enter cells more effectively than the free drug, and this was subsequently proven by flow cytometry showing that the PEG-modified bundles achieved an uptake greater than the doxorubicin alone. Remarkably, the nanotubes also outperformed the free doxorubicin on a multidrug-resistant cell line, validating that this class of materials is worth investigation given the wide range of aggressive diseases for which traditional small molecule drug discovery sometimes struggles to cater.
More recently, our group has undertaken a considerable effort in studying cyclic peptide–polymer conjugate assemblies for drug delivery, largely by attaching biocompatible RAFT polymers with a range of functionalities that act as a handle for drug loading of different organometallic drugs. Because of a lack of defined hydrophilic or hydrophobic properties, organometallics are a challenging class of drugs to use in the most common nanoparticle delivery methods since these systems usually involve encapsulation of drug cargo. Therefore, drug loading via covalent bonds or ligand binding can overcome these issues. The first report of a singular cyclic peptide–polymer conjugate nanotubes by Blunden et al. employed RAFT polymerization to generate a statistical copolymer of poly(2-hydroxyethyl acrylate) (pHEA) and poly(2-chloroethyl methyl acrylate) (pCEMA) to compose the corona around a central cyclic peptide by attaching onto two azide functional handles within the sequence (Figure 49).131 The latter monomer moiety underwent a halogen exchange to iodide to enable rapid substitution with an amine group present on organometallic drug RAPTA-C, which is highly selective and effective toward in vivo metastatic cancers. After preparation of the final nanotubes in water (20 nm in diameter and ranging from 200 to 500 nm in length) administration to ovarian A2780 cells and the cisplatin-resistant equivalent, compared the activity against the pure drug and a 10-fold increase in cytotoxicity (IC50) for the macromolecular drug complex was found in each case. Given that the drug was covalently bound to the carrier, this is even more remarkable since there may be less opportunity for the RAPTA-C to detach and reach its target site.
Figure 49.

(a) Chemical structure of the drug-loaded cyclic peptide–polymer conjugate 154. (b) Scheme of the nanotube loaded with organometallic drug RAPTA-C. Reproduced with permission from ref (131). Copyright 2014 John Wiley and Sons.
As the first case of singular nanotubes affording a significant improvement in a therapeutic in vitro context, this consolidated the hypothesis that these materials might be a strong candidate for integrating nanomedicine and polymer chemistry to generate a nanoscale supramolecular carrier. Larnaudie et al. advanced knowledge regarding the in vitro behavior of the nanotubes by investigating the cytotoxicity, intracellular drug fate, and carrier selectivity of these conjugates.173 An organoiridium drug was complexed onto a 2-arm conjugate 156 using pHPMA as an example of a biocompatible polymer of clinical relevance. This was copolymerized with 5% of pyridine based 2-(3-(pyridin-4-ylmethyl)ureido)ethyl)methacrylate (PUEMA) monomer for ligation of a novel organoiridium drug and upon attachment to cyclic peptide and assembly in water (see structure in Figure 53), assessed for antiproliferative activity against A2780 cell line (comparing to the free drug and a polymer–drug conjugate control). Figure 50 shows the improved cytotoxicity and, more interestingly, the strong selectivity for cancerous cells over healthy human cells. Furthermore, poor delivery of the drug carrier into cells when incubating at low temperatures confirmed an energy-dependent endocytosis pathway for entry.
Figure 53.

Pharmacokinetic profile comparing the overall plasma concentration of conjugate (156) vs equivalent free polymer (155) in vivo, indicating that nanotubes are cleared less rapidly and remain in circulation at a high concentration for longer. Reproduced with permission from ref (158). Copyright 2018 Elsevier.
Figure 50.

Cytotoxicity profile for the iridium drug-loaded conjugate 156 compared with free drug and drug-bearing polymer 155 (a) and the antiproliferative effects on cancerous cells vs healthy cells (b), which suggest a favorable selectivity index. Reproduced with permission from ref (173). Copyright 2018 American Chemical Society.
While the nanotube carrier dose afforded the most significant effect on the cells, it was found to have lower uptake than the free drug or polymer carrier, which could mean it delivers by a more efficient mechanism once encapsulated into the endosome. Additionally, confocal microscopy colocalization of rhodamine stained nanotubes and lysotracker green corroborates the likelihood of endocytic uptake pathways and implies the advantageous intracellular behavior of nanotubes where they have achieved endosomal escape and arrival in lysosomal compartments capable of processing drug cargo.158
Figure 51.

Confocal microscopy imaging as evidence of the intracellular localization of rhodamine labeled nanotubes in the lysosomal compartments of human PC3 cells. Reproduced with permission from ref (158). Copyright 2018 Elsevier.
The tubisome nanoparticles introduced in section 4.3.1 have also shown positive results for anticancer treatment in vitro.132 Amphiphilic tubisomes 157 synthesized by Yang et al. encapsulated doxorubicin in the core and exhibited controlled release behavior through fine-tuning of a UV-responsive polymer as the hydrophobic polymer arm. Figure 52 illustrates the design of the cyclic peptide−polymer conjugates, which through synergy of hydrogen bonding and hydrophobic effect, assemble into cylindrical micelle type particles which can later disassemble upon photocleavage of the NMBA. Doxorubicin was loaded into the central cavity (10.3 wt %), and upon incubation with MDA-MB-231 cells, with UV irradiation for 20 min, a cytotoxicity profile similar to that of the free drug was determined. Although the cytotoxicity was not improved upon, the responsive nature of the nanoparticle could be beneficial where the time-dependent release behavior exhibited a burst release that is favorable for some treatment types.
Figure 52.

Chemical structure of asymmetric conjugate 157 and the trigger responsive assembly into tubisomes which can successfully deliver anticancer drug doxorubicin into cells. Reproduced with permission from ref (132). Copyright 2020 John Wiley and Sons.
While the in vitro performance of cyclic peptide-based nanostructures continues to be explored, the in vivo stability profile is also being considered in parallel to evaluate the basic behaviors prior to further optimizations. The nondrug loaded nanotubes composed of the cyclic peptide–polymer conjugate 156, and the nonassembling polymer counterpart 155 previously used by Lanaurdie et al. were subjected to radioactive labeling with [14C]-ethanolamine and injected into rats for assessment.158 The circulatory retention of the nanotubes was higher than linear polymer over the same time period (Figure 53), which Lanaurdie et al. attribute to less rapid renal clearance on account of high aspect ratio, larger molecular weight, and enhanced structural integrity.
Next, having established that higher aspect ratio nanotubes outperform the singular polymeric coils as a drug delivery vector, future research might wish to look at the impact of having a dynamic supramolecular architecture for this carrier. A significant limitation of nanoparticles is that the pharmacodynamic effect is still under scrutiny by the field since many materials end up accumulating in organs indefinitely. The extent of this phenomenon widely varies depending on the material, and a rigid definition of what constitutes a “safe” material is yet to be established. In the meantime, efforts could navigate toward ensuring a balance between retention and elimination of the nanoparticles. Since cyclic peptide nanostructures are held together by weak intermolecular interactions, they theoretically should gradually disassemble under biological conditions as each unimeric conjugate is “sheared” away over time. Comparison of supramolecular nanotubes against covalently bound materials of similar morphology would help elucidate the importance of dynamic behaviors for the in vivo efficacy.
In conclusion, supramolecular cyclic peptide nanostructures have demonstrated good proof-of-concept as delivery vectors in vitro and in vivo; they can enhance the uptake and activity of different drugs which traditionally struggle as therapeutics; some evidence for their mode of action has surfaced, although a more thorough inspection will help to inform the design in future trials. Additionally, the biocompatibility and strong pharmacokinetic profiles indicate that supramolecular chemistry is a resource that should be pursued as a necessity for modular design of exciting architectures that can be easily manipulated. However, a number of factors still need to be addressed, such as the size distribution of the nanotubes and assurance of sample uniformity for each administration into a live subject. In addition, a more comprehensive trial of the carrier biocompatibility over an extended time frame should be undertaken prior to introducing drug-loaded materials on tumor grafted in vivo models.
6.4. Gene Delivery
In recent years, gene therapy has emerged as a powerful universal method of reprogramming genetic material to cure multiple diseases such as cancer, viral infection, or inherited disorders.220 By introducing new genetic code, or “knocking out” mutative genes at the cell nuclei, a positive impact on regulation of normal cellular functions can be observed. However, this therapeutic technique faces considerable challenges. It is highly dependent on the vector composition to preserve the integrity of the load, evade the immune response and target on a cellular level.221 To date, most nanotubular vectors for genetic transfer have been relatively large in size and use intermolecular binding between genetic material and different amino acid moieties to construct a loaded vector. Since peptide nanotubes are composed of closely stacked unimers, the presence of repulsive electrostatic forces is unwelcome for producing supramolecular structures and, therefore, presents a significant difficulty when attempting to complex charged genetic loads. To date, a select number of cyclic peptide species have been studied for gene delivery applications (Table 4).
Table 4. Library of Information for Compounds 158–160 and the Specific Genetic Material Being Delivered for the Application in Different Models.
| delivery |
||||||
|---|---|---|---|---|---|---|
| compound number | peptide sequence | gene | administration | model | organ expression | ref |
| 158 | cyclo-[(d-Ala-l-Lys)4-]* | pF143-GFP | ND | HeLa cells | ND | (222) |
| ND | HEK-293 cells | ND | ||||
| 159 | cyclo-[(d-Trp-l-Tyr)4-] | pCMV-LacZ | oral | mice | stomach, duodenum, liver, kidney | (223) |
| pCMV-hRluc | Oral | Mice | ||||
| 160 | cyclo-[(d-Trp-l-Tyr)4-] | pCMV-bcl-xl-eGFP | eye | mice | cornea | (224) |
| CAP3 pRFP-C-RS | eye | mice | ||||
One of the Lys side-chains was functionalized with a guanidiniocarbonylpyrrole moiety (GCP).
A nonassembling cationic cyclic octapeptide 158 synthesized by Li et al. was modified with a novel arginine analogue rationalized by molecular modeling to partake in enhanced binding with adjacent peptide backbones and overcome lysine residue charge repulsion (Figure 54). This was hypothesized to stabilize the assembly and form distinctive tubular structures in solution (pH 7.4).222 Complexation with calf thymus DNA did not alter the nanoparticle integrity (as confirmed by TEM and dynamic light scattering (DLS)) and demonstrated the utility of the design for carrier purposes. Furthermore, transfection efficiency assay using green fluorescent protein (GFP) reporter labeling determined that transfection of HeLa and HEK-293 cell lines by SCPNs was on a comparable level to the commercially used polyethylene imine but occurred independently of active endocytosis pathways and was notably less cytotoxic.
Figure 54.

Chemical structure of the cationic cyclic octapeptide 158 and illustration of the assembling cationic nanotubes binding ctDNA and the process of fluorescent labeling with GFP to track cellular uptake and transfection efficiencies. Reproduced with permission from ref (222). Copyright 2015 John Wiley and Sons.
Therefore, proper consideration of SCPNs for gene delivery therapies is surely a worthwhile avenue given their potential to resolve the setbacks of current medicines by avoiding endosomal entrapment and exhibiting fewer side effects on healthy cells. For instance, Panigrahi et al. reported stable peptide nanotubes capable of delivering siRNA at low concentrations to HCT-116 colorectal cancer cells (via caveolae and clathrin-mediated endocytosis) with capability matching that of lipofectamine, but further benefit from negligible cytotoxicity.225 Hsieh et al. proposed that assembled cyclo-[(d-Trp-l-Tyr)4-] nanotubes between 1 and 20 μm in length can act as an oral gene delivery vector to the stomach, duodenum, liver, or kidney where a strong binding constant (3.2 × 108 M–1) was calculated between plasmid DNA (pCMV-lacZ and pCMV-hRluc) and tyrosine peptide residues using fluorescence spectroscopy (Note: although this publication does not specify the size of the CP ring, based on the data provided we suggest it is an octapeptide).223 The shielding properties of the nanotubes were evident from the stability of plasmid to DNase 1, acidic conditions (pH 2 gastric acid simulation) and bile digestion for 50, 60, and 180 min respectively, indicating a broad potential for in vivo targets. Southern blot analysis and TM-rhodamine labeled DNA tracing determined that the aforementioned organs had elevated expression of mRNA for the lacZ reporter genes, possibly due to the higher permeability of nanotubes into these regions with intact genetic loads. The effect of this was apparent from β-galactose expression increasing by 41% in the kidney at 48 h, and 49%, 63%, and 46% at 72 h in the stomach, duodenum and liver respectively and bioluminescence imaging of ex vivo mouse organs gave confirmation of these accumulations (Figure 55).
Figure 55.

Ex vivo bioluminescence imaging of mice organs after oral administration of pCMV-hRluc: cyclo-[(d-Trp-l-Tyr)4-] nanotubes for 48 h (kidney) or 72 h (stomach, duodenum, liver). Reproduced with permission from ref (223). Copyright 2012 American Chemical Society.
Using this same peptide species, Lee et al. investigated further gene delivery properties for treatment of corneal injury via a topical eye drop formulation administering the caspase 3 silencing shRNA to artificially induced epithelial debridement on nude mice.224 The known high aspect ratio and fast internalization rates of nanotubes are rationalized to be crucial features for this therapy, requiring good penetration through tightly packed layers of the epithelial matrix. Histological analysis of ThT stained nanotubes demonstrated that delivery into the epithelial layers and stroma had been achieved just 180 min after the first dosage. Notably, the material had even reached the nuclear region of corneal keratocytes, despite previous studies suggesting a delayed DNA release. The successful uptake of the carrier could be translated to accomplishing a biological impact where caspase 3 apoptotic activity was calculated to be significantly lower in the treated tissue compared to native corneal wound control subjects. This result could be explored in the future for the prevention of permanent corneal damage and vision deterioration.
6.5. Antiviral Applications
Transfer and replication of viral material is highly dependent on the low pH of endocytic vesicles, which trigger conformational protein changes needed for membrane fusion, lysis, and escape from these compartments prior to any degradation by other organelles. The appeal of cyclic peptide nanotubes due to their capacity to embed into membrane models is therefore amplified by the evidence of antiviral effects which Horne et al. first discovered upon the rational design of octapeptide nanotubes, which could prevent the growth of adenovirus infection. A cyclo-[l-Ser-d-His-l-Lys-d-Arg-l-Lys-d-Trp-l-Leu-d-Trp-] peptide demonstrated dose-dependent inhibition of adenovirus against HeLa cells (IC50 = 5 μM). Horne et al. hypothesized that nanotubes block the development of acidic pH in endosomes due to transmembrane channel ion transport, preventing viral escape from these capsules (Figure 56).226
Figure 56.

Illustration of the proposed mechanism of action for peptides 161–171 during the early phases of adenovirus infection cycle. In the absence of peptide (a), viral material is taken up via clathrin-mediated endocytosis, and the acidic environment can allow disassembly and escape. Administration of peptide (b) prevents the formation of an acidic endosomal environment and therefore limits the escape of the virus. Reproduced with permission from ref (226). Copyright 2005 Elsevier.
This was demonstrated by an experiment in which incubation of the peptide 4 h post Ad5-GFP infection did not prevent the escalation of the virus. This implied that the peptide does not impact transcription or translation processes but was vital for disrupting earlier processes like cellular entry or endosome formation. The influence of the assembled nanotube upon endosomal pH during infection was evaluated by fluorescence microscopy and imaging of carboxy-fluorescein labeled virus, namely the defective Ad2-tsl which has similar receptor properties to wild type adenovirus but cannot escape the endosome. The analysis revealed that the intracellular location of the virus remained the same in the presence or absence of the peptide, but the relative fluorescence was significantly different. Calculation of relative pH revealed the untreated cells tended toward stronger acidity (pH 5.3) compared to treated cells (pH 6.8), which corroborates the initial hypothesis of endosomal acidification being a primary target for nanotubes.
In addition, since Ghadiri and co-workers’ initial investigation of cyclic peptide nanotubes in 1993, over 1200 octapeptide sequences have been amassed throughout the known literature. Of these, 144 were screened by Montero et al. against HCV in an ELISA assay to afford the 9 most potent amphiphilic peptides, best described by having overall neutrality, positive charges adjacent to hydrophobic moieties, and Glu, Asp or Gln residues filling the cyclic ring.227 Further modifications to these compounds were made (Table 5) and compared to their linear analogues which had a very poor antiviral performance, as did N-methylated nonassembling cyclic peptides, corroborating previous thought that insertion of the elongated supramolecular structure into membranes is an essential component of the antiviral response. The screening also elaborated that while self-assembly is fundamental, this alone was insufficient for pronounced effects, and hence sequence specificity is also key to impart a balance between anti-HCV behavior and low toxicity to mammalian cells. Montero et al. performed a time-dependent inoculation experiment and observed that antiviral activity was only possible when peptides were present during initial viral exposure or throughout the entire inoculation period, implying that a crucial early stage of cell entry was obstructed.227 Subsequently, more proof of the inhibitory mechanism occurred when postbinding of the virus to cellular membranes was garnered by comparing the degree of infection at 4 °C, which freezes any energy-dependent uptake mechanisms. Peptide dosage was kept constant and added to varying concentrations of HCV load in infected Huh-7 cells, either during or post inoculation period and susceptibility of binding versus postbinding events to inhibition was compared to DMSO controls. A significant reduction in infectivity was clear during postbinding processes, which corroborates the hypothesis that degradation of endosomal compartments is responsible for limiting viral spread. The potential of the peptides to properly limit viral spread was clear from infection of Huh-7 cells with a low viral load: large numbers of HCV-positive antigen cells were observed by immunofluorescence (anti E2) for control DMSO cultures (Figure 57), compared to almost no evidence for this in the peptide treated cells (compounds 182 and 183 significantly reduced HCV RNA accumulation, indicating there was no spread of viral material between cells). This remarkable feature to control viral spread suggests that this class of compounds could be used as part of a synergistic combination therapy with other antiviral compounds.
Table 5. Library of Data for Compounds 161–190 with Antiviral Activity against Influenza A or Hepatitis C.
| antiviral
activity |
|||||
|---|---|---|---|---|---|
| IC50 (μM) |
LD50 (μM) | ||||
| compound number | peptide sequence | influenza A | hepatitis C | HeLa cell | ref |
| 161 | l-Ser-d-His-l-Lys-d-Arg-l-Lys-d-Trp-l-Leu-d-Trp | 5 | ND | 57 | (226) |
| 162 | d-Trp-l-Leu-d-Trp-l-Lys-d-Arg-l-Lys-d-His-l-Ser | 5 | ND | 35 | |
| 163 | d-Ser-l-His-d-Lys-l-Arg-d-Lys-l-Trp-d-Leu-l-Trp | 7 | ND | 63 | |
| 164 | l-Ser-d-His-l-Lys-d-Arg-l-Lys-d-Ala-l-Leu-d-Trp | >30 | ND | >100 | |
| 165 | l-Ser-d-His-l-Lys-d-Arg-l-Lys-d-Trp-l-Leu-d-Ala | >30 | ND | >100 | |
| 166 | l-Ala-d-His-l-Lys-d-Arg-l-Lys-d-Trp-l-Leu-d-Trp | 8 | ND | 54 | |
| 167 | l-Ala-d-Ser-l-Lys-d-Arg-l-Lys-d-Trp-l-Leu-d-Trp | 13 | ND | 70 | |
| 168 | l-Ser-d-His-l-Ala-d-Arg-l-Lys-d-Trp-l-Leu-d-Trp | 19 | ND | >100 | |
| 169 | l-Ser-d-His-l-Lys-d-Ala-l-Lys-d-Trp-l-Leu-d-Trp | 7 | ND | 65 | |
| 170 | l-Ser-d-His-l-Lys-d-Arg-l-Ala-d-Trp-l-Leu-d-Trp | 6 | ND | 43 | |
| 171 | l-Ser-d-His-l-Lys-d-Arg-l-Lys-d-Trp-l-Ala-d-Trp | 17 | ND | 100 | |
| 172 | d-Trp-l-Leu-d-Trp-l-Ser-d-Glu-l-Gln-d-Ser-l-Lys | ND | 3.3 ± 0.5 | 65 | (227) |
| 173 | d-Trp-l-Leu-d-Trp-l-Ser-d-Glu-l-Lys-d-Asn-l-Ser | ND | 7.7 ± 3.8 | >100 | |
| 174 | d-Trp-l-Leu-d-Trp-l-Ser-d-Ser-l-Lys-d-Glu-l-Asn | ND | 7.9 ± 2.1 | >100 | |
| 175 | d-Trp-l-Leu-d-Trp-l-Ser-d-Glu-l-Gln-d-Ser-l-Orn | ND | 10.3 ± 3.4 | >100 | |
| 176 | d-Trp-l-Leu-d-Trp-l-Arg-d-Glu-l-Gln-d-Glu-l-Arg | ND | 15.5 ± 2.8 | >100 | |
| 177 | d-Trp-l-Leu-d-Tyr-l-Lys-d-Arg-l-Glu-d-Asp-l-Tyr | ND | 8.9 ± 1.6 | >100 | |
| 178 | d-Trp-l-Leu-d-Trp-l-DPA-d-Glu-l-Gln-d-Glu-l-Z | ND | 6.9 ± 3.4 | 80 | |
| 179 | d-Trp-l-Leu-d-Trp-l-Arg-d-Glu-l-Ser-d-Gln-l-Lys | ND | 3.4 ± 0.4 | 90 | |
| 180 | d-Trp-l-Leu-d-Trp-l-Orn-d-Glu-l-Asn-d-Glu-l-Lys | ND | 11 ± 2.8 | >100 | |
| 181 | d-Trp-l-Leu-d-Trp-l-Glu-d-Orn-l-Asn-d-Lys-l-Glu | ND | >50 | >100 | |
| 182 | d-Trp-l-Leu-d-Trp-l-Ser-d-Glu-l-Asn-d-Ser-l-Lys | ND | 6.5 ± 0.8 | >100 | |
| 183 | d-Trp-l-Leu-d-Trp-l-Orn-d-Ser-l-Asn-d-Glu-l-Ser | ND | 7.8 ± 0.3 | >100 | |
| 184 | d-Trp-l-Ile-d-Trp-l-Orn-d-Glu-l-Asn-d-Glu-l-Lys | ND | 5.5 ± 1.2 | 45 | |
| 185 | d-Trp-l-Leu-d-Trp-l-Glu-d-Orn-l-Asn-d-Glu-l-Lys | ND | 18.4 ± 3.6 | >100 | |
| 186 | Ac-Trp-l-Leu-d-Trp-l-Orn-d-Glu-l-Asp-d-Glu-l-Lys-NH2 | ND | >50 | ND | |
| 187 | Ac-Asp-d-Glu-l-Lys-d-Trp-l-Leu-d-Trp-l-Orn-d-Glu-NH2 | ND | >50 | ND | |
| 188 | Ac-Leu-d-Trp-l-Orn-d-Glu-l-Asp-d-Glu-l-Lys-d-Trp-NH2 | ND | >50 | ND | |
| 189 | d-Trp-l-Leu(Me)-d-Trp-l-Ser-d-Glu-l-Asp-d-Ser(Me)-l-Lys | ND | >50 | ND | |
| 190 | d-Trp-l-Leu(Me)-d-Trp-l-Ser(Me)-d-Glu-l-Asp-d-Ser-l-Lys | ND | >50 | ND | |
Figure 57.
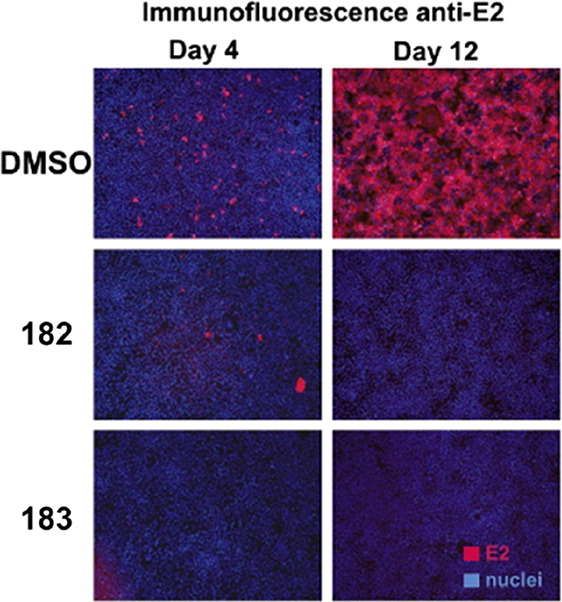
Imaging to demonstrate the growth of HCV in the Huh-7 cell line for a DMSO control treatment (strong immunofluorescence) and the successful suppression of the virus upon incubation with peptides 182 and 183. Reproduced with permission from ref (227). Copyright 2011 Elsevier.
6.6. Summary
To date, self-assembling cyclic peptides have demonstrated strong potential for a broad range of biological applications, and in many cases, might even be able to outperform or replace current highly regarded therapeutics. However, most of the materials studied for the aforementioned applications are cyclic α-alt(d,l)-peptides, and while the literature shows extensive optimization and the generation of large peptide libraries, future research should also explore the structures described in the early sections of this Review, and how they might benefit existing design motifs. Additionally, although there is some strong evidence of the biocompatibility of peptides and peptide–polymer conjugates, further tests are needed to assess long-term side effects of the materials in vivo and establish their chances of clinical success. These materials are an exciting opportunity for researchers to work across various disciplines and their synthetic pliability opens up a huge potential; the dual-purpose design of having both (1) specific peptide sequences capable of inducing targeted effects on peptide sensitive antibacterial or antiviral strains and (2) capability for extended supramolecular assemblies into nanoparticles that carry different types of cargo, have allowed these materials to merge the fields of small molecule therapies and macromolecular chemistry.
7. Conclusion and Perspective
Since their first report in the mid 1970s, self-assembling cyclic peptides have been shown to be versatile functional building blocks from which a wide range of functional supramolecular nanomaterials can be formed. The versatility of peptide synthesis provides access to an infinite range of functionalities on both the outer ring and the inner ring. Upon self-assembly, this highly adaptable design feature enables us to impart the desired functionality on the outer shell or the inner channel of a nanotube. The wide range of peptides available also enables the design of an infinite range of functional materials. In the last 10 years, the functionalization of cyclic peptides with polymers has opened a new range of opportunities for these materials. In addition to adding functionality, the conjugation of polymers has vastly improved the solubility of these nano-objects and led to exciting applications particularly in the areas of biology and medicine. Thus far, these cyclic peptides have been shown to form transmembrane channels, be antimicrobial and promising drug delivery vectors.
The huge strides in synthesis and characterization of the SCPNs have made these systems easier to produce and manipulate; in turn, making them increasingly more desirable to study and commercialize. It is this recent development that will catalyze the further applications of SCNPs and enable them to become readily accessible by the wider scientific community.
Acknowledgments
The authors acknowledge funding from the European Research Council (S.P., Q.S., S.C.L.H.; TUSUPO 647106), the Molecular Analytical Science Centre for Doctoral Training (M.K.), and the Engineering and Physical Science Research and Astra Zeneca (S.K.H.).
Biographies
Qiao Song received his Bachelor’s degree from Department of Chemical Engineering, Tsinghua University, China, in 2012. He, then, completed his Ph.D. under the supervision of Prof. Xi Zhang in the Department of Chemistry, Tsinghua University, China, in 2017. After a 3-year postdoctoral research in Prof. Sébastien Perrier’s group at the University of Warwick, UK, he started his academic career as a research assistant professor at Shenzhen Grubbs Institute, Southern University of Science and Technology, China, from 2020. His research interests focus on highly ordered functional supramolecular assemblies and nanomaterials, as well as noncovalently engineered macromolecules.
Zihe Cheng received her BS degree in Applied Chemistry from Nanjing University of Information Science and Technology, China, in 2017. She undertook her Msc degree in Polymer Chemistry at the University of Warwick in 2019. She is currently working towards her PhD in Chemistry under the supervision of Prof. Sébastien Perrier. Her research focuses on the design, synthesis, and application of self-assembling amphiphilic cyclic peptide–polymer conjugates.
Maria Kariuki’s academic pathway began at Keele University, where she graduated with a BSc degree in Forensic Science. She, then, attended the University of Warwick in 2017 to pursue a masters in Analytical Sciences. During this time, she joined the Sébastien Perrier group to research the effects of sample treatments on the stability of cyclic peptide polymer conjugates through chromatographic and light scattering characterization. On completing her masters, she stayed with the group to undertake a Ph.D. still centered on the characterization of cyclic peptide conjugates but predominantly by asymmetric field flow fractionation. Besides her academic responsibilities, Maria is keen on promoting diversity in STEM through her initiatives as a committee member of the University of Warwick’s Network for Ethnic Minorities (NEMP).
Stephen C. L. Hall graduated from the University of Birmingham in 2014, before undertaking his PhD in Biophysics under the supervision of Prof. Tim Dafforn at the University of Birmingham and Dr. Thomas Arnold at the Diamond Light Source, the UK synchrotron. In 2018, he took up a postdoctoral position at the University of Warwick with Prof. Sébastien Perrier focusing on the use of neutron scattering techniques for characterizing self-assembled macromolecular materials. In 2020, Stephen joined the ISIS Neutron and Muon Source as an Instrument Scientist in Neutron Reflectometry, investigating the structure and interfacial behavior of self-assembled polymeric and biomimetic systems.
Sophie K. Hill graduated in 2018 with an MChem degree in Chemistry with Medicinal Chemistry from The University of Manchester. Her final year project was completed under the supervision of Dr. Simon Webb and investigated the self-assembly of sulfur based lipids in prebiotic chemistry. Following this, she began her Ph.D. under supervision of Prof. Sébastien Perrier at the University of Warwick, focusing on cyclic peptide–polymer conjugate nanotubes for delivery of anticancer drugs.
Julia Y. Rho graduated from the University of Warwick (England, UK) in 2015. She completed her Ph.D. in Chemistry at the same institution, graduation in 2019, under the supervision of Prof. Sebastien Perrier. Since then, she has conducted postdoctoral research at the George and Josephine Butler Polymer Research Laboratory at the University of Florida (Florida, USA) under the supervision of Prof. Brent Sumerlin. Her research interests include supramolecular chemistry, controlled radical polymerization and peptide–polymer conjugation. Recently, she was awarded the 2020 Macro Group UK Jon Weaver PhD prize and was selected as a PMSE Future Faculty Scholar (2020).
Sébastien Perrier graduated from the Ecole National Supérieure de Chimie de Montpellier, France, in 1998. He undertook his PhD at the University of Warwick, England, and spent one year as a postdoctoral fellow at the University of New South Wales, Australia. He started his academic career at Leeds in 2002 as a lecturer and then moved to the University of Sydney in 2007, as director of the Key Centre for Polymers & Colloids. In October 2013, Sébastien was appointed as the Monash–Warwick Alliance Chair in Polymer Chemistry, a joint appointment between the Chemistry Department and the Medical School at the University of Warwick, UK, and the Faculty of Pharmacy at Monash University, Australia. Sébastien’s team focuses on the use of macromolecular engineering to design functional nanostructured materials, with applications ranging from material science to nanotechnology and nanomedicine. He is a member of the editorial boards of Soft Matter, Macromolecules, European Polymer Journal, Polymers, Click Chemistry, ACS Macro Letters, Chemical Communications, and Chemical Society Reviews and is an Associate Editor of Polymer Chemistry.
Author Contributions
Q.S., Z.C., M.K., S.C.L.H., and S.K.H. contributed equally to this work.
The authors declare no competing financial interest.
References
- Lehn J.-M. Supramolecular Polymer Chemistry—Scope and Perspectives. Polym. Int. 2002, 51, 825–839. 10.1002/pi.852. [DOI] [Google Scholar]
- Moore T. S.; Winmill T. F. CLXXVII.—The State of Amines in Aqueous Solution. J. Chem. Soc., Trans. 1912, 101, 1635–1676. 10.1039/CT9120101635. [DOI] [Google Scholar]
- Jorgensen W. L.; Pranata J. Importance of Secondary Interactions in Triply Hydrogen Bonded Complexes: Guanine-Cytosine vs Uracil-2,6-Diaminopyridine. J. Am. Chem. Soc. 1990, 112, 2008–2010. 10.1021/ja00161a061. [DOI] [Google Scholar]
- Murray T. J.; Zimmerman S. C. New Triply Hydrogen Bonded Complexes with Highly Variable Stabilities. J. Am. Chem. Soc. 1992, 114, 4010–4011. 10.1021/ja00036a079. [DOI] [Google Scholar]
- Sijbesma R. P.; Meijer E. W. Quadruple Hydrogen Bonded Systems. Chem. Commun. 2003, 5–16. 10.1039/b205873c. [DOI] [PubMed] [Google Scholar]
- Corbin P. S.; Zimmerman S. C. Self-Association without Regard to Prototropy. A Heterocycle That Forms Extremely Stable Quadruply Hydrogen-Bonded Dimers. J. Am. Chem. Soc. 1998, 120, 9710–9711. 10.1021/ja981884d. [DOI] [Google Scholar]
- Chang S. K.; Hamilton A. D. Molecular Recognition of Biologically Interesting Substrates: Synthesis of an Artificial Receptor for Barbiturates Employing Six Hydrogen Bonds. J. Am. Chem. Soc. 1988, 110, 1318–1319. 10.1021/ja00212a065. [DOI] [Google Scholar]
- Corbin P. S.; Zimmerman S. C. Complexation-Induced Unfolding of Heterocyclic Ureas: A Hydrogen-Bonded, Sheetlike Heterodimer. J. Am. Chem. Soc. 2000, 122, 3779–3780. 10.1021/ja992830m. [DOI] [PubMed] [Google Scholar]
- Zeng H.; Miller R. S.; Flowers R. A.; Gong B. A Highly Stable, Six-Hydrogen-Bonded Molecular Duplex. J. Am. Chem. Soc. 2000, 122, 2635–2644. 10.1021/ja9942742. [DOI] [Google Scholar]
- Sijbesma R. P.; Beijer F. H.; Brunsveld L.; Folmer B. J. B.; Hirschberg J. H. K. K.; Lange R. F. M.; Lowe J. K. L.; Meijer E. W. Reversible Polymers Formed from Self-Complementary Monomers Using Quadruple Hydrogen Bonding. Science 1997, 278, 1601–1604. 10.1126/science.278.5343.1601. [DOI] [PubMed] [Google Scholar]
- Krieg E.; Bastings M. M. C.; Besenius P.; Rybtchinski B. Supramolecular Polymers in Aqueous Media. Chem. Rev. 2016, 116, 2414–2477. 10.1021/acs.chemrev.5b00369. [DOI] [PubMed] [Google Scholar]
- Sato K.; Hendricks M. P.; Palmer L. C.; Stupp S. I. Peptide Supramolecular Materials for Therapeutics. Chem. Soc. Rev. 2018, 47, 7539–7551. 10.1039/C7CS00735C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin C. K.; Hamblin G. D.; Sleiman H. F. Supramolecular DNA Assembly. Chem. Soc. Rev. 2011, 40, 5647–5656. 10.1039/c1cs15253j. [DOI] [PubMed] [Google Scholar]
- Shimizu T.; Ding W.; Kameta N. Soft-Matter Nanotubes: A Platform for Diverse Functions and Applications. Chem. Rev. 2020, 120, 2347–2407. 10.1021/acs.chemrev.9b00509. [DOI] [PubMed] [Google Scholar]
- Hamley I. W. Peptide Nanotubes. Angew. Chem., Int. Ed. 2014, 53, 6866–6881. 10.1002/anie.201310006. [DOI] [PubMed] [Google Scholar]
- Gao X.; Matsui H. Peptide-Based Nanotubes and Their Applications in Bionanotechnology. Adv. Mater. 2005, 17, 2037–2050. 10.1002/adma.200401849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringsdorf H. Structure and Properties of Pharmacologically Active Polymers. J. Polym. Sci., Part C: Polym. Symp. 1975, 51, 135–153. 10.1002/polc.5070510111. [DOI] [Google Scholar]
- Chen C.; Ng D. Y. W.; Weil T. Polymer Bioconjugates: Modern Design Concepts toward Precision Hybrid Materials. Prog. Polym. Sci. 2020, 105, 101241. 10.1016/j.progpolymsci.2020.101241. [DOI] [Google Scholar]
- Otter R.; Besenius P. Supramolecular Assembly of Functional Peptide-Polymer Conjugates. Org. Biomol. Chem. 2019, 17, 6719–6734. 10.1039/C9OB01191A. [DOI] [PubMed] [Google Scholar]
- Olson R. A.; Korpusik A. B.; Sumerlin B. S. Enlightening Advances in Polymer Bioconjugate Chemistry: Light-based Techniques for Grafting to and from Biomacromolecules. Chem. Sci. 2020, 11, 5142–5156. 10.1039/D0SC01544J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman A. S.; Stayton P. S. Conjugates of Stimuli-Responsive Polymers and Proteins. Prog. Polym. Sci. 2007, 32, 922–932. 10.1016/j.progpolymsci.2007.05.005. [DOI] [Google Scholar]
- Lutz J.-F.; Börner H. G. Modern Trends in Polymer Bioconjugates Design. Prog. Polym. Sci. 2008, 33, 1–39. 10.1016/j.progpolymsci.2007.07.005. [DOI] [Google Scholar]
- Messina M. S.; Messina K. M. M.; Bhattacharya A.; Montgomery H. R.; Maynard H. D. Preparation of Biomolecule-Polymer Conjugates by Grafting-from Using ATRP, RAFT, or ROMP. Prog. Polym. Sci. 2020, 100, 101186. 10.1016/j.progpolymsci.2019.101186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Santis P.; Morosetti S.; Rizzo R. Conformational Analysis of Regular Enantiomeric Sequences. Macromolecules 1974, 7, 52–58. 10.1021/ma60037a011.4837982 [DOI] [Google Scholar]
- Tomasic L.; Lorenzi G. P. Some Cyclic Oligopeptide with S2n Symmetry. Helv. Chim. Acta 1987, 70, 1012–1016. 10.1002/hlca.19870700413. [DOI] [Google Scholar]
- Saviano M.; Zaccaro L.; Lombardi A.; Pedone C.; Di Blasio B.; Sun X.; Lorenzi G. P. A Structural Two-ring Version of a Tubular Stack of β-rings in Crystals of a Cyclic D,L-Hexapeptide. J. Inclusion Phenom. Mol. Recognit. Chem. 1994, 18, 27–36. 10.1007/BF00706936. [DOI] [Google Scholar]
- Ghadiri M. R.; Granja J. R.; Milligan R. A.; McRee D. E.; Khazanovich N. Self-assembling Organic Nanotubes Based on a Cyclic Peptide Architecture. Nature 1993, 366, 324–327. 10.1038/366324a0. [DOI] [PubMed] [Google Scholar]
- Engels M.; Bashford D.; Ghadiri M. R. Structure and Dynamics of Self-Assembling Peptide Nanotubes and the Channel-Mediated Water Organization and Self-Diffusion. A Molecular Dynamics Study. J. Am. Chem. Soc. 1995, 117, 9151–9158. 10.1021/ja00141a005. [DOI] [Google Scholar]
- Ghadiri M. R.; Granja J. R.; Buehler L. K. Artificial Transmembrane Ion Channels from Self-assembling Peptide Nanotubes. Nature 1994, 369, 301–304. 10.1038/369301a0. [DOI] [PubMed] [Google Scholar]
- Khazanovich N.; Granja J. R.; McRee D. E.; Milligan R. A.; Ghadiri M. R. Nanoscale Tubular Ensembles with Specified Internal Diameters. Design of a Self-Assembled Nanotube with a 13 Å Pore. J. Am. Chem. Soc. 1994, 116, 6011–6012. 10.1021/ja00092a079. [DOI] [Google Scholar]
- Hartgerink J. D.; Granja J. R.; Milligan R. A.; Ghadiri M. R. Self-Assembling Peptide Nanotubes. J. Am. Chem. Soc. 1996, 118, 43–50. 10.1021/ja953070s. [DOI] [Google Scholar]
- Insua I.; Montenegro J. 1D to 2D Self Assembly of Cyclic Peptides. J. Am. Chem. Soc. 2020, 142, 300–307. 10.1021/jacs.9b10582. [DOI] [PubMed] [Google Scholar]
- Diaz S.; Insua I.; Bhak G.; Montenegro J. Sequence Decoding of 1D to 2D Self-Assembling Cyclic Peptides. Chem. - Eur. J. 2020, 26, 14765–14770. 10.1002/chem.202003265. [DOI] [PubMed] [Google Scholar]
- Silk M. R.; Newman J.; Ratcliffe J. C.; White J. F.; Caradoc-Davies T.; Price J. R.; Perrier S.; Thompson P. E.; Chalmers D. K. Parallel and Antiparallel Cyclic D/L Peptide Nanotubes. Chem. Commun. 2017, 53, 6613–6616. 10.1039/C7CC00846E. [DOI] [PubMed] [Google Scholar]
- Rosenthal-Aizman K.; Svensson G.; Undén A. Self-Assembling Peptide Nanotubes from Enantiomeric Pairs of Cyclic Peptides with Alternating D and L Amino Acid Residues. J. Am. Chem. Soc. 2004, 126, 3372–3373. 10.1021/ja0372659. [DOI] [PubMed] [Google Scholar]
- Sun X.; Lorenzi G. P. On the Stacking of β-Rings: The Solution Self-Association Behavior of Two Partially N-methylated Cyclo(hexaleucines). Helv. Chim. Acta 1994, 77, 1520–1526. 10.1002/hlca.19940770607. [DOI] [Google Scholar]
- Ghadiri M. R.; Kobayashi K.; Granja J. R.; Chadha R. K.; McRee D. E. The Structural and Thermodynamic Basis for the Formation of Self-Assembled Peptide Nanotubes. Angew. Chem., Int. Ed. Engl. 1995, 34, 93–95. 10.1002/anie.199500931. [DOI] [Google Scholar]
- Kobayashi K.; Granja J. R.; Ghadiri M. R. β-Sheet Peptide Architecture: Measuring the Relative Stability of Parallel vs. Antiparallel β-Sheets. Angew. Chem., Int. Ed. Engl. 1995, 34, 95–98. 10.1002/anie.199500951. [DOI] [Google Scholar]
- Clark T. D.; Buriak J. M.; Kobayashi K.; Isler M. P.; McRee D. E.; Ghadiri M. R. Cylindrical β-Sheet Peptide Assemblies. J. Am. Chem. Soc. 1998, 120, 8949–8962. 10.1021/ja981485i. [DOI] [Google Scholar]
- Rubin D. J.; Amini S.; Zhou F.; Su H.; Miserez A.; Joshi N. S. Structural, Nanomechanical, and Computational Characterization of D,L-Cyclic Peptide Assemblies. ACS Nano 2015, 9, 3360–3368. 10.1021/acsnano.5b00672. [DOI] [PubMed] [Google Scholar]
- Rubin D. J.; Nia H. T.; Desire T.; Nguyen P. Q.; Gevelber M.; Ortiz C.; Joshi N. S. Mechanical Reinforcement of Polymeric Fibers through Peptide Nanotube Incorporation. Biomacromolecules 2013, 14, 3370–3375. 10.1021/bm4008293. [DOI] [PubMed] [Google Scholar]
- Shaikh H.; Rho J. Y.; Macdougall L. J.; Gurnani P.; Lunn A. M.; Yang J.; Huband S.; Mansfield E. D. H.; Peltier R.; Perrier S. Hydrogel and Organogel Formation by Hierarchical Self-Assembly of Cyclic Peptides Nanotubes. Chem. - Eur. J. 2018, 24, 19066–19074. 10.1002/chem.201804576. [DOI] [PubMed] [Google Scholar]
- Méndez-Ardoy A.; Granja J. R.; Montenegro J. pH-Triggered Self-Assembly and Hydrogelation of Cyclic Peptide Nanotubes Confined in Water Micro-Droplets. Nanoscale Horiz. 2018, 3, 391–396. 10.1039/C8NH00009C. [DOI] [PubMed] [Google Scholar]
- Méndez-Ardoy A.; Bayón-Fernández A.; Yu Z.; Abell C.; Granja J. R.; Montenegro J. Spatially Controlled Supramolecular Polymerization of Peptide Nanotubes by Microfluidics. Angew. Chem., Int. Ed. 2020, 59, 6902–6908. 10.1002/anie.202000103. [DOI] [PubMed] [Google Scholar]
- Mattia E.; Otto S. Supramolecular Systems Chemistry. Nat. Nanotechnol. 2015, 10, 111–119. 10.1038/nnano.2014.337. [DOI] [PubMed] [Google Scholar]
- Giuseppone N. Toward Self-Constructing Materials: A Systems Chemistry Approach. Acc. Chem. Res. 2012, 45, 2178–2188. 10.1021/ar2002655. [DOI] [PubMed] [Google Scholar]
- Ashkenasy G.; Hermans T. M.; Otto S.; Taylor A. F. Systems Chemistry. Chem. Soc. Rev. 2017, 46, 2543–2554. 10.1039/C7CS00117G. [DOI] [PubMed] [Google Scholar]
- Cissé N.; Kudernac T. Light-Fuelled Self-Assembly of Cyclic Peptides into Supramolecular Tubules. ChemSystemsChem 2020, 2, e2000012 10.1002/syst.202000012. [DOI] [Google Scholar]
- Seebach D.; Matthews J. L.; Meden A.; Wessels T.; Baerlocher C.; McCusker L. B. Cyclo-β-Peptides: Structure and Tubular Stacking of Cyclic Tetramers of 3-Aminobutanoic Acid as Determined from Powder Diffraction Data. Helv. Chim. Acta 1997, 80, 173–182. 10.1002/hlca.19970800116. [DOI] [Google Scholar]
- Clark T. D.; Buehler L. K.; Ghadiri M. R. Self-Assembling Cyclic β3-Peptide Nanotubes as Artificial Transmembrane Ion Channels. J. Am. Chem. Soc. 1998, 120, 651–656. 10.1021/ja972786f. [DOI] [Google Scholar]
- Fujimura F.; Hirata T.; Morita T.; Kimura S.; Horikawa Y.; Sugiyama J. Columnar Assembly of Cyclic β-Amino Acid Functionalized with Pyranose Rings. Biomacromolecules 2006, 7, 2394–2400. 10.1021/bm060415y. [DOI] [PubMed] [Google Scholar]
- Hirata T.; Fujimura F.; Horikawa Y.; Sugiyama J.; Morita T.; Kimura S. Molecular Assembly Formation of Cyclic Hexa-β-Peptide Composed of Acetylated Glycosamino Acids. Biopolymers 2007, 88, 150–156. 10.1002/bip.20694. [DOI] [PubMed] [Google Scholar]
- Fujimura F.; Horikawa Y.; Morita T.; Sugiyama J.; Kimura S. Double Assembly Composed of Lectin Association with Columnar Molecular Assembly of Cyclic Tri-β-peptide Having Sugar Units. Biomacromolecules 2007, 8, 611–616. 10.1021/bm060862d. [DOI] [PubMed] [Google Scholar]
- Hirata T.; Fujimura F.; Kimura S. A Novel Polypseudorotaxane Composed of Cyclic β-Peptide as Bead Component. Chem. Commun. 2007, 1023–1025. 10.1039/B617768A. [DOI] [PubMed] [Google Scholar]
- Ishihara Y.; Kimura S. Nanofiber Formation of Amphiphilic Cyclic Tri-β-Peptide. J. Pept. Sci. 2010, 16, 110–114. 10.1002/psc.1206. [DOI] [PubMed] [Google Scholar]
- Amorín M.; Castedo L.; Granja J. R. New Cyclic Peptide Assemblies with Hydrophobic Cavities: The Structural and Thermodynamic Basis of a New Class of Peptide Nanotubes. J. Am. Chem. Soc. 2003, 125, 2844–2845. 10.1021/ja0296273. [DOI] [PubMed] [Google Scholar]
- Calvelo M.; Lamas A.; Guerra A.; Amorín M.; Garcia-Fandino R.; Granja J. R. Parallel Versus Antiparallel β-Sheet Structure in Cyclic Peptide Hybrids Containing γ- or δ-Cyclic Amino Acids. Chem. - Eur. J. 2020, 26, 5846–5858. 10.1002/chem.201905554. [DOI] [PubMed] [Google Scholar]
- García-Fandiño R.; Castedo L.; Granja J. R.; Vázquez S. A. Interaction and Dimerization Energies in Methyl-Blocked α,γ-Peptide Nanotube Segments. J. Phys. Chem. B 2010, 114, 4973–4983. 10.1021/jp910919k. [DOI] [PubMed] [Google Scholar]
- Amorín M.; Villaverde V.; Castedo L.; Granja J. R. New α,γ-Peptide Tubulets. J. Drug Delivery Sci. Technol. 2005, 15, 87–92. 10.1016/S1773-2247(05)50011-X. [DOI] [Google Scholar]
- Amorín M.; Castedo L.; Granja J. R. Self-Assembled Peptide Tubelets with 7 Å Pores. Chem. - Eur. J. 2005, 11, 6543–6551. 10.1002/chem.200500478. [DOI] [PubMed] [Google Scholar]
- Amorín M.; Brea R. J.; Castedo L.; Granja J. R. The Smallest α,γ-Peptide Nanotubulet Segments: Cyclic α,γ-Tetrapeptide Dimers. Org. Lett. 2005, 7, 4681–4684. 10.1021/ol0518885. [DOI] [PubMed] [Google Scholar]
- Brea R. J.; Castedo L.; Granja J. R. Large-Diameter Self-Assembled Dimers of α,γ-Cyclic Peptides, with the Nanotubular Solid-State Structure of cyclo-[(L-Leu-D-MeN-γ-Acp)4-]·4CHCl2COOH. Chem. Commun. 2007, 3267–3269. 10.1039/b703659k. [DOI] [PubMed] [Google Scholar]
- Brea R. J.; Amorín M.; Castedo L.; Granja J. R. Methyl-Blocked Dimeric α,γ-Peptide Nanotube Segments: Formation of a Peptide Heterodimer through Backbone-Backbone Interactions. Angew. Chem., Int. Ed. 2005, 44, 5710–5713. 10.1002/anie.200501555. [DOI] [PubMed] [Google Scholar]
- Pizzi A.; Ozores H. L.; Calvelo M.; García-Fandiño R.; Amorín M.; Demitri N.; Terraneo G.; Bracco S.; Comotti A.; Sozzani P.; Bezuidenhout C. X.; Metrangolo P.; Granja J. R. Tight Xenon Confinement in a Crystalline Sandwich-like Hydrogen-Bonded Dimeric Capsule of a Cyclic Peptide. Angew. Chem., Int. Ed. 2019, 58, 14472–14476. 10.1002/anie.201906599. [DOI] [PubMed] [Google Scholar]
- Reiriz C.; Castedo L.; Granja J. R. New α,γ-Cyclic Peptides-Nanotube Molecular Caps Using α,α-Dialkylated α-Amino Acids. J. Pept. Sci. 2008, 14, 241–249. 10.1002/psc.984. [DOI] [PubMed] [Google Scholar]
- García-Fandiño R.; Granja J. R.; D’Abramo M.; Orozco M. Theoretical Characterization of the Dynamical Behavior and Transport Properties of α,γ-Peptide Nanotubes in Solution. J. Am. Chem. Soc. 2009, 131, 15678–15686. 10.1021/ja903400n. [DOI] [PubMed] [Google Scholar]
- Reiriz C.; Brea R. J.; Arranz R.; Carrascosa J. L.; Garibotti A.; Manning B.; Valpuesta J. M.; Eritja R.; Castedo L.; Granja J. R. α,γ-Peptide Nanotube Templating of One-Dimensional Parallel Fullerene Arrangements. J. Am. Chem. Soc. 2009, 131, 11335–11337. 10.1021/ja904548q. [DOI] [PubMed] [Google Scholar]
- García-Fandiño R.; Amorín M.; Castedo L.; Granja J. R. Transmembrane Ion Transport by Self-Assembling α,γ-Peptide Nanotubes. Chem. Sci. 2012, 3, 3280–3285. 10.1039/c2sc21068a. [DOI] [Google Scholar]
- Amorín M.; Castedo L.; Granja J. R. Folding Control in Cyclic Peptides through N-Methylation Pattern Selection: Formation of Antiparallel β-Sheet Dimers, Double Reverse Turns and Supramolecular Helices by 3α,γ Cyclic Peptides. Chem. - Eur. J. 2008, 14, 2100–2111. 10.1002/chem.200701059. [DOI] [PubMed] [Google Scholar]
- Hourani R.; Zhang C.; van der Weegen R.; Ruiz L.; Li C.; Keten S.; Helms B. A.; Xu T. Processable Cyclic Peptide Nanotubes with Tunable Interiors. J. Am. Chem. Soc. 2011, 133, 15296–15299. 10.1021/ja2063082. [DOI] [PubMed] [Google Scholar]
- Ruiz L.; Benjamin A.; Sullivan M.; Keten S. Regulating Ion Transport in Peptide Nanotubes by Tailoring the Nanotube Lumen Chemistry. J. Phys. Chem. Lett. 2015, 6, 1514–1520. 10.1021/acs.jpclett.5b00252. [DOI] [PubMed] [Google Scholar]
- Li L.; Zhan H.; Duan P.; Liao J.; Quan J.; Hu Y.; Chen Z.; Zhu J.; Liu M.; Wu Y.-D.; Deng J. Self-Assembling Nanotubes Consisting of Rigid Cyclic γ-Peptides. Adv. Funct. Mater. 2012, 22, 3051–3056. 10.1002/adfm.201200488. [DOI] [Google Scholar]
- Guerra A.; Brea R. J.; Amorín M.; Castedo L.; Granja J. R. Self-Assembling Properties of All γ-Cyclic Peptides Containing Sugar Amino Acid Residues. Org. Biomol. Chem. 2012, 10, 8762–8766. 10.1039/c2ob26612a. [DOI] [PubMed] [Google Scholar]
- Chen J.; Li Q.; Wu P.; Liu J.; Wang D.; Yuan X.; Zheng R.; Sun R.; Li L. Cyclic γ-Peptides with Transmembrane Water Channel Properties. Front. Chem. 2020, 8, 368. 10.3389/fchem.2020.00368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvelo M.; Vázquez S.; García-Fandiño R. Molecular Dynamics Simulations for Designing Biomimetic Pores Based on Internally Functionalized Self-Assembling α,γ-Peptide Nanotubes. Phys. Chem. Chem. Phys. 2015, 17, 28586–28601. 10.1039/C5CP04200C. [DOI] [PubMed] [Google Scholar]
- Calvelo M.; Granja J. R.; Garcia-Fandino R. Competitive Double-Switched Self-Assembled Cyclic Peptide Nanotubes: A Dual Internal and External Control. Phys. Chem. Chem. Phys. 2019, 21, 20750–20756. 10.1039/C9CP02327E. [DOI] [PubMed] [Google Scholar]
- Gauthier D.; Baillargeon P.; Drouin M.; Dory Y. L. Self-Assembly of Cyclic Peptides into Nanotubes and Then into Highly Anisotropic Crystalline Materials. Angew. Chem., Int. Ed. 2001, 40, 4635–4638. . [DOI] [PubMed] [Google Scholar]
- Leclair S.; Baillargeon P.; Skouta R.; Gauthier D.; Zhao Y.; Dory Y. L. Micrometer-Sized Hexagonal Tubes Self-Assembled by a Cyclic Peptide in a Liquid Crystal. Angew. Chem., Int. Ed. 2004, 43, 349–353. 10.1002/anie.200352259. [DOI] [PubMed] [Google Scholar]
- Bélanger D.; Tong X.; Soumaré S.; Dory Y. L.; Zhao Y. Cyclic Peptide-Polymer Complexes and Their Self-Assembly. Chem. - Eur. J. 2009, 15, 4428–4436. 10.1002/chem.200802337. [DOI] [PubMed] [Google Scholar]
- Lamas A.; Guerra A.; Amorín M.; Granja J. R. New Self-Assembling Peptide Nanotubes of Large Diameter Using δ-Amino Acids. Chem. Sci. 2018, 9, 8228–8233. 10.1039/C8SC02276C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horne W. S.; Stout C. D.; Ghadiri M. R. A Heterocyclic Peptide Nanotube. J. Am. Chem. Soc. 2003, 125, 9372–9376. 10.1021/ja034358h. [DOI] [PubMed] [Google Scholar]
- van Maarseveen J. H.; Horne W. S.; Ghadiri M. R. Efficient Route to C2 Symmetric Heterocyclic Backbone Modified Cyclic Peptides. Org. Lett. 2005, 7, 4503–4506. 10.1021/ol0518028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghorai A.; Gayen A.; Kulsi G.; Padmanaban E.; Laskar A.; Achari B.; Mukhopadhyay C.; Chattopadhyay P. Simultaneous Parallel and Antiparallel Self-Assembly in a Triazole/Amide Macrocycle Conformationally Homologous to D-,L-α-Amino Acid Based Cyclic Peptides: NMR and Molecular Modeling Study. Org. Lett. 2011, 13, 5512–5515. 10.1021/ol2022356. [DOI] [PubMed] [Google Scholar]
- Ghorai A.; Padmanaban E.; Mukhopadhyay C.; Achari B.; Chattopadhyay P. Design and Synthesis of Regioisomeric Triazole Based Peptidomimetic Macrocycles and Their Dipole Moment Controlled Self-Assembly. Chem. Commun. 2012, 48, 11975–11977. 10.1039/c2cc36566a. [DOI] [PubMed] [Google Scholar]
- Kulsi G.; Ghorai A.; Achari B.; Chattopadhyay P. Design and Synthesis of Conformationally Homogeneous Pseudo Cyclic Peptides Through Amino Acid Insertion: Investigations on Their Self Assembly. RSC Adv. 2015, 5, 64675–64681. 10.1039/C5RA11850F. [DOI] [Google Scholar]
- Chow H. Y.; Zhang Y.; Matheson E.; Li X. Ligation Technologies for the Synthesis of Cyclic Peptides. Chem. Rev. 2019, 119, 9971–10001. 10.1021/acs.chemrev.8b00657. [DOI] [PubMed] [Google Scholar]
- Rovero P.; Quartara L.; Fabbri G. Synthesis of Cyclic Peptides on Solid Support. Tetrahedron Lett. 1991, 32, 2639–2642. 10.1016/S0040-4039(00)78806-X. [DOI] [Google Scholar]
- Chen J.; Zhang B.; Xie C.; Lu Y.; Wu W. Synthesis of a Highly Hydrophobic Cyclic Decapeptide by Solid-Phase Synthesis of Linear Peptide and Cyclization in Solution. Chin. Chem. Lett. 2010, 21, 391–394. 10.1016/j.cclet.2009.11.026. [DOI] [Google Scholar]
- Beierle J. M.; Horne W. S.; van Maarseveen J. H.; Waser B.; Reubi J. C.; Ghadiri M. R. Conformationally Homogeneous Heterocyclic Pseudotetrapeptides as Three-Dimensional Scaffolds for Rational Drug Design: Receptor-Selective Somatostatin Analogues. Angew. Chem., Int. Ed. 2009, 48, 4725–4729. 10.1002/anie.200805901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horne W. S.; Olsen C. A.; Beierle J. M.; Montero A.; Ghadiri M. R. Probing the Bioactive Conformation of an Archetypal Natural Product HDAC Inhibitor with Conformationally Homogeneous Triazole-Modified Cyclic Tetrapeptides. Angew. Chem., Int. Ed. 2009, 48, 4718–4724. 10.1002/anie.200805900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiriz C.; Amorín M.; García-Fandiño R.; Castedo L.; Granja J. R. α,γ-Cyclic Peptide Ensembles with a Hydroxylated Cavity. Org. Biomol. Chem. 2009, 7, 4358–4361. 10.1039/b911247m. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Vázquez N.; García-Fandiño R.; Amorín M.; Granja J. R. Self-Assembling α,γ-Cyclic Peptides that Generate Cavities with Tunable Properties. Chem. Sci. 2016, 7, 183–187. 10.1039/C5SC03187G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Vázquez N.; García-Fandiño R.; Aldegunde M. J.; Brea J.; Loza M. I.; Amorín M.; Granja J. R. cis-Platinum Complex Encapsulated in Self-Assembling Cyclic Peptide Dimers. Org. Lett. 2017, 19, 2560–2563. 10.1021/acs.orglett.7b00871. [DOI] [PubMed] [Google Scholar]
- Burade S. S.; Saha T.; Bhuma N.; Kumbhar N.; Kotmale A.; Rajamohanan P. R.; Gonnade R. G.; Talukdar P.; Dhavale D. D. Self-Assembly of Fluorinated Sugar Amino Acid Derived α,γ-Cyclic Peptides into Transmembrane Anion Transport. Org. Lett. 2017, 19, 5948–5951. 10.1021/acs.orglett.7b02942. [DOI] [PubMed] [Google Scholar]
- Darnall S. M.; Li C.; Dunbar M.; Alsina M.; Keten S.; Helms B. A.; Xu T. Organic Nanotube with Subnanometer, pH-Responsive Lumen. J. Am. Chem. Soc. 2019, 141, 10953–10957. 10.1021/jacs.9b03732. [DOI] [PubMed] [Google Scholar]
- Wang D.; Guo L.; Zhang J.; Roeske R. W.; Jones L. R.; Chen Z.; Pritchard C. Artificial Ion Channels Formed by a Synthetic Cyclic Peptide. J. Pept. Res. 2001, 57, 301–306. 10.1046/j.1397-002X.2000.00823.x. [DOI] [PubMed] [Google Scholar]
- Horne W. S.; Ashkenasy N.; Ghadiri M. R. Modulating Charge Transfer through Cyclic D,L-α-Peptide Self-Assembly. Chem. - Eur. J. 2005, 11, 1137–1144. 10.1002/chem.200400923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkenasy N.; Horne W. S.; Ghadiri M. R. Design of Self-Assembling Peptide Nanotubes with Delocalized Electronic States. Small 2006, 2, 99–102. 10.1002/smll.200500252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uji H.; Kim H.; Imai T.; Mitani S.; Sugiyama J.; Kimura S. Electronic Properties of Tetrathiafulvalene-Modified Cyclic-β-Peptide Nanotube. Biopolymers 2016, 106, 275–282. 10.1002/bip.22850. [DOI] [PubMed] [Google Scholar]
- Tabata Y.; Kamano Y.; Kimura S.; Uji H. Engineering pH-Responsive Switching of Donor-π-Acceptor Chromophore Alignments Along a Peptide Nanotube Scaffold. RSC Adv. 2020, 10, 3588–3592. 10.1039/D0RA00231C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamano Y.; Tabata Y.; Uji H.; Kimura S. Chiral and Random Arrangements of Flavin Chromophores Along Cyclic Peptide Nanotubes on Gold Influencing Differently on Surface Potential and Piezoelectricity. RSC Adv. 2019, 9, 3618–3624. 10.1039/C8RA10466B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabata Y.; Mitani S.; Uji H.; Imai T.; Kimura S. The Effect of Macrodipole Orientation on the Piezoelectric Response of Cyclic β-Peptide Nanotube Bundles on Gold Substrates. Polym. J. 2019, 51, 601–609. 10.1038/s41428-019-0169-4. [DOI] [Google Scholar]
- Ishihara Y.; Kimura S. Four-Peptide-Nanotube Bundle Formation by Self-Assembling of Cyclic Tetra-β-Peptide Using G-Quartet Motif. Biopolymers 2013, 100, 141–147. 10.1002/bip.22177. [DOI] [PubMed] [Google Scholar]
- Tabata Y.; Kamano Y.; Uji H.; Imai T.; Kimura S. Electronic Properties of Cyclic β-Peptide Nanotube Bundles Reflecting Structural Arrangement. Chem. Lett. 2019, 48, 322–324. 10.1246/cl.181007. [DOI] [Google Scholar]
- Ishihara Y.; Kimura S. Peptide Nanotube Composed of Cyclic Tetra-β-Peptide Having Polydiacetylene. Biopolymers 2012, 98, 155–160. 10.1002/bip.22029. [DOI] [PubMed] [Google Scholar]
- Fujimura F.; Kimura S. Columnar Assembly Formation and Metal Binding of Cyclic Tri-β-peptides Having Terpyridine Ligands. Org. Lett. 2007, 9, 793–796. 10.1021/ol0629622. [DOI] [PubMed] [Google Scholar]
- Tabata Y.; Mitani S.; Kimura S. Peptide Nanotube Aligning Side Chains onto One Side. J. Pept. Sci. 2016, 22, 391–396. 10.1002/psc.2881. [DOI] [PubMed] [Google Scholar]
- Montenegro J.; Vázquez-Vázquez C.; Kalinin A.; Geckeler K. E.; Granja J. R. Coupling of Carbon and Peptide Nanotubes. J. Am. Chem. Soc. 2014, 136, 2484–2491. 10.1021/ja410901r. [DOI] [PubMed] [Google Scholar]
- Cuerva M.; García-Fandiño R.; Vázquez-Vázquez C.; López-Quintela M. A.; Montenegro J.; Granja J. R. Self-Assembly of Silver Metal Clusters of Small Atomicity on Cyclic Peptide Nanotubes. ACS Nano 2015, 9, 10834–10843. 10.1021/acsnano.5b03445. [DOI] [PubMed] [Google Scholar]
- Hernández-Eguía L. P.; Brea R. J.; Castedo L.; Ballester P.; Granja J. R. Regioisomeric Control Induced by DABCO Coordination to Rotatable Self-Assembled Bis- and Tetraporphyrin α,γ-Cyclic Octapeptide Dimers. Chem. - Eur. J. 2011, 17, 1220–1229. 10.1002/chem.201002271. [DOI] [PubMed] [Google Scholar]
- Panciera M.; Amorín M.; Castedo L.; Granja J. R. Design of Stable β-Sheet-Based Cyclic Peptide Assemblies Assisted by Metal Coordination: Selective Homo- and Heterodimer Formation. Chem. - Eur. J. 2013, 19, 4826–4834. 10.1002/chem.201203213. [DOI] [PubMed] [Google Scholar]
- Panciera M.; Amorín M.; Granja J. R. Molecular Pom Poms from Self-Assembling α,γ-Cyclic Peptides. Chem. - Eur. J. 2014, 20, 10260–10265. 10.1002/chem.201403319. [DOI] [PubMed] [Google Scholar]
- Panciera M.; González-Freire E.; Calvelo M.; Amorín M.; Granja J. R. Induced α,γ-Cyclic Peptide Rotodimer Recognition by Nucleobase Scaffolds. Pept. Sci. 2020, 112, e24132 10.1002/pep2.24132. [DOI] [Google Scholar]
- Brea R. J.; Castedo L.; Granja J. R.; Herranz M. Á.; Sánchez L.; Martín N.; Seitz W.; Guldi D. M. Electron Transfer in Me-Blocked Heterodimeric α,γ-Peptide Nanotubular Donor-Acceptor Hybrids. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 5291–5294. 10.1073/pnas.0609506104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragay G.; Ventura B.; Guerra A.; Pintre I.; Chiorboli C.; García-Fandiño R.; Flamigni L.; Granja J. R.; Ballester P. Self-Sorting of Cyclic Peptide Homodimers into a Heterodimeric Assembly Featuring an Efficient Photoinduced Intramolecular Electron-Transfer Process. Chem. - Eur. J. 2014, 20, 3427–3438. 10.1002/chem.201304200. [DOI] [PubMed] [Google Scholar]
- Brea R. J.; Vázquez M. E.; Mosquera M.; Castedo L.; Granja J. R. Controlling Multiple Fluorescent Signal Output in Cyclic Peptide-Based Supramolecular Systems. J. Am. Chem. Soc. 2007, 129, 1653–1657. 10.1021/ja066885h. [DOI] [PubMed] [Google Scholar]
- Brea R. J.; Pérez-Alvite M. J.; Panciera M.; Mosquera M.; Castedo L.; Granja J. R. Highly Efficient and Directional Homo- and Heterodimeric Energy Transfer Materials Based on Fluorescently Derivatized α,γ-Cyclic Octapeptides. Chem. - Asian J. 2011, 6, 110–121. 10.1002/asia.201000545. [DOI] [PubMed] [Google Scholar]
- Couet J.; Samuel J. D. J. S.; Kopyshev A.; Santer S.; Biesalski M. Peptide-Polymer Hybrid Nanotubes. Angew. Chem., Int. Ed. 2005, 44, 3297–3301. 10.1002/anie.200462993. [DOI] [PubMed] [Google Scholar]
- Couet J.; Biesalski M. Surface-Initiated ATRP of N-Isopropylacrylamide from Initiator-Modified Self-Assembled Peptide Nanotubes. Macromolecules 2006, 39, 7258–7268. 10.1021/ma061200j. [DOI] [Google Scholar]
- Couet J.; Biesalski M. Polymer-Wrapped Peptide Nanotubes: Peptide-Grafted Polymer Mass Impacts Length and Diameter. Small 2008, 4, 1008–1016. 10.1002/smll.200700967. [DOI] [PubMed] [Google Scholar]
- Larnaudie S. C.; Brendel J. C.; Jolliffe K. A.; Perrier S. Cyclic Peptide-Polymer Conjugates: Grafting-to vs Grafting-from. J. Polym. Sci., Part A: Polym. Chem. 2016, 54, 1003–1011. 10.1002/pola.27937. [DOI] [Google Scholar]
- Chapman R.; Jolliffe K. A.; Perrier S. Synthesis of Self-Assembling Cyclic Peptide-Polymer Conjugates Using Click Chemistry. Aust. J. Chem. 2010, 63, 1169–1172. 10.1071/CH10128. [DOI] [Google Scholar]
- Chapman R.; Jolliffe K. A.; Perrier S. Modular Design for the Controlled Production of Polymeric Nanotubes from Polymer/Peptide Conjugates. Polym. Chem. 2011, 2, 1956–1963. 10.1039/c1py00202c. [DOI] [Google Scholar]
- Poon C. K.; Chapman R.; Jolliffe K. A.; Perrier S. Pushing the Limits of Copper Mediated Azide-Alkyne Cycloaddition (CuAAC) to Conjugate Polymeric Chains to Cyclic Peptides. Polym. Chem. 2012, 3, 1820–1826. 10.1039/c2py00510g. [DOI] [Google Scholar]
- Chapman R.; Bouten P. J. M.; Hoogenboom R.; Jolliffe K. A.; Perrier S. Thermoresponsive Cyclic Peptide - Poly(2-ethyl-2-oxazoline) Conjugate Nanotubes. Chem. Commun. 2013, 49, 6522–6524. 10.1039/c3cc42327a. [DOI] [PubMed] [Google Scholar]
- Chapman R.; Jolliffe K. A.; Perrier S. Multi-Shell Soft Nanotubes from Cyclic Peptide Templates. Adv. Mater. 2013, 25, 1170–1172. 10.1002/adma.201204094. [DOI] [PubMed] [Google Scholar]
- Chapman R.; Koh M. L.; Warr G. G.; Jolliffe K. A.; Perrier S. Structure Elucidation and Control of Cyclic Peptide-Derived Nanotube Assemblies in Solution. Chem. Sci. 2013, 4, 2581–2589. 10.1039/c3sc00064h. [DOI] [Google Scholar]
- Chapman R.; Warr G. G.; Perrier S.; Jolliffe K. A. Water-Soluble and pH-Responsive Polymeric Nanotubes from Cyclic Peptide Templates. Chem. - Eur. J. 2013, 19, 1955–1961. 10.1002/chem.201203602. [DOI] [PubMed] [Google Scholar]
- Danial M.; My-Nhi Tran C.; Young P. G.; Perrier S.; Jolliffe K. A. Janus Cyclic Peptide-Polymer Nanotubes. Nat. Commun. 2013, 4, 2780. 10.1038/ncomms3780. [DOI] [PubMed] [Google Scholar]
- Song Q.; Yang J.; Rho J. Y.; Perrier S. Supramolecular Switching of the Self-Assembly of Cyclic Peptide-Polymer Conjugates via Host-Guest Chemistry. Chem. Commun. 2019, 55, 5291–5294. 10.1039/C9CC01914F. [DOI] [PubMed] [Google Scholar]
- Blunden B. M.; Chapman R.; Danial M.; Lu H.; Jolliffe K. A.; Perrier S.; Stenzel M. H. Drug Conjugation to Cyclic Peptide-Polymer Self-Assembling Nanotubes. Chem. - Eur. J. 2014, 20, 12745–12749. 10.1002/chem.201403130. [DOI] [PubMed] [Google Scholar]
- Yang J.; Song J.-I.; Song Q.; Rho J. Y.; Mansfield E. D. H.; Hall S. C. L.; Sambrook M.; Huang F.; Perrier S. Hierarchical Self-Assembled Photo-Responsive Tubisomes from a Cyclic Peptide-Bridged Amphiphilic Block Copolymer. Angew. Chem., Int. Ed. 2020, 59, 8860–8863. 10.1002/anie.201916111. [DOI] [PubMed] [Google Scholar]
- Brendel J. C.; Sanchis J.; Catrouillet S.; Czuba E.; Chen M. Z.; Long B. M.; Nowell C.; Johnston A.; Jolliffe K. A.; Perrier S. Secondary Self-Assembly of Supramolecular Nanotubes into Tubisomes and Their Activity on Cells. Angew. Chem., Int. Ed. 2018, 57, 16678–16682. 10.1002/anie.201808543. [DOI] [PubMed] [Google Scholar]
- Brendel J. C.; Catrouillet S.; Sanchis J.; Jolliffe K. A.; Perrier S. Shaping Block Copolymer Micelles by Supramolecular Polymerization: Making ‘Tubisomes’. Polym. Chem. 2019, 10, 2616–2625. 10.1039/C9PY00179D. [DOI] [Google Scholar]
- Danial M.; Tran C. M. N.; Jolliffe K. A.; Perrier S. Thermal Gating in Lipid Membranes Using Thermoresponsive Cyclic Peptide-Polymer Conjugates. J. Am. Chem. Soc. 2014, 136, 8018–8026. 10.1021/ja5024699. [DOI] [PubMed] [Google Scholar]
- Rho J. Y.; Brendel J. C.; MacFarlane L. R.; Mansfield E. D. H.; Peltier R.; Rogers S.; Hartlieb M.; Perrier S. Probing the Dynamic Nature of Self-Assembling Cyclic Peptide-Polymer Nanotubes in Solution and in Mammalian Cells. Adv. Funct. Mater. 2018, 28, 1704569. 10.1002/adfm.201704569. [DOI] [Google Scholar]
- Song Q.; Yang J.; Hall S. C. L.; Gurnani P.; Perrier S. Pyridyl Disulfide Reaction Chemistry: An Efficient Strategy toward Redox-Responsive Cyclic Peptide-Polymer Conjugates. ACS Macro Lett. 2019, 8, 1347–1352. 10.1021/acsmacrolett.9b00538. [DOI] [PubMed] [Google Scholar]
- Larnaudie S. C.; Brendel J. C.; Jolliffe K. A.; Perrier S. pH-Responsive, Amphiphilic Core-Shell Supramolecular Polymer Brushes from Cyclic Peptide-Polymer Conjugates. ACS Macro Lett. 2017, 6, 1347–1351. 10.1021/acsmacrolett.7b00728. [DOI] [PubMed] [Google Scholar]
- Mansfield E. D. H.; Hartlieb M.; Catrouillet S.; Rho J. Y.; Larnaudie S. C.; Rogers S. E.; Sanchis J.; Brendel J. C.; Perrier S. Systematic Study of the Structural Parameters Affecting the Self-Assembly of Cyclic Peptide-Poly(Ethylene Glycol) Conjugates. Soft Matter 2018, 14, 6320–6326. 10.1039/C8SM01133H. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Wang Z.; Zhang X. Characterization of Supramolecular Polymers. Chem. Soc. Rev. 2012, 41, 5922–5932. 10.1039/c2cs35084j. [DOI] [PubMed] [Google Scholar]
- Rho J. Y.; Cox H.; Mansfield E. D. H.; Ellacott S. H.; Peltier R.; Brendel J. C.; Hartlieb M.; Waigh T. A.; Perrier S. Dual Self-Assembly of Supramolecular Peptide Nanotubes to Provide Stabilisation in Water. Nat. Commun. 2019, 10, 4708. 10.1038/s41467-019-12586-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai Y.; Eisenberg A. Self-assembly of Block Copolymers. Chem. Soc. Rev. 2012, 41, 5969–5985. 10.1039/c2cs35115c. [DOI] [PubMed] [Google Scholar]
- Wei M.; Gao Y.; Li X.; Serpe M. J. Stimuli-Responsive Polymers and Their Applications. Polym. Chem. 2017, 8, 127–143. 10.1039/C6PY01585A. [DOI] [Google Scholar]
- Hu L.; Zhang Q.; Li X.; Serpe M. J. Stimuli-Responsive Polymers for Sensing and Actuation. Mater. Horiz. 2019, 6, 1774–1793. 10.1039/C9MH00490D. [DOI] [Google Scholar]
- Hu L.; Wan Y.; Zhang Q.; Serpe M. J. Harnessing the Power of Stimuli-Responsive Polymers for Actuation. Adv. Funct. Mater. 2020, 30, 1903471. 10.1002/adfm.201903471. [DOI] [Google Scholar]
- Theato P.; Sumerlin B. S.; O’Reilly R. K.; Epps T. H. III Stimuli Responsive Materials. Chem. Soc. Rev. 2013, 42, 7055–7056. 10.1039/c3cs90057f. [DOI] [PubMed] [Google Scholar]
- Ma X.; Tian H. Stimuli-Responsive Supramolecular Polymers in Aqueous Solution. Acc. Chem. Res. 2014, 47, 1971–1981. 10.1021/ar500033n. [DOI] [PubMed] [Google Scholar]
- Catrouillet S.; Brendel J. C.; Larnaudie S.; Barlow T.; Jolliffe K. A.; Perrier S. Tunable Length of Cyclic Peptide-Polymer Conjugate Self-Assemblies in Water. ACS Macro Lett. 2016, 5, 1119–1123. 10.1021/acsmacrolett.6b00586. [DOI] [PubMed] [Google Scholar]
- Gil E. S.; Hudson S. M. Stimuli-Reponsive Polymers and Their Bioconjugates. Prog. Polym. Sci. 2004, 29, 1173–1222. 10.1016/j.progpolymsci.2004.08.003. [DOI] [Google Scholar]
- Cobo I.; Li M.; Sumerlin B. S.; Perrier S. Smart Hybrid Materials by Conjugation of Responsive Polymers to Biomacromolecules. Nat. Mater. 2015, 14, 143–159. 10.1038/nmat4106. [DOI] [PubMed] [Google Scholar]
- Leriche G.; Chisholm L.; Wagner A. Cleavable Linkers in Chemical Biology. Bioorg. Med. Chem. 2012, 20, 571–582. 10.1016/j.bmc.2011.07.048. [DOI] [PubMed] [Google Scholar]
- Bargh J. D.; Isidro-Llobet A.; Parker J. S.; Spring D. R. Cleavable Linkers in Antibody-Drug Conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. 10.1039/C8CS00676H. [DOI] [PubMed] [Google Scholar]
- Hartlieb M.; Catrouillet S.; Kuroki A.; Sanchez-Cano C.; Peltier R.; Perrier S. Stimuli-Responsive Membrane Activity of Cyclic-Peptide-Polymer Conjugates. Chem. Sci. 2019, 10, 5476–5483. 10.1039/C9SC00756C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer C.; Liu J.; Wong L.; Tippett M.; Bulmus V.; Davis T. P. Stability and Utility of Pyridyl Disulfide Functionality in RAFT and Conventional Radical Polymerizations. J. Polym. Sci., Part A: Polym. Chem. 2008, 46, 7207–7224. 10.1002/pola.23028. [DOI] [Google Scholar]
- Boyer C.; Liu J.; Bulmus V.; Davis T. P.; Barner-Kowollik C.; Stenzel M. H. Direct Synthesis of Well-Defined Heterotelechelic Polymers for Bioconjugations. Macromolecules 2008, 41, 5641–5650. 10.1021/ma800289u. [DOI] [Google Scholar]
- Wolfe L. S.; Calabrese M. F.; Nath A.; Blaho D. V.; Miranker A. D.; Xiong Y. Protein-Induced Photophysical Changes to the Amyloid Indicator Dye Thioflavin T. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 16863–16868. 10.1073/pnas.1002867107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L.; Fan Z.; Wang Y.; Huang Y.; Schmidt M.; Zhang M. Tunable Synthesis of Self-Assembled Cyclic Peptide Nanotubes and Nanoparticles. Soft Matter 2015, 11, 3822–3832. 10.1039/C5SM00533G. [DOI] [PubMed] [Google Scholar]
- Larnaudie S. C.; Sanchis J.; Nguyen T. H.; Peltier R.; Catrouillet S.; Brendel J. C.; Porter C. J. H.; Jolliffe K. A.; Perrier S. Cyclic Peptide-Poly(HPMA) Nanotubes as Drug Delivery Vectors: In Vitro Assessment, Pharmacokinetics and Biodistribution. Biomaterials 2018, 178, 570–582. 10.1016/j.biomaterials.2018.03.047. [DOI] [PubMed] [Google Scholar]
- Modena M.; Rühle B.; Burg T. P.; Wuttke S. Nanoparticle Characterization: Nanoparticle Characterization: What to Measure?. Adv. Mater. 2019, 31, 1901556. 10.1002/adma.201901556. [DOI] [PubMed] [Google Scholar]
- Egerton R. F.Physical Principles of Electron Microscopy: An introduction to TEM, SEM, and AEM, 2nd ed.; Springer: Boston, 2016. [Google Scholar]
- Sawyer L. C.; Grubb D. T.; Meyers G. F.. Polymer Microscopy, 3rd ed.; Springer: New York, 2008. [Google Scholar]
- Zhang C.; Xu T. Co-Assembly of Cyclic Peptide Nanotubes and Block Copolymers in Thin Films: Controlling the Kinetic Pathway. Nanoscale 2015, 7, 15117–15121. 10.1039/C5NR03915K. [DOI] [PubMed] [Google Scholar]
- ten Cate M. G. J.; Severin N.; Börner H. G. Self-Assembling Peptide-Polymer Conjugates Comprising (D-alt-L)-Cyclopeptides as Aggregator Domains. Macromolecules 2006, 39, 7831–7838. 10.1021/ma061738p. [DOI] [Google Scholar]
- Tam J.; Merino D. Stochastic Optical Reconstruction Microscopy (STORM) in Comparison with Stimulated Emission Depletion (STED) and Other Imaging Methods. J. Neurochem. 2015, 135, 643–658. 10.1111/jnc.13257. [DOI] [PubMed] [Google Scholar]
- Yuste R.Imaging: A Laboratory Manual; Cold Spring Harbor Laboratory Press: New York, 2011. [Google Scholar]
- Xu J.; Ma H.; Liu Y. Stochastic Optical Reconstruction Microscopy (STORM). Curr. Protoc. Cytom. 2017, 81, 12.46.1–12.46.27. 10.1002/cpcy.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust M. J.; Bates M.; Zhuang X. Sub-Diffraction-Limit Imaging by Stochastic Optical Reconstruction Microscopy (STORM). Nat. Methods 2006, 3, 793–796. 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson A.-L.; Dastoor P. C.; Flynn J.; Palmer W.; Martin A.; Smith D. W.; Woldu A.; Hua S. Advantages and Limitations of Current Imaging Techniques for Characterizing Liposome Morphology. Front. Pharmacol. 2018, 9, 80. 10.3389/fphar.2018.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson D. B.; Evans J. E.. Low-Dose Imaging Techniques for Transmission Electron Microscopy; Intech Open: Croatia, 2012. [Google Scholar]
- Pu S.; Gong C.; Robertson A. W. Liquid Cell Transmission Electron Microscopy and its Applications. R. Soc. Open Sci. 2020, 7, 191204. 10.1098/rsos.191204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt P. J. Light Scattering and the Absolute Characterization of Macromolecules. Anal. Chim. Acta 1993, 272, 1–40. 10.1016/0003-2670(93)80373-S. [DOI] [Google Scholar]
- Zimm B. H. The Scattering of Light and the Radial Distribution Function of High Polymer Solutions. J. Chem. Phys. 1948, 16, 1093–1099. 10.1063/1.1746738. [DOI] [Google Scholar]
- Larnaudie S. C.; Brendel J. C.; Romero-Canelon I.; Sanchez-Cano C.; Catrouillet S.; Sanchis J.; Coverdale J. P. C.; Song J.-I.; Habtemariam A.; Sadler P. J.; Jolliffe K. A.; Perrier S. Cyclic Peptide-Polymer Nanotubes as Efficient and Highly Potent Drug Delivery Systems for Organometallic Anticancer Complexes. Biomacromolecules 2018, 19, 239–247. 10.1021/acs.biomac.7b01491. [DOI] [PubMed] [Google Scholar]
- Groves P. Diffusion Ordered Spectroscopy (DOSY) as Applied to Polymers. Polym. Chem. 2017, 8, 6700–6708. 10.1039/C7PY01577A. [DOI] [Google Scholar]
- Viel S.; Capitani D.; Mannina L.; Segre A. Diffusion-Ordered NMR Spectroscopy: A Versatile Tool for the Molecular Weight Determination of Uncharged Polysaccharides. Biomacromolecules 2003, 4, 1843–1847. 10.1021/bm0342638. [DOI] [PubMed] [Google Scholar]
- Zayed J. M.; Biedermann F.; Rauwald U.; Scherman O. A. Probing Cucurbit[8]uril-Mediated Supramolecular Block Copolymer Assembly in Water Using Diffusion NMR. Polym. Chem. 2010, 1, 1434–1436. 10.1039/c0py00197j. [DOI] [Google Scholar]
- Huang Z.; Yang L.; Liu Y.; Wang Z.; Scherman O. A.; Zhang X. Supramolecular Polymerization Promoted and Controlled through Self-Sorting. Angew. Chem., Int. Ed. 2014, 53, 5351–5355. 10.1002/anie.201402817. [DOI] [PubMed] [Google Scholar]
- Sivia D. S.Elementary Scattering Theory: For X-ray and Neutron Users; Oxford University Press: New York, 2011. [Google Scholar]
- Brumberger H.Modern Aspects of Small-Angle Scattering; Springer: Netherlands, 2010; Vol. 451. [Google Scholar]
- Ballauff M. SAXS and SANS Studies of Polymer Colloids. Curr. Opin. Colloid Interface Sci. 2001, 6, 132–139. 10.1016/S1359-0294(01)00072-3. [DOI] [Google Scholar]
- Hyland L. L.; Taraban M. B.; Yu Y. B. Using Small-Angle Scattering Techniques to Understand Mechanical Properties of Biopolymer-Based Biomaterials. Soft Matter 2013, 9, 10218–10228. 10.1039/c3sm51209f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifton L. A.; Hall S. C. L.; Mahmoudi N.; Knowles T. J.; Heinrich F.; Lakey J. H.. Structural Investigations of Protein–Lipid Complexes Using Neutron Scattering. In Lipid–Protein Interactions: Methods and Protocols; Kleinschmidt J. H., Eds.; Springer: New York, 2019; p 201–251. [DOI] [PubMed] [Google Scholar]
- Rapaport H.; Kim H. S.; Kjaer K.; Howes P. B.; Cohen S.; Als-Nielsen J.; Ghadiri M. R.; Leiserowitz L.; Lahav M. Crystalline Cyclic Peptide Nanotubes at Interfaces. J. Am. Chem. Soc. 1999, 121, 1186–1191. 10.1021/ja982420i. [DOI] [Google Scholar]
- Podzimek S. Truths and Myths About the Determination of Molar Mass Distribution of Synthetic and Natural Polymers by Size Exclusion Chromatography. J. Appl. Polym. Sci. 2014, 131, 40111–40120. 10.1002/app.40111. [DOI] [Google Scholar]
- Ward T. C. Molecular Weight and Molecular Weight Distributions in Synthetic Polymers. J. Chem. Educ. 1981, 58, 867–879. 10.1021/ed058p867. [DOI] [Google Scholar]
- Doncom K. E. B.; Blackman L. D.; Wright D. B.; Gibson M. I.; O’Reilly R. K. Dispersity Effects in Polymer Self-Assemblies: A Matter of Hierarchical Control. Chem. Soc. Rev. 2017, 46, 4119–4134. 10.1039/C6CS00818F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plog J. P.; Kulicke W. M.; Clasen C. Influence of the Molar Mass Distribution on the Elongational Behaviour of Polymer Solutions in Capillary Breakup. Appl. Rheol. 2005, 15, 28–37. 10.1515/arh-2005-0002. [DOI] [Google Scholar]
- Oberlerchner J. T.; Rosenau T.; Potthast A. Overview of Methods for the Direct Molar Mass Determination of Cellulose. Molecules 2015, 20, 10313–10341. 10.3390/molecules200610313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C. S.Handbook of Size Exclusion Chromatography and Related Techniques; Marcel Dekker Inc.: New York, 2004. [Google Scholar]
- Yang L.; Tan X.; Wang Z.; Zhang X. Supramolecular Polymers: Historical Development, Preparation, Characterization, and Functions. Chem. Rev. 2015, 115, 7196–7239. 10.1021/cr500633b. [DOI] [PubMed] [Google Scholar]
- Striegel A. M.; Isenberg S. L.; Côté G. L. An SEC/MALS Study of Alternan Degradation During Size-Exclusion Chromatographic Analysis. Anal. Bioanal. Chem. 2009, 394, 1887–1893. 10.1007/s00216-009-2895-5. [DOI] [PubMed] [Google Scholar]
- Schimpf M. E.; Caldwell K.; Giddings J. C.. Field-Flow Fractionation Handbook; John Wiley & Sons Inc.: Canada, 2000. [Google Scholar]
- Wahlund K. G.; Nilsson L.. Flow FFF—Basics and Key Applications. In Field-Flow Fractionation in Biopolymer Analysis; Williams S. K. R., Caldwell K. D., Eds.; Springer: Vienna, 2011; pp 1–21. [Google Scholar]
- Yang N. J.; Hinner M. J. Getting Across the Cell Membrane: An Overview for Small Molecules, Peptides, and Proteins. Methods Mol. Biol. 2015, 1266, 29–53. 10.1007/978-1-4939-2272-7_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. S.; Ghadiri M. R.; Hartgerink J. D. Oriented Self-Assembly of Cyclic Peptide Nanotubes in Lipid Membranes. J. Am. Chem. Soc. 1998, 120, 4417–4424. 10.1021/ja9735315. [DOI] [Google Scholar]
- Sanchez-Quesada J.; Kim H. S.; Ghadiri M. R. A Synthetic Pore-Mediated Transmembrane Transport of Glutamic Acid. Angew. Chem., Int. Ed. 2001, 40, 2503–2506. . [DOI] [PubMed] [Google Scholar]
- Liu J.; Fan J.; Tang M.; Cen M.; Yan J.; Liu Z.; Zhou W. Water Diffusion Behaviors and Transportation Properties in Transmembrane Cyclic Hexa-, Octa- and Decapeptide Nanotube. J. Phys. Chem. B 2010, 114, 12183–12192. 10.1021/jp1039207. [DOI] [PubMed] [Google Scholar]
- Liu J.; Fan J.; Cen M.; Song X.; Liu D.; Zhou W.; Liu Z.; Yan J. Dependences of Water Permeation through Cyclic Octa-Peptide Nanotubes on Channel Length and Membrane Thickness. J. Chem. Inf. Model. 2012, 52, 2132–2138. 10.1021/ci300185c. [DOI] [PubMed] [Google Scholar]
- Yan X.; Fan J.; Yu Y.; Xu J.; Zhang M. Transport Behavior of a Single Ca2+, K+, and Na+ in a Water-Filled Transmembrane Cyclic Peptide Nanotube. J. Chem. Inf. Model. 2015, 55, 998–1011. 10.1021/acs.jcim.5b00025. [DOI] [PubMed] [Google Scholar]
- Carvajal-Diaz J. A.; Cagin T. Electrophoretic Transport of Na+ and K+ Ions Within Cyclic Peptide Nanotubes. J. Phys. Chem. B 2016, 120, 7872–7879. 10.1021/acs.jpcb.6b02884. [DOI] [PubMed] [Google Scholar]
- Zhang M.; Fan J.; Xu J.; Weng P.; Lin H. Different Transport Behaviors of NH4+ and NH3 in Transmembrane Cyclic Peptide Nanotubes. J. Mol. Model. 2016, 22, 233. 10.1007/s00894-016-3081-2. [DOI] [PubMed] [Google Scholar]
- Danial M.; Perrier S.; Jolliffe K. A. Effect of the Amino Acid Composition of Cyclic Peptides on Their Self-Assembly in Lipid Bilayers. Org. Biomol. Chem. 2015, 13, 2464–2473. 10.1039/C4OB02041C. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Leman L. J.; Search D. J.; Garcia R. A.; Gordon D. A.; Maryanoff B. E.; Ghadiri M. R. Self-Assembling Cyclic D,L-α-Peptides as Modulators of Plasma HDL Function. A Supramolecular Approach toward Antiatherosclerotic Agents. ACS Cent. Sci. 2017, 3, 639–646. 10.1021/acscentsci.7b00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemerovski-Glikman M.; Richman M.; Rahimipour S. Structure-Based Study of Antiamyloidogenic Cyclic D,L-α-Peptides. Tetrahedron 2014, 70, 7639–7644. 10.1016/j.tet.2014.07.097. [DOI] [Google Scholar]
- Chemerovski-Glikman M.; Rozentur-Shkop E.; Richman M.; Grupi A.; Getler A.; Cohen H. Y.; Shaked H.; Wallin C.; Warmlander S. K.; Haas E.; Graslund A.; Chill J. H.; Rahimipour S. Self-Assembled Cyclic D,L-α-Peptides as Generic Conformational Inhibitors of the α-Synuclein Aggregation and Toxicity: In Vitro and Mechanistic Studies. Chem. - Eur. J. 2016, 22, 14236–14246. 10.1002/chem.201601830. [DOI] [PubMed] [Google Scholar]
- Richman M.; Wilk S.; Chemerovski M.; Warmlander S. K.; Wahlstrom A.; Graslund A.; Rahimipour S. In Vitro and Mechanistic Studies of an Antiamyloidogenic Self-Assembled Cyclic D,L-α-Peptide Architecture. J. Am. Chem. Soc. 2013, 135, 3474–3484. 10.1021/ja310064v. [DOI] [PubMed] [Google Scholar]
- Theuretzbacher U. Global Antibacterial Resistance: The Never-Ending Story. J. Glob. Antimicrob. Resist. 2013, 1, 63–69. 10.1016/j.jgar.2013.03.010. [DOI] [PubMed] [Google Scholar]
- Fernandez-Lopez S.; Kim H.-S.; Choi E. C.; Delgado M.; Granja J. R.; Khasanov A.; Kraehenbuehl K.; Long G.; Weinberger D. A.; Wilcoxen K. M.; Ghadiri M. R. Antibacterial Agents Based on the Cyclic D,L-α-Peptide Architecture. Nature 2001, 412, 452–455. 10.1038/35086601. [DOI] [PubMed] [Google Scholar]
- Dartois V. r.; Sanchez-Quesada J.; Cabezas E.; Chi E.; Dubbelde C.; Dunn C.; Granja J.; Gritzen C.; Weinberger D.; Ghadiri M. R.; Parr T. R. Systemic Antibacterial Activity of Novel Synthetic Cyclic Peptides. Antimicrob. Agents Chemother. 2005, 49, 3302–3310. 10.1128/AAC.49.8.3302-3310.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher J. T.; Finlay J. A.; Callow M. E.; Callow J. A.; Ghadiri M. R. A Combinatorial Approach to the Discovery of Biocidal Six-Residue Cyclic D,L-α-Peptides Against the Bacteria Methicillin-Resistant Staphylococcus aureus (MRSA) and E. coli and the Biofouling Algae Ulva linza and Navicula perminuta. Chem. - Eur. J. 2007, 13, 4008–4013. 10.1002/chem.200601583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claro B.; González-Freire E.; Calvelo M.; Bessa L. J.; Goormaghtigh E.; Amorín M.; Granja J. R.; Garcia-Fandiño R.; Bastos M. Membrane Targeting Antimicrobial Cyclic Peptide Nanotubes - An Experimental and Computational Study. Colloids Surf., B 2020, 196, 111349. 10.1016/j.colsurfb.2020.111349. [DOI] [PubMed] [Google Scholar]
- Motiei L.; Rahimipour S.; Thayer D. A.; Wong C.-H.; Ghadiri M. R. Antibacterial Cyclic D,L-α-Glycopeptides. Chem. Commun. 2009, 3693–3695. 10.1039/b902455g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh W.-H.; Liaw J. Applications of Cyclic Peptide Nanotubes (cPNTs). J. Food Drug Anal. 2019, 27, 32–47. 10.1016/j.jfda.2018.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battistella C.; Klok H.-A. Controlling and Monitoring Intracellular Delivery of Anticancer Polymer Nanomedicines. Macromol. Biosci. 2017, 17, 1700022. 10.1002/mabi.201700022. [DOI] [PubMed] [Google Scholar]
- Tran S.; DeGiovanni P. J.; Piel B.; Rai P. Cancer Nanomedicine: A Review of Recent Success in Drug Delivery. Clin. Trans. Med. 2017, 6, 44. 10.1186/s40169-017-0175-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X.; Vo C.; Taylor M.; Smith B. R. Non-Spherical Micro- and Nanoparticles in Nanomedicine. Mater. Horiz. 2019, 6, 1094–1121. 10.1039/C8MH01527A. [DOI] [Google Scholar]
- Bauer L. A.; Birenbaum N. S.; Meyer G. J. Biological Applications of High Aspect Ratio Nanoparticles. J. Mater. Chem. 2004, 14, 517–526. 10.1039/b312655b. [DOI] [Google Scholar]
- Chen J.; Zhang B.; Xia F.; Xie Y.; Jiang S.; Su R.; Lu Y.; Wu W. Transmembrane Delivery of Anticancer Drugs Through Self-Assembly of Cyclic Peptide Nanotubes. Nanoscale 2016, 8, 7127–7136. 10.1039/C5NR06804E. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Yi S.; Sun L.; Huang Y.; Lenaghan S. C.; Zhang M. Doxorubicin-Loaded Cyclic Peptide Nanotube Bundles Overcome Chemoresistance in Breast Cancer Cells. J. Biomed. Nanotechnol. 2014, 10, 445–454. 10.1166/jbn.2014.1724. [DOI] [PubMed] [Google Scholar]
- Sung Y. K.; Kim S. W. Recent Advances in the Development of Gene Delivery Systems. Biomater. Res. 2019, 23, 8. 10.1186/s40824-019-0156-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi B.; Zheng M.; Tao W.; Chung R.; Jin D.; Ghaffari D.; Farokhzad O. C. Challenges in DNA Delivery and Recent Advances in Multifunctional Polymeric DNA Delivery Systems. Biomacromolecules 2017, 18, 2231–2246. 10.1021/acs.biomac.7b00803. [DOI] [PubMed] [Google Scholar]
- Li M.; Ehlers M.; Schlesiger S.; Zellermann E.; Knauer S. K.; Schmuck C. Incorporation of a Non-Natural Arginine Analogue into a Cyclic Peptide Leads to Formation of Positively Charged Nanofibers Capable of Gene Transfection. Angew. Chem., Int. Ed. 2016, 55, 598–601. 10.1002/anie.201508714. [DOI] [PubMed] [Google Scholar]
- Hsieh W.-H.; Chang S.-F.; Chen H.-M.; Chen J.-H.; Liaw J. Oral Gene Delivery with cyclo-(D-Trp-Tyr) Peptide Nanotubes. Mol. Pharmaceutics 2012, 9, 1231–1249. 10.1021/mp200523n. [DOI] [PubMed] [Google Scholar]
- Lee Y.-H.; Chang S.-F.; Liaw J. Anti-Apoptotic Gene Delivery with cyclo-(D-Trp-Tyr) Peptide Nanotube via Eye Drop Following Corneal Epithelial Debridement. Pharmaceutics 2015, 7, 122–136. 10.3390/pharmaceutics7030122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahi B.; Singh R. K.; Mishra S.; Mandal D. Cyclic Peptide-Based Nanostructures as Efficient siRNA Carriers. Artif. Cells, Nanomed., Biotechnol. 2018, 46, S763–S773. 10.1080/21691401.2018.1511574. [DOI] [PubMed] [Google Scholar]
- Horne W. S.; Wiethoff C. M.; Cui C.; Wilcoxen K. M.; Amorín M.; Ghadiri M. R.; Nemerow G. R. Antiviral Cyclic D,L-α-Peptides: Targeting a General Biochemical Pathway in Virus Infections. Bioorg. Med. Chem. 2005, 13, 5145–5153. 10.1016/j.bmc.2005.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero A.; Gastaminza P.; Law M.; Cheng G.; Chisari F. V.; Ghadiri M. R. Self-Assembling Peptide Nanotubes with Antiviral Activity against Hepatitis C Virus. Chem. Biol. 2011, 18, 1453–1462. 10.1016/j.chembiol.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]